

Keywords: hypertension, inflammation, NLRP3, placental ischemia, preeclampsia
Abstract
Preeclampsia (PE) is a pregnancy-specific hypertensive disorder with end-organ damage that presents after 20 wk of gestation. PE pathophysiology often includes vascular dysfunction and increased inflammation that continues to damage patient health even after PE resolves. Currently, there is no cure for PE beyond delivery of the fetal-placental unit. Previous clinical studies have identified elevated placental NLRP3 expression in patients with PE and suggest NLRP3 as a potential therapeutic target. In this study, we examined the effect of NLRP3 inhibition on PE pathophysiology in the reduced uterine perfusion pressure (RUPP) model rat using MCC950 (20 mg/kg/day) or esomeprazole (3.5 mg/kg/day). We hypothesized that increased NLRP3 in response to placental ischemia impairs anti-inflammatory IL-33 signaling to induce T-helper 17 cell (TH17) and cytolytic NK cell (cNK) activation, which is known to mediate oxidative stress and vascular dysfunction leading to maternal HTN and intrauterine growth restriction. RUPP rats had significantly higher placental NLRP3 expression, maternal blood pressure, fetal reabsorption rate, vascular resistance, oxidative stress, cNKs and TH17s, and decreased IL-33 compared with normal pregnant (NP) rats. NLRP3 inhibition, with either treatment, significantly reduced placental NLRP3 expression, maternal blood pressure, fetal reabsorption rates, vascular resistance, oxidative stress, cNK, and TH17 populations in RUPP rats. Based on our findings, NLRP3 inhibition reduces PE pathophysiology and esomeprazole may be a potential therapeutic for PE treatment.
INTRODUCTION
Preeclampsia (PE) is a pregnancy-specific maternal syndrome characterized by development of hypertension (HTN) in combination with end-organ dysfunction after 20 wk of gestation (1). In the United States, PE continuously ranks among the top 10 leading causes of pregnancy-related mortality, and this trend has continued to rise over the past 20 years (2). Not only is PE a significant factor in maternal and fetal morbidity, it has also been recognized as a significant risk factor for developing cardiovascular disease later in life in both the mother and child (3). Despite the serious health risks PE poses, there remains no effective cure beyond delivery of the fetal-placenta unit. Although PE etiology is still poorly understood (4, 5), the most widely proposed mechanism implicates shallow trophoblast invasion and insufficient uterine spiral artery remodeling in causing placental ischemia, resulting in chronic immune activation, endothelial dysfunction, and the manifestation of PE (1, 6).
Women with PE are in a state of chronic immune activation that includes the release of inflammatory cytokines, anti-angiogenic factors, and increased oxidative stress (7–9). Immune cell populations also undergo a shift from a fetal-tolerant state toward an effector function state, with increased populations of effector cells including proinflammatory T-helper 17 cells (TH17s) and cytolytic NK cells (cNKs) and decreased populations of immune-tolerant cells (10–13). The reduced uterine perfusion pressure (RUPP) preclinical model of placental ischemia utilized in this study recapitulates many of these characteristics of PE (14) and is widely used to study the immunological and pathophysiological changes associated with PE (15–19).
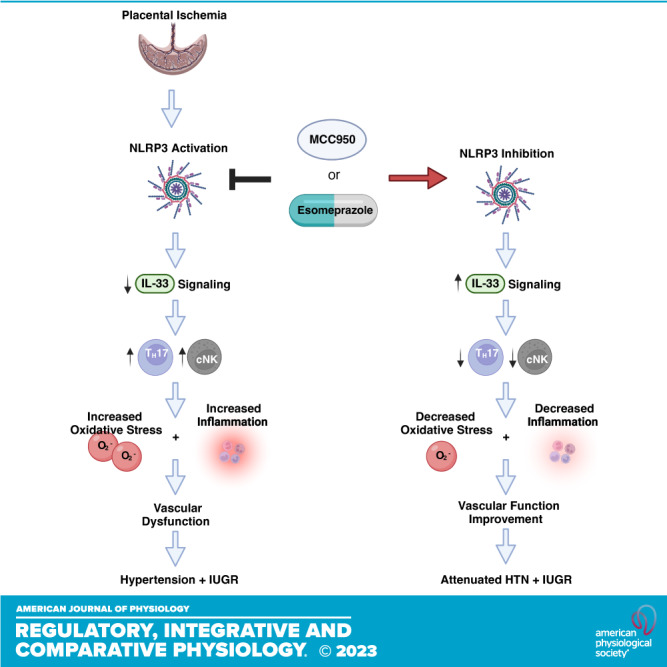
Placental NOD-like receptor and pyrin domain-containing protein 3 (NLRP3) expression is increased in women with PE (20). Thus, NLRP3 inflammasome has been suggested as a potential therapeutic target for PE (21). The NLRP3 inflammasome is an innate immune component that initiates pyroptosis and triggers proinflammatory cytokine release (22–24). NLRP3 inflammasome activation is associated with activation of TH17s, and cNKs (25–28), effector immune cells that are significantly increased in PE and are known to mediate pathophysiology of placental ischemia. Yet the mechanism by which this occurs is unknown. One potential mechanism of NLRP3 inflammasome-mediated activation of TH17s and cNKs is inhibition of IL-33 signaling (29–32). In other inflammatory diseases, NLRP3 has been shown to inhibit IL-33 signaling (32, 33), and studies of human PE samples have exhibited decreased IL-33 signaling (34, 35). IL-33 is a pleiotropic cytokine that modulates immune response and regulates immune cells, to promote a type 2 immunity response through its binding to membrane-bound ST2 receptors (36–40), which are expressed by TH17s and NKs.
Based on this evidence, we hypothesized that in PE, NLRP3 inhibition improves IL-33 signaling to reduce TH17s and cNK activation. The reduction in TH17s and cNK activation will yield decreased inflammation and reactive oxygen species and improve maternal vascular function, to attenuate HTN and IUGR. Using the RUPP rat model of placental ischemia, we inhibited NLRP3 inflammasome activation and assessed TH17s and cNK activation, IL-33 signaling molecules, and PE pathophysiology.
METHODS
Animals
Gestation day (GD) 10 or 11 Sprague-Dawley rats were purchased from Envigo (Indianapolis, IN). Animals were randomly assigned to experimental groups and housed in the Center for Comparative Research at the University of Mississippi Medical Center (UMMC). All protocols in this study were in accordance with the National Institutes of Health guidelines for the use and care of animals. Protocols (no. 1475 A) were approved by the UMMC Institutional Animal Care and Use Committee.
Reduced Uterine Perfusion Pressure Surgery
On GD14, a subset of pregnant rats underwent the RUPP surgery under isoflurane anesthesia as previously described (41, 42). After a midline incision was made, the lower abdominal aorta was isolated. A silver clip (0.203 mm) was applied to the aorta superior to the iliac bifurcation to restrict blood flow to the uterine horn. Restrictive clips (0.100 mm) were also applied to branches of the ovarian arteries to reduce compensatory ovarian circulation to the uterus. Another subset of pregnant rats also underwent a sham surgery of the RUPP in which clips were not placed (NP). Animals that had complete fetal resorption were excluded from the study as dictated by our protocol. The experimental timeline, design, and animal subsets are detailed in Supplemental Fig. S4 (all Supplemental material is available at https://doi.org/10.6084/m9.figshare.21675140).
Drug Administration
MCC950 (20 mg/kg/day; S7809, Selleck Chemicals, Houston, TX), a direct inhibitor of NLRP3 (43), was dissolved in saline and administered intraperitoneally or esomeprazole (3.5 mg/kg/day; 217087-09-7, MilliporeSigma, Burlington, MA), a FDA-approved proton pump inhibitor that is safe for use during pregnancy and previously shown to attenuate NLRP3 inflammasome activation (44), was administered via oral gavage in a 1% methyl cellulose (9004-67-5, MilliporeSigma, Burlington, MA) solution immediately after surgery and every 24 h following from GD 14–18. Control animals received saline intraperitoneal injections and were orally gavaged with 1% methyl cellulose. Dosing was chosen based on previously published studies in rat models of inflammatory and cardiovascular diseases (45–47). No changes in body weight, behavior, or food intake were observed between vehicle and drug-treated animals.
Uterine Artery Resistance Index Measurement
On GD18, rats underwent Doppler sonography under isoflurane anesthesia and the uterine artery resistance index (UARI) was measured. A Vevo 770 unit (Visual Sonics, Toronto, Canada) with a 30-Hz transducer (model no. 710B) was used to take Doppler velocimetry measurements on the uterine arteries. One placenta from each uterine horn was imaged to capture waveforms representing the peak systolic velocity (PSV) and end-diastolic flow velocity (EDV). The placenta visualized was the placenta midway between the ovary and cervix on both sides. Three waveforms were measured for velocities per frame. UARI was calculated using the equation UARI = (PSV – EDV)/PSV.
Mean Arterial Pressure Measurement
On GD18, catheters constructed from V3 tubing (BB21785, Scientific Commodities, Lake Havasu City, AZ) were inserted into the carotid arteries and tunneled to the back of the neck under isoflurane anesthesia. On GD19, rats were placed in individual restrainers. Conscious mean arterial pressure (MAP) was monitored with a pressure transducer (Powerlab, ADInstruments, Colorado Springs, CO) and recorded for 30 min following a 30-min stabilization period.
