

Keywords: acute lung inflammation, interstitial macrophage, macrophage subsets, RNA sequencing
Abstract
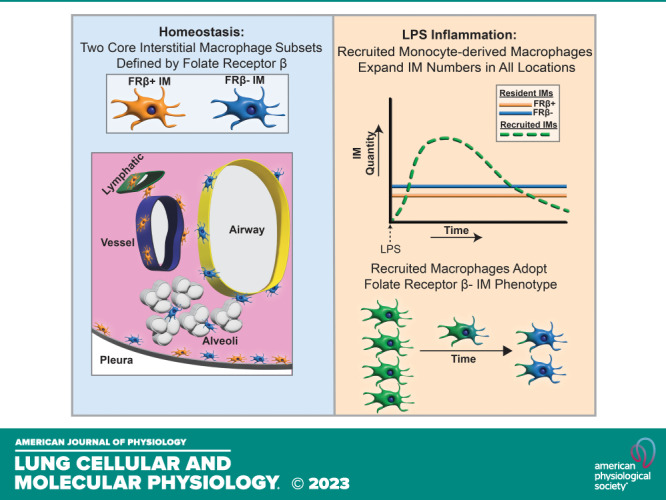
Interstitial macrophages (IMs) reside in the lung tissue surrounding key structures including airways, vessels, and alveoli. Recent work has described IM heterogeneity during homeostasis, however, there are limited data on IMs during inflammation. We sought to characterize IM origin, subsets, and transcriptomic profiles during homeostasis and lipopolysaccharide (LPS) induced acute lung inflammation. During homeostasis, we used three complementary methods, spectral flow cytometry, single-cell RNA-sequencing, and gene regulatory network enrichment, to demonstrate that IMs can be divided into two core subsets distinguished by surface and transcriptional expression of folate receptor β (Folr2/FRβ). These subsets inhabited distinct niches within the lung interstitium. Within FRβ+ IMs we identified a subpopulation marked by coexpression of LYVE1. During acute LPS-induced inflammation, lung IM numbers expand. Lineage tracing revealed IM expansion was due to recruitment of monocyte-derived IMs. At the peak of inflammation, recruited IMs were comprised two unique subsets defined by expression of genes associated with interferon signaling and glycolytic pathways. As recruited IMs matured, they adopted the overall transcriptional state of FRβ− resident IMs but retained expression in several origin-specific genes, such as IL-1β. FRβ+ IMs were of near-pure resident origin. Taken together our data show that during LPS-induced inflammation, there are distinct populations of IMs that likely have unique functions. FRΒ+ IMs comprise a stable, resident population, whereas FRβ− ΙΜs represent a mixed population of resident and recruited IMs.
INTRODUCTION
Macrophages serve crucial roles by providing host defense, clearing debris, regulating inflammatory responses, and promoting tissue repair (1). In the lung, macrophages can be broadly defined as airspace macrophages (AMs) versus. interstitial macrophages (IMs) based on the compartments in which they reside. During health, resident AM and IM pools are maintained in constant numbers via self-replication with minimal contribution from the bone marrow and circulation. However, during inflammation, both AM and IM numbers increase substantially (2–4). In the airspace, this is driven by recruited monocyte-derived AMs that are transcriptionally and functionally distinct from resident AMs (5–7). However, it is unclear whether the IM expansion during inflammation results from recruitment of monocyte-derived macrophages, proliferative expansion of resident IMs, or both.
It is clear that subsets of lung IMs are present during health (2, 8–11). However, our ability to study the functions of these cells has been hampered by lack of consensus on the best identification strategy (12). Whether novel IM subsets with unique functions arise during acute inflammation is unknown. Thus, an improved understanding of IM heterogeneity during inflammation would help to understand subset-specific roles and could lead to novel therapeutic targets for inflammatory lung diseases.
Recent methodologic advances allow us to study lung IMs during inflammation. This enables us to 1) distinguish recruited IMs using a lineage tracing model (13) and 2) separate IMs from recruited AMs using intratracheal antibody instillation (14). In addition, innovations in RNA-sequencing and spectral flow cytometry allow us to assess for multiple defining IM population markers simultaneously. Here we leverage these tools to understand IM origin and heterogeneity during homeostasis and acute inflammation after intratracheal instillation of lipopolysaccharide (LPS).
Given the lack of consensus regarding IM subpopulations at homeostasis, we initially evaluated IMs during their steady state. Clustering algorithms applied to spectral flow cytometry and single-cell RNA-seq (scRNA-seq) based platforms identified two major IM subgroups during homeostasis. Although a number of molecules distinguished these subsets, the most robust was folate receptor β (FRβ). Using a lineage tracing system, we found that IM expansion after intratracheal LPS was driven by accumulation of monocyte-derived recruited IMs. Resident IMs remained constant in number. ScRNA-seq demonstrated that recruited IMs had unique programming during peak inflammation, which included gene expression of many monocyte-related genes like Ly6c2 and unique interferon-responsive gene expression. The recruited IMs rapidly adopted the transcriptional state of FRβ− resident IMs as inflammation resolved and did not convert to FRβ+ IMs. In sum, our data define the heterogeneity of IMs in both health and acute inflammation and present a framework by which IM subsets may be identified for further study.
MATERIALS AND METHODS
Animals
All experiments were approved by the Institutional Animal Care and Use Committee at National Jewish Health. Mice were obtained from Jackson Laboratories (Bar Harbor, ME) and bred at National Jewish Health. Wild-type C57BL/6 mice were used for spectral flow cytometry experiments. For all other experiments, CX3CR1-ERT2 (B6.129P2(C)-CX3CR1tm2.1(cre/ERT2)Jung/J) mice were crossed with R26-stopfl/fl-TdTomato (B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J) mice. Experimental animals from this cross were hemizygous for both genes. Both male and female mice were used in these experiments unless otherwise stated in figure legends.
Tamoxifen Administration
Tamoxifen (Sigma Pharmaceuticals, North Liberty, IA) was suspended in corn oil and administered at 0.25 mg/g of body weight by intraperitoneal injection on days 1, 4, and 7 (pulse) at 10–12 wk of age for lineage tracing experiments. Experiments were performed after a 4-wk wait period.
LPS Administration
LPS (Escherichia coli O55:B5; List Biological Laboratories, Campbell, CA) was administered intratracheally using a 200 μL pipet tip at a dose of 20 μg in 50 μL of PBS with direct laryngoscopy performed under sedation with isoflurane (Baxter, Deerfield IL).
Tissue Harvest for Flow Cytometry
Mice were euthanized with intraperitoneal Fatal Plus (Vortech Pharmaceuticals, Dearborn, MI). To label cells in the airspaces, anti-CD45 antibody (1 mL total at 2 μg/mL concentration) was instilled via 18-gauge catheter in the trachea immediately after euthanasia. After 4 min of dwell time, anti-CD45 solution was aspirated and bronchoalveolar lavage (BAL) was performed with 5 consecutive lavages of 1 mL of PBS containing 0.5 mM EDTA. BAL fluid was pelleted and resuspended in HBSS containing 0.3% BSA and 0.3 mM EDTA. Lungs were perfused by injecting 10 mL of PBS through the right ventricle during which lungs visibly blanched. Lung tissue was finely chopped with a razor blade and then incubated at 37 C in 1 mL of 0.4 mg/mL Liberase TM (Roche, Indianapolis, IN) for 25 min. Following incubation, tissue was pipetted rapidly up and down to further disaggregate, then filtered through 100 μm filters. Lung cells were pelleted and resuspended in then filter through 100 μm and 70 μm filters then resuspended HBSS containing 0.3% BSA and 0.3 mM EDTA.
