

Keywords: ATF4, CD36, lipotoxicity, palmitate, PPARγ
Abstract
Hepatic lipotoxicity plays a central role in the pathogenesis of nonalcoholic fatty liver disease; however, the underlying mechanisms remain elusive. Here, using both cultured hepatocytes (AML-12 cells and primary mouse hepatocytes) and the liver-specific gene knockout mice, we investigated the mechanisms underlying palmitate-elicited upregulation of CD36, a class B scavenger receptor mediating long-chain fatty acids uptake, and its role in palmitate-induced hepatolipotoxicity. We found that palmitate upregulates hepatic CD36 expression. Despite being a well-established target gene of PPARγ transactivation, our data demonstrated that the palmitate-induced CD36 upregulation in hepatocytes is in fact PPARγ-independent. We previously reported that the activation of ATF4, one of three canonical pathways activated upon endoplasmic reticulum (ER) stress induction, contributes to palmitate-triggered lipotoxicity in hepatocytes. In this study, our data revealed for the first time that ATF4 plays a critical role in mediating hepatic CD36 expression. Genetic inhibition of ATF4 attenuated CD36 upregulation induced by either palmitate or ER stress inducer tunicamycin in hepatocytes. In mice, tunicamycin upregulates liver CD36 expression, whereas hepatocyte-specific ATF4 knockout mice manifest lower hepatic CD36 expression when compared with control animals. Furthermore, we demonstrated that CD36 upregulation upon palmitate exposure represents a feedforward mechanism in that siRNA knockdown of CD36 in hepatocytes blunted ATF4 activation induced by both palmitate and tunicamycin. Finally, we confirmed that the ATF4-CD36 pathway activation contributes to palmitate-induced hepatolipotoxicity as genetic inhibition of either ATF4 or CD36 alleviated cell death and intracellular triacylglycerol accumulation. Collectively, our data demonstrate that CD36 upregulation by ATF4 activation contributes to palmitate-induced hepatic lipotoxicity.
NEW & NOTEWORTHY We provided the initial evidence that ATF4 is a principal transcription factor mediating hepatic CD36 expression in that both palmitate- and ER stress-elicited CD36 upregulation was blunted by ATF4 gene knockdown in hepatocytes, and hepatocyte-specific ATF4 knockout mice manifested lower hepatic CD36 expression. We further confirmed that the ATF4-CD36 pathway activation contributes to palmitate-induced hepatolipotoxicity as genetic inhibition of either ATF4 or CD36 alleviated cell death and intracellular triacylglycerol accumulation in response to exogenous palmitate exposure.
INTRODUCTION
Lipotoxicity, a term developed to describe the detrimental effects caused by intracellular accumulation of active lipid species in nonadipose tissues/organs, plays a central role in the initiation and progression of nonalcoholic fatty liver disease (NAFLD) (1). Various active lipid mediators, including diacylglycerol (DAG), free fatty acids (FFAs), cholesterol, lysophosphatidylcholine (LPC), among others, have been documented to contribute to lipotoxicity (2). Of FFAs, the saturated fatty acids (SFAs) trigger cellular dysfunction and cell death in many cells, including hepatocytes (3–5). Palmitate is the most abundant SFA in human circulation. Notably, patients with metabolic disorders, including insulin resistance and obesity, show elevated circulatory palmitate levels (6). Although palmitate-induced lipotoxicity has been well documented in various cell types and considered to contribute to the pathogenesis of many obesity-related metabolic disorders, including NAFLD (7, 8), the exact mechanisms by which palmitate triggers cell death and cellular dysfunction in hepatocytes remain elusive.
Endoplasmic reticulum (ER) stress contributes to palmitate-induced lipotoxicity in many cell types (3, 9). ER stress occurs upon the accumulation of unfolded or misfolded proteins inside ER lumen, leading to the activation of a signaling network termed the unfolded protein response (UPR), which aims to restore ER homeostasis. Three major signaling pathways comprise the UPR, including RNA-dependent protein kinase-like ER kinase (PERK), inositol-requiring enzyme 1-α (IRE1α), and activating transcription 6 (ATF6) (10). Of the UPR pathways, PERK activation phosphorylates the eukaryotic translation initiation factor 2α (eIF2α), leading to the activation of ATF4, a transcription factor that regulates the expression of many downstream genes, primarily to help the cell cope with the ER stress (11). Moreover, ATF4 has been reported to mediate many physiological and pathological events, including both carbohydrate (12) and lipid metabolism (13). Importantly, ATF4-null mice are protected from the development of steatosis and hypertriglyceridemia when fed a high-fat diet (14). We previously reported that hepatocyte-specific ATF4 knockout mice are resistant to the development of alcoholic liver disease (15), and in hepatocytes, ATF4 upregulation contributes to palmitate-induced cell death (16). These previous studies altogether suggest that ATF4 plays a mechanistic role in the pathogenesis of NAFLD.
CD36 (fatty acid translocase) is a multifunctional transmembrane glycoprotein belonging to the scavenger family with a high affinity for long-chain FAs. Abundantly expressed in many cells and tissues, including the intestine, adipose tissue, heart, skeletal muscle, and macrophages, CD36 plays a vital role in regulating cellular FA uptake (17–19). Physiologically, CD36 expression in hepatocytes is relatively low; however, under certain pathophysiological conditions, hepatic CD36 is upregulated, contributing to fatty liver development (19). It has been reported that although increased CD36 expression in the liver by either pharmacological activation of liver X receptor (LXR) or adenovirus-mediated overexpression leads to hepatic steatosis (20, 21), hepatocyte-specific disruption of CD36 ameliorates fatty liver development and improves insulin sensitivity in mice fed with high-fat diet (22). Notably, liver tissues obtained from patients with NAFLD demonstrate significantly elevated CD36 expression (20, 21).
Palmitate exposure upregulates CD36 expression in several cell types, including macrophages (5) and cardiomyocytes (23), with distinct molecular mechanisms involved. However, the questions such as how palmitate affects CD36 expression in hepatocytes and if it contributes to palmitate-induced hepatolipotoxicity remain unknown.
In this study, both in vitro cell culture and in vivo animal studies were performed. We discovered that in hepatocytes, palmitate upregulates CD36 expression by activating ATF4, one of three canonical pathways activated upon UPR, and genetic knockdown of CD36 expression blunted palmitate-induced hepatocyte cell death and intracellular triacylglycerol (24) accumulation.
MATERIALS AND METHODS
Reagents
Cell culture and reagents were purchased from Gibco (Grand Island, NY). AML-12 cells were purchased from the American Type Culture Collection (Manassas, VA). Palmitate, GSK1940029, and tunicamycin were obtained from Sigma-Aldrich (St. Louis, MO). Torin-1, SBI0206965, and rapamycin were purchased from APExBIO Technology LLC (Houston, TX). For Western blot analysis, primary antibodies for ATF4 (Product No. 11815), CD36 (Product No. 14347), PPARγ (Product No. 2430), and β-actin (Product No. 4970) were purchased from Cell Signaling Technology (Beverly, CA). The secondary antibody, IRDye 800CW secondary antibody, was purchased from LI-COR (Lincoln, NE). Antibodies were validated by either siRNA knockdown (ATF4, CD36, and PPARγ) or pharmacological inhibition of target proteins (pS6).
