

Background:
Hyperlipidemia (hypercholesterolemia and/or hypertriglyceridemia) is a risk factor for atherosclerosis. Nogo-B receptor (NgBR) plays important roles in hepatic steatosis and cholesterol transport. However, the effect of NgBR overexpression on atherosclerosis remains unknown.
Materials and Methods:
Apolipoprotein E deficient (ApoE-/-) mice infected with adeno-associated virus (AAV)-NgBR expression vector were fed a high-fat diet for 12 weeks, followed by determination of atherosclerosis and the involved mechanisms.
Results:
We determined that high expression of NgBR by AAV injection mainly occurs in the liver and it can substantially inhibit en face and aortic root sinus lesions. NgBR overexpression also reduced levels of inflammatory factors in the aortic root and serum, and levels of cholesterol, triglyceride, and free fatty acids in the liver and serum. Mechanistically, NgBR overexpression increased the expression of scavenger receptor type BI and the genes for bile acid synthesis, and decreased the expression of cholesterol synthesis genes by reducing sterol regulatory element-binding protein 2 maturation in the liver, thereby reducing hypercholesterolemia. In addition, NgBR overexpression activated AMP-activated protein kinase α via the Ca2+ signaling pathway, which inhibited fat synthesis and improved hypertriglyceridemia.
Conclusions:
Taken together, our study demonstrates that overexpression of NgBR enhanced cholesterol metabolism and inhibited cholesterol/fatty acid synthesis to reduce hyperlipidemia, and reduced vascular inflammation, thereby inhibiting atherosclerosis in ApoE-/- mice. Our study indicates that NgBR might be a potential target for atherosclerosis treatment.
INTRODUCTION
Atherosclerosis is the most common underlying pathology of coronary heart disease. Various factors affect the development of atherosclerosis, such as hyperlipidemia (hypercholesterolemia and/or hypertriglyceridemia), hypertension, inflammation, diabetes, and smoking.1 Among them, hyperlipidemia plays a major role in the development of atherogenesis.
In the circulation system, excessive LDL cholesterol (LDL-C) can cause endothelial dysfunction and increase the secretion of proinflammatory factors which may promote the adhesion of monocytes to the endothelium layer. The monocytes infiltrating to the underneath of endothelium layer will differentiate into macrophages which can bind and internalize modified, particularly the oxidatively modified LDL, to form lipid-laden foam cells, the prominent part of atherosclerotic lesions.2,3 Clinically, lowering serum total cholesterol (CHO), particularly LDL-C levels, is the main strategy for atherosclerosis treatment. Statins inhibit the activity of 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR), the rate-limiting enzyme for cholesterol synthesis, and upregulate expression of LDL receptor (LDLR) for LDL-C clearance. Therefore, statins potently reduce CHO and LDL-C levels.4
In general, the NAFLD patients accompanied by hypertriglyceridemia are also more likely to develop atherosclerosis.5 Excessive triglyceride (TG) and free fatty acids (FFAs) have vascular toxicity and may enter the artery wall to activate endothelial cells and induce inflammation, thereby exacerbating atherosclerosis.6 Glucagon-like peptide-1 receptor agonists, thyroid hormone receptor beta-agonists, and other compounds that are being developed to alleviate NAFLD have been demonstrated anti-atherogenesis properties.7 In addition, many studies including ours suggest that anti-hypertriglyceridemia can make substantial contributions to the inhibition of atherosclerosis. For instance, treatment of high-fat diet (HFD)-fed apoE deficient (ApoE-/-) with the combination of a MEK1/2 inhibitor and a liver X receptor (LXR) agonist or LXR agonist encapsulated in a nanofiber hydrogel antagonized LXR agonist-induced lipogenesis/hypertriglyceridemia, therefore, it further enhanced LXR agonist-inhibited atherosclerosis.8–10
Reticulon 4B (Nogo-B) receptor (NgBR) is a transmembrane protein initially thought to interact with Nogo-B in endothelial cell tube formation.11 Nogo-B has been shown to play an important role in the control of vascular function and blood pressure.12,13 However, many studies have shown that Nogo-B and NgBR can function independently and have no physiological interaction. NgBR interacts with the Niemann Pick C2 protein to regulate cholesterol transport.14 Adeno-associated virus (AAV)-mediated hepatic NgBR overexpression improved lipid metabolism and insulin sensitivity in a mouse model of type 2 diabetes.15 Reactive oxygen species overload suppresses NgBR expression in endothelial cells and smooth muscle cells in pulmonary hypertension models, however, pulmonary hypertension is generally unassociated with atherosclerosis.16–19 Meanwhile, hepatic NgBR is involved in the de novo synthesis of lipids and the hypolipidemic effects of statins,20,21 suggesting a possible influence of NgBR on the blood lipid levels. Dyslipidemia is a key factor in atherosclerosis, and the liver is a central site for lipid metabolism. Therefore, we focused on the effect of hepatic rather than vascular NgBR on atherosclerosis, due to the possibility that hepatic NgBR may affect cholesterol and lipid metabolism. In this study, we overexpressed NgBR by infecting ApoE-/- mice with AAV-NgBR expression vector (AAV-NgBR) to determine if NgBR overexpression can inhibit HFD-induced atherosclerosis and the involved mechanisms.
MATERIALS AND METHODS
In vivo studies
Animal studies were reported in compliance with the ARRIVE guidelines. The care of animals and experimental projects conformed to the “Guide for the Care and Use of Laboratory Animals” of the National Institute of Health (NIH Publications No. 8023, revised 1978) were approved by the Ethics Committee of Hefei University of Technology (HFUT20190601002).
Male ApoE-/- mice (~8-wk-old, ~28 g) with C57BL/6J background were purchased from the Animal Center of Nanjing University (Nanjing, China). The animals were hosted at 23±1 °C, with a relative humidity of 60%–70% and 12 hours of light/dark cycles. All mice were free to access water and food.
NgBR expression was driven by the CMV promoter, in fusion with the Flag tag (GFP) at the C-terminus. AAV vectors were serotyped with AAV2/9 coat protein and packaged by Hanbio Biotechnology Co. (Shanghai, China). The viral titer was ~4.5×1011 vg/mL for AAV2/9-CMV-m-NgBR-3xflag-GFP vector (named as AAV-NgBR) or ~2.5×1013 vg/mL for AAV2/9-GFP empty vector (named as AAV-GFP). After acclimation to the housing environment for >7 days, ApoE-/- mice randomly received a tail-vein injection of 1×1012 vg AAV-GFP/mouse as the control group (HFD) or 1×1012 vg AAV-NgBR/mouse as the NgBR overexpression group (HFD-NgBR) (10 mice/group). After the virus injection, all the animals were started HFD (21% fat, 0.5% cholesterol) feeding to induce atherosclerosis. During the treatment, we recorded the body weight, food intake, and external appearance, and observed no difference between the 2 groups. Twelve weeks later, all the mice were euthanized, followed by the collection of blood and tissue samples individually. Initially, the infection efficiency was confirmed by determining NgBR mRNA expression by quantitative real-time PCR with total RNA extracted from liver samples. Totally, 8 mice with clear NgBR overexpression (>3-fold in mRNA levels) in HFD-NgBR group were continued all the rest experiments, and 2 mice with no substantial NgBR overexpression were abandoned.
After preparation, activities of alanine aminotransferase, aspartate transaminase, alkaline phosphatase, and levels of TG, CHO, HDL cholesterol (HDL-C) and LDL-C in serum were determined by an automatic biochemical analyzer (3100, Hitachi High-Technologies Corporation). The level of VLDL cholesterol (VLDL-C) was determined by the ELISA kit which was purchased from J&L Biological (Shanghai, China).