Flow Cytometry
Flow cytometry was performed on single-cell suspensions as described previously (8, 13, 14). Lung digestates were run separately from BAL cells for flow cytometry experiments. Lung digestate and BAL were pooled together for fluorescence-activated cell sorting (FACS) before single-cell RNA-sequencing experiments. Cells were protected from light and incubations were performed on ice. Single-cell suspensions were treated with unlabeled CD16/CD32 for 15 min to block non-specific FcyR-mediated binding then were stained with surface antibody panels for 1 h, and then washed. The gating strategies are outlined in Supplemental Figs. S1A and S4A (all Supplemental material is available at https://doi.org/10.6084/m9.figshare.20315406.v1). CD88 was used to distinguish macrophages from dendritic cells (15). Antibody manufacturer, clone numbers, and concentrations are available in Supplemental Table S1. Spectral cytometry was performed using the Aurora cytometer (Cytek Biosciences, Fremont, CA), conventional flow cytometry experiments using the LSRFortessa cytometer (BD Biosciences, San Jose, CA), and FACS using the SY3200 cell sorter (Sony Biotechnology, Champaign, IL). All cytometry data were analyzed using FlowJo software (Tree Star, Ashland, OR). Uniform Manifold Approximation and Projection (UMAP) plug-in for FlowJo was used for dimensional reduction of spectral cytometry data with Euclidean metrics, nearest neighbors set at 15, minimum distance of 0.5 using the following parameters: FRβ, CD206, LYVE1, CCR2, CD11c, CD11b, CX3CR1, MHCII, and Ly6C. Antibody specificity was not independently validated for these commercially available antibodies, but unstained, single-stain, and fluorescence minus one controls were used to define gating for positive and negative populations.
Single-Cell RNA Isolation and Sequencing
Cells from mice (n = 3–4/timepoint) were individually sorted using FACS into three bins: 1) CD45+ cells (live, CD45+), 2) resident IMs (live, CD45+, CD64+, CD88+, IT CD45−, tdT+), or 3) recruited IM (live, CD45+, CD64+, CD88+, IT CD45−, tdT−). Cells were manually counted using a hemocytometer and resuspended at 1 × 106 cells/mL in 0.06% BSA in PBS solution.
Single cells were captured using a Chromium Box (10× Genomics, Pleasanton, CA). Libraries were sequenced using the Illumina NovaSEQ 6000 sequencer (Illumina, Inc., San Diego, CA). Sequencing was targeted to 5,000 cells in the CD45+ library and 3,000 cells in each of the IM sorts with sequencing depth of 100,000 reads/cell. The transcriptional data are publicly accessible in the Gene Expression Omnibus (GSE218884).
Single-Cell RNA-Sequencing Data Analysis
Clustering was performed using Seurat (16) and single-cell regulatory network inference and clustering (SCENIC) (17). Specifics of scRNA-seq computational analysis and data normalization are described in the appendix.
Immunofluorescence of Frozen Lung Tissue
Male pulse-wait resident IM reporter mice (CX3CR1ERT2-Cre x R26Stop(fl/fl)tdTomato) were euthanized at homeostasis or 6 days after LPS (n = 3/group). Lungs were perfused and lavaged as described under Tissue Harvest for Flow Cytometry and were inflated with 1 mL of 1% PFA, 12.5% sucrose solution, and 50% optimal cutting temperature (OCT) compound (Fisher Healthcare, Houston TX), as previously described (8). Tracheas were tied off and lungs were removed, mounted in OCT, and frozen. 10-μm sections were stained with primary antibodies overnight at 4°C then incubated with secondary antibodies for 1 h at room temperature. TrueVIEW (Vector, Newark CA) was applied before mounting. Whole slide scans were performed on the VS200 Slide Scanner (Olympus, Tokyo, Japan) at ×20 magnification. Confocal images were obtained on the Zeiss LSM 700-Laser Scanning Confocal (Zeiss, Oberkochen, Germany) using z-stacking with 1-μm sections. Image brightness and contrast were adjusted in Olympus OlyVIA software (for images on VS200) and on ImageJ (https://imagej.net). All images were compared with corresponding isotype controls. Antibody manufacturer and clone are listed in Supplemental Table S1.
Statistical Analysis of Conventional Flow Cytometry
Statistical analysis was performed using Prism software (GraphPad Software, Inc., San Diego, CA). Statistical analyses were performed using two-tailed ANOVA and Tukey’s multiple comparisons test. A P value less than 0.05 was considered statistically significant. All bar graphs are shown as means (±SE).
RESULTS
Flow Cytometry Distinguishes Two Core IM Subsets at Homeostasis Defined by Folate Receptor β Surface Expression
To determine an optimal set of cell surface markers to distinguish IM subtypes during homeostasis, we assessed expression of eight putative cell surface markers identified from literature (2, 8–12) using spectral flow cytometry. The panel was applied to lung digestates and data from four mice were concatenated (Supplemental Fig. S1A). Dimensional reduction of IMs was performed with Uniform Manifold Approximation and Projection (UMAP) to probe for subpopulations. UMAP plots revealed distinct populations of IMs (Fig. 1A).
Figure 1.
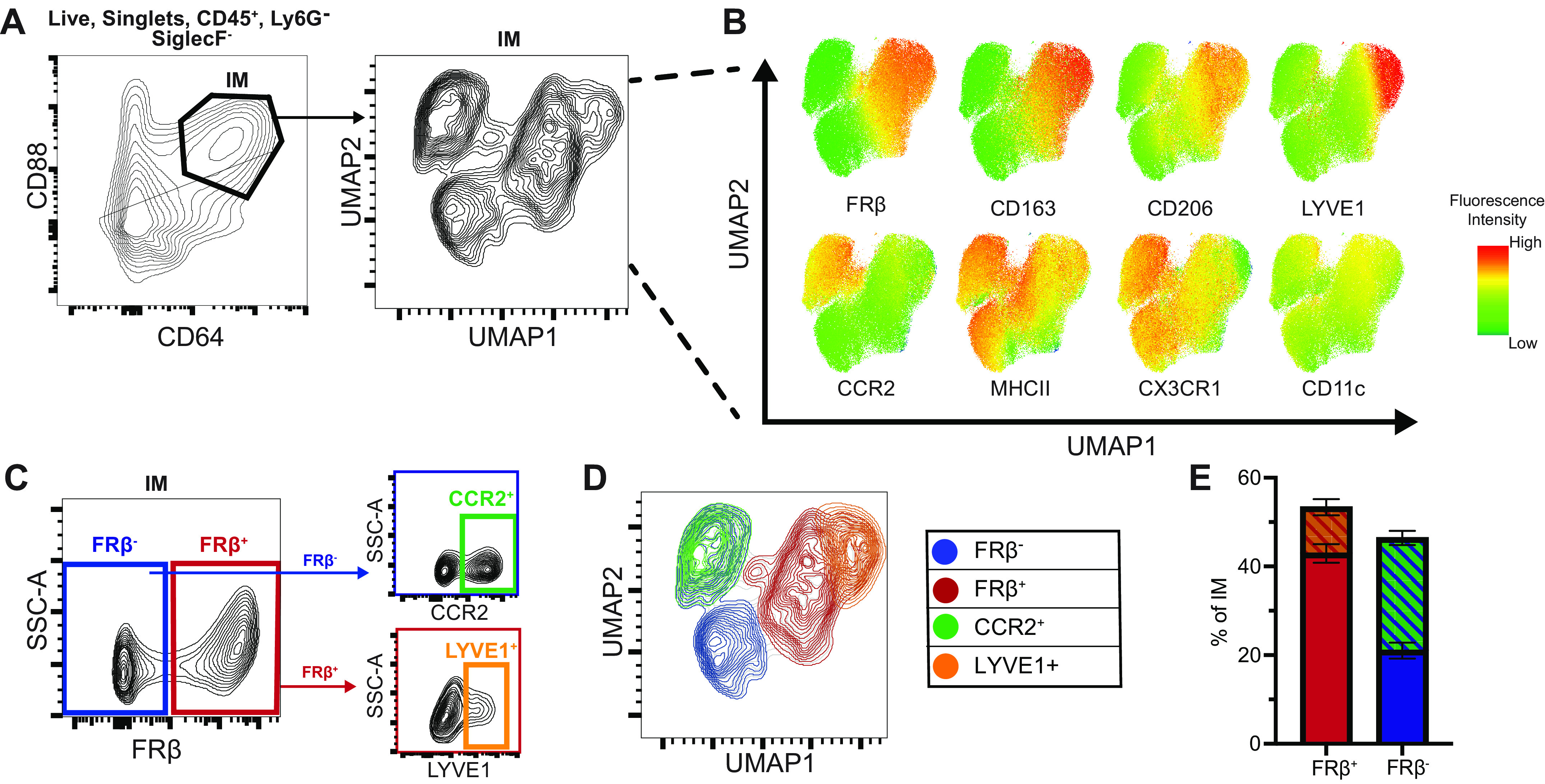
Spectral flow cytometry of murine lung IMs at homeostasis identifies core binary identity defined by FRβ expression. Lung digests from 10-wk-old male mice (n = 4) were assessed by spectral flow cytometry and concatenated for analysis. A: 93,000 IMs underwent dimensional reduction using Uniform Manifold Approximation and Projection (UMAP) of eight macrophage markers. B: fluorescence intensity heatmaps of individual surface markers overlaid on UMAP plot. C: gating strategy to identify FRβ+ and FRβ− IMs, FRβ−CCR2+ IMs and FRβ+LYVE1+ IMs. D: four populations from C overlaid on UMAP from A. E: proportions of total IMs for each subpopulation identified in C. FRβ, folate receptor β; IM, interstitial macrophage.
