

Keywords: Alzheimer’s disease, amyloid-β, expression, heparan sulfate proteoglycan, metabolism
Abstract
Alzheimer’s disease (AD) is the most common form of dementia. Currently, there is no effective treatment for AD, as its etiology remains poorly understood. Mounting evidence suggests that the accumulation and aggregation of amyloid-β peptides (Aβ), which constitute amyloid plaques in the brain, is critical for initiating and accelerating AD pathogenesis. Considerable efforts have been dedicated to shedding light on the molecular basis and fundamental origins of the impaired Aβ metabolism in AD. Heparan sulfate (HS), a linear polysaccharide of the glycosaminoglycan family, co-deposits with Aβ in plaques in the AD brain, directly binds and accelerates Aβ aggregation, and mediates Aβ internalization and cytotoxicity. Mouse model studies demonstrate that HS regulates Aβ clearance and neuroinflammation in vivo. Previous reviews have extensively explored these discoveries. Here, this review focuses on the recent advancements in understanding abnormal HS expression in the AD brain, the structural aspects of HS-Aβ interaction, and the molecules involved in modulating Aβ metabolism through HS interaction. Furthermore, this review presents a perspective on the potential effects of abnormal HS expression on Aβ metabolism and AD pathogenesis. In addition, the review highlights the importance of conducting further research to differentiate the spatiotemporal components of HS structure and function in the brain and AD pathogenesis.
INTRODUCTION
Alzheimer’s disease (AD) is the most prevalent type of dementia, severely affecting patients’ quality of life (1). Currently, limited treatments are available to slow disease progression (2), but often lead to notable side effects (3). AD is marked by pathological lesions that include the accumulation of amyloid plaques in the cerebral parenchyma and vasculature (cerebral amyloid angiopathy, CAA), the formation of neurofibrillary tangles (NFT) in the neurons, and neuroinflammation (1, 4, 5). Mounting evidence supports the amyloid cascade hypothesis, which states that the accumulation and aggregation of amyloid-β peptides (Aβ) are crucial factors responsible for the initiation and acceleration of AD pathogenesis (6, 7). Compelling evidence suggests that amyloid plaques develop due to an imbalance between Aβ production and clearance from the brain. The equilibrium of Aβ production and clearance is determined by the expression and processing of the amyloid precursor protein (APP), the distribution and degradation of Aβ within the brain, and the elimination of Aβ via the cerebral blood vasculature (8). Therefore, a comprehensive understanding of the molecular mechanisms underlying amyloid plaque formation is essential for developing effective therapies to combat this devastating disease (9).
APP is a single-pass transmembrane protein that consists of a large N-terminal ectodomain and a shorter C-terminus that contains a crucial YENPTY sequence required for intracellular protein-sorting (Fig. 1) (10, 11). APP undergoes alternative splicing, generating eight isoforms, of which the 695 amino acid isoform is expressed predominantly in the central nervous system (CNS), whereas the 751 and 770 amino acid isoforms are more ubiquitously expressed (10, 11). APP is transported through secretory and endocytic pathways and undergoes sequential processing, leading to nonamyloidogenic and amyloidogenic end-products (Fig. 1). The nonamyloidogenic pathway begins with α-secretase cleaving the APP ectodomain to release soluble APPα (sAPPα) into the extracellular space. The remaining C-terminal fragment of 83 amino acids (CTF83) remains embedded in the plasma membrane. Cleavage of CTF83 by γ-secretase releases a small p3 fragment into the extracellular space and the APP intracellular domain (AICD) into the cytoplasm. The amyloidogenic pathway starts with β-secretase cleaving APP, releasing sAPPβ into the extracellular space. The remaining C-terminal fragment contains 99 amino acids (CTF99) and is further cleaved by γ-secretase to release Aβ into the extracellular space and AICD into the cytoplasm (10–14). The Aβ peptides with 40 or 42 residues in length (Aβ40 and Aβ42, respectively) are found in the most pathogenic plaques in AD. Aβ is primarily cleared through the vasculature via multiple pathways in the brain, including transport through the blood-brain barrier (BBB) to peripheral blood circulation, local degradation by cerebral vascular smooth muscle cells and pericytes, and diffuse drainage through perivascular space and the meningeal lymphatic system (15–22). In addition, Aβ is degraded locally in parenchyma by microglia, neurons, and extracellular proteolytic enzymes, including neprilysin, insulin-degrading enzyme, and matrix metalloproteinases 2 and 9 (23, 24). The brain coexists with different forms of Aβ, including nontoxic monomers, toxic and soluble oligomers, and aggregated amyloid fibrils (25). Nontoxic monomers and toxic soluble oligomers are present in a dynamic equilibrium, whereas aggregated fibrils are irreversible once formed (25). Excess Aβ production or disruption of Aβ clearance leads to the accumulation of toxic soluble Aβ oligomers and insoluble, aggregated Aβ fibrils, ultimately forming amyloid plaques in AD. The molecular mechanisms and root causes responsible for Aβ accumulation and deposition in AD are not entirely understood.
Figure 1.
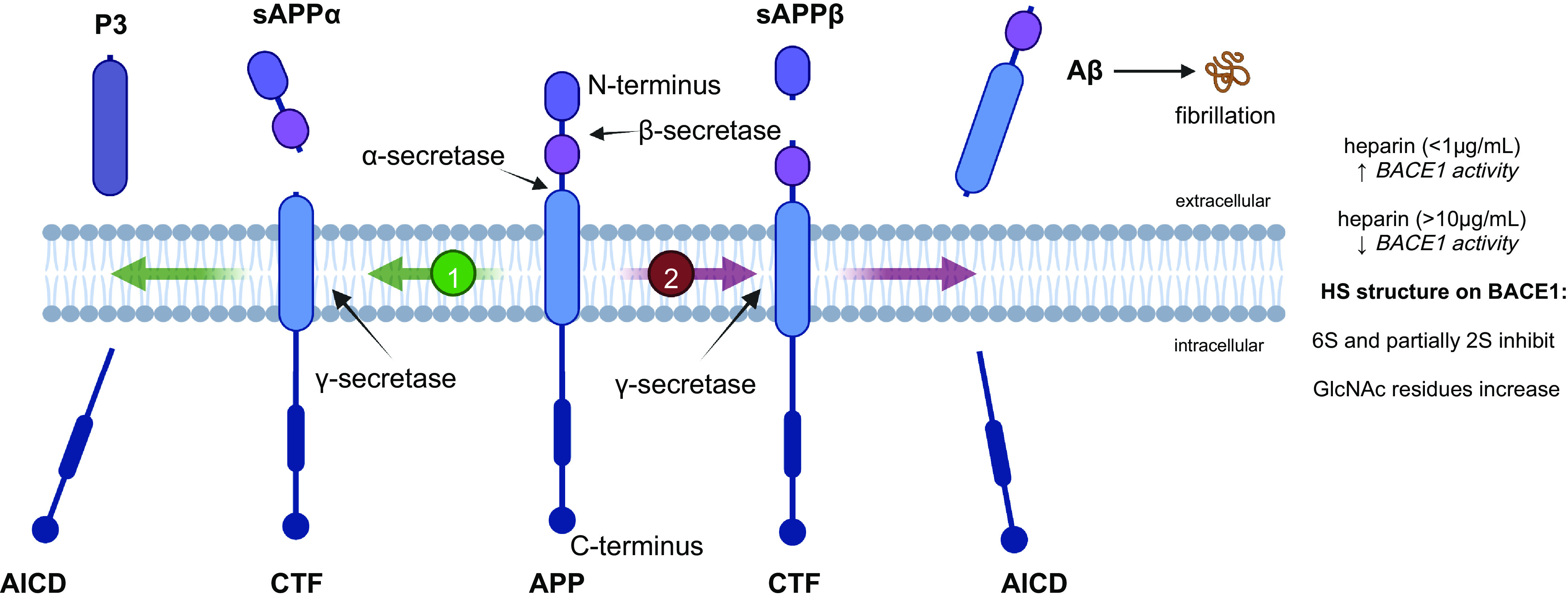
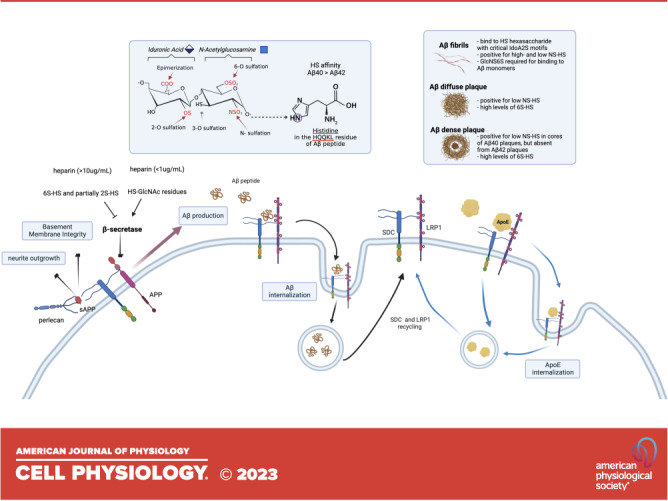
Amyloid precursor protein (APP) cleavage by α-, β-, and γ-secretase. APP undergoes sequential cleavage following the nonamyloidogenic pathway (1) or the pro-amyloidogenic pathway (2). The nonamyloidogenic pathway is mediated by α- and γ-secretase, which produce the fragments soluble (s)APPα, P3, and amyloid precursor protein intracellular domain (AICD). The pro-amyloidogenic pathway is processed by β- and γ-secretase, which yields the fragments sAPPβ, amyloid β (Aβ), and AICD. The Aβ monomers aggregate to form oligomers and fibrils. Aβ, sAPPα and sAPPβ bind heparan sulfate (HS). The γ-secretase (beta-site APP cleaving enzyme 1, BACE1) binds HS and heparin, and this binding depends on 6S and 2S modifications and the level of N-acetylglucosamine (GlcNAc) residues. CTF, C-terminal fragment.
Heparan sulfate (HS) is a linear polysaccharide that belongs to the glycosaminoglycan (GAG) family (Fig. 2) (26, 27). HS is synthesized in the Golgi apparatus through a multiple-step process involving multiple enzymes (26, 27). The synthesis of HS starts with Exostosin-like 3 (EXTL3), which adds an N-acetylglucosamine residue (GlcNAc) to the glucuronic acid (GlcA) residue of a tetrasaccharide linker. This linker consists of GlcA-galactose (Gal)-Gal-xylose (Xyl) and attaches to protein cores to form heparan sulfate proteoglycans (HSPG). Subsequently, the heterodimer Exostosin-1/2 (EXT1/2) co-polymerases elongate the HS chain by adding GlcA and GlcNAc alternatively. The growing HS chain undergoes various modifications, including the initial N-sulfation (NS) of GlcNAc by N-deacetylase/N-sulfotransferase-1–4 (NDST1-4), and the following conversion of GlcA to iduronic acid (IdoA) by GlcA epimerase (GLCE), 2-O-sulfation (2S) of IdoA and, less frequently, GlcA by HS 2-O-sulfotransferase (HS2ST) and 6-O (6S) and 3-O (3S) sulfation of N-sulfated GlcNAc (GlcNS) by HS 6-O-sulfotransferase1-3 (HS6ST1-3) and HS 3-O-sulfotransferase1-6 (HS3ST1-6), respectively. After its biosynthesis, HS can undergo remodeling on the cell surface and in the extracellular matrix (ECM) by the 6-O-endosulfatases (SULF1-2), which remove 6S from GlcNS and causes changes in the structure of HS. HS can also be cleaved by heparanase (HPSE, not shown in Fig. 2), which breaks down the HS chain into smaller fragments (28–32). The abundance and structure of HS expression are determined by the enzymes responsible for its biosynthesis, remodeling, and degradation.
Figure 2.
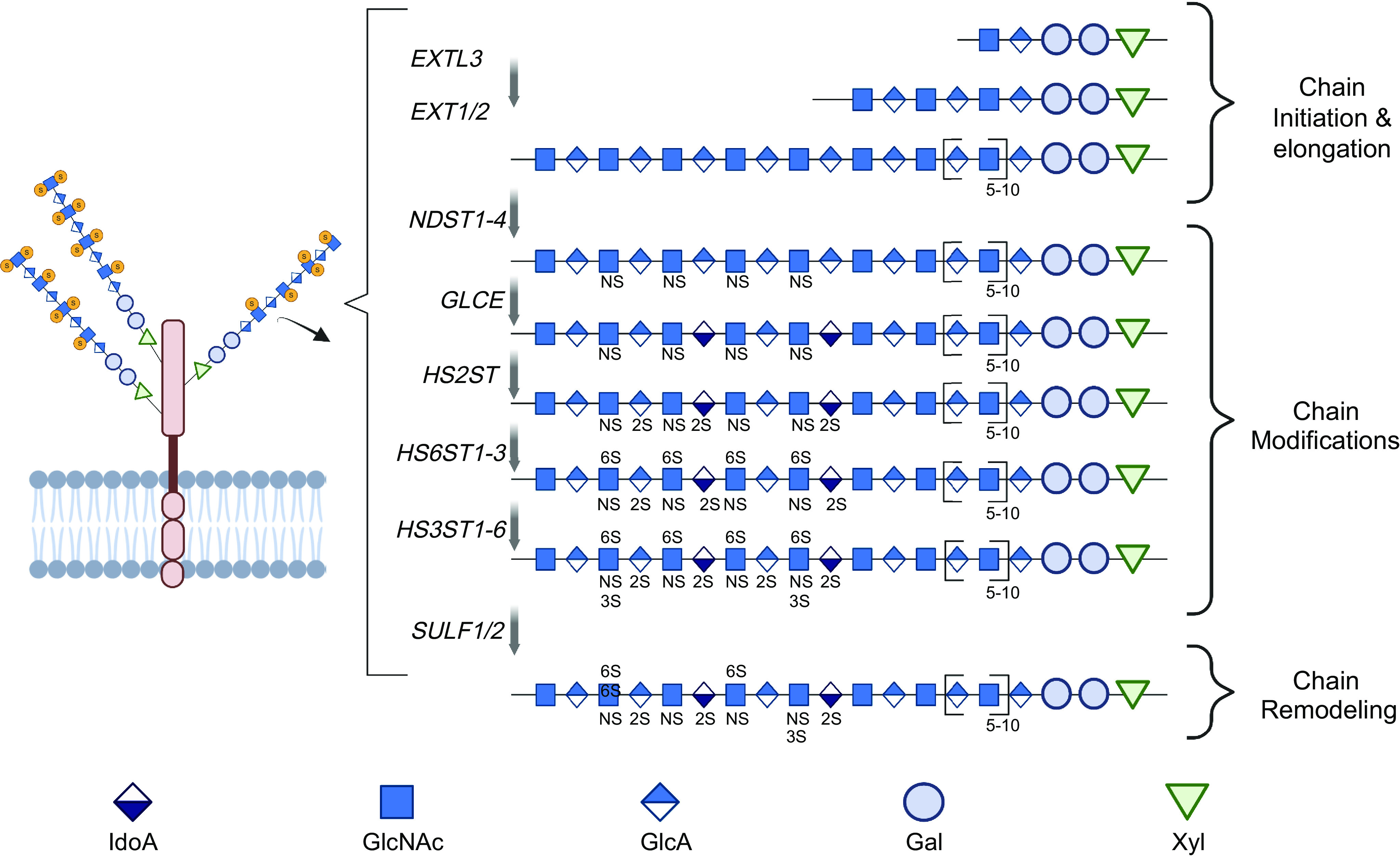
Heparan sulfate proteoglycan (HSPG) and heparan sulfate (HS) biosynthesis and remodeling. HS is covalently attached to core proteins to form HSPG via the Xyl-Gal-Gal-GlcA tetrasaccharide linkage. HS biosynthesis includes chain initiation and elongation catalyzed by exostosin-like glycosyltransferase 3 (EXTL3) and exostosin glycosyltransferase 1/2 (EXT1/2), respectively, and concurrent chain modification catalyzed by N-deacetylase/N-sulfotransferase 1–4 (NDST1-4), glucuronic acid epimerase (GLCE), heparan sulfate 2-O-sulfotransferase (HS2ST), heparan sulfate 6-O-sulfotransferase 1–3 (HS6ST1-3), and heparan sulfate 3-O-sulfotransferase 1–6 (HS3ST1-6). Expressed HS can be remodeled through removal of 6S by 6-O-endosulfatase 1–2 (SULF1-2) and cleavage by heparanase (HPSE) (not shown).