Sample Collection and Protein Isolation
Rats were anesthetized for blood and tissue collection after MAP measurement. Total litter size and number of live fetuses per litter were recorded. Placentas and fetuses were weighed and recorded for each dam and averaged. Randomly selected placenta tissues (the decidua, basal zone, and labyrinth zone included) were immediately frozen in liquid nitrogen and stored at −80°C until future analyses. Placenta and kidney samples were homogenized in T-PER Tissue Protein Extraction Reagent (78510, Thermo Fisher Scientific, Waltham, MA) containing 1 mM activated vanadate (BP-440, Boston BioProducts, Ashland, MA), protease inhibitor cocktail (P8340, MilliporeSigma, Burlington, MA), and 1X HALT protease and phosphatase inhibitor (78441, Thermo Fisher Scientific, Waltham, MA).
Renal Hemodynamics
On GD19, rats were anesthetized with ketamine (30 mg/kg; 00143950810, Hikma, Berkley Heights, NJ) and inactin (50 mg/kg; T133, MilliporeSigma, Burlington, MA) and surgically prepared for measurement of glomerular filtration rate (GFR) and renal blood flow (RBF). A catheter was inserted into the jugular vein for intravenous infusion of 0.9% NaCl solution, FITC-labeled inulin (2 mg/mL; F3272, MilliporeSigma, Burlington, MA), and para-aminohippuric acid (PAH) (4 mg/mL; A1422, MilliporeSigma, Burlington, MA) at a rate of 100 μL/min. The carotid artery was catheterized to monitor MAP. A catheter was inserted into the bladder to collect urine. After a 15-min equilibration period, urine and plasma samples were collected during a 30-min period. At the end of each experiment, the kidneys were removed and weighed. The concentration of FITC-labeled inulin in urine and plasma samples was determined using a plate reader fluorometer to measure GFR. The concentration of PAH in urine and plasma samples was determined using the PAH Assay Kit (MAK101-1KT, MilliporeSigma, Burlington, MA) to assess RBF. Renal vascular resistance (RVR) was calculated as MAP/RBF.
Real-Time Quantitative PCR
RNA was isolated from placenta and kidney samples using the Qiagen RNeasy Plus Mini Kit (74134, Qiagen, Germantown, MD), according to the manufacturer’s protocol. RNA samples were checked for purity on a BioTek Synergy H1 microplate reader with a Take3 multivolume plate (BTTAKE3TRIO, Thermo Fisher Scientific, Waltham, MA) and an A260/A280 ratio of 1.8–2.2 was confirmed before cDNA conversion. Samples were converted to cDNA using the iScript Reverse Transcription Supermix for RT-qPCR (1708841, Bio-Rad, Hercules, CA). Real-time quantitative PCR (RT-qPCR) was carried out using the Bio-Rad SYBR Green Supermix (1725271), the PrimePCR SYBR Green Assay for rat NLRP3 (10025641) and GAPDH (10025636), and iCycler (Bio-Rad, Hercules, CA). Samples were activated at 95°C for 2 min for 1 cycle. They were denatured at 95°C for 5 s and extended at 60°C for 30 s for 40 cycles. qPCR products were verified to be single amplicons using melt curve analysis, carried out from 65°C to 95°C in 0.5°C increment at 5 s/step. NLRP3 expression levels were normalized to GAPDH and quantified using the comparative CT method (ΔΔCT).
Bio-Plex Multiplex Assay
IL-1β levels were quantified in homogenized placenta and kidney samples using a Bio-Plex multiplex immunoassay (17008988, Bio-Rad, Hercules, CA) and analyzed on a Bio-Rad Bio-Plex 200 system according to the manufacturer’s instructions. Samples were not diluted for the assay. Data were normalized to protein concentration, determined via the Pierce BCA Protein Assay Kit (23225, Thermo Fisher Scientific, Waltham, MA) and expressed as pg/mg.
Flow Cytometry
Single-cell suspensions of leukocytes were prepared from blood, placenta, and kidney as previously described (41, 48) and blocked with 10% goat and mouse serum before staining for flow cytometry. Antibodies used to stain for NK cells (defined as CD3−ANK61+ cells for total NK cells and CD3−ANK61+ ANK44+ cells for activated NK cells) were as follows: adenomatous polyposis coli protein (APC)-conjugated anti-CD3 (1:10, Miltenyi Biotec; Cat. No. 130-102-679, RRID:AB_2657097), mouse anti-rat ANK61 (1:50, Abcam; Cat. No. ab36392, RRID:AB_776652), mouse anti-rat ANK44 (1:100, Abcam; Cat. No. ab36388, RRID:AB_776651), goat anti-mouse IgG FITC (1:50, Abcam; Cat. No. ab6785, RRID:AB_955241), and rabbit anti-mouse IgG AlexaFluor405 (1:100, Abcam; Cat. No. ab175651, RRID:AB_2923541). Antibodies used to stain for TH17 cells (defined as CD4+ CD25−RORγ+) were as follows: FITC-conjugated anti-CD4 antibody (1:10, Miltenyi Biotec; Cat. No. 130-107-623, RRID:AB_2657928), phycoerythrin (PE)-conjugated anti-CD25 (1:50, BD Biosciences; Cat. No. 554866, RRID:AB_395564), and peridinin-chlorophyll-protein (PerCP)-conjugated anti-RORγt (1:5, R and D Systems; Cat. No. IC6006C, RRID:AB_10571437). Flow cytometry was carried out on a Miltenyi MACSQuant Analyzer 10 (Miltenyi Biotec, San Diego, CA) and analyzed using FlowLogic software (Innovai, Sydney, Australia). After dead cell exclusion using Viobility Fixable dye staining (130-109-814, Miltenyi Biotec, San Diego, CA), lymphocytes were gated in the forward and side scatter plots and analyzed with fluorescence minus one (FMO) controls after doublet exclusion. Gating strategies for NK cells (Supplemental Fig. S8) and TH17 cells (Supplemental Fig. S9) can be found in the supplemental material. Data are expressed as the percentage of live cells in the gated lymphocyte population.
Oxidative Stress Measurement
Superoxide, a reactive oxygen species molecule, was measured in homogenized placenta and kidney samples, using lucigenin as previously described by our laboratory (41, 42). Homogenized samples were incubated with lucigenin (5 µM) and allowed to equilibrate at 37°C for 15 min in the dark. Luminescence was then measured for 10 s with a BioTek Plate Reader. An assay blank with only lucigenin was subtracted from the reading. Data were normalized to protein concentration, determined via the Pierce BCA Protein Assay Kit (23225, Thermo Fisher Scientific, Waltham, MA) and expressed as RLU per minute per milligram protein.
IL-33 and Soluble ST2 Quantification
Plasma, placental, and renal levels of interleukin-33 (IL-33) were quantified by ELISA (M3300, R&D Systems, Minneapolis, MN), and plasma levels of soluble interleukin 1 receptor-like 1 (ST2) were quantified by ELISA (MBS2702253, MyBioSource, San Diego, CA) according to the manufacturer’s protocol. Undiluted plasma and placental samples were used. Tissue IL-33 levels were normalized to protein concentration, determined via the Pierce BCA Protein Assay Kit (23225, Thermo Fisher Scientific, Waltham, MA; expressed as pg/mg).
Statistical Analysis
All data were presented as mean ± SD. Statistical analyses were performed using a two-way ANOVA followed by the Tukey’s multiple comparison test in GraphPad Prism 8 (GraphPad Software, San Diego, CA). For the two-way ANOVA analyses, row factors were defined as NP and RUPP and column factors were defined as vehicle treatment, MCC950 treatment, and esomeprazole treatment. Data were evaluated for normality using the Shapiro–Wilk test and for outliers using the ROUT method. A P value < 0.05 was considered statistically significant.
RESULTS
MCC950 and Esomeprazole Decrease Placental NLRP3 mRNA Expression and NLRP3 Activation
Using RT-qPCR, we measured NLRP3 mRNA expression in the placenta (Fig. 1A) and kidney (Fig. 1B) of dams from each experimental group. Placental NLRP3 expression was 5.579 ± 3.147-fold higher in RUPP compared with NP (P = 0.0004), but expression levels in the kidney were not significantly different between RUPP and NP. MCC950 and esomeprazole treatment reduced placental NLRP3 mRNA expression. MCC950 treatment reduced placental NLRP3 mRNA expression levels to a 1.861 ± 1.131-fold increase over NP rats (P = 0.0047). Esomeprazole treatment reduced placental NLRP3 mRNA expression levels to a 1.769 ± 1.448-fold increase over NP rats (P = 0.0036). Corresponding IHC images (Supplemental Fig. S5) of NLRP-stained placental slides show more intense NLRP3 protein expression in the decidua of RUPP rats compared with NP rats. Quantification of staining demonstrated a slight but insignificant increase of NLRP3 protein in placentas of RUPP. NLRP3 expression in the treated placentas from both the NP and RUPP groups were similar to vehicle-treated NP rats (Supplemental Fig. S5, all supplemental material is available at https://doi.org/10.6084/m9.figshare.21675140). We saw no significant differences in NLRP3 expression in the kidneys. We measured IL-1β levels using a Bio-Plex multiplex immunoassay in the placenta (Fig. 1C) and kidney (Fig. 1D) and found that placental IL-1β levels were decreased after NLRP3 inhibition in RUPP rats (MCC950, P = 0.0157; esomeprazole, P = 0.0237) but no difference was seen with renal IL-1β levels.
Figure 1.

MCC950 and esomeprazole inhibition decreased placental NLRP3 mRNA expression and activation but had no significant effect on renal mRNA NLRP3 expression and activation in placental ischemic rats. NLRP3 inhibition also differentially mediates IL33 levels in circulation and in the placenta. Placental NLRP3 mRNA expression (A), renal NLRP3 mRNA (B), placental IL-1β levels (C), and renal IL-1β levels (D). NLRP3 expression was measured using quantitative real-time-PCR and IL-1β levels were measured using a Bio-Plex assay. IL-33 levels were measured in plasma (E) and placenta using an ELISA (F). G: soluble ST2 levels were measured in plasma using ELISA. Data were presented as means ± SD. *P < 0.05 vs. NP + Vehicle, #P < 0.05 vs. RUPP + Vehicle, n = 5–8. NP, normal pregnant; RUPP, reduced uterine perfusion pressure.