Folate receptor β (FRβ) strongly defined UMAP1, dichotomizing IMs into two major subsets (Fig. 1B). FRβ+ IMs (53.3% ± 3.1%) could be further divided into LYVE1− (42.9% ± 2.1%) and a rare LYVE1+ (10.4% ± 1.8%) subset, whereas FRβ− IMs (46.7% ± 3.1%) could be divided based on CCR2 (CCR2+ 25.4% ± 1.6%, CCR2− 21.0% ± 1.8%) (Fig. 1C). The gating strategies used to identify these four subsets are shown in Fig. 1, C–E.
Expression of CD163 and high expression of CD206 were also efficacious in distinguishing the same subsets as FRβ (Fig. 1B, Supplemental Fig. S1, C–E). However, we favor FRβ because CD206 has some expression on all IMs making it more difficult to define a positive and negative population and CD163 can be cleaved from the cell surface by ADAM17 (18, 19), which is known to increase during acute inflammation. The overlap of CD163 and FRβ with LYVE1 and CCR2 subsets is shown in Supplemental Fig. S1, H and I.
To evaluate how these populations aligned with earlier reported IM subset gating strategies, we overlaid the FRβ+ population with previously defined IM1 (CD206hiMHCIIlo)/IM2 (CD206hiMHCIIhi)/IM3 (CD11chiCD206lo) (8), CD206hi (2), LYVE1+ (9, 11), and CCR2+ (11) subsets on the IM UMAP projection. The FRβ+ population had a similar distribution to IM1/IM2 and CD206hi IMs (Supplemental Fig. S1, B, E, and F) (2, 8). FRβ+ IMs also correlated with the recently described TIM4, LYVE1, and/or FRβ IMs (TLF+) (11). However, the TLF schema masked that LYVE1 marked a unique subpopulation within FRβ+ IMs encompassing a small portion of IMs (Fig. 1E) (9). Finally, CCR2 expression marked a population within FRβ− cells agreeing with prior reports (11). Collectively our data show that surface FRβ expression strongly defines two core IM subsets. Subpopulations expressing LYVE1 or CCR2 can be found within FRβ+ or FRβ- IMs, respectively.
ScRNA-Seq Identifies Two Core IM Subsets during Homeostasis That Can Be Distinguished Based on Folr2 (FRβ) Gene Expression
As a complement to flow cytometry, unbiased assessment of IM subpopulations was performed with scRNA-seq. Because we planned to evaluate IM origin during inflammation in future experiments, we used a well-characterized resident IM lineage tracing model (CX3CR1ERT2-Cre x R26Stop(fl/fl)tdTomato pulse-wait) in which a pulse of tamoxifen followed by a wait period of 4 wk efficiently labels resident IMs with tdTomato (tdT), without labeling Ly6Chi monocytes (13, 20, 21). From each mouse (n = 3), cells were sorted into three separate lanes (Supplemental Fig. S2). The first lane included all leukocytes by sorting CD45+ cells. These CD45+ lanes assisted with aligning replicates and ensured that our IM enrichment strategy did not exclude any IM subpopulations. The next two lanes were IM enriched sorts that were separated based on expression of tdT. Our previous characterization of this reporter system demonstrated that a small fraction of resident IMs fails to express tdT. Collecting tdT− IMs enabled us to evaluate for differences between tdT+ and tdT− resident IM populations. Each lane underwent sequencing, quality control, and batch correction before reintegration for data analysis (Fig. 2A).
Figure 2.
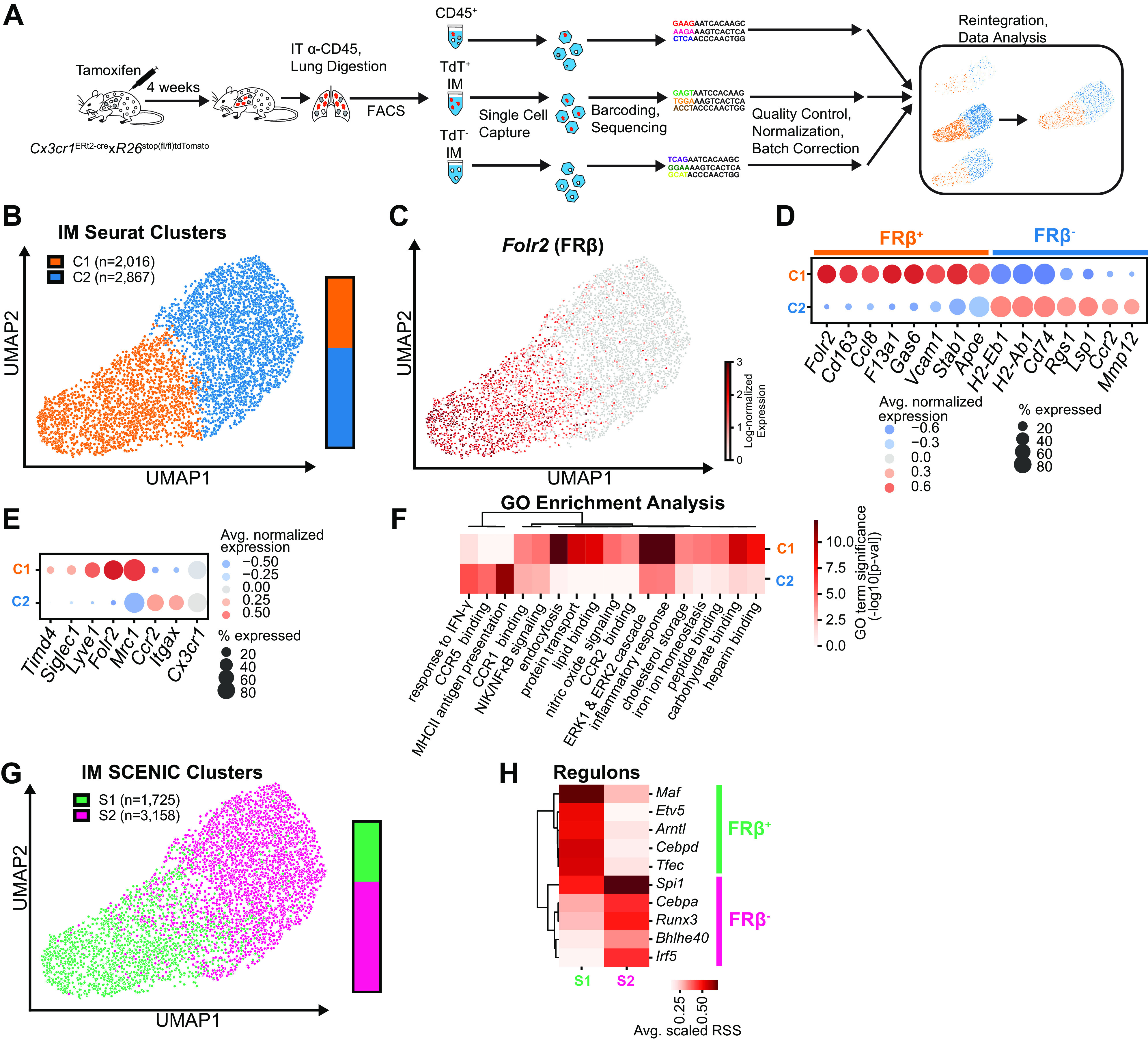
Single-cell RNA-sequencing of murine lung IMs at homeostasis identifies core binary identity defined by Folr2 expression. In anticipation of sequencing IMs during inflammation, A: sequencing was performed on three resident IM lineage tracing male mice (n = 3). CD45+ cells, tdΤ+ IMs, and tdT− IMs were sorted from lung digestates from individual mice, sequenced in parallel, and reintegrated for data analysis. Data from all mice were pooled before clustering and expression analysis. B: UMAP projection of Seurat clustering of all IMs demonstrated two core IM clusters. C: feature plot of Folr2 expression. D: dot plot of highly differentially expressed genes between C1 and C2. X-axis depicts genes and the Y-axis refers to clusters from B. E: dot plot of gene expression in C1 vs. C2 of previously described IM surface markers. F: heatmap of functional enrichment analysis of Gene Ontology (GO) terms comparing C1 vs. C2. G: comparison of primary clusters identified by SCENIC vs. Seurat clustering, with SCENIC clusters overlaid on Seurat UMAP. H: heatmap of top five regulons defining each SCENIC cluster. Avg, average; FACS, fluorescence-activated cell sorting; FRβ, folate receptor β; IM, interstitial macrophage; IT, intratracheal; RSS, regulon specificity score; SCENIC, single-cell regulatory network inference and clustering; tdT, tdTomato; UMAP, Uniform Manifold Approximation and Projection.