Cell Culture
AML-12 cells were cultured as monolayers in Dulbecco’s modified Eagle’s medium (DMEM/F12) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, 0.1 mg/mL streptomycin, 40 ng/mL dexamethasone, and insulin transferrin selenium (ITS) (containing 5 mg/L insulin, 5 mg/L transferrin, and 5 μg/L selenium). Cells were grown in 75-cm2 flasks and maintained in an incubator at 37°C with a humidified atmosphere of 5% CO2. Unless otherwise stated, cells were grown to a confluence of 80% before any treatments. Primary mouse hepatocytes were isolated and cultured as previously described by Lee et al. (25).
Animal Studies
Animal studies were approved by the Institutional Animal Care and Use Committee at the University of Illinois at Chicago and consistent with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Ten-week-old C57BL/6N male mice (Charles River Lab, Chicago, IL) were used to model ER stress. Mice were injected intraperitoneally with either tunicamycin (2 mg/kg body wt), a well-established ER stress inducer, isovolumic vehicle control (150 mM dextrose), and livers were harvested 16 h later. Hepatocyte-specific ATF4 knockout mice (ALKO) were generated by crossing ATF4flox/flox(f/f) mice (donated by Dr. Christopher M. Adams at the University of Iowa) and C57Bl/6J mice and albumin-cre (Jackson Laboratory, Bar Harbor, ME). The liver samples from Ppargflox/flox (used as control) and liver-specific Pparg knockout (LKO-Pparg) mice were provided by Dr. Jose Cordoba-Chacon at the University of Illinois at Chicago.
siRNA Transfection
Cultured AML-12 cells were transfected with mouse siRNAs for ATF4, PPARγ, and CD36 (Santa Cruz, Dallas, TX, no. sc-41446, no. sc-29455; no. sc-37245). Cells were plated in six-well plates or 24-well plates, which were allowed to adhere for 24 h before being transfected with siRNA. Transfection was performed according to Invitrogen’s Lipofectamin 2000 Transfection Reagent (Carlsbad, CA) protocol. Briefly, AML-12 cells were cultured to 60%–70% confluence in a six-well plate and transfected with 8-μL siRNA (10 µM) using 8-μL Lipo 2000 or a 24-well plate transfected with 2-μL siRNA (20 μM) using 2-μL Lipo 2000. Cells were transfected with scramble RNA under the same conditions as a control. The transfection results were identified by real-time qPCR after a 24-h siRNA transfection.
LDH Release Assay
Lactate dehydrogenase (LDH) release assay was performed to determine cell death. Cells were seeded at 1 × 105 cells/mL in a 24-well plate, incubated, and allowed to grow overnight. Cells were treated with indicated treatments, cell supernatant was collected, and LDH release was detected using CyQUANT LDH Cytotoxicity Assay (Invitrogen, Eugene, OR) according to the manufacturer’s instructions. Absorption was measured at 490 nm and 680 nm using a microplate spectrophotometer, SPECTRAmax 340PC (Molecular Devices Corp., Sunnyvale, CA). The relative LDH release levels were scaled as folds of the control group.
Cellular TG Assay
Cells were seeded at 1 × 105 in 24-well plates overnight. Cells were treated with indicated treatments, then washed with PBS. NaOH (0.25 M) was added to cells and mixed for 10 min. Hexane/isopropanol (1 mL; 3:2) was added to cells for 1 h at room temperature with frequent vortex. Samples were centrifuged for 10 min, and TAG was extracted and dried thoroughly. The reagent for TAG assay (Pointe Scientific, Canton, MI) was added. After a brief heating and vortex, absorption was measured at OD500 nm using a microplate spectrophotometer, SPECTRAmax 340PC (Molecular Devices Corp., Sunnyvale, CA). The relative cellular TG levels were scaled as folds of protein level compared with control cells.
Total Protein Extraction and Western Blot Analysis
Total protein from cell culture and mice liver samples were obtained using RIPA lysis buffer system from Santa Cruz Biotechnology (Dallas, TX). Samples were incubated on ice with frequent vortex for 15 min, then centrifuged for 10 min at 12,000 r/min. Protein concentration of samples was determined using Enhanced BCA Protein Assay Kit (Pierce Biotechnology, Rockford, IL) and performed according to the manufacturer’s instructions. Equal amounts of protein (30 μg) were loaded onto 10% or 8% SDS-PAGE gel, depending on the molecular weight of the desired proteins. They were transferred to a LI-COR Odyssey nitrocellulose transfer membrane (Lincoln, NE). After the transfer, membranes were blocked in 5% (wt/vol) nonfat dry milk in PBS and probed with indicated primary antibodies. After incubation with primary antibodies, membranes were washed in PBST (PBS-0.1% Tween 20), then incubated with LI-COR IRDye 800CW secondary antibody (Lincoln, NE) at 1:10,000 dilution in blocking buffer at room temperature on a rocker for 1 h, followed by final washing in PBST. Immunoreactive bands of predicted molecular weights were visualized using the LI-COR Odyssey CLx system (Lincoln, NE) and quantified with Imagine Studio, version 4.
ELISA Assay for CD36 Protein Abundance
AML-12 cells were seeded and cultured in 12-well plates and total proteins were extracted with RIPA buffer containing proteinase inhibitors. Cellular CD36 protein abundance was measured using a commercially available mouse CD36 ELISA kit (Cat. No. EMCD36) from Invitrogen (Carlsbad, CA) according to the manufacturer’s instructions. All results were standardized by total protein content.
Total RNA Isolation and Quantitative Real-Time PCR
Total RNA from cultured and treated cells was isolated via the Invitrogen TRIzol reagent (Carlsbad, CA) extraction method. For each sample, 1 μg of total RNA was reverse transcribed using a high-capacity cDNA reverse transcription kit (Applied Biosystem, Foster City, CA). The cDNA was then amplified in a MicroAmp Optical 96-well reaction plate with an SYBR Green PCR Master Mix (Applied Biosystems, Warrington, UK) in an ABI7500 FAST sequence detection system (Applied Biosystem, Foster City, CA). After normalization, relative gene expression was calculated by a housekeeping gene (mouse GAPDH and 18S mRNA).