NLRP3 Inhibition Differentially Mediates IL-33 Levels in Circulation and Placenta
Similar to what is reported in human studies (35), placental levels of IL-33 were higher in NP rats compared with untreated RUPP (17.503 ± 10.272 pg/mg of protein vs. 4.355 ± 2.595 pg/mg of protein, P = 0.0010; Fig. 1E). Neither treatment with MCC950 nor esomeprazole changed placental IL-33 levels in RUPP. IL-33 levels in plasma (Fig. 1F) did not significantly differ between NP and RUPP rats. Circulating IL-33 levels were elevated in treated RUPP rats (Fig. 1F), with 1568.493 ± 574.026 pg/mL in MCC950-treated RUPP rats and 1115.286 ± 238.465 pg/mL in esomeprazole-treated rats compared with 1.172 ± 1.978 pg/mL in untreated RUPP rats. Soluble ST2 levels were significantly increased in plasma of RUPP rats compared with NP rats (P = 0.0006; Fig. 1G), with 1662.494 ± 1284.825 pg/mL in RUPP rats compared with 386.727 ± 189.913 pg/mL in NP rats. Treatment with MCC950 reduced soluble ST2 levels in RUPP rats to 149.912 ± 139.564 pg/mL (P < 0.0001). Treatment with esomeprazole significantly reduced soluble ST2 levels in RUPP rats to 106.936 ± 61.344 pg/mL (P < 0.0001). ST2 expression was also evaluated in the placenta using Western blots though no significant difference was observed among the different groups (Supplemental Fig. S6).
Maternal Blood Pressure and Fetal Outcomes Improve with NLRP3 Inhibition
RUPP rats had higher MAP compared with NP rats (133 ± 3 mmHg vs. 109 ± 5 mmHg, P < 0.0001; Fig. 2A). MCC950 and esomeprazole treatment reduced MAP in RUPP rats (P < 0.0001). Treatment in RUPP rats achieved a MAP reduction of 22 ± 4 mmHg with MCC950 and a MAP reduction of 21 ± 7 mmHg with esomeprazole.
Figure 2.

NLRP3 inhibition decreased maternal MAP and fetal reabsorption rates in placenta ischemic rats. A: maternal MAP as measured by direct carotid catheterization. B: placenta weight, values expressed as average placenta weight in each litter. C: fetal weight, values expressed as average pup weight in each litter. D: fetal reabsorption rate, calculated as number of reabsorbed fetuses divided by total litter size. Data were presented as means ± SD. *P < 0.05 vs. NP + Vehicle, #P < 0.05 vs. RUPP + Vehicle, n = 7–10. MAP, mean arterial pressure; NP, normal pregnant; RUPP, reduced uterine perfusion pressure.
RUPP rats had lower placenta weights (0.445 ± 0.100 g) and fetal weights (2.071 ± 0.127 g) compared with NP rats (placenta weight = 0.611 ± 0.113 g, fetal weight = 2.449 ± 0.161 g). However, MCC950 and esomeprazole treatment did not improve placental weight (Fig. 2B) though esomeprazole treatment trended to improve fetal weights (P = 0.05; Fig. 2C). RUPP rats had a fetal reabsorption rate of 70.421 ± 18.464% in comparison with NP rats, which have a fetal reabsorption rate of 4.153 ± 4.309%. Moreover, NLRP3 inflammasome inhibition with MCC950 or esomeprazole treatment both lowered fetal reabsorption rates in RUPP rats (P < 0.0001; Fig. 2D).
Renal Vasculature Function and Uterine Artery Function Improve with NLRP3 Inhibition
We assessed uterine and renal vascular function in each group. RUPP rats had higher UARI compared with NP rats (0.714 ± 0.074 compared with 0.560 ± 0.036, P = 0.0017; Fig. 3A). In RUPP rats, esomeprazole treatment trended toward a reduction in UARI (0.608 ± 0.063, P = 0.05), whereas MCC950 treatment significantly reduced UARI (0.478 ± 0.035, P < 0.0001). No significant differences in renal blood flow (RBF) (Fig. 3B) or renal vascular resistance (RVR; Fig. 3C) were observed between RUPP and NP. However, RVR decreased with esomeprazole treatment (11.888 ± 10.028 mmHg/mL/min vs. 26.070 ± 13.079 mmHg/mL/min, P = 0.0497) and trended toward reduction with MCC950 treatment (12.208 ± 4.558 mmHg/mL/min vs. 26.070 ± 13.079 mmHg/mL/min, P = 0.05) in RUPP compared with vehicle-treated RUPP rats.
Figure 3.

NLRP3 inhibition decreased renal vascular resistance in placental ischemic rats and improved uterine artery function. Results of UARI (A) as measured via Doppler sonography, renal blood flow measurements (B), and renal vascular resistance measurements (C). NLRP3 inhibition did not improve kidney injury in placental ischemic rats. E: glomerular filtration rate was measured via FITC sinistrin clearance. Results of glomerular injury (F) and renal fibrosis (G) scored in periodic acid Schiff (PAS) stained, paraffin-embedded kidney sections. Data were presented as means ± SD. *P < 0.05 vs. NP + Vehicle, #P < 0.05 vs. RUPP + Vehicle, n = 4–9. NP, normal pregnant; RUPP, reduced uterine perfusion pressure; UARI, uterine artery resistance index.
We assessed kidney injury in each treatment group by measuring GFR, scoring glomerular injury, and renal fibrosis. There was no significant difference between NP and RUPP rats in glomerular filtration rate (Fig. 3E), glomerular injury (Fig. 3F), or renal fibrosis (Fig. 3G).
Oxidative Stress and Inflammation Are Reduced in NLRP3-Inhibited Placental Ischemic Rats
RUPP rats had higher levels of oxidative stress (ROS) in both the placenta and kidney compared with NP rats (P = 0.001). ROS levels in the placenta (Fig. 4A) were reduced by both MCC950 (P = 0.0019) and esomeprazole treatment in RUPP rats (P < 0.0001). NP rats had placental ROS levels of 158.049 ± 81.081 RLU/min/mg protein compared with 490.523 ± 220.622 RLU/min/mg protein in RUPP rats. MCC950 treatment in RUPP rats decreased placental ROS levels to 206.150 ± 111.608 RLU/min/mg protein and esomeprazole treatment in RUPP decreased placental ROS levels to 48.431 ± 26.116 RLU/min/mg protein.
Figure 4.

NLRP3 inhibition decreased placental and renal oxidative stress in placental ischemic rats. Placental ROS (A) and renal ROS (B) were measured by the lucigenin assay. Data were presented as mean ± SD. *P < 0.05 vs. NP + Vehicle, #P < 0.05 vs. RUPP + Vehicle, n = 5–10. NP, normal pregnant; ROS, reactive oxygen species; RUPP, reduced uterine perfusion pressure.
NP rats had renal ROS levels (Fig. 4B) of 151.000 ± 34.346 RLU/min/mg protein compared with 392.796 ± 142.440 RLU/min/mg protein in RUPP rats (P = 0.0134). MCC950 treatment in RUPP rats was able to decrease renal ROS levels to 107.233 ± 74.177 RLU/min/mg protein (P = 0.0014), and esomeprazole treatment in RUPP rats made no significant difference in renal ROS, with ROS levels at 475.184 ± 168.942 RLU/min/mg protein.
RUPP rats had higher activated cNKs and TH17s in the circulation and placenta compared with NP rats. The activated cNK population was 3.296% higher in RUPP rats compared with NP rats in circulation (P = 0.0001; Fig. 5A). In the placenta, activated cNKs were 2.013% higher in RUPP rats compared with NP rats (P = 0.0002; Fig. 5B). The activated cNK population was not significantly higher in RUPP rats compared with NP rats in the kidney (Fig. 5C). Treatment with MCC950 and esomeprazole reduced activated cNK populations in circulation (MCC950, P < 0.0001; esomeprazole, P < 0.0001), the placenta (MCC950, P = 0.0013; esomeprazole, P = 0.0009), and the kidney (MCC950, P < 0.0001; esomeprazole, P < 0.0001) of RUPP rats similar to or below NP levels.
Figure 5.

cNK activation in the circulation, placenta, and kidney were reduced after NLRP3 inhibition. NLRP3 inhibition also reduced circulating and placental TH17 populations. cNK cell populations were measured via flow cytometry in circulation (A), placenta (B), and kidney (C). TH17 cell populations were measured via flow cytometry in circulation (D), placenta (E), and kidney (F). Data were presented as mean ± SD. *P < 0.05 vs. NP + Vehicle, #P < 0.05 vs. RUPP + Vehicle, n = 4–8. NP, normal pregnant; RUPP, reduced uterine perfusion pressure.
TH17 populations in circulation were 1.947% higher in RUPP rats compared with NP rats (P = 0.0227, Fig. 5D). Placental TH17 populations were 6.898% higher in RUPP rats compared with NP rats (P = 0.0027; Fig. 5E). Renal TH17 populations were not significantly different between NP and RUPP rats. Only treatment with MCC950 reduced circulating TH17 populations in RUPP rats (P = 0.0032). Treatment with MCC950 (P < 0.0001) and esomeprazole (P = 0.0002) also reduced placental TH17s in RUPP rats to levels equivalent to NP rats.