A total of 24,023 cells underwent Seurat clustering (Supplemental Fig. S3A). Our use of a generous IM gating strategy during FACS and inclusion of a CD45+ sort meant that other cell types were present in the initial clusters including other myeloid, lymphoid, endothelial, epithelial, and stromal cells (Supplemental Fig. S3B). We eliminated cells from non-IM clusters and reclustered 4,883 IMs by isolating cells with high expression of known IM genes (e.g., C1qa, Mertk, Adgre1, C5ar1) and low expression of alveolar macrophage genes (e.g., Siglecf, Marco), dendritic cell genes (e.g., Zbtb46, Xcr1), or actively replicating genes (e.g., Pclaf2, Top2a) (Supplemental Fig. S3, C–E). TdT− IMs were rare and did not form unique clusters but rather clustered with tdT+ IMs (Supplemental Fig. S3G).
Seurat clustering demonstrated 2 principal IM clusters, which could be distinguished by expression of Folr2 (FRβ) (Fig. 2, B and C). The FRβ+ cluster (C1) had high expression of Cd163 and F13a1 whereas the FRβ− cluster (C2) had higher expression of MHCII-related genes, as well as Ccr2 and Mmp12 (Fig. 2D). We compared our clusters to subset-defining genes published by other groups and found that the FRβ+ cluster (C1) had high expression of CD206+ (Mrc1) (Fig. 2E) (2). We further found that expression of Lyve1 (9, 11), Timd4 (11), and Siglec1 (CD169) (10) were all highly specific to the FRβ+ IM cluster (C1) but were only expressed by a small proportion of cells, suggesting that these markers identify additional subgroups within the FRβ+ population.
There were 1,184 significantly differentially expressed genes (DEGs) between the two IM clusters (Fig. 2D, Supplemental Table S2). Functional enrichment analysis revealed that FRβ− IMs (C2) had amplification in genes associated with antigen presentation (Fig. 2F, Supplemental Table S3). In comparison, FRβ+ IMs (C1) were enriched in pathways associated with endocytosis, chemokine signaling, and binding of proteins, lipids, and carbohydrates (Fig. 2F). Moreover, the two clusters demonstrated differential enrichment in inflammatory response genes and chemokine signaling genes, highlighting that the lung has active immune-related signatures even at homeostasis.
As a complementary method for clustering IMs, we evaluated gene regulatory networks using SCENIC. SCENIC combines expression of transcription factors with genes that have an adjacent binding-motif into a “regulon,” which can be used for clustering of populations (17). Using SCENIC, we identified two clusters of IMs (Fig. 2G). These two clusters correlated with the Seurat clusters and continued to correspond to FRβ expression. The FRβ+ cluster (S1) was defined by high activity of Maf, Etv5, and Cebpd regulons and the FRβ− cluster (S2) was defined by the Irf5, Cebpa, and Runx3 regulons (Fig. 2H, Supplemental Table S4). These may represent lineage-determining transcription factors that define the respective subsets (22).
Although it is well established that IMs arise during embryogenesis and are capable of self-replication (20, 23), growing evidence supports that some IMs are replaced over time by circulating monocytes (2, 8, 11). To determine whether rates of replacement varied between subsets, we took advantage of the fact that tdT expression is equivalent across IM subsets immediately following our tamoxifen pulse regimen (13). After the 4-wk wait period, tdT− IMs were rare, but had a slight predominance within the FRβ− cluster (68% of tdT− in C2 vs. 57% of tdT+ in C2; Supplemental Fig. S3G). In addition, tdT− IMs in the FRβ− cluster (C2) had higher expression of CCR2. This supports prior literature that analogous subsets (IM3, CD206lo, CCR2+) are replaced more readily by circulating monocytes.
Smaller Subsets Can Be Identified within FRβ+ and FRβ− IM Populations
Because we identified four possible IM populations by flow cytometry (Fig. 1D), we increased the resolution of Seurat clustering to evaluate for additional IM heterogeneity (Fig. 3A). When clustering resolution was increased, the FRβ+ cluster was split into an additional cluster (cluster c1), which correlated with Lyve1 gene expression (Fig. 3, A and B).
Figure 3.
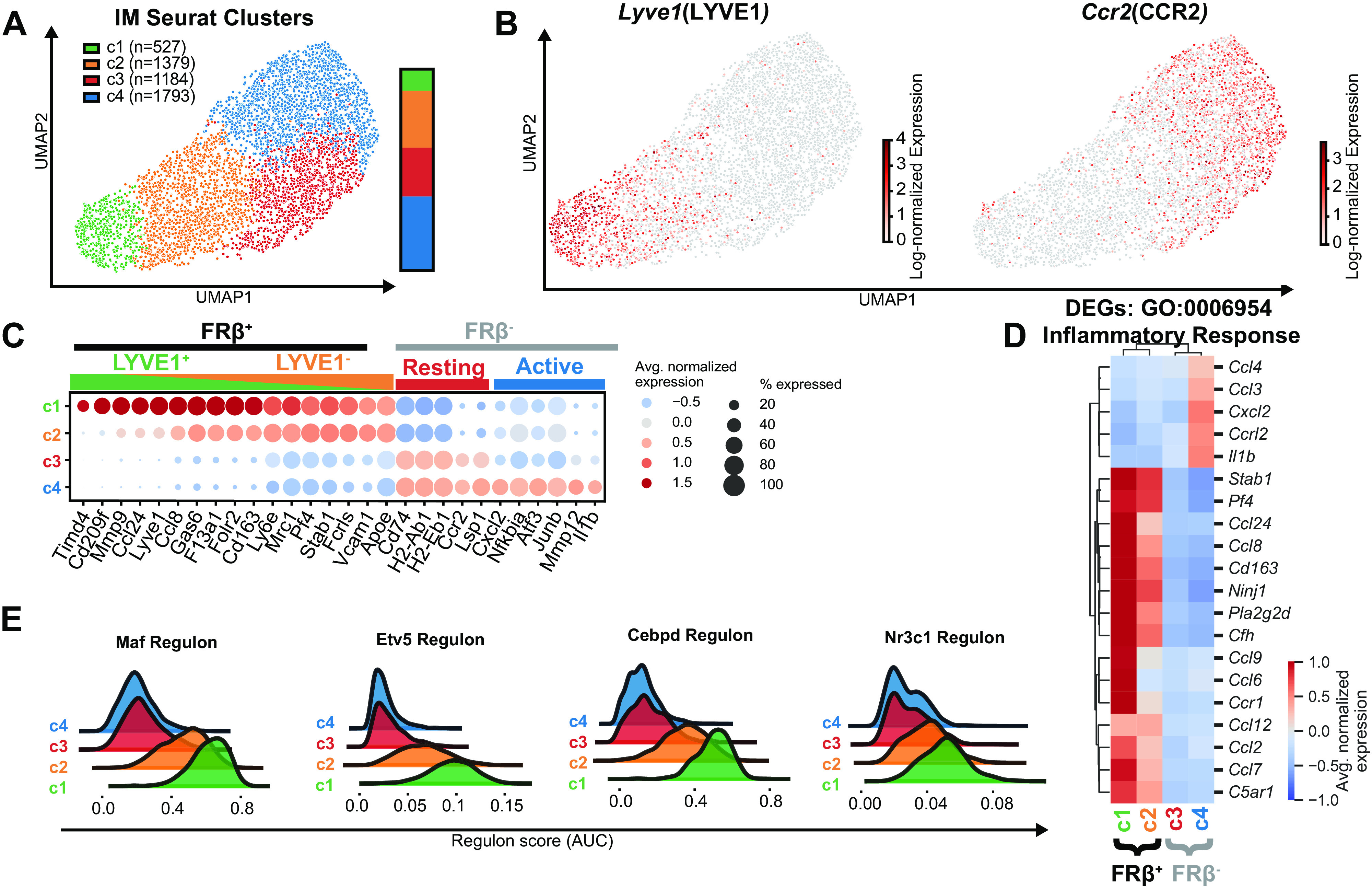
Reclustering murine lung IMs during homeostasis with increased resolution identifies additional IM clusters within FRβ+ and FRβ− subsets. A: four IM subsets identified by clustering with increased resolution. B: feature plot of Lyve1 and Ccr2 expression. C: dot plot of highly differentially expressed genes between the four clusters. D: heatmap of top differentially expressed genes within Gene Ontology (GO) 00069543 Inflammatory Response pathway. E: overlapping histograms of selected SCENIC regulons that define clusters c1 and c2. AUC, area under the curve; Avg, average; DEGs, differentially expressed genes; FRβ, folate receptor β; SCENIC, single-cell regulatory network inference and clustering; UMAP, Uniform Manifold Approximation and Projection.