Primers
The following primers were followed. Gapdh: Forward: 5′- ATGCCAGTGAGCTTCCCGTTCAG-3′, Reverse: 5′- CATCACTGCCACCCAGAAGACTG-3′; 18sRNA: Forward: 5′- GTAACCCGTTGAACCCCATT-3′, Reverse: 5′- CCATCCAATCGGTAGTAGCG-3′; Atf4: Forward: 5′- AACCTCATGGGTTCTCCAGCGA-3′, Reverse: 5′- CTCCAACATCCAATCTGTCCCG-3′; Pparg: Forward: 5′- GTACTGTCGGTTTCAGAAGTGCC-3′, Reverse: 5′- ATCTCCGCCAACAGCTTCTCCT-3′; Cd36: Forward: 5′- GGACATTGAGATTCTTTTCCTCTG-3′, Reverse: 5′- GCAAAGGCATTGGCTGGAAGAAC-3′; Cidec: Forward: 5′- TCGGAAGGTTCGCAAAGGCATC-3′, Reverse: 5′- CTCCACGATTGTGCCATCTTCC-3′; Plin2: Forward: 5′- GAAGGATGGAGGAAAGACTGC-3′, Reverse: 5′- GGTAGTCGTCACCACATCCTTC-3′.
Statistical Analysis
All data are expressed as means ± SD. Statistical analysis was performed using a Student’s t test to compare two groups or one-way ANOVA for three or more groups. GraphPad Prism software (version 9) was used to calculate statistical significance.
RESULTS
Exogenous Palmitate Exposure Induces Cell Death and Upregulates CD36 Expression in Hepatocytes
The effects of exogenous palmitate exposure on hepatocyte cell death and hepatic CD36 expression were examined in both AML-12 and primary mouse hepatocytes. Palmitate-induced hepatotoxicity was evaluated by LDH release assay. In AML-12 cells, exogenous palmitate addition to the media increased LDH release in a dose-dependent manner (Fig. 1A). Although palmitate at 0.2 mM only subtly increased LDH release, more than four times that were observed when palmitate was increased to 0.6 mM. Similar results were observed in primary mouse hepatocytes in which palmitate exposure (0.6 mM) led to a significant elevation of LDH release (Fig. 1B), confirming that exogenous palmitate exposure causes hepatocyte cell death. CD36 gene expression was determined by real-time qPCR, and protein abundance was detected by ELISA. In AML-12 cells, palmitate increased CD36 expression at both mRNA and protein (Fig. 1, C and D) levels. In accordance, in primary mouse hepatocytes, palmitate exposure at 0.6 mM upregulates CD36 expression (Fig. 1, E and F).
Figure 1.

Exogenous palmitate exposure induces cell death and upregulates CD36 expression in hepatocytes. A: AML-12 cells were treated with the indicated concentrations of palmitate for 16 h. Cell death was determined via LDH release assay. Data are expressed as means ± SD, n = 4 separate experiments. Differences between groups were determined using one-way ANOVA analysis. Bars with different lowercase letters differ significantly (*P < 0.05). B: primary mouse hepatocytes were treated with 0.6 mM palmitate for 16 h. Cell death was determined via LDH release assay. Data are expressed as means ± SD, n = 4 separate experiments. Student’s t test was used for statistical evaluation (****P < 0.0001 vs. control). C: AML-12 cells were treated with the indicated concentrations of palmitate for 16 h. Total RNA was extracted and CD36 mRNA expression was determined by real-time-qPCR. Data are expressed as means ± SD, n = 3 separate experiments. Differences between groups were determined using one-way ANOVA analysis. Bars with different lowercase letters differ significantly (*P < 0.05). D: AML-12 cells were treated with 0.4 mM palmitate for 16 h. Total protein was extracted and CD36 protein abundance was measured by ELISA kit. Data are expressed as means ± SD, n = 3 separate experiments. Student’s t test was used for statistical evaluation (**P < 0.01 vs. control). E and F: primary mouse hepatocytes were treated with 0.6 mM palmitate for 16 h. Total RNA and protein were extracted and CD36 expression was determined by real-time qPCR and Western blotting, respectively. Data are expressed as means ± SD, n = 4 separate experiments. Student’s t test was used for statistical evaluation (*P < 0.05 vs. control). CD36, cluster of differentiation 36.
Palmitate-Induced CD36 Upregulation Is PPARγ-Independent in Hepatocytes
The CD36 gene contains a peroxisome-proliferator response element, and PPARγ is the best-characterized transcription factor mediating CD36 expression (26). To determine whether PPARγ activation is mechanistically involved in palmitate-mediated CD36 upregulation in hepatocytes, we first examined the effects of palmitate exposure on PPARγ transactivation in both AML-12 cells and primary mouse hepatocytes via detecting the expression of signature target genes of PPARγ, including Cidec, Plin2, and Fabp4. As shown in Fig. 2, A and B, in both AML-12 cells and primary mouse hepatocytes, all these genes were upregulated by palmitate exposure, indicative of PPARγ activation in response to palmitate exposure in hepatocytes. We then transfected AML-12 cells with either control (scrambled) siRNA or siRNA for PPARγ overnight, followed by palmitate (0.4 mM) treatment for 16 h. As shown in Fig. 2, C–E, PPARγ siRNA transfection dramatically reduced PPARγ expression and activity in both control and palmitate-treated cells, confirming the transfection efficiency. Unexpectedly, the genetic PPARγ knockdown failed to affect palmitate-induced CD36 upregulation (Fig. 2, F and G), suggesting that palmitate upregulates CD36 expression in a PPARγ-independent mechanism in hepatocytes. This notion was further validated by the observations that in AML-12 hepatocytes, GW1929, a potent PPARγ agonist, did not affect Cd36 gene expression (Fig. 2H), and in mice, hepatocyte-specific PPARγ ablation was not associated with altered hepatic Cd36 expression (Fig. 2, I and J).
Figure 2.

Palmitate-induced CD36 upregulation is PPARγ-independent in hepatocytes. Primary mouse hepatocytes (A) and AML-12 cells (B) were treated with palmitate at 0.6 mM and 0.4 mM, respectively, for 16 h. PPARγ transactivation was determined by detecting its downstream target genes using real-time-qPCR. Data are expressed as means ± SD, n = 3 or 4 separate experiments. Student’s t test was used for statistical evaluation (*P < 0.05; ***P < 0.001). AML-12 cells were transfected with either scramble siRNA (si-Ctrl) or Pparg siRNA (si-Pparg) for 24 h before palmitate exposure (0.4 mM) for 16 h. Pparg expression (C and D), transactivation (E), and CD36 (F and G) expression were determined by real-time qPCR, Western blotting, and ELISA, and data were expressed as means ± SD, n = 3 separate experiments. Student's t test was used for statistical evaluation (*P < 0.05; **P < 0.01; ***P < 0.001). H: AML-12 cells were treated with PPARγ agonist, GW1929 (10 µM), for 16 h. PPARγ transactivation was determined by detecting its downstream target genes using real-time qPCR. Data are expressed as means ± SD, n = 3 separate experiments. Student’s t test was used for statistical evaluation (*P < 0.05). I and J: total RNA was extracted from the liver tissues of control and hepatocyte-specific Pparg knockout (LKO-Pparg) mice and subjected to real-time qPCR for CD36 gene expression. Data are expressed as means ± SD (n = 5 or 6 mice/group). CD36, cluster of differentiation 36; PPARγ, peroxisome proliferator-activated receptor γ.