Liver and serum FFA levels were determined using the FFA assay kit (BC0595, Solarbio, Beijing, China). Liver TG or CHO levels were determined using the LabAssay triglyceride kit (290-63701, WAKO, Japan) and tissue CHO assay kit (E1015, Applygen, Beijing, China). Lipid accumulation in and structure of the liver were determined by oil red O and hematoxylin-eosin (H&E) staining of liver frozen and paraffin sections, respectively. Hepatic fibrosis was determined by picrosirius red staining of liver paraffin sections.8
The entire aortas and cross cryosections of the aortic root (5-μm) were conducted oil red O staining to determine lesions as described.8 The cross cryosections were also used to determine necrotic cores and collagen content in atherosclerotic plaques by H&E and picrosirius red staining, respectively.9,22
Cell culture
LO2 cells, a human hepatic cell line purchased from ATCC (Manassas, VA, USA), were cultured in complete RPMI 1640 medium (Biological Industries) supplemented with 10% (vol/vol) FBS, 50 μg/mL penicillin/streptomycin and 2 mmol/L glutamine. Cells were switched to a serum-free medium and received treatment when the confluence was 70%–80%.
Statistics analysis
All the data were obtained by repeating at least 5 times in each experiment, and the representative results are presented. All the data analysis was carried out by technicians who were blinded to which samples/animals represent treatments and controls. All values (control and test) are normalized to the mean of the experimental control group and expressed as means±SEM. All the data were initially analyzed for normality and equal variance as a justification for using parametric or nonparametric analyses using Prism software. Statistical significance was evaluated using the unpaired 2-tailed Student t test for 2 groups or 1-way ANOVA with post hoc test among more than 2 groups. Data including 2 variables were analyzed with the 2-way ANOVA and post hoc test. The significant difference was considered if p<0.05.
The list of other reagents and other experimental procedures were presented in the “Supplemental Materials (http://links.lww.com/HC9/A119).”
RESULTS
Overexpression of NgBR inhibits atherosclerosis and increases plaque stability in ApoE-/- mice
Both clinical and basic research demonstrate that hypertension, obesity, and dyslipidemia are the risk factors for the development of atherosclerosis, the major pathogen for cardiovascular diseases. We believed that abnormal liver NgBR expression may be linked to these risk factors to impact the development of atherosclerosis. Thus, we initially determined the changes of NgBR expression in the liver of patients or mice with hypertension, obesity, and dyslipidemia. Based on a search of the NCBI gene expression omnibus database, we selected three relevant data (GEO access: GSE19817, GSE32095, GSE15653) and confirmed NgBR expression in the liver. We found lower hepatic NgBR transcript levels in hypertension mice than in normotension mice (Figure S1A, http://links.lww.com/HC9/A119). HFD feeding to wild type mice for 11 weeks reduced NgBR transcript levels in the liver (Figure S1B, http://links.lww.com/HC9/A119). While the reduced NgBR transcript levels in the livers of obese patients were observed (Figure S1C, http://links.lww.com/HC9/A119). The differences in hepatic NgBR levels in these models suggest that NgBR may be involved in the pathophysiological status of metabolic diseases including atherosclerosis.
To determine the effect of NgBR on atherosclerosis in the context of hypercholesterolemia, we detected NgBR protein expression in the liver of normal chow-fed C57BL/6J mice and HFD-fed ApoE-/- mice, or in normal chow-fed and HFD-fed ApoE-/- mice. Compared with normal chow-fed C57BL/6J mice or ApoE-/- mice, hepatic NgBR expression was significantly decreased in HFD-fed ApoE-/- mouse liver (Figure 1A, B). Consistently, similar results in LDLR deficient (LDLR-/-) mice were observed (Figure 1C, D). These preliminary results suggest the association of NgBR expression in the liver with the development of atherosclerosis.
FIGURE 1.
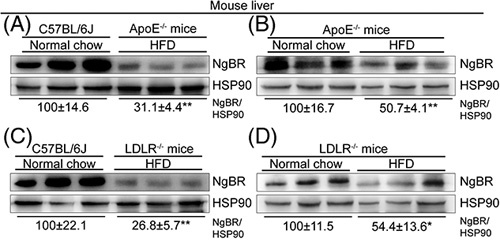
NgBR expression is reduced in the liver of atherosclerotic mice. (A–D) C57BL/6J, ApoE−/− and LDLR−/− mice (~8-week-old, male) were fed normal chow or HFD as indicated for 12 weeks, followed by determination of liver NgBR expression by Western blot. **p<0.01, n=3. Abbreviation: HFD, high-fat diet.
Based on the results above, we further speculated that NgBR overexpression can inhibit atherosclerosis in ApoE-/- mice. Currently, no available NgBR chemical activators have been developed, thus, we employed an AAV system to manipulate NgBR expression in vivo, particularly in the liver. We injected ApoE-/- mice AAV empty vector (HFD group) or AAV-NgBR expression vector (HFD-NgBR group), and then fed them HFD for 12 weeks (Figure 2A). At the end of the experiment, we initially confirmed that 2 of 10 mice in HFD-NgBR group had no changes in NgBR expression in the liver, and had to abandon these 2 mice for the rest experiments. Although most natural AAV coat proteins are able to trigger efficient transgene expression in the liver, we still determined if the high NgBR expression by AAV injection can occur in other tissues than the liver. As shown in Figure S2A–F (http://links.lww.com/HC9/A119), AAV injection potently increased NgBR expression in the liver, while having little effect on NgBR expression in mouse kidney, spleen, pancreas, skeletal muscle, and aortas, suggesting that at the dose we used, injection of AAV-NgBR caused high NgBR expression mainly in the liver. Meanwhile, we found NgBR overexpression had little effect on Nogo-B expression in mouse liver or LO2 cells either (Figure S2G–J, http://links.lww.com/HC9/A119).
FIGURE 2.
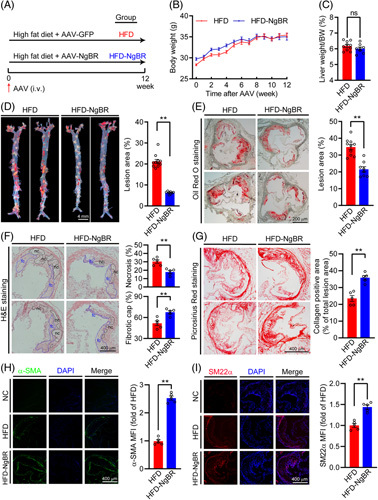
Overexpression of NgBR inhibits lesion development and increases plaque stability in ApoE−/− mice. A (experimental design): ApoE−/− mice (~8-week-old, male) were randomly divided into 2 groups and fed HFD. One group was injected AAV empty vector (HFD group) and another group AAV-NgBR expression vector (HFD-NgBR group) from tail vein, each at the same dose of 1012 vg/mouse. At the end of 12-week experiment, mouse tissue samples were collected. Expression of NgBR mRNA among the all mice was screened by quantitative real-time PCR. All the mice in HFD group (n=10) and 8 mice in HFD-NgBR group with confirmed NgBR overexpression by quantitative real-time PCR (n=8) were used for the following assays. (B) mouse bodyweight monitored weekly. (C) Ratio of liver weight to bodyweight. (D and E) Lesions in en face aortas and aortic root cross sections determined by oil red O staining. (F) H&E staining of aortic root cross sections with quantitative analysis of necrotic cores and fibrous cap areas. nc: necrotic cores marked with black dotted line; fc: fibrotic cap marked with blue dotted line. (G) Collagen content determined by picrosirius red staining of aortic root cross sections with quantitative analysis. (B–G) ns, not significant; **P<0.01, n=10 (HFD group), n=8 (HFD-NgBR group). (H and I) Smooth muscle cells content detected by immunofluorescent staining of the aortic root cross sections with anti-α-SMA and SM22α antibody. NC (negative control): primary antibody was replaced by rabbit normal IgG. **p<0.01, n=5. Abbreviations: AAV, adeno-associated virus; HFD, high-fat diet.
During the treatment, mouse body weight was routinely checked and found to be close between the 2 groups (Figure 2B). The ratio of liver weight to body weight was not changed either by NgBR overexpression (Figure 2C). However, compared with the HFD group, en face aortic lesions and sinus lesions in the aortic root were inhibited by ~68% and 45% (Figure 2D, E), suggesting overexpression of NgBR can substantially inhibit atherosclerosis.