In addition to expressing its own set of distinct genes (e.g., Timd4, Retnla, CD209f, and Fgfr1), the LYVE1+ subset (c1) had augmented expression of several FRβ+ subset-specific genes (e.g., Gas6 and F13a1) compared with the LYVE1− group(c2) (Fig. 3C, Supplemental Table S5). Enrichment analysis demonstrated that the LYVE1+ population (c1) had amplified gene expression in most of the FRβ+ pathways whereas LYVE1−FRβ+ IMs (c2) had few unique pathways of their own (Supplemental Fig. S3J). For instance, an in-depth evaluation of the Gene Ontology (GO) “inflammatory response” pathway demonstrated that the LYVE1+ cluster (c1) had enhanced expression of chemokine genes compared with the LYVE1−FRβ+ group (c2) (Fig. 3D). Similarly, the LYVE1+ cluster (c1) demonstrated increased activity of FRβ+ defining regulons (e.g., Maf, Cebpd, Etv5, and Nr3c1) when compared with the LYVE1−FRβ+ cluster (c2) (Fig. 3E), but no novel regulons. This gradient of expression of FRβ+ defining genes, pathways, and regulons suggests that the LYVE1+ cluster may represent a more transcriptionally active population of FRβ+ cells.
Unexpectedly, the additional FRβ− cluster (c4) was not defined by Ccr2 as observed with flow cytometry and as described by Dick et al. (11) (Fig. 3, A and B). Instead, clustering appeared to define a FRβ− subset (c4) with higher expression of genes associated with inflammation and matrix remodeling (e.g., Il1b, Nfkbia, Atf3, and Mmp12) and a subset (c3) with few DEGs of its own. Although we have not performed functional studies on these two subsets, for ease we have termed these clusters “active” and “resting,” respectively. Equal and incomplete expression of Ccr2 was observed in both “active” (c4) and “resting” (c3) FRβ− clusters with 50%–60% of cells in each cluster expressing the gene (Fig. 3C). Thus, although CCR2 was useful as a surface marker to define a FRβ− IM population on flow cytometry (Fig. 1C), it did not distinguish a population with unique transcriptional programming using scRNA-seq.
Tissue digestion has been associated with upregulation of stress genes in various cell types. When compared with the “resting” FRβ− cluster (c3), the “active” FRβ− cluster (c4) had upregulation of 36 out of 58 genes that have been demonstrated to increase during warm tissue digestion (Supplemental Fig. S4 and Supplemental Table S6) (24). Although this raises concern that the “active” cluster (c4) may have arisen from technical aspects of IM isolation, many of these stress genes have important immune-related functions and may serve true purposes in homeostatic IMs.
FRβ+ IMs Are Adjacent to Vessels and Lymphatics Whereas FRβ− ΙΜs Are Adjacent to Alveoli and Airways
Given that macrophage programming is influenced by local microenvironment (25), we sought to establish whether different IM subsets inhabited unique locations within the lung. We performed immunofluorescence microscopy of pulse-wait resident IM reporter mice (13). Because CD206 is expressed at some level by all IMs during homeostasis (Supplemental Fig. S3F), resident IMs were identified by coexpression of CD206 and tdT reporter. IMs were abundant in the bronchovascular bundle and the subpleural spaces and less dense in the interalveolar septa (Supplemental Fig. S5, A and B). Although anti-FRβ readily stained FRβ+ IMs by flow cytometry, the same clone was ineffective in our preserved tissue sections. Because CD163 expression closely aligns with FRβ expression (Fig. 1B), we immunostained with CD163 as a surrogate for FRβ+ ΙΜs. CD163+ ΙΜs were abundant adjacent to vessels and lymphatics and were not identified in the alveolar interstitium. In contrast, CD163− IMs were found near airways and in the alveolar interstitium. Both subsets were present in the subpleural regions (Fig. 4, A–F).
Figure 4.
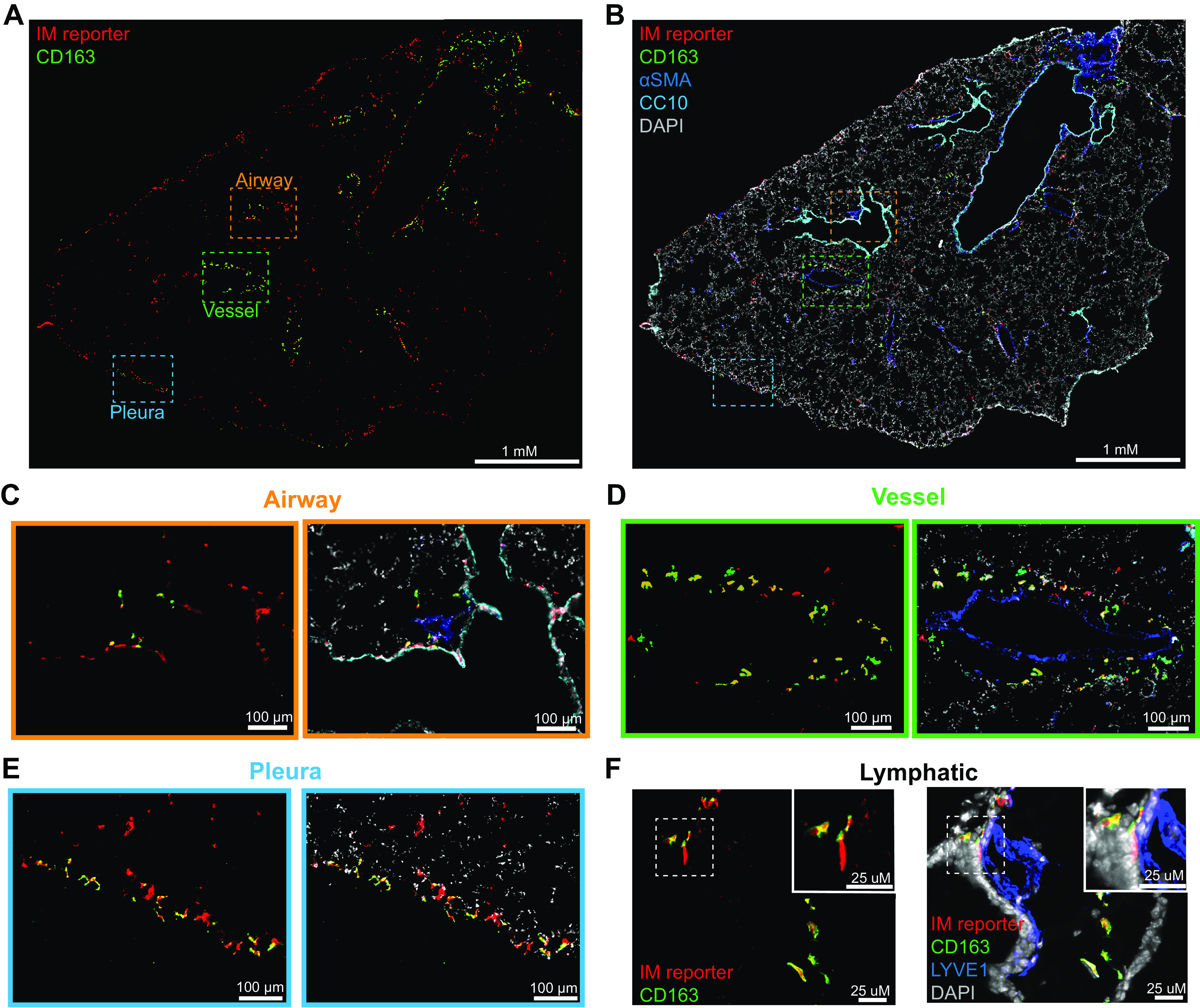
CD163+ IMs are located near vessels, lymphatics, and pleura. CD163− IMs are adjacent to airways, alveoli and pleura. Immunofluorescence microscopy was performed on 10-μm thick frozen sections from male and female pulse-wait resident IM reporter mice (CX3CR1ERT2-Cre x R26Stop(fl/fl)tdTomato) at homeostasis (male mouse shown). CD163 was used to identify FRβ+ IMs. A: whole lobe projection of reporter positive IMs (red) and CD163 (green) imaged at ×20 on scanning microscope from a male mouse. B: airways (CC10/cyan) and vessels (αSMA/blue) and nuclei (DAPI/gray) are highlighted on whole lobe projection. Zoomed projection of IMs near an airway (C), a vessel (D), and the pleura (E). F: maximal intensity projection of 1 μM thick images of CD163 (green) IMs (red) and lymphatics (LYVE1/blue) using confocal microscopy. αSMA, α-smooth muscle actin; CC10, Clara Cell 10 kDa Protein; DAPI, 4′,6-diamidino-2-phenylindole; FRβ, folate receptor β; IM, interstitial macrophage.