ATF4 Activation Contributes to Palmitate-Induced CD36 Upregulation in Hepatocytes
We previously reported that palmitate exposure activated ATF4 in AML-12 cells (16). Here, using primary mouse hepatocytes, we further confirmed the notion that palmitate upregulates/activates ATF4 in hepatocytes, evidenced by increased expression of ATF4 (Fig. 3, A and B) and VLDLr (Fig. 3A), a well-established target gene of ATF4 activation (27). To determine whether ATF4 activation plays a regulatory role in palmitate-induced CD36 upregulation, we genetically knocked down ATF4 in AML-12 cells by transfecting cells with ATF4 siRNA overnight before palmitate (0.4 mM) exposure for 16 h. ATF4 siRNA transfection significantly reduced ATF4 expression (Fig. 3, C and D) and activity (Fig. 3E) in both untreated and palmitate-treated AML-12 cells. Importantly, our data unraveled that ATF4 knockdown reduced not only basal CD36 expression but also prevented palmitate-induced CD36 upregulation (Fig. 3, F and G), indicating that in hepatocytes, ATF4 activation plays a mechanistic role in palmitate-induced CD36 upregulation.
Figure 3.

ATF4 activation contributes to palmitate-induced CD36 upregulation in hepatocytes. A: primary mouse hepatocytes were treated with palmitate at 0.6 mM for 16 h. mRNA levels of Atf4 and Vldlr, an ATF4 target gene, were measured using real-time qPCR. Data are expressed as means ± SD, n = 4 separate experiments. Student’s t test was used for statistical evaluation (**P < 0.01; ***P < 0.001). B: primary mouse hepatocytes were treated with palmitate at 0.6 mM for 16 h. ATF4 protein was detected by Western blotting. The same loading control (actin) was used as presented in Fig. 1F as they were from the same Western blot membrane. AML-12 cells were transfected with either scramble siRNA (si-Ctrl) or Atf4 siRNA (si-Atf4) for 24 h before a 16-h palmitate exposure (0.4 mM). ATF4 expression (C and D), activity (E), and CD36 expression (F and G) were determined by real-time qPCR, Western blotting, and ELISA, respectively, and data were expressed as means ± SD, n = 4 separate experiments. Student’s t test was used for statistical evaluation (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001). ATF4, activating transcription factor 4; CD36, cluster of differentiation 36.
ATF4 Activation Mediates ER Stress-Triggered CD36 Upregulation in Hepatocytes
The PERK/ATF4 pathway is one of three canonical pathways activated upon ER stress induction (11). To assess the generality of the notion that ATF4 plays a vital role in mediating CD36 expression in hepatocytes, we first treated AML-12 hepatocytes with a well-established ER stress inducer, tunicamycin (5 μM), for 16 h and examined the effects of tunicamycin on PPARγ activation by measuring the expression of several signature PPARγ target genes including Pparg, CD36, Cidec, and plin2. As shown in Fig. 4A, tunicamycin activated PPARγ in AML-12 hepatocytes in that all four PPARγ target genes were significantly upregulated. To determine whether PPARγ activation contributes to ER stress-induced CD36 upregulation in hepatocytes, we genetically knocked down PPARγ expression in AML-12 cells via PPARγ siRNA transfection before tunicamycin treatment. Genetic knockdown of PPARγ (Fig. 4, B and C) did not affect tunicamycin-induced CD36 upregulation (Fig. 4D). Moreover, PPARγ siRNA knockdown did not affect ATF4 upregulation in response to ER stress induction (Fig. 4, C and E). However, when ATF4 siRNA was exploited to knock down cellular ATF4 expression (Fig. 4F), tunicamycin-induced CD36 upregulation was prevented entirely (Fig. 4G), confirming our notion that the activation of ATF4, not PPARγ, contributes to ER stress-induced CD36 overexpression. To validate the in vivo relevance of our observations from in vitro cell culture, male C57 BL/6 mice were intraperitoneally injected with either vehicle control or tunicamycin (2 mg/kg body wt), and liver samples were harvested 16 h later. As shown in Fig. 4, H and I, ER stress induction by tunicamycin resulted in upregulated hepatic CD36 expression. In accordance, when compared with age- and sex-matched control mice, liver-specific ATF4 knockout mice manifested reduced hepatic CD36 expression (Fig. 4J), indicative of the critical involvement of ATF4 in regulating hepatic CD36 expression.
Figure 4.

ATF4 activation mediates ER stress-triggered CD36 upregulation in hepatocytes. A: AML-12 cells were treated with tunicamycin (5 µM), a chemical ER stress inducer, for 16 h. PPARγ transactivation was determined by detecting its downstream target genes using real-time qPCR. Data are expressed as means ± SD, n = 4 separate experiments. Student’s t test was used for statistical evaluation (*P < 0.05; **P < 0.01 vs. corresponding control). B–E: AML-12 cells were transfected with either scramble siRNA (si-Ctrl) or Pparg siRNA (si-Pparg) for 24 h before tunicamycin treatment (5 µM) for 16 h. PPARγ expression (B and C), CD36 gene expression (D), and ATF4 expression (C and E) were determined by real-time qPCR and Western blotting, respectively. Data were expressed as means ± SD, n = 4 separate experiments. Student’s t test was used for statistical evaluation (**P < 0.01; ***P < 0.001). F and G: AML-12 cells were transfected with either scramble siRNA (si-Ctrl) or Atf4 siRNA (si-Atf4) for 24 h before tunicamycin treatment (5 µM) for 16 h. Gene expression of Atf4 (F) and CD36 (G) was determined by real-time qPCR and data were expressed as means ± SD, n = 3 separate experiments. Student’s t test was used for statistical evaluation (**P < 0.01; ***P < 0.001). H and I: male C57 BL/6 mice were intraperitoneally injected with either vehicle or tunicamycin (2 mg/kg body wt). The liver samples were harvested 16 h later. Hepatic CD36 expression was determined by real-time qPCR and Western blotting, respectively. Data were expressed as means ± SD, n = 3 mice. Differences between the two groups were determined using Student’s t test (**P < 0.01 vs. control group). J: total RNA was extracted from the liver tissues of control and hepatocyte-specific Atf4 knockout (LsATF4-KO) mice and subjected to real-time qPCR for CD36 gene expression. Data were expressed as means ± SD, n = 4 mice/group. Differences between the two groups were determined using Student’s t test (*P < 0.05 vs. control group). ATF4, activating transcription factor 4; CD36, cluster of differentiation 36; ER, endoplasmic reticulum; PPARγ, peroxisome proliferator-activated receptor γ.