NLRP3 Inhibition Did Not Significantly Impact Caspase-1 Activity or Gasdermin D Levels
In addition to NLRP3 expression and immune cell population changes, we also measured some downstream mediators of NLRP3 signaling such as caspase-1 activity and Gasdermin D (GSDMD) levels. There was no significant difference in caspase-1 activity between NP and RUPP rats in the placenta or the kidney (Supplemental Fig. S1). There were also no significant differences in GSDMD protein expression in the placenta (Supplemental Fig. S7) though GSDMD expression trended higher in RUPP compared with NP.
DISCUSSION
Elevated NLRP3 expression has been reported in preeclampsia, in addition to other pregnancy disorders and hypertensive diseases; and studies suggest an important role for NLRP3 inflammasome in mediating the inflammation and vascular dysfunction associated with these conditions (49–53). We report for the first time that the RUPP rat model has elevated placental levels of NLRP3 expression and alterations in IL-33 signaling molecules that are associated with increased inflammation and maternal vascular resistance, analogous to clinical characteristics of patients with PE. Although MCC950 and esomeprazole had differential effects on various characteristics of PE, both treatments significantly improve maternal MAP and fetal survival. Since esomeprazole is a proton pump inhibitor, its highly significant effect on fetal reabsorption may be due to a reduction in mitochondrial ROS, a factor that can activate NLRP3 (54). Reduction of mitochondrial ROS using esomeprazole has been previously demonstrated by Hastie et al. (55) to reduce sFlt-1 secretion, a pathway normally overactive in preeclampsia. Reduction of mitochondrial ROS has also been shown to mediate hypertension and slightly decrease fetal resorption in the RUPP rat model (56).
The importance of NLRP3-mediated activation in causing endothelial dysfunction and vascular damage as a mechanism of hypertension has been shown by multiple studies. Bruder-Nascimento et al. (57) demonstrated that aldosterone-induced vascular damage is dependent on the ROS-induced activation of NLRP3 and the subsequent processing and release of IL-1β. Aberrant ROS production has also been shown to lead to activation of the NLRP3 inflammasome, promoting the release of IL-1β and induction of a proinflammatory response in endothelial cells (58). In addition to its roles in contributing to endothelial inflammation, NLRP3-mediated pyroptosis has been identified as a potential cause of endothelial cell death and NLRP3 activation is linked to endothelial barrier dysfunction (59). Though we observed no changes in caspase-1 activity with NLRP3 inhibition, caspase-1-mediated pyroptotic death can be induced through the activation of other inflammasomes such as NLRP1 and NLRC4 (60). We and others have previously demonstrated the role of increased ROS and inflammation in contributing to endothelial dysfunction and hypertension in PE (11, 56, 61–64). Direct inhibition of ROS with tempol, mito-Tempol and MitoQ all attenuated maternal hypertension in RUPP rats (18, 56). In addition, direct inhibition of inflammatory cytokines including TNF, IFN, and IL-17 significantly improved maternal blood pressure and decreased oxidative stress in RUPP rats (65–68). Improvements in placental oxidative stress and systemic inflammation after NLRP3 inhibition may be a mechanism behind the improvement in UARI and RVR we see in treated RUPP rats. Though fetal and placental weights were not significantly improved by treatment, these reductions in oxidative stress and inflammation from NLRP3 inhibition may yield benefits in clinical PE.
We found increases in TH17 and cNK populations as well as in ROS levels in RUPP compared with NP rats that were ameliorated by NLRP3 inhibition. NLRP3 activation has been linked to increased activation in TH17 and cNK populations in other diseases, whereas IL-33 signaling has been inversely linked to TH17 and cNK activation (26, 69–73). Previous studies from our laboratory and others have clearly demonstrated roles for TH17 and cNK activation in promoting generation of mitochondrial ROS, localized tissue ROS, inflammation and vascular function (13, 41, 74, 75). The decrease in ROS observed after NLRP3 inhibition may be due to the decreases in TH17 and cNK populations. Our recent investigation into the role of cNK cells in placental ischemia-induced vascular dysfunction demonstrated that RUPP cNK cells induce excessive ROS and decrease nitric oxide bioavailability in pregnant rats (76). Excess ROS production decreases bioavailability of nitric oxide, an important regulator of vascular resistance and function (77) and therefore, decreasing ROS production improves vascular function. We observed a slight, but insignificant, increase in renal ROS with esomeprazole treatment. However, recent studies report associations between proton pump inhibitor use and adverse kidney outcomes such as acute kidney injury, acute interstitial nephritis, and chronic kidney disease (78, 79). Thus, the lack of an improvement in renal oxidative stress with esomeprazole treatment is probable. The difference in improvement that we observed in uterine artery resistance between MCC950 and esomeprazole treatment may be due to the difference in reduction of placental TH17s between the two treatments.
We hypothesized that NLRP3 inhibition may be reducing TH17s and cNKs through increasing IL-33 signaling. NLRP3 activation has been linked to increased activation in TH17 and cNK populations in other diseases, whereas IL-33 signaling has been inversely linked to TH17 and cNK activation (26, 69–73). IL-33 can play either a pro- or anti-inflammatory role in immune response (80–82). However, studies consistently report that in cardiovascular diseases, IL-33 signaling is considered cardio-protective (83). IL-33 signaling takes place when IL-33 binds to the membrane-bound form of the ST2 receptor and is regulated by changes in the soluble form of the ST2 receptor, which acts as a decoy to inhibit signaling (34). Clinical studies have found decreased IL-33 signaling in patients with PE (34, 35); and preclinical studies show that NLRP3 inflammasome inhibits IL-33 signaling in other inflammatory diseases by caspase-mediated cleavage or by IL-1β -mediated mechanisms (32, 33). Our data show that placental levels of IL-33 are decreased in RUPP rats compared with NP rats and plasma levels of IL-33 increase after NLRP3 inflammasome inhibition. Importantly, plasma levels of soluble ST2 receptor in RUPP rats were significantly higher compared with NP rats. After NLRP3 was inhibited, soluble ST2 was significantly decreased in treated RUPP rats. This suggests that during placental ischemia, IL-33 signaling may be impaired in the plasma due to increased levels of soluble ST2 receptor and impaired in the placenta due to decreased levels of IL-33. Though we saw no difference in ST2 expression in the placenta, the currently available methods for evaluating ST2 in rats do not allow us to distinguish between the soluble and membrane-bound forms of ST2.
The ST2 receptor is expressed on the surfaces of TH17 and NK cells. In studies on gut inflammation, IL-33 binding to ST2 receptors on TH17 cells induces an immunosuppressive regulatory phenotype and IL-33 mediates expansion of the TReg population (84–86). This supports the idea that impaired IL-33 signaling may contribute to the proinflammatory state seen in preeclampsia through its modulatory effects on the TReg population, where sufficient TReg populations are needed for maternal-fetal tolerance during normal pregnancy (87, 88). In PE, there is a shift from a TReg dominant population toward a more TH17 dominant population, which our laboratory has previously shown to directly activate cNK cells (13, 74). We will further examine the contributions of the IL-33 and ST2 signaling pathway to PE pathophysiology in future studies.
Inhibition of NLRP3 by treatment with MCC950 has been tested in animal models of pregnancy disorders and shown benefit. For example, MCC950 treatment reduced preterm birth and fetal mortality in a mouse model of intraamniotic inflammation (69). Furthermore, crystal-induced placental inflammation was inhibited by MCC950 treatment in an in vitro placental explant model of PE (89). Thus, targeting NLRP3 may be a therapeutic strategy applicable in various inflammation induced pregnancy-associated disorders. However, use of MCC950 is not practical due to the liver toxicity observed in a clinical trial of rheumatoid arthritis and its limited pharmacokinetic properties (90, 91). Other candidate drugs for specific inhibition of NLRP3 are currently under investigation and may have potential as clinical interventions in PE.
Previous studies using in vivo and in vitro models of PE including the l-NAME mouse model, the l-NAME rat model, the sFlt-1 mouse model, and in vitro LPS model have shown improvements in various characteristics of PE such as reductions in maternal hypertension, sFlt-1 secretion, soluble endoglin secretion, and endothelial dysfunction-induced autophagy after esomeprazole treatment (46, 92–94). Although these studies demonstrated improvements in PE pathophysiology, no mechanistic explanation for esomeprazole’s effects was explored. Based on data from this current study, it is possible that esomeprazole is exerting its beneficial effects through inhibition of NLRP3. Esomeprazole treatment was able to recapitulate nearly all of the improvements observed after MCC950 treatment in RUPP rats. Esomeprazole has been proposed as a potential therapeutic for treatment/prevention of PE (95). A South African clinical trial administering 40 mg of esomeprazole, daily to women with early-onset PE found no improvements in maternal or fetal outcome markers, although time to delivery was extended by ∼3 days in the esomeprazole-treated group (96). Furthermore, a more recent clinical trial reported that addition of esomeprazole to standard care significantly increased time to delivery and decreased the incidence of postpartum complications in women with early-onset PE compared with women who received standard treatment only (97). Our study, which used a comparable dose of esomeprazole, suggests therapeutic potential for this drug in late-onset PE. However, to our knowledge, there have been no clinical studies investigating the therapeutic potential of esomeprazole in late-onset PE. This knowledge gap should be addressed in future clinical studies.