The tetrasaccharide linker of HS covalently binds to selected serine residues of protein cores via O-glycosylation to form HSPGs that are expressed on the cell surfaces, in the ECM, and in intracellular secretory vesicles (26, 27). The HS chain of HSPGs interacts with numerous protein ligands, including growth factors, growth factor receptors, morphogens, cytokines, matrix proteins, and others. Through these interactions, HS modulates various physiological and pathological processes, such as organ development, leukocyte trafficking, tumorigenesis, and lipid metabolism (28, 33–40). HS binds protein ligands through electrostatic interactions imposed primarily by negatively charged sulfate residues and unique binding sites composed of varying sulfation and epimerization modifications in specific arrangements (29, 30, 32, 39, 41–44). In patients with AD and mouse models, HS co-deposits with Aβ in plaques in the cerebral parenchyma and vasculature (45–53). Biochemical and in vitro cell studies demonstrate that HS binds to Aβ, accelerating Aβ aggregation (54–59) and mediating Aβ cellular internalization and cytotoxicity (60–64). In AD mouse models, HS reduction through overexpression of HPSE, mainly in neurons and astrocytes, or neuronal HS ablation attenuates neuroinflammation and lowers Aβ burden in the brain, demonstrating that HS functionally promotes amyloidopathy in AD (24, 65, 66). Furthermore, the accumulation of HS in neurons at birth in a mouse model for a severe lysosomal storage disease called mucopolysaccharidosis type IIIA results in fibril formation and aggregation of Aβ as well as other amyloidogenic proteins, including α-synuclein, prion protein, and Tau, suggesting that HS may be a causative factor in Aβ fibrillization in AD (67). Therefore, inhibiting the Aβ-HS interaction may be a promising therapeutic strategy for AD treatment. These key findings have been discussed in detail in reviews by Snow et al. (52, 68), Ariga et al. (25), Zhang et al. (69), and Jin et al. (70). This review discusses recent studies reporting aberrant HS expression in patients with AD, the structural characteristics of HS-Aβ interaction, and the molecules with which HS interacts to modulate Aβ metabolism. Our perspective on the potential implications of aberrant HS expression in Aβ metabolism and AD pathogenesis is also provided.
ABERRANT HS EXPRESSION IN AD
Several studies documented clear aberrant expression of HS in the AD brain. Shimizu et al. (45) analyzed 25 patients with AD and found a 9.3- and 6.6-fold increase of HS in the hippocampus and the superior frontal gyrus, respectively. Compared with nondemented elderly subjects, HS in the AD brain is more densely concentrated in the thickened basement membrane adjacent to the endothelial cells of capillary vessels but not inside the endothelial cells and in the core of amyloid plaques (45, 59). These observations suggest that the marked HS increase in AD brains is mainly from the capillary basement membrane and senile plaques (45). Huynh et al. (46) and Wang et al. (71) confirmed the elevation of HS in hippocampus and frontal cortex of patients with AD, respectively. Despite an earlier study observing no alteration in HS structure in AD (72), Huynh et al. observed that GAGs from the AD hippocampus, including HS and chondroitin sulfate (CS), had altered growth factor binding capacities. AD GAGs showed decreased capacities to bind fibroblast growth factor-1 and -2 and vascular endothelial growth factor 165, and increased capabilities to bind heparin-binding epidermal growth factor-like growth factor, pleiotrophin, and tau, indicating a structural alteration in AD HS (46). Very recently, Wang et al. (71) analyzed the composition of AD HS and detected a significant increase in multiple sulfated disaccharides (ΔUA2S-GlcNS, ΔUA2S-GlcNAc, ΔUA-GlcNAc6S, and ΔUA2S-GlcNAc6S) and a tetrasaccharide with rare 3S (ΔUA-GlcNAc6S-GlcA-GlcNS3S6S) in human AD frontal cortex. These studies establish aberrant HS expression in the AD brain, affecting both HS expression levels and structure.
The composition, sulfation pattern, remodeling, and length of HS in tissue are determined by the expression profiles of HS biosynthetic and remodeling enzymes (Fig. 2). One study analyzed the postmortem hippocampus of 8 control and 8 patients with AD and found that multiple HS-regulated genes, including NDST2, GLCE, HS2ST, HS6ST1, HS6ST2, HS3ST2, HS3ST3A1, HS3ST3B1, and HPSE, were upregulated in AD (73). Another study examined 5 AD and 5 control human brains and observed similar results, reporting upregulated expression of NDST2, HS3ST2, HS3ST4, GLCE, and HPSE in the AD brains (46). Immunostaining of SULF1 and SULF2 in the brains of 20 patients with AD and 20 age-and gender-matched cognitively normal controls revealed that SULF2 expression in AD cases was significantly decreased in the parahippocampal gyrus and the frontal lobe gray matter (74). There were no differences in SULF1 expression in the parahippocampus or frontal lobe of AD cases and controls. In addition, there were no differences in SULF1 or SULF2 expression in the white matter of AD cases compared with cognitively normal controls (74). A recent study examined the transcriptomic expression of enzymes responsible for HS biosynthesis and modification in different brain regions in 18 patients with AD grouped at mild, moderate, or severe stages (6 per stage) and compared with six healthy individuals (75). The examined brain regions include the amygdala, anterior hippocampus, posterior hippocampus, claustrum, calcarine fissure, globus pallidus, and cerebellum. Several genes were downregulated across the three AD stages and in multiple brain regions, including EXT1, NDST2, HS2ST, HS6ST3, and SULF2. The number of gene alterations was higher in moderate AD compared with mild AD (Fig. 3A). In severe AD, there were fewer alterations of genes related to the early stages of HS biosynthesis. However, several genes involved in the late steps of HS biosynthesis, namely HS3STs, HS6STs, and SULF1, were upregulated (Fig. 3A). The alterations in HS gene expression correlated with progressive brain atrophy, with the cerebellum having the highest number of affected genes (Fig. 3B) (75). These results indicate that the alterations in HS biosynthetic and remodeling gene expression depend on the brain region and stage of AD pathology (Fig. 3, A and B). Interestingly, the altered transcriptional expression in the hippocampus reported by Perez-Lopez et al. differed significantly from what was observed earlier by Huynh et al. (Table 1). Despite this, these studies have consistently observed upregulated expression of HS3ST genes (Fig. 3 and Table 1) (46, 73, 75), which is in agreement with the elevated HS 3-O sulfation in AD reported by Wang et al. (46, 71, 75). Notably, a recent study utilizing multilayer analysis of patients with AD revealed that the proteopathic nature of AD is not reflected at an RNA level (77), which may explain discrepancies in studies using immunostaining versus transcript quantification. When grouped into 14 modules, the genes related to the cell-ECM interaction module were preserved at an RNA level. In contrast, the module containing matrisome-related genes showed an elevation only at the protein level in patients with AD. This change was observed more prominently in the temporal cortex than in the frontal cortex. As many proteins in the matrisome module bind HS or are HSPGs (77), suggesting that HS might critically regulate the functions of matrisome and its dysregulation could contribute to AD pathogenesis.
Figure 3.
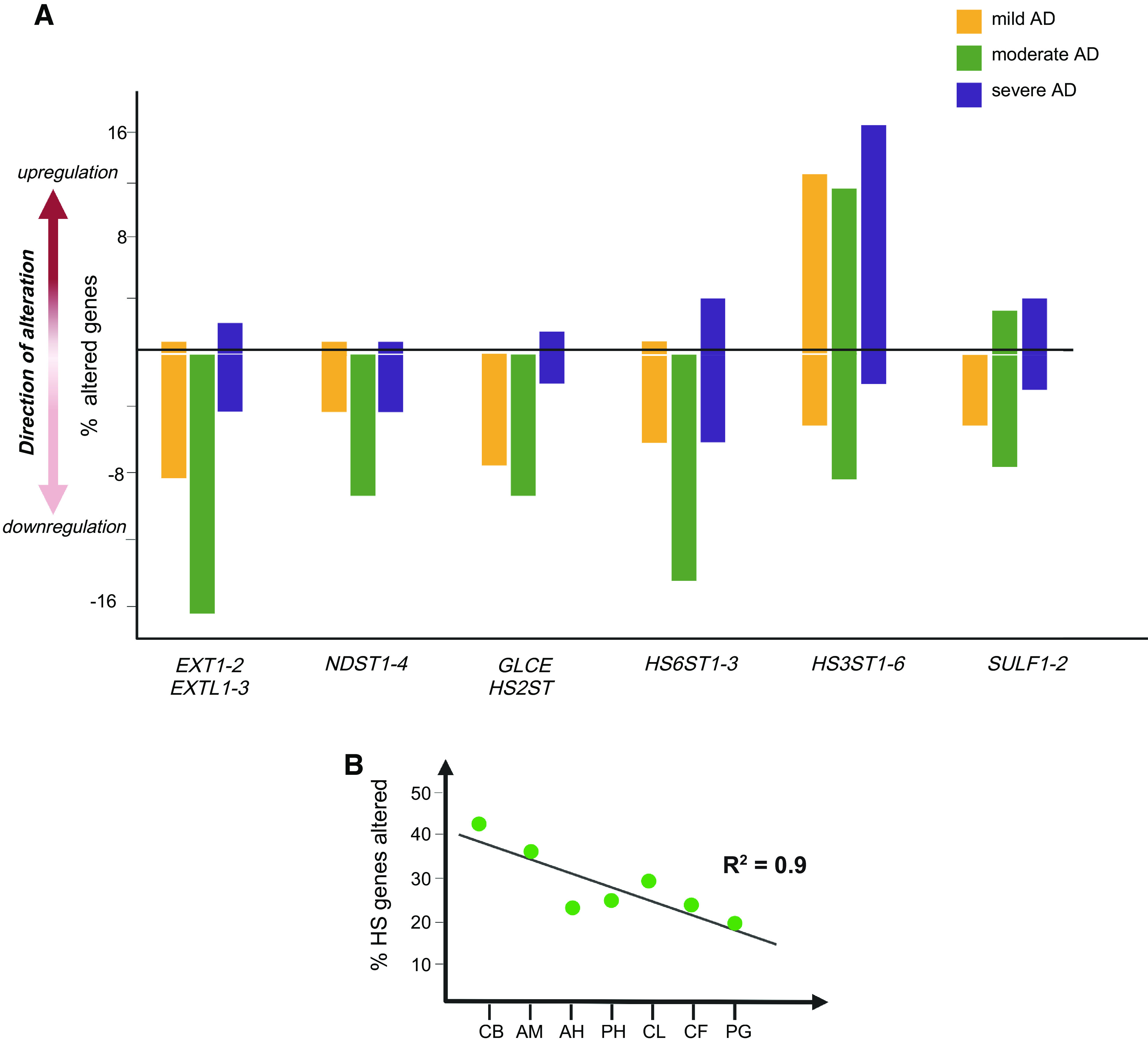
Dysregulation of genes responsible for heparan sulfate (HS) synthesis and remodeling in human Alzheimer’s disease (AD) brains. Summarized from the study reported by Perez-Lopez et al. (75). The expression of HS biosynthesis and 6-O-endosulfatase (SULF) genes was altered in human AD brains, with the degree of dysregulation varying depending on the gene family, the disease stage, and the brain region. A: all HS-related gene families had greater dysregulation in moderate AD compared to mild AD. Genes responsible for early- and middle-stage-HS biosynthesis, including Exostosins (EXTs), N-deacetylase/N-sulfotransferases (NDSTs), glucuronic acid epimerase (GLCE), and heparan sulfate 2-O-sulfotransferase (HS2ST) families, were downregulated in subjects with AD, while genes involved in later-stage HS biosynthesis, such as heparan sulfate 6-O-sulfotransferases (HS6STs) and heparan sulfate 3-O-Sulfotransferases (HS3STs), and the remodeling gene SULFs, tend to be upregulated in AD. B: the cerebellum (CB) exhibited the highest number of gene perturbations, and the numbers of gene perturbations and the brain regions fit a linear regression following a spatiotemporal pattern of the amygdala (AM), anterior hippocampus (AH), posterior hippocampus (PH), claustrum (CL), calcarine fissure (CF) and globus pallidus (PG).
Table 1.
Dysregulated expression of HS biosynthesis genes in AD hippocampus and temporal cortex reported by Huynh et al., Perez-Lopez et al., and Rosenthal et al.
| Sepulveda-Diaz et al. |
Huynh et al. |
Perez-Lopez et al. |
Rosenthal et al. |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Gene | Hippocampus | Hippocampus | Anterior Hippocampus Mild Moderate Severe | Posterior Hippocampus Mild Moderate Severe | Temporal Cortex and Cerebellum | ||||
| EXTL2 | ↓ | ||||||||
| EXTL3 | ↓ | ↓ | ↓ | ||||||
| EXT1 | ↓ | ↓ | |||||||
| EXT2 | ↓ | ↓ | |||||||
| NDST1 | ↓ | ↓ | |||||||
| NDST2 | ↑ | ↑ | ↓ | ↓ | |||||
| NDST3 | — | — | — | — | — | — | |||
| NDST4 | ↑ | ||||||||
| GLCE | ↑ | ↑ | ↑ | ↓ | |||||
| HS2ST | ↑ | ↓ | ↓ | ||||||
| HS6ST1 | ↑ | ↑ | |||||||
| HS6ST2 | ↑ | ↑ | ↓ | ↓ | ↓ | ||||
| HS6ST3 | ↓ | ↓ | ↓ | ↓ | |||||
| HS3ST1 | ↓ | ↓ | ↓ | ||||||
| HS3ST2 | ↑ | ↑ | ↓ | ↓ | |||||
| HS3ST3A1 | ↑ | ↑ | |||||||
| HS3ST1B1 | ↑ | ↑ | ↑ | ↑ | |||||
| HS3ST4 | ↑ | ↑ | ↓ | ↓ | |||||
| HS3ST5 | ↑ | ↑ | ↑ | ↑ | ↑ | ↓ | |||
| HS3ST6 | ↓ | ↓ | |||||||
Downward arrows indicate downregulation of gene expression; upward arrows indicate upregulation of gene expression; dashes indicate no detected transcripts. Sepulveda-Diaz et al. (73) analyzed the postmortem hippocampus from eight control and eight patients with AD (mean age = 76.8 ± 3.5 yr). Huynh et al. (46) analyzed the postmortem hippocampus from five control and five patients with AD (mean age = 72.3 ± 7.0 yr). Perez-Lopez et al. (75) analyzed the postmortem anterior and posterior hippocampus from six control and 18 patients with AD (6 mild, 6 moderate, and 6 severe AD) (mean age = 84 ± 11.0 yr). The gene transcript data from other brain regions are not included here (75). Rosenthal et al. (76) mapped the gene network landscape of AD by integrating genomics and transcriptomics data and observed altered HS gene expression in AD patients’ temporal cortex and cerebellum from a GWAS of 455,258 individuals with 71,880 proxy cases and 383,378 controls. EXT, exostosin glycosyltransferase; EXTL, exostosin like glycosyltransferase; GLCE, glucuronic acid epimerase; HS, heparan sulfate; HS2ST, heparan sulfate 2-O-sulfotransferase; HS3ST, heparan sulfate 3-O-sulfotransferase; HS6ST, heparan sulfate 6-O-sulfotransferase; NDST, N-deacetylase/N-sulfotransferase.
HEPARAN SULFATE-Aβ INTERACTION
Biochemical studies have shown that Aβ directly binds to HS and heparin, a highly sulfated HS analog. The major HS binding motif within Aβ has been identified as V12HHQKL17 (Fig. 4A) (49, 50, 68, 78–85). HS-Aβ binding depends on electrostatic interactions involving the imidazole side chain of the histidine residues of Aβ. This interaction is strengthened by increased sulfation and the size of HS and heparin (78, 86–89). Interestingly, HS binds to Aβ40 with higher affinity than Aβ42 (87, 90) and HS induces Aβ40, not Aβ42, to form maltese-cross spherical congophilic plaques identical to those seen in AD brain (68). Anti-HS phage display antibody staining has identified specific structures of HS that co-deposit with Aβ in the AD brain. The phage display antibodies EV3C3 and HS4C3 recognize fully N-sulfated motifs in the HS chain, whereas RB4EA12 and HS4E4 recognize HS regions with a lower degree of NS. The EV3C3 and HS4C3 antibodies stained strongly in both fibrillar and nonfibrillar Aβ plaques, whereas the RB4EA12 and HS4E4 antibodies stained fibrillar Aβ plaques only (Fig. 4B) (80). HS4E4 epitopes preferentially colocalized with Aβ40 in the cores of senile plaques and are absent from Aβ42-rich diffuse deposits in sporadic AD brains (Fig. 4B) (91). In agreement with the observations by Snow et al. (51), HSPGs were not found in diffuse deposits in the cerebellum but in the cores of plaque deposits in the hippocampus and cortex of AD brains. These results suggest that differently structured HS co-deposits with Aβ plaques. The HS structures that co-deposit with nonfibrillar Aβ plaques are more restricted, consisting of highly N-sulfated structures (80). In addition, the anti-HS phage display antibody RB4CD12 recognizes highly sulfated HS domains (92) and strongly stained diffuse and neurotic Aβ plaques in human AD and transgenic AD mouse models (93). The RB4CD12 staining diminished when the tissues were pretreated with extracellular sulfatases SULF1 and SULF2, suggesting that the Aβ-binding HS moieties contain 6-O-sulfation (Fig. 4A) (93). These immunostaining studies indicate that the Aβ-binding HS moieties within Aβ plaques. have common NS and 6S modifications in vivo.
Figure 4.
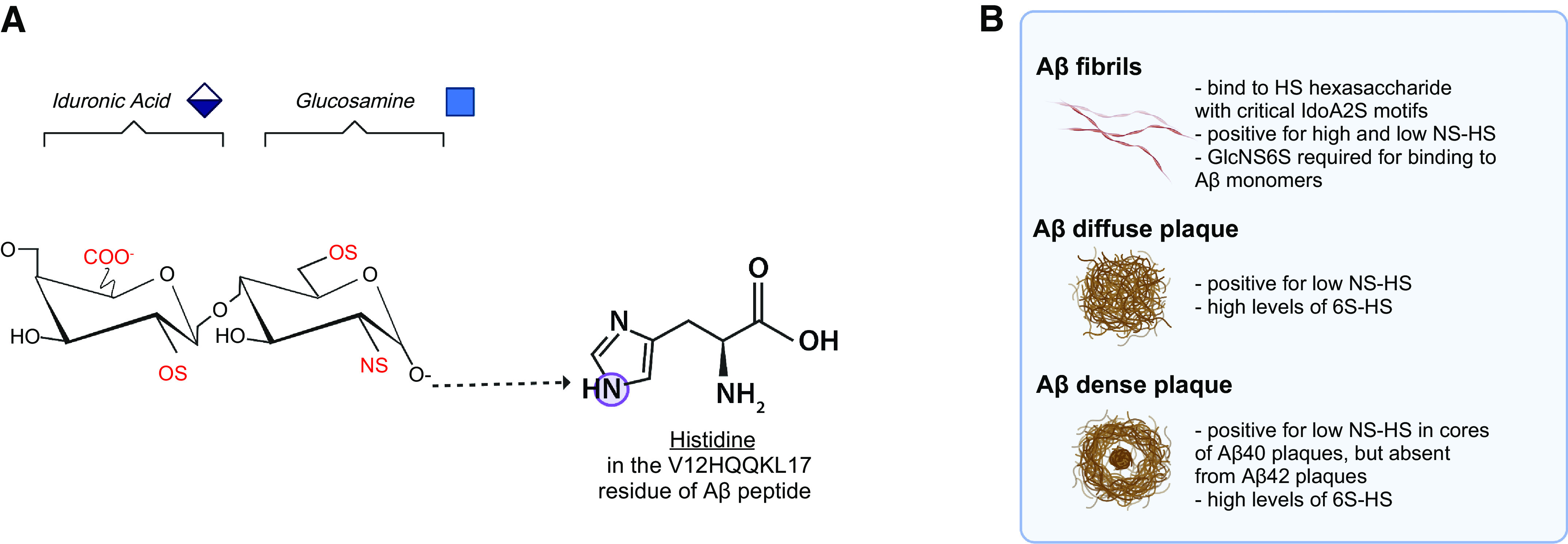
Interaction between heparan sulfate (HS) and amyloid beta (Aβ). A: the modifications highlighted in red within HS are necessary for high-affinity binding to Aβ, and the histidine residues within Aβ participate in the HS-Aβ interaction. B: the molecular interactions between HS and Aβ depend on both HS modifications and Aβ structure, as demonstrated by co-staining of Aβ with anti-HS phage display antibodies and analysis of affinity-purified Aβ-binding HS hexasaccharide from the human cerebral cortex. Additional data also indicate that heparin binding to Aβ aggregates depends primarily on sulfation level rather than a specific type of sulfate moiety (not shown here).
Biochemical and cell-based studies have gained additional insights into HS structural features critical for Aβ binding and reported conflicting findings. For instance, maltese-cross amyloid plaque core formation can be induced by N-desulfated, N-acetylated heparin, but not the completely desulfated heparin with or without N-acetylation, suggesting O-sulfation, not N-sulfation, is required for HS GAG to bind Aβ40 (68, 94). Analysis of HS oligosaccharides from the human cerebral cortex has determined that Aβ fibrils bind to N-sulfated hexasaccharide domains that contain critical IdoA2S residues, with GlcNS6S being an additional requirement for Aβ monomer binding (Fig. 4A) (54). A study with synthesized HS polysaccharides determined that NS and 6S are essential for Aβ binding beyond electrostatic attraction (Fig. 4A) (87). Mapping and molecular modeling of heparin oligosaccharides using two-dimensional (2-D) NMR have determined the tetrasaccharide HexA-GlcNS-IdoA2S-GlcNS6S as the Aβ-binding motif of heparin (Fig. 4A) (78). IdoA, NS, and 6S are essential modifications for high-affinity Aβ binding, while 3-O sulfation is not directly involved, although it may cause steric hindrance (87). Furthermore, a study utilizing a heparinoid-immobilized microarray chip assay determined that the binding of heparin to Aβ aggregates was not significantly altered after removing NS, 6S, or 2S (62). In the same study, heparin significantly inhibited the cellular uptake of Aβ aggregates. The inhibition was abolished when NS was removed but only moderately reduced upon removal of 6S or 2S, showing specifically NS and the overall level of sulfation are critical for HS to bind to aggregated Aβ (62). Taken together, current data indicate that the sulfation level and modification pattern of HS and the isoform and fibrillation state of Aβ determine the interactions between HS and Aβ. However, further clarification, particularly with respect to the related HS structures, is needed with advanced techniques.
THE MOLECULES THAT HS INTERACTS WITH TO MODULATE Aβ METABOLISM
HS plays a complex role in Aβ metabolism and related pathogenesis of AD. Besides direct binding to accelerate Aβ fibril and aggregate formation and internalization (54–56), HS also regulates Aβ generation and clearance via various molecular mechanisms. In the following sections, we discuss some of the most reported proteins that interact with HS and critically impact Aβ metabolism.
Secreted APPs
APP is an integral transmembrane protein and acts as a cell surface receptor to regulate various functions, including neurite growth, neuronal adhesion, axonogenesis, cell motility, and gene expression. APP undergoes amyloidogenic and nonamyloidogenic processing, which results in the generation of extracellular sAPP fragments (Fig. 1). Interestingly, sAPP is necessary and sufficient to rescue prominent abnormalities of APP-knockout mice, such as the impairment in spatial learning and long-term potentiation, indicating that the primary biological function of APP is mediated by its ectodomain (95–100). sAPP has been found to bind heparin (96) and cell membrane-anchored HSPGs, including glypicans and syndecans (101). It is unknown whether cell-surface HSPGs regulate APP functions, and if any alteration of this interaction contributes to AD pathogenesis. sAPP catalyzes the nitric oxide-dependent deaminative release of HS chains from the HSPG Glypican-1 (102). The Sortilin-related receptor 1 (SORT1) involves endosomal trafficking of APP and other targets in neurons and is genetically associated with AD (103). The extracellular HSPG perlecan is partly responsible for the ubiquitination and degradation of SORT1, suggesting that perlecan may inhibit SORT1 function to disturb APP processing, leading to APP accumulation in AD (103). Perlecan also binds to sAPP (104). Since perlecan accumulates before amyloid deposition, its interaction with sAPP and SORT1 may affect Aβ production (105, 106). In fact, perlecan was isolated and examined as a model for Aβ co-deposition and fibril formation before developing Aβ aggregation-driven mouse models of AD (107). Moreover, the interaction between sAPP and ECM is critical for signal transduction in neurons (96, 98). ECM-bound sAPP, but not free sAPP, stimulates neurite outgrowth in cultured chick sympathetic and mouse hippocampal neurons through HS binding (96, 98). One possible molecular mechanism underlying the accentuated neurite outgrowth may be attributed to binding to the ECM, which shields sAPP from degradation by proteases (99). Interestingly, not all forms of HSPG accentuate neurite outgrowth. HSPG purified from the postnatal day 3 mouse brain stimulated neurite outgrowth, whereas HSPGs from the embryonic day 10 mouse brain (a developmental stage before the major period of neurite outgrowth) or HSPGs isolated from the liver did not induce neurite outgrowth. These observations suggest that developmentally regulated HSPGs, most likely the specific HS structures, are required for sAPP to stimulate neurite outgrowth (96). Dysregulated HS structures in AD may disturb the vital neurite outgrowth-promoting function of sAPP, leading to the loss of synapses and neurites in AD. Furthermore, heparin binding to sAPP stimulates APP synthesis and secretion through the amyloidogenic pathway (108). Therefore, elevated HS levels in AD might promote APP expression to enhance Aβ production, contributing to AD pathogenesis. Other ECM proteins, specifically those associated with the basement membrane, such as collagen IV, laminin, fibronectin, and entactin, can also bind to sAPP (96, 104). Under physiological conditions, these interactions may function to form and maintain a healthy basement membrane. In AD, HS, perlecan, laminin, collagen IV, and fibronectin co-deposit with amyloid plaques and APP (109). The aberrant expression of HS in AD may disrupt the interaction between ECM HSPGs, sAPP, and other ECM proteins, destabilizing leading to basement membrane and, consequently, leading to basement membrane rupture and loss of neurons in AD (104).
β-Site APP Cleaving Enzyme 1
β-Site APP cleaving enzyme 1 (BACE1) is the primary β-secretase in the brain, responsible for cleaving and generating Aβ (14) (Fig. 1). Endogenous HS immunoprecipitated with BACE1 from cell lysates and colocalizes with BACE1 in the Golgi complex and on the cell surface, two of its putative sites of the enzyme action. HS and heparin bind BACE1 directly and inhibit BACE1 enzyme activity in cell culture, acting as an endogenous inhibitor of BACE1 to suppress Aβ generation (110–113). HS inhibition of BACE1 depends on HS size and specific structural characteristics; 6S is essential, 2S is moderately required, whereas additional GlcNAc residues enhance BACE1 activity. Oversulfation of HS chains reduces HS inhibition of BACE1 (110–113). These results indicate that the interaction of heparin and HS with BACE1 requires specific HS structures, not just sulfation levels. One study suggested that α-secretase functions by releasing sAPPα and HS to inhibit BACE1 and Aβ production (102). The downregulated expression of EXTL2, EXT1, EXT2, and HS6ST1-3 in AD (75) may reduce cell surface HS expression and the level of 6S, leading to attenuated HS suppression of BACE1 and a subsequent increase in Aβ production. In addition, reduced NDST expression in AD may increase GlcNAc residues, enhancing BACE1 activity to increase Aβ production. Therefore, aberrant HS expression may significantly increase BACE1 activity, representing an important mechanism to switch from a physiological nonamyloidogenic process to a pathological amyloidogenic process underlying enhanced Aβ production in AD. Furthermore, BACE1 influences HS expression as well. Pharmacological inhibition of BACE1 reduced HS6ST3 secretion and increased the cellular accumulation of 6-O sulfated HS (114), revealing a positive regulatory feedback mechanism. Therefore, BACE1 may promote HS6ST3 secretion and reduce cellular HS 6S to enhance its activity, thereby increasing Aβ production.
Apolipoprotein E
Apolipoprotein E (ApoE) is a secreted protein and is a critical player in lipid transport within the brain. It is abundantly expressed in astrocytes, microglia, vascular mural cells, and choroid plexus cells, and to a lesser extent in stressed neurons (115). There are three allelic variants of ApoE: ε2, ε3, and ε4. The ApoE ε4 variant is associated with an increased risk of AD, whereas the ApoE ε2 variant is associated with a lower AD risk compared with the more common ApoE ε3 variant (116–120). A growing body of evidence suggests that ApoE ε4 results in a higher AD risk by inhibiting Aβ clearance, promoting Aβ aggregation, and suppressing Aβ cellular uptake and metabolism, leading to extracellular Aβ accumulation (9, 121–125). On the other hand, ApoE ε2 decreases Aβ aggregation and promotes cellular uptake (126). In addition, ApoE ε4 is associated with the attenuated release of HS from Glypican-1 in endosomes, which leads to decreased suppression of BACE1 and increased Aβ production (102). ApoE stimulates neuronal APP transcription and Aβ production in a variant-dependent manner, with ε4 > ε3 > ε2 (127). Earlier studies suggested that the three ApoE variants bind HS with similar affinities (128–130). However, recent studies have determined that ApoE ε4 has a threefold higher affinity for HS than ε2 and ε3 (131, 132). Interestingly, the ApoE ε3 Christchurch R136S mutation impairs binding to HS and significantly delays the onset of cognitive impairment of a PSEN1 mutation carrier (133). Meanwhile, multiple approaches, including 13C nuclear magnetic resonance, molecular modeling, and testing with N-desulfated heparin, have determined that NS and 6S of glucosamine residues are required for high-affinity HS binding to ApoE (130). ApoE modulates HS-mediated Aβ cell surface binding and internalization, possibly in a lipidation-dependent manner. O’Callaghan et al. (134) observed that delipidated human ApoE increases cell association of Aβ40 through HS. On the contrary, Fu et al. (132) reported that lipidated ApoE, secreted from immortalized astrocytes, significantly suppresses Aβ binding to the cell surface and cellular Aβ uptake through cell surface HS, and this effect is ApoE variant-independent. Further studies with delipidated and lipidated ApoE variants in the same experimental setting may clarify this lipidation and ApoE variant dependence. Meanwhile, it would be interesting to test if the aberrant HS expression in AD alters HS-ApoE interaction contributing to dysregulated Aβ metabolism in AD, and whether the increased AD risk of ApoE ε4 variant is mediated by its higher binding affinity for HS.
Low-Density Lipoprotein Receptor-Associated Protein-1
Lipoprotein receptor-associated protein-1 (LRP1) is an endocytic receptor that has a repertoire of more than 40-ligand repertoire, including ApoE and Aβ. In the brain, LRP1 plays a major role in maintaining Aβ homeostasis and is expressed abundantly in various cell types, including neurons, vascular cells, and glial cells (124). LRP1-mediated endocytosis regulates cellular Aβ uptake by binding to Aβ directly or indirectly through its co-receptors or ligands. LRP1 and HSPG form an immunoprecipitated complex on the cell surface that mediates lipid metabolism (135). The HSPG and LRP1 complex partially mediates ApoE uptake (136). These findings suggest that HS and LRP1 have a cooperative function on the cell surface. Functional studies have shown that heparin and HS deficiency impair LRP1-mediated Aβ uptake in mouse neuronal cells and mouse embryonic fibroblasts, indicating that LRP1 requires cell surface HS to internalize Aβ (137). As Aβ directly binds to HS and LRP1, the current literature suggests that HS may act as a co-receptor to facilitate LRP1-mediated Aβ uptake. Similarly, the downregulation of EXTL2, EXT1, EXT2, NDST1-2, and HS6ST1-3 in AD may reduce cell surface HS expression, which could attenuate LRP1-mediated Aβ uptake. This may represent one important molecular mechanisms underlying increased Aβ accumulation in AD.
PERTURBATIONS IN HSPGs AND OTHER GLYCOSAMINOGLYCANS IN AD
In addition to alterations in HS chains in AD, changes in HSPG expression and function, including their core proteins, have also been reported (52). HSPGs include the ECM-associated perlecan, agrin, and collagen XVIII, the cell-membrane-anchored syndecans and glypicans, and serglycin in the secretory vesicle. First demonstrated by cationic dyes, including Ruthenium red and Cuprolinic blue (48), several human tissue staining studies have observed HSPGs in amyloid plaques in AD, which have been outlined in Table 2. In addition, with increasing neuroinflammation, immune cells also synthesize more HSPGs in AD (147).
Table 2.
Summary of HSPGs detected by immunolabeling in human patients with AD
| Immunopositively Labeled Areas |
Antibody | Source of Tissue Sample | Subjects Analyzed Mixed Genders | References | |||||
|---|---|---|---|---|---|---|---|---|---|
| Normal Vessels | CAA Vessel | NP/SP | Primitive Plaque | Other | |||||
| Syndecans | ✓ | ✓ | ✓ | 10H4(SDC2 ectodomain) | F, P, T, O | 7 AD/CAA, 3 controls | (138) | ||
| ✓ | 2E9 (SDC1,3 cytoplasmic domain) | ||||||||
| ✓ | 1C7 (SDC3 ectodomain) | ||||||||
| ✓ | Fibrillar plaques only | 10H4 (SDC2 ectodomain) | CB | 8 AD, 3 controls | (139) | ||||
| ✓ | 2E9 (SDC1,3 cytoplasmic domain) | ||||||||
| ✓ | 1C7 (SDC3 ectodomain) | ||||||||
| ✓ | Classic plaques, not primitive plaques | 10H4 (SDC2) | N/A | 4 AD, 4 controls | (140) | ||||
| ✓ | Some neurons and NFTs | Anti-SDC1 | TE, EH, PH, CL, CS, PL, CB | 18 AD, 6 controls | (141) | ||||
| ✓ | ✓ | Anti-SDC4 | |||||||
| Glypicans | ✓ | ✓ | ✓ | S1 (GPC1) | F, P, T, O | 7 AD/CAA, 3 controls | (138) | ||
| ✓ | ✓ | ✓ | S1 (GPC1) | CB, F | 9 AD, 3 controls | (139) | |||
| ✓ | ✓ | ✓ | fibrillar plaque only | S1 (GPC1) | N/A | 4 AD, 4 controls | (140) | ||
| ✓ | ✓ | ✓ | neurons and neuropils with NFTs | Anti-GPC4 Anti-GPC6 |
TE, EH, PH, CL, CS, PL, CB | 18 AD, 6 controls | (141) | ||
| Agrin | ✓ | ✓ | ✓ | ✓ | highest of all HSPGs | JM72 | F, T, H | 11 AD, 3 controls | (142) |
| ✓ | ✓ | ✓ | ✓ | Higher in AD, concentrated in plaques and neurons | Agrin antiserum | PFC, VC, T, H, A | 16 AD, 11 control cases | (143) | |
| ✓ | ✓ | ✓ | ✓ | Higher in AD, in plaques and neurons | Agrin antiserum | PFC, H | 22 AD, 12 controls | (144) | |
| ✓ | ✓ | ✓ | ✓ | Agrin antiserum | N/A | N/A | (145) | ||
| ✓ | ✓ | ✓ | ✓ | The highest of all HSPGs | JMJ2 (Agrin peptide) | F, P, T, O | 7 AD/CAA, 3 controls | (138) | |
| BI31 (Agrin core protein) | |||||||||
| ✓ | ✓ | ✓ | ✓ | The highest of all HSPGs | JMJ2 (Agrin peptide) | CB, F | 9 AD, 3 controls | (139) | |
| BI31 (Agrin core protein) | |||||||||
| Collagen XVIII | ✓ | ✓ | ✓ | QH48.18 | H, F, O | 11 AD/CAA, 5 AD only, 3 control cases | (139) | ||
| QH1415.7 | |||||||||
| Perlecan | ✓ | ✓ | Not associated with plaques | 1948P, 95J10,HK-102 | F, T, H | 11 AD, 3 control cases | (142) | ||
| ✓ | ✓ | Not associated with plaques | 1948P | F, P, T, O | 7 AD/CAA, 3 controls | (138) | |||
| 95J10 | |||||||||
| ✓ | ✓ | increased in areas with plaques in Braak >2 | 1948 P | CB, F | 9 AD, 3 controls | (139) | |||
| 95J10 | |||||||||
| ✓ | ✓ | Not associated with plaques | mAb (ZYMED) | N/A | 4 AD, 4 controls | (140) | |||
| ✓ | ✓ | Increased staining intensity with Braak stage | Abcam (ab2501) | SF, IT | 8 AD, 10 mild AD 12 controls | (146) | |||
| Serglycin | ✓ | ✓ | ✓ | ✓ | Anti-SRGN | TE, EH, PH, CL, CS, PL, CB | 18 AD, 6 controls | (141) | |
The table shows different immunopositively labeled areas, antibodies used, source of tissue samples, and subjects analyzed in various studies related to the investigation of heparan sulfate proteoglycans (HSPGs) in Alzheimer’s disease (AD) and cerebral amyloid angiopathy (CAA). The presence and distribution of these proteoglycans were investigated in different areas of the brain, including normal blood vessels, primitive plaques, fibrillar plaques, classic plaques, neurons, and neuropils with neurofibrillary tangles (NFTs). The number of subjects analyzed varied among the studies. GPC, glypican; NP/SP, neuritic plaque/senile plaque; SDC, syndecan. The areas of the brain are indicated by letters as follows: H, hippocampus; AH, anterior hippocampus; PH, posterior hippocampus; A, amygdala; CB, cerebellum; C, claustrum; CS, calcarine sulcus; F, frontal cortex; G, globus palludus; P, parietal cortex; PL, pallidum; T, temporal cortex; TE, trans entorhinal region; ST, superior temporal cortex; O, occipital cortex; PFC, prefrontal cortex.
Functional studies have indicated that perlecan is involved in AD pathology. Perlecan, along with Aβ40, is the only HSPG reported to induce congophilic maltese-cross formation of Aβ similar to that in clinical AD (94). Perlecan hinders Aβ from degradation for 2 wk in vitro (57) and causes fibrillar Aβ persistence in the brain (148). Perlecan is the primary HSPG in Matrigel (148), which was used in the “AD in a dish” model developed by Choi et al. (53). In this model, Matrigel was used to coat and grow neuronal cells, and plaque-like deposits were formed in a dish followed by tangle formation a few weeks later, suggesting that perlecan is required for the spontaneous development of Aβ plaques and Tau tangles in a neuronal cell context (53). In addition, HS induces the microtube-binding protein Tau to form paired helical filaments in neurons, another hallmark of AD pathology (4, 5, 43, 149, 150). These studies support the crucial role of HS and HSPGs in AD disease development as hypothesized by Snow et al. (68).
Alterations in HSPG expression in patients with AD have also been investigated, as outlined in Table 3. Notably, Lorente-Gea et al. (141) reported that HSPG expression in AD differs significantly with respect to the disease severity and spatiotemporal distribution. Recently, Johnson et al. (77) reported that the matrisome module was significantly elevated in patients with AD, and these proteomic changes are not reflected at the RNA level. The plaque and tangle-associated proteins with the highest eigenvalue correlations with the matrisome module were ApoE, APP, Syndecan 4, and Glypican 5 (77). In agreement with this finding, Lorente-Gea et al. (141) reported elevated Syndecan 5 and Serglycin expression throughout the AD brains. Further functional studies of the specific HSPGs are needed to better understand how they contribute to AD pathogenesis.
Table 3.
Summary of the expression profiles, impact on Aβ, and affected molecular pathways of HSPGs in patients with AD
| HSPG | Expression in AD | Impact on Aβ | Affected Molecular Pathway |
|---|---|---|---|
| Perlecan (HSPG2) |
|
|
|
| Agrin (AGRN) | |||
| Glypicans (GPC1–6) |
|
||
| Syndecans (SDC1-4) |
|
||
| Collagen XVIII (COL18A1) |
|
|
|
| Serglycin (SRGN) |
|
|
|
| Testicans (SPOCK1-3) |
|
|
|
|
The table lists six heparan sulfate proteoglycans (HSPGs), namely, Perlecan (HSPG2), Agrin (AGRN), Glypicans (GPC1–6), Syndecans (SDC1–4), Collagen XVIII (COL18A1), and Serglycin (SRGN) and their different effects on Aβ aggregation and involvement in various molecular pathways. Perlecan is reported to induce congophilic maltese-cross formation of amyloid beta (Aβ) and slows Aβ degradation. Agrin, abundant in Alzheimer’s disease (AD) and insoluble Aβ plaques, binds to Aβ, accelerates its fibrillization and stabilizes formed Aβ fibrils. Glypicans promote Aβ multimerization, internalization, and toxicity, whereas syndecans are involved in hippocampal long-term potentiation and endocytosis of Aβ. Collagen XVIII and Serglycin are overexpressed in most brain areas in patients with AD, with their exact role still being explored. Finally, testicans (SPOCK1-3) are elevated in patients with AD and suggested to drive neuroinflammation in conjunction with Aβ production. APP, amyloid precursor protein; BBB, blood-brain barrier; CSF, cerebrospinal fluid; LRP1, low density lipoprotein-related protein 1; SPOCK, testican.
In addition, other GAGs, including CS and dermatan sulfate, are associated with Aβ plaques and NFT in AD (79, 175). Perineuronal nets are CS-containing matrices that enmesh neural networks involved in memory and cognition. CS sulfation patterns in perineural nets are altered in AD, and these alterations occur before the development of AD clinicopathology (176). Changing the CS sulfation pattern may contribute to the initiation and progression of the underlying degenerative processes, suggesting the alteration of CS sulfation pattern as a critical player in the development of AD clinicopathology (176). A newly discovered type of CS/HS proteoglycan, named testicans 1–3 (SPOCK1-3), was identified in patients with AD in proteomic analyses (173). Abundant in the ECM, SPOCK1 is trafficked by APP, affecting Aβ production (173), and SPOCK2 is elevated in patients with AD (151) and an transgenic AD mouse model (174), and it may drive neuroinflammation in conjunction with Aβ in AD pathogenesis (174). Furthermore, cerebral keratan sulfate is selectively lost in AD (177).
CONCLUSIONS AND PERSPECTIVE
Ample evidence has established that HS is aberrantly expressed in AD, including changes in its expression level, structure, and the enzymes responsible for its biosynthesis, remodeling, and degradation. However, the HS level and structural alterations detected in AD brains do not correlate well with changes in the expression of genes responsible for HS synthesis and remodeling (46, 71, 73, 75). The downregulation of HS genes responsible for its biosynthetic initiation and elongation include EXTL3, EXT1, and EXT2 (Table 1) (75), indicating that active HS biosynthesis is reduced in cells in the AD brain. However, biochemical analyses detected elevated levels of HS in AD brain (45, 46, 71). These observations suggest that the HS elevation in the AD brain is due to its accumulation rather than active HS biosynthesis. HS immunostaining studies have found HS to be more densely concentrated in the thickened basement membrane adjacent to the endothelial cells of capillary vessels (where HS co-deposition with Aβ is commonly seen) and amyloid plaques in AD brains (45). Meanwhile, Aβ binding has been shown to protect HS from degradation by HPSE (81), which may facilitate HS accumulation in AD brains. Taken together, these transcriptional, biochemical, and immunostaining data suggest that the elevation in HS detected in AD brains may reflect accumulated HS that co-deposits with Aβ, rather than newly biosynthesized HS, such as the cell surface HS. Cell surface HS essentially mediates Aβ binding and cellular uptake in AD. The downregulation of HS expression on cell surface may impair Aβ clearance, representing a major pathway failure contributing to Aβ accumulation in AD.
The gene transcription data from several studies conducted to date have been inconsistent (46, 73, 75). The discrepancy is likely due to the heterogeneity of AD etiology, as well as variations in disease severity, genetic background, brain regions and cell types analyzed, and differences in postmortem tissue collection. To improve our understanding of AD, new studies should be conducted with larger cohorts of patients with AD grouped based on systematic pathological analysis and with sufficient controls. In addition, these reported studies are based on tissue analysis in bulk. Single-cell RNA sequencing has revealed that the expression pattern of HS-related genes is unique for brain cell types (178), supporting the idea that the structure of HS is cell type-specific (179). Further studies using high-resolution single-cell and spatial multiomics techniques could advance our understanding and provide a clearer picture of the correlation between HS-related gene expression and HS structural alterations seen in AD. Specifically, these studies could answer how these changes might be associated with AD initiation and progression (77). Furthermore, a study that specially targeted HS expression in neurons in a mutant APP transgenic mouse model determined the roles of neuronal HS expression in Aβ metabolism and neuroinflammation (24). Similar studies are needed to determine the roles of HS expressed in other brain cell types in Aβ metabolism and AD-related pathology. Systematic in vivo studies could advance our understanding of the spatiotemporal roles of aberrant HS expression and its role in the disrupted Aβ metabolism as seen in AD. Moreover, these findings might facilitate the development of highly efficient HS-targeting therapeutics for AD treatment.
HS plays crucial role in regulating Aβ metabolism through direct and indirect mechanisms. The sulfation level and modification pattern of HS, along with the isoform and fibrillation state of Aβ, determine the binding of HS to Aβ. Direct binding of HS accelerates Aβ aggregation, fibrillization, and deposition in the brain. The indirect effects of HS function are mediated through interactions with distinct HS-binding proteins, including sAPP, BACE1, LRP1, and ApoE. These interactions may suppress Aβ production, enhance Aβ uptake, and modulate Aβ clearance (180). Downregulated expression of EXTL3, EXT1, EXT2, NDST2, and HS6STs suggests that brain cells reduce HS expression in AD (75). This reduction might disrupt the HS-modulated sAPP, BACE1, LRP1, and ApoE pathways, leading to increased Aβ production and suppressed Aβ clearance, thus exacerbating Aβ deposition and amyloid plaque formation in AD. Furthermore, HS may also affect other yet-to-be-revealed molecular pathways associated with Aβ metabolism, such as the triggering receptor expressed on myeloid cell 2 (TREM2) pathway. TREM2 is an extracellular innate immune receptor expressed on myeloid lineage cells, such as dendritic cells and resident tissue macrophages, including osteoclasts and microglia. TREM2 modulates microglial function, Aβ phagocytosis, and suppresses Aβ-induced inflammatory response (181–184). TREM2 binds to and inhibits Aβ polymerization (183, 185, 186). Cell surface HS binds to TREM2 likely to cluster or orient TREM2 for its ligand binding (182, 183). Genome-wide association studies linked TREM2 variants to late-onset AD, particularly the R47H and R62H variants (187–189). Interestingly, the heterozygous R47H variant carries roughly the same risk as a copy of the ApoE ε4 allele (190). The R47H and R62H variants have reduced binding to HS on the cell surface and to ligands such as phospholipids and lipid particles. There is the possibility that decreased binding of the TREM2 variants to HS may disrupt TREM2’s ability to recognize and signal in response to Aβ. Aberrant expression of HS in AD may alter this pathway to impair Aβ clearance, thus exacerbating Aβ deposition in the brain. Addressing these emerging questions in future studies will advance the mechanistic understanding of HS in Aβ metabolism.
GRANTS
This work was made possible through funding from several sources, including NIH Grants 7R01HL093339, 5U01CA225784, 1RF1AG074289, and 1RF1AG069039, and the Swedish Research Council Grant 2021-01094.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
I.O.M. and L.W. prepared figures; I.O.M. and L.W. drafted manuscript; I.O.M., J-P.L., and L.W. edited and revised manuscript; I.O.M., J-P.L., and L.W. approved final version of manuscript.