Intracellular Metabolism Is Required for Palmitate-Elicited ATF4 Activation
The role of palmitate intracellular metabolism in activating ATF4 was subsequently determined. Acyl-CoA synthetase catalyzes the conversion of palmitate to palmitoyl-CoA, the first step of intracellular palmitate metabolism. To examine whether the acyl-CoA synthetase activity is required for palmitate-triggered ATF4 activation as well as CD36 upregulation, AML-12 cells were pretreated with Triacsin C (10 µM), an inhibitor of long fatty acid acyl-CoA synthetase, for 2 h before a 16-h palmitate exposure. ATF4 expression was determined. As shown in Fig. 5, A and B, Triacsin C impeded palmitate-induced ATF4 upregulation, which was concomitant with alleviated palmitate-induced CD36 upregulation (Fig. 5, C and D). Stearoyl-CoA desaturase (SCD)-1 catalyzes saturated fatty acid desaturation. To assess the role of SCD-1-catalyzed cellular desaturation reaction in palmitate-induced ATF4 activation, we pretreated AML-12 cells with a specific SCD-1 inhibitor, GSK-1940029 (20 µM), for 2 h before palmitate exposure for 16 more hours. As shown in Fig. 5, E and F, SCD-1 inhibition increased basal ATF4 expression and exacerbated palmitate-induced ATF4 upregulation. In accordance, a significant elevation of basal CD36 expression, at both mRNA and protein levels, was observed upon SCD-1 inhibitor treatment (Fig. 5, G and H). Although SCD-1 inhibition aggravated palmitate-induced CD36 mRNA increase, it did not further enhance CD36 protein upregulation in response to palmitate exposure (Fig. 5H).
Figure 5.

Intracellular metabolism is required for palmitate-elicited ATF4 activation. A–D: AML-12 cells were pretreated with Triacsin C, an inhibitor of long fatty acid acyl-CoA synthetase, for 2 h before a 16-h palmitate exposure. ATF4 and CD36 expression was determined by real-time qPCR, Western blotting, and ELISA, respectively. Data were expressed as means ± SD, n = 4 separate experiments. Student’s t test was used for statistical evaluation (**P < 0.01). E–H: AML-12 cells were pretreated with GSK-1940029 (20 µM), a specific SCD-1 inhibitor, for 2 h before palmitate exposure for 16 more hours. ATF4 and CD36 expression was determined by real-time qPCR, Western blotting, and ELISA, respectively. Data were expressed as means ± SD, n = 4 separate experiments. Student’s t test was used for statistical evaluation (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001). ATF4, activating transcription factor 4; CD36, cluster of differentiation 36.
CD36 Expression Is Required for ATF4 Activation upon Palmitate Exposure and ER Stress Induction in Hepatocytes
CD36 facilitates cellular fatty acid (FA) uptake (26). Our observations that exogenous palmitate exposure upregulates CD36 expression via activating ATF4 suggest that the ATF4-CD36 pathway represents a feedforward mechanism to promote palmitate uptake and lipotoxicity induction. To further understand the role of CD36 upregulation in palmitate-induced ATF4 activation, the genetic knockdown of CD36 was achieved via siRNA transfection (Fig. 6A) before either palmitate (0.4 mM) or tunicamycin (5 µm) exposure. As expected, CD36 gene knockdown blunted palmitate-triggered ATF4 upregulation (Fig. 6, B and D). Unexpectedly, tunicamycin-induced ATF4 upregulation was also attenuated by CD36 gene knockdown (Fig. 6, C and D), suggesting that CD36 is required for ER stress-induced ATF4 activation.
Figure 6.

CD36 expression is required for ATF4 activation upon palmitate exposure and ER stress induction in hepatocytes. A and B: AML-12 cells were transfected with either scramble siRNA (si-Ctrl) or Cd36 siRNA (si-Cd36) for 24 h before palmitate exposure (0.4 mM) for 16 h. Gene expression of Cd36 (A) and Atf4 (B) was determined by real-time qPCR and data were expressed as means ± SD, n = 4 separate experiments. Student’s t test was used for statistical evaluation (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001). C: AML-12 cells were transfected with either scramble siRNA (si-Ctrl) or Cd36 siRNA (si-Cd36) for 24 h before tunicamycin treatment (5 µM) for 16 h. Gene expression of Atf4 was determined by real-time qPCR and data were expressed as means ± SD, n = 4 separate experiments. Student’s t test was used for statistical evaluation (***P < 0.001; ****P < 0.0001). D: AML-12 cells were transfected with either scramble siRNA (si-Ctrl) or Cd36 siRNA (si-Cd36) for 24 h before palmitate exposure (0.4 mM) or tunicamycin treatment (5 µM) for 16 h. Protein abundance of ATF6 was detected by Western blotting. ATF4, activating transcription factor 4; CD36, cluster of differentiation 36; ER, endoplasmic reticulum.
The ATF4-CD36 Pathway Activation Contributes to Palmitate-Induced Lipotoxicity in Hepatocytes
We established that palmitate exposure activates the ATF4-CD36 pathway. To determine the role of this pathway in palmitate-induced lipotoxicity in hepatocytes, AML-12 cells were first transfected with ATF4 siRNA for overnight to knock down ATF4 gene expression, followed by 16-h palmitate exposure. As shown in Fig. 7, A and B, ATF4 knockdown attenuated cell death and intracellular TAG accumulation induced by palmitate exposure. To determine whether CD36 upregulation also contributes to palmitate-induced hepatotoxicity, we then transfected AML-12 cells with CD36 siRNA overnight before a 16-h palmitate exposure. CD36 siRNA transfection efficiently downregulated CD36 expression (Fig. 7C). As expected, CD36 knockdown dramatically ameliorated palmitate-induced incremental LDH release (Fig. 7D). Unexpectedly, CD36 knockdown only slightly reduced palmitate-induced intracellular TAG elevation (Fig. 7E). Moreover, CD36 knockdown did not affect palmitate-elicited de novo lipogenesis pathway activation (Fig. 7E).
Figure 7.

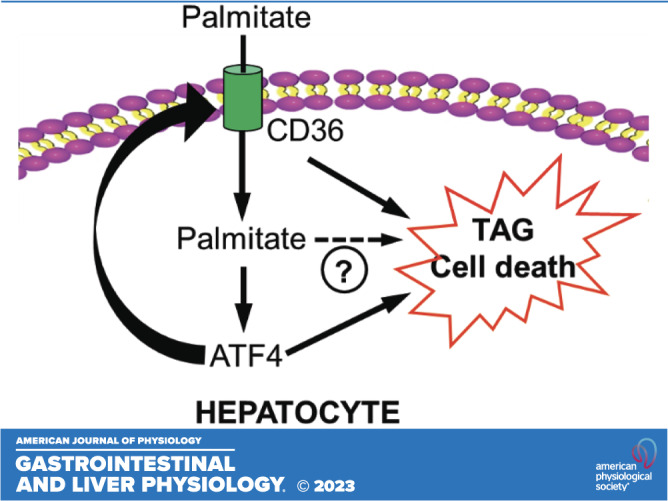
The ATF4-CD36 pathway activation contributes to palmitate-induced lipotoxicity in hepatocytes. A and B: AML-12 cells were transfected with either scramble siRNA (si-Ctrl) or Atf4 siRNA (si-Atf4) for 24 h before palmitate exposure (0.4 mM) for 16 h. LDH release (A) and intracellular TAG contents (B) were measured. Student’s t test was used for statistical evaluation (**P < 0.01; ****P < 0.0001). C–F: AML-12 cells were transfected with either scramble siRNA (si-Ctrl) or Cd36 siRNA (si-Cd36) for 24 h before palmitate exposure (0.4 mM) for 16 h. Cd36 gene expression (C), LDH release (D), intracellular TAG contents (E), and de novo lipogenic genes (F) were measured. Student’s t test was used for statistical evaluation (**P < 0.01; ***P < 0.001; ****P < 0.0001). G: schematic illustration of the role of the ATF4-CD36 pathway activation in palmitate-induced hepatocyte lipotoxicity. ATF4, activating transcription factor 4; CD36, cluster of differentiation 36; TAG, triacylglycerol.