Though we found no significant difference in kidney injury as examined in kidney histology in MCC950 and esomeprazole-treated rats, these findings are not necessarily surprising as there was no difference between untreated RUPP and NP rats similar to previously published research (98). Previously published studies have demonstrated significant reductions in GFR in RUPP rats compared with NP rats though we saw no changes in GFR in our study (16, 99). However, this may be contributed to the difference in methods as those studies used conscious rats for their renal hemodynamic measurements while we used unconscious rats. Though esomeprazole treatment decreased renal vascular resistance in RUPP rats, the lack of effect on renal TH17 populations and renal oxidative stress implies that this improvement in renal vascular resistance may be from effects on other inflammatory or molecular pathways. The lack of significant difference in renal NLRP3 expression, activation, or caspase-1 activity, suggests that NLRP3 inflammasome-dependent mechanisms may not be important in contributing to renal function in the pathophysiology of PE.
NLRP3 can exert biological change in either its inflammasome-dependent form or its inflammasome-independent form. The inflammasome-dependent form has been more widely studied and has been identified as a crucial player in triggering immune responses by activating caspase-1 and IL-1β (100). However, emerging studies have identified roles for inflammasome-independent NLRP3 in mediating renal changes, specifically TGF-β-induced epithelial-mesenchymal transition and renal fibrosis in the unilateral ureteral obstruction model of renal fibrosis (101). The inflammasome-independent NLRP3 mechanisms and their contributions to renal injury in PE require further exploration. However, this is beyond the scope of our study.
In this study, we demonstrate that inhibition of NLRP3, whether by administration of MCC950 or by administration of esomeprazole, reduces many of the pathophysiological characteristics associated with PE. With the established safety of esomeprazole use during pregnancy coupled with the potential for positive impact in the few clinical studies that have been performed, we believe this could represent an important advance in pharmacotherapeutic management of PE.
Perspectives and Significance
This study is the first to show that inhibition of NLRP3 with MCC950 attenuates increased inflammation, oxidative stress, and vascular dysfunction using an in vivo model of placental ischemia. Our findings on esomeprazole-mediated NLRP3 inhibition in placental ischemia lead to exciting possibilities for novel PE therapies. Current pharmacotherapies for the management of patients with PE are limited and repurposing a drug with an accepted safety profile in pregnancy would lower the time and cost barriers to patient use.
We demonstrate NLRP3 inhibition can lead to increased IL-33 signaling and improve PE-associated characteristics such as maternal hypertension, oxidative stress, impaired vascular function, and inflammation. We also demonstrate that increased NLRP3 expression and decreased IL-33 levels have been observed in placentas of women with PE. Inhibition of NLRP3, improved IL-33 signaling which was associated with decreases in proinflammatory TH17s and cNK cells, which are known to contribute to PE pathophysiology.
Summary
The data presented in this paper suggest a promising role for targeting NLRP3 in the treatment of PE. Esomeprazole, an FDA-approved category C drug that is safe in pregnancy, may have therapeutic potential to be added to existing pharmacotherapy for the management of patients with PE.
DATA AVAILABILITY
Data will be made available upon reasonable request.
SUPPLEMENTAL DATA
Supplemental Figs. S1–S9: https://doi.org/10.6084/m9.figshare.21675140.
GRANTS
This work was supported by the National Institutes of Health Grants F31-HL165852 to X. Wang, F31-HL149257 to O. K. Travis, T32-HL105324 to C. A. Shields, R01-DK109133 to J. M. Williams, and R00-HL130456 and R01-HL151407 to D. C. Cornelius.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.M.W. and D.C.C. conceived and designed research; X.W., O.K.T., C.A.S., G.A.T., C.G., C.W.N., H.L.G., O.G.C., T.D., R.T., and D.C.C. performed experiments; X.W., O.K.T., C.A.S., G.A.T., C.G., C.W.N., H.L.G., R.T., and D.C.C. analyzed data; X.W., O.G.C., J.M.W., and D.C.C. interpreted results of experiments; X.W., C.W.N., H.L.G., T.D., and D.C.C. prepared figures; X.W. drafted manuscript; X.W., J.M.W., and D.C.C. edited and revised manuscript; X.W., O.K.T., C.A.S., G.A.T., C.G., C.W.N., H.L.G., O.G.C., T.D., R.T., J.M.W., and D.C.C. approved final version of manuscript.
REFERENCES
- 1. Phipps EA, Thadhani R, Benzing T, Karumanchi SA. Pre-eclampsia: pathogenesis, novel diagnostics and therapies. Nat Rev Nephrol 15: 275–289, 2019. [Erratum in Nat Rev Nephrol 15: 386, 2019]. doi: 10.1038/s41581-019-0119-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Collier AY, Molina RL. Maternal mortality in the United States: updates on trends, causes, and solutions. Neoreviews 20: e561–e574, 2019. doi: 10.1542/neo.20-10-e561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mosca L, Benjamin EJ, Berra K, Bezanson JL, Dolor RJ, Lloyd-Jones DM, Newby LK, Piña IL, Roger VL, Shaw LJ, Zhao D, Beckie TM, Bushnell C, D'Armiento J, Kris-Etherton PM, Fang J, Ganiats TG, Gomes AS, Gracia CR, Haan CK, Jackson EA, Judelson DR, Kelepouris E, Lavie CJ, Moore A, Nussmeier NA, Ofili E, Oparil S, Ouyang P, Pinn VW. Effectiveness-based guidelines for the prevention of cardiovascular disease in women–2011 update: a guideline from the American Heart Association. Circulation 123: 1243–1262, 2011. [Erratum in Circulation 123: e624, 2011]. doi: 10.1161/CIR.0b013e31820faaf8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hladunewich M, Karumanchi SA, Lafayette R. Pathophysiology of the clinical manifestations of preeclampsia. Clin J Am Soc Nephrol 2: 543–549, 2007. doi: 10.2215/CJN.03761106. [DOI] [PubMed] [Google Scholar]
- 5. Uzan J, Carbonnel M, Piconne O, Asmar R, Ayoubi JM. Pre-eclampsia: pathophysiology, diagnosis, and management. Vasc Health Risk Manag 7: 467–474, 2011. doi: 10.2147/VHRM.S20181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gathiram P, Moodley J. Pre-eclampsia: its pathogenesis and pathophysiolgy. Cardiovasc J Afr 27: 71–78, 2016. doi: 10.5830/CVJA-2016-009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Aggarwal R, Jain AK, Mittal P, Kohli M, Jawanjal P, Rath G. Association of pro- and anti-inflammatory cytokines in preeclampsia. J Clin Lab Anal 33: e22834, 2019. doi: 10.1002/jcla.22834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Conrad KP, Benyo DF. Placental cytokines and the pathogenesis of preeclampsia. Am J Reprod Immunol 37: 240–249, 1997. doi: 10.1111/j.1600-0897.1997.tb00222.x. [DOI] [PubMed] [Google Scholar]
- 9. Phoswa WN, Khaliq OP. The role of oxidative stress in hypertensive disorders of pregnancy (preeclampsia, gestational hypertension) and metabolic disorder of pregnancy (gestational diabetes mellitus). Oxid Med Cell Longev 2021: 5581570, 2021. doi: 10.1155/2021/5581570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aneman I, Pienaar D, Suvakov S, Simic TP, Garovic VD, McClements L. Mechanisms of key innate immune cells in early- and late-onset preeclampsia. Front Immunol 11: 1864, 2020. doi: 10.3389/fimmu.2020.01864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cornelius DC, Cottrell J, Amaral LM, LaMarca B. Inflammatory mediators: A causal link to hypertension during preeclampsia. Br J Pharmacol 176: 1914–1921, 2019. doi: 10.1111/bph.14466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. LaMarca B. The role of immune activation in contributing to vascular dysfunction and the pathophysiology of hypertension during preeclampsia. Minerva Ginecol 62: 105–120, 2010. [PMC free article] [PubMed] [Google Scholar]
- 13. Shields CA, McCalmon M, Ibrahim T, White DL, Williams JM, LaMarca B, Cornelius DC. Placental ischemia-stimulated t-helper 17 cells induce preeclampsia-associated cytolytic natural killer cells during pregnancy. Am J Physiol Regul Integr Comp Physiol 315: R336–R343, 2018. doi: 10.1152/ajpregu.00061.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li J, LaMarca B, Reckelhoff JF. A model of preeclampsia in rats: The reduced uterine perfusion pressure (rupp) model. Am J Physiol Heart Circ Physiol 303: H1–H8, 2012. doi: 10.1152/ajpheart.00117.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Amaral LM, Faulkner JL, Elfarra J, Cornelius DC, Cunningham MW, Ibrahim T, Vaka VR, McKenzie J, LaMarca B. Continued investigation into 17-ohpc: results from the preclinical rupp rat model of preeclampsia. Hypertension 70: 1250–1255, 2017. doi: 10.1161/HYPERTENSIONAHA.117.09969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alexander BT, Kassab SE, Miller MT, Abram SR, Reckelhoff JF, Bennett WA, Granger JP. Reduced uterine perfusion pressure during pregnancy in the rat is associated with increases in arterial pressure and changes in renal nitric oxide. Hypertension 37: 1191–1195, 2001. doi: 10.1161/01.hyp.37.4.1191. [DOI] [PubMed] [Google Scholar]
