

Abstract
Patients with autosomal recessive (AR) IL-12p40 or IL-12Rβ1 deficiency display Mendelian susceptibility to mycobacterial disease (MSMD) due to impaired IFN-γ production and, less commonly, chronic mucocutaneous candidiasis (CMC) due to impaired IL-17A/F production. We report six patients from four kindreds with AR IL-23R deficiency. These patients are homozygous for one of four different loss-of-function IL23R variants. All six patients have a history of MSMD but only two suffered from CMC. We show that IL-23 induces IL-17A only in MAIT cells, possibly contributing to the incomplete penetrance of CMC in patients unresponsive to IL-23. By contrast, IL-23 is required for both baseline and Mycobacterium-inducible IFN-γ immunity in both Vδ2+ γδ T and MAIT cells, probably contributing to the higher penetrance of MSMD in these patients. Human IL-23 appears to contribute to IL-17A/F-dependent immunity to Candida in a single lymphocyte subset, but is required for IFN-γ-dependent immunity to Mycobacterium in at least two lymphocyte subsets.
One-Sentence Summary:
IL-23 signaling is required for baseline and IL-23-inducible IFN-γ immunity in both Vδ2+ γδ T and MAIT cells.
INTRODUCTION
Life-threatening disease during primary infection in otherwise healthy individuals can result from monogenic inborn errors of immunity (IEI) (1, 2). Studies of such IEIs can shed light on the essential and redundant roles of the corresponding human genes in host defense in natura, while clarifying mechanisms of disease (3–6). Mendelian susceptibility to mycobacterial disease (MSMD) is characterized by selective susceptibility to clinical disease caused by weakly virulent mycobacteria, such as Mycobacterium bovis-Bacillus Calmette-Guérin (BCG) vaccine and environmental mycobacteria (7). Patients with MSMD are also prone to bona fide tuberculosis (TB), and to severe disease caused by Salmonella and, more rarely, other intra-macrophagic bacteria, fungi, and parasites (8). The genetic dissection of MSMD has led to the discovery of 37 genetic disorders, involving 19 genes, with allelic heterogeneity. All but one of these genetic disorders clearly affect interferon-γ (IFN-γ)-mediated immunity, the exception being ZNFX1 deficiency, the pathogenic mechanism of which remains unclear (7, 9–14).
The two most common etiologies of MSMD are autosomal recessive (AR) complete IL-12Rβ1 and IL-12p40 deficiencies. Since 1998, these deficiencies have been reported in more than 400 and 100 patients, respectively (15–24). AR complete IL-12p40 deficiency is a clinical phenocopy of IL-12Rβ1 deficiency (19, 24–27). IL-12p40 dimerizes with IL-12p35 to form IL-12, or with IL-23p19 to form IL-23 (28, 29). IL-12Rβ1 dimerizes with IL-12Rβ2 to form the IL-12 receptor, or with IL-23R to form the IL-23 receptor (30, 31). Consequently, both IL-12- and IL-23-mediated immunity are abolished in patients with IL-12Rβ1 or IL-12p40 deficiency (11). Patients with IL-12Rβ1 or IL-12p40 deficiency display incomplete penetrance for MSMD, with about a third of adult patients remaining MSMD-free; the penetrance for TB in endemic areas is probably higher, as Mycobacterium tuberculosis is at least 1,000 times more virulent than BCG and environmental mycobacteria (7, 8, 18, 19, 22, 32–34).
About 25% of patients with IL-12Rβ1 or IL-12p40 deficiency also suffer from chronic mucocutaneous candidiasis (CMC) (8, 18, 19, 35, 36). CMC is characterized by recurrent lesions of the skin, nails, and oral and genital mucosae caused by Candida spp. (36). This condition is not observed in patients with most other genetic etiologies of MSMD, including those in whom IFN-γ-mediated immunity is completely abolished (7, 37–40). The occurrence of isolated CMC in patients with autosomal dominant (AD) IL-17F deficiency or inborn errors of the IL-17-responsive pathway, such as AR IL-17RA, IL-17RC, and ACT1, and AD JNK1 deficiencies, revealed the essential role of IL-17A/F in mediating mucocutaneous immunity to Candida albicans (C. albicans) (41–45). IL-12Rβ1-deficient patients have low counts of circulating T helper 17 (TH17) cells, which has been attributed to the abolition of IL-23-dependent IL-17 cytokine induction in these patients (46). Likewise, patients with RORγT deficiency display impaired IFN-γ and IL-17A/F production and suffer from both MSMD and CMC (47).
In 2018, we reported AR IL-12Rb2 deficiency in three individuals from the same family: one with MSMD, another with TB, and an asymptomatic individual (11). More surprisingly, we reported AR IL-23R deficiency in two siblings with MSMD but not CMC (11). Together with the report that homozygosity for the common P1104A TYK2 allele selectively impairs cellular responses to IL-23 and underlies TB and MSMD with high and low penetrance, respectively (9, 12, 48), these findings suggested that human IL-23 is required for optimal IFN-γ-dependent immunity to mycobacteria, but redundant for optimal IL-17-dependent immunity to C. albicans (11). This finding is intriguing in two ways, as the TH1/TH17 dichotomy had long revolved around the idea that IL-12 is a signature inducer of IFN-γ, whereas IL-23 is a signature inducer of IL-17A/F (49–52). We investigated the role of human IL-23 in host defense further, by searching for new patients with AR IL-23R deficiency in a cohort of >15,000 patients with diverse infectious diseases.
RESULTS
Six patients from four kindreds homozygous for private IL23R variants
We searched for non-synonymous, essential splice site, or copy number variants of IL23R in a cohort of >15,000 patients with highly diverse infectious diseases, including 1,618 patients with MSMD, 1,454 with TB, and 730 with CMC. The cohorts overlap due to the occurrence of syndromic forms of infection. We selected rare variants (MAF<0.01) that were homozygous or potentially compound heterozygous. Six patients from four kindreds were homozygous for private IL23R variants (NM_144701.2) (Figure 1A). P1 and P2 from Kindred A, reported in previous studies (11, 22), are homozygous for the c.344G>A missense variant, which results in the p.C115Y substitution in a key domain of IL-23R (Figure 1B) (11, 53). P3, from Kindred B, is homozygous for the c.367+1G>A variant, which is predicted to alter the canonical donor splice site of exon 3 (Figure 1A and 1B). P4 from Kindred C, is homozygous for the c.1149–1G>A variant, which is predicted to alter the canonical acceptor splice site of exon 10 (Figure 1A and 1B). Finally, P5 and P6, from Kindred D, are homozygous for the nonsense c.805G>T variant (p.E269*) upstream from the segment encoding the transmembrane domain of IL-23R (Figure 1A and 1B) (53). All heterozygous or homozygous wild-type (WT) relatives of these patients were healthy (Figure 1A). The four IL23R variants found in kindreds A-D are not reported in any individuals of the Genome Aggregation Database (gnomAD) v2.1.1 (https://gnomad.broadinstitute.org/), ATAVDB (http://atavdb.org), Great Middle East Database (http://igm.ucsd.edu/gme), the Iranome database (http://www.iranome.com/), or in any other individuals from our in-house database (>15,000 individuals). Finally, these four IL23R variants are proven (p.C115Y) or predicted to be loss-of-function (pLOF) (c.367+1G>A, c.1149–1G>A, p.E269*), with combined annotation-dependent depletion (CADD) scores well above the mutation significance cutoff (MSC) of IL23R (Figure 1C) (54–56). These data suggest that these six patients have AR IL-23R deficiency.
Figure 1: Six patients from four kindreds with private homozygous IL23R variants.

(A) Pedigrees of the four kindreds studied here. The IL23 variants are indicated below the kindred name. Solid black symbols indicate patients with MSMD, and black diagonal stripes indicate CMC. Symbols linked with a double line indicate consanguinity. The genotype is indicated under each symbol, with M corresponding to the variant found in each kindred, and WT indicating wild-type. Arrows indicate the index case in each family. (B) Schematic representation of the WT IL-23R gene/protein, the colorless area represents the extracellular domain, the black area the transmembrane domain and the gray area the intracellular domain of the protein. The positions of the variants found in the patients are indicated by arrows. (C) All homozygous variants found in gnomAD V2.1.1 (in black) for IL23R are plotted according to their combined annotation-dependent depletion (CADD score; y axis) and minor allele frequency (MAF; x axis). The dashed line indicates the mutation significance cutoff (MSC) for IL23R. (D) Gene14 level negative selection. IL23R is not under negative selection, like other genes for which mutations underlie AR inborn errors of immunity (IEI), as determined by CoNeS. (E) Schematic representation of the impact of the different variants of the patients (P1-P6) on IL- 23R.
Six patients with MSMD, including two with CMC
The six patients belonged to four unrelated families originating from and living in Iran. Principal component analysis on whole-exome sequencing (WES) data confirmed the Iranian ancestry of P5 and P6, whereas P1 to P4 were closer to the European population (Figure S1A) (57). The high rate of homozygosity in these patients was suggestive of familial consanguinity (Figure S1B) (58), and their kinship coefficients suggested that the four kindreds were not related to each other (59). P1 (Kindred A), born in 1994 to consanguineous parents, was vaccinated with BCG in early infancy and developed BCG-adenitis (BCG-itis), which persisted for one year before spontaneously resolving (Table S1). Her brother, P2, born in 2006, was also vaccinated with BCG early in infancy, leading to BCG-itis, which progressed to disseminated BCG disease (BCG-osis) despite antibiotic treatment, resulting in the death of this patient at the age of eight years. P3 (Kindred B), born in 2010 to consanguineous parents, was vaccinated with BCG and developed BCG-osis (22). P4 (Kindred C), born in 2008 to consanguineous parents, was vaccinated with BCG and developed BCG-itis, which persisted for three years before spontaneously resolving. P4 has also suffered from oral and esophageal candidiasis, which persisted between the ages of three months and ten years. He was treated with fluconazole, and no recurrence has been observed on prophylaxis. P5 (Kindred D), born in 2015 to consanguineous parents, was vaccinated with BCG and developed axillary BCG adenitis (BCG-itis) at eight months, with spontaneous resolution at 12 months. His brother P6, born in 2019, was vaccinated with BCG and developed recurrent axillary BCG-itis, which resolved at 12 months after abscess drainage and antimycobacterial therapy until the age of 18 months. P6 has also presented one episode of oral candidiasis, which resolved on fluconazole. No infections with other intramacrophagic pathogens, such as environmental mycobacteria and Salmonella, were observed in any of the six patients, despite such infections being common in patients with IL-12Rβ1 and IL-12p40 deficiencies. Clinical penetrance was complete for BCG disease, but incomplete for CMC (two of the six patients) (Table S1). All six patients have been infected with diverse viruses and bacteria (Figure S1C), without severe clinical consequences. WES analysis of the six patients found no other homozygous candidate variants of known MSMD- or CMC-causing genes. Other rare homozygous or hemizygous coding and splice-site (including essential splice site) variants were present, with CADD scores above the corresponding MSC scores, and these variants were not present in the homozygous or hemizygous state in GnomAD v2.1.1. However, none of these variants were related to IFN-γ or IL-17 immunity (Figure S1B and Table S2). Overall, the six patients with a putative diagnosis of AR IL-23R deficiency had an MSMD phenotype, and two also had CMC.
Population genetics of the IL23R locus
No IL23R pLOF variants were found in the homozygous state in the general population. Only 13 in-frame variants were found in the homozygous state; all were missense variants with MAFs ranging from 5.3 × 10−5 to 8.8 × 10−1 (Figure 1C). We reported all of these variants to be neutral in an overexpression assay in a previous study (11). A revised analysis of the population genetics of IL23R showed that the situation had not changed since 2018. The IL23R locus is subject to negative selection, with a consensus-based measure of negative selection (CoNeS) score of −0.08, consistent with an AR IEI (Figure 1D) (60), and an intermediate gene damage index (GDI) of 3.6 (61). Similarly, we found no pLOF variants and only one in-frame variant of IL23A in the homozygous state in the general population. Overall, we found six MSMD patients from four kindreds homozygous for an experimentally proven missense LOF (eLOF) (kindred A) or pLOF (kindreds B, C, D) variant. A significant enrichment in homozygosity for such variants, which were found exclusively in patients with MSMD, was observed in the 802 MSMD patients with no identified genetic etiology relative to >3,000 patients with diverse infectious diseases from our database and the general population (p=5.7 × 10−4 and p=3.5 × 10−9, respectively). The absence of homozygous pLOF variants in the TB cohort, which includes patients without MSMD from countries in which BCG vaccination is mandatory, further suggests that AR IL-23R deficiency displays complete penetrance for BCG disease. Collectively, these findings strongly suggest that the six patients have MSMD (and CMC, for two of the patients) because of AR IL-23 deficiency.