DISCUSSION
Exploiting palmitate exposure of cultured AML-12 hepatocytes and primary mouse hepatocytes as our in vitro hepatolipotoxicity model, we demonstrated that CD36 upregulation contributes to palmitate-induced hepatocyte cell death and cellular TAG increment. Importantly, we identified ATF4, one of three canonic pathways activated during ER stress, as a critical transcription factor mediating CD36 expression in hepatocytes. In mice, administration of tunicamycin, a chemical ER stress inducer, increased hepatic CD36 expression. In contrast, hepatocyte-specific ATF4 knockout mice manifested lowered hepatic CD36 expression when compared with both age- and sex-matched control mice. In AML-12 cells, both palmitate and tunicamycin activated ATF4 and upregulated CD36 expression. Remarkably, genetic knockdown of ATF4 impeded CD36 increase by both palmitate and tunicamycin. Although both palmitate and tunicamycin triggered PPARγ transactivation in hepatocytes, unlike adipocytes and macrophages in which PPARγ plays a critical role in regulating CD36 expression, CD36 expression in hepatocytes is PPARγ-independent as genetic inhibition of PPARγ activation failed to alter CD36 overexpression in response to palmitate or tunicamycin treatment. Finally, we demonstrated that the ATF4-CD36 pathway activation contributes to palmitate-induced lipotoxicity in hepatocytes as siRNA for either ATF4 or CD36 confers protection against palmitate-induced cell death and intracellular TAG accumulation. Together, our results indicate that ATF4 activation-induced CD36 upregulation contributes to lipotoxicity in hepatocytes (Fig. 7G).
CD36 is a well-documented long-chain FA transporter and is abundantly expressed in adipocytes, myocytes, and macrophages, among others (26). CD36-null mice demonstrate impaired FA uptake into adipose tissues, muscle, and heart, leading to a significant increase in plasma FFAs and TG (28). Although CD36 expression in hepatocytes is weak under physiological conditions, fatty liver development is associated with increased hepatic CD36 expression (29). Importantly, hepatocyte-specific CD36 ablation protected mice from fatty liver development in the setting of HFD feeding (22), suggesting CD36 plays a mechanistic role in NAFLD pathogenesis. Obesity plays a central role in NAFLD development. Adipose tissue insulin resistance is intimately related to obesity, where one of the principal hallmarks is incremental circulatory FFA levels due to unrestrained adipose tissue lipolysis (6). Palmitate is the most abundant SFA found in human circulation (1). Elevated plasma concentrations of palmitate have been implicated in NAFLD initiation and development (6). Palmitate exposure has been reported to increase CD36 expression in several cell types with distinct functions proposed (26, 30). Interestingly, few studies examined whether and how palmitate exposure affects hepatocyte CD36 expression (31). In this study, using both AML-12 cells and primary mouse hepatocytes, our data confirmed that exogenous palmitate exposure upregulates hepatic CD36 expression at both mRNA and protein levels.
PPARγ, which plays a critical role in regulating lipid and glucose metabolism (26), is a well-established transcriptional regulator of CD36 expression in various cells and tissues, including macrophages, adipose tissue as well as heart, and has been shown to promote FA storage, TAG synthesis, and glucose uptake (32). Intriguingly, it remains ambiguous whether PPARγ is similarly an essential regulator of hepatic CD36 expression. In the present study, we initially posited that PPARγ transactivation contributed to palmitate-induced CD36 upregulation in AML-12 hepatocytes. In line with previous reports in various cell types, including hepatocytes (33), palmitate exposure of hepatocytes leads to PPARγ transactivation. Unexpectedly, we showed that palmitate-induced CD36 upregulation was independent of PPARγ activation in hepatocytes in that genetic knockdown of PPARγ by siRNA transfection failed to attenuate CD36 increase upon palmitate exposure. These observations altogether suggest that the cell type-specific regulatory mechanisms for CD36 expression exist, and in hepatocytes, PPARγ is not a significant regulator of CD36 expression. Indeed, a similar scenario has been reported in monocytes in which retinoic acid-induced CD36 expression was shown to be independent of the PPARγ transactivation (34).
SFAs, such as palmitate, induce cellular dysfunction/cell death in various cell types, including hepatocytes (3, 5), and accumulated evidence, including our studies, supports that ER stress induction is a central mechanism underlying SFA-induced lipotoxicity. The PERK-ATF4 pathway is one of the three major canonical UPR pathways activated during ER stress. Under conditions of ER stress, the PERK pathway is activated and phosphorylates eIF2α, which selectively induces ATF4 transactivation (11). We have reported that ATF4 activation contributed to palmitate-induced lipotoxicity in AML-12 cells through upregulating expression of nicotinamide N-methyltransferase (NNMT), an enzyme catabolizing nicotinamide methylation/degradation (16). In line with our previous report, in this study, our result demonstrates that palmitate exposure resulted in ATF4 activation. Although the regulatory role of ATF4 activation on hepatic CD36 expression has been documented, the results remain controversial. For instance, a study conducted by Yeh et al. (35) reported that overexpression of ATF4 increased CD36 in the livers of zebrafish; however, in another study, Li et al. (36) documented that, compared with control animals, the liver-specific ATF4 knockout mice did not show altered hepatic CD36 expression. Here, we provided new evidence confirming that ATF4 is an important transcription factor mediating CD36 expression in hepatocytes. In AML-12 hepatocytes, both palmitate-induced and ER stress inducer tunicamycin-instigated CD36 upregulation was blunted when ATF4 gene was genetically knocked down by siRNA transfection. Furthermore, in mice, tunicamycin administration increased hepatic CD36 expression, whereas hepatocyte-specific ATF4 knockout mice manifested lower basal CD36 expression in the liver. These data altogether indicate that the distinct regulatory systems for CD36 exist in hepatocytes, and ATF4 plays a vital role in mediating hepatic CD36 expression.
Given the critical role of CD36 in cellular FA uptake, it seems that palmitate-induced CD36 upregulation represents a feedforward mechanism facilitating palmitate uptake by the cell, which can further activate ATF4. Our observation confirmed this notion that genetic CD36 inhibition blunted ATF4 activation induced by palmitate exposure. Intriguingly, when CD36 was knocked down by siRNA transfection, tunicamycin-induced ATF4 activation was also compromised. Although the observation was very unexpected, it implies that other than serving as a long-chain FA transporter, certain unknown functions of CD36 in the cellular response to ER stress exist, which warrant further investigations.