- 17. LaMarca B, Wallukat G, Llinas M, Herse F, Dechend R, Granger JP. Autoantibodies to the angiotensin type i receptor in response to placental ischemia and tumor necrosis factor alpha in pregnant rats. Hypertension 52: 1168–1172, 2008. doi: 10.1161/HYPERTENSIONAHA.108.120576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sedeek M, Gilbert JS, LaMarca BB, Sholook M, Chandler DL, Wang Y, Granger JP. Role of reactive oxygen species in hypertension produced by reduced uterine perfusion in pregnant rats. Am J Hypertens 21: 1152–1156, 2008. doi: 10.1038/ajh.2008.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sholook MM, Gilbert JS, Sedeek MH, Huang M, Hester RL, Granger JP. Systemic hemodynamic and regional blood flow changes in response to chronic reductions in uterine perfusion pressure in pregnant rats. Am J Physiol Heart Circ Physiol 293: H2080–H2084, 2007. doi: 10.1152/ajpheart.00667.2007. [DOI] [PubMed] [Google Scholar]
- 20. C Weel I, Romão-Veiga M, Matias ML, Fioratti EG, Peraçoli JC, Borges VT, Araujo JP, Peraçoli MT. Increased expression of nlrp3 inflammasome in placentas from pregnant women with severe preeclampsia. J Reprod Immunol 123: 40–47, 2017. doi: 10.1016/j.jri.2017.09.002. [DOI] [PubMed] [Google Scholar]
- 21. Shirasuna K, Karasawa T, Takahashi M. Role of the NLRP3 inflammasome in preeclampsia. Front Endocrinol (Lausanne) 11: 80, 2020. doi: 10.3389/fendo.2020.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci 20: 3328, 2019. doi: 10.3390/ijms20133328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pellegrini C, Fornai M, Colucci R, Lopez-Castejon G, Cocco M, Garella D, Bertinaria M, Blandizzi C, Antonioli L. Targeting of NLRP3 inflammasome with a novel selective inhibitor as a suitable strategy for the pharmacological treatment of bowel inflammation. Gastroenterology 150: S968–S969, 2016. doi: 10.1016/S0016-5085(16)33282-6. [DOI] [Google Scholar]
- 24. Zahid A, Li BF, Kombe AJK, Jin TC, Tao JH. Pharmacological inhibitors of the NLRP3 inflammasome. Front Immunol 10: 2538, 2019. doi: 10.3389/fimmu.2019.02538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dupaul-Chicoine J, Arabzadeh A, Dagenais M, Douglas T, Champagne C, Morizot A, Rodrigue-Gervais IG, Breton V, Colpitts SL, Beauchemin N, Saleh M. The Nlrp3 inflammasome suppresses colorectal cancer metastatic growth in the liver by promoting natural killer cell tumoricidal activity. Immunity 43: 751–763, 2015. doi: 10.1016/j.immuni.2015.08.013. [DOI] [PubMed] [Google Scholar]
- 26. Zhao C, Gu Y, Zeng X, Wang J. NLRP3 inflammasome regulates Th17 differentiation in rheumatoid arthritis. Clin Immunol 197: 154–160, 2018. doi: 10.1016/j.clim.2018.09.007. [DOI] [PubMed] [Google Scholar]
- 27. Hatscher L, Lehmann CHK, Purbojo A, Onderka C, Liang C, Hartmann A, Cesnjevar R, Bruns H, Gross O, Nimmerjahn F, Ivanović-Burmazović I, Kunz M, Heger L, Dudziak D. Select hyperactivating nlrp3 ligands enhance the th1- and th17-inducing potential of human type 2 conventional dendritic cells. Sci Signal 14: eabe1757, 2021. doi: 10.1126/scisignal.abe1757. [DOI] [PubMed] [Google Scholar]
- 28. Chen TTW, Cheng PC, Chang KC, Cao JP, Feng JL, Chen CC, Lam HYP, Peng SY. Activation of the nlrp3 and aim2 inflammasomes in a mouse model of schistosoma mansoni infection. J Helminthol 94: e72, 2019. doi: 10.1017/S0022149X19000622. [DOI] [PubMed] [Google Scholar]
- 29. Jovanovic IP, Pejnovic NN, Radosavljevic GD, Pantic JM, Milovanovic MZ, Arsenijevic NN, Lukic ML. Interleukin-33/st2 axis promotes breast cancer growth and metastases by facilitating intratumoral accumulation of immunosuppressive and innate lymphoid cells. Int J Cancer 134: 1669–1682, 2014. doi: 10.1002/ijc.28481. [DOI] [PubMed] [Google Scholar]
- 30. Schuijs MJ, Png S, Richard AC, Tsyben A, Hamm G, Stockis J, Garcia C, Pinaud S, Nicholls A, Ros XR, Su J, Eldridge MD, Riedel A, Serrao EM, Rodewald HR, Mack M, Shields JD, Cohen ES, McKenzie ANJ, Goodwin RJA, Brindle KM, Marioni JC, Halim TYF. Ilc2-driven innate immune checkpoint mechanism antagonizes nk cell antimetastatic function in the lung. Nat Immunol 21: 998–1009, 2020. doi: 10.1038/s41590-020-0745-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Han M, Ishikawa T, Bermick JR, Rajput C, Lei J, Goldsmith AM, Jarman CR, Lee J, Bentley JK, Hershenson MB. Il-1β prevents ILC2 expansion, type 2 cytokine secretion, and mucus metaplasia in response to early-life rhinovirus infection in mice. Allergy 75: 2005–2019, 2020. doi: 10.1111/all.14241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Madouri F, Guillou N, Fauconnier L, Marchiol T, Rouxel N, Chenuet P, Ledru A, Apetoh L, Ghiringhelli F, Chamaillard M, Zheng SG, Trovero F, Quesniaux VF, Ryffel B, Togbe D. Caspase-1 activation by NLRP3 inflammasome dampens il-33-dependent house dust mite-induced allergic lung inflammation. J Mol Cell Biol 7: 351–365, 2015. doi: 10.1093/jmcb/mjv012. [DOI] [PubMed] [Google Scholar]
- 33. Strangward P, Haley MJ, Albornoz MG, Barrington J, Shaw T, Dookie R, Zeef L, Baker SM, Winter E, Tzeng TC, Golenbock DT, Cruickshank SM, Allan SM, Craig A, Liew FY, Brough D, Couper KN. Targeting the IL33-NLRP3 axis improves therapy for experimental cerebral malaria. Proc Natl Acad Sci USA 115: 7404–7409, 2018. doi: 10.1073/pnas.1801737115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Granne I, Southcombe JH, Snider JV, Tannetta DS, Child T, Redman CW, Sargent IL. ST2 and IL-33 in pregnancy and pre-eclampsia. PLoS One 6: e24463, 2011. doi: 10.1371/journal.pone.0024463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen H, Zhou X, Han T-L, Baker PN, Qi H, Zhang H. Decreased il-33 production contributes to trophoblast cell dysfunction in pregnancies with preeclampsia. Mediators Inflamm 2018: 9787239, 2018. doi: 10.1155/2018/9787239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Afferni C, Buccione C, Andreone S, Galdiero MR, Varricchi G, Marone G, Mattei F, Schiavoni G. The pleiotropic immunomodulatory functions of il-33 and its implications in tumor immunity. Front Immunol 9: 2601, 2018. doi: 10.3389/fimmu.2018.02601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen Z, Hu Y, Gong Y, Zhang X, Cui L, Chen R, Yu Y, Yu Q, Chen Y, Diao H, Chen J, Wang Y, Shi Y. Interleukin-33 alleviates psoriatic inflammation by suppressing the t helper type 17 immune response. Immunology 160: 382–392, 2020. doi: 10.1111/imm.13203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mehraj V, Ponte R, Routy JP. The dynamic role of the il-33/st2 axis in chronic viral-infections: alarming and adjuvanting the immune response. EBioMedicine 9: 37–44, 2016. doi: 10.1016/j.ebiom.2016.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xu H, Turnquist HR, Hoffman R, Billiar TR. Role of the IL-33-ST2 axis in sepsis. Mil Med Res 4: 3, 2017. doi: 10.1186/s40779-017-0115-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Neumann K, Schiller B, Tiegs G. NLRP3 inflammasome and IL-33: Novel players in sterile liver inflammation. Int J Mol Sci 19: 2732, 2018. doi: 10.3390/ijms19092732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Travis OK, White D, Baik C, Giachelli C, Thompson W, Stubbs C, Greer M, Lemon JP, Williams JM, Cornelius DC. Interleukin-17 signaling mediates cytolytic natural killer cell activation in response to placental ischemia. Am J Physiol Regul Integr Comp Physiol 318: R1036–R1046, 2020. doi: 10.1152/ajpregu.00285.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Travis OK, White D, Pierce WA, Ge Y, Stubbs CY, Spradley FT, Williams JM, Cornelius DC. Chronic infusion of interleukin-17 promotes hypertension, activation of cytolytic natural killer cells, and vascular dysfunction in pregnant rats. Physiol Rep 7: e14038, 2019. doi: 10.14814/phy2.14038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz A, Croker DE, Butler MS, Haneklaus M, Sutton CE, Núñez G, Latz E, Kastner DL, Mills KH, Masters SL, Schroder K, Cooper MA, O'Neill LA. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21: 248–255, 2015. doi: 10.1038/nm.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Balza E, Piccioli P, Carta S, Lavieri R, Gattorno M, Semino C, Castellani P, Rubartelli A. Proton pump inhibitors protect mice from acute systemic inflammation and induce long-term cross-tolerance. Cell Death Dis 7: e2304, 2016. doi: 10.1038/cddis.2016.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ren P, Wu D, Appel R, Zhang L, Zhang C, Luo W, Robertson AAB, Cooper MA, Coselli JS, Milewicz DM, Shen YH, LeMaire SA. Targeting the NLRP3 inflammasome with inhibitor mcc950 prevents aortic aneurysms and dissections in mice. J Am Heart Assoc 9: e014044, 2020. doi: 10.1161/JAHA.119.014044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shafik AN, Khattab MA, Osman AH. Magnesium sulfate versus esomeprazole impact on the neonates of preeclamptic rats. Eur J Obstet Gynecol Reprod Biol 225: 236–242, 2018. doi: 10.1016/j.ejogrb.2018.05.004. [DOI] [PubMed] [Google Scholar]