ACKNOWLEDGMENTS
We express our gratitude to Joseph D. Mcmillan for language editing of the manuscript. All figures and graphical abstract were created with BioRender and published with permission. This article is part of the special collection “Deciphering the Role of Proteoglycans and Glycosaminoglycans in Health and Disease.” Dr. Liliana Schaefer served as Guest Editor of this collection.
REFERENCES
- 1. Long JM, Holtzman DM. Alzheimer disease: an update on pathobiology and treatment strategies. Cell 179: 312–339, 2019. doi: 10.1016/j.cell.2019.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, Kanekiyo M, Li D, Reyderman L, Cohen S, Froelich L, Katayama S, Sabbagh M, Vellas B, Watson D, Dhadda S, Irizarry M, Kramer LD, Iwatsubo T. Lecanemab in early Alzheimer's disease. N Engl J Med 388: 9–21, 2023. doi: 10.1056/NEJMoa2212948. [DOI] [PubMed] [Google Scholar]
- 3. Salloway S, Chalkias S, Barkhof F, Burkett P, Barakos J, Purcell D, Suhy J, Forrestal F, Tian Y, Umans K, Wang G, Singhal P, Budd Haeberlein S, Smirnakis K. Amyloid-related imaging abnormalities in 2 phase 3 studies evaluating aducanumab in patients with early Alzheimer disease. JAMA Neurol 79: 13–21, 2022. doi: 10.1001/jamaneurol.2021.4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhu Y, Gandy L, Zhang F, Liu J, Wang C, Blair LJ, Linhardt RJ, Wang L. Heparan sulfate proteoglycans in tauopathy. Biomolecules 12: 1792, 2022. doi: 10.3390/biom12121792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mah D, Zhao J, Liu X, Zhang F, Liu J, Wang L, Linhardt R, Wang C. The sulfation code of tauopathies: heparan sulfate proteoglycans in the prion like spread of tau pathology. Front Mol Biosci 8: 671458, 2021. doi: 10.3389/fmolb.2021.671458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297: 353–356, 2002. [Erratum in Science 297: 2209, 2002]. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 7. Selkoe DJ. Deciphering the genesis and fate of amyloid beta-protein yields novel therapies for Alzheimer disease. J Clin Invest 110: 1375–1381, 2002. doi: 10.1172/JCI16783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hampel H, Hardy J, Blennow K, Chen C, Perry G, Kim SH, Villemagne VL, Aisen P, Vendruscolo M, Iwatsubo T, Masters CL, Cho M, Lannfelt L, Cummings JL, Vergallo A. The amyloid-beta pathway in Alzheimer’s disease. Mol Psychiatry 26: 5481–5503, 2021. doi: 10.1038/s41380-021-01249-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 330: 1774, 2010. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bayer TA, Cappai R, Masters CL, Beyreuther K, Multhaup G. It all sticks together—the APP-related family of proteins and Alzheimer's disease. Mol Psychiatry 4: 524–528, 1999. doi: 10.1038/sj.mp.4000552. [DOI] [PubMed] [Google Scholar]
- 11. O'Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer's disease. Annu Rev Neurosci 34: 185–204, 2011. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286: 735–741, 1999. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 13. Kimberly WT, LaVoie MJ, Ostaszewski BL, Ye W, Wolfe MS, Selkoe DJ. Gamma-secretase is a membrane protein complex comprised of presenilin, nicastrin, Aph-1, and Pen-2. Proc Natl Acad Sci USA 100: 6382–6387, 2003. doi: 10.1073/pnas.1037392100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chow VW, Mattson MP, Wong PC, Gleichmann M. An overview of APP processing enzymes and products. Neuromolecular Med 12: 1–12, 2010. doi: 10.1007/s12017-009-8104-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV. Clearance of Alzheimer's amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest 106: 1489–1499, 2000. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Storck SE, Meister S, Nahrath J, Meißner JN, Schubert N, Di Spiezio A, Baches S, Vandenbroucke RE, Bouter Y, Prikulis I, Korth C, Weggen S, Heimann A, Schwaninger M, Bayer TA, Pietrzik CU. Endothelial LRP1 transports amyloid-beta(1–42) across the blood-brain barrier. J Clin Invest 126: 123–136, 2015. doi: 10.1172/JCI81108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bell RD, Deane R, Chow N, Long X, Sagare A, Singh I, Streb JW, Guo H, Rubio A, Van Nostrand W, Miano JM, Zlokovic BV. SRF and myocardin regulate LRP-mediated amyloid-beta clearance in brain vascular cells. Nat Cell Biol 11: 143–153, 2009. doi: 10.1038/ncb1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kanekiyo T, Liu CC, Shinohara M, Li J, Bu G. LRP1 in brain vascular smooth muscle cells mediates local clearance of Alzheimer's amyloid-beta. J Neurosci 32: 16458–16465, 2012. doi: 10.1523/JNEUROSCI.3987-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ma Q, Zhao Z, Sagare AP, Wu Y, Wang M, Owens NC, Verghese PB, Herz J, Holtzman DM, Zlokovic BV. Blood-brain barrier-associated pericytes internalize and clear aggregated amyloid-beta42 by LRP1-dependent apolipoprotein E isoform-specific mechanism. Mol Neurodegener 13: 57, 2018. [Erratum in Mol Neurodegener 17: 71, 2022]. doi: 10.1186/s13024-018-0286-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Roberts KF, Elbert DL, Kasten TP, Patterson BW, Sigurdson WC, Connors RE, Ovod V, Munsell LY, Mawuenyega KG, Miller-Thomas MM, Moran CJ, Cross DT 3rd, Derdeyn CP, Bateman RJ. Amyloid-beta efflux from the central nervous system into the plasma. Ann Neurol 76: 837–844, 2014. doi: 10.1002/ana.24270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bell RD, Sagare AP, Friedman AE, Bedi GS, Holtzman DM, Deane R, Zlokovic BV. Transport pathways for clearance of human Alzheimer's amyloid beta-peptide and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow Metab 27: 909–918, 2007. doi: 10.1038/sj.jcbfm.9600419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wardlaw JM, Benveniste H, Nedergaard M, Zlokovic BV, Mestre H, Lee H, Doubal FN, Brown R, Ramirez J, MacIntosh BJ, Tannenbaum A, Ballerini L, Rungta RL, Boido D, Sweeney M, Montagne A, Charpak S, Joutel A, Smith KJ, Black SE; colleagues from the Fondation Leducq Transatlantic Network of Excellence on the Role of the Perivascular Space in Cerebral Small Vessel Disease. Perivascular spaces in the brain: anatomy, physiology and pathology. Nat Rev Neurol 16: 137–153, 2020. doi: 10.1038/s41582-020-0312-z. [DOI] [PubMed] [Google Scholar]
- 23. Saido T, Leissring MA. Proteolytic degradation of amyloid beta-protein. Cold Spring Harb Perspect Med 2: a006379, 2012. doi: 10.1101/cshperspect.a006379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu CC, Zhao N, Yamaguchi Y, Cirrito JR, Kanekiyo T, Holtzman DM, Bu G. Neuronal heparan sulfates promote amyloid pathology by modulating brain amyloid-beta clearance and aggregation in Alzheimer's disease. Sci Transl Med 8: 332ra344, 2016. doi: 10.1126/scitranslmed.aad3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ariga T, Miyatake T, Yu RK. Role of proteoglycans and glycosaminoglycans in the pathogenesis of Alzheimer's disease and related disorders: amyloidogenesis and therapeutic strategies–a review. J Neurosci Res 88: 2303–2315, 2010. doi: 10.1002/jnr.22393. [DOI] [PubMed] [Google Scholar]
- 26. Sarrazin S, Lamanna WC, Esko JD. Heparan sulfate proteoglycans. Cold Spring Harb Perspect Biol 3: a004952, 2011. doi: 10.1101/cshperspect.a004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Qiu H, Shi S, Yue J, Xin M, Nairn AV, Lin L, Liu X, Li G, Archer-Hartmann SA, Dela Rosa M, Galizzi M, Wang S, Zhang F, Azadi P, van Kuppevelt TH, Cardoso WV, Kimata K, Ai X, Moremen KW, Esko JD, Linhardt RJ, Wang L. A mutant-cell library for systematic analysis of heparan sulfate structure-function relationships. Nat Methods 15: 889–899, 2018. doi: 10.1038/s41592-018-0189-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 446: 1030–1037, 2007. doi: 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
- 29. Esko JD, Selleck SB. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem 71: 435–471, 2002. doi: 10.1146/annurev.biochem.71.110601.135458. [DOI] [PubMed] [Google Scholar]
- 30. Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem 68: 729–777, 1999. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- 31. Kjellen L. Glucosaminyl N-deacetylase/N-sulphotransferases in heparan sulphate biosynthesis and biology. Biochem Soc Trans 31: 340–342, 2003. doi: 10.1042/bst0310340. [DOI] [PubMed] [Google Scholar]
- 32. Esko JD, Lindahl U. Molecular diversity of heparan sulfate. J Clin Invest 108: 169–173, 2001. doi: 10.1172/JCI13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li JP, Kusche-Gullberg M. Heparan sulfate: biosynthesis, structure, and function. Int Rev Cell Mol Biol 325: 215–273, 2016. doi: 10.1016/bs.ircmb.2016.02.009. [DOI] [PubMed] [Google Scholar]
- 34. Kraushaar DC, Yamaguchi Y, Wang L. Heparan sulfate is required for embryonic stem cells to exit from self-renewal. J Biol Chem 285: 5907–5916, 2010. [Erratum in J Biol Chem 290: 23023, 2015]. doi: 10.1074/jbc.M109.066837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kraushaar DC, Rai S, Condac E, Nairn A, Zhang S, Yamaguchi Y, Moremen K, Dalton S, Wang L. Heparan sulfate facilitates FGF and BMP signaling to drive mesoderm differentiation of mouse embryonic stem cells. J Biol Chem 287: 22691–22700, 2012. doi: 10.1074/jbc.M112.368241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang B, Xiao W, Qiu H, Zhang F, Moniz HA, Jaworski A, Condac E, Gutierrez-Sanchez G, Heiss C, Clugston RD, Azadi P, Greer JJ, Bergmann C, Moremen KW, Li D, Linhardt RJ, Esko JD, Wang L. Heparan sulfate deficiency disrupts developmental angiogenesis and causes congenital diaphragmatic hernia. J Clin Invest 124: 209–221, 2014. doi: 10.1172/JCI71090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rai S, Alsaidan OA, Yang H, Cai H, Wang L. Heparan sulfate inhibits transforming growth factor beta signaling and functions in cis and in trans to regulate prostate stem/progenitor cell activities. Glycobiology 30: 381–395, 2020. doi: 10.1093/glycob/cwz103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang L, Fuster M, Sriramarao P, Esko JD. Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat Immunol 6: 902–910, 2005. doi: 10.1038/ni1233. [DOI] [PubMed] [Google Scholar]
- 39. Wang L, Brown JR, Varki A, Esko JD. Heparin's anti-inflammatory effects require glucosamine 6-O-sulfation and are mediated by blockade of L- and P-selectins. J Clin Invest 110: 127–136, 2002. doi: 10.1172/JCI14996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Talsma DT, Katta K, Ettema MAB, Kel B, Kusche-Gullberg M, Daha MR, Stegeman CA, van den Born J, Wang L. Endothelial heparan sulfate deficiency reduces inflammation and fibrosis in murine diabetic nephropathy. Lab Invest 98: 427–438, 2018. doi: 10.1038/s41374-017-0015-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yue J, Jin W, Yang H, Faulkner J, Song X, Qiu H, Teng M, Azadi P, Zhang F, Linhardt RJ, Wang L. Heparan sulfate facilitates spike protein-mediated SARS-CoV-2 host cell invasion and contributes to increased infection of SARS-CoV-2 G614 mutant and in lung cancer. Front Mol Biosci 8: 649575, 2021. doi: 10.3389/fmolb.2021.649575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zong C, Huang R, Condac E, Chiu Y, Xiao W, Li X, Lu W, Ishihara M, Wang S, Ramiah A, Stickney M, Azadi P, Amster IJ, Moremen KW, Wang L, Sharp JS, Boons GJ. Integrated approach to identify heparan sulfate ligand requirements of Robo1. J Am Chem Soc 138: 13059–13067, 2016. doi: 10.1021/jacs.6b08161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhao J, Zhu Y, Song X, Xiao Y, Su G, Liu X, Wang Z, Xu Y, Liu J, Eliezer D, Ramlall TF, Lippens G, Gibson J, Zhang F, Linhardt RJ, Wang L, Wang C. 3-O-Sulfation of heparan sulfate enhances tau interaction and cellular uptake. Angew Chem Int Ed Engl 59: 1818–1827, 2020. doi: 10.1002/anie.201913029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zong C, Venot A, Li X, Lu W, Xiao W, Wilkes JL, Salanga CL, Handel TM, Wang L, Wolfert MA, Boons GJ. Heparan sulfate microarray reveals that heparan sulfate-protein binding exhibits different ligand requirements. J Am Chem Soc 139: 9534–9543, 2017. doi: 10.1021/jacs.7b01399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shimizu H, Ghazizadeh M, Sato S, Oguro T, Kawanami O. Interaction between beta-amyloid protein and heparan sulfate proteoglycans from the cerebral capillary basement membrane in Alzheimer's disease. J Clin Neurosci 16: 277–282, 2009. doi: 10.1016/j.jocn.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 46. Huynh MB, Ouidja MO, Chantepie S, Carpentier G, Maiza A, Zhang G, Vilares J, Raisman-Vozari R, Papy-Garcia D. Glycosaminoglycans from Alzheimer's disease hippocampus have altered capacities to bind and regulate growth factors activities and to bind tau. PLoS One 14: e0209573, 2019. doi: 10.1371/journal.pone.0209573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hosono-Fukao T, Ohtake-Niimi S, Nishitsuji K, Hossain MM, van Kuppevelt TH, Michikawa M, Uchimura K. RB4CD12 epitope expression and heparan sulfate disaccharide composition in brain vasculature. J Neurosci Res 89: 1840–1848, 2011. doi: 10.1002/jnr.22690. [DOI] [PubMed] [Google Scholar]
- 48. Snow AD, Lara S, Nochlin D, Wight TN. Cationic dyes reveal proteoglycans structurally integrated within the characteristic lesions of Alzheimer's disease. Acta Neuropathol 78: 113–123, 1989. doi: 10.1007/BF00688198. [DOI] [PubMed] [Google Scholar]
- 49. Snow AD, Mar H, Nochlin D, Kimata K, Kato M, Suzuki S, Hassell J, Wight TN. The presence of heparan sulfate proteoglycans in the neuritic plaques and congophilic angiopathy in Alzheimer's disease. Am J Pathol 133: 456–463, 1988. [PMC free article] [PubMed] [Google Scholar]
- 50. Snow AD, Mar H, Nochlin D, Sekiguchi RT, Kimata K, Koike Y, Wight TN. Early accumulation of heparan sulfate in neurons and in the beta-amyloid protein-containing lesions of Alzheimer's disease and Down's syndrome. Am J Pathol 137: 1253–1270, 1990. [PMC free article] [PubMed] [Google Scholar]
- 51. Snow AD, Sekiguchi RT, Nochlin D, Kalaria RN, Kimata K. Heparan sulfate proteoglycan in diffuse plaques of hippocampus but not of cerebellum in Alzheimer's disease brain. Am J Pathol 144: 337–347, 1994. [PMC free article] [PubMed] [Google Scholar]
- 52. Snow AD, Wight TN. Proteoglycans in the pathogenesis of Alzheimer's disease and other amyloidoses. Neurobiol Aging 10: 481–497, 1989. doi: 10.1016/0197-4580(89)90108-5. [DOI] [PubMed] [Google Scholar]
- 53. Choi SH, Kim YH, Hebisch M, Sliwinski C, Lee S, D'Avanzo C, Chen H, Hooli B, Asselin C, Muffat J, Klee JB, Zhang C, Wainger BJ, Peitz M, Kovacs DM, Woolf CJ, Wagner SL, Tanzi RE, Kim DY. A three-dimensional human neural cell culture model of Alzheimer's disease. Nature 515: 274–278, 2014. doi: 10.1038/nature13800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lindahl B, Westling C, Gimenez-Gallego G, Lindahl U, Salmivirta M. Common binding sites for beta-amyloid fibrils and fibroblast growth factor-2 in heparan sulfate from human cerebral cortex. J Biol Chem 274: 30631–30635, 1999. doi: 10.1074/jbc.274.43.30631. [DOI] [PubMed] [Google Scholar]
- 55. Cui H, Hung AC, Freeman C, Narkowicz C, Jacobson GA, Small DH. Size and sulfation are critical for the effect of heparin on APP processing and Abeta production. J Neurochem 123: 447–457, 2012. doi: 10.1111/j.1471-4159.2012.07929.x. [DOI] [PubMed] [Google Scholar]
- 56. Watson DJ, Lander AD, Selkoe DJ. Heparin-binding properties of the amyloidogenic peptides Abeta and amylin. Dependence on aggregation state and inhibition by Congo red. J Biol Chem 272: 31617–31624, 1997. doi: 10.1074/jbc.272.50.31617. [DOI] [PubMed] [Google Scholar]
- 57. Castillo GM, Lukito W, Wight TN, Snow AD. The sulfate moieties of glycosaminoglycans are critical for the enhancement of beta-amyloid protein fibril formation. J Neurochem 72: 1681–1687, 1999. doi: 10.1046/j.1471-4159.1999.721681.x. [DOI] [PubMed] [Google Scholar]
- 58. Perry G, Siedlak SL, Richey P, Kawai M, Cras P, Kalaria RN, Galloway PG, Scardina JM, Cordell B, Greenberg BD. and Association of heparan sulfate proteoglycan with the neurofibrillary tangles of Alzheimer's disease. J Neurosci 11: 3679–3683, 1991. doi: 10.1523/JNEUROSCI.11-11-03679.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Perlmutter LS, Chui HC, Saperia D, Athanikar J. Microangiopathy and the colocalization of heparan sulfate proteoglycan with amyloid in senile plaques of Alzheimer's disease. Brain Res 508: 13–19, 1990. doi: 10.1016/0006-8993(90)91111-s. [DOI] [PubMed] [Google Scholar]
- 60. Sandwall E, O'Callaghan P, Zhang X, Lindahl U, Lannfelt L, Li JP. Heparan sulfate mediates amyloid-beta internalization and cytotoxicity. Glycobiology 20: 533–541, 2010. doi: 10.1093/glycob/cwp205. [DOI] [PubMed] [Google Scholar]
- 61. Kuwabara K, Nishitsuji K, Uchimura K, Hung SC, Mizuguchi M, Nakajima H, Mikawa S, Kobayashi N, Saito H, Sakashita N. Cellular interaction and cytotoxicity of the iowa mutation of apolipoprotein A-I (ApoA-IIowa) amyloid mediated by sulfate moieties of heparan sulfate. J Biol Chem 290: 24210–24221, 2015. doi: 10.1074/jbc.M115.652545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Stopschinski BE, Holmes BB, Miller GM, Manon VA, Vaquer-Alicea J, Prueitt WL, Hsieh-Wilson LC, Diamond MI. Specific glycosaminoglycan chain length and sulfation patterns are required for cell uptake of tau versus alpha-synuclein and beta-amyloid aggregates. J Biol Chem 293: 10826–10840, 2018. doi: 10.1074/jbc.RA117.000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chen L, Sanderson RD. Heparanase regulates levels of syndecan-1 in the nucleus. PLoS One 4: e4947, 2009. doi: 10.1371/journal.pone.0004947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Clausen TM, Sandoval DR, Spliid CB, Pihl J, Perrett HR, Painter CD, et al. SARS-CoV-2 infection depends on cellular heparan sulfate and ACE2. Cell 183: 1043–1057.e15, 2020. doi: 10.1016/j.cell.2020.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhang X, Wang B, O'Callaghan P, Hjertstrom E, Jia J, Gong F, Zcharia E, Nilsson LN, Lannfelt L, Vlodavsky I, Lindahl U, Li JP. Heparanase overexpression impairs inflammatory response and macrophage-mediated clearance of amyloid-beta in murine brain. Acta Neuropathol 124: 465–478, 2012. doi: 10.1007/s00401-012-0997-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jendresen CB, Cui H, Zhang X, Vlodavsky I, Nilsson LN, Li JP. Overexpression of heparanase lowers the amyloid burden in amyloid-beta precursor protein transgenic mice. J Biol Chem 290: 5053–5064, 2015. doi: 10.1074/jbc.M114.600569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Monaco A, Maffia V, Sorrentino NC, Sambri I, Ezhova Y, Giuliano T, Cacace V, Nusco E, De Risi M, De Leonibus E, Schrader T, Klarner FG, Bitan G, Fraldi A. The amyloid inhibitor CLR01 relieves autophagy and ameliorates neuropathology in a severe lysosomal storage disease. Mol Ther 28: 1167–1176, 2020. [Erratum in Mol Ther 30: 3499, 2022]. doi: 10.1016/j.ymthe.2020.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Snow AD, Cummings JA, Lake T. The unifying hypothesis of Alzheimer’s disease: heparan sulfate proteoglycans/glycosaminoglycans are key as first hypothesized over 30 years ago. Front Aging Neurosci 13: 710683, 2021. doi: 10.3389/fnagi.2021.710683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhang GL, Zhang X, Wang XM, Li JP. Towards understanding the roles of heparan sulfate proteoglycans in Alzheimer's disease. Biomed Res Int 2014: 516028, 2014. doi: 10.1155/2014/516028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Jin W, Zhang F, Linhardt RJ. Glycosaminoglycans in neurodegenerative diseases. Adv Exp Med Biol 1325: 189–204, 2021. doi: 10.1007/978-3-030-70115-4_9. [DOI] [PubMed] [Google Scholar]
- 71. Wang Z, Arnold K, Dhurandahare VM, Xu Y, Pagadala V, Labra E, Jeske W, Fareed J, Gearing M, Liu J. Analysis of 3-O-sulfated heparan sulfate using isotopically labeled oligosaccharide calibrants. Anal Chem 94: 2950–2957, 2022. [Erratum in Anal Chem 94: 4134, 2022]. doi: 10.1021/acs.analchem.1c04965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lindahl B, Lindahl U. Amyloid-specific heparan sulfate from human liver and spleen. J Biol Chem 272: 26091–26094, 1997. doi: 10.1074/jbc.272.42.26091. [DOI] [PubMed] [Google Scholar]
- 73. Sepulveda-Diaz JE, Alavi Naini SM, Huynh MB, Ouidja MO, Yanicostas C, Chantepie S, Villares J, Lamari F, Jospin E, van Kuppevelt TH, Mensah-Nyagan AG, Raisman-Vozari R, Soussi-Yanicostas N, Papy-Garcia D. HS3ST2 expression is critical for the abnormal phosphorylation of tau in Alzheimer's disease-related tau pathology. Brain 138: 1339–1354, 2015. doi: 10.1093/brain/awv056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Roberts RO, Kang YN, Hu C, Moser CD, Wang S, Moore MJ, Graham RP, Lai JP, Petersen RC, Roberts LR. Decreased expression of sulfatase 2 in the brains of Alzheimer's disease patients: implications for regulation of neuronal cell signaling. Alzheimers Dis Rep 1: 115–124, 2017. doi: 10.3233/ADR-170028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Perez-Lopez N, Martin C, Garcia B, Solis-Hernandez MP, Rodriguez D, Alcalde I, Merayo J, Fernandez-Vega I, Quiros LM. Alterations in the expression of the genes responsible for the synthesis of heparan sulfate in brains with Alzheimer disease. J Neuropathol Exp Neurol 80: 446–456, 2021. doi: 10.1093/jnen/nlab028. [DOI] [PubMed] [Google Scholar]
- 76. Rosenthal SB, Wang H, Shi D, Liu C, Abagyan R, McEvoy LK, Chen CH. Mapping the gene network landscape of Alzheimer's disease through integrating genomics and transcriptomics. PLoS Comput Biol 18: e1009903, 2022. doi: 10.1371/journal.pcbi.1009903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Johnson ECB, Carter EK, Dammer EB, Duong DM, Gerasimov ES, Liu Y, Liu J, Betarbet R, Ping L, Yin L, Serrano GE, Beach TG, Peng J, De Jager PL, Haroutunian V, Zhang B, Gaiteri C, Bennett DA, Gearing M, Wingo TS, Wingo AP, Lah JJ, Levey AI, Seyfried NT. Large-scale deep multi-layer analysis of Alzheimer's disease brain reveals strong proteomic disease-related changes not observed at the RNA level. Nat Neurosci 25: 213–225, 2022. doi: 10.1038/s41593-021-00999-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zhou X, Wang Y, Zheng W, Deng G, Wang F, Jin L. Characterizing heparin tetrasaccharides binding to amyloid-beta peptide. Front Mol Biosci 9: 824146, 2022. doi: 10.3389/fmolb.2022.824146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. DeWitt DA, Silver J, Canning DR, Perry G. Chondroitin sulfate proteoglycans are associated with the lesions of Alzheimer's disease. Exp Neurol 121: 149–152, 1993. doi: 10.1006/exnr.1993.1081. [DOI] [PubMed] [Google Scholar]
- 80. Bruinsma IB, Te Riet L, Gevers T, ten Dam GB, van Kuppevelt TH, David G, Kusters B, de Waal RM, Verbeek MM. Sulfation of heparan sulfate associated with amyloid-beta plaques in patients with Alzheimer's disease. Acta Neuropathol 119: 211–220, 2010. doi: 10.1007/s00401-009-0577-1. [DOI] [PubMed] [Google Scholar]
- 81. Bame KJ, Danda J, Hassall A, Tumova S. Abeta(1-40) prevents heparanase-catalyzed degradation of heparan sulfate glycosaminoglycans and proteoglycans in vitro. A role for heparan sulfate proteoglycan turnover in Alzheimer's disease. J Biol Chem 272: 17005–17011, 1997. doi: 10.1074/jbc.272.27.17005. [DOI] [PubMed] [Google Scholar]
- 82. Bergamaschini L, Rossi E, Storini C, Pizzimenti S, Distaso M, Perego C, De Luigi A, Vergani C, De Simoni MG. Peripheral treatment with enoxaparin, a low molecular weight heparin, reduces plaques and beta-amyloid accumulation in a mouse model of Alzheimer's disease. J Neurosci 24: 4181–4186, 2004. doi: 10.1523/JNEUROSCI.0550-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Brunden KR, Richter-Cook NJ, Chaturvedi N, Frederickson RC. pH-dependent binding of synthetic beta-amyloid peptides to glycosaminoglycans. J Neurochem 61: 2147–2154, 1993. doi: 10.1111/j.1471-4159.1993.tb07453.x. [DOI] [PubMed] [Google Scholar]
- 84. Genedani S, Agnati LF, Leo G, Buzzega D, Maccari F, Carone C, Andreoli N, Filaferro M, Volpi N. beta-Amyloid fibrillation and/or hyperhomocysteinemia modify striatal patterns of hyaluronic acid and dermatan sulfate: possible role in the pathogenesis of Alzheimer's disease. Curr Alzheimer Res 7: 150–157, 2010. doi: 10.2174/156720510790691074. [DOI] [PubMed] [Google Scholar]
- 85. Giulian D, Haverkamp LJ, Yu J, Karshin W, Tom D, Li J, Kazanskaia A, Kirkpatrick J, Roher AE. The HHQK domain of β-amyloid provides a structural basis for the immunopathology of Alzheimer's disease. J Biol Chem 273: 29719–29726, 1998. doi: 10.1074/jbc.273.45.29719. [DOI] [PubMed] [Google Scholar]
- 86. Nguyen K, Rabenstein DL. Interaction of the heparin-binding consensus sequence of β-amyloid peptides with heparin and heparin-derived oligosaccharides. J Phys Chem B 120: 2187–2197, 2016. doi: 10.1021/acs.jpcb.5b12235. [DOI] [PubMed] [Google Scholar]
- 87. Madine J, Pandya MJ, Hicks MR, Rodger A, Yates EA, Radford SE, Middleton DA. Site-specific identification of an abeta fibril-heparin interaction site by using solid-state NMR spectroscopy. Angew Chem Int Ed Engl 51: 13140–13143, 2012. doi: 10.1002/anie.201204459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Stewart KL, Hughes E, Yates EA, Akien GR, Huang TY, Lima MA, Rudd TR, Guerrini M, Hung SC, Radford SE, Middleton DA. Atomic details of the interactions of glycosaminoglycans with amyloid-beta fibrils. J Am Chem Soc 138: 8328–8331, 2016. doi: 10.1021/jacs.6b02816. [DOI] [PubMed] [Google Scholar]
- 89. Stewart KL, Hughes E, Yates EA, Middleton DA, Radford SE. Molecular Origins of the Compatibility between glycosaminoglycans and Abeta40 amyloid fibrils. J Mol Biol 429: 2449–2462, 2017. doi: 10.1016/j.jmb.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Madine J, Clayton JC, Yates EA, Middleton DA. Exploiting a (13)C-labelled heparin analogue for in situ solid-state NMR investigations of peptide-glycan interactions within amyloid fibrils. Org Biomol Chem 7: 2414–2420, 2009. doi: 10.1039/b820808e. [DOI] [PubMed] [Google Scholar]
- 91. O'Callaghan P, Sandwall E, Li JP, Yu H, Ravid R, Guan ZZ, van Kuppevelt TH, Nilsson LN, Ingelsson M, Hyman BT, Kalimo H, Lindahl U, Lannfelt L, Zhang X. Heparan sulfate accumulation with Abeta deposits in Alzheimer's disease and Tg2576 mice is contributed by glial cells. Brain Pathol 18: 548–561, 2008. doi: 10.1111/j.1750-3639.2008.00152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Dennissen MA, Jenniskens GJ, Pieffers M, Versteeg EM, Petitou M, Veerkamp JH, van Kuppevelt TH. Large, tissue-regulated domain diversity of heparan sulfates demonstrated by phage display antibodies. J Biol Chem 277: 10982–10986, 2002. doi: 10.1074/jbc.M104852200. [DOI] [PubMed] [Google Scholar]
- 93. Hosono-Fukao T, Ohtake-Niimi S, Hoshino H, Britschgi M, Akatsu H, Hossain MM, Nishitsuji K, van Kuppevelt TH, Kimata K, Michikawa M, Wyss-Coray T, Uchimura K. Heparan sulfate subdomains that are degraded by Sulf accumulate in cerebral amyloid ss plaques of Alzheimer's disease: evidence from mouse models and patients. Am J Pathol 180: 2056–2067, 2012. doi: 10.1016/j.ajpath.2012.01.015. [DOI] [PubMed] [Google Scholar]
- 94. Castillo GM, Snow AD. In Vitro formation of congophilic maltese-cross amyloid plaques to identify anti-plaque therapeutics for the treatment of Alzheimer’s disease and prion diseases. US Patent 7,148,001 B2. Dec 12, 2006. [Google Scholar]
- 95. Ring S, Weyer SW, Kilian SB, Waldron E, Pietrzik CU, Filippov MA, Herms J, Buchholz C, Eckman CB, Korte M, Wolfer DP, Muller UC. The secreted beta-amyloid precursor protein ectodomain APPs alpha is sufficient to rescue the anatomical, behavioral, and electrophysiological abnormalities of APP-deficient mice. J Neurosci 27: 7817–7826, 2007. doi: 10.1523/JNEUROSCI.1026-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Small DH, Nurcombe V, Reed G, Clarris H, Moir R, Beyreuther K, Masters CL. A heparin-binding domain in the amyloid protein precursor of Alzheimer's disease is involved in the regulation of neurite outgrowth. J Neurosci 14: 2117–2127, 1994. doi: 10.1523/JNEUROSCI.14-04-02117.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Williamson TG, Mok SS, Henry A, Cappai R, Lander AD, Nurcombe V, Beyreuther K, Masters CL, Small DH. Secreted glypican binds to the amyloid precursor protein of Alzheimer's disease (APP) and inhibits APP-induced neurite outgrowth. J Biol Chem 271: 31215–31221, 1996. doi: 10.1074/jbc.271.49.31215. [DOI] [PubMed] [Google Scholar]
- 98. Small DH, Williamson T, Reed G, Clarris H, Beyreuther K, Masters CL, Nurcombe V. The role of heparan sulfate proteoglycans in the pathogenesis of Alzheimer's disease. Ann N Y Acad Sci 777: 316–321, 1996. doi: 10.1111/j.1749-6632.1996.tb34439.x. [DOI] [PubMed] [Google Scholar]
- 99. Small DH, Nurcombe V, Moir R, Michaelson S, Monard D, Beyreuther K, Masters CL. Association and release of the amyloid protein precursor of Alzheimer's disease from chick brain extracellular matrix. J Neurosci 12: 4143–4150, 1992. doi: 10.1523/JNEUROSCI.12-11-04143.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Schubert D, LaCorbiere M, Saitoh T, Cole G. Characterization of an amyloid beta precursor protein that binds heparin and contains tyrosine sulfate. Proc Natl Acad Sci USA 86: 2066–2069, 1989. doi: 10.1073/pnas.86.6.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Reinhard C, Borgers M, David G, De Strooper B. Soluble amyloid-beta precursor protein binds its cell surface receptor in a cooperative fashion with glypican and syndecan proteoglycans. J Cell Sci 126: 4856–4861, 2013. doi: 10.1242/jcs.137919. [DOI] [PubMed] [Google Scholar]
- 102. Cheng F, Fransson LA, Mani K. Reversal of apolipoprotein E4-dependent or chemical-induced accumulation of APP degradation products by vitamin C-induced release of heparan sulfate from glypican-1. Glycobiology 31: 800–811, 2021. doi: 10.1093/glycob/cwaa120. [DOI] [PubMed] [Google Scholar]
- 103. Tanimoto R, Palladino C, Xu SQ, Buraschi S, Neill T, Gomella LG, Peiper SC, Belfiore A, Iozzo RV, Morrione A. The perlecan-interacting growth factor progranulin regulates ubiquitination, sorting, and lysosomal degradation of sortilin. Matrix Biol 64: 27–39, 2017. doi: 10.1016/j.matbio.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Narindrasorasak S, Altman RA, Gonzalez-DeWhitt P, Greenberg BD, Kisilevsky R. An interaction between basement membrane and Alzheimer amyloid precursor proteins suggests a role in the pathogenesis of Alzheimer's disease. Lab Invest 72: 272–282, 1995. [PubMed] [Google Scholar]
- 105. Li L, Liu F, Welser-Alves JV, McCullough LD, Milner R. Upregulation of fibronectin and the alpha5beta1 and alphavbeta3 integrins on blood vessels within the cerebral ischemic penumbra. Exp Neurol 233: 283–291, 2012. doi: 10.1016/j.expneurol.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Trout AL, Rutkai I, Biose IJ, Bix GJ. Review of alterations in Perlecan-associated vascular risk factors in dementia. Int J Mol Sci 21: 679, 2020. doi: 10.3390/ijms21020679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Snow AD, Sekiguchi R, Nochlin D, Fraser P, Kimata K, Mizutani A, Arai M, Schreier WA, Morgan DG. An important role of heparan sulfate proteoglycan (Perlecan) in a model system for the deposition and persistence of fibrillar A beta-amyloid in rat brain. Neuron 12: 219–234, 1994. doi: 10.1016/0896-6273(94)90165-1. [DOI] [PubMed] [Google Scholar]
- 108. Leveugle B, Ding W, Laurence F, Dehouck MP, Scanameo A, Cecchelli R, Fillit H. Heparin oligosaccharides that pass the blood-brain barrier inhibit beta-amyloid precursor protein secretion and heparin binding to beta-amyloid peptide. J Neurochem 70: 736–744, 1998. doi: 10.1046/j.1471-4159.1998.70020736.x. [DOI] [PubMed] [Google Scholar]
- 109. Jorda-Siquier T, Petrel M, Kouskoff V, Smailovic U, Cordelieres F, Frykman S, Muller U, Mulle C, Barthet G. APP accumulates with presynaptic proteins around amyloid plaques: a role for presynaptic mechanisms in Alzheimer's disease? Alzheimers Dement 18: 2099–2116, 2022. doi: 10.1002/alz.12546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Patey SJ, Edwards EA, Yates EA, Turnbull JE. Heparin derivatives as inhibitors of BACE-1, the Alzheimer's beta-secretase, with reduced activity against factor Xa and other proteases. J Med Chem 49: 6129–6132, 2006. doi: 10.1021/jm051221o. [DOI] [PubMed] [Google Scholar]
- 111. Leveugle B, Ding W, Durkin JT, Mistretta S, Eisle J, Matic M, Siman R, Greenberg BD, Fillit HM. Heparin promotes beta-secretase cleavage of the Alzheimer's amyloid precursor protein. Neurochem Int 30: 543–548, 1997. doi: 10.1016/s0197-0186(96)00103-9. [DOI] [PubMed] [Google Scholar]
- 112. Scholefield Z, Yates EA, Wayne G, Amour A, McDowell W, Turnbull JE. Heparan sulfate regulates amyloid precursor protein processing by BACE1, the Alzheimer's beta-secretase. J Cell Biol 163: 97–107, 2003. doi: 10.1083/jcb.200303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Klaver DW, Wilce MC, Gasperini R, Freeman C, Juliano JP, Parish C, Foa L, Aguilar MI, Small DH. Glycosaminoglycan-induced activation of the beta-secretase (BACE1) of Alzheimer's disease. J Neurochem 112: 1552–1561, 2010. doi: 10.1111/j.1471-4159.2010.06571.x. [DOI] [PubMed] [Google Scholar]
- 114. Nagai N, Habuchi H, Kitazume S, Toyoda H, Hashimoto Y, Kimata K. Regulation of heparan sulfate 6-O-sulfation by beta-secretase activity. J Biol Chem 282: 14942–14951, 2007. doi: 10.1074/jbc.M610691200. [DOI] [PubMed] [Google Scholar]
- 115. Mahley RW. Apolipoprotein E: remarkable protein sheds light on cardiovascular and neurological diseases. Clin Chem 63: 14–20, 2017. doi: 10.1373/clinchem.2016.255695. [DOI] [PubMed] [Google Scholar]
- 116. Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, Rosi BL, Gusella JF, Crapper-MacLachlan DR, Alberts MJ. and Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer's disease. Neurology 43: 1467–1472, 1993. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 117. Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261: 921–923, 1993. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 118. Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, van Duijn CM. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 278: 1349–1356, 1997. doi: 10.1001/jama.1997.03550160069041. [DOI] [PubMed] [Google Scholar]
- 119. Kok E, Haikonen S, Luoto T, Huhtala H, Goebeler S, Haapasalo H, Karhunen PJ. Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Ann Neurol 65: 650–657, 2009. doi: 10.1002/ana.21696. [DOI] [PubMed] [Google Scholar]
- 120. Morris JC, Roe CM, Xiong C, Fagan AM, Goate AM, Holtzman DM, Mintun MA. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol 67: 122–131, 2010. doi: 10.1002/ana.21843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Hawkes CA, Sullivan PM, Hands S, Weller RO, Nicoll JA, Carare RO. Disruption of arterial perivascular drainage of amyloid-beta from the brains of mice expressing the human APOE epsilon4 allele. PLoS One 7: e41636, 2012. doi: 10.1371/journal.pone.0041636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Robert J, Button EB, Yuen B, Gilmour M, Kang K, Bahrabadi A, Stukas S, Zhao W, Kulic I, Wellington CL. Clearance of beta-amyloid is facilitated by apolipoprotein E and circulating high-density lipoproteins in bioengineered human vessels. eLife 6: e29595, 2017. doi: 10.7554/eLife.29595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. DeMattos RB, Cirrito JR, Parsadanian M, May PC, O'Dell MA, Taylor JW, Harmony JA, Aronow BJ, Bales KR, Paul SM, Holtzman DM. ApoE and clusterin cooperatively suppress Abeta levels and deposition: evidence that ApoE regulates extracellular Abeta metabolism in vivo. Neuron 41: 193–202, 2004. doi: 10.1016/s0896-6273(03)00850-x. [DOI] [PubMed] [Google Scholar]
- 124. Kanekiyo T, Bu G. The low-density lipoprotein receptor-related protein 1 and amyloid-beta clearance in Alzheimer's disease. Front Aging Neurosci 6: 93, 2014. doi: 10.3389/fnagi.2014.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Li Y, Rusinek H, Butler T, Glodzik L, Pirraglia E, Babich J, Mozley PD, Nehmeh S, Pahlajani S, Wang X, Tanzi EB, Zhou L, Strauss S, Carare RO, Theise N, Okamura N, de Leon MJ. Decreased CSF clearance and increased brain amyloid in Alzheimer's disease. Fluids Barriers CNS 19: 21, 2022. doi: 10.1186/s12987-022-00318-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Hudak A, Josvay K, Domonkos I, Letoha A, Szilak L, Letoha T. The interplay of apoes with syndecans in influencing key cellular events of amyloid pathology. Int J Mol Sci 22: 7070, 2021. doi: 10.3390/ijms22137070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Huang YA, Zhou B, Wernig M, Sudhof TC. ApoE2, ApoE3, and ApoE4 differentially stimulate APP transcription and Abeta secretion. Cell 168: 427–441.e21, 2017. doi: 10.1016/j.cell.2016.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Saito H, Dhanasekaran P, Nguyen D, Baldwin F, Weisgraber KH, Wehrli S, Phillips MC, Lund-Katz S. Characterization of the heparin binding sites in human apolipoprotein E. J Biol Chem 278: 14782–14787, 2003. doi: 10.1074/jbc.M213207200. [DOI] [PubMed] [Google Scholar]
- 129. Futamura M, Dhanasekaran P, Handa T, Phillips MC, Lund-Katz S, Saito H. Two-step mechanism of binding of apolipoprotein E to heparin: implications for the kinetics of apolipoprotein E-heparan sulfate proteoglycan complex formation on cell surfaces. J Biol Chem 280: 5414–5422, 2005. doi: 10.1074/jbc.M411719200. [DOI] [PubMed] [Google Scholar]
- 130. Libeu CP, Lund-Katz S, Phillips MC, Wehrli S, Hernaiz MJ, Capila I, Linhardt RJ, Raffai RL, Newhouse YM, Zhou F, Weisgraber KH. New insights into the heparan sulfate proteoglycan-binding activity of apolipoprotein E. J Biol Chem 276: 39138–39144, 2001. doi: 10.1074/jbc.M104746200. [DOI] [PubMed] [Google Scholar]
- 131. Yamauchi Y, Deguchi N, Takagi C, Tanaka M, Dhanasekaran P, Nakano M, Handa T, Phillips MC, Lund-Katz S, Saito H. Role of the N- and C-terminal domains in binding of apolipoprotein E isoforms to heparan sulfate and dermatan sulfate: a surface plasmon resonance study. Biochemistry 47: 6702–6710, 2008. doi: 10.1021/bi8003999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Fu Y, Zhao J, Atagi Y, Nielsen HM, Liu CC, Zheng H, Shinohara M, Kanekiyo T, Bu G. Apolipoprotein E lipoprotein particles inhibit amyloid-beta uptake through cell surface heparan sulphate proteoglycan. Mol Neurodegener 11: 37, 2016. doi: 10.1186/s13024-016-0099-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Arboleda-Velasquez JF, Lopera F, O’Hare M, Delgado-Tirado S, Marino C, et al. Resistance to autosomal dominant Alzheimer's disease in an APOE3 Christchurch homozygote: a case report. Nat Med 25: 1680–1683, 2019. doi: 10.1038/s41591-019-0611-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. O'Callaghan P, Noborn F, Sehlin D, Li JP, Lannfelt L, Lindahl U, Zhang X. Apolipoprotein E increases cell association of amyloid-beta 40 through heparan sulfate and LRP1 dependent pathways. Amyloid 21: 76–87, 2014. doi: 10.3109/13506129.2013.879643. [DOI] [PubMed] [Google Scholar]
- 135. Wilsie LC, Orlando RA. The low density lipoprotein receptor-related protein complexes with cell surface heparan sulfate proteoglycans to regulate proteoglycan-mediated lipoprotein catabolism. J Biol Chem 278: 15758–15764, 2003. doi: 10.1074/jbc.M208786200. [DOI] [PubMed] [Google Scholar]
- 136. Mahley RW, Ji ZS. Remnant lipoprotein metabolism: key pathways involving cell-surface heparan sulfate proteoglycans and apolipoprotein E. J Lipid Res 40: 1–16, 1999. [PubMed] [Google Scholar]
- 137. Kanekiyo T, Zhang J, Liu Q, Liu CC, Zhang L, Bu G. Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal amyloid-beta uptake. J Neurosci 31: 1644–1651, 2011. doi: 10.1523/JNEUROSCI.5491-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. van Horssen J, Otte-Holler I, David G, Maat-Schieman ML, van den Heuvel LP, Wesseling P, de Waal RM, Verbeek MM. Heparan sulfate proteoglycan expression in cerebrovascular amyloid beta deposits in Alzheimer's disease and hereditary cerebral hemorrhage with amyloidosis (Dutch) brains. Acta Neuropathol 102: 604–614, 2001. doi: 10.1007/s004010100414. [DOI] [PubMed] [Google Scholar]
- 139. van Horssen J, Kleinnijenhuis J, Maass CN, Rensink AA, Otte-Holler I, David G, van den Heuvel LP, Wesseling P, de Waal RM, Verbeek MM. Accumulation of heparan sulfate proteoglycans in cerebellar senile plaques. Neurobiol Aging 23: 537–545, 2002. doi: 10.1016/s0197-4580(02)00010-6. [DOI] [PubMed] [Google Scholar]
- 140. Watanabe N, Araki W, Chui DH, Makifuchi T, Ihara Y, Tabira T. Glypican-1 as an Abeta binding HSPG in the human brain: its localization in DIG domains and possible roles in the pathogenesis of Alzheimer's disease. FASEB J 18: 1013–1015, 2004. doi: 10.1096/fj.03-1040fje. [DOI] [PubMed] [Google Scholar]
- 141. Lorente-Gea L, Garcia B, Martin C, Ordiales H, Garcia-Suarez O, Pina-Batista KM, Merayo-Lloves J, Quiros LM, Fernandez-Vega I. Heparan sulfate proteoglycans undergo differential expression alterations in Alzheimer disease brains. J Neuropathol Exp Neurol 79: 474–483, 2020. doi: 10.1093/jnen/nlaa016. [DOI] [PubMed] [Google Scholar]
- 142. Verbeek MM, Otte-Höller I, van den Born J, van den Heuvel LPWJ, David G, Wesseling P, de Waal RMW. Agrin is a major heparan sulfate proteoglycan accumulating in Alzheimer's disease brain. Am J Pathol 155: 2115–2125, 1999. doi: 10.1016/S0002-9440(10)65529-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Donahue JE, Berzin TM, Rafii MS, Glass DJ, Yancopoulos GD, Fallon JR, Stopa EG. Agrin in Alzheimer's disease: altered solubility and abnormal distribution within microvasculature and brain parenchyma. Proc Natl Acad Sci USA 96: 6468–6472, 1999. doi: 10.1073/pnas.96.11.6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Berzin TM, Zipser BD, Rafii MS, Kuo-Leblanc V, Yancopoulos GD, Glass DJ, Fallon JR, Stopa EG. Agrin and microvascular damage in Alzheimer's disease. Neurobiol Aging 21: 349–355, 2000. doi: 10.1016/s0197-4580(00)00121-4. [DOI] [PubMed] [Google Scholar]
- 145. Cotman SL, Halfter W, Cole GJ. Agrin binds to beta-amyloid (Abeta), accelerates abeta fibril formation, and is localized to Abeta deposits in Alzheimer's disease brain. Mol Cell Neurosci 15: 183–198, 2000. doi: 10.1006/mcne.1999.0816. [DOI] [PubMed] [Google Scholar]
- 146. Lepelletier FX, Mann DM, Robinson AC, Pinteaux E, Boutin H. Early changes in extracellular matrix in Alzheimer's disease. Neuropathol Appl Neurobiol 43: 167–182, 2017. doi: 10.1111/nan.12295. [DOI] [PubMed] [Google Scholar]
- 147. Miller JD, Cummings J, Maresh GA, Walker DG, Castillo GM, Ngo C, Kimata K, Kinsella MG, Wight TN, Snow AD. Localization of perlecan (or a perlecan-related macromolecule) to isolated microglia in vitro and to microglia/macrophages following infusion of beta-amyloid protein into rodent hippocampus. Glia 21: 228–243, 1997. doi:. [DOI] [PubMed] [Google Scholar]
- 148. Castillo GM, Cummings JA, Ngo C, Yang W, Snow AD. Novel purification and detailed characterization of perlecan isolated from the Engelbreth-Holm-Swarm tumor for use in an animal model of fibrillar A beta amyloid persistence in brain. J Biochem 120: 433–444, 1996. doi: 10.1093/oxfordjournals.jbchem.a021430. [DOI] [PubMed] [Google Scholar]
- 149. Arrasate M, Perez M, Valpuesta JM, Avila J. Role of glycosaminoglycans in determining the helicity of paired helical filaments. Am J Pathol 151: 1115–1122, 1997. [PMC free article] [PubMed] [Google Scholar]
- 150. Goedert M, Jakes R, Spillantini MG, Hasegawa M, Smith MJ, Crowther RA. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 383: 550–553, 1996. doi: 10.1038/383550a0. [DOI] [PubMed] [Google Scholar]
- 151. Zhang Q, Ma C, Chin LS, Li L. Integrative glycoproteomics reveals protein N-glycosylation aberrations and glycoproteomic network alterations in Alzheimer's disease. Sci Adv 6: eabc5802, 2020. doi: 10.1126/sciadv.abc5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Trout AL, Kahle MP, Roberts JM, Marcelo A, de Hoog L, Boychuk JA, Grupke SL, Berretta A, Gowing EK, Boychuk CR, Gorman AA, Edwards DN, Rutkai I, Biose IJ, Ishibashi-Ueda H, Ihara M, Smith BN, Clarkson AN, Bix GJ. Perlecan domain-V enhances neurogenic brain repair after stroke in mice. Transl Stroke Res 12: 72–86, 2021. doi: 10.1007/s12975-020-00800-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Bix G, Fu J, Gonzalez EM, Macro L, Barker A, Campbell S, Zutter MM, Santoro SA, Kim JK, Hook M, Reed CC, Iozzo RV. Endorepellin causes endothelial cell disassembly of actin cytoskeleton and focal adhesions through alpha2beta1 integrin. J Cell Biol 166: 97–109, 2004. [Erratum in J Cell Biol 201: 641, 2013]. doi: 10.1083/jcb.200401150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Lee B, Clarke D, Al Ahmad A, Kahle M, Parham C, Auckland L, Shaw C, Fidanboylu M, Orr AW, Ogunshola O, Fertala A, Thomas SA, Bix GJ. Perlecan domain V is neuroprotective and proangiogenic following ischemic stroke in rodents. J Clin Invest 121: 3005–3023, 2011. [Erratum in J Clin Invest 122: 777, 2012]. doi: 10.1172/JCI46358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Parham C, Auckland L, Rachwal J, Clarke D, Bix G. Perlecan domain V inhibits amyloid-beta induced brain endothelial cell toxicity and restores angiogenic function. J Alzheimers Dis 38: 415–423, 2014. doi: 10.3233/JAD-130683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Parham CL, Shaw C, Auckland LD, Dickeson SK, Griswold-Prenner I, Bix G. Perlecan domain V inhibits amyloid-beta induced activation of the alpha2beta1 integrin-mediated neurotoxic signaling cascade. J Alzheimers Dis 54: 1629–1647, 2016. doi: 10.3233/JAD-160290. [DOI] [PubMed] [Google Scholar]
- 157. Siegel G, Malmsten M, Ermilov E. Anionic biopolyelectrolytes of the syndecan/perlecan superfamily: physicochemical properties and medical significance. Adv Colloid Interface Sci 205: 275–318, 2014. doi: 10.1016/j.cis.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 158. Verbeek MM, Otte-Holler I, Ruiter DJ, de Waal RM. Human brain pericytes as a model system to study the pathogenesis of cerebrovascular amyloidosis in Alzheimer's disease. Cell Mol Biol (Noisy-le-grand ) 45: 37–46, 1999. [PubMed] [Google Scholar]
- 159. Timmer NM, van Horssen J, Otte-Holler I, Wilhelmus MM, David G, van Beers J, de Waal RM, Verbeek MM. Amyloid beta induces cellular relocalization and production of agrin and glypican-1. Brain Res 1260: 38–46, 2009. doi: 10.1016/j.brainres.2008.12.063. [DOI] [PubMed] [Google Scholar]
- 160. Rauch SM, Huen K, Miller MC, Chaudry H, Lau M, Sanes JR, Johanson CE, Stopa EG, Burgess RW. Changes in brain beta-amyloid deposition and aquaporin 4 levels in response to altered agrin expression in mice. J Neuropathol Exp Neurol 70: 1124–1137, 2011. doi: 10.1097/NEN.0b013e31823b0b12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Del Campo Milan M, Zuroff L, Jimenez CR, Scheltens P, Teunissen CE. Can agrin cerebrospinal fluid concentration be used as an early biomarker for Alzheimer's disease? Alzheimers Dement (Amst) 1: 75–80, 2015. doi: 10.1016/j.dadm.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Gingras J, Rassadi S, Cooper E, Ferns M. Agrin plays an organizing role in the formation of sympathetic synapses. J Cell Biol 158: 1109–1118, 2002. doi: 10.1083/jcb.200203012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Zhang X, Han X, Xia K, Xu Y, Yang Y, Oshima K, Haeger SM, Perez MJ, McMurtry SA, Hippensteel JA, Ford JA, Herson PS, Liu J, Schmidt EP, Linhardt RJ. Circulating heparin oligosaccharides rapidly target the hippocampus in sepsis, potentially impacting cognitive functions. Proc Natl Acad Sci USA 116: 9208–9213, 2019. doi: 10.1073/pnas.1902227116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Hagino S, Iseki K, Mori T, Zhang Y, Hikake T, Yokoya S, Takeuchi M, Hasimoto H, Kikuchi S, Wanaka A. Slit and glypican-1 mRNAs are coexpressed in the reactive astrocytes of the injured adult brain. Glia 42: 130–138, 2003. doi: 10.1002/glia.10207. [DOI] [PubMed] [Google Scholar]
- 165. Doan RN, Bae BI, Cubelos B, Chang C, Hossain AA, Al-Saad S, Mukaddes NM, Oner O, Al-Saffar M, Balkhy S, Gascon GG, Homozygosity Mapping Consortium For A, Nieto M, Walsh CA; Homozygosity Mapping Consortium for Autism. Mutations in human accelerated regions disrupt cognition and social behavior. Cell 167: 341–354.e12, 2016. e312, doi: 10.1016/j.cell.2016.08.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Cheng F, Ruscher K, Fransson LA, Mani K. Non-toxic amyloid beta formed in the presence of glypican-1 or its deaminatively generated heparan sulfate degradation products. Glycobiology 23: 1510–1519, 2013. doi: 10.1093/glycob/cwt079. [DOI] [PubMed] [Google Scholar]
- 167. Ma K, Xing S, Luan Y, Zhang C, Liu Y, Fei Y, Zhang Z, Liu Y, Chen X. Glypican 4 regulates Abeta internalization in neural stem cells partly via low-density lipoprotein receptor-related protein 1. Front Cell Neurosci 15: 732429, 2021. doi: 10.3389/fncel.2021.732429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Bartosch AMW, Mathews R, Mahmoud MM, Cancel LM, Haq ZS, Tarbell JM. Heparan sulfate proteoglycan glypican-1 and PECAM-1 cooperate in shear-induced endothelial nitric oxide production. Sci Rep 11: 11386, 2021. doi: 10.1038/s41598-021-90941-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Letoha T, Hudak A, Kusz E, Pettko-Szandtner A, Domonkos I, Josvay K, Hofmann-Apitius M, Szilak L. Contribution of syndecans to cellular internalization and fibrillation of amyloid-beta(1-42). Sci Rep 9: 1393, 2019. doi: 10.1038/s41598-018-37476-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Lauri SE, Kaukinen S, Kinnunen T, Ylinen A, Imai S, Kaila K, Taira T, Rauvala H. Reg1ulatory role and molecular interactions of a cell-surface heparan sulfate proteoglycan (N-syndecan) in hippocampal long-term potentiation. J Neurosci 19: 1226–1235, 1999. doi: 10.1523/JNEUROSCI.19-04-01226.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Lark DS, LaRocca TJ. Expression of exosome biogenesis genes is differentially altered by aging in the mouse and in the human brain during Alzheimer's disease. J Gerontol A Biol Sci Med Sci 77: 659–663, 2021. doi: 10.1093/gerona/glab322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Zhang Q, Ma C, Gearing M, Wang PG, Chin LS, Li L. Integrated proteomics and network analysis identifies protein hubs and network alterations in Alzheimer's disease. Acta Neuropathol Commun 6: 19, 2018. doi: 10.1186/s40478-018-0524-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Barrera-Ocampo A, Arlt S, Matschke J, Hartmann U, Puig B, Ferrer I, Zurbig P, Glatzel M, Sepulveda-Falla D, Jahn H. Amyloid-beta precursor protein modulates the sorting of testican-1 and contributes to its accumulation in brain tissue and cerebrospinal fluid from patients with Alzheimer disease. J Neuropathol Exp Neurol 75: 903–916, 2016. doi: 10.1093/jnen/nlw065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Blasco Tavares Pereira Lopes F, Schlatzer D, Wang R, Li X, Feng E, Koyuturk M, Qi X, Chance MR. Temporal and sex-linked protein expression dynamics in a familial model of Alzheimer's disease. Mol Cell Proteomics 21: 100280, 2022. doi: 10.1016/j.mcpro.2022.100280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Snow AD, Mar H, Nochlin D, Kresse H, Wight TN. Peripheral distribution of dermatan sulfate proteoglycans (decorin) in amyloid-containing plaques and their presence in neurofibrillary tangles of Alzheimer's disease. J Histochem Cytochem 40: 105–113, 1992. doi: 10.1177/40.1.1370306. [DOI] [PubMed] [Google Scholar]
- 176. Logsdon AF, Francis KL, Richardson NE, Hu SJ, Faber CL, Phan BA, Nguyen V, Setthavongsack N, Banks WA, Woltjer RL, Keene CD, Latimer CS, Schwartz MW, Scarlett JM, Alonge KM. Decoding perineuronal net glycan sulfation patterns in the Alzheimer's disease brain. Alzheimers Dement 18: 942–954, 2022. doi: 10.1002/alz.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Lindahl B, Eriksson L, Spillmann D, Caterson B, Lindahl U. Selective loss of cerebral keratan sulfate in Alzheimer's disease. J Biol Chem 271: 16991–16994, 1996. doi: 10.1074/jbc.271.29.16991. [DOI] [PubMed] [Google Scholar]
- 178. Moon S, Zhao YT. Spatial, temporal and cell-type-specific expression profiles of genes encoding heparan sulfate biosynthesis enzymes and proteoglycan core proteins. Glycobiology 31: 1308–1318, 2021. doi: 10.1093/glycob/cwab054. [DOI] [PubMed] [Google Scholar]
- 179. Kato M, Wang H, Bernfield M, Gallagher JT, Turnbull JE. Cell surface syndecan-1 on distinct cell types differs in fine structure and ligand binding of its heparan sulfate chains. J Biol Chem 269: 18881–18890, 1994. [PubMed] [Google Scholar]
- 180. Zhang X, O’Callaghan P, Li H, Tan Y, Zhang G, Barash U, Wang X, Lannfelt L, Vlodavsky I, Lindahl U, Li J-P. Heparanase overexpression impedes perivascular clearance of amyloid-beta from murine brain: relevance to Alzheimer's disease. Acta Neuropathol Commun 9: 84, 2021. doi: 10.1186/s40478-021-01182-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med 201: 647–657, 2005. doi: 10.1084/jem.20041611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Kober DL, Alexander-Brett JM, Karch CM, Cruchaga C, Colonna M, Holtzman MJ, Brett TJ. Neurodegenerative disease mutations in TREM2 reveal a functional surface and distinct loss-of-function mechanisms. Elife 5: e20391, 2016. doi: 10.7554/eLife.20391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Kober DL, Brett TJ. TREM2-ligand interactions in health and disease. J Mol Biol 429: 1607–1629, 2017. doi: 10.1016/j.jmb.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Bhute S, Sarmah D, Datta A, Rane P, Shard A, Goswami A, Borah A, Kalia K, Dave KR, Bhattacharya P. Molecular pathogenesis and interventional strategies for Alzheimer's disease: promises and pitfalls. ACS Pharmacol Transl Sci 3: 472–488, 2020. doi: 10.1021/acsptsci.9b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Kober DL, Stuchell-Brereton MD, Kluender CE, Dean HB, Strickland MR, Steinberg DF, Nelson SS, Baban B, Holtzman DM, Frieden C, Alexander-Brett J, Roberson ED, Song Y, Brett TJ. Functional insights from biophysical study of TREM2 interactions with apoE and Aβ1-42. Alzheimers Dement. 17: 475–488, 2021. doi: 10.1002/alz.12194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Sudom A, Talreja S, Danao J, Bragg E, Kegel R, Min X, Richardson J, Zhang Z, Sharkov N, Marcora E, Thibault S, Bradley J, Wood S, Lim AC, Chen H, Wang S, Foltz IN, Sambashivan S, Wang Z. Molecular basis for the loss-of-function effects of the Alzheimer's disease-associated R47H variant of the immune receptor TREM2. J Biol Chem 293: 12634–12646, 2018. doi: 10.1074/jbc.RA118.002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Jin SC, Benitez BA, Karch CM, Cooper B, Skorupa T, Carrell D, Norton JB, Hsu S, Harari O, Cai Y, Bertelsen S, Goate AM, Cruchaga C. Coding variants in TREM2 increase risk for Alzheimer's disease. Hum Mol Genet 23: 5838–5846, 2014. doi: 10.1093/hmg/ddu277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Lill CM, Rengmark A, Pihlstrom L, Fogh I, Shatunov A, Sleiman PM, Wang LS, Liu T, Lassen CF, Meissner E, Alexopoulos P, Calvo A, Chio A, Dizdar N, Faltraco F, Forsgren L, Kirchheiner J, Kurz A, Larsen JP, Liebsch M, Linder J, Morrison KE, Nissbrandt H, Otto M, Pahnke J, Partch A, Restagno G, Rujescu D, Schnack C, Shaw CE, SLAGEN Consortium, et al. The role of TREM2 R47H as a risk factor for Alzheimer's disease, frontotemporal lobar degeneration, amyotrophic lateral sclerosis, and Parkinson's disease. Alzheimers Dement 11: 1407–1416, 2015. doi: 10.1016/j.jalz.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Jin SC, Carrasquillo MM, Benitez BA, Skorupa T, Carrell D, Patel D, Lincoln S, Krishnan S, Kachadoorian M, Reitz C, Mayeux R, Wingo TS, Lah JJ, Levey AI, Murrell J, Hendrie H, Foroud T, Graff-Radford NR, Goate AM, Cruchaga C, Ertekin-Taner N. TREM2 is associated with increased risk for Alzheimer's disease in African Americans. Mol Neurodegener 10: 19, 2015. doi: 10.1186/s13024-015-0016-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Golde TE, Streit WJ, Chakrabarty P. Alzheimer's disease risk alleles in TREM2 illuminate innate immunity in Alzheimer's disease. Alzheimers Res Ther 5: 24, 2013. doi: 10.1186/alzrt178. [DOI] [PMC free article] [PubMed] [Google Scholar]