Our data corroborate previous reports of subsets similar to CD163+ IMs being located around vessels and lymphatics (2, 9). However, we did not observe CD163+ IMs to be frequent around airways as reported by others (2). Instead CD163− IM predominated around airways, which correlates with previous reports of LYVE1− IMs location adjacent to neurons near airways (9). We could find no prior reports describing IMs at the pleural surface, where we observed both CD163+ and CD163− IMs.
IM Expansion after IT LPS Is Driven by Recruited Monocyte-Derived IMs
Having explored IM heterogeneity during homeostasis, we sought to evaluate whether the underlying mechanism of IM expansion after IT LPS-induced inflammation was due to recruitment of monocyte-derived IMs or expansion of resident IM subsets. To achieve this, we again used our pulse-wait resident IM lineage tracing model (13). To segregate macrophages in the airspaces from those in the interstitium, fluorescently labeled anti-CD45 antibodies were instilled into the airspaces immediately after euthanasia (14). IMs were defined based on their expression of CD64 and CD88 and the absence of intratracheal anti-CD45 labeling (Fig. 5A, Supplemental Fig. S6, A–D).
Figure 5.

Quantification of murine IMs after intratracheal lipopolysaccharide (LP) shows expansion is driven by recruited IMs. Flow cytometry was performed on male and female resident IM reporter mice (CX3CR1ERT2-Cre x R26Stop(fl/fl)tdTomato “pulse-wait”). A: IMs were identified as IT anti-CD45− macrophages. B: representative gating showing separation of resident and recruited IMs by tdT reporter label in IMs. C: bar graph of recruited monocyte-derived (tdT−) IM numbers over time. D: bar graph of resident (tdT+) IM numbers over time. Values shown are means ± SE. Data were analyzed with ANOVA and Tukey’s multiple comparisons test. *P < 0.05. IM, interstitial macrophage; IT, intratracheal; Rec, recruited; Res, resident; tdT, tdTomato.
Recruited IMs (tdT−) predominated in the lung 3 days after LPS and decreased throughout the course of inflammation (Fig. 5B). In comparison resident IMs (tdT+) remained relatively constant in number (Fig. 5C). In these experiments we tracked CD163 surface expression, which aligns closely with FRβ+ expression (Fig. 1B). Over 15 days, we found that recruited IMs almost exclusively remained negative for CD163 (Supplemental Fig. S6, E and F). During inflammation, the percentage of CD163+ resident IMs decreased slightly. Spectral flow cytometry performed on wild-type mice 3 days after LPS demonstrated that almost all IMs express CCR2 whereas FRβ is present on a small population of IMs (Supplemental Fig. S6G). Overall, these data demonstrate that IM expansion after LPS is the result of recruitment of monocytes that mature into macrophages phenotypically similar to FRβ− IMs.
To assess the location of recruited IMs, we performed immunofluorescence on pulse-wait resident IM reporter mice 6 days after LPS. Recruited IMs, identified as CD206+, tdT− cells within the interstitium, were present at multiple locations within the lung. This included the alveolar interstitium, and adjacent to vessels, airways, and pleura (Supplemental Fig. S7).
Sc-RNA-Seq Demonstrates that Recruited IMs Populate Distinct Clusters
To assess transcriptional differences between recruited monocyte-derived IMs and resident IMs during inflammation we performed scRNA-seq on IMs isolated from the pulse-wait lineage tracing model. Reporter mice had lungs harvested 3 and 6 days after LPS. IMs were purified by FACS and sorted into resident and recruited populations based on tdT (Supplemental Fig. S2). Combined with our day 0 data (homeostasis), 79,430 cells were available for analysis after quality control. 21,619 IMs were identified and underwent reclustering, which revealed five unique populations (LPS clusters L1–L5) (Fig. 6A, Supplemental Fig. S8).
Figure 6.