- 47. Swamy V, Setty R, Shankaraiah M, Jyothi T, Rajendra S. A study on drug-drug interaction of esomeprazole and anti-diabetic drugs. J Young Pharm 2: 424–427, 2010. doi: 10.4103/0975-1483.71624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Poudel B, Shields CA, Brown AK, Ekperikpe U, Johnson T, Cornelius DC, Williams JM. Depletion of macrophages slows the early progression of renal injury in obese dahl salt-sensitive leptin receptor mutant rats. Am J Physiol Renal Physiol 318: F1489–F1499, 2020. doi: 10.1152/ajprenal.00100.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. De Miguel C, Pelegrín P, Baroja-Mazo A, Cuevas S. . Emerging role of the inflammasome and pyroptosis in hypertension. Int J Mol Sci 22: 1064, 2021. doi: 10.3390/ijms22031064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu Y, Yin HL, Li C, Jiang F, Zhang SJ, Zhang XR, Li YL. Sinapine thiocyanate ameliorates vascular endothelial dysfunction in hypertension by inhibiting activation of the nlrp3 inflammasome. Front Pharmacol 11: 620159, 2020. doi: 10.3389/fphar.2020.620159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gao P, Zha Y, Gong X, Qiao F, Liu H. The role of maternal-foetal interface inflammation mediated by nlrp3 inflammasome in the pathogenesis of recurrent spontaneous abortion. Placenta 101: 221–229, 2020. doi: 10.1016/j.placenta.2020.09.067. [DOI] [PubMed] [Google Scholar]
- 52. Lu M, Ma F, Xiao J, Yang L, Li N, Chen D. . NLRP3 inflammasome as the potential target mechanism and therapy in recurrent spontaneous abortions. Mol Med Rep 19: 1935–1941, 2019. doi: 10.3892/mmr.2019.9829. [DOI] [PubMed] [Google Scholar]
- 53. Zhu D, Zou H, Liu J, Wang J, Ma C, Yin J, Peng X, Li D, Yang Y, Ren Y, Zhang Z, Zhou P, Wang X, Cao Y, Xu X. Inhibition of hmgb1 ameliorates the maternal-fetal interface destruction in unexplained recurrent spontaneous abortion by suppressing pyroptosis activation. Front Immunol 12: 782792, 2021. doi: 10.3389/fimmu.2021.782792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xu M, Wang L, Wang M, Wang H, Zhang H, Chen Y, Wang X, Gong J, Zhang JJ, Adcock IM, Chung KF, Li F. Mitochondrial ROS and NLRP3 inflammasome in acute ozone-induced murine model of airway inflammation and bronchial hyperresponsiveness. Free Radic Res 53: 780–790, 2019. doi: 10.1080/10715762.2019.1630735. [DOI] [PubMed] [Google Scholar]
- 55. Hastie R, Brownfoot FC, Pritchard N, Hannan NJ, Cannon P, Nguyen V, Palmer K, Beard S, Tong S, Kaitu'u-Lino TJ. EGFR (epidermal growth factor receptor) signaling and the mitochondria regulate sFlt-1 (soluble FMS-like tyrosine kinase-1) secretion. Hypertension 73: 659–670, 2019. doi: 10.1161/HYPERTENSIONAHA.118.12300. [DOI] [PubMed] [Google Scholar]
- 56. Vaka VR, McMaster KM, Cunningham MW Jr, Ibrahim T, Hazlewood R, Usry N, Cornelius DC, Amaral LM, LaMarca B. Role of mitochondrial dysfunction and reactive oxygen species in mediating hypertension in the reduced uterine perfusion pressure rat model of preeclampsia. Hypertension 72: 703–711, 2018. doi: 10.1161/HYPERTENSIONAHA.118.11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bruder-Nascimento T, Ferreira NS, Zanotto CZ, Ramalho F, Pequeno IO, Olivon VC, Neves KB, Alves-Lopes R, Campos E, Silva CA, Fazan R, Carlos D, Mestriner FL, Prado D, Pereira FV, Braga T, Luiz JP, Cau SB, Elias PC, Moreira AC, Câmara NO, Zamboni DS, Alves-Filho JC, Tostes RC. Nlrp3 inflammasome mediates aldosterone-induced vascular damage. Circulation 134: 1866–1880, 2016. doi: 10.1161/CIRCULATIONAHA.116.024369. [DOI] [PubMed] [Google Scholar]
- 58. Grebe A, Hoss F, Latz E. Nlrp3 inflammasome and the il-1 pathway in atherosclerosis. Circ Res 122: 1722–1740, 2018. doi: 10.1161/CIRCRESAHA.118.311362. [DOI] [PubMed] [Google Scholar]
- 59. Bai BC, Yang YY, Wang Q, Li M, Tian C, Liu Y, Aung LHH, Li PF, Yu T, Chu XM. Nlrp3 inflammasome in endothelial dysfunction. Cell Death Dis 11: 776, 2020. doi: 10.1038/s41419-020-02985-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 7: 99–109, 2009. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wallace K, Cornelius DC, Scott J, Heath J, Moseley J, Chatman K, LaMarca B. Cd4+ t cells are important mediators of oxidative stress that cause hypertension in response to placental ischemia. Hypertension 64: 1151–1158, 2014. doi: 10.1161/HYPERTENSIONAHA.114.03590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sanchez-Aranguren LC, Prada CE, Riaño-Medina CE, Lopez M. Endothelial dysfunction and preeclampsia: Role of oxidative stress. Front Physiol 5: 372, 2014. doi: 10.3389/fphys.2014.00372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Matsubara K, Higaki T, Matsubara Y, Nawa A. Nitric oxide and reactive oxygen species in the pathogenesis of preeclampsia. Int J Mol Sci 16: 4600–4614, 2015. doi: 10.3390/ijms16034600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Guerby P, Tasta O, Swiader A, Pont F, Bujold E, Parant O, Vayssiere C, Salvayre R, Negre-Salvayre A. Role of oxidative stress in the dysfunction of the placental endothelial nitric oxide synthase in preeclampsia. Redox Biol 40: 101861, 2021. doi: 10.1016/j.redox.2021.101861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cornelius DC, Hogg JP, Scott J, Wallace K, Herse F, Moseley J, Wallukat G, Dechend R, LaMarca B. Administration of interleukin-17 soluble receptor c suppresses TH17 cells, oxidative stress, and hypertension in response to placental ischemia during pregnancy. Hypertension 62: 1068–1073, 2013. doi: 10.1161/HYPERTENSIONAHA.113.01514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cunningham MW, Jayaram A, Deer E, Amaral LM, Vaka VR, Ibrahim T, Cornelius DC, LaMarca B. Tumor necrosis factor alpha (tnf-alpha) blockade improves natural killer cell (nk) activation, hypertension, and mitochondrial oxidative stress in a preclinical rat model of preeclampsia. Hypertens Pregnancy 39: 399–404, 2020. doi: 10.1080/10641955.2020.1793999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Travis OK, Tardo GA, Giachelli C, Siddiq S, Nguyen HT, Crosby MT, Johnson T, Brown AK, Williams JM, Cornelius DC. Tumor necrosis factor-alpha blockade improves uterine artery resistance, maternal blood pressure, and fetal growth in placental ischemic rats. Pregnancy Hypertens 25: 39–47, 2021. doi: 10.1016/j.preghy.2021.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Travis OK, Tardo GA, Giachelli C, Siddiq S, Nguyen HT, Crosby MT, Johnson TD, Brown AK, Booz GW, Smith AN, Williams JM, Cornelius DC. Interferon gamma neutralization reduces blood pressure, uterine artery resistance index, and placental oxidative stress in placental ischemic rats. Am J Physiol Regul Integr Comp Physiol 321: R112–R124, 2021. doi: 10.1152/ajpregu.00349.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Clark SE, Schmidt RL, McDermott DS, Lenz LL. A batf3/nlrp3/il-18 axis promotes natural killer cell il-10 production during listeria monocytogenes infection. Cell Rep 23: 2582–2594, 2018. doi: 10.1016/j.celrep.2018.04.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Dagenais M, Saleh M. Linking cancer-induced nlrp3 inflammasome activation to efficient nk cell-mediated immunosurveillance. Oncoimmunology 5: e1129484, 2016. doi: 10.1080/2162402X.2015.1129484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Morales Y, Miller EA, Shecter I, Fitzgerald KA, Golenbock DT, Stadecker M, Kalantari P. NLRP3 and AIM2 inflammasome-triggered TH17 cell response promotes severe immunopathology in schistosomiasis. J Immunol 206: 112.07, 2021. doi: 10.4049/jimmunol.206.Supp.112.07. [DOI] [Google Scholar]
- 72. Ohayon DE, Ali A, Alarcon PC, Krishnamurthy D, Osterburg AR, Borchers MT, Waggoner SN. Interleukin-33 modulates human natural killer cell responses. J Immunol 200: 121, 2018. doi: 10.4049/jimmunol.200.Supp.164.21. [DOI] [Google Scholar]
