

Abstract
Accumulating evidence shows how diverse physiological functions, such as metabolism, immunity, tissue homeostasis, and hematopoiesis, are intricately and profoundly intertwined at multiple levels. This brief review will present evidence from a rapidly expanding field of immunometabolism, highlighting how cells that are relevant to processes at play in determining vascular health and disease can be programmed by changes in their metabolic environment. It will focus on how such changes can be imprinted or trained, particularly through epigenetic modifications, such that adaptations driven by metabolic signals can cause persistent changes in cell function, even after the original stimulus has been corrected or removed. Recognition of these processes and elucidation of the mechanisms underlying them stand to have far-reaching implications for the diagnosis and treatment of diabetes and related metabolic states.
Keywords: hematopoiesis, homeostasis, immunity, macrophages, metabolism
Graphical Abstract
A graphic abstract is available for this article.

Responding to changes in the metabolic and nutritive environment is a fundamental requirement of life, and apparently, more complex activities, including immune function, have evolved with the necessary backdrop of cellular metabolism.1 Moreover, despite reductionist tendencies to compartmentalize functions, such as metabolism, immunity, vascular biology, and hematopoiesis, emerging data show that they are intricately and profoundly intertwined at multiple levels. Furthermore, the conventional functional attributions of certain cells, not least macrophages, clearly underestimate their broader roles in tissue homeostasis.2 This brief review will focus on ways in which cells that are influential in processes of significance to vascular health are modulated by changes in their metabolic environment and the relevance of these to cardiovascular diseases and their treatment. It will focus on how such changes can be imprinted or trained, particularly through epigenetic modifications, such that adaptations driven by metabolic signals can cause persistent changes in cell function, even after the original stimulus has been corrected or removed.
CLINICAL CONTEXT
Metabolic perturbations, most prominently type 1 and type 2 diabetes, hypercholesterolemia, and obesity adversely affect cardiovascular health. Diabetes is a special case because although hyperglycemia is a cardinal feature in both type 1 and type 2 diabetes, and accelerates atherosclerosis and its complications,3 conventional treatments aimed at reducing blood glucose do not translate straightforwardly to reduced vascular risk. Emerging evidence suggest that this paradox may be linked to trained immunity, which will be discussed in detail here.
EFFECTS OF HYPERGLYCEMIA ON THE ATHEROSCLEROTIC PROCESS
Elevated glucose can exacerbate atherosclerosis through direct mechanisms at the level of the plaque, for example, (1) nonenzymatic glycation of proteins and lipoproteins and (2) by driving oxidative stress.4 The glycation of lipoproteins renders them more atherogenic5 and prone to promote inflammatory processes through activation of pattern recognition receptors for advanced glycation end products,6,7 whereas oxidation of lipoproteins renders them susceptible to uptake by scavenger receptors. Accordingly, atherosclerotic plaques from coronary arteries of patients with diabetes show a greater abundance of lipid-rich atheroma with greater macrophage content.8
In addition, there has been a proliferation of recent evidence showing that hyperglycemia can drive atherosclerosis through remote effects on bone marrow.9 Diabetic mice have increased numbers of circulating neutrophils and Ly6-Chi monocytes, reflecting hyperglycemia-induced proliferation and expansion of bone marrow myeloid progenitors with ensuing release of monocytes into the circulation.10 Furthermore, the pattern of hyperglycemia is also important with transient episodes of hyperglycemia driving myelopoiesis more intensely. Under conditions of hyperglycemia, glucose uptake via the glucose transporter GLUT1 (glucose transporter 1; encoded by the Slc2a1 gene) and enhanced glycolysis in neutrophils promote the production of S100a8/a9, which induces myelopoiesis by binding receptors for advanced glycation end product in bone marrow precursors. S100a8/a9 are Ca2+ binding proteins and comprise ≈45% of the cytoplasmic proteins in neutrophils. They exist in the form of heterodimer with S100a8/a9 being constitutively expressed in neutrophils and monocytes as a Ca2+ sensor, participating in cytoskeleton rearrangement and arachidonic acid metabolism. During inflammation, S100a8/a9 is released and plays a role in modulating the inflammatory response by stimulating leukocyte recruitment and inducing cytokine secretion.11 Myeloid-specific deletion of Slc2a1 or pharmacological inhibition of S100a8/a9 reduced transient hyperglycemia-induced myelopoiesis and atherosclerosis.12
In addition to driving myeloid cell production, hyperglycemia exacerbates atherosclerosis progression by altering cell function. It is associated with increased expression of proinflammatory genes and resistance to induction of M2-associated gene expression13,14 which leads to retarded plaque regression.13 In other words, hyperglycemia changes monocyte/macrophage functions in ways that promote the progression of atherosclerosis and impede processes that mediate its regression or repair.15
In keeping with the abundant evidence that hyperglycemia drives atherosclerosis progression and impairs its regression, treatments to prevent the vascular complications of diabetes have largely focused on lowering blood glucose. However, this approach has proven remarkably ineffective at reducing cardiovascular risk.16 Intensive glucose-lowering therapy is effective in lowering vascular risk in patients with type 1 diabetes, but the effect is deferred for several years and temporally disconnected from the control of blood glucose.17 In type 2 diabetes, glucose reduction is associated with a reduction in microvascular complications but has either shown no or only modest effects on atherosclerosis-related vascular outcomes such as acute myocardial infarction.16,18 Persistent risk of cardiovascular complications, even after glucose-lowering, has been termed metabolic memory, but the underlying mechanisms have, until recently, been largely obscure.
MECHANISMS OF IMMUNE-MEDIATED LEGACY EFFECT
Multiple emerging strands of evidence suggest that epigenetic mechanisms could underlie this phenomenon. Chen et al19,20 have shown several hundred changes in DNA (hypo- and hyper-) methylation at functionally significant sites in relation to hyperglycemia and its complications. These changes, detected in peripheral monocytes, can persist for years, despite intensive glucose management.
In the context of atherosclerosis and diabetic vascular disease, roles for both cells of the innate immune system (Edgar et al, unpublished data, 2021) and vascular endothelial cells21,22 are already becoming apparent in this regard. Cells of the innate immune system had conventionally been considered to respond to stimuli de novo, without the ability to recall prior functionally important exposures. However, recent groundbreaking studies have demonstrated that after exposure to certain antigens (eg, Bacillus Calmette-Guérin or β-glucan), macrophages undergo reprogramming, such that subsequent exposure to those (or selected other) immune stimuli evoke an enhanced (or modified) response. Significantly, given the relatively short life of peripheral myeloid cells, these changes are programmed at the level of bone marrow progenitor cells.23 For this review, two further observations are key (1) although the initial exposure was of immune nature, the signal transduction was, in part, mediated through alterations in cellular metabolic pathways, such as glycolysis and the tricarboxylic acid (TCA) cycle and (2) that changes in cellular metabolites can impact epigenetic programming through chromatin modifications, which can be regulated by metabolic pathways (eg, glycolysis, TCA cycle, fatty acid, and amino acid metabolism).24 Importantly, some metabolites from these pathways (eg, fumarate and acetyl-CoA [acetyl coenzyme A]) can serve as signaling molecules, substrates, and cofactors for chromatin-modifying enzymes, such as the histone demethylase KDM5 (lysine-specific demethylase 5) or histone acetyltransferases, leading to specific changes in histone methylation and acetylation of genes involved in the innate immune responses.25–29
This can be exemplified by studies in monocytes where β-glucan exposure initiates a signaling pathway involving dectin 1 (a β-glucan receptor), Akt (protein kinase B; a serine/threonine-specific protein kinase), mTOR (mechanistic target of rapamycin), and HIFlα (hypoxia-inducible factor 1α). This switches cellular metabolism from oxidative phosphorylation to glycolysis resulting in increased glucose consumption and higher lactate production.25 Integrated metabolomic and transcriptomic analyses of human β-glucan–trained monocytes revealed increased glycolysis (over the TCA cycle) but with concomitant replenishment of TCA cycle intermediates as a result of glutaminolysis leading to accumulation of succinate, malate, and fumarate, the last of which influenced epigenetic programming by downregulating the activity of KDM5 histone demethylases (which are responsible for H3K4 demethylation, see below).30
This new knowledge, in respect of immune function regulated by metabolic intermediaries, immediately raises the possibility that disorders of a primarily metabolic nature, which are also associated with disease states where innate immune mechanisms are active, might be susceptible to persistent immunomodulatory effects through chromatin modification in response to metabolic alterations.31,32 This possibility is further enhanced by the knowledge that (1) macrophages switch from oxidative phosphorylation to glycolysis in response to proinflammatory stimuli (associated with so-called M1 status) and that (2) interventions that promote glycolysis lead, in turn, to macrophage activation. In monocytes and macrophages, cellular energetic pathways are governed by an interplay between cell-intrinsic and environmental stimuli which, in turn, markedly affect immune-related function and gene expression.33 Thus, in response to IL (interleukin)-4, M2 macrophages primarily metabolize fatty acids and rely on oxidative phosphorylation,34 whereas M1 macrophages require glucose and shift to aerobic glycolysis, similar to the Warburg effect in cancer.35 Altogether, these metabolic changes occur as a consequence of cytokine stimulation but can also themselves determine macrophage function.36
Elevating glucose results in a shift towards glycolysis with a reduction in oxidative phosphorylation.37 Furthermore, hyperglycemia prompts a transition to a more proinflammatory state in which M1-type responses are promoted and M2-type responses are suppressed.13 Significantly, these changes persist even after the cells are subsequently maintained in a normal physiological glucose environment. Bone marrow obtained from mice in which hyperglycemia was induced by streptozotocin and maintained for 6 weeks before transplantation into normoglycemic Ldlr−/− mice was also able to retain a memory of its previous hyperglycemic environment and drive accelerated atherosclerosis (Edgar et al, unpublished data, 2021). Examination of the chromatin landscape of bone marrow–derived macrophages from diabetic mice showed hundreds of areas of differential chromatin accessibility. In basal conditions, the cell transcriptome was minimally different in bone marrow–derived macrophages between diabetic and wild-type mice, but proinflammatory stimulation resulted in marked differences, indicating that diabetes had induced a trained state in which cells were primed to respond more exuberantly after a secondary stimulus (Edgar et al, unpublished data, 2021); in other words, a type of hyperglycemia-induced trained immunity could be detected. These effects were associated with patterns of histone methylation that have been linked to transcriptional activation states in prior examples of trained immunity.38 Peripheral blood myeloid cells are short-lived and, therefore, not likely to be capable of both receiving and maintaining at length any training stimulus. As with the induction of trained immunity with high-fat diet,39 reprogramming at the level of bone marrow progenitor cells, could transfer training to their progeny.23,40
These newly described interfaces between metabolism and immunity are intriguing and several details remain to be clarified. More detailed consideration of the molecular processes involved, including the expanding repertoire of enzymes involved in writing, erasing, and reading epigenetic marks are provided in recent comprehensive reviews.29,41 Previous studies have identified chromatin modifications that are associated with trained immunity phenotypes. Two key epigenetic marks are the acquisition of histone 3 lysine 27 acetylation marks at distal enhancers and histone 3 lysine 4 trimethylation marks at the promoters of stimulated genes.29 Systematic evaluation of the acquisition of open chromatin as measured by assay for transposase-accessible chromatin using sequencing and chromatin immunoprecipitation followed by sequencing will start to identify patterning among the hundreds of modified sites across the genome. The mechanistic questions will shift one step back: to try to understand what makes the modifiable sites (either at the promoters of stimulated genes or at distal regulatory elements) targets and by what processes are they rendered susceptible to modification.42 Eventually, long noncoding RNAs, transcription factors, downstream mediators, and other potentially tunable therapeutic targets should emerge.
Although these types of modifications are highly plausible mediators of enhanced atherosclerosis and hyperglycemic memory in diabetes, additional important issues remain to be clarified. Experiments in mice suggest that the hyperglycemic epigenetic memory can persist for at least 3 months. As mentioned above, the clinical trial data suggest that some form of hyperglycemic memory state can persist for several years. However, systematic assessments of the dynamics of trained immunity are currently lacking. Unresolved questions relate to the degree of hyperglycemia required for these effects. Are the training effects more susceptible to chronically or transiently elevated glucose? Are patients in prediabetic states susceptible? How long does the training persist, and can it be removed physiologically—if so, over what time frame and under what influences? What are the effects of insulin and other antidiabetic therapeutics?
Furthermore, additional mechanisms may explain prolonged immune activation despite glucose-lowering, including the persistence of proinflammatory glycated moieties that activate receptors for advanced glycation end product. In this theory of metabolic memory, AGEs (advanced glycation endproducts) are proposed to accumulate chronically in a variety of tissues, including the arterial wall, providing long-term activation of receptors for advanced glycation end product and the downstream immune-activating consequences reflective of a previous hyperglycemic state.43
EFFECTS OF HIGH GLUCOSE ON ENDOTHELIAL CELLS
Clearly, in hyperglycemia, all tissues are exposed to the elevated blood glucose, raising the possibility that other cell types relevant to vascular health and disease might also be reprogrammed. Endothelial dysfunction is well described in diabetes and as an early event in atherogenesis. High-glucose conditions mediate chromatin changes underlying altered gene expression patterns in endothelial cells. Epigenome-wide analysis of aortic endothelial cells stimulated with high glucose identified acetylation of H3K9/K14 (histone H3K9/K14) and DNA methylation patterns correlating with transcriptional activation of various gene pathways closely associated with metabolic and cardiovascular disease.44 Of particular relevance to pathways of atherogenesis and vascular inflammation, histone methyltransferase Set7 (a lysine-specific SET-domain methyltransferase) mediates high-glucose–induced inflammation via epigenetic regulation of the transcription factor NF-κB (nuclear factor κB).21 Paneni et al21 showed that Set7 expression was also increased in peripheral blood mononuclear cells from patients with type 2 diabetes when compared with controls. Patients with type 2 diabetes showed Set7-dependent monomethylation of lysine 4 of histone 3 on the NF-κB p65 promoter. This epigenetic signature was associated with upregulation of NF-κB, subsequent transcription of oxidant/inflammatory genes, and increased plasma levels of intercellular cell adhesion molecule-1 and monocyte chemoattractant protein-1, which were also associated with markers of oxidative stress and endothelial dysfunction. Similarly, the adaptor protein p66Shc (adapter protein p66Shc) is a driver of mitochondrial oxidative stress and vascular damage in experimental diabetes, and epigenetic remodeling of p66Shc is responsible for the persistence of endothelial dysfunction in diabetic mice despite glycemic control with insulin.22 Moreover, in human diabetic patients subject to continuous glucose monitoring, plasma glucose variability expressed by the mean amplitude of glycemic excursions and postprandial incremental area under the curve were related to DNA hypomethylation and histone 3 acetylation on the p66Shc promoter in monocytes, whereas HbA1c (glycated hemoglobin) was not, suggesting that glucose fluctuations contribute to chromatin remodeling.45
BEYOND GLUCOSE AND POSSIBLE EXTRAVASCULAR EFFECTS
Glutamine (the most abundant amino acid in the plasma of humans and many other mammals) is effective in inducing polarization of M2 macrophages through the glutamine-UDP-N-acetylglucosamine pathway and α-ketoglutarate, produced via glutaminolysis, whereas succinate synthesized via glutamine-dependent anaplerosis (ie, replenishment of TCA cycle intermediates that have been extracted for other purposes) or the γ-aminobutyric acid shunt promotes polarization of M1 macrophages.46 The anti-inflammatory effects of glutamine on immune cells (eg, neutrophils, macrophages, and T cells) have been extensively studied (reviewed in Bettencourt and Powell47), and the clinical relevance is being considered. Given that excess white adipose tissue (WAT) mass confers an increased cardiovascular risk, it is also possible that changes in WAT metabolism may directly or indirectly link atherosclerosis and cardiovascular disease. A recent study compared metabolites released from WAT of obese and nonobese women. Glutamine was found to be the most significantly downregulated metabolite in obesity and inversely associated with a pernicious WAT phenotype. Glutamine administration in vitro and in vivo attenuated both proinflammatory processes and glycolysis in adipocytes and reduced UDP N-acetylglucosamine levels. UDP N-acetylglucosamine is the substrate for the post-translational modification O-linked β-N-acetylglucosamine mediated by the enzyme O-linked β-N-acetylglucosamine transferase. Functional studies in human adipocytes established an epigenetic link between reduced glutamine, O-linked β-N-acetylglucosamineylation of nuclear proteins, and a proinflammatory transcriptional response.48 Although the effects on atherosclerosis were not investigated, future studies may help delineate whether disturbed glutamine metabolism in WAT may facilitate the atherosclerotic process. In any case, this demonstrates the close relationship between inflammation and metabolism not only in vessels but also in metabolic tissues causally linked to type 2 diabetes.
CONCLUDING COMMENTS AND NEW OPPORTUNITIES
A picture emerges in which diabetes is not only a metabolic disease, with vascular complications but one with profound and sustained effects on immune cell function. This new appreciation should bring opportunities to target disease prevention and to develop new disease-modifying therapies. The immunologic phenotype of trained immunity is likely to be variable depending on the depth of training, which might, in turn, depend on the patterns, degree, and duration of the provoking factors. Furthermore, type 2 diabetes, in particular, is a complex metabolic state in which other factors, such as insulin and changes in insulin sensitivity may have a bearing.
Observational evidence in diabetes suggests that the reprogramming can last months, even years.19 However, in other contexts, trained immunity is generally reversible and shorter-lived than classical epitope-specific adaptive immunity.29 This is clinically relevant because it implies opportunities for retraining, perhaps through fasting; alterations to diet and exercise; patterns of substrate provision; and, of course, pharmacological interventions. That said, all of these possibilities remain open to further inquiry. Pharmacological interventions to modify trained immunity may be less straightforward and will require a better understanding of the patterns of epigenetic modifications that confer benefit versus harm. As with other immunomodulatory interventions, treatments that are nonselective may be detrimental to countering infection. Strategies that target particular (pathogenic) macrophage niches are reviewed elsewhere in this series may be more amenable.49 Nonetheless, appreciation of these disease-relevant immune processed and the ability to both characterize and modify them offer clear opportunities to address longstanding clinical challenges in the management of diabetes and its complications.
Figure. Diabetes-related hyperglycemia alters cellular metabolism and drives proinflammatory, epigenetic cellular reprogramming.
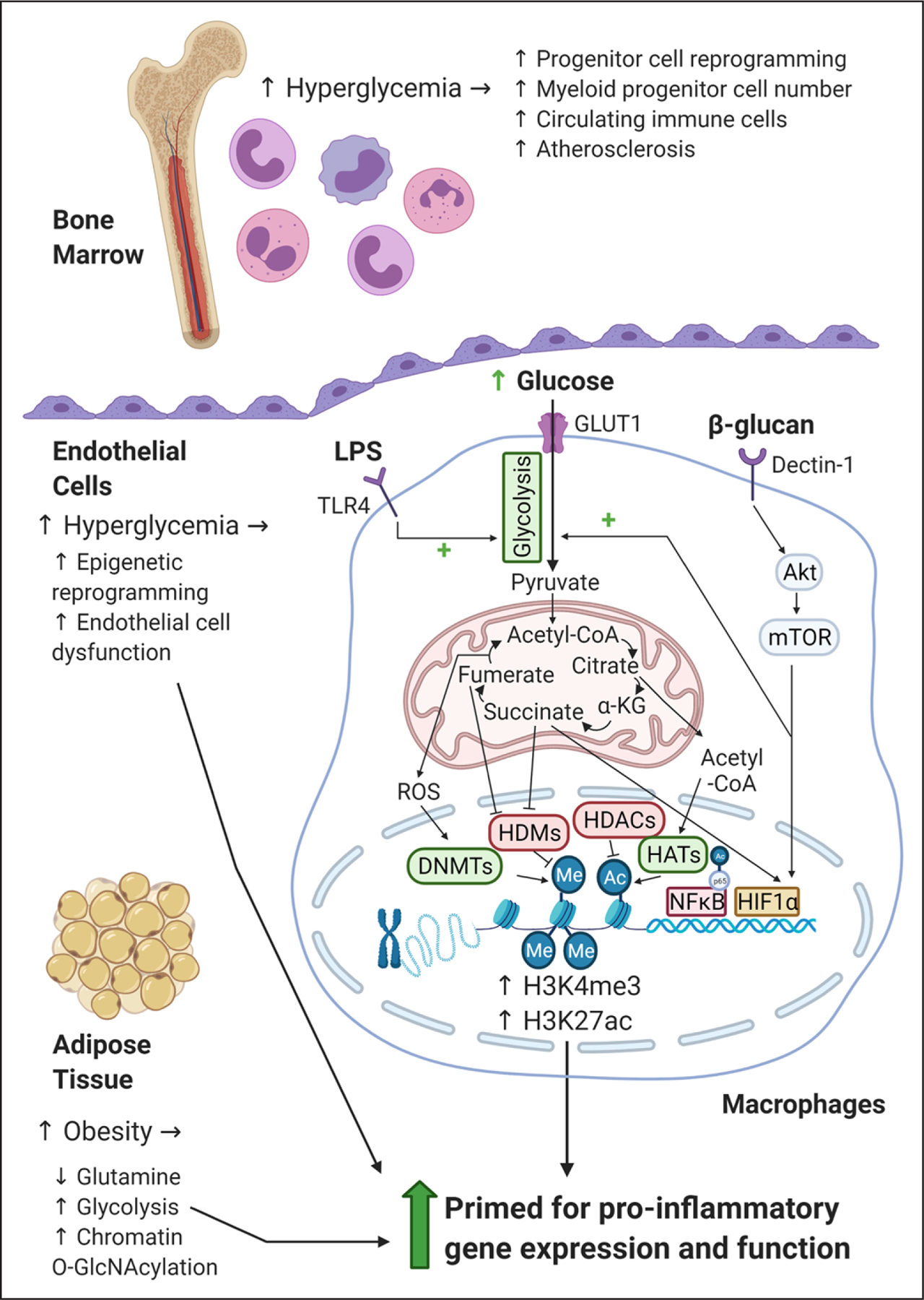
Illustrative summary of how hyperglycemia drives changes in bone marrow, endothelial cells, monocytes, macrophages, and adipocytes. In macrophages, increased levels of glucose, or proinflammatory stimulation with lipopolysaccharide (LPS), increase rates of glycolysis and drive a broken tricarboxylic acid cycle leading to the accumulation of reactive oxygen species (ROS) and metabolites such as succinate and acetyl-CoA (acetyl coenzyme A). Changes in metabolite levels alter gene expression and cell function directly, for example, succinate can stabilize the transcription factor HIF1 α (hypoxia-inducible factor 1 α); or they can prime cells through epigenetic modifications, such as histone 3 lysine 4 trimethylation (H3K4me3) and histone 3 lysine 27 acetylation (H3K27ac), by influencing the enzymes that control their levels: DNMTs (DNA methyltransferases), HDMs (histone demethylases), HDACs (histone deacetylases), and HATs (histone acetyltransferases). Glutamine is downregulated in adipose tissue in obesity and is inversely associated with a pernicious white adipose tissue phenotype. In human adipocytes reduced glutamine leads to O-GlcNAcylation of nuclear proteins, and a proinflammatory transcriptional response. α-KG indicates α-ketoglutarate; Akt, protein kinase B; GLUT1, glucose transporter 1; mTOR, mechanistic target of rapamycin; NF-κB, nuclear factor κB; O-GlcNAc, O-linked β-N-acetylglucosamine; and TLR, toll-like receptor.
Highlights.
Diabetes is not only a metabolic disease, with vascular complications, but one with profound and sustained effects on immune cell function, including through trained immunity.
This new appreciation should bring opportunities to target disease prevention and to develop new disease-modifying therapies.
The immunologic phenotype of trained immunity is likely to be variable depending on the depth of training, which might, in turn, depend on the patterns, degree, and duration of the provoking factors.
Sources of Funding
R.P. Choudhury and M. Ryden receive support as part of the NovoNordisk Foundation award Metabolite-related inflammation in diabetes spectrum diseases: Me-RIAD; grant number NNF 0064142. R.P. Choudhury acknowledges the support of the British Heart Foundation Centre for Research Excellence, Oxford, and the NIHR Biomedical Research Centre, Oxford. L. Edgar is an employee of NovoNordisk.
Nonstandard Abbreviations and Acronyms
- acetyl-CoA
acety coenzyme A
- HIFlα
hypoxia-inducible factor 1α
- IL
interleukin
- KDM5
lysine-specific demethylase 5
- mTOR
mechanistic target of rapamycin
- NF-κB
nuclear factor κB
- TCA
tricarboxylic acid
- WAT
white adipose tissue
Footnotes
Disclosures
None.
Contributor Information
Robin P. Choudhury, Radcliffe Department of Medicine, University of Oxford, United Kingdom
Laurienne Edgar, Radcliffe Department of Medicine, University of Oxford, United Kingdom, Novo Nordisk A/S, Gatwick, United Kingdom.
Mikael Rydén, Department of Medicine (H7), Karolinska Institute, C2-94, Karolinska University Hospital, Huddinge, Stockholm, Sweden.
Edward A. Fisher, Department of Medicine, NYU Grossman School of Medicine
REFERENCES
- 1.Penkov S, Mitroulis I, Hajishengallis G, Chavakis T. Immunometabolic crosstalk: an ancestral principle of trained immunity? Trends Immunol 2019;40:1–11. doi: 10.1016/j.it.2018.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature 2013;496:445–455. doi: 10.1038/nature12034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laakso M. Hyperglycemia and cardiovascular disease in type 2 diabetes. Diabetes 1999;48:937–942. doi: 10.2337/diabetes.48.5.937 [DOI] [PubMed] [Google Scholar]
- 4.Guzik TJ, Mussa S, Gastaldi D, Sadowski J, Ratnatunga C, Pillai R, Channon KM. Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation 2002;105:1656–1662. doi: 10.1161/01.cir.0000012748.58444.08 [DOI] [PubMed] [Google Scholar]
- 5.Brownlee M, Cerami A, Vlassara H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N Engl J Med 1988;318:1315–1321. doi: 10.1056/NEJM198805193182007 [DOI] [PubMed] [Google Scholar]
- 6.Lin RY, Choudhury RP, Cai W, Lu M, Fallon JT, Fisher EA, Vlassara H. Dietary glycotoxins promote diabetic atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis 2003;168:213–220. doi: 10.1016/s0021-9150(03)00050-9 [DOI] [PubMed] [Google Scholar]
- 7.Harja E, Bu DX, Hudson BI, Chang JS, Shen X, Hallam K, Kalea AZ, Lu Y, Rosario RH, Oruganti S, et al. Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE−/− mice. J Clin Invest 2008;118:183–194. doi: 10.1172/JCI32703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moreno PR, Murcia AM, Palacios IF, Leon MN, Bernardi VH, Fuster V, Fallon JT. Coronary composition and macrophage infiltration in atherectomy specimens from patients with diabetes mellitus. Circulation 2000;102: 2180–2184. doi: 10.1161/01.cir.102.18.2180 [DOI] [PubMed] [Google Scholar]
- 9.Barrett TJ, Murphy AJ, Goldberg IJ, Fisher EA. Diabetes-mediated myelopoiesis and the relationship to cardiovascular risk. Ann N Y Acad Sci 2017;1402:31–42. doi: 10.1111/nyas.13462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagareddy PR, Murphy AJ, Stirzaker RA, Hu Y, Yu S, Miller RG, Ramkhelawon B, Distel E, Westerterp M, Huang LS, et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab 2013;17:695–708. doi: 10.1016/j.cmet.2013.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang S, Song R, Wang Z, Jing Z, Wang S, Ma J. S100A8/A9 in Inflammation. Front Immunol 2018;9:1298. doi: 10.3389/fimmu.2018.01298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flynn MC, Kraakman MJ, Tikellis C, Lee MKS, Hanssen NMJ, Kammoun HL, Pickering RJ, Dragoljevic D, Al-Sharea A, Barrett TJ, et al. Transient intermittent hyperglycemia accelerates atherosclerosis by promoting myelopoiesis. Circ Res 2020;127:877–892. doi: 10.1161/CIRCRESAHA.120.316653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parathath S, Grauer L, Huang LS, Sanson M, Distel E, Goldberg IJ, Fisher EA. Diabetes adversely affects macrophages during atherosclerotic plaque regression in mice. Diabetes 2011;60:1759–1769. doi: 10.2337/db10-0778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shanmugam N, Reddy MA, Guha M, Natarajan R. High glucose-induced expression of proinflammatory cytokine and chemokine genes in monocytic cells. Diabetes 2003;52:1256–1264. doi: 10.2337/diabetes.52.5.1256 [DOI] [PubMed] [Google Scholar]
- 15.Barman PK, Urao N, Koh TJ. Diabetes induces myeloid bias in bone marrow progenitors associated with enhanced wound macrophage accumulation and impaired healing. J Pathol 2019;249:435–446. doi: 10.1002/path.5330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Newman JD, Vani AK, Aleman JO, Weintraub HS, Berger JS, Schwartzbard AZ. The changing landscape of diabetes therapy for cardiovascular risk reduction: JACC state-of-the-art review. J Am Coll Cardiol 2018;72:1856–1869. doi: 10.1016/j.jacc.2018.07.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nathan DM, Cleary PA, Backlund JY, Genuth SM, Lachin JM, Orchard TJ, Raskin P, Zinman B; Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study Research Group. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 2005;353:2643–2653. doi: 10.1056/NEJMoa052187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gerstein HC, Miller ME, Genuth S, Ismail-Beigi F, Buse JB, Goff DC Jr, Probstfield JL, Cushman WC, Ginsberg HN, Bigger JT, et al. ; ACCORD Study Group. Long-term effects of intensive glucose lowering on cardiovascular outcomes. N Engl J Med 2011;364:818–828. doi: 10.1056/NEJMoa1006524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Z, Miao F, Paterson AD, Lachin JM, Zhang L, Schones DE, Wu X, Wang J, Tompkins JD, Genuth S, et al. ; DCCT/EDIC Research Group. Epigenomic profiling reveals an association between persistence of DNA methylation and metabolic memory in the DCCT/EDIC type 1 diabetes cohort. Proc Natl Acad Sci U S A 2016;113:E3002–E3011. doi: 10.1073/pnas.1603712113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Z, Miao F, Braffett BH, Lachin JM, Zhang L, Wu X, Roshandel D, Carless M, Li XA, Tompkins JD, et al. ; DCCT/EDIC Study Group. DNA methylation mediates development of HbA1c-associated complications in type 1 diabetes. Nat Metab 2020;2:744–762. doi: 10.1038/s42255-020-0231-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paneni F, Costantino S, Battista R, Castello L, Capretti G, Chiandotto S, Scavone G, Villano A, Pitocco D, Lanza G, et al. Adverse epigenetic signatures by histone methyltransferase Set7 contribute to vascular dysfunction in patients with type 2 diabetes mellitus. Circ Cardiovasc Genet 2015;8:150–158. doi: 10.1161/CIRCGENETICS.114.000671 [DOI] [PubMed] [Google Scholar]
- 22.Paneni F, Mocharla P, Akhmedov A, Costantino S, Osto E, Volpe M, Lüscher TF, Cosentino F. Gene silencing of the mitochondrial adaptor p66(Shc) suppresses vascular hyperglycemic memory in diabetes. Circ Res 2012;111:278–289. doi: 10.1161/CIRCRESAHA.112.266593 [DOI] [PubMed] [Google Scholar]
- 23.Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T, Eugster A, Troullinaki M, Palladini A, Kourtzelis I, et al. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell 2018;172:147–161.e12. doi: 10.1016/j.cell.2017.11.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Donohoe DR, Bultman SJ. Metaboloepigenetics: interrelationships between energy metabolism and epigenetic control of gene expression. J Cell Physiol 2012;227:3169–3177. doi: 10.1002/jcp.24054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, Giamarellos-Bourboulis EJ, Martens JH, Rao NA, Aghajanirefah A, et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 2014;345:1250684. doi: 10.1126/science.1250684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saeed S, Quintin J, Kerstens HH, Rao NA, Aghajanirefah A, Matarese F, Cheng SC, Ratter J, Berentsen K, van der Ent MA, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 2014;345:1251086. doi: 10.1126/science.1251086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bekkering S, Arts RJW, Novakovic B, Kourtzelis I, van der Heijden CDCC, Li Y, Popa CD, Ter Horst R, van Tuijl J, Netea-Maier RT, et al. Metabolic Induction of Trained Immunity through the Mevalonate Pathway. Cell 2018;172:135–146.e9. doi: 10.1016/j.cell.2017.11.025 [DOI] [PubMed] [Google Scholar]
- 28.Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonça LE, Pacis A, Tzelepis F, Pernet E, Dumaine A, Grenier JC, et al. BCG educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell 2018;172:176–190.e19. doi: 10.1016/j.cell.2017.12.031 [DOI] [PubMed] [Google Scholar]
- 29.Netea MG, Domínguez-Andrés J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, Joosten LAB, van der Meer JWM, Mhlanga MM, Mulder WJM, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol 2020;20:375–388. doi: 10.1038/s41577-020-0285-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arts RJ, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, Lachmandas E, Rodrigues F, Silvestre R, Cheng SC, Wang SY, et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab 2016;24:807–819. doi: 10.1016/j.cmet.2016.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Diepen JA, Thiem K, Stienstra R, Riksen NP, Tack CJ, Netea MG. Diabetes propels the risk for cardiovascular disease: sweet monocytes becoming aggressive? Cell Mol Life Sci 2016;73:4675–4684. doi: 10.1007/s00018-016-2316-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thiem K, Stienstra R, Riksen NP, Keating ST. Trained immunity and diabetic vascular disease. Clin Sci (Lond) 2019;133:195–203. doi: 10.1042/CS20180905 [DOI] [PubMed] [Google Scholar]
- 33.O’Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol 2016;16:553–565. doi: 10.1038/nri.2016.70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, Wagner RA, Greaves DR, Murray PJ, Chawla A. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab 2006;4:13–24. doi: 10.1016/j.cmet.2006.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Renner K, Singer K, Koehl GE, Geissler EK, Peter K, Siska PJ, Kreutz M. Metabolic hallmarks of tumor and immune cells in the tumor microenvironment. Front Immunol 2017;8:248. doi: 10.3389/fimmu.2017.00248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stienstra R, Netea-Maier RT, Riksen NP, Joosten LAB, Netea MG. Specific and complex reprogramming of cellular metabolism in myeloid cells during innate immune responses. Cell Metab 2017;26:142–156. doi: 10.1016/j.cmet.2017.06.001 [DOI] [PubMed] [Google Scholar]
- 37.Ayala TS, Tessaro FHG, Jannuzzi GP, Bella LM, Ferreira KS, Martins JO. High glucose environments interfere with bone marrow-derived macrophage inflammatory mediator release, the TLR4 pathway and glucose metabolism. Sci Rep 2019;9:11447. doi: 10.1038/s41598-019-47836-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, O’Neill LA, Xavier RJ. Trained immunity: a program of innate immune memory in health and disease. Science 2016;352:aaf1098. doi: 10.1126/science.aaf1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Christ A, Günther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, Scholz CJ, Oosting M, Haendler K, Baßler K, et al. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell 2018;172:162–175.e14. doi: 10.1016/j.cell.2017.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang F, Higgins JM. Histone modifications and mitosis: countermarks, landmarks, and bookmarks. Trends Cell Biol 2013;23:175–184. doi: 10.1016/j.tcb.2012.11.005 [DOI] [PubMed] [Google Scholar]
- 41.Keating ST, Plutzky J, El-Osta A. Epigenetic changes in diabetes and cardiovascular risk. Circ Res 2016;118:1706–1722. doi: 10.1161/CIRCRESAHA.116.306819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Domínguez-Andrés J, Fanucchi S, Joosten LAB, Mhlanga MM, Netea MG. Advances in understanding molecular regulation of innate immune memory. Curr Opin Cell Biol 2020;63:68–75. doi: 10.1016/j.ceb.2019.12.006 [DOI] [PubMed] [Google Scholar]
- 43.Yan SF, Ramasamy R, Bucciarelli LG, Wendt T, Lee LK, Hudson BI, Stern DM, Lalla E, DU Yan S, Rong LL, et al. RAGE and its ligands: a lasting memory in diabetic complications? Diab Vasc Dis Res 2004;1:10–20. doi: 10.3132/dvdr.2004.001 [DOI] [PubMed] [Google Scholar]
- 44.Pirola L, Balcerczyk A, Tothill RW, Haviv I, Kaspi A, Lunke S, Ziemann M, Karagiannis T, Tonna S, Kowalczyk A, et al. Genome-wide analysis distinguishes hyperglycemia regulated epigenetic signatures of primary vascular cells. Genome Res 2011;21:1601–1615. doi: 10.1101/gr.116095.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Costantino S, Paneni F, Battista R, Castello L, Capretti G, Chiandotto S, Tanese L, Russo G, Pitocco D, Lanza GA, et al. Impact of glycemic variability on chromatin remodeling, oxidative stress, and endothelial dysfunction in patients with type 2 diabetes and with target HbA1c levels. Diabetes 2017;66:2472–2482. doi: 10.2337/db17-0294 [DOI] [PubMed] [Google Scholar]
- 46.Liu PS, Wang H, Li X, Chao T, Teav T, Christen S, Di Conza G, Cheng WC, Chou CH, Vavakova M, et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol 2017;18:985–994. doi: 10.1038/ni.3796 [DOI] [PubMed] [Google Scholar]
- 47.Bettencourt IA, Powell JD. Targeting metabolism as a novel therapeutic approach to autoimmunity, inflammation, and transplantation. J Immunol 2017;198:999–1005. doi: 10.4049/jimmunol.1601318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Petrus P, Lecoutre S, Dollet L, Wiel C, Sulen A, Gao H, Tavira B, Laurencikiene J, Rooyackers O, Checa A, et al. Glutamine links obesity to inflammation in human white adipose tissue. Cell Metab 2020;31:375–390.e11. doi: 10.1016/j.cmet.2019.11.019 [DOI] [PubMed] [Google Scholar]
- 49.Mulder WJM, Ochando J, Joosten LAB, Fayad ZA, Netea MG. Therapeutic targeting of trained immunity. Nat Rev Drug Discov 2019;18:553–566. doi: 10.1038/s41573-019-0025-4 [DOI] [PMC free article] [PubMed] [Google Scholar]