In this current study, we demonstrated that the ATF4-CD36 pathway contributes to palmitate-induced lipotoxicity in AML-12 cells as genetic inhibition of either ATF4 or CD36 confers protection against hepatocyte cell death and intracellular TAG accumulation in response to exogenous palmitate exposure. However, it is noteworthy that although the protective effects on cell death were comparable between ATF4 and CD36 inhibition, unlike this with ATF4 inhibition, CD36 siRNA knockdown only slightly, although significantly, reduced palmitate-induced intracellular TAG accumulation. We reasoned that this observation might result from activated de novo lipogenesis and/or suppressed fatty acid β-oxidation upon genetic CD36 knockdown which compensated for decreased fatty acid uptake due to CD36 downregulation. Our data did not support the notion that CD36 knockdown was associated with enhanced de novo lipogenesis (Fig. 7F); however, we cannot exclude the potential effect of CD36 knockdown on fatty acid β-oxidation.
In conclusion, this study provides evidence that the ATF4-CD36 pathway plays a mechanistic role in palmitate-induced hepatolipotoxicity. We demonstrate that palmitate exposure activates both ATF4 and PPARγ and upregulates CD36 expression in hepatocytes. Importantly, we identify the transcription factor ATF4 as a major regulator mediating CD36 expression in hepatocytes and mouse liver, and inhibition of either ATF4 or CD36 provides protection against palmitate-induced cell death and intracellular TAG accumulation. These results altogether suggest the clinically beneficial potential of CD36 inhibitors in the treatment of a variety of metabolic disorders including NAFLD.
DATA AVAILABILITY
Data will be made available upon reasonable request.
GRANTS
This work was in part funded by National Institute on Alcohol Abuse and Alcoholism Grant R01AA026603 (to Z.S.) and National Institute of Diabetes and Digestive and Kidney Disease Grant R01DK131038 (to J.C-C.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Z.S. conceived and designed research; A.G., J.W., Q.S., S.M.L., and J.C-C. performed experiments; A.G., J.W., and Q.S. analyzed data; A.G., J.W., S.M.L., J.C-C., and Z.S. interpreted results of experiments; A.G., J.W., and Q.S. prepared figures; A.G. and J.W. drafted manuscript; Z.S. edited and revised manuscript; Z.S. approved final version of manuscript.
REFERENCES
- 1. Ogawa Y, Imajo K, Honda Y, Kessoku T, Tomeno W, Kato S, Fujita K, Yoneda M, Saito S, Saigusa Y, Hyogo H, Sumida Y, Itoh Y, Eguchi K, Yamanaka T, Wada K, Nakajima A. Palmitate-induced lipotoxicity is crucial for the pathogenesis of nonalcoholic fatty liver disease in cooperation with gut-derived endotoxin. Sci Rep 8: 11365, 2018. doi: 10.1038/s41598-018-29735-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Liangpunsakul S, Chalasani N. Lipid mediators of liver injury in nonalcoholic fatty liver disease. Am J Physiol Gastrointest Liver Physiol 316: G75–G81, 2019. doi: 10.1152/ajpgi.00170.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen Y-Y, Sun L-Q, Wang B-A, Zou X-M, Mu Y-M, Lu J-M. Palmitate induces autophagy in pancreatic β-cells via endoplasmic reticulum stress and its downstream JNK pathway. Int J Mol Med 32: 1401–1406, 2013. doi: 10.3892/ijmm.2013.1530. [DOI] [PubMed] [Google Scholar]
- 4. Guo W, Wong S, Xie W, Lei T, Luo Z. Palmitate modulates intracellular signaling, induces endoplasmic reticulum stress, and causes apoptosis in mouse 3T3-L1 and rat primary preadipocytes. Am J Physiol Endocrin Metab 293: E576–E586, 2007. doi: 10.1152/ajpendo.00523.2006. [DOI] [PubMed] [Google Scholar]
- 5. Kim DH, Cho YM, Lee KH, Jeong S-W, Kwon O-J. Oleate protects macrophages from palmitate-induced apoptosis through the downregulation of CD36 expression. Biochem Biophys Res Commun 488: 477–482, 2017. doi: 10.1016/j.bbrc.2017.05.066. [DOI] [PubMed] [Google Scholar]
- 6. Wrzosek M, Zawadzka Z, Sawicka A, Bobrowska-Korczak B, Białek A. Impact of fatty acids on obesity-associated diseases and radical weight reduction. Obes Surg 32: 428–440, 2022. doi: 10.1007/s11695-021-05789-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Karmi A, Iozzo P, Viljanen A, Hirvonen J, Fielding BA, Virtanen K, Oikonen V, Kemppainen J, Viljanen T, Guiducci L, Haaparanta-Solin M, Någren K, Solin O, Nuutila P. Increased brain fatty acid uptake in metabolic syndrome. Diabetes 59: 2171–2177, 2010. doi: 10.2337/db09-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Malhi H, Gores GJ. Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin Liver Dis 28: 360–369, 2008. doi: 10.1055/s-0028-1091980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Karaskov E, Scott C, Zhang L, Teodoro T, Ravazzola M, Volchuk A. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic beta-cell apoptosis. Endocrinology 147: 3398–3407, 2006. doi: 10.1210/en.2005-1494. [DOI] [PubMed] [Google Scholar]
- 10. Liu X, Green RM. Endoplasmic reticulum stress and liver diseases. Liver Res 3: 55–64, 2019. doi: 10.1016/j.livres.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rozpedek W, Pytel D, Mucha B, Leszczynska H, Diehl JA, Majsterek I. The role of the PERK/eIF2α/ATF4/CHOP signaling pathway in tumor progression during endoplasmic reticulum stress. Curr Mol Med 16: 533–544, 2016. doi: 10.2174/1566524016666160523143937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ren L-P, Yu X, Song G-Y, Zhang P, Sun L-N, Chen S-C, Hu Z-J, Zhang X-M. Impact of activating transcription factor 4 signaling on lipogenesis in HepG2 cells. Mol Med Rep 14: 1649–1658, 2016. doi: 10.3892/mmr.2016.5453. [DOI] [PubMed] [Google Scholar]
- 13. Wang C, Huang Z, Du Y, Cheng Y, Chen S, Guo F. ATF4 regulates lipid metabolism and thermogenesis. Cell Res 20: 174–184, 2010. doi: 10.1038/cr.2010.4. [DOI] [PubMed] [Google Scholar]