- 73. Palmieri V, Ebel J-F, Ngo Thi Phuong N, Klopfleisch R, Vu VP, Adamczyk A, Zöller J, Riedel C, Buer J, Krebs P, Hansen W, Pastille E, Westendorf AM. Interleukin-33 signaling exacerbates experimental infectious colitis by enhancing gut permeability and inhibiting protective th17 immunity. Mucosal Immunology 14: 923–936, 2021. doi: 10.1038/s41385-021-00386-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cornelius DC, Amaral LM, Wallace K, Campbell N, Thomas AJ, Scott J, Herse F, Wallukat G, Dechend R, LaMarca B. Reduced uterine perfusion pressure t-helper 17 cells cause pathophysiology associated with preeclampsia during pregnancy. Am J Physiol Regul Integr Comp Physiol 311: R1192–R1199, 2016. doi: 10.1152/ajpregu.00117.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Vaka VR, McMaster KM, Cornelius DC, Ibrahim T, Jayaram A, Usry N, Cunningham MW Jr, Amaral LM, LaMarca B. Natural killer cells contribute to mitochondrial dysfunction in response to placental ischemia in reduced uterine perfusion pressure rats. Am J Physiol Regul Integr Comp Physiol 316: R441–R447, 2019. doi: 10.1152/ajpregu.00279.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Travis OK, Baik C, Tardo GA, Amaral L, Jackson C, Greer M, Giachelli C, Ibrahim T, Herrock OT, Williams JM, Cornelius DC. Adoptive transfer of placental ischemia-stimulated natural killer cells causes a preeclampsia-like phenotype in pregnant rats. Am J Reprod Immunol 85: e13386, 2021. doi: 10.1111/aji.13386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Nunes PR, Mattioli SV, Sandrim VC. NLRP3 activation and its relationship to endothelial dysfunction and oxidative stress: implications for preeclampsia and pharmacological interventions. Cells 10: 2828, 2021. doi: 10.3390/cells10112828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Al-Aly Z, Maddukuri G, Xie Y. Proton pump inhibitors and the kidney: Implications of current evidence for clinical practice and when and how to deprescribe. Am J Kidney Dis 75: 497–507, 2020. doi: 10.1053/j.ajkd.2019.07.012. [DOI] [PubMed] [Google Scholar]
- 79. Peng YC, Lin CL, Yeh HZ, Chang CS, Wu YL, Kao CH. Association between the use of proton pump inhibitors and the risk of esrd in renal diseases: a population-based, case-control study. Medicine (Baltimore) 95: e3363, 2016. doi: 10.1097/MD.0000000000003363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Cayrol C, Girard JP. Interleukin-33 (il-33): a nuclear cytokine from the il-1 family. Immunol Rev 281: 154–168, 2018. doi: 10.1111/imr.12619. [DOI] [PubMed] [Google Scholar]
- 81. Dwyer GK, D'Cruz LM, Turnquist HR. Emerging functions of IL-33 in homeostasis and immunity. Annu Rev Immunol 40: 15–43, 2022. doi: 10.1146/annurev-immunol-101320-124243. [DOI] [PubMed] [Google Scholar]
- 82. Shakerian L, Kolahdooz H, Garousi M, Keyvani V, Kamal Kheder R, Abdulsattar Faraj T, Yazdanpanah E, Esmaeili S-A. Il-33/st2 axis in autoimmune disease. Cytokine 158: 156015, 2022. doi: 10.1016/j.cyto.2022.156015. [DOI] [PubMed] [Google Scholar]
- 83. Miller AM. Role of il-33 in inflammation and disease. J Inflamm (Lond) 8: 22, 2011. doi: 10.1186/1476-9255-8-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Matta BM, Lott JM, Mathews LR, Liu Q, Rosborough BR, Blazar BR, Turnquist HR. IL-33 is an unconventional alarmin that stimulates il-2 secretion by dendritic cells to selectively expand il-33r/st2+ regulatory t cells. J Immunol 193: 4010–4020, 2014. doi: 10.4049/jimmunol.1400481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Pascual-Reguant A, Bayat Sarmadi J, Baumann C, Noster R, Cirera-Salinas D, Curato C, Pelczar P, Huber S, Zielinski CE, Löhning M, Hauser AE, Esplugues E. Th17 cells express St2 and are controlled by the alarmin il-33 in the small intestine. Mucosal Immunol 10: 1431–1442, 2017. doi: 10.1038/mi.2017.5. [DOI] [PubMed] [Google Scholar]
- 86. Schiering C, Krausgruber T, Chomka A, Fröhlich A, Adelmann K, Wohlfert EA, Pott J, Griseri T, Bollrath J, Hegazy AN, Harrison OJ, Owens BMJ, Löhning M, Belkaid Y, Fallon PG, Powrie F. The alarmin il-33 promotes regulatory t-cell function in the intestine. Nature 513: 564–568, 2014. doi: 10.1038/nature13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Guerin LR, Prins JR, Robertson SA. Regulatory t-cells and immune tolerance in pregnancy: a new target for infertility treatment? Hum Reprod Update 15: 517–535, 2009. doi: 10.1093/humupd/dmp004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wang W, Sung N, Gilman-Sachs A, Kwak-Kim J. T helper (th) cell profiles in pregnancy and recurrent pregnancy losses: Th1/th2/th9/th17/th22/tfh cells. Front Immunol 11: 2025, 2020. doi: 10.3389/fimmu.2020.02025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Stodle GS, Silva GB, Tangeras LH, Gierman LM, Nervik I, Dahlberg UE, Sun C, Aune MH, Thomsen LCV, Bjørge L, Iversen AC. Placental inflammation in pre-eclampsia by nod-like receptor protein (nlrp)3 inflammasome activation in trophoblasts. Clin Exp Immunol 193: 84–94, 2018. doi: 10.1111/cei.13130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Mullard A. Nlrp3 inhibitors stoke anti-inflammatory ambitions. Nat Rev Drug Discov 18: 405–407, 2019. doi: 10.1038/d41573-019-00086-9. [DOI] [PubMed] [Google Scholar]
- 91. Corcoran SE, Halai R, Cooper MA. Pharmacological inhibition of the nod-like receptor family pyrin domain containing 3 inflammasome with mcc950. Pharmacol Rev 73: 968–1000, 2021. doi: 10.1124/pharmrev.120.000171. [DOI] [PubMed] [Google Scholar]
- 92. Binder NK, Brownfoot FC, Beard S, Cannon P, Nguyen TV, Tong S, Kaitu'u-Lino TJ, Hannan NJ. Esomeprazole and sulfasalazine in combination additively reduce sFlt-1 secretion and diminish endothelial dysfunction: Potential for a combination treatment for preeclampsia. Pregnancy Hypertens 22: 86–92, 2020. doi: 10.1016/j.preghy.2020.07.013. [DOI] [PubMed] [Google Scholar]
- 93. Gu S, Zhou C, Pei J, Wu Y, Wan S, Zhao X, Hu J, Hua X. Esomeprazole inhibits hypoxia/endothelial dysfunction-induced autophagy in preeclampsia. Cell Tissue Res 388: 181–194, 2022. doi: 10.1007/s00441-022-03587-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Onda K, Tong S, Beard S, Binder N, Muto M, Senadheera SN, Parry L, Dilworth M, Renshall L, Brownfoot F, Hastie R, Tuohey L, Palmer K, Hirano T, Ikawa M, Kaitu'u-Lino T, Hannan NJ. Proton pump inhibitors decrease soluble FMS-like tyrosine kinase-1 and soluble endoglin secretion, decrease hypertension, and rescue endothelial dysfunction. Hypertension 69: 457–468, 2017. [Erratum in: Hypertension 71: e11, 2018]. doi: 10.1161/HYPERTENSIONAHA.116.08408. [DOI] [PubMed] [Google Scholar]
- 95. Hastie R, Bergman L, Cluver CA, Wikman A, Hannan NJ, Walker SP, Wikström AK, Tong S, Hesselman S. Proton pump inhibitors and preeclampsia risk among 157 720 women. Hypertension 73: 1097–1103, 2019. doi: 10.1161/HYPERTENSIONAHA.118.12547. [DOI] [PubMed] [Google Scholar]
- 96. Cluver CA, Hannan NJ, van Papendorp E, Hiscock R, Beard S, Mol BW, Theron GB, Hall DR, Decloedt EH, Stander M, Adams KT, Rensburg M, Schubert P, Walker SP, Tong S. Esomeprazole to treat women with preterm preeclampsia: A randomized placebo controlled trial. Am J Obstet Gynecol 219: 388.e1–388.e17, 2018. doi: 10.1016/j.ajog.2018.07.019. [DOI] [PubMed] [Google Scholar]
- 97. Atwa KA, Ibrahim ZM, Elshaer M, Taha OT, Aboelroose AA. Role of esomeprazole in early preeclampsia: A randomized controlled trial. Austin J Obstet Gynecol 8: 1189, 2021. doi: 10.26420/austiniobstetgynecol.2021.1189. [DOI] [Google Scholar]
- 98. Paauw ND, Joles JA, Spradley FT, Bakrania B, Zsengeller ZK, Franx A, Verhaar MC, Granger JP, Lely AT. Exposure to placental ischemia impairs postpartum maternal renal and cardiac function in rats. Am J Physiol Regul Integr Comp Physiol 312: R664–R670, 2017. doi: 10.1152/ajpregu.00510.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Llinás MT, Alexander BT, Capparelli MF, Carroll MA, Granger JP. Cytochrome p-450 inhibition attenuates hypertension induced by reductions in uterine perfusion pressure in pregnant rats. Hypertension 43: 623–628, 2004. doi: 10.1161/01.HYP.0000117721.83371.9f. [DOI] [PubMed] [Google Scholar]
- 100. He Y, Hara H, Nunez G. Mechanism and regulation of nlrp3 inflammasome activation. Trends Biochem Sci 41: 1012–1021, 2016. doi: 10.1016/j.tibs.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Kim YG, Kim SM, Kim KP, Lee SH, Moon JY. The role of inflammasome-dependent and inflammasome-independent nlrp3 in the kidney. Cells 8: 1389, 2019. doi: 10.3390/cells8111389. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figs. S1–S9: https://doi.org/10.6084/m9.figshare.21675140.
Data Availability Statement
Data will be made available upon reasonable request.