Two mutant alleles of IL23R disrupt mRNA splicing
The c.367+1G>A and c.1149–1G>A IL23R variants affect essential splice sites. We therefore hypothesized that they would produce aberrantly spliced transcripts. We first tested their effect on IL23R mRNA splicing by performing exon trapping. Both c.367+1G>A and c.1149–1G>A IL23R mutant mRNAs were aberrant, as shown by comparison with cells transfected with wild-type (WT) IL23R (Figure S1D and S1E). We then used mRNA extracted from Epstein-Barr virus-immortalized B lymphocytes (EBV-B cells) from a healthy control and P3, and from activated and expanded T cells (T-blasts) from a healthy control and P4, to generate and amplify IL23R cDNAs from exons 2 to 5 (healthy control and P3) or exons 8 to 11 (healthy control and P4). Sanger sequencing of the PCR products followed by TOPO-TA cloning showed a match to the canonical transcript for >90% of cDNAs from the mRNA isolated from healthy control cells, but none of those from the mRNA from patient cells (Figure S1F and S1G). Three transcripts were detected in P3’s EBV-B cells. Two of these transcripts predominated and were predicted to encode a protein with an in-frame partial deletion of the extracellular domain of IL-23R (G45_Y123delinsD, G24_Y123delinsD). The minor transcript was predicted to encode a truncated protein (G45Ffs*16) (Figure 1E). All four transcripts observed in P4 T-blasts were predicted to encode truncated proteins (Figure 1E). Thus, c.367+1 G>A and c.1149–1G>A led to aberrantly spliced and pLOF IL23R mRNAs in P3 and P4, respectively, at least in the cell types tested, with no leakiness. These findings suggest that these patients had AR IL-23R deficiency.
Four loss-of-function IL23R alleles
We then investigated the expression and function of the four mutant alleles. As a surrogate for these alleles, we generated cDNAs corresponding to the variants of P1/P2 (C115Y), and P5/P6 (E269*) and to the transcripts observed in the cells of P3 (G45_Y123delinsD, G24_Y123delinsD and G45Ffs*16) and P4 (I384Kfs*6, I384Lfs*8, D349Gfs*3, and N350*). We used lentivirus-mediated gene transfer to ensure the stable expression of WT or mutant IL23R cDNA in an EBV-B cell line that endogenously expressed STAT3 and IL-12Rβ1, but not IL-23R, and had been engineered to express STAT4 in a stable manner (referred to here as EBV-BSTAT4) (11, 62). Transduction efficiency was assessed by measuring the cell-surface expression of ΔNGFR (CD271, incorporated into the backbone of the lentiviral plasmid) (Figure S2A)(63). EBV-BSTAT4 cells transduced with the WT or mutant IL23R cDNAs produced similar amounts of IL23R mRNA (Figure S2B). However, relative to EBV-BSTAT4 cells transduced with WT IL23R cDNA, EBV-BSTAT4 cells transduced with mutant IL23R cDNA had similar (G24_Y123delinsD or G45_Y123delinsD), weaker (C115Y, I384Kfs*6, I384Lfs*8), or no (G45Ffs*16, N349Gfs*3, N350*, E269*) cell-surface expression of IL-23R (Figure 2A and S2C). We then stimulated EBV-BSTAT4 cells expressing WT or mutant IL-23R proteins with IL-23, or IFN-α2b as a control. Human IL-23-dependent signaling via IL-12Rβ1/IL-23R in these cells preferentially results in STAT3 activation. EBV-BSTAT4 cells expressing WT, but not mutant IL-23R phosphorylated STAT3 in response to IL-23, whereas all cells phosphorylated STAT3 in response to IFN-α2b (Figure 2B and S2D). All the IL23R mutant alleles were, therefore, LOF for responses to IL-23. These findings strongly suggest that the patients had AR complete IL-23R deficiency.
Figure 2: Four loss-of-function IL23R alleles, and six patients with AR complete IL23R Deficiency.
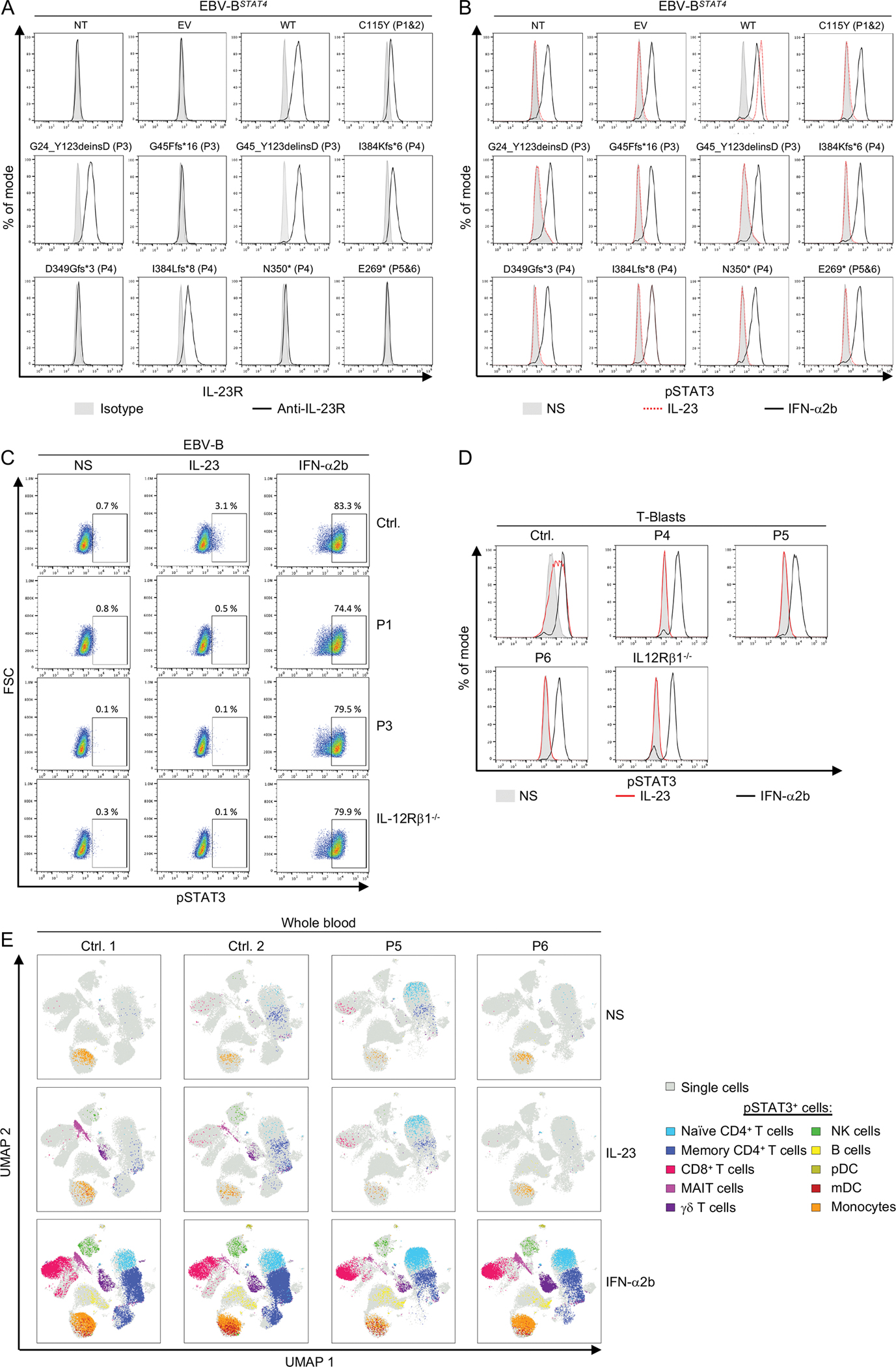
(A-B) EBV-BSTAT4 cells were either left non-transduced (NT) or were transduced with lentiviruses generated with an empty vector (EV) or with vectors containing the WT or the mutated IL23R cDNA. Cell-surface IL-23R expression was assessed by flow cytometry (A). EBV-BSTAT4 cells were left unstimulated (NS), or were stimulated with IL-23 or IFN-α2b as a positive control. STAT3 phosphorylation was assessed by flow cytometry (B). (C) EBV-B cells from a healthy control (Ctrl.), P1, P3 and an IL-12Rβ1-deficient patient were left unstimulated (NS), or were stimulated with IL-23 or IFN-α2b as a positive control. STAT3 phosphorylation was assessed by flow cytometry. The results shown in A-C are representative of three independent experiments. (D) T-blasts from a healthy control (Ctrl.), P4, P5, P6, and an IL-12Rβ1-deficient patient were left unstimulated (NS), or were stimulated with IL-23 or IFN-α2b as a positive control. STAT3 phosphorylation was assessed by flow cytometry. (E) Whole blood from two healthy controls, P5, and P6 was left unstimulated (NS), or was stimulated with IL-23 or IFN-α2b as a positive control. STAT3 phosphorylation in the various cell subsets was assessed by pSTAT CyTOF and the pSTAT3-positive cells were depicted in a uniform manifold approximation and projection (UMAP) visualization.
Six patients with AR complete IL-23R deficiency
Our next set of experiments used patient-derived cells to capture the effects of the patients’ full genotypes at these loci in the context of their own genome. We assessed the cell-surface expression and function of IL-23R in EBV-B cells from P1 and P3. Relative to EBV-B cells from a healthy control or an IL-12Rβ1-deficient patient, cell-surface IL-23R expression was normal in P1 and much weaker in P3 (Figure S2E). These EBV-B cells were then left unstimulated or stimulated with IL-23 or IFN-α2b. EBV-B cells from the healthy control responded to IL-23 by phosphorylating STAT3, whereas EBV-B cells from P1, P3, and an IL-12Rβ1-deficient patient did not. By contrast, all cells phosphorylated STAT3 in response to IFN-α2b (Figure 2C). We then assessed the function of IL-23R in T-blasts. T-blasts from healthy controls (including P4’s sister, who is WT for IL-23R), P4, P5, and P6, and an IL-12Rβ1-deficient patient were left unstimulated, or were stimulated with IL-23 or IFN-α2b. T-blasts from healthy controls responded to IL-23 by phosphorylating STAT3, whereas T-blasts from P4, P5, P6, and the IL12Rβ1-deficient patient did not (Figure 2D and Figure S2F). T-blasts from healthy controls, P4, P5, P6, and an IL-12Rβ1 deficient patient responded to IFN-α2b by phosphorylating STAT3 or STAT4, and to IL-12 by phosphorylating STAT4, with the exception of IL-12Rβ1 deficient T-blasts (Figure 2D, and Figure S2F and S2G). Finally, whole blood from two healthy individuals, including a travel control, P5 and P6, was left unstimulated, or stimulated with IL-23 or IFN-α2b, and STAT phosphorylation was assessed by mass cytometry by time-of-flight (CyTOF) (Figure 2E and Figure S2H). In cells from healthy donors, we observed an induction of STAT3 phosphorylation, and, to a much lesser extent, an induction of STAT5 phosphorylation in response to IL-23, predominantly in mucosal-associated invariant T (MAIT) and γδ T cells, and, to a lesser extent, in natural killer (NK) and memory CD4+ T cells (Figure 2E and Figure S2H). In P5 and P6, no STAT phosphorylation was induced upon IL-23 stimulation in any of the leukocyte subsets tested (Figure 2E and Figure S2H). By contrast, STAT phosphorylation in response to IFN-α2b was similar in cells from P5 and P6 and in the healthy controls (Figure 2E and Figure S2H). The six patients, thus, had a complete form of AR IL-23R deficiency.
Impaired basal IFN-γ immunity ex vivo in inherited IL-23R deficiency
As a first approach to deciphering the cellular basis of MSMD and CMC in patients with inherited IL-23R deficiency, we compared the frequency of leukocyte subsets in the whole blood of healthy controls, five IL-23R-deficient patients, three IL-12Rβ1-deficient patients, and four patients heterozygous for STAT1 gain-of-function (GOF) variants (64), by CyTOF (Figure 3A and Figure S3A), and in peripheral blood mononuclear cells (PBMCs), by spectral flow cytometry (Figure S3B). The counts and frequencies of myeloid cells, NK cells, MAIT cells, γδ T cells, innate lymphoid cells (ILCs), total CD4+ αβ T cells, total CD8+ αβ T cells, and memory TH1, TH2, TH1*, and TH17 cells in IL-23R-deficient patients were within the range of healthy controls (Figure 3A and Figure S3A–S3B). We then analyzed PBMC samples from P3, P4, and P5 by single-cell RNA sequencing (scRNA-seq). We simultaneously analyzed PBMCs from two IL-12Rβ1- and two IL-12Rβ2-deficient patients, to investigate IL-23-dependent but IL-12-independent cellular phenotypes underlying MSMD, and from one STAT1 GOF patient, to investigate IL-23-dependent cellular phenotypes underlying CMC. Batch-corrected unsupervised clustering identified 22 different leukocyte subsets (Figure 3B, S3C and S3D). Pseudobulk principal component analysis (PCA) revealed the transcriptional phenotypes of different leukocyte subsets of IL-23R-, IL-12Rβ1-, and IL-12Rβ2-deficient patients, the patient with a STAT1 GOF variant, and healthy adults and children (Figure S3E). Gene set enrichment analysis (GSEA) revealed a downregulation of genes regulated by IFN-γ in most clusters identified, except for plasma cells, type 1 conventional dendritic cells and plasmacytoid dendritic cells, in IL-23R-deficient patients relative to healthy children (Figure 3C and D). These baseline transcriptional alterations may be associated with an impairment of intercellular communication, as suggested by the probabilistic inference of intercellular communications in CellChat analysis (65) (Figure 3E). Interestingly, GSEA identified the high mobility group box 1 (HMGB1)-regulated geneset as the only transcription factor target geneset jointly downregulated in IL-23R-deficient and STAT1 GOF TH17 cells (Figure 3F), suggesting that a deficiency of HMGB1 activity may underlie impaired TH17-dependent immunity in both IL-23R-deficient and STAT1 GOF patients. Overall, these results suggest that IL-23R deficiency impairs IFN-γ immunity in at least 19 leukocyte subsets in vivo, without affecting their development per se.
Figure 3: Impaired basal IFN-γ signaling in vivo in patients with inherited IL-23R Deficiency.
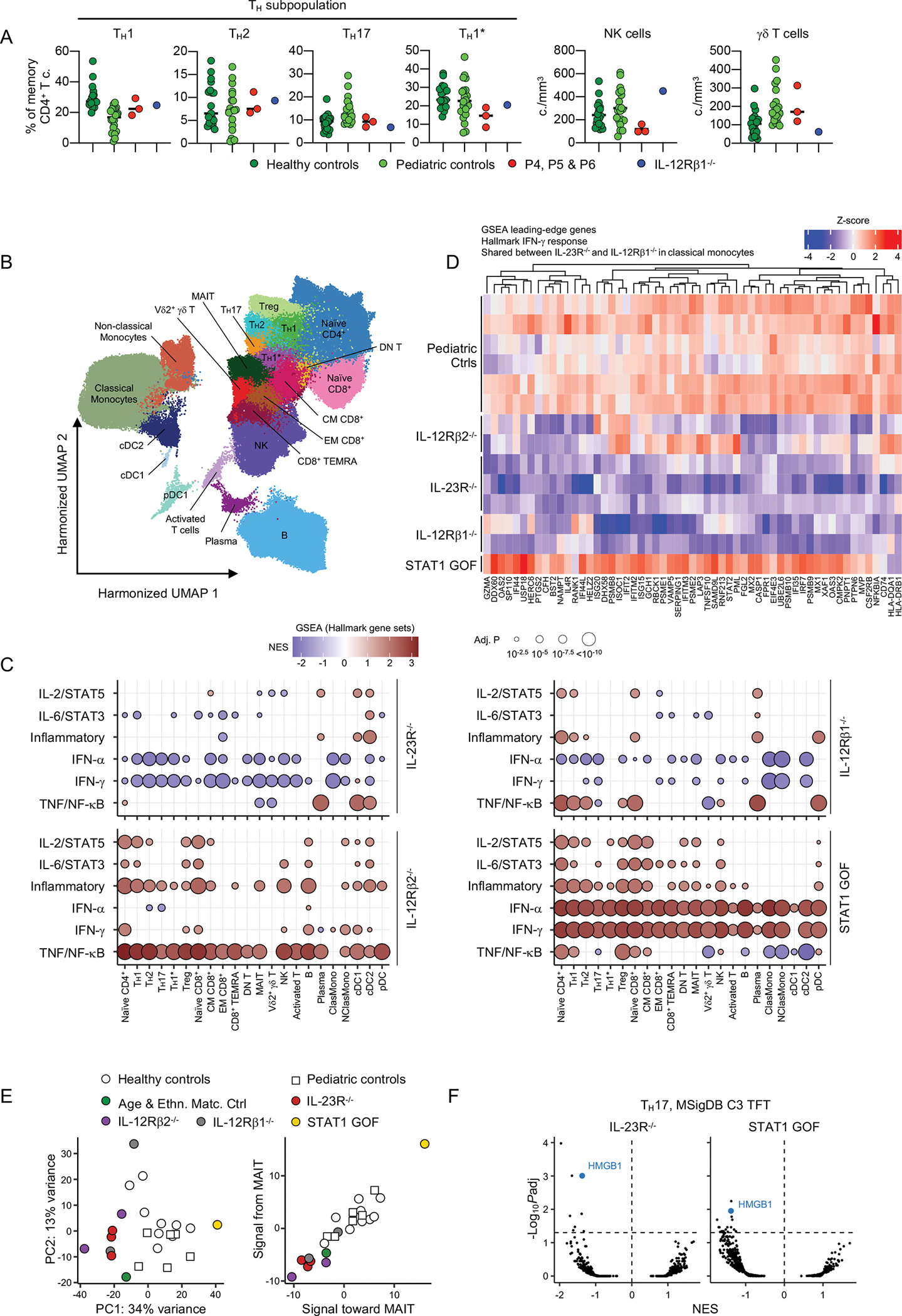
(A) Frequency of memory CD4+ T cells (TH1, TH2, TH17, TH1*) and absolute counts of NK and γδ T cells in 19 healthy adult controls, 20 healthy pediatric controls, and patients (P4, P5 and P6). (B-F) Single-cell transcriptome analysis. Cryopreserved peripheral blood mononuclear cells (PBMCs) from P3, P4 and P6 were analyzed with cryopreserved PBMCs from healthy adult and pediatric controls, two IL-12Rβ1- and two IL-12Rβ2-deficient patients, and one patient heterozygous for a STAT1 gain-of-function (GOF) variant. (B) Clustering analysis. After batch-correction with Harmony (28), clusters were identified manually with the aid of the SingleR pipeline (78), guided by the MonacoImmuneDataset (79). (C) Pseudobulk differential expression analysis. IL-23R-, IL-12Rβ1-, and IL-12Rβ2-deficient and STAT1-GOF patients were compared with healthy controls (adults and children combined). Gene set enrichment analysis (GSEA) was conducted on the fold-change ranking against the Hallmark gene sets (http://www.gseamsigdb.org/gsea/msigdb/genesets.jsp?collection=H). Only immune-related pathways are shown. (D) Differential gene expression in classical monocytes between adult and pediatric control cells and IL-23R-, IL12Rβ1-, and IL-12Rβ2-deficient and STAT1-GOF cells. Colors reflect Z-transformed normalized pseudobulk read counts. (E) Intercellular communication analysis with CellChat (65). Principal component analysis (PCA) of the CellChat-predicted signaling probability of communication between all cell-to-cell pairs and all receptor-ligand pairs (left panel), and PCA on CellChat-predicted probability of signaling to and from MAIT cells (right panel). (F) GSEA was conducted on the fold-change ranking against all transcription factor target gene sets (http://www.gsea-msigdb.org/gsea/msigdb/genesets.jsp?collection=TFT) of IL-23R-deficient and STAT1-GOF TH17 cells. NES, normalized enrichment score.
IL-23 induces IFNG in NK, MAIT and Vδ2+ γδ T cells
We delineated IL-23-dependent cellular responses further by analyzing PBMCs from two IL-23R-deficient (P4 and P6) patients, one IL-12Rβ1-deficient patient, one STAT1 GOF patient, and six healthy individuals (including P4’s sister), by scRNA-seq, after incubation with or without IL-23 for six hours. Clustering analysis identified 18 leukocyte subsets (Figure 4A and S4A). GSEA identified IFN-γ-regulated genes with IL-23-driven induction in healthy controls, the expression of which was weaker in several leukocyte subsets in IL-23R- and IL-12Rβ1-deficient patients, including NK cells, Vδ2+ γδ T cells, monocytes, and myeloid dendritic cells (mDC). (Figure 4B and S4B). This observation suggested that IL-23 stimulation directly induces IFNG expression. Indeed, upon IL-23 stimulation, we observed an increase in the percentage of NK, MAIT, and Vδ2+ γδ T cells expressing IFNG in healthy controls and the STAT1 GOF patient, but not in IL-23R- or IL-12Rβ1-deficient patients (Figure 4C). In comparisons of IL-23R- or IL-12Rβ1-deficient patients with healthy controls, IFNG was the gene for which IL-23-driven induction was most strikingly impaired in Vδ2+ γδ T, MAIT, and NK cells (Figure 4D). By contrast, no induction of IL17A or IL17F was detected in IL-23-stimulated cells from healthy controls. Thus, human IL-23 acts predominantly as an early IFN-γ-inducing cytokine in innate-like adaptive (Vδ2+ γδ T, MAIT) and innate (NK) lymphocytes, which, in turn, induce IFN-γ-dependent transcriptional programs in both lymphoid and myeloid leukocyte subsets.
Figure 4: Impaired ex vivo IL-23-mediated production of IFN-γ in patients with inherited IL-23R deficiency.

(A-D) Single-cell RNA sequencing. We analyzed PBMCs from two IL-23R-deficient (P4 and P6), one IL-12RbR1-deficient patient, and one STAT1-GOF patient, together with six healthy controls (including P4’s sister). Cells were either left unstimulated or were stimulated with IL-23 for 6 hours. (A) Clustering analysis. (B) Pseudobulk differential expression (DE) analysis. Log2 fold-changes in expression were estimated with DESeq2 for the interaction between stimulation (non-stim. vs. IL-23) and genotype (WT vs. IL-23R-deficient, IL-12Rβ1-deficient or STAT1 GOF). GSEA for the Hallmark gene sets was performed on the basis of log2 fold-change ranking. Gene sets with FDR-adjusted P values below 0.05 for at least one cell subset are shown. (C) Percentage of cells expressing IFNG among each cell subset, without (left panel), or with (right panel) IL-23 stimulation. (D) Two-dimensional plot, log2 fold-change (FC) for the interaction between IL-23R deficiency and stimulation (x axis) and the interaction between IL-12Rβ1 deficiency and stimulation (y axis) in TH1, TH2/TH17, TH1*, CD8 EM, MAIT, Vδ2+ γδ T, and NK cells (red, IFNG. blue, TNF). (E) Percentages of IFN-γ+ cells, on intracellular flow cytometry, for the cell subsets indicated, following stimulation with and without live M. bovis-BCG in the presence and absence of IL-12 and IL-23, or PMA/ionomycin (P/I). Nonparametric Mann-Whitney tests were used for analysis (E), with *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Impaired ex vivo IL-23-mediated induction of IFN-γ in inherited IL-23R deficiency
Human IFN-γ is essential for antimycobacterial immunity, as at least 36 of the 37 known genetic etiologies of MSMD affect the production of, or response to IFN-γ. Given the IL-23-dependent induction of IFNG at mRNA level observed on scRNA-seq, we performed ex vivo experiments to test the hypothesis that inherited IL-23R deficiency impairs IFN-γ production in response to mycobacteria. We used spectral flow cytometry to assess intracellular IFN-γ production by lymphocyte subsets. Following IL-23 stimulation, the frequency of IFN-γ-expressing NK, MAIT, Vδ2+ γδ T, and to a lesser extent CD4+ T cells in healthy controls increased relative to unstimulated cells, whereas no induction of intracellular IFN-γ was observed in IL-23R-deficient cells (Figure 4E). Following infection with BCG, intracellular IFN-γ induction was mildly impaired in CD4+ T cells and more severely in MAIT and Vδ2+ γδ T cells of IL-23R-deficient patients, and this impairment was not rescued by exogenous IL-12 (Figure 4E). Intracellular IFN-γ production in response to BCG infection was not significantly impaired in the NK of the IL-23R-deficient patient (Figure 4E). These results are consistent with the IL-23-dependent induction of IFNG expression observed in MAIT and Vδ2+ γδ T cells from healthy controls on scRNAseq. Overall, IL-23R deficiency impairs IFN-γ production upon mycobacterial infection ex vivo; this impairment involves at least MAIT and Vδ2+ γδ T cells, and leads to insufficient IFN-γ-dependent protective immunity to mycobacteria.
Normal development of BCG-specific CD4+ memory T cells in IL-23R-deficient patients
Our results suggested that IFN-γ induction by MAIT and Vδ2+ γδ T cells was deficient in IL-23R-deficient patients, but we could not rule out a contribution of impaired memory CD4+ T cell function to the mycobacterial disease in these patients. We therefore sorted naïve and memory CD4+ T cells from P4, P5, and healthy controls, and cultured them for five days under TH0 (T-cell activation and expansion [TAE] beads only) or TH1 (TAE beads plus IL-12) polarizing conditions. IFN-γ was induced in TH1-stimulated naïve CD4+ T cells from the IL-23R-deficients patients, but the amounts in which it was produced and secreted (assessed by determining the percentage of IFN-γ+ cells and the concentration of IFN-γ in the supernatant) were at or below the lower end of the range for naïve CD4+ T cells from healthy donors (Figure 5A and S5A). Similar results were obtained for IL23R-deficient memory CD4+ T cells in that these cells were able to produce and secrete IFN-γ, but in smaller amounts than CD4+ T cells from healthy donors (Figure 5B and S5B). We then assessed the impact of IL-23R-deficiency on the proportion of BCG-reactive memory CD4+ T cells and their IFN-γ production. Following stimulation with tuberculin purified protein derivative (PPD), the proportion of IFN-γ+ cells among CD40L+/CD69+ reactive memory CD4+ T cells was similar between healthy controls and the IL-23R-deficient patients (Figure S5C). Following polyclonal stimulation of T cells with antibody complexes binding CD2, CD3, and CD28, the proportion of CD40L+/CD69+ activated memory CD4+ T cells, including that of IFN-γ+ cells, was lower in IL-23R-deficient patients, when compared with healthy controls (Figure S5C). Finally, we assessed the reactivity to BCG of memory CD4+ T cells expanded in vitro with PHA, IL-2 and irradiated allogeneic PBMCs; the frequency of BCG-specific memory CD4+ T cells was similar between healthy controls and IL-23R-deficient patients (Figure 5C and Figure S5D and S5E). Most of these cells belonged to the CCR6+ compartment (Figure 5D and Figure S5F), suggesting that IL-23R-deficiency does not impair the development of BCG-specific TH1* cells (66). IFN-γ production by BCG-specific memory CD4+ T cells was also similar between IL-23R-deficient patients and healthy controls (Figure 5E). Overall, IL-23R deficiency slightly decreased the capacity of CD4+ T cells to differentiate into IFN-γ-producing cells, but did not appear to affect the development of BCG-specific memory CD4+ T cells. The normal frequency of BCG-specific CD4+ memory T cells and their intact ability to produce IFN-γ relative to healthy individuals may account for the absence of mycobacterial disease recurrence in the seven IL-23R-deficient patients described to date (11, 67) (see Table S1).
Figure 5: Normal development of BCG- and C. albicans specific memory CD4+ T cells in patients with inherited IL-23R deficiency.
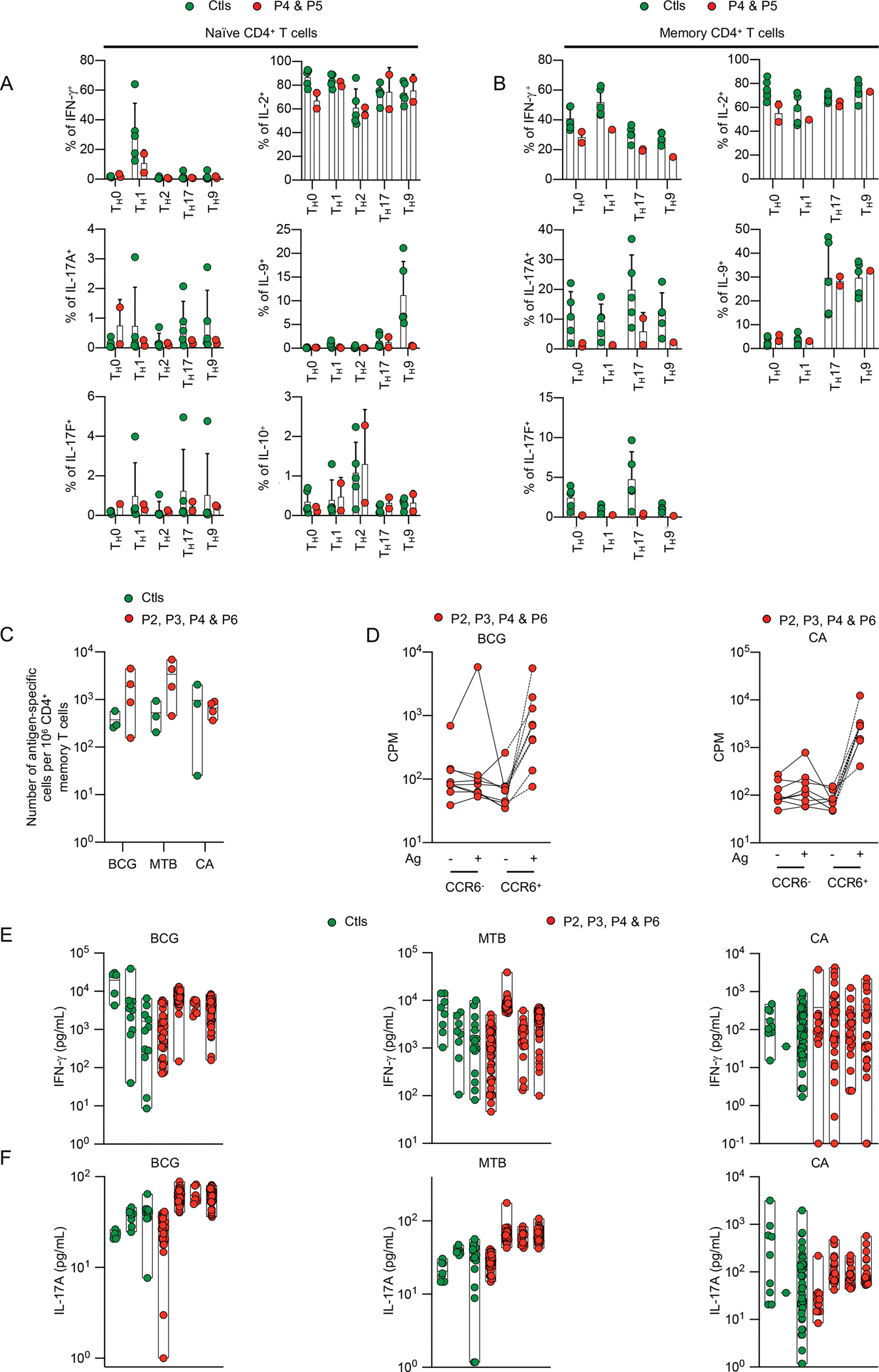
(A-B) Naïve (A) or memory (B) CD4+ T cells from healthy controls (Ctls), P4, and P5 were either kept under TH0 (T-cell activation/expansion [TAE] beads) conditions, or were polarized under TH1 (TAE beads + IL-12), TH2 (TAE beads + IL-4), TH17 (TAE beads + IL-1/IL-6/IL-21/IL-23/TGF-β), or TH9 (TAE beads + IL-9) conditions. After five days of culture, the percentages of IFN-γ+, IL-17A+, and IL-17F+ cells were assessed intracellularly by flow cytometry. (C-F) Multiple memory CD4+ T cell lines were generated by stimulation with PHA, IL-2 and irradiated allogeneic PBMCs from the sorted memory CD4+ T cells of three healthy controls, P2, P3, P4, and P6. Lines were screened for reactivity with peptide pools covering antigens from BCG, Mycobacterium tuberculosis (MTB), or C. albicans (CA). (C) The proliferation of memory CD4+ T-cell lines after stimulation with autologous B cells pulsed with BCG, MTB, or CA peptide pools was measured by determining 3H-thymidine incorporation, and the frequencies (mean number per million CD4+ memory T cells) of BCG-, MTB-, and CA-specific memory CD4+ T cells were estimated assuming a Poisson distribution. (D) Proliferation, measured by 3H-thymidine incorporation, of memory CD4+ CCR6− or CD4+ CCR6+ T-cell lines, stimulated with autologous B cells pulsed with BCG or CA peptide pools. (E-F) BCG, MTB, or CA reactive memory CD4+ T-cell lines from each individual were selected, and the concentrations of IFN-γ (E) and IL-17A (F) in the supernatant were determined with a Luminex assay. Each dot on the graph corresponds to the value for a single antigen-reactive T-cell line.
Impaired ex vivo IL-23-mediated production of IL-17 cytokines in inherited IL-23R deficiency
IL-17A/F are cytokines essential for anti-Candida mucosal immunity, as all 14 known genetic etiologies of CMC (isolated or syndromic) affect IL-17-mediated immunity (36, 68). We therefore performed ex vivo experiments to assess the impact of inherited IL-23R deficiency on IL-17-dependent immunity to Candida. We first assessed the responses of PBMCs from P3, P4, and P5 to ex vivo stimulation for eight hours with phorbol 12-myristate 13-acetate and ionomycin (P/I). We measured intracellular IL-17A production in different lymphocyte subsets by spectral flow cytometry. Following P/I stimulation, the frequencies of IL-17A+ MAIT and CD4+ T cells were significantly lower in IL-23R-deficient patients than in healthy controls (Figure 6A). We assessed the response of IL-23R-deficient PBMCs to ex vivo stimulation with IL-23 for 48 hours. We observed no increase in the frequency of IL-17A+ MAIT cells upon IL-23 stimulation in IL-23R-deficient patients, contrary to our findings for healthy controls (Figure 6A). Frequencies of IL-17A+ Vδ2+ γδ T, NK, and CD4+, cells did not increase upon IL-23 stimulation in healthy controls. We then assessed the responses of IL-23R-deficient PBMCs to ex vivo stimulation with heat-killed C. albicans (HKCA). HKCA stimulation induced the secretion of IL-17A/F and IL-22 in healthy control PBMCs, but not in PBMCs from P3, P4, P5, and P6 (Figure 6B and Figures S6A and B). Finally, we stimulated PBMCs from healthy individuals and IL-23R-deficient patients with IL-23, with or without IL-1β, in the presence or absence of HKCA. We found that the stimulation of control PBMCs with IL-23 in the presence of IL-1β enhanced the production of IL-17A, IL-17F and IL-22 relative to stimulation with IL-1β or IL-23 alone, and that IL-23 further induced IL-17F production in the presence of HKCA. By contrast, under the same stimulation conditions, no IL-17A, IL-17F, or IL-22 was detected in the supernatant of PBMCs from IL-23R-deficient patients (Figure 6B and Figure S6B). Overall, MAIT cells from IL-23R-deficient patients, like those of IL-12Rβ1-deficient patients or STAT1 GOF patients, displayed impaired IL-17A induction upon IL-23 stimulation, and PBMCs from IL-23R- and IL-12Rβ1-deficient patients displayed impaired IL-17A/F production in response to heat-killed C. albicans, possibly accounting for their risk of CMC (41–47).
Figure 6: Impaired ex vivo IL-23-mediated production of IL-17 cytokines in patients with inherited IL-23R deficiency.

(A) Percentages of IL-17A+ cells on intracellular flow cytometry for the indicated cell subsets, from the indicated individuals (healthy controls, Ctrls; patients, P3, P4, P6; one IL-12Rβ1 deficient patient, and one STAT1-GOF patient) unstimulated (–), or stimulated with IL-23 or PMA/ionomycin (P/I). (B) IL-17A and IL-17F secretion by PBMCs, assessed by Legendplex assays on the supernatants, for the indicated individuals (healthy controls, Ctrls; patients P5, P6; and one IL-12Rβ1-deficient patient). The cells were cultured for five days, and were then either left unstimulated (–), or stimulated with IL-1β (+), and/or IL-23 (+), in the presence (+) or absence (–) of heat-killed C. albicans (HKCA), or anti-CD2/CD3/CD28 mAb-coated beads (CD2/3/28). Nonparametric Mann-Whitney tests were used for analysis in panels A-B
(*p<0.05, **p<0.01, ****p<0.0001).
Normal development of C. albicans-specific CD4+ memory T cells in IL-23R-deficient patients
CMC displays incomplete penetrance in IL-23R deficiency, suggesting that IL-17 production may be preserved in some cell subsets. We therefore studied the impact of IL-23R deficiency on the development of TH17 cells. We sorted naïve and memory CD4+ T cells from P4, P5, and healthy individuals. After five days of culture under TH17 polarizing (TAE beads plus TGF-β, IL-1β, IL-6, IL-21 and IL-23) conditions, the production of IL-17A and IL-17F (assessed by determining the percentage of IL-17A+ and IL-17F+ cells among naïve and memory CD4+ T cells, and the concentration of IL-17A and IL-17F secreted by naïve and memory CD4+ T cells) by IL-23R-deficient cells was impaired relative to that of healthy controls, but not entirely abolished (Figure 5A and B, and Figure S5A and B). We then assessed the reactivity to C. albicans of in vitro-expanded CD4+ memory T cells from P2, P3, P4, P6 and healthy controls; the frequency of C. albicans-specific cells was similar for healthy controls and IL-23R-deficient patients (Figure 5C and Figure S5D). Most of these cells belonged to the CCR6+ compartment (Figure 5D), suggesting that IL-23R-deficiency does not impair the development of C. albicans-specific TH17 cells (66). Moreover, IL-17A production by C. albicans-specific CD4+ memory T cells was similar for IL-23R-deficient patients and healthy controls (Figure 5F). Overall, these results suggest that IL-23R deficiency slightly decreases the capacity of CD4+ T cells to differentiate into TH17-producing cells but does not impair the development of C. albicans-specific CD4+ memory T cells. The normal frequency of C. albicans-specific CD4+ memory T cells and the conservation of their capacity to produce IL-17A may be sufficient to protect against CMC, in some patients, potentially accounting for the incomplete penetrance of CMC in IL-23R-deficient patients (36, 41–44).
DISCUSSION
We report the characterization of six patients from four unrelated Iranian kindreds with AR complete IL-23R deficiency. These patients have normal numbers of circulating leukocytes from various subsets, including cell types that normally express IL-23R, such as NK, iNKT, MAIT, Vδ2+ γδ T, TH1, TH17 and TH1* cells (11, 69). The vulnerability of some IL-12p40-, IL-12Rβ1-, and IL-23R-deficient patients to CMC suggests that human IL-23 plays a crucial role in IL-17A/F-mediated defense against mucocutaneous infections with Candida spp. in a subset of affected individuals. Indeed, CMC is seen in ~ 25 to 35% of patients with any of these three disorders, implying that IL-23 is redundant for defense against Candida in most individuals. We observed an IL-23-driven induction of IL-17A ex vivo only for MAIT cells from healthy individuals, this induction being abolished in IL23R-deficient patients. We also found impaired IL-17A/F secretion by IL-23R-deficient leukocytes in response heat-killed C. albicans, possibly due to the impaired IL-23 responses of MAIT cells. In a mouse model of experimental autoimmune encephalomyelitis (EAE), IL-23 drives the expansion of the IL-17A+ CD4+ T-cell population (70). IL-23-deficient mice (Il23p19−/−) have a higher C. albicans burden than WT mice in models of oropharyngeal candidiasis (OPC) and cutaneous C. albicans infection (71, 72). The molecular and cellular basis of CMC in patients with IL-23R deficiency is probably similar to that in IL-12p40- and IL-12Rβ1-deficient patients, whose leukocytes also display no IL-23 signaling. In patients with any of these three disorders, CMC displays incomplete penetrance, which does not seem to be explained by the interindividual variability of IL-17A/F production by leukocyte subsets. Incomplete penetrance may reflect the observations that the development of C. albicans-specific memory CD4+ T cells is preserved in IL-23R-deficient patients, and that IL-17A induction is controlled by IL-23 in only one lymphocyte subset (MAIT cells), at least ex vivo.
By contrast, human IL-23/IL-23R signaling was surprisingly found to be essential for IFN-γ-mediated immunity to mycobacteria in all IL-23R-deficient patients identified. Our previous report of kindred A in 2018 was suggestive, but not conclusive, as only two siblings with MSMD were reported, and the cellular basis of the disease was not characterized (11). The data reported here, together with the recent description of disseminated environmental mycobacterial disease in an IL-23R-deficient patient (67), further suggest that human IL-23 is essential for host defense against even weakly virulent mycobacteria, as IL-23R deficiency underlies MSMD with complete penetrance in five unrelated kindreds with different and private IL23R genotypes. An ascertainment bias is unlikely to have affected this assessment, as two of the six MSMD patients are siblings of index cases. We also deciphered the cellular basis of mycobacterial disease. We showed, by scRNAseq, that inherited IL-23R deficiency impairs IFN-γ-dependent signaling in MAIT, Vδ2+ γδ T, TH1, TH17, and TH1* cells in the basal state in vivo. We speculate that various environmental stimuli continuously trigger low-level IFN-γ signaling in an IL-23-dependent manner in the basal state in healthy individuals. This model is further supported by the observations that IL-23R-deficient MAIT and Vδ2+ γδ cells display impaired induction of IFNG mRNA and IFN-γ protein upon stimulation with IL-23 and live BCG mycobacteria ex vivo. However, these findings do not exclude the possible contribution of other cellular subsets to IL-23-dependent immunity to mycobacteria, such as IL-23R-deficient CD4+ T cells, which showed decreased production of IFN-γ under some conditions. Exogenous IL-12 did not completely rescue the defective IFN-γ production of IL-23R-deficient cells, indicating that IL-12 and IL-23 control IFN-γ production in different ways. Consistently, we previously showed that a partial and selective defect of IL-23 signaling due to a common homozygous missense variant of TYK2 (P1104A) predisposes patients to TB (9, 12). The physiological significance of this finding is supported by its evolutionary impact; the frequency of the P1104A TYK2 allele has decreased in Europeans over the last 2,000 years, probably due to the negative selection exerted by TB (and possibly, to a lesser extent, by other intramacrophagic pathogens) (73). In this context, our report further attests to the essential contribution of human IL-23 to antimycobacterial immunity.
Our findings therefore suggest that human IL-12 and IL-23 are functionally more closely related, via their induction of IFN-γ, than previously thought based on the classical TH1/TH17 paradigm established in mice (49–51, 74). The discovery of IL-23 and IL-23R in the early 2000s was associated with the definition of TH17 cells as a distinct T-cell lineage, different from TH1 and TH2 cells (49–51, 74). In this view, IL-12 is an IFN-γ-inducing signature cytokine, whereas IL-23 is an IL-17-inducing signature cytokine (49–51). The observation that the administration of IFN-γ — the archetypal TH1 cytokine — protects mice from EAE, whereas Ifng deletion worsens EAE suggested that the TH1/TH2 dichotomy was insufficient (50, 74). Subsequent findings that IL-23 promotes the expansion of a T-cell population that produces IL-17, IL-6, and TNF (70), and EAE resistance in IL-23-deficient mice paved the way for the definition of TH17 cells (75). This led to the IL-12/IFN-γ-TH1 and IL-23/IL-17-TH17 dichotomy (50, 51). The experiment of Nature reported here suggests that this dichotomy holds only partially in humans, and that human IL-23 is instead primarily an antimycobacterial IFN-γ-inducing cytokine. IL-12 is essential for IFN-γ production by antigen-specific CD4+ αβ T cells (66), which are purely adaptive T cells, but IL-23 is essential for IFN-γ production primarily by innate-like adaptive cells (MAIT and Vδ2+ γδ T). We, thus, speculate that IL-23 may also be the phylogenetically ancestral IFN-γ-inducing cytokine. Our findings further suggest that the mechanisms underlying the therapeutic efficacy of IL-23p19 blockers may involve an inhibition not only of excessive IL-17 immunity, if indeed this immunity is inhibited at all, but also, and perhaps predominantly or exclusively, of excessive IFN-γ immunity (51, 74). They also challenge the involvement of IL-17 immunity inferred from IL-23R signals detected in genome-wide association studies (51, 76, 77).
MATERIALS AND METHODS
Study design
We performed WES on a cohort of 802 patients with MSMD of unidentified genetic etiology. We identified six patients homozygous for one out of four different eLOF or pLOF IL23R variants. All six patients had a history of MSMD and two suffered from CMC. We examined the consequences of these mutations both in isogenic overexpression experiments and in the patients’ cells. We found that the four IL23R variants abolish cell response to IL-23. We performed ex vivo and in vitro experiments using PBMCs from the patients and controls, to investigate the impact of IL-23R deficiency on IFN-γ-dependent immunity to mycobacteria and IL-17-dependent immunity to C. albicans. Conclusions were drawn from analyzing the results from aforementioned approaches collectively.
Supplementary Material
Supplementary Figure 1: Private homozygous IL23R variants in four Iranian kindreds
Supplementary Figure 2: Loss-of-function IL23R alleles and AR complete IL23R deficiency
Supplementary Figure 3: Development of peripheral mononuclear hematopoietic cells in IL-23R-deficient patients
Supplementary Figure 4: Impaired ex vivo IL-23-mediated production of IFN-γ in cells from patients with IL-23R deficiency
Supplementary Figure 5: Normal development of BCG- and C. albicans-specific memory CD4+ T cells in patients with inherited IL-23R deficiency
Supplementary Figure 6: Impaired ex vivo IL-23-mediated production of IL-17 cytokines in patients with inherited IL-23R deficiency
Table S1: Summary of the medical history of patients with inherited IL-23R deficiency
Table S2: List of all homozygous coding, essential-splicing site and splice-site variants with a CADD above the MSC not present in the homozygous state in GnomAD but detected in the analyses of the exomes of P1 to P6
Table S3: Genotypes of IL-12Rβ1- and IL-12Rb2-deficient patients and STAT1-GOF patients included as controls
Table S4: Gating strategy for the assessment of STAT phosphorylation by mass cytometry (CyTOF).
Table S5: Gating strategy for deep immunophenotyping by mass cytometry (CyTOF).
Table S6: Gating strategy for deep immunophenotyping by spectral flow cytometry.
Table S7: Gating strategy for the ex vivo evaluation of IFN-γ+ and IL-17A+ cells after PBMC stimulation with IL-23 or IL-12, in the presence or absence of BCG infection.
Table S8: Gating strategy for the ex vivo evaluation of BCG-reactive memory CD4+ T cells.
Table S9 : Sequence of primers used for this study
Acknowledgments
We thank the patients and their families for placing their trust in us. We thank the members of both branches of the Laboratory of Human Genetics of Infectious Diseases. We thank Janet Markle and Ruben Martinez-Barricarte for helpful discussions. We thank Yelena Nemirovskaya, Mark Woollett, Dana Liu, Soraya Boucherit, Erin Williams, Christine Rivalain, Maya Chrabieh, and Lazaro Lorenzo for administrative assistance. We thank the National Institutes of Health (NIH) Tetramer Core Facility (NTCF) for providing the 5-OP-RU-loaded MR1 tetramer, which was developed jointly with Dr. James McCluskey, Dr. Jamie Rossjohn, and Dr. David Fairlie. The following reagent was obtained through BEI Resources, NIAID, NIH: Mycobacterium tuberculosis, strain H37Rv, whole-cell lysate, NR-14822.
Funding
The HGMI laboratory was funded in part by the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH) (grant numbers R01AI095983 to J.-L.C. and J.Bu and R01AI127564 to J.-L.C. and A.P.), the National Center for Research Resources and the National Center for Advancing Sciences, NIH (grant number 8UL1TR000043 for J.-L.C.), The Rockefeller University, the St. Giles Foundation, Institut National de la Santé et de la Recherche Médicale (INSERM), Paris Cité University, the Integrative Biology of Emerging Infectious Diseases Laboratory of Excellence (ANR-10-LABX-62-IBEID), the French National Research Agency (ANR) under the “Investments for the future” program (ANR-10-IAHU-01), Cross Lab Imagine Grant (The inborn errors of the keratinocyte-leukocyte cross-talk in humans to A.P.), ANR-GENMSMD (ANR-16-CE17-0005-01 to J.Bu.), ANR FNS LTh-MSMD-CMCD (grant no. ANR-18-CE93-0008-01 to F.S. and A.P.), ANRS project ECTZ170784-ANRS0073, ANRS project ECTZ170784-ANRS0073 to SBD, the French Foundation for Medical Research (FRM) (EQU201903007798), the European Union’s Horizon 2020 research and innovation program under grant agreement on. 824110 (EASI-genomics), the Square Foundation, Grandir - Fonds de solidarité pour l’enfance, and the SCOR Corporate Foundation for Science, and the Swiss National Science Foundation (n. 310030L_182728 to F.S). Q.P was supported by the Assistance Publique Hôpitaux de Paris (Année Recherche) and is supported by the MD-PhD program of INSERM (Ecole de l’INSERM Liliane Bettencourt). J.Bo. is supported by the EMBO fellowship program. J.R. was supported by the INSERM PhD program for doctors of pharmacy (poste d’accueil INSERM). J.R. and T.L.V. are supported by the Bettencourt-Schueller Foundation and the MD-PhD program of the Imagine Institute. A-L. N is supported by the Bettencourt Schueller Foundation and the International PhD program of the Imagine Institute. M.O. is supported by the David Rockefeller Graduate Program, the New York Hideyo Noguchi Memorial Society (HNMS), the Funai Foundation for Information Technology (FFIT), and the Honjo International Scholarship Foundation (HISF), and the National Cancer Institute (NCI) F99 Award (F99CA274708). A.A.A. and C.A.F. are supported by Ministerio de Ciencia Tecnología e Innovación MINCIENCIAS, Colombia (111574455633/CT 713-2016 and 111584467551/CT 415-2020), the Movilidad Académica ECOS-Nord/MINCIENCIAS, Colombia (CT 806-2018/046-2019) and the Comité para el Desarrollo de la Investigación, CODI - UdeA, Colombia (CT 2017-16003). R.Y. was supported by the Immune Deficiency Foundation and the Stony Wold-Herbert Fund.
Footnotes
Conflicts of interest/Competing interests
The authors have no conflict of interest to declare.
Availability of data and materials
All raw and processed data and biological materials, including immortalized cell lines from patients, are available upon request from the corresponding authors under a material/data transfer agreement.
References
- 1.Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, Klein C, Morio T, Oksenhendler E, Picard C, Puel A, Puck J, Seppänen MRJ, Somech R, Su HC, Sullivan KE, Torgerson TR, Meyts I, The Ever-Increasing Array of Novel Inborn Errors of Immunity: an Interim Update by the IUIS Committee. J Clin Immunol. 41, 666–679 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, Klein C, Morio T, Oksenhendler E, Picard C, Puel A, Puck J, Seppänen MRJ, Somech R, Su HC, Sullivan KE, Torgerson TR, Meyts I, Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol (2022), doi: 10.1007/s10875-022-01289-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Casanova J-L, Abel L, The human genetic determinism of life-threatening infectious diseases: genetic heterogeneity and physiological homogeneity? Hum Genet. 139, 681–694 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Casanova J-L, Abel L, Lethal Infectious Diseases as Inborn Errors of Immunity: Toward a Synthesis of the Germ and Genetic Theories. Annu Rev Pathol. 16, 23–50 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Casanova J-L, Abel L, Mechanisms of viral inflammation and disease in humans. Science. 374, 1080–1086 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Casanova J-L, Abel L, From rare disorders of immunity to common determinants of infection: Following the mechanistic thread. Cell. 185, 3086–3103 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bustamante J, Mendelian susceptibility to mycobacterial disease: recent discoveries. Hum Genet. 139, 993–1000 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bustamante J, Boisson-Dupuis S, Abel L, Casanova J-L, Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-γ immunity. Semin Immunol. 26, 454–470 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kerner G, Ramirez-Alejo N, Seeleuthner Y, Yang R, Ogishi M, Cobat A, Patin E, Quintana-Murci L, Boisson-Dupuis S, Casanova J-L, Abel L, Homozygosity for TYK2 P1104A underlies tuberculosis in about 1% of patients in a cohort of European ancestry. Proc Natl Acad Sci U S A. 116, 10430–10434 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang R, Mele F, Worley L, Langlais D, Rosain J, Benhsaien I, Elarabi H, Croft CA, Doisne J-M, Zhang P, Weisshaar M, Jarrossay D, Latorre D, Shen Y, Han J, Ogishi M, Gruber C, Markle J, Al Ali F, Rahman M, Khan T, Seeleuthner Y, Kerner G, Husquin LT, Maclsaac JL, Jeljeli M, Errami A, Ailal F, Kobor MS, Oleaga-Quintas C, Roynard M, Bourgey M, El Baghdadi J, Boisson-Dupuis S, Puel A, Batteux F, Rozenberg F, Marr N, Pan-Hammarström Q, Bogunovic D, Quintana-Murci L, Carroll T, Ma CS, Abel L, Bousfiha A, Di Santo JP, Glimcher LH, Gros P, Tangye SG, Sallusto F, Bustamante J, Casanova J-L, Human T-bet Governs Innate and Innate-like Adaptive IFN-γ Immunity against Mycobacteria. Cell. 183, 1826–1847.e31 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martínez-Barricarte R, Markle JG, Ma CS, Deenick EK, Ramírez-Alejo N, Mele F, Latorre D, Mahdaviani SA, Aytekin C, Mansouri D, Bryant VL, Jabot-Hanin F, Deswarte C, Nieto-Patlán A, Surace L, Kerner G, Itan Y, Jovic S, Avery DT, Wong N, Rao G, Patin E, Okada S, Bigio B, Boisson B, Rapaport F, Seeleuthner Y, Schmidt M, Ikinciogullari A, Dogu F, Tanir G, Tabarsi P, Bloursaz MR, Joseph JK, Heer A, Kong X-F, Migaud M, Lazarov T, Geissmann F, Fleckenstein B, Arlehamn CL, Sette A, Puel A, Emile J-F, van de Vosse E, Quintana-Murci L, Di Santo JP, Abel L, Boisson-Dupuis S, Bustamante J, Tangye SG, Sallusto F, Casanova J-L, Human IFN-γ immunity to mycobacteria is governed by both IL-12 and IL-23. Sci Immunol. 3, eaau6759 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boisson-Dupuis S, Ramirez-Alejo N, Li Z, Patin E, Rao G, Kerner G, Lim CK, Krementsov DN, Hernandez N, Ma CS, Zhang Q, Markle J, Martinez-Barricarte R, Payne K, Fisch R, Deswarte C, Halpern J, Bouaziz M, Mulwa J, Sivanesan D, Lazarov T, Naves R, Garcia P, Itan Y, Boisson B, Checchi A, Jabot-Hanin F, Cobat A, Guennoun A, Jackson CC, Pekcan S, Caliskaner Z, Inostroza J, Costa-Carvalho BT, de Albuquerque JAT, Garcia-Ortiz H, Orozco L, Ozcelik T, Abid A, Rhorfi IA, Souhi H, Amrani HN, Zegmout A, Geissmann F, Michnick SW, Muller-Fleckenstein I, Fleckenstein B, Puel A, Ciancanelli MJ, Marr N, Abolhassani H, Balcells ME, Condino-Neto A, Strickler A, Abarca K, Teuscher C, Ochs HD, Reisli I, Sayar EH, El-Baghdadi J, Bustamante J, Hammarström L, Tangye SG, Pellegrini S, Quintana-Murci L, Abel L, Casanova J-L, Tuberculosis and impaired IL-23-dependent IFN-γ immunity in humans homozygous for a common TYK2 missense variant. Sci Immunol. 3, eaau8714 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Le Voyer T, Neehus A-L, Yang R, Ogishi M, Rosain J, Alroqi F, Alshalan M, Blumental S, Al Ali F, Khan T, Ata M, Rozen L, Demulder A, Bastard P, Gruber C, Roynard M, Seeleuthener Y, Rapaport F, Bigio B, Chrabieh M, Sng D, Berteloot L, Boddaert N, Rozenberg F, Al-Muhsen S, Bertoli-Avella A, Abel L, Bogunovic D, Marr N, Mansouri D, Al Mutairi F, Béziat V, Weil D, Mahdaviani SA, Ferster A, Zhang S-Y, Reversade B, Boisson-Dupuis S, Casanova J-L, Bustamante J, Inherited deficiency of stress granule ZNFX1 in patients with monocytosis and mycobacterial disease. Proc Natl Acad Sci U S A. 118, e2102804118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin-Fernandez M, Buta S, Le Voyer T, Li Z, Dynesen LT, Vuillier F, Franklin L, Ailal F, Muglia Amancio A, Malle L, Gruber C, Benhsaien I, Altman J, Taft J, Deswarte C, Roynard M, Nieto-Patlan A, Moriya K, Rosain J, Boddaert N, Bousfiha A, Crow YJ, Jankovic D, Sher A, Casanova J-L, Pellegrini S, Bustamante J, Bogunovic D, A partial form of inherited human USP18 deficiency underlies infection and inflammation. J Exp Med. 219, e20211273 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Altare F, Durandy A, Lammas D, Emile JF, Lamhamedi S, Le Deist F, Drysdale P, Jouanguy E, Döffinger R, Bernaudin F, Jeppsson O, Gollob JA, Meinl E, Segal AW, Fischer A, Kumararatne D, Casanova JL, Impairment of mycobacterial immunity in human interleukin-12 receptor deficiency. Science. 280, 1432–1435 (1998). [DOI] [PubMed] [Google Scholar]
- 16.de Jong R, Altare F, Haagen IA, Elferink DG, Boer T, van Breda Vriesman PJ, Kabel PJ, Draaisma JM, van Dissel JT, Kroon FP, Casanova JL, Ottenhoff TH, Severe mycobacterial and Salmonella infections in interleukin-12 receptor-deficient patients. Science. 280, 1435–1438 (1998). [DOI] [PubMed] [Google Scholar]
- 17.Altare F, Lammas D, Revy P, Jouanguy E, Döffinger R, Lamhamedi S, Drysdale P, Scheel-Toellner D, Girdlestone J, Darbyshire P, Wadhwa M, Dockrell H, Salmon M, Fischer A, Durandy A, Casanova JL, Kumararatne DS, Inherited interleukin 12 deficiency in a child with bacille Calmette-Guérin and Salmonella enteritidis disseminated infection. J Clin Invest. 102, 2035–2040 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Beaucoudrey L, Samarina A, Bustamante J, Cobat A, Boisson-Dupuis S, Feinberg J, Al-Muhsen S, Jannière L, Rose Y, de Suremain M, Kong X-F, Filipe-Santos O, Chapgier A, Picard C, Fischer A, Dogu F, Ikinciogullari A, Tanir G, Al-Hajjar S, Al-Jumaah S, Frayha HH, AlSum Z, Al-Ajaji S, Alangari A, Al-Ghonaium A, Adimi P, Mansouri D, Ben-Mustapha I, Yancoski J, Garty B-Z, Rodriguez-Gallego C, Caragol I, Kutukculer N, Kumararatne DS, Patel S, Doffinger R, Exley A, Jeppsson O, Reichenbach J, Nadal D, Boyko Y, Pietrucha B, Anderson S, Levin M, Schandené L, Schepers K, Efira A, Mascart F, Matsuoka M, Sakai T, Siegrist C-A, Frecerova K, Blüetters-Sawatzki R, Bernhöft J, Freihorst J, Baumann U, Richter D, Haerynck F, De Baets F, Novelli V, Lammas D, Vermylen C, Tuerlinckx D, Nieuwhof C, Pac M, Haas WH, Müller-Fleckenstein I, Fleckenstein B, Levy J, Raj R, Cohen AC, Lewis DB, Holland SM, Yang KD, Wang X, Wang X, Jiang L, Yang X, Zhu C, Xie Y, Lee PPW, Chan KW, Chen T-X, Castro G, Natera I, Codoceo A, King A, Bezrodnik L, Di Giovani D, Gaillard MI, de Moraes-Vasconcelos D, Grumach AS, da Silva Duarte AJ, Aldana R, Espinosa-Rosales FJ, Bejaoui M, Bousfiha AA, Baghdadi JE, Özbek N, Aksu G, Keser M, Somer A, Hatipoglu N, Aydogmus Ç, Asilsoy S, Camcioglu Y, Gülle S, Ozgur TT, Ozen M, Oleastro M, Bernasconi A, Mamishi S, Parvaneh N, Rosenzweig S, Barbouche R, Pedraza S, Lau YL, Ehlayel MS, Fieschi C, Abel L, Sanal O, Casanova J-L, Revisiting human IL-12Rβ1 deficiency: a survey of 141 patients from 30 countries. Medicine (Baltimore). 89, 381–402 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prando C, Samarina A, Bustamante J, Boisson-Dupuis S, Cobat A, Picard C, AlSum Z, Al-Jumaah S, Al-Hajjar S, Frayha H, Al-Mousa H, Ben-Mustapha I, Adimi P, Feinberg J, de Suremain M, Jannière L, Filipe-Santos O, Mansouri N, Stephan J-L, Nallusamy R, Kumararatne DS, Bloorsaz MR, Ben-Ali M, Elloumi-Zghal H, Chemli J, Bouguila J, Bejaoui M, Alaki E, AlFawaz TS, Al Idrissi E, ElGhazali G, Pollard AJ, Murugasu B, Wah Lee B, Halwani R, Al-Zahrani M, Al Shehri MA, Al-Zahrani M, Bin-Hussain I, Mahdaviani SA, Parvaneh N, Abel L, Mansouri D, Barbouche R, Al-Muhsen S, Casanova J-L, Inherited IL-12p40 deficiency: genetic, immunologic, and clinical features of 49 patients from 30 kindreds. Medicine (Baltimore). 92, 109–122 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blancas-Galicia L, Peñafiel-Vicuña AK, Scheffler-Mendoza S, Rojas-Maruri M, Rivas-Larrauri F, Rodríguez-Lozano AL, Bustamante J, Yamazaki-Nakashimada MA, Recurrent Salmonella Infections and Nephritis Complicating IgA Vasculitis in a Patient with IL12-RB1 Deficiency. J Investig Allergol Clin Immunol, 0 (2021). [DOI] [PubMed] [Google Scholar]
- 21.Rosain J, Oleaga-Quintas C, Deswarte C, Verdin H, Marot S, Syridou G, Mansouri M, Mahdaviani SA, Venegas-Montoya E, Tsolia M, Mesdaghi M, Chernyshova L, Stepanovskiy Y, Parvaneh N, Mansouri D, Pedraza-Sánchez S, Bondarenko A, Espinosa-Padilla SE, Yamazaki-Nakashimada MA, Nieto-Patlán A, Kerner G, Lambert N, Jacques C, Corvilain E, Migaud M, Grandin V, Herrera MT, Jabot-Hanin F, Boisson-Dupuis S, Picard C, Nitschke P, Puel A, Tores F, Abel L, Blancas-Galicia L, De Baere E, Bole-Feysot C, Casanova J-L, Bustamante J, A Variety of Alu-Mediated Copy Number Variations Can Underlie IL-12Rβ1 Deficiency. J Clin Immunol. 38, 617–627 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mahdaviani SA, Mansouri D, Jamee M, Zaki-Dizaji M, Aghdam KR, Mortaz E, Khorasanizadeh M, Eskian M, Movahedi M, Ghaffaripour H, Baghaie N, Hassanzad M, Chavoshzadeh Z, Mansouri M, Mesdaghi M, Ghaini M, Noori F, Eskandarzadeh S, Kahkooi S, Poorabdolah M, Tabarsi P, Moniri A, Farnia P, Karimi A, Boisson-Dupuis S, Rezaei N, Marjani M, Casanova J-L, Bustamante J, Velayati AA, Mendelian Susceptibility to Mycobacterial Disease (MSMD): Clinical and Genetic Features of 32 Iranian Patients. J Clin Immunol. 40, 872–882 (2020). [DOI] [PubMed] [Google Scholar]
- 23.Lashkari HP, Madkaikar M, Dalvi A, Gupta M, Bustamante J, Sharma M, Rawat A, Bhatia P, Bhat KG, Rao S, Kamath N, Moideen F, Latour S, Winter S, Bhavani GS, Girisha KM, Clinical and Genetic Spectrum of Inborn Errors of Immunity in a Tertiary Care Center in Southern India. Indian J Pediatr. 89, 233–242 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taur PD, Gowri V, Pandrowala AA, Iyengar VV, Chougule A, Golwala Z, Chandak S, Agarwal R, Keni P, Dighe N, Bodhanwala M, Prabhu S, George B, Fouzia NA, Edison ES, Arunachalam AK, Madkaikar MR, Dalvi AD, Yadav RM, Bargir UA, Kambli PM, Rawat A, Das J, Joshi V, Pilania RK, Jindal AK, Bhat S, Bhattad S, Unni J, Radhakrishnan N, Raj R, Uppuluri R, Patel S, Lashkari HP, Aggarwal A, Kalra M, Udwadia Z, Bafna VS, Kanade T, Puel A, Bustamante J, Casanova JL, Desai MM, Clinical and Molecular Findings in Mendelian Susceptibility to Mycobacterial Diseases: Experience From India. Front Immunol. 12, 631298 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mahdaviani SA, Marjani M, Jamee M, Khavandegar A, Ghaffaripour H, Eslamian G, Ghaini M, Eskandarzadeh S, Casanova J-L, Bustamante J, Mansouri D, Velayati AA, Disseminated Mycobacterium simiae Infection in a Patient with Complete IL-12p40 Deficiency. Iran J Allergy Asthma Immunol. 20, 376–381 (2021). [DOI] [PubMed] [Google Scholar]
- 26.Alodayani AN, Al-Otaibi AM, Deswarte C, Frayha HH, Bouaziz M, AlHelale M, Le Voyer T, Nieto-Patlan A, Rattina V, AlZahrani M, Halwani R, Al Sohime F, Al-Mousa H, Al-Muhsen S, Alhajjar SH, Dhayhi NS, Abel L, Casanova J-L, Bin-Hussain I, AlBarrak MS, Al-Jumaah SA, Bustamante J, Mendelian Susceptibility to Mycobacterial Disease Caused by a Novel Founder IL12B Mutation in Saudi Arabia. J Clin Immunol. 38, 278–282 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharifinejad N, Mahdaviani SA, Jamee M, Daneshmandi Z, Moniri A, Marjani M, Tabarsi P, Farnia P, Rekabi M, Fallahi M, Hashemimoghaddam SA, Mohkam M, Bustamante J, Casanova J-L, Mansouri D, Velayati AA, Leukocytoclastic vasculitis in patients with IL12B or IL12RB1 deficiency: case report and review of the literature. Pediatr Rheumatol Online J. 19, 121 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kobayashi M, Fitz L, Ryan M, Hewick RM, Clark SC, Chan S, Loudon R, Sherman F, Perussia B, Trinchieri G, Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. J Exp Med. 170, 827–845 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, Zonin F, Vaisberg E, Churakova T, Liu M, Gorman D, Wagner J, Zurawski S, Liu Y, Abrams JS, Moore KW, Rennick D, de Waal-Malefyt R, Hannum C, Bazan JF, Kastelein RA, Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 13, 715–725 (2000). [DOI] [PubMed] [Google Scholar]
- 30.Presky DH, Yang H, Minetti LJ, Chua AO, Nabavi N, Wu CY, Gately MK, Gubler U, A functional interleukin 12 receptor complex is composed of two beta-type cytokine receptor subunits. Proc Natl Acad Sci U S A. 93, 14002–14007 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, Pflanz S, Zhang R, Singh KP, Vega F, To W, Wagner J, O’Farrell A-M, McClanahan T, Zurawski S, Hannum C, Gorman D, Rennick DM, Kastelein RA, de Waal Malefyt R, Moore KW, A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J Immunol. 168, 5699–5708 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Fieschi C, Dupuis S, Catherinot E, Feinberg J, Bustamante J, Breiman A, Altare F, Baretto R, Le Deist F, Kayal S, Koch H, Richter D, Brezina M, Aksu G, Wood P, Al-Jumaah S, Raspall M, Da Silva Duarte AJ, Tuerlinckx D, Virelizier J-L, Fischer A, Enright A, Bernhöft J, Cleary AM, Vermylen C, Rodriguez-Gallego C, Davies G, Blütters-Sawatzki R, Siegrist C-A, Ehlayel MS, Novelli V, Haas WH, Levy J, Freihorst J, Al-Hajjar S, Nadal D, De Moraes Vasconcelos D, Jeppsson O, Kutukculer N, Frecerova K, Caragol I, Lammas D, Kumararatne DS, Abel L, Casanova J-L, Low penetrance, broad resistance, and favorable outcome of interleukin 12 receptor beta1 deficiency: medical and immunological implications. J Exp Med. 197, 527–535 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boisson-Dupuis S, El Baghdadi J, Parvaneh N, Bousfiha A, Bustamante J, Feinberg J, Samarina A, Grant AV, Janniere L, El Hafidi N, Hassani A, Nolan D, Najib J, Camcioglu Y, Hatipoglu N, Aydogmus C, Tanir G, Aytekin C, Keser M, Somer A, Aksu G, Kutukculer N, Mansouri D, Mahdaviani A, Mamishi S, Alcais A, Abel L, Casanova J-L, IL-12Rβ1 deficiency in two of fifty children with severe tuberculosis from Iran, Morocco, and Turkey. PLoS One. 6, e18524 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boisson-Dupuis S, The monogenic basis of human tuberculosis. Hum Genet. 139, 1001–1009 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ouederni M, Sanal O, Ikinciogullari A, Tezcan I, Dogu F, Sologuren I, Pedraza-Sánchez S, Keser M, Tanir G, Nieuwhof C, Colino E, Kumararatne D, Levy J, Kutukculer N, Aytekin C, Herrera-Ramos E, Bhatti M, Karaca N, Barbouche R, Broides A, Goudouris E, Franco JL, Parvaneh N, Reisli I, Strickler A, Shcherbina A, Somer A, Segal A, Angel-Moreno A, Lezana-Fernandez JL, Bejaoui M, Bobadilla-Del Valle M, Kachboura S, Sentongo T, Ben-Mustapha I, Bustamante J, Picard C, Puel A, Boisson-Dupuis S, Abel L, Casanova J-L, Rodríguez-Gallego C, Clinical features of Candidiasis in patients with inherited interleukin 12 receptor β1 deficiency. Clin Infect Dis. 58, 204–213 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Puel A, Human inborn errors of immunity underlying superficial or invasive candidiasis. Hum Genet. 139, 1011–1022 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kerner G, Rosain J, Guérin A, Al-Khabaz A, Oleaga-Quintas C, Rapaport F, Massaad MJ, Ding J-Y, Khan T, Ali FA, Rahman M, Deswarte C, Martinez-Barricarte R, Geha RS, Jeanne-Julien V, Garcia D, Chi C-Y, Yang R, Roynard M, Fleckenstein B, Rozenberg F, Boisson-Dupuis S, Ku C-L, Seeleuthner Y, Béziat V, Marr N, Abel L, Al-Herz W, Casanova J-L, Bustamante J, Inherited human IFN-γ deficiency underlies mycobacterial disease. J Clin Invest. 130, 3158–3171 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jouanguy E, Altare F, Lamhamedi S, Revy P, Emile JF, Newport M, Levin M, Blanche S, Seboun E, Fischer A, Casanova JL, Interferon-gamma-receptor deficiency in an infant with fatal bacille Calmette-Guérin infection. N Engl J Med. 335, 1956–1961 (1996). [DOI] [PubMed] [Google Scholar]
- 39.Newport MJ, Huxley CM, Huston S, Hawrylowicz CM, Oostra BA, Williamson R, Levin M, A mutation in the interferon-gamma-receptor gene and susceptibility to mycobacterial infection. N Engl J Med. 335, 1941–1949 (1996). [DOI] [PubMed] [Google Scholar]
- 40.Dorman SE, Holland SM, Mutation in the signal-transducing chain of the interferon-gamma receptor and susceptibility to mycobacterial infection. J Clin Invest. 101, 2364–2369 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lévy R, Okada S, Béziat V, Moriya K, Liu C, Chai LYA, Migaud M, Hauck F, Al Ali A, Cyrus C, Vatte C, Patiroglu T, Unal E, Ferneiny M, Hyakuna N, Nepesov S, Oleastro M, Ikinciogullari A, Dogu F, Asano T, Ohara O, Yun L, Della Mina E, Bronnimann D, Itan Y, Gothe F, Bustamante J, Boisson-Dupuis S, Tahuil N, Aytekin C, Salhi A, Al Muhsen S, Kobayashi M, Toubiana J, Abel L, Li X, Camcioglu Y, Celmeli F, Klein C, AlKhater SA, Casanova J-L, Puel A, Genetic, immunological, and clinical features of patients with bacterial and fungal infections due to inherited IL-17RA deficiency. Proc Natl Acad Sci U S A. 113, E8277–E8285 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, Migaud M, Israel L, Chrabieh M, Audry M, Gumbleton M, Toulon A, Bodemer C, El-Baghdadi J, Whitters M, Paradis T, Brooks J, Collins M, Wolfman NM, Al-Muhsen S, Galicchio M, Abel L, Picard C, Casanova J-L, Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science. 332, 65–68 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ling Y, Cypowyj S, Aytekin C, Galicchio M, Camcioglu Y, Nepesov S, Ikinciogullari A, Dogu F, Belkadi A, Levy R, Migaud M, Boisson B, Bolze A, Itan Y, Goudin N, Cottineau J, Picard C, Abel L, Bustamante J, Casanova J-L, Puel A, Inherited IL-17RC deficiency in patients with chronic mucocutaneous candidiasis. J Exp Med. 212, 619–631 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boisson B, Wang C, Pedergnana V, Wu L, Cypowyj S, Rybojad M, Belkadi A, Picard C, Abel L, Fieschi C, Puel A, Li X, Casanova J-L, An ACT1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis. Immunity. 39, 676–686 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li J, Ritelli M, Ma CS, Rao G, Habib T, Corvilain E, Bougarn S, Cypowyj S, Grodecká L, Lévy R, Béziat V, Shang L, Payne K, Avery DT, Migaud M, Boucherit S, Boughorbel S, Guennoun A, Chrabieh M, Rapaport F, Bigio B, Itan Y, Boisson B, Cormier-Daire V, Syx D, Malfait F, Zoppi N, Abel L, Freiberger T, Dietz HC, Marr N, Tangye SG, Colombi M, Casanova J-L, Puel A, Chronic mucocutaneous candidiasis and connective tissue disorder in humans with impaired JNK1-dependent responses to IL-17A/F and TGF-β. Sci Immunol. 4, eaax7965 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Beaucoudrey L, Puel A, Filipe-Santos O, Cobat A, Ghandil P, Chrabieh M, Feinberg J, von Bernuth H, Samarina A, Jannière L, Fieschi C, Stéphan J-L, Boileau C, Lyonnet S, Jondeau G, Cormier-Daire V, Le Merrer M, Hoarau C, Lebranchu Y, Lortholary O, Chandesris M-O, Tron F, Gambineri E, Bianchi L, Rodriguez-Gallego C, Zitnik SE, Vasconcelos J, Guedes M, Vitor AB, Marodi L, Chapel H, Reid B, Roifman C, Nadal D, Reichenbach J, Caragol I, Garty B-Z, Dogu F, Camcioglu Y, Gülle S, Sanal O, Fischer A, Abel L, Stockinger B, Picard C, Casanova J-L, Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. J Exp Med. 205, 1543–1550 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Okada S, Markle JG, Deenick EK, Mele F, Averbuch D, Lagos M, Alzahrani M, Al-Muhsen S, Halwani R, Ma CS, Wong N, Soudais C, Henderson LA, Marzouqa H, Shamma J, Gonzalez M, Martinez-Barricarte R, Okada C, Avery DT, Latorre D, Deswarte C, Jabot-Hanin F, Torrado E, Fountain J, Belkadi A, Itan Y, Boisson B, Migaud M, Arlehamn CSL, Sette A, Breton S, McCluskey J, Rossjohn J, de Villartay J-P, Moshous D, Hambleton S, Latour S, Arkwright PD, Picard C, Lantz O, Engelhard D, Kobayashi M, Abel L, Cooper AM, Notarangelo LD, Boisson-Dupuis S, Puel A, Sallusto F, Bustamante J, Tangye SG, Casanova J-L, IMMUNODEFICIENCIES. Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science. 349, 606–613 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kerner G, Laval G, Patin E, Boisson-Dupuis S, Abel L, Casanova J-L, Quintana-Murci L, Human ancient DNA analyses reveal the high burden of tuberculosis in Europeans over the last 2,000 years. Am J Hum Genet. 108, 517–524 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fieschi C, Casanova J-L, The role of interleukin-12 in human infectious diseases: only a faint signature. Eur J Immunol. 33, 1461–1464 (2003). [DOI] [PubMed] [Google Scholar]
- 50.Steinman L, A brief history of T(H)17, the first major revision in the T(H)1/T(H)2 hypothesis of T cell-mediated tissue damage. Nat Med. 13, 139–145 (2007). [DOI] [PubMed] [Google Scholar]
- 51.Teng MWL, Bowman EP, McElwee JJ, Smyth MJ, Casanova J-L, Cooper AM, Cua DJ, IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nat Med. 21, 719–729 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Stritesky GL, Yeh N, Kaplan MH, IL-23 promotes maintenance but not commitment to the Th17 lineage. J Immunol. 181, 5948–5955 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bloch Y, Bouchareychas L, Merceron R, Składanowska K, Van den Bossche L, Detry S, Govindarajan S, Elewaut D, Haerynck F, Dullaers M, Adamopoulos IE, Savvides SN, Structural Activation of Pro-inflammatory Human Cytokine IL-23 by Cognate IL-23 Receptor Enables Recruitment of the Shared Receptor IL-12Rβ1. Immunity. 48, 45–58.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J, A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 46, 310–315 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Itan Y, Shang L, Boisson B, Ciancanelli MJ, Markle JG, Martinez-Barricarte R, Scott E, Shah I, Stenson PD, Gleeson J, Cooper DN, Quintana-Murci L, Zhang S-Y, Abel L, Casanova J-L, The mutation significance cutoff: gene-level thresholds for variant predictions. Nat Methods. 13, 109–110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang P, Bigio B, Rapaport F, Zhang S-Y, Casanova J-L, Abel L, Boisson B, Itan Y, PopViz: a webserver for visualizing minor allele frequencies and damage prediction scores of human genetic variations. Bioinformatics. 34, 4307–4309 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Belkadi A, Pedergnana V, Cobat A, Itan Y, Vincent QB, Abhyankar A, Shang L, El Baghdadi J, Bousfiha A, Exome/Array Consortium, Alcais A, Boisson B, Casanova J-L, Abel L, Whole-exome sequencing to analyze population structure, parental inbreeding, and familial linkage. Proc Natl Acad Sci U S A. 113, 6713–6718 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Woods CG, Cox J, Springell K, Hampshire DJ, Mohamed MD, McKibbin M, Stern R, Raymond FL, Sandford R, Malik Sharif S, Karbani G, Ahmed M, Bond J, Clayton D, Inglehearn CF, Quantification of homozygosity in consanguineous individuals with autosomal recessive disease. Am J Hum Genet. 78, 889–896 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thompson EA, The estimation of pairwise relationships. Ann Hum Genet. 39, 173–188 (1975). [DOI] [PubMed] [Google Scholar]
- 60.Rapaport F, Boisson B, Gregor A, Béziat V, Boisson-Dupuis S, Bustamante J, Jouanguy E, Puel A, Rosain J, Zhang Q, Zhang S-Y, Gleeson JG, Quintana-Murci L, Casanova J-L, Abel L, Patin E, Negative selection on human genes underlying inborn errors depends on disease outcome and both the mode and mechanism of inheritance. Proc Natl Acad Sci U S A. 118, e2001248118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Itan Y, Shang L, Boisson B, Patin E, Bolze A, Moncada-Vélez M, Scott E, Ciancanelli MJ, Lafaille FG, Markle JG, Martinez-Barricarte R, de Jong SJ, Kong X-F, Nitschke P, Belkadi A, Bustamante J, Puel A, Boisson-Dupuis S, Stenson PD, Gleeson JG, Cooper DN, Quintana-Murci L, Claverie J-M, Zhang S-Y, Abel L, Casanova J-L, The human gene damage index as a gene-level approach to prioritizing exome variants. Proc Natl Acad Sci U S A. 112, 13615–13620 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.de Paus RA, Geilenkirchen MA, van Riet S, van Dissel JT, van de Vosse E, Differential expression and function of human IL-12Rβ2 polymorphic variants. Mol Immunol. 56, 380–389 (2013). [DOI] [PubMed] [Google Scholar]