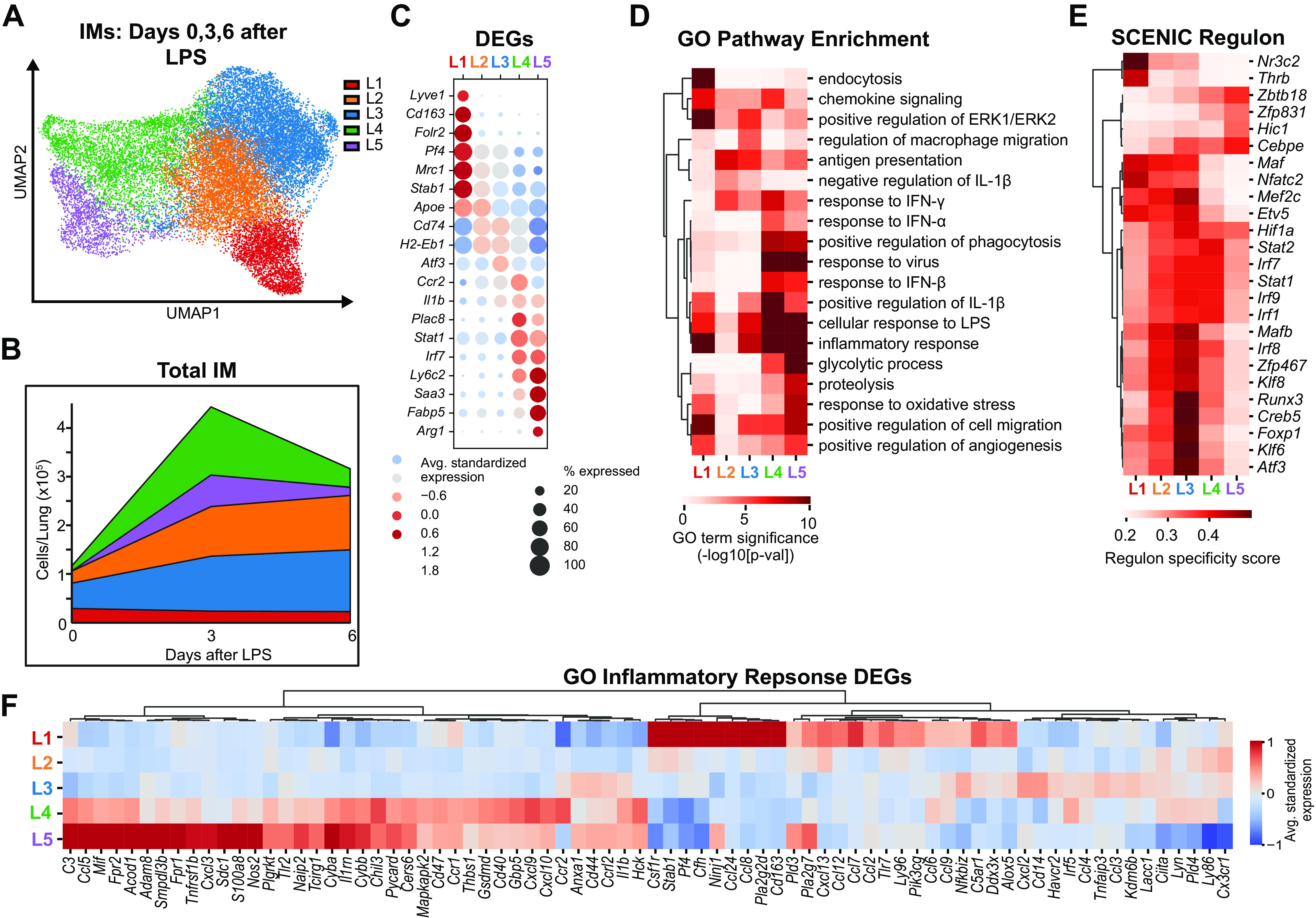
Single-cell RNA-sequencing of lung IMs after intratracheal LPS identifies new clusters. Sequencing was performed on three populations sorted from resident IM lineage tracing male mice at homeostasis (n = 3), 3 (n = 4), and 6 days (n = 3) after LPS, using the scheme outlined in Fig. 2A A: UMAP plot of Seurat clustering of IMs demonstrates five IM clusters. B: total IMs per cluster were calculated by multiplying each cluster proportion by the mean cell count from flow cytometry experiments in Fig. 4, B and C. C: dot plot of highly differentially expressed genes defining each cluster. D: heat map of differential pathway enrichment analysis between the five clusters. E: heat map of differential activity of defining regulons identified by SCENIC analysis between the five clusters. F: differential genes in Gene Ontology (GO) 00069543 Inflammatory Response pathway highlights differences in inflammatory related transcripts between the clusters. DEG, differential expressed gene; IM, interstitial macrophage; LPS, lipopolysaccharide; SCENIC, single-cell regulatory network inference and clustering; UMAP, Uniform Manifold Approximation and Projection.
The total number of cells per lung from each cluster was calculated by multiplying the cluster proportions by mean cell number from our LPS flow cytometry kinetic in Fig. 5, B and C. At homeostasis IMs primarily resided in clusters L1, L2, and L3 (Fig. 6B). At the peak of inflammation (3 days after LPS), there was a dramatic increase in the number of cells in two new clusters, L4 and L5. Clusters L2 and L3 increased in quantity throughout inflammation and resolution whereas cluster L1 remained relatively constant throughout.
Analysis of DEGs between the clusters revealed that cluster L1 had high expression of Folr2, corresponding to the FRβ+ subset (C1) identified in our homeostasis analysis (Fig. 6C, Supplemental Table S7). Clusters L2 and L3 lacked Folr2 and had similar defining genes to the FRβ− “resting” (c3) and “active” clusters (c4) seen in our expanded homeostasis analysis. Clusters L4 and L5 included high expression of monocyte associated genes Ly6c2, Plac8, and Ms4a4c that were not seen in any cluster during homeostasis, suggesting that these clusters contained immature monocyte-derived IMs.
GO pathway enrichment analysis highlighted that both immature clusters (L4 and L5) were defined by elevated expression of interferon-responsive genes (Ifitm3, Irf7, and Stat1) and genes associated with CXCR3 signaling (Cxcl9 and Cxcl10). This was particularly enhanced in cluster L4 (Fig. 6, C and D; Supplemental Table S8). Cluster L5 had high expression of redox, proteolysis, and glycolysis related genes previously reported to be increased in a subset of recruited AMs (5) including Saa3, Ccl5, Nos2, Ly6i, AA467197, Hp, Acod1, and Arg1. Notably, all IM clusters showed potential to contribute to the inflammatory response, with expression of specific cytokines and chemokines restricted among individual IM subsets (Fig. 6F).
SCENIC analysis identified clear gene regulatory network differences between clusters (Fig. 6E, Supplemental Table S9). Clusters L4 and L5 had relatively low activity of the Maf and Mafb regulons providing additional evidence of their immature macrophage state. Cluster L4 also had increased activity in many regulons associated with interferon signaling including Stat1, Stat2, Irf7, and Irf9. Cluster L5 demonstrated increased activity of the regulon Cebpe. Regulons for clusters L1, L2, and L3 closely aligned with those discussed previously during homeostasis with a few notable additions during inflammation. The “active” FRβ− cluster (L3) had high expression of several regulons with known roles in mediating inflammation, for example, Klf6, when compared with its naïve state. The FRβ+ cluster (L1) had strong activation of the Nr3c2 (mineralocorticoid receptor) and Thrb (thyroid hormone receptor β) regulons. Unique signal-dependent transcription factor activity highlights distinctive responses between IM subsets and suggests discrete roles for these subsets during inflammation.
Recruited IMs Have Unique Programming Early and Adopt the Transcriptional Profile of FRβ− Resident IMs over Time
We next asked whether tdT expression (IM origin) or time explained any of our clustering. Resident (tdT+) IMs at homeostasis primarily occupied clusters L1, L2, and L3 (Fig. 7A). They remained largely within these clusters at similar ratios over the course of inflammation. In contrast, at peak inflammation (3 days after LPS), most recruited IMs populated the distinct immature IM clusters (L4 and L5), proving that this signature indeed reflects new monocyte origin. Some recruited IMs were also found in L2 and L3, overlapping with FRβ− resident IM clusters. At 6 days after LPS, most recruited IMs overlayed the FRβ− resident IM clusters (L2 and L3) (Fig. 7, A and B). Taken together, this suggests that recruited IMs have distinct programming early during inflammation and adopt the FRβ− subset programming as resolution progresses, whereas resident IMs remain relatively constant.
Figure 7.