The size of necrotic cores, the thickness of fibrous cap, and collagen content or smooth muscle cell phenotype are essential factors for the stability of atherosclerotic plaques.23 The results of H&E staining of aortic root lesions demonstrate that the areas of necrosis cores were reduced in HFD-NgBR group. Meanwhile, the thickness of the fibrotic cap of HFD-NgBR group was increased compared with HFD group (Figure 2F). The picrosirius red staining shows that the collagen content in HFD-NgBR group was increased significantly (~1.5-fold of that in HFD group) (Figure 2G). In addition, smooth muscle cell content was increased in HFD-NgBR group, characterized as increased contractile smooth muscle cells [α-smooth muscle actin (α-SMA) and transgelin (SM22α) positive cells] in lesions (Figure 2H, I). Taken together, these data suggest that NgBR overexpression can ameliorate atherosclerosis complex in HFD-fed ApoE-/- mice.
Overexpression of NgBR reduces hepatic lipid accumulation, improves hyperlipidemia, and diminishes aortic inflammatory response
Associated with the development of atherosclerosis, HFD also induces hyperlipidemia and fatty liver. Overexpression of NgBR blocked the color changes of the liver induced by HFD (Figure 3A). The results of oil red O and H&E (Figure 3A) staining demonstrate severe lipid accumulation, many vacuolar droplets, and infiltration of inflammatory cells in the liver of HFD group mice. However, all of these changes were substantially reduced in mice in the HFD-NgBR group. Compared with mice in the HFD group, the results of the lipid quantitative assay show that levels of CHO, TG, and FFA in the mouse liver of HFD-NgBR group were clearly reduced (Figure 3B–D). The long-term steatohepatitis can cause liver fibrosis. However, NgBR overexpression reduced expression of the genes for collagen formation (Figure S3A, http://links.lww.com/HC9/A119), demonstrating its anti-liver fibrosis function which was further confirmed by picrosirius red staining of liver sections (Figure S3B, http://links.lww.com/HC9/A119).
FIGURE 3.
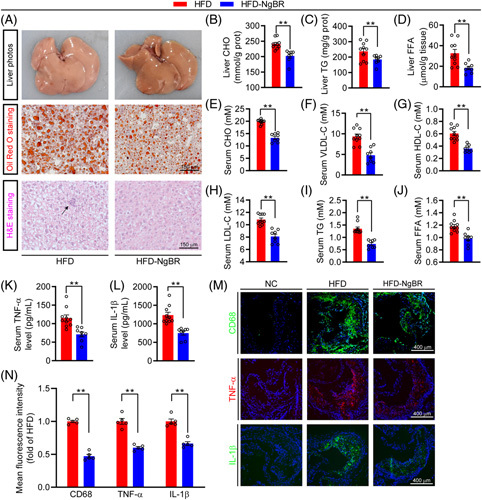
NgBR overexpression improves NAFLD, hyperlipidemia and inflammatory response. Liver samples of mice in Figure 2 were used to complete the following assays: liver photos, oil red O staining of liver frozen sections and H&E staining of liver paraffin sections with an arrow indicates inflammatory cells in the liver (A). Quantitative analysis of liver CHO (B), TG (C), and FFA (D) with total liver lipid extract. Mouse serum samples in Figure 2 were used to determine levels of CHO (E), VLDL-C (F), HDL-C (G), LDL-C (H), TG (I) and FFA (J). Serum TNF-α (K) and IL-1β (L) levels were determined by Elisa. (B–L) *p<0.05, **p<0.01, n=10 (HFD group), n=8 (HFD-NgBR group). (M and N) Expression of CD68, TNF-α, and IL-1β in aortic root cross sections were detected by immunofluorescent staining with the corresponding antibody. NC: primary antibody was replaced by rabbit normal IgG. **p<0.01, n=5. Abbreviation: HFD, high-fat diet.
Compared with HFD group, NgBR overexpression reduced serum levels of CHO, VLDL-C, HDL-C, and LDL-C by ~33.5%, 49.0%, 20.0%, and 29.4% (Figure 3E–H), respectively. Similarly, levels of serum TG and FFA in HFD-NgBR group were decreased by ~45% and 20% (Figure 3I, J). Thus, overexpression of NgBR clearly reduces HFD-induced hepatic lipid accumulation, hypercholesterolemia, and hypertriglyceridemia.
The chronic inflammation is another risk factor for atherosclerosis. NgBR overexpression clearly reduced serum TNF-α and IL-1β levels (Figure 3K, L). Meanwhile, levels of CD68, TNF-α, and IL-1β in atherosclerotic plaques were decreased in HFD-NgBR group (Figure 3M, N), indicating reduced macrophage/foam cell accumulation and inflammation in lesions by NgBR overexpression. Alkaline phosphatase is a biomarker for vascular calcification, inflammation and endothelial dysfunction in renal and cardiovascular diseases. Although NgBR overexpression had little effect on serum alanine aminotransferase and aspartate transaminase levels, it clearly decreased serum alkaline phosphatase levels (Figure S3C–E, http://links.lww.com/HC9/A119). Taken together, the data above suggest that overexpression of NgBR can improve hyperlipidemia and reduce the inflammatory response.
The anti-hypercholesterolemia mechanisms of NgBR overexpression
The liver is the main tissue for cholesterol synthesis and metabolism. Determination of mRNA expression of the genes for cholesterol synthesis and metabolism indicates that NgBR overexpression reduced expression of the genes for cholesterol synthesis, such as HMGCR and 3-hydroxysteroid-24 reductase (DHCR24) (Figure 4B). In contrast, NgBR overexpression substantially increased the expression of scavenger receptor type BI (SR-BI) (Figure 4B), an anti-atherogenic molecule by mediating reverse cholesterol transport through uptake of HDL-C in the liver for routing to bile synthesis.24 Meanwhile, NgBR overexpression induced expression of cytochrome P450 family 7 subfamily A member 1 (CYP7A1) (Figure 4B), the rate-limiting enzyme for cholesterol catabolism/bile acid synthesis. The results of Western blot also confirm that mice with NgBR overexpression had reduced HMGCR, DHCR24 and LDLR protein expression, and increased SR-BI protein expression (Figures 4C, S7A, http://links.lww.com/HC9/A119). The immunohistochemical staining further confirms NgBR overexpression reduced HMGCR while increased SR-BI expression in the liver (Figure 4D).
FIGURE 4.
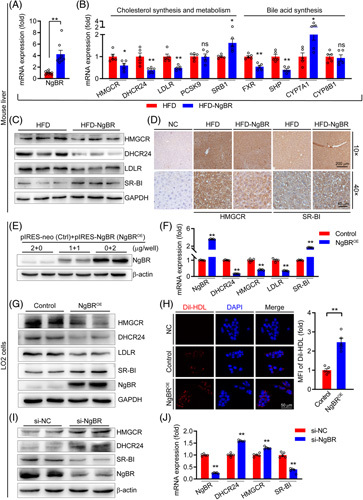
NgBR overexpression inhibits hypercholesterolemia. Total RNA, protein and sections were prepared with liver samples from mice used in Figure 2, and used for the following determinations. Expression of NgBR mRNA [A, **p<0.01, n=10 (HFD group), n=8 (HFD-NgBR group)], mRNA expression of the genes for cholesterol synthesis/metabolism and bile acid synthesis (B, *p<0.05, **p<0.01, n=5) by quantitative real-time PCR (qPCR). (C) Protein expression of NgBR, HMGCR, DHCR24, LDLR, and SR-BI by Western blot. (D) Expression of HMGCR and SR-BI by immunohistochemical staining with anti-SR-BI antibody. NC: primary antibody was replaced by rabbit normal IgG. (E): LO2 cells were transfected with empty vector (Control, Ctrl) or NgBR expression vector (NgBROE) followed by determination of NgBR by Western blot. (F and G) Expression of NgBR, DHCR24, HMGCR, LDLR, SR-BI in control and NgBR overexpressing LO2 cells were detected by qPCR (**p<0.01, n=5) and Western blot. (H) LO2 cells with or without NgBR overexpression were seeded in confocal dishes. Cells were then switched to serum-free medium containing Dil-HDL (20 µg/ml) and incubated for 4 hours at 37 °C, followed by photographed with a fluorescence microscope (**p<0.01, n=5). (I and J) LO2 cells were transfected with scrambled siRNA (si-NC) or NGBR siRNA (si-NGBR) at 50 nmol/L for 48 hours, followed by determination of HMGCR, DHCR24, SR-BI, and NgBR protein and mRNA expression by Western blot and qPCR. The statistical results of band density analysis were presented in Figure S7, (http://links.lww.com/HC9/A119). Abbreviations: HFD, HFD, high-fat diet; ns: not significant.
In vitro, transfection of LO2 cells with an NgBR expression vector (Figures 4E, S7B, http://links.lww.com/HC9/A119) regulated expression of the molecules for cholesterol synthesis/metabolism at protein and transcript levels (Figures 4F, G, S7C, http://links.lww.com/HC9/A119) in a similar pattern to that in vivo by AAV-NgBR injection. We further tested the ability of LO2 cells to take up Dil-HDL, and found that NgBR overexpression significantly increased HDL uptake by LO2 cells, suggesting that NgBR overexpression may activate SR-BI and reverse cholesterol transport (Figure 4H). Meanwhile, we observed that ApoA1, a major component of HDL, was significantly reduced in the serum (Figure S4A, http://links.lww.com/HC9/A119). Pregnane X receptor (PXR) acts as a negative regulator of SR-BI.25 Correspondingly, NgBR overexpression reduced PXR transcript in vivo and in vitro (Figure S4B, http://links.lww.com/HC9/A119), confirming the activation of SR-BI pathway. In contrast, inhibition of NgBR expression by siRNA increased the expression of HMGCR and DHCR24, and reduced the expression of SR-BI in LO2 cells (Figures 4I, J, S7D, http://links.lww.com/HC9/A119). Taken together, these results suggest that the anti-hypercholesterolemia by NgBR overexpression is completed through inhibition of cholesterol synthesis and activation of cholesterol metabolism in hepatocytes/liver.
Overexpression of NgBR restrains SREBP2 maturation
SREBP2 is an intracellular cholesterol sensor by regulating the expression of the genes for cholesterol synthesis, particularly HMGCR. We found that NgBR overexpression decreased SREBP2 protein in the liver (Figure 5A) without effect on SREBP2 transcript (Figure 5B). Furthermore, NgBR overexpression had little effect on the expression of SREBP2 precursor [SREBP2(p)] but clearly reduced the active form of SREBP2 [SREBP2(m)] in mouse liver and LO2 cells (Figures 5C, D, S7E, F, http://links.lww.com/HC9/A119), suggesting SREBP2 maturation/function is reduced.
FIGURE 5.
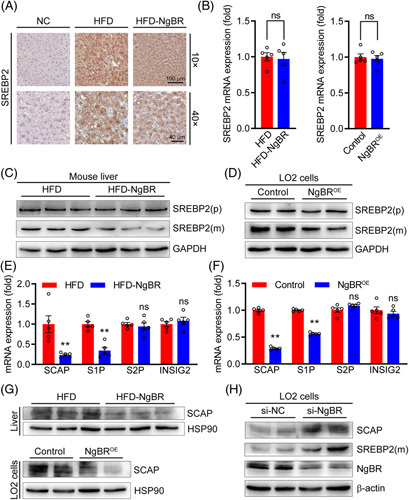
Overexpression of NgBR reduces SREBP2 maturation. (A) Expression of SREBP2 in the liver was detected by immunohistochemical staining with anti-SREBP2 antibody. NC: primary antibody was replaced by rabbit normal IgG. (B) mRNA expression of SREBP2 in liver and LO2 cells was detected by quantitative real-time PCR (qPCR). n=5. (C and D) Protein expression of SREBP2 precursor [SREBP2(p)] and mature SREBP2 [SREBP2(m)] in liver and LO2 cells were detected by Western blot. (E and F) mRNA expression of SCAP, S1P, S2P, and INSIG2 in liver and LO2 cells were detected by qPCR. **p<0.01, n=5. (G) Expression of SCAP protein in liver and LO2 cells were detected by Western blot. (H) LO2 cells were transfected with 50 nmol/L control siRNA (si-NC) or NgBR siRNA (si-NGBR) for 48 hours, followed by determination of SCAP, SREBP2(m), and NgBR protein expression by Western blot. The statistical results of band density analysis were presented in Figure S7 (http://links.lww.com/HC9/A119). Abbreviations: HFD, high-fat diet; ns: not significant.
The complex composed of site 1 proteases (S1P), S2P and SREBP cleavage activating protein (SCAP) is responsible for cleavage or maturation of SREBP2 into the active form. Insulin-induced gene 2 (INSIG2) acts as a switch to release SCAP-SREBP2 complex from the endoplasmic reticulum (ER) and cleaves it in the Golgi apparatus.26 In ApoE-/- mouse liver and LO2 cells, NgBR overexpression reduced expression of SCAP and S1P while having little effect on S2P and INSIG2 (Figures 5E–G, S7G, http://links.lww.com/HC9/A119), suggesting reduced SCAP expression may play a critical role in NgBR-inhibited SREBP2 maturation. Reciprocally, reduced NgBR expression by si-NgBR increased both SCAP and SREBP2(m) (Figures 5H, S7H, http://links.lww.com/HC9/A119), associated with enhanced SCAP and S1P but not SREBP2 mRNA expression (Figure S4C, http://links.lww.com/HC9/A119).
Taken together, the reduction of cholesterol levels by NgBR overexpression might be related to the inhibition of HMGCR and DHCR24 expression by inactivating the SCAP-SREBP2 maturation pathway.
The underlying mechanisms for anti-hypertriglyceridemia by NgBR overexpression
Associated with reduced lipid accumulation in mouse liver and TG and FFA levels in mouse serum (Figure 3A, C, D, I, J), we determined that NgBR overexpression reduced expression of the genes for lipogenesis (fatty acid or TG synthesis) while enhanced expression of the genes for TG hydrolysis at both transcript and protein levels (Figures 6A, B, S8A, http://links.lww.com/HC9/A119) in mouse liver. Similar results were obtained with NgBR overexpressing LO2 cells (Figures 6C, D, S7B, http://links.lww.com/HC9/A119).
FIGURE 6.
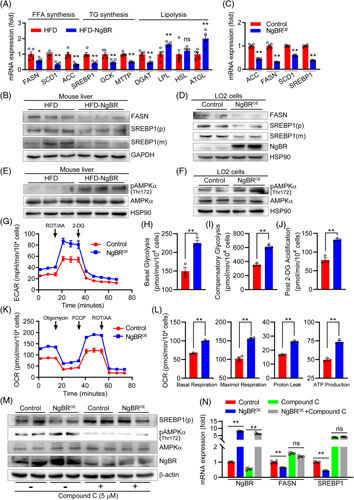
Overexpression of NgBR reduces lipid synthesis and actives AMPKα⊡ mRNA expression of the genes for lipogenesis and lipolysis in mouse liver were determined by quantitative real-time PCR (qPCR) (A, *p<0.05, **p<0.01, n=5). Protein expression of FASN, SREBP1 precursor (p) and mature form (m) were determined by Western blot (B). (C and D) Expression of the genes for lipogenesis were detected by qPCR and Western blot as indicated. **p<0.01, n=5. (E and F) Expression of pAMPKα and AMPKα in mouse liver and LO2 cells were detected by Western blot. After transfected with control or NgBR expression vectors, LO2 cells were seeded in plates at ~1×104 cells/well and cultured overnight. Then extracellular acidification rate was monitored over the sequential injection of Rot/AA (0.5 μM) and 2-deoxyglucose (50 mM) (G–J). The OCR was monitored over the sequential injection of oligomycin (1 μM), FCCP (1.5 μM) and Rot/AA (0.5 μM) (K, L). (H–J, L) **p<0.01, n=3. LO2 cells transfected with control or NgBR expression vector were treated with compound C (5 μM) for 24 hours, followed by determination of SREBP1(p), pAMPKα, and AMPKα protein expression by Western blot (M), and NgBR, FASN, and SREBP1 mRNA expression by qPCR (N, **p<0.01, n=5) as indicated. The statistical results of band density analysis were presented in Figure S8 (http://links.lww.com/HC9/A119). Abbreviations: HFD, high-fat diet; ns: not significant.
Activation of AMPK redirects metabolism toward increased catabolism and decreased anabolism, improving lipid homeostasis, glycolysis, and mitochondrial homeostasis.27 We previously demonstrated that hepatic NgBR deficiency induced lipogenesis by inhibiting AMPKα in C57BL/6J mice.20 Reciprocally, in this study, we determined that NgBR overexpression increased phosphorylated AMPKα (pAMPKα, the active form of AMPKα) without effect on AMPKα expression in the liver of HFD-fed ApoE-/- mice, the animals have totally different pathological context from C57BL/6J mice, and LO2 cells (Figures 6E, F, S8C, D, http://links.lww.com/HC9/A119).
To further determine the effect of NgBR on AMPKα activity and the underlying mechanisms, we utilized the Seahorse Extracellular Flux XFp analyzer to determine extracellular acidification rate (the process which can reflect the ability of glycolysis and be activated by AMPKα in LO2 cells. The results in Figure 6G–J clearly show that NgBR overexpression strengthened glycolysis in cells. Subsequently, the results of oxygen consumption rate assay demonstrate the activation of energy metabolism by NgBR-enhanced glycolysis since the basal respiration, maximal respiration, proton leak and ATP production were increased in NgBR overexpressing cells (Figure 6K, L). Similar to in vivo study, associated with AMPKα activation, NgBR overexpression reduced expression of SREBP1 and FASN. However, the inactivation of AMPKα by Compound C substantially attenuated NgBR overexpression-reduced SREBP1 and FASN (Figures 6M, N; S8M, http://links.lww.com/HC9/A119). Thus, these results suggest the anti-hypertriglyceridemia of NgBR overexpression is mainly through reduction of lipogenesis and activation of energy metabolism with involvement of AMPKα activation.
NgBR overexpression activates AMPKα via Ca2+/CaM/CAMKK2 pathway
The precise mechanism by which NgBR influences AMPKα pathway is unclear and should be further investigated. The intracellular Ca2+ can bind to calmodulin (CaM) to form the Ca2+-CaM complex to activate calcium/CaM-dependent protein kinase kinase 2 (CAMKK2), which in turn activates AMPKα by phosphorylating it.27 In contrast, inhibition of mitochondrial Ca2+ uptake can reduce oxidative phosphorylation to increase lipid accumulation by dephosphorylating AMPKα28. To determine the role of Ca2+ signaling pathway in NgBR-activated AMPKα, we initially conducted a flow cytometry assay on the fluorescence of Ca2+ labeled with fluo-4/AM, and observed that NgBR overexpression increased intracellular Ca2+ concentrations (Figure 7A). The increased intracellular Ca2+ concentration in NgBR overexpressing LO2 cells was further confirmed by the experiment determining the rate of Ca2+ intake from the extracellular Ca2+ source. Indeed, NgBR overexpressing LO2 cells took in less extracellular Ca2+ than control cells since in the context of the high intracellular Ca2+ concentration, the intracellular Ca2+ homeostasis limited Ca2+ uptake extracellularly by the cells (Figure 7B, C).
FIGURE 7.
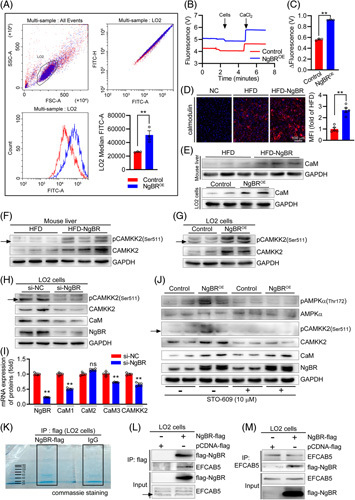
Overexpression of NgBR activates AMPKα by Ca2+/CaM/CAMKK2 pathway. (A) LO2 cells transfected with control or NgBR expression vector were labeled with fluo-4/AM, followed by determination of Ca2+ fluorescence by flow cytometry. **p<0.01, n=3. (B and C) Extracellular Ca2+ concentrations were measured by Oroboros 2K-fluorimetry which reflect the capacity of Ca2+ intake by cells indirectly. Cells: LO2 cell transfected with control or NgBR expression vector; CaCl2: 1 μM. The value of fluorescence change (ΔFluorescence) was calculated before and after addition of CaCl2 when the baseline was stable. The more fluorescence change reflects the higher extracellular Ca2+ concentration, indicating reduced Ca2+ intake by cells. **p<0.01, n=3. (D) CaM expression in mouse liver was detected by immunofluorescent staining of liver sections with anti-CaM antibody. NC: primary antibody was replaced by rabbit normal IgG. **p<0.01, n=5. (E–G) Expression of CaM, pCAMKK2, and CAMKK2 in mouse liver or LO2 cells as indicated were determined by Western blot. (H and I) LO2 cells were transfected with scrambled siRNA (si-NC) or NgBR siRNA (si-NgBR) at 50 nM for 48 hours, followed by determination of pCAMKK2, CAMKK2, CaM, NgBR protein expression by Western blot (H), and CAMKK2, CaM1/2/3, NgBR mRNA expression by quantitative real-time PCR (qPCR) (I). **p<0.01, n=5. (J) The transfected cells were treated with STO-609 (10 μM) as indicated. Protein expression of pAMPKα, AMPKα, pCAMKK2, CAMKK2, CaM, and NgBR were detected by Western blot. (K) Total cellular proteins (500 μg) extracted from pcDNA-flag NgBR-transfected cells were conducted IP with anti-flag antibody (NgBR-flag) and rabbit normal IgG (IgG), respectively. The precipitated proteins were then subjected to SDS-PAGE. After electrophoresis, the gel was stained with Coomassie brilliant blue and photographed. (L and M) Total cellular proteins extracted from pcDNA-flag-transfected (pcDNA-flag) and pcDNA-flag NgBR-transfected (NgBR-flag) cells were conducted IP with anti-flag (L) or anti-EFCAB5 antibody (M), followed by Western blot with anti-flag or anti-NgBR antibody. The statistical results of band density analysis in Western blots were presented in Figure S8 (http://links.lww.com/HC9/A119). Abbreviations: HFD, high-fat diet; ns: not significant.
As a consequence of increased cellular Ca2+ concentration, increased CaM expression was observed in NgBR overexpressing mouse liver and LO2 cells (Figures 7D, E; S6C, D; S8F, http://links.lww.com/HC9/A119), which in turn activated CAMKK2 expression and phosphorylation (pCAMKK2) (Figures 7F, G, S8G, H, http://links.lww.com/HC9/A119). Reciprocally, inhibition of NgBR expression by siRNA decreased the expression of CaM, CAMKK2, and pCAMKK2 in LO2 cells (Figures 7H, S8I, http://links.lww.com/HC9/A119). Furthermore, inhibition of CAMKK2 by a chemical inhibitor, STO-609, had little effect on NgBR-induced CaM expression, but blocked NgBR-activated AMPKα (Figures 7J, S8J, http://links.lww.com/HC9/A119), confirming that the Ca2+/CaM/CAMKK2 pathway is the important mediator for activation of AMPKα by NgBR overexpression.
To further dissect the molecular mechanism by which NgBR regulates Ca2+ signaling, we transfected LO2 cells with pcDNA3.1-3×Flag vector (pcDNA-flag-transfected cells) or pcDNA3.1-3×Flag NgBR expression vector (pcDNA-flag-NgBR-transfected cells). Initially, the total cellular proteins extracted from pcDNA-flag-NgBR-transfected cells were conducted immunoprecipitation (IP) with rabbit normal IgG or anti-flag antibody, followed by SDS-PAGE (Figure 7K) and MS assay to identify the candidate protein that can interact with NgBR and regulate Ca2+ signaling pathway. Indeed, MS assay identified some different proteins between the normal IgG and anti-flag-precipitated samples (Figure S6A, http://links.lww.com/HC9/A119). The further screening assay on these proteins indicated EF-hand calcium binding domain 5-containing protein (EFCAB5, a member of the EFCAB family with involvement of several Ca2+ signaling events 29 is rich in anti-flag-precipitated sample, implying EFCAB5 can interact with flag-NgBR (Figure S6B, http://links.lww.com/HC9/A119).
To further verify the interaction between NgBR and EFCAB5, the total cellular proteins extracted from pcDNA-flag-transfected and pcDNA-flag NgBR-transfected cells were conducted IP with anti-EFCAB5 or anti-flag antibody, followed by Western blot assay with anti-NgBR or anti-EFCAB5 antibody. In anti-flag antibody-precipitated proteins, both flag-NgBR and EFCAB5 were detected in pcDNA-flag NgBR-transfected cells, not in pcDNA-flag-transfected cells (Figure 7L). In anti-EFCAB5-precipitated proteins, flag-NgBR was detected in pcDNA-flag NgBR-transfected cells only (Figure 7M). Thus, these results strongly suggest the interaction between NgBR and EFCAB5, and such interaction may consequently activate Ca2+/CaM/CAMKK2 pathway to reduce lipogenesis and activate energy metabolism, the important mechanisms for amelioration of HFD-induced hypertriglyceridemia by NgBR overexpression.
DISCUSSION
Reduction of cholesterol levels can effectively reduce the risk of cardiovascular diseases. SR-BI profoundly influences cholesterol metabolism and the development of atherosclerosis in animal models.24 Hepatic overexpression of SR-BI reduced atherosclerosis in LDLR-/- mice,30 while inhibition of SR-BI expression enhanced atherosclerosis by increasing LDL-C levels in circulation.31
In this study, we determined that NgBR overexpression increased SR-BI expression and increased HDL uptake, which should enhance reverse cholesterol transport (Figure 4B–H) to ameliorate hypercholesterolemia (Figure 3E–H). In addition, expression of ABCA1 and ABCG5/8 (Figure S4D-F, http://links.lww.com/HC9/A119) were not influenced, indicating the decreased HDL-C is mainly regulated by increased SR-BI. Multiple factors can positively and negatively regulate SR-BI expression, respectively.32 We determined that PXR, the negative regulator, was significantly reduced by NgBR overexpression (Figure S4A, http://links.lww.com/HC9/A119). Interestingly, studies have shown that activation of AMPKα inhibited PXR expression.33,34 Thus, NgBR overexpression may inhibit PXR, thereby increasing the expression of SR-BI through AMPKα activation. Moreover, we found that NgBR overexpression increased CYP7A1 which can promote bile acid synthesis to improve hypercholesteremia (Figure 4B). Studies have shown that activation of AMPK inhibits the transcriptional activity of FXR and downregulates SHP.35,36 Thus, the changes we observed in the FXR-SHP-CYP7A1 pathway may be attributed to activation of AMPK by NgBR overexpression. Taken together, we determined the mechanism by which NgBR regulates AMPKα activation and the NgBR overexpression-activated SR-BI and bile acid synthesis pathways may be regulated by AMPKα activation.
Cholesterol synthesis is another important factor influencing cholesterol levels. Reduced cellular cholesterol levels enhances SCAP-mediated SREBP2 maturation to trigger HMGCR expression and cholesterol synthesis.37 Our results suggest that overexpression of NgBR inhibits SCAP expression, thereby inhibiting SREBP2 maturation (Figure 5G, H) to reduce cholesterol synthesis. NgBR has been reported to stabilize Niemann Pick C2 protein to facilitate the translocation of cholesterol out of the lysosome, from where it is eventually released into the ER for processing and utilization.14,38–41 At the same time, SCAP is localized to the ER and can respond to cholesterol levels. Therefore, NgBR overexpression may increase the delivery of lysosomal-derived cholesterol to the ER, thereby inhibiting SCAP expression in a feedback manner.
Reduced LDLR expression (Figure 4B, C), another SREBP2 target, further confirms the effect of NgBR overexpression on SREBP2 maturation/function and cholesterol synthesis. Activation of LDLR expression can enhance LDL-C clearance in the liver, which reduces circulating LDL-C levels and benefits atherosclerosis. However, overexpression of NgBR simultaneously reduced LDLR expression (Figure 4B, C, F, G) and serum TC, VLDL-C, and LDL-C levels (Figure 3E, H). The contradictory effects of NgBR overexpression on LDLR expression and circulating LDL-C levels indicate that the contribution of reduced LDLR expression to circulating LDL-C levels would be overwhelmed by that of NgBR-inhibited expression of molecules for cholesterol synthesis. Therefore, reduction of cholesterol synthesis should be another important mechanism for NgBR-ameliorated hypercholesterolemia.
Studies have demonstrated that ectopic fat deposition in the liver can make substantial contributions to atherosclerosis.5 Recent genetic studies have revealed an important relationship between plasma TG levels and the risk of atherosclerotic cardiovascular disease.42 Bastian Ramms et al.43 found a significant improvement in atherosclerosis after lowering plasma TG by administration of an anti-oligonucleotide of ApoC-III. In our study, we determined that serum and liver TG and FFA levels were decreased by NgBR overexpression in pro-atherogenic mice (Figure 3C, D; I, J). Combining the results in our previous study,20 we believed that NgBR overexpression reduces transcription of fatty acid synthesis genes by inactivating LXRα. Indeed, overexpression of NgBR reduced nuclear LXRα but not LXRβ levels (Figure S5, http://links.lww.com/HC9/A119). Since FFAs lipolyzed from TG-rich lipoproteins (mainly VLDL) can directly activate a variety of proinflammatory, proapoptotic, and procoagulant pathways to aggravate the development of atherosclerotic cardiovascular disease,44,45 the reduced levels of inflammatory cytokines in both atherosclerotic plaques and serum (Figure 3K–N) should be attributed to reduced TG, VLDL, and FFA levels by NgBR overexpression (Figure 3C, D, F, I, J).
Activation of AMPKα plays an important role in lipid metabolism.27 In this study, we found overexpression of NgBR increased pAMPKα (Figure 6E, F) and glycolysis (Figure 6G–J), which further supports that AMPKα activation is another important mechanism for anti-hypertriglyceridemia by NgBR. Furthermore, we unveiled that NgBR overexpression activates AMPKα through the regulation of Ca2+ signaling pathway, in which NgBR overexpression increased intracellular Ca2+ concentrations (Figure 7A, B). Meanwhile, we found that NgBR overexpression promoted cellular respiratory capacity and expression of the genes related to mitochondrial fusion and division, indicating NgBR overexpression enhanced mitochondrial dynamics (Figure S6E, F, http://links.lww.com/HC9/A119) to increase mitochondrial abundance.46 In addition, it has been reported that increased cytosolic Ca2+ can promote ATP production and mitochondrial abundance,47,48 suggesting that the increased mitochondrial abundance, extracellular acidification rate and oxygen consumption rate may be also associated with increased Ca2+ concentrations.
More importantly, the Ca2+/CaM/CAMKK2/AMPKα pathway plays an important role in lipogenesis. For instance, hepatocytes isolated from CAMKK2-/- mice have increased de novo lipogenesis.49 S100 calcium-binding protein A16 competes with CAMKK2 to bind CaM, thereby inactivating Ca2+/CaM/CAMKK2/AMPKα and enhancing hepatic lipogenesis.50 Reciprocally, in this study, we demonstrated that associated with the activation of Ca2+/CaM/CAMKK2/AMPKα pathway (Figure 7), NgBR overexpression reduced lipogenesis. In addition, we demonstrated that increase of Ca2+ concentration by NgBR overexpression is related to the interaction between NgBR and EFCAB5 (Figures 7, S6, http://links.lww.com/HC9/A119), a protein closely associated with RYRs regulating intracellular Ca2+ release/functions.
CONCLUSIONS
Our study demonstrates that NgBR overexpression plays an important role in the regulation of lipid homeostasis. It potently ameliorates hypercholesterolemia by activating the SR-BI pathway to enhance cholesterol metabolism and inactivating the SREBP2 pathway to reduce cholesterol synthesis. Meanwhile, NgBR overexpression activates AMPKα via Ca2+/CaM/CAMKK2 pathway through interaction with EFCAB5 to inhibit lipogenesis and activate energy metabolism, thereby reducing hypertriglyceridemia and FFA-induced inflammation. These functions together result in that NgBR overexpression inhibits HFD-induced atherosclerosis in ApoE-/- mice, suggesting NgBR might be a potential target for atherosclerosis treatment.
Supplementary Material
AUTHOR CONTRIBUTIONS
Ke Gong, Yajun Duan, Jihong Han, Wenquan Hu, Qing R Miao, and Yasuko Iwakiri designed the study, drafted and edited the manuscript. Ke Gong and Mengyao Wang performed most of the experiments. Dandan Wang, Yongyao Gao, Likun Ma, Xiaoxiao Yang, Xinran Zhu, Shasha Chen, Huaxin Li, Mengxue Zhang, Yuanli Chen, and Chenzhong Liao assisted with the experimental operation or data collection.
FUNDING INFORMATION
This study was supported by the China NSFC grants 82173807 to Yajun Duan, 81973316 to Jihong Han, and 81870192 to Likun Ma; Tianjin Municipal Science and Technology Commission of China Grant 20JCZDJC00710 and the Fundamental Research Funds for the Central Universities (Nankai University) 63211045 to Jihong Han.
CONFLIT OF INTEREST
The authors have no conflicts to report.
DATA AVAILABILITY STATEMENT
Data will be made available on reasonable request.
Footnotes
Abbreviations: AAV, adeno-associated virus; ACC, acetyl-CoA carboxylase; AMPKα, AMP-activated protein kinase α; ApoE-/-, apolipoprotein E deficiency; CaM, calmodulin; ABCG1/5/8, ATP-banding cassette G1 or G5 or G8; CAMKK2, calcium/calmodulin-dependent protein kinase kinase 2; IP, immunoprecipitation; CHO, total cholesterol; DHCR24, 24-dehydrocholesterol reductase; EFCAB5, EF-hand calcium binding domain-containing protein 5; ER, endoplasmic reticulum; FASN, fatty acid synthase; FFA, free fatty acid; H&E, hematoxylin-eosin; HDL-C, high-density lipoprotein cholesterol; HFD, high-fat diet; HMGCR, 3-hydroxy-3-methylglutaryl-coenzyme A reductase; LDL-C, LDL cholesterol; LDLR, LDL receptor; LXR, liver X receptor; NgBR, Nogo-B receptor; Nogo-B, reticulon 4B; pAMPKα, phosphorylated AMPKα; pCAMKK2, phosphorylated CAMKK2; PXR, pregnane X receptor; qPCR, quantitative real-time PCR; SCAP, SREBP cleavage activating protein; SR-BI, scavenger receptor class B type I; SREBP1/2, sterol regulatory element-binding protein 1 or 2; TG, triglyceride; VLDL-C, VLDL cholesterol.
Ke Gong and Mengyao Wang made equal contributions to this work.
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website, www.hepcommjournal.com
Contributor Information
Ke Gong, Email: 18326051470@163.com.
Mengyao Wang, Email: 965488614@qq.com.
Dandan Wang, Email: 18256505275@163.com.
Likun Ma, Email: lkma@ustc.edu.cn.
Xiaoxiao Yang, Email: yangxiaoxiao@hfut.edu.cn.
Xinran Zhu, Email: 17805599656@163.com.
Shasha Chen, Email: 18209685258@163.com.
Mengxue Zhang, Email: 1511857236@qq.com.
Huaxin Li, Email: 1594159323@qq.com.
Yuanli Chen, Email: chenyuanli@hfut.edu.cn.
Wenquan Hu, Email: huwenquan2002@163.com.
Qing R. Miao, Email: Qing.Miao@nyulangone.org.
Yasuko Iwakiri, Email: yasuko.iwakiri@yale.edu.
Chenzhong Liao, Email: czliao@hfut.edu.cn.
Yajun Duan, Email: yduan@hfut.edu.cn.
Jihong Han, Email: jihonghan2008@nankai.edu.cn.
REFERENCES
- 1.Nadruz W, Jr, Goncalves A, Claggett B, Querejeta Roca G, Shah AM, Cheng S, et al. Influence of cigarette smoking on cardiac biomarkers: the Atherosclerosis Risk in Communities (ARIC) Study. Eur J Heart Fail. 2016;18:629–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gimbrone MA, Jr, Garcia-Cardena G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118:620–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Catapano AL, Pirillo A, Norata GD. Vascular inflammation and low-density lipoproteins: is cholesterol the link? A lesson from the clinical trials. Br J Pharmacol. 2017;174:3973–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rozman D, Monostory K. Perspectives of the non-statin hypolipidemic agents. Pharmacol Ther. 2010;127:19–40. [DOI] [PubMed] [Google Scholar]
- 5.Kapuria D, Takyar VK, Etzion O, Surana P, O'Keefe JH, Koh C. Association of hepatic steatosis with subclinical atherosclerosis: systematic review and meta-analysis. Hepatol Commun. 2018;2:873–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldberg IJ. 2017 George Lyman Duff Memorial Lecture: fat in the blood, fat in the artery, fat in the heart: triglyceride in physiology and disease. Arterioscler Thromb Vasc Biol. 2018;38:700–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stols-Goncalves D, Hovingh GK, Nieuwdorp M, Holleboom AG. NAFLD and atherosclerosis: two sides of the same dysmetabolic coin? Trends Endocrinol Metab. 2019;30:891–902. [DOI] [PubMed] [Google Scholar]
- 8.Chen Y, Duan Y, Yang X, Sun L, Liu M, Wang Q, et al. Inhibition of ERK1/2 and activation of LXR synergistically reduce atherosclerotic lesions in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2015;35:948–59. [DOI] [PubMed] [Google Scholar]
- 9.Liu Y, Wei Z, Ma X, Yang X, Chen Y, Sun L, et al. 25-Hydroxycholesterol activates the expression of cholesterol 25-hydroxylase in an LXR-dependent mechanism. J Lipid Res. 2018;59:439–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma C, Feng K, Yang X, Yang Z, Wang Z, Shang Y, et al. Targeting macrophage liver X receptors by hydrogel-encapsulated T0901317 reduces atherosclerosis without effect on hepatic lipogenesis. Br J Pharmacol. 2021;178:1620–38. [DOI] [PubMed] [Google Scholar]
- 11.Miao RQ, Gao Y, Harrison KD, Prendergast J, Acevedo LM, Yu J, et al. Identification of a receptor necessary for Nogo-B stimulated chemotaxis and morphogenesis of endothelial cells. Proc Natl Acad Sci U S A. 2006;103:10997–11002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edwards JK. Hypertension: Nogo-B regulates BP homeostasis. Nat Rev Nephrol. 2015;11:631. [DOI] [PubMed] [Google Scholar]
- 13.Cantalupo A, Zhang Y, Kothiya M, Galvani S, Obinata H, Bucci M, et al. Nogo-B regulates endothelial sphingolipid homeostasis to control vascular function and blood pressure. Nat Med. 2015;21:1028–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harrison KD, Miao RQ, Fernandez-Hernando C, Suarez Y, Davalos A, Sessa WC. Nogo-B receptor stabilizes Niemann-Pick type C2 protein and regulates intracellular cholesterol trafficking. Cell Metab. 2009;10:208–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Y, Hu W, Li Q, Zhao S, Zhao D, Zhang S, et al. NGBR is required to ameliorate type 2 diabetes in mice by enhancing insulin sensitivity. J Biol Chem. 2021;296:100624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poch D, Mandel J. Pulmonary hypertension. Ann Intern Med. 2021;174:ITC49–ITC64. [DOI] [PubMed] [Google Scholar]
- 17.Yang YD, Li MM, Xu G, Feng L, Zhang EL, Chen J, et al. Nogo-B receptor directs mitochondria-associated membranes to regulate vascular smooth muscle cell proliferation. Int J Mol Sci. 2019;20:2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tadokoro KS, Rana U, Jing X, Konduri GG, Miao QR, Teng RJ. Nogo-B receptor modulates pulmonary artery smooth muscle cell function in developing lungs. Am J Respir Cell Mol Biol. 2016;54:892–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teng RJ, Rana U, Afolayan AJ, Zhao B, Miao QR, Konduri GG. Nogo-B receptor modulates angiogenesis response of pulmonary artery endothelial cells through eNOS coupling. Am J Respir Cell Mol Biol. 2014;51:169–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu W, Zhang W, Chen Y, Rana U, Teng RJ, Duan Y, et al. Nogo-B receptor deficiency increases liver X receptor alpha nuclear translocation and hepatic lipogenesis through an adenosine monophosphate-activated protein kinase alpha-dependent pathway. Hepatology. 2016;64:1559–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang W, Yang X, Chen Y, Hu W, Liu L, Zhang X, et al. Activation of hepatic Nogo-B receptor expression—a new anti-liver steatosis mechanism of statins. Biochim Biophys Acta Mol Cell Biol Lipids. 2018;1863:177–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu L, Zeng P, Yang X, Duan Y, Zhang W, Ma C, et al. Inhibition of vascular calcification. Arterioscler Thromb Vasc Biol. 2018;38:2382–95. [DOI] [PubMed] [Google Scholar]
- 23.Fishbein MC. The vulnerable and unstable atherosclerotic plaque. Cardiovasc Pathol. 2010;19:6–11. [DOI] [PubMed] [Google Scholar]
- 24.Linton MF, Tao H, Linton EF, Yancey PG. SR-BI: a multifunctional receptor in cholesterol homeostasis and atherosclerosis. Trends Endocrinol Metab. 2017;28:461–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sporstol M, Tapia G, Malerod L, Mousavi SA, Berg T. Pregnane X receptor-agonists down-regulate hepatic ATP-binding cassette transporter A1 and scavenger receptor class B type I. Biochem Biophys Res Commun. 2005;331:1533–41. [DOI] [PubMed] [Google Scholar]
- 26.Xu D, Wang Z, Xia Y, Shao F, Xia W, Wei Y, et al. The gluconeogenic enzyme PCK1 phosphorylates INSIG1/2 for lipogenesis. Nature. 2020;580:530–55. [DOI] [PubMed] [Google Scholar]
- 27.Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018;19:121–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tomar D, Jana F, Dong Z, Quinn WJ, III, Jadiya P, Breves SL, et al. Blockade of MCU-mediated Ca(2+) uptake perturbs lipid metabolism via PP4-dependent AMPK dephosphorylation. Cell Rep. 2019;26:3709–25 e3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chazin WJ. Relating form and function of EF-hand calcium binding proteins. Acc Chem Res. 2011;44:171–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kozarsky KF, Donahee MH, Glick JM, Krieger M, Rader DJ. Gene transfer and hepatic overexpression of the HDL receptor SR-BI reduces atherosclerosis in the cholesterol-fed LDL receptor-deficient mouse. Arterioscler Thromb Vasc Biol. 2000;20:721–77. [DOI] [PubMed] [Google Scholar]
- 31.Huszar D, Varban ML, Rinninger F, Feeley R, Arai T, Fairchild-Huntress V, et al. Increased LDL cholesterol and atherosclerosis in LDL receptor-deficient mice with attenuated expression of scavenger receptor B1. Arterioscler Thromb Vasc Biol. 2000;20:1068–73. [DOI] [PubMed] [Google Scholar]
- 32.Shen WJ, Azhar S, Kraemer FB. SR-B1: a unique multifunctional receptor for cholesterol influx and efflux. Annu Rev Physiol. 2018;80:95–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oladimeji PO, Lin W, Brewer CT, Chen T. Glucose-dependent regulation of pregnane X receptor is modulated by AMP-activated protein kinase. Sci Rep. 2017;7:46751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krausova L, Stejskalova L, Wang H, Vrzal R, Dvorak Z, Mani S, et al. Metformin suppresses pregnane X receptor (PXR)-regulated transactivation of CYP3A4 gene. Biochem Pharmacol. 2011;82:1771–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lien F, Berthier A, Bouchaert E, Gheeraert C, Alexandre J, Porez G, et al. Metformin interferes with bile acid homeostasis through AMPK-FXR crosstalk. J Clin Invest. 2014;124:1037–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhai Z, Niu KM, Liu H, Lin C, Tu Y, Liu Y, et al. Policosanol alleviates hepatic lipid accumulation by regulating bile acids metabolism in C57BL6/mice through AMPK-FXR-TGR5 cross-talk. J Food Sci. 2021;86:5466–78. [DOI] [PubMed] [Google Scholar]
- 37.Ouyang N, Gan H, He Q, Lei H, Wang SY, Liu Q, et al. Dysfunction of cholesterol sensor SCAP promotes inflammation activation in THP-1 macrophages. Exp Cell Res. 2018;367:162–9. [DOI] [PubMed] [Google Scholar]
- 38.Yu SH, Wang T, Wiggins K, Louie RJ, Merino EF, Skinner C, et al. Lysosomal cholesterol accumulation contributes to the movement phenotypes associated with NUS1 haploinsufficiency. Genet Med. 2021;23:1305–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xue J, Zhu Y, Wei L, Huang H, Li G, Huang W, et al. Loss of Drosophila NUS1 results in cholesterol accumulation and Parkinson’s disease-related neurodegeneration. FASEB J. 2022;36:e22411. [DOI] [PubMed] [Google Scholar]
- 40.Chang TY, Chang CC, Ohgami N, Yamauchi Y. Cholesterol sensing, trafficking, and esterification. Annu Rev Cell Dev Biol. 2006;22:129–57. [DOI] [PubMed] [Google Scholar]
- 41.Meng Y, Heybrock S, Neculai D, Saftig P. Cholesterol handling in lysosomes and beyond. Trends Cell Biol. 2020;30:452–66. [DOI] [PubMed] [Google Scholar]
- 42.Boren J, Taskinen MR, Bjornson E, Packard CJ. Metabolism of triglyceride-rich lipoproteins in health and dyslipidaemia. Nat Rev Cardiol. 2022;19:577–92. [DOI] [PubMed] [Google Scholar]
- 43.Ramms B, Patel S, Sun X, Pessentheiner AR, Ducasa GM, Mullick AE, et al. Interventional hepatic apoC-III knockdown improves atherosclerotic plaque stability and remodeling by triglyceride lowering. JCI Insight. 2022;7:e15414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Basu D, Bornfeldt KE. Hypertriglyceridemia and atherosclerosis: using human research to guide mechanistic studies in animal models. Front Endocrinol (Lausanne). 2020;11:504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Toth PP. Triglyceride-rich lipoproteins as a causal factor for cardiovascular disease. Vasc Health Risk Manag. 2016;12:171–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qu Z, Liu A, Liu C, Tang Q, Zhan L, Xiao W, et al. Theaflavin promotes mitochondrial abundance and glucose absorption in myotubes by activating the CaMKK2-AMPK signal axis via calcium-ion influx. J Agric Food Chem. 2021;69:8144–59. [DOI] [PubMed] [Google Scholar]
- 48.Di Benedetto G, Scalzotto E, Mongillo M, Pozzan T. Mitochondrial Ca(2)(+) uptake induces cyclic AMP generation in the matrix and modulates organelle ATP levels. Cell Metab. 2013;17:965–75. [DOI] [PubMed] [Google Scholar]
- 49.Racioppi L, Means AR. Calcium/calmodulin-dependent protein kinase kinase 2: roles in signaling and pathophysiology. J Biol Chem. 2012;287:31658–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kan J, Zhao C, Lu S, Shen G, Yang J, Tong P, et al. S100A16, a novel lipogenesis promoting factor in livers of mice and hepatocytes in vitro. J Cell Physiol. 2019;234:21395–406. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available on reasonable request.