- 63.Martínez-Barricarte R, de Jong SJ, Markle J, de Paus R, Boisson-Dupuis S, Bustamante J, van de Vosse E, Fleckenstein B, Casanova J-L, Curr Protoc Immunol, in press, doi: 10.1002/cpim.15. [DOI] [PubMed] [Google Scholar]
- 64.Liu L, Okada S, Kong X-F, Kreins AY, Cypowyj S, Abhyankar A, Toubiana J, Itan Y, Audry M, Nitschke P, Masson C, Toth B, Flatot J, Migaud M, Chrabieh M, Kochetkov T, Bolze A, Borghesi A, Toulon A, Hiller J, Eyerich S, Eyerich K, Gulácsy V, Chernyshova L, Chernyshov V, Bondarenko A, Grimaldo RMC, Blancas-Galicia L, Beas IMM, Roesler J, Magdorf K, Engelhard D, Thumerelle C, Burgel P-R, Hoernes M, Drexel B, Seger R, Kusuma T, Jansson AF, Sawalle-Belohradsky J, Belohradsky B, Jouanguy E, Bustamante J, Bué M, Karin N, Wildbaum G, Bodemer C, Lortholary O, Fischer A, Blanche S, Al-Muhsen S, Reichenbach J, Kobayashi M, Rosales FE, Lozano CT, Kilic SS, Oleastro M, Etzioni A, Traidl-Hoffmann C, Renner ED, Abel L, Picard C, Maródi L, Boisson-Dupuis S, Puel A, Casanova J-L, Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med. 208, 1635–1648 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan C-H, Myung P, Plikus MV, Nie Q, Inference and analysis of cell-cell communication using CellChat. Nat Commun. 12, 1088 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sallusto F, Heterogeneity of Human CD4(+) T Cells Against Microbes. Annu Rev Immunol. 34, 317–334 (2016). [DOI] [PubMed] [Google Scholar]
- 67.Staels F, Lorenzetti F, De Keukeleere K, Willemsen M, Gerbaux M, Neumann J, Tousseyn T, Pasciuto E, De Munter P, Bossuyt X, Gijsbers R, Liston A, Humblet-Baron S, Schrijvers R, A Novel Homozygous Stop Mutation in IL23R Causes Mendelian Susceptibility to Mycobacterial Disease. J Clin Immunol (2022), doi: 10.1007/s10875-022-01320-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lévy R, Langlais D, Béziat V, Rapaport F, Rao G, Lazarov T, Bourgey M, Zhou YJ, Briand C, Moriya K, Ailal F, Avery DT, Markle J, Lim AI, Ogishi M, Yang R, Pelham S, Emam M, Migaud M, Deswarte C, Habib T, Saraiva LR, Moussa EA, Guennoun A, Boisson B, Belkaya S, Martinez-Barricarte R, Rosain J, Belkadi A, Breton S, Payne K, Benhsaien I, Plebani A, Lougaris V, Di Santo JP, Neven B, Abel L, Ma CS, Bousfiha AA, Marr N, Bustamante J, Liu K, Gros P, Geissmann F, Tangye SG, Casanova J-L, Puel A, Inherited human c-Rel deficiency disrupts myeloid and lymphoid immunity to multiple infectious agents. J Clin Invest. 131, e150143 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, Parente E, Filì L, Ferri S, Frosali F, Giudici F, Romagnani P, Parronchi P, Tonelli F, Maggi E, Romagnani S, Phenotypic and functional features of human Th17 cells. J Exp Med. 204, 1849–1861 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ, IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 201, 233–240 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, Ho AW, Hai JH, Yu JJ, Jung JW, Filler SG, Masso-Welch P, Edgerton M, Gaffen SL, Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med. 206, 299–311 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kagami S, Rizzo HL, Kurtz SE, Miller LS, Blauvelt A, IL-23 and IL-17A, but not IL-12 and IL-22, are required for optimal skin host defense against Candida albicans. J Immunol. 185, 5453–5462 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kerner G, Laval G, Patin E, Boisson-Dupuis S, Abel L, Casanova J-L, Quintana-Murci L, Human ancient DNA analyses reveal the high burden of tuberculosis in Europeans over the last 2,000 years. Am J Hum Genet. 108, 517–524 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gaffen SL, Jain R, Garg AV, Cua DJ, The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. 14, 585–600 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD, Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 421, 744–748 (2003). [DOI] [PubMed] [Google Scholar]
- 76.Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, Steinhart AH, Abraham C, Regueiro M, Griffiths A, Dassopoulos T, Bitton A, Yang H, Targan S, Datta LW, Kistner EO, Schumm LP, Lee AT, Gregersen PK, Barmada MM, Rotter JI, Nicolae DL, Cho JH, A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 314, 1461–1463 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, Ripke S, Lee JC, Jostins L, Shah T, Abedian S, Cheon JH, Cho J, Dayani NE, Franke L, Fuyuno Y, Hart A, Juyal RC, Juyal G, Kim WH, Morris AP, Poustchi H, Newman WG, Midha V, Orchard TR, Vahedi H, Sood A, Sung JY, Malekzadeh R, Westra H-J, Yamazaki K, Yang S-K, International Multiple Sclerosis Genetics Consortium, International IBD Genetics Consortium, Barrett JC, Alizadeh BZ, Parkes M, Bk T, Daly MJ, Kubo M, Anderson CA, Weersma RK, Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 47, 979–986 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, Butte AJ, Bhattacharya M, Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol. 20, 163–172 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Monaco G, Lee B, Xu W, Mustafah S, Hwang YY, Carré C, Burdin N, Visan L, Ceccarelli M, Poidinger M, Zippelius A, Pedro de Magalhães J, Larbi A, RNA-Seq Signatures Normalized by mRNA Abundance Allow Absolute Deconvolution of Human Immune Cell Types. Cell Rep. 26, 1627–1640.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Firth D, Bias reduction of maximum likelihood estimates. Biometrika. 80, 27–38 (1993). [Google Scholar]
- 81.logistf package (https://cran.r-project.org/web/packages/logistf/index.html). [Google Scholar]
- 82.Khan T, Rahman M, Ahmed I, Al Ali F, Jithesh PV, Marr N, Human leukocyte antigen class II gene diversity tunes antibody repertoires to common pathogens. Front Immunol. 13, 856497 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mina MJ, Kula T, Leng Y, Li M, de Vries RD, Knip M, Siljander H, Rewers M, Choy DF, Wilson MS, Larman HB, Nelson AN, Griffin DE, de Swart RL, Elledge SJ, Measles virus infection diminishes preexisting antibodies that offer protection from other pathogens. Science. 366, 599–606 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xu GJ, Kula T, Xu Q, Li MZ, Vernon SD, Ndung’u T, Ruxrungtham K, Sanchez J, Brander C, Chung RT, O’Connor KC, Walker B, Larman HB, Elledge SJ, Viral immunology. Comprehensive serological profiling of human populations using a synthetic human virome. Science. 348, aaa0698 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mohan D, Wansley DL, Sie BM, Noon MS, Baer AN, Laserson U, Larman HB, Publisher Correction: PhIP-Seq characterization of serum antibodies using oligonucleotide-encoded peptidomes. Nat Protoc. 14, 2596 (2019). [DOI] [PubMed] [Google Scholar]
- 86.Khan T, Rahman M, Ali FA, Huang SSY, Ata M, Zhang Q, Bastard P, Liu Z, Jouanguy E, Béziat V, Cobat A, Nasrallah GK, Yassine HM, Smatti MK, Saeed A, Vandernoot I, Goffard J-C, Smits G, Migeotte I, Haerynck F, Meyts I, Abel L, Casanova J-L, Hasan MR, Marr N, Distinct antibody repertoires against endemic human coronaviruses in children and adults. JCI Insight. 6, 144499 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ogishi M, Arias AA, Yang R, Han JE, Zhang P, Rinchai D, Halpern J, Mulwa J, Keating N, Chrabieh M, Lainé C, Seeleuthner Y, Ramírez-Alejo N, Nekooie-Marnany N, Guennoun A, Muller-Fleckenstein I, Fleckenstein B, Kilic SS, Minegishi Y, Ehl S, Kaiser-Labusch P, Kendir-Demirkol Y, Rozenberg F, Errami A, Zhang S-Y, Zhang Q, Bohlen J, Philippot Q, Puel A, Jouanguy E, Pourmoghaddas Z, Bakhtiar S, Willasch AM, Horneff G, Llanora G, Shek LP, Chai LYA, Tay SH, Rahimi HH, Mahdaviani SA, Nepesov S, Bousfiha AA, Erdeniz EH, Karbuz A, Marr N, Navarrete C, Adeli M, Hammarstrom L, Abolhassani H, Parvaneh N, Al Muhsen S, Alosaimi MF, Alsohime F, Nourizadeh M, Moin M, Arnaout R, Alshareef S, El-Baghdadi J, Genel F, Sherkat R, Kiykim A, Yücel E, Keles S, Bustamante J, Abel L, Casanova J-L, Boisson-Dupuis S, Impaired IL-23-dependent induction of IFN-γ underlies mycobacterial disease in patients with inherited TYK2 deficiency. J Exp Med. 219, e20220094 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, ru Loh P, Raychaudhuri S, Fast, sensitive and accurate integration of single-cell data with Harmony. Nature Methods. 16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, Hoffman P, Stoeckius M, Papalexi E, Mimitou EP, Jain J, Srivastava A, Stuart T, Fleming LM, Yeung B, Rogers AJ, McElrath JM, Blish CA, Gottardo R, Smibert P, Satija R, Integrated analysis of multimodal single-cell data. Cell. 184, 3573–3587.e29 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology. 15, 1–21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK, limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan C-H, Myung P, V Plikus M, Nie Q, Inference and analysis of cell-cell communication using CellChat. Nature Communications. 12, 1088 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.R Core Team R: A Language and Environment for Statistical Computing (2018). [Google Scholar]
- 94.Monaco G, Lee B, Xu W, Mustafah S, Hwang YY, Carré C, Burdin N, Visan L, Ceccarelli M, Poidinger M, Zippelius A, Pedro de Magalhães J, Larbi A, RNA-Seq Signatures Normalized by mRNA Abundance Allow Absolute Deconvolution of Human Immune Cell Types. Cell reports. 26, 1627–1640.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ma CS, Avery DT, Chan A, Batten M, Bustamante J, Boisson-Dupuis S, Arkwright PD, Kreins AY, Averbuch D, Engelhard D, Magdorf K, Kilic SS, Minegishi Y, Nonoyama S, French MA, Choo S, Smart JM, Peake J, Wong M, Gray P, Cook MC, Fulcher DA, Casanova J-L, Deenick EK, Tangye SG, Functional STAT3 deficiency compromises the generation of human T follicular helper cells. Blood. 119, 3997–4008 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ma CS, Chew GYJ, Simpson N, Priyadarshi A, Wong M, Grimbacher B, Fulcher DA, Tangye SG, Cook MC, Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med. 205, 1551–1557 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Geiger R, Duhen T, Lanzavecchia A, Sallusto F, Human naive and memory CD4+ T cell repertoires specific for naturally processed antigens analyzed using libraries of amplified T cells. J Exp Med. 206, 1525–1534 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Private homozygous IL23R variants in four Iranian kindreds
Supplementary Figure 2: Loss-of-function IL23R alleles and AR complete IL23R deficiency
Supplementary Figure 3: Development of peripheral mononuclear hematopoietic cells in IL-23R-deficient patients
Supplementary Figure 4: Impaired ex vivo IL-23-mediated production of IFN-γ in cells from patients with IL-23R deficiency
Supplementary Figure 5: Normal development of BCG- and C. albicans-specific memory CD4+ T cells in patients with inherited IL-23R deficiency
Supplementary Figure 6: Impaired ex vivo IL-23-mediated production of IL-17 cytokines in patients with inherited IL-23R deficiency
Table S1: Summary of the medical history of patients with inherited IL-23R deficiency
Table S2: List of all homozygous coding, essential-splicing site and splice-site variants with a CADD above the MSC not present in the homozygous state in GnomAD but detected in the analyses of the exomes of P1 to P6
Table S3: Genotypes of IL-12Rβ1- and IL-12Rb2-deficient patients and STAT1-GOF patients included as controls
Table S4: Gating strategy for the assessment of STAT phosphorylation by mass cytometry (CyTOF).
Table S5: Gating strategy for deep immunophenotyping by mass cytometry (CyTOF).
Table S6: Gating strategy for deep immunophenotyping by spectral flow cytometry.
Table S7: Gating strategy for the ex vivo evaluation of IFN-γ+ and IL-17A+ cells after PBMC stimulation with IL-23 or IL-12, in the presence or absence of BCG infection.
Table S8: Gating strategy for the ex vivo evaluation of BCG-reactive memory CD4+ T cells.
Table S9 : Sequence of primers used for this study
Data Availability Statement
All raw and processed data and biological materials, including immortalized cell lines from patients, are available upon request from the corresponding authors under a material/data transfer agreement.