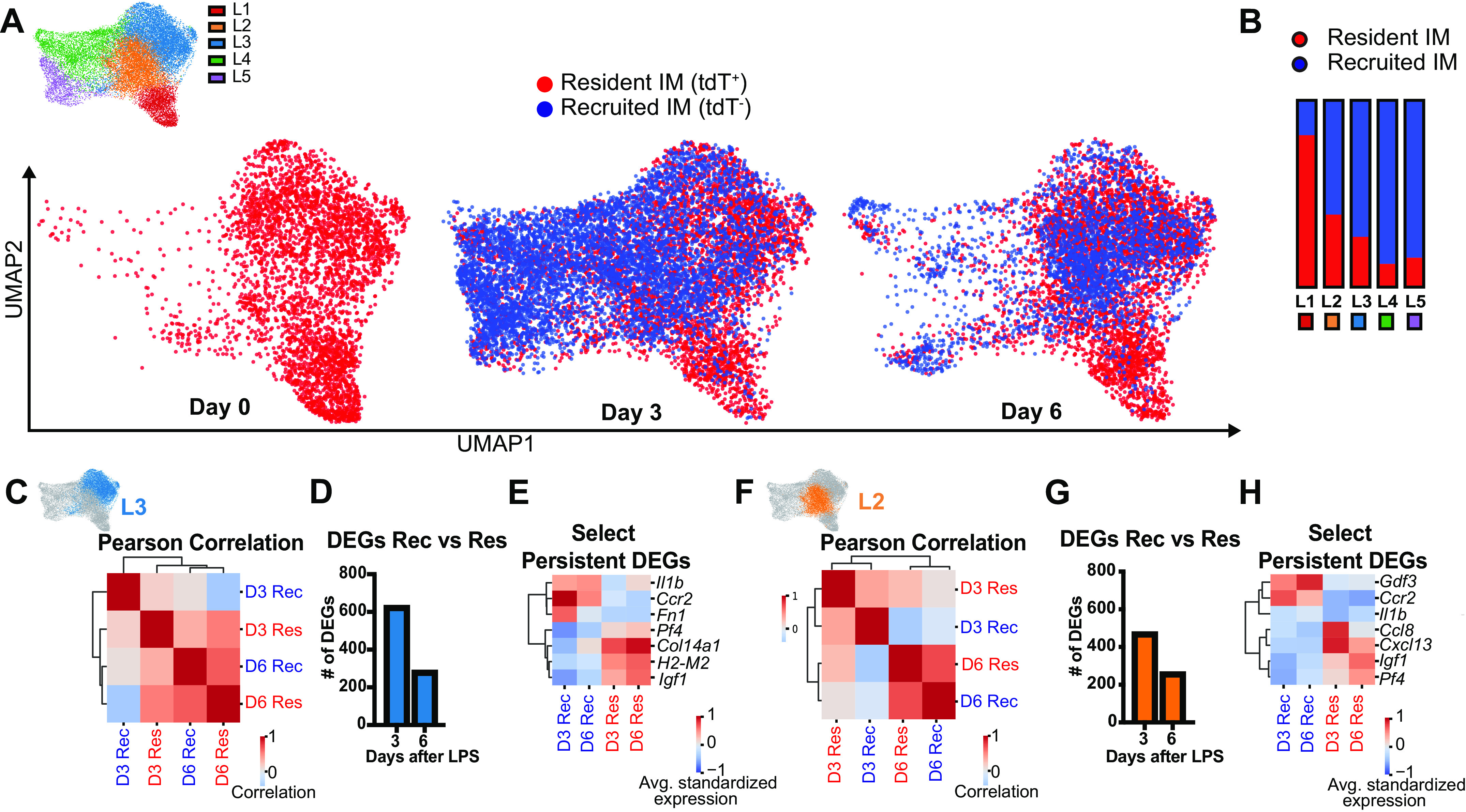
Time and cell origin determine IM clustering after LPS-induced lung inflammation. A: UMAP plot of IMs showing resident vs. recruited origin at each time point. B: proportion of resident vs. recruited IMs by cluster. C and D: within cluster L3, increasing correlation between recruited and resident IMs over time by Pearson’s correlation (C), and number of significant DEGs (D). E: select DEGs in cluster L3 that remain segregated by cell origin at day 6. F and G: within cluster L2, increasing correlation between recruited and resident IMs over time by Pearson’s correlation (F), and number of significant DEGs (G). H: select DEGs in cluster L2 that remain segregated by cell origin at day 6. DEG, differential expressed gene; IM, interstitial macrophage; LPS, lipopolysaccharide; UMAP, Uniform Manifold Approximation and Projection.
Because recruited IMs began to exhibit greater overlap with the FRβ− clusters (L2 and L3) as inflammation resolved (Fig. 7A), we evaluated whether recruited IMs became more transcriptionally similar to resident IMs over time. Pearson’s correlation coefficient of top 1,000 variably expressed genes demonstrated that recruited and resident IMs were more similar to each other at day 6 than day 3 in both FRβ− clusters (L2 and L3) (Fig. 7, C and F). This correlated with a reduction in the total number of DEGs between recruited and resident IMs from day 3 to day 6 (Fig. 7, D and G). However, there were specific genes whose expression were more confined to cell origin. For example, recruited IMs expressed more Il1b, Ccr2, and Gdf3 whereas resident IMs had increased expression of Igf1 and Pf4 (Fig. 7, E and H, Supplemental Table S10). Taken together these data suggest that as recruited IMs mature, they become similar to the FRβ− resident IMs although some genes with potentially important functions are retained from the cell’s origin.
DISCUSSION
The primary objectives of our work were to carefully define subpopulations of pulmonary IMs during homeostasis and to determine how the IM population changes during acute inflammation. Our data build on previous reports that pulmonary IMs exist in distinct subsets during homeostasis and show that FRβ is a reliable marker to separate IM subsets into two main groups. These subsets inhabit discrete niches within the lung interstitium. Our data further show that during acute inflammation from LPS, monocytes are recruited to the interstitium and mature into IMs. Early in inflammation these recruited IMs have unique programming that suggests they enhance the inflammatory response. As inflammation resolves, recruited IMs that remain adopt the transcriptional profile of FRβ− resident IMs.
There is no clear consensus on the optimal strategy to distinguish IM subsets at homeostasis (12). Using three complementary methods, we show that FRβ serves as a highly reliable marker that defines resident IM core identity and that FRβ+ IMs fully encompass previously described IM1/2, CD206+, LYVE1+, CD169+, and TLF+ IM subsets (2, 8, 9, 11). Moreover, we find that FRβ is an improvement over these strategies as it robustly and broadly marks the primary division in transcriptional profiles and exhibits clear positive and negative surface expression on flow cytometry. In comparison, CD206 is expressed by all IMs on a continuous gradient, whereas LYVE1, TIM4, and CD169 all mark subsets within FRβ+ IM population that share the core FRβ+ IM identity. Although CD163 is coexpressed by all FRβ+ IMs transcriptionally, a minor portion of FRβ+ cells were found to lack surface CD163 expression during flow cytometry, which may relate to surface shedding in vivo (18, 19) or during tissue processing. Lastly, although CCR2 protein expression can identify a subset within FRβ− IMs, this does not correspond to a transcriptionally unique subset of FRβ− cells. Furthermore, CCR2 can be induced in resident IMs during inflammation. Thus, the value of CCR2 as a marker for IM subsets with distinct programming is unclear. Collectively, FRβ expression at the mRNA and protein level is the best marker we identified to separate resident IMs into their two major subsets.
The roles played by FRβ+ IMs during homeostasis have not been fully defined. We found that FRβ+ IMs predominate in the adventitia of pulmonary vessels, an area proposed to contain regional immune hubs (26). FRβ+ IMs express CXCL13 and CCL8, which are key chemokines for homing B cells, T helper 2 cells, group 2 innate lymphoid cells, and CD103+ dendritic cells (27–29). This suggests that FRβ+ IMs may be involved in maintaining the local immune environment. FRβ+ IMs also highly express the growth factor IGF-1 (30), which could help maintain the local fibroblast pool, perhaps as part of a circuit where fibroblast-secreted CSF1 maintains IMs (31). FRβ+ IMs may also interact with nerves (9, 10), lymphatics (32), and adjacent blood vessels (33). Finally, FRβ+ IMs highly express several scavenger receptors and have enrichment in endocytosis pathways related to clearance of debris and apoptotic cells. Collectively, FRβ+ IMs transcriptional profiles suggest that these cells may play a key role in maintaining the complex immune and mesenchymal niche of the bronchovascular bundle.
In comparison to FRβ+ IMs, FRβ− IMs are defined by higher expression of antigen processing and antigen presentation genes. As this subset was found adjacent to airways and alveoli, these cells may be programmed for surveillance and presentation of pathogens that have crossed from the airspaces into the interstitium. In addition, the “active” FRβ− subpopulation showed high expression of IL-1β, which has been shown to stimulate alveolar epithelial regeneration (34, 35). Collectively, our data suggest that FRβ− IMs may be playing roles in protecting and maintaining the bronchial and alveolar niches.
During LPS-induced acute inflammation, monocyte-derived macrophages are recruited to the lung interstitium and do not express FRβ. At the peak of inflammation, most recruited IMs reside in two unique “immature” clusters (L4 and L5) that share enrichment of transcripts in inflammation and metabolism-related pathways. One of the subsets has high expression genes downstream from interferon signaling, including Stat1, Stat3, and Irf9. Interferon-inducible chemokines CXCL9 and CXCL10, which are known chemoattractants for T cells, neutrophils, and macrophages (36, 37), were also highly upregulated in this cluster. CXCL9 and CXCL10 also play roles in angiogenesis (38), suggesting that this subset may simultaneously be involved in regulation of vascular repair while augmenting inflammatory cell recruitment. The other immature recruited IM cluster is enriched in redox and glycolytic pathways and is notably characterized by genes previously described in recruited AM populations (5). The interstitium has been posited as a transition site for eventual alveolar macrophages (39). Hence, this subset of IMs may represent tissue precursors for recruited AMs. Additional lineage tracing studies would need to be undertaken to test this hypothesis.
As inflammation begins to resolve, recruited IMs decrease in number. Those that remain obtain similