

Abstract
Neuronal injury can cause mitochondrial damage, leading to reduced energy production, decreased Ca2+ storage capacity, and increased reactive oxygen species. A new study reveals a mechanism to trigger the axonal transport of previously anchored mitochondria and promote neuroprotection and axon regeneration by replacing damaged with functional mitochondria.
The nervous system uses large amounts of ATP, much more than other organs, and neurons constitute the main energy consumers. Mature neurons are highly polarized cells, typically exhibiting an axon and dendrites that emerge from the cell body. The polarized nature of neurons requires that organelles, proteins, and RNAs are delivered to distal sites, often long distances from the neuronal cell soma. Directed transport of cargoes is driven by motor proteins, with microtubule-dependent motor proteins facilitating long-distance axonal transport. The microtubule plus-end directed kinesin motor proteins are used for anterograde transport in axons, and for every 8 nm step that a kinesin motor takes along a microtubule, one ATP molecule is consumed — in large neurons, this amounts to the hydrolysis of millions of ATP molecules when moving cargoes from soma to axon tip in rodents. Even more energy is consumed in larger organisms, such as humans. The motor protein for retrograde axon transport, dynein, has similar energy requirements as kinesins. Thus, both soma-to-axon and axon-to-soma transport are energetically very expensive. While some vesicles have been shown to have actively glycolytic cargoes that produce ATP during their transport along axons1, mitochondrial respiration is likely the major source of ATP generation in axons. Upon neuronal injury, mitochondria are damaged, and at the same time ATP demands are very high during the repair process, potentially triggering an energy crisis. A new study in this issue of Current Biology from Huang et al.2 uncovers a neuron-intrinsic mechanism — actually an axon-intrinsic mechanism — that ensures a neuron can meet its elevated energy needs after ischemic and traumatic injury. Injured mitochondria not only fail to generate ATP but also lose the capacity to store Ca2+ and typically release reactive oxygen species (ROS), further emphasizing the need to carefully regulate mitochondrial functions in injured axons.
Microtubule-dependent mitochondrial transport requires a heterodimeric complex of Miro (also known as RHOT1/2) and Milton (also known as TRAK1/2) proteins for linking a mitochondrion to the kinesin and dynein motors. Motility of mitochondria in axons is regulated by syntaphilin (SNPH), a protein that anchors mitochondria to microtubules and thus helps to keep them in place. Neuronal SNPH expression increases as neurons advance from the growth phase and acquire their mature form, suggesting a potential mechanistic link for decreased mitochondrial motility in mature neurons. In the new work, Huang et al.2 asked whether the reduction in mitochondrial transport can be reversed in mature neurons. SNPH’s microtubule-anchoring function is Ca2+ dependent and regulated by neuronal activity. Neuronal firing leads to elevated Ca2+ and ‘trapping’ of mitochondria at presynaptic sites3, presumably to supply energy in the form of ATP that is needed for proper synaptic function. In developing neurons, SNPH is required to anchor mitochondria at emerging branch sites in the axon4. These and other studies point to SNPH as an important anchor molecule in developing and mature axons that holds mitochondria in place, thereby ensuring a localized source of energy. Indeed, collateral branching of sensory axons in response to neurotrophin exposure requires mitochondrial respiration and local mRNA translation adjacent to incipient branch points5. While ‘on demand’ anchoring of mitochondria to sites of high energy consumption provides many advantages for neuron function, anchoring of mitochondria appears to interfere with axon regeneration, a highly energy-demanding process, as demonstrated in Snph knockout mice subjected to spinal cord injury. After injury of the mutant mice, die-back of severed corticospinal tract axons was reduced, axons grew past the injury site and motor recovery was improved compared with wild-type mice6. The new study by Huang et al.2 is significant, as it now reveals a novel mechanism for how the anchor function of SNPH can be regulated: phosphorylation of SNPH by AKT locally activates p21-associated kinase 5 (PAK5) and PAK5 inactivates SNPH to increase mitochondria mobility.
Anchoring of mitochondria along axons likely occurs at energy-intensive locations. But what happens to those anchored mitochondria when the axon is injured? Damaged mitochondria are sequestered and eliminated through a process referred to as mitophagy. Both intra-axonal mitophagy and somal mitophagy, which would require retrograde transport of damaged mitochondria, have been reported7. Regardless of whether this elimination occurs at local sites or centrally in the soma, the damaged mitochondria in ischemic and injured axons need to be replenished by healthy mitochondria for energy supply and to limit further axonal damage (for example, by buffering axoplasmic Ca2+ and attenuating oxidative stress). Also, the capacity for neurons to regenerate severed axons in non-permissive environments, such as the adult central nervous system, has been correlated with their ability to transport mitochondria along axons8,9. Huang et al.2 show that PAK5 localizes to the outer mitochondrial membrane in injured axons, where it phosphorylates a cluster of evolutionarily conserved serine and threonine residues in SNPH, thereby turning off SNPH’s anchoring activity and enabling mitochondrial transport. This, along with the engagement of kinesin and dynein motors through the Miro–Milton complex, would facilitate the delivery of functional mitochondria from proximal axonal regions and the removal of damaged mitochondria from the injured region (Figure 1). Notably, the Miro–Milton complex is also Ca2+ sensitive, with elevated Ca2+ causing dissociation of Miro from Milton and releasing the mitochondrion from motor proteins10. So, loss of Ca2+-buffering capacity as a result of mitochondrial dysfunction would both increase SNPH anchoring activity and restrict mitochondrial engagement with motor proteins.
Figure 1. A combination of axonal translation of Pak5 mRNA and AKT activation reprograms mitochondrial motility in injured axons.
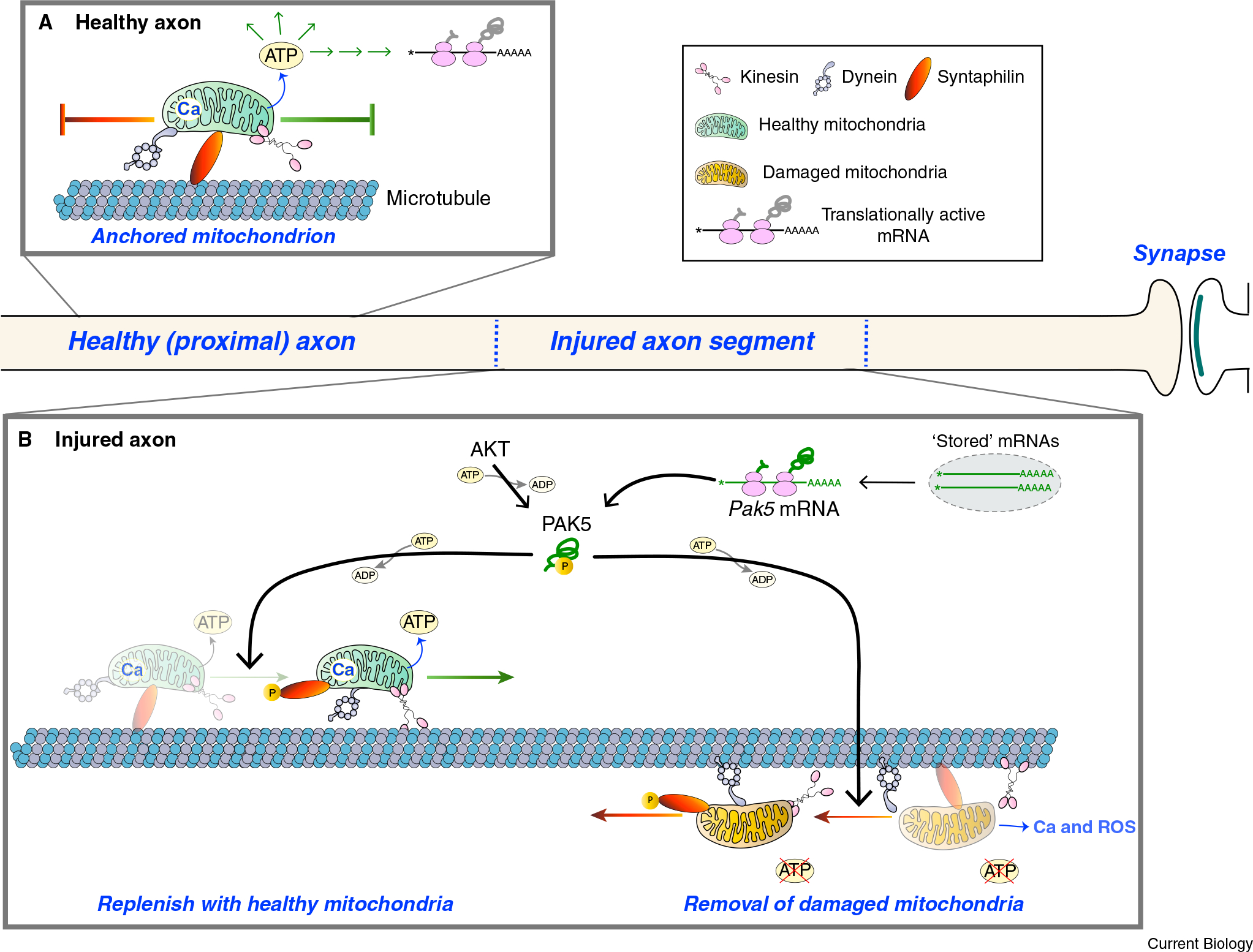
(A) In a mature uninjured axon, most mitochondria are anchored in place on microtubules by SNPH. This provides a localized source of energy (ATP) and Ca2+ (Ca) storage. (B) Axonal injury damages mitochondria by reducing oxidative phosphorylation so that the local source of ATP is at risk. This also results in the release of Ca2+ from mitochondria and a localized increase in reactive oxygen species (ROS). The Miro–Milton complex (not shown) provides an adapter for the association of mitochondria with kinesin and dynein motor proteins, and release of Ca2+ from mitochondria into the axoplasm would further impact mitochondrial motility by promoting the dissociation of Miro from Milton. Huang et al.2 show that injury can trigger the translation of Pak5 mRNA, with AKT activating the nascent PAK5 protein, and activated PAK5 phosphorylating SNPH to release the SNPH mitochondrial anchor. This brings to the injury site functional mitochondria that promote both neuroprotection and axon regeneration, as well as removing damaged mitochondria that can impede neuroprotection and neural repair.
In the PAK family of protein kinases, PAK4, PAK5 and PAK6 are group II PAKs that bind to Rho-family GTPases, particularly CDC42, with GTPase interaction impacting their subcellular localization11. Ribosome–mRNA co-immunoprecipitations from optic nerve axons pointed to translation of Pak5 mRNA in axons12, but neither the regulation of its intra-axonal translation nor the functional outcomes of that translation had been investigated. Similar to CDC42’s effect on neuritogenesis, PAK5 activity has been linked to neurite growth, but surprisingly deletion of the murine PAK5 gene resulted in only a modest behavioral phenotype with no apparent morphological changes in the nervous system13. Thus, the finding that PAK5 supports neuronal survival and regeneration after axonal injury is quite novel2 and raises the possibility that a defective injury response could be a phenotypic trait of PAK5−/− mice.
Many different functional outcomes have been ascribed to axonally synthesized proteins to date, including effects locally in the axon as well as axon-to-soma signaling. Mitochondrial function in axons has been shown to be supported by intra-axonal translation of mRNAs encoding nuclear-encoded mitochondrial proteins14. Although RNAs are packaged into membraneless granules for transport into axons, some RNA transport granules bind to vesicles for their transport into axons15. An axonopathy-associated mutation of Rab7, a protein required for maturation of late endosomes, impairs axonal mitochondrial function in a manner dependent on protein synthesis16. Further evidence to support the impact of localized translation on mitochondria comes from observations that disrupting axonal delivery of Bcl-w mRNA, which encodes a pro-survival Bcl-2 family protein that localizes to mitochondria, triggers axonal degeneration17. Analogous to the finding that Pak5 mRNA translation inhibits SNPH’s anchoring activity, as uncovered by Huang et al.2, these studies together link mRNA transport and localized translation with axonal mitochondrial function and axon health. However, the findings from Huang et al.2 regarding intra-axonal translation of Pak5 mRNA also connect injury-induced translational regulation in axons to mitochondrial function (Figure 1).
Traumatic injury of axons is known to locally elevate axoplasmic Ca2+ in the proximal axon tip. Elevated Ca2+ activates proteases necessary for growth cone formation18 and in addition activates translation of several axonal mRNAs to synthesize proteins that orchestrate the injury response. Indeed, some axonal mRNAs are stored locally in stress-granule-like structures, with injury releasing the mRNAs for translation19. These stored mRNAs include those encoding proteins and transcription factors that are used for axon-to-soma signaling to initiate injury responses in the cell body, as well as endoplasmic reticulum chaperone proteins and mTOR, which can function locally to orchestrate the axon’s temporal response to axotomy19. The present work from Huang et al.2 points to Pak5 mRNA as an injury-response mRNA that is translated following axonal injury to subsequently restore local energy production by mobilizing the transport of mitochondria after axotomy. This would also increase Ca2+-storage capacity in the proximal axon and attenuate ROS production. With evidence that mitochondria are the energy source for localized mRNA translation in dendrites and axons5,20, this Pak5–SNPH pathway also supports regeneration by enabling localized protein synthesis in axons (Figure 1). In addition, these new findings for the Pak5–SNPH pathway importantly reveal common molecular responses to the physical severing of axons seen following spinal cord injury and to the ischemic injury of oxygen glucose deprivation. Indeed, the findings from Huang et al.2 raise the exciting possibility that intra-axonally translated mRNAs can also provide neuroprotection after ischemic injury.
REFERENCES
- 1.Zala D, Hinckelmann MV, Yu H, Lyra da Cunha MM, Liot G, Cordelieres FP, Marco S, and Saudou F (2013). Vesicular glycolysis provides on-board energy for fast axonal transport. Cell 152, 479–491. [DOI] [PubMed] [Google Scholar]
- 2.Huang N, Li S, Xie Y, Han Q, Xu X-M, and Sheng Z-H (2021). Reprogramming an energetic AKT-PAK5 axis boosts axon energy supply and facilitates neuron survival and regeneration after injury and ischemia. Curr. Biol. 31, 3098–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen Y, and Sheng ZH (2013). Kinesin-1-syntaphilin coupling mediates activity-dependent regulation of axonal mitochondrial transport. J. Cell Biol. 202, 351–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Courchet J, Lewis TL Jr., Lee S, Courchet V, Liou DY, Aizawa S, and Polleux F (2013). Terminal axon branching is regulated by the LKB1-NUAK1 kinase pathway via presynaptic mitochondrial capture. Cell 153, 1510–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spillane M, Ketschek A, Donnelly CJ, Pacheco A, Twiss JL, and Gallo G (2012). Nerve growth factor-induced formation of axonal filopodia and collateral branches involves the intra-axonal synthesis of regulators of the actin-nucleating Arp2/3 complex. J. Neurosci. 32, 17671–17689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Han Q, Xie Y, Ordaz JD, Huh AJ, Huang N, Wu W, Liu N, Chamberlain KA, Sheng ZH, and Xu XM (2020). Restoring cellular energetics promotes axonal regeneration and functional recovery after spinal cord injury. Cell Metab. 31, 623–641.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stavoe AKH, and Holzbaur ELF (2019). Autophagy in neurons. Annu. Rev. Cell Dev. Biol. 35, 477–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cartoni R, Pekkurnaz G, Wang C, Schwarz TL, and He Z (2017). A high mitochondrial transport rate characterizes CNS neurons with high axonal regeneration capacity. PLoS One 12, e0184672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalinski AL, Kar AN, Craver J, Tosolini AP, Sleigh JN, Lee SJ, Hawthorne A, Brito-Vargas P, Miller-Randolph S, Passino R, et al. (2019). Deacetylation of Miro1 by HDAC6 blocks mitochondrial transport and mediates axon growth inhibition. J. Cell Biol. 218, 1871–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cai Q, and Sheng ZH (2009). Moving or stopping mitochondria: Miro as a traffic cop by sensing calcium. Neuron 61, 493–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ha BH, Morse EM, Turk BE, and Boggon TJ (2015). Signaling, regulation, and specificity of the type II p21-activated kinases. J. Biol. Chem. 290, 12975–12983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shigeoka T, Jung H, Jung J, Turner-Bridger B, Ohk J, Lin JQ, Amieux PS, and Holt CE (2016). Dynamic axonal translation in developing and mature visual circuits. Cell 166, 181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Furnari MA, Jobes ML, Nekrasova T, Minden A, and Wagner GC (2013). Functional deficits in PAK5, PAK6 and PAK5/PAK6 knockout mice. PLoS One 8, e61321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gale JR, Aschrafi A, Gioio AE, and Kaplan BB (2018). Nuclear-encoded mitochondrial mRNAs: a powerful force in axonal growth and development. Neuroscientist 24, 142–155. [DOI] [PubMed] [Google Scholar]
- 15.Liao YC, Fernandopulle MS, Wang G, Choi H, Hao L, Drerup CM, Patel R, Qamar S, Nixon-Abell J, Shen Y, et al. (2019). RNA granules hitchhike on lysosomes for long-distance transport, using annexin A11 as a molecular tether. Cell 179, 147–164.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cioni JM, Lin JQ, Holtermann AV, Koppers M, Jakobs MAH, Azizi A, Turner-Bridger B, Shigeoka T, Franze K, Harris WA, et al. (2019). Late endosomes act as mRNA translation platforms and sustain mitochondria in axons. Cell 176, 56–72.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cosker KE, Fenstermacher SJ, Pazyra-Murphy MF, Elliott HL, and Segal RA (2016). The RNA-binding protein SFPQ orchestrates an RNA regulon to promote axon viability. Nat. Neurosci. 19, 690–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bradke F, Fawcett JW, and Spira ME (2012). Assembly of a new growth cone after axotomy: the precursor to axon regeneration. Nat. Rev. Neurosci. 13, 183–193. [DOI] [PubMed] [Google Scholar]
- 19.Dalla Costa I, Buchanan C, Zdradzinski MD, Sahoo PK, Smith TP, Thames E, Kar AN, and Twiss JL (2020). Functional platforms for organizing axonal mRNA transport and translation. Nat. Rev. Neurosci. 22, 77–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rangaraju V, Lauterbach M, and Schuman EM (2019). Spatially stable mitochondrial compartments fuel local translation during plasticity. Cell 176, 73–84. [DOI] [PubMed] [Google Scholar]