- 14. Seo J, Fortuno ES III, Suh JM, Stenesen D, Tang W, Parks EJ, Adams CM, Townes T, Graff JM. Atf4 regulates obesity, glucose homeostasis, and energy expenditure. Diabetes 58: 2565–2573, 2009. doi: 10.2337/db09-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Song Q, Chen Y, Wang J, Hao L, Huang C, Griffiths A, Sun Z, Zhou Z, Song Z. ER stress-induced upregulation of NNMT contributes to alcohol-related fatty liver development. J Hepatol 73: 783–793, 2020. doi: 10.1016/j.jhep.2020.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Griffiths A, Wang J, Song Q, Iyamu ID, Liu L, Park J, Jiang Y, Huang R, Song Z. Nicotinamide N-methyltransferase upregulation via the mTORC1-ATF4 pathway activation contributes to palmitate-induced lipotoxicity in hepatocytes. Am J Physiol Cell Physiol 321: C585–C595, 2021. doi: 10.1152/ajpcell.00195.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Coburn CT, Knapp FF Jr, Febbraio M, Beets AL, Silverstein RL, Abumrad NA. Defective uptake and utilization of long chain fatty acids in muscle and adipose tissues of CD36 knockout mice. J Biol Chem 275: 32523–32529, 2000. doi: 10.1074/jbc.M003826200. [DOI] [PubMed] [Google Scholar]
- 18. Hajri T, Abumrad NA. Fatty acid transport across membranes: relevance to nutrition and metabolic pathology. Annu Rev Nutr 22: 383–415, 2002. doi: 10.1146/annurev.nutr.22.020402.130846. [DOI] [PubMed] [Google Scholar]
- 19. Su X, Abumrad NA. Cellular fatty acid uptake: a pathway under construction. Trends Endocrinol Metab 20: 72–77, 2009. doi: 10.1016/j.tem.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Koonen DPY, Jacobs RL, Febbraio M, Young ME, Soltys C-LM, Ong H, Vance DE, Dyck JRB. Increased hepatic CD36 expression contributes to dyslipidemia associated with diet-induced obesity. Diabetes 56: 2863–2871, 2007. doi: 10.2337/db07-0907. [DOI] [PubMed] [Google Scholar]
- 21. Zhou J, Febbraio M, Wada T, Zhai Y, Kuruba R, He J, Lee JH, Khadem S, Ren S, Li S, Silverstein RL, Xie W. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosis. Gastroenterology 134: 556–567, 2008. doi: 10.1053/j.gastro.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 22. Wilson CG, Tran JL, Erion DM, Vera NB, Febbraio M, Weiss EJ. Hepatocyte-specific disruption of CD36 attenuates fatty liver and improves insulin sensitivity in HFD-fed mice. Endocrinology 157: 570–585, 2016. doi: 10.1210/en.2015-1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Adrian L, Lenski M, Tödter K, Heeren J, Böhm M, Laufs U. AMPK prevents palmitic acid-induced apoptosis and lipid accumulation in cardiomyocytes. Lipids 52: 737–750, 2017. doi: 10.1007/s11745-017-4285-7. [DOI] [PubMed] [Google Scholar]
- 24. Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol 47: 571–579, 2007. doi: 10.1016/j.jhep.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee SM, Pusec CM, Norris GH, De Jesus A, Diaz-Ruiz A, Muratalla J, Sarmento-Cabral A, Guzman G, Layden BT, Cordoba-Chacon J. Hepatocyte-specific loss of PPARγ protects mice from NASH and increases the therapeutic effects of rosiglitazone in the liver. Cell Mol Gastroenterol Hepatol 11: 1291–1311, 2021. doi: 10.1016/j.jcmgh.2021.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maréchal L, Laviolette M, Rodrigue-Way A, Sow B, Brochu M, Caron V, Tremblay A. The CD36-PPARγ pathway in metabolic disorders. Int J Mol Sci 19: 1529, 2018. doi: 10.3390/ijms19051529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jo H, Choe SS, Shin KC, Jang H, Lee JH, Seong JK, Back SH, Kim JB. Endoplasmic reticulum stress induces hepatic steatosis via increased expression of the hepatic very low-density lipoprotein receptor. Hepatology 57: 1366–1377, 2013. doi: 10.1002/hep.26126. [DOI] [PubMed] [Google Scholar]
- 28. Goudriaan JR, Dahlmans VE, Teusink B, Ouwens DM, Febbraio M, Maassen JA, Romijn JA, Havekes LM, Voshol PJ. CD36 deficiency increases insulin sensitivity in muscle, but induces insulin resistance in the liver in mice. J Lipid Res 44: 2270–2277, 2003. doi: 10.1194/jlr.M300143-JLR200. [DOI] [PubMed] [Google Scholar]
- 29. Rada P, González-Rodríguez Á, García-Monzón C, Valverde ÁM. Understanding lipotoxicity in NAFLD pathogenesis: is CD36 a key driver? Cell Death Dis 11: 802, 2020. doi: 10.1038/s41419-020-03003-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pan J, Fan Z, Wang Z, Dai Q, Xiang Z, Yuan F, Yan M, Zhu Z, Liu B, Li C. CD36 mediates palmitate acid-induced metastasis of gastric cancer via AKT/GSK-3β/β-catenin pathway. J Exp Clin Cancer Res 38: 52, 2019. doi: 10.1186/s13046-019-1049-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu J, Yang P, Zuo G, He S, Tan W, Zhang X, Su C, Zhao L, Wei L, Chen Y, Ruan X, Chen Y. Long-chain fatty acid activates hepatocytes through CD36 mediated oxidative stress. Lipids Health Dis 17: 153, 2018. doi: 10.1186/s12944-018-0790-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang X, Zhang W, Chen Y, Li Y, Sun L, Liu Y, Liu M, Yu M, Li X, Han J, Duan Y. Activation of Peroxisome Proliferator-activated Receptor γ (PPARγ) and CD36 Protein Expression: THE DUAL PATHOPHYSIOLOGICAL ROLES OF PROGESTERONE. J Biol Chem 291: 15108–15118, 2016. doi: 10.1074/jbc.m116.726737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Morán-Salvador E, López-Parra M, García-Alonso V, Titos E, Martínez-Clemente M, González-Périz A, López-Vicario C, Barak Y, Arroyo V, Clària J. Role for PPARγ in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J 25: 2538–2550, 2011. doi: 10.1096/fj.10-173716. [DOI] [PubMed] [Google Scholar]
- 34. Han S, Sidell N. Peroxisome-proliferator-activated-receptor gamma (PPARgamma) independent induction of CD36 in THP-1 monocytes by retinoic acid. Immunology 106: 53–59, 2002. doi: 10.1046/j.1365-2567.2002.01404.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yeh K-Y, Lai C-Y, Lin C-Y, Hsu C-C, Lo C-P, Her GM. ATF4 overexpression induces early onset of hyperlipidaemia and hepatic steatosis and enhances adipogenesis in zebrafish. Sci Rep 7: 16362, 2017. doi: 10.1038/s41598-017-16587-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li K, Xiao Y, Yu J, Xia T, Liu B, Guo Y, Deng J, Chen S, Wang C, Guo F. Liver-specific gene inactivation of the transcription factor ATF4 alleviates alcoholic liver steatosis in mice. J Biol Chem 29: 18536–18546, 2016. doi: 10.1074/jbc.m116.726836. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available upon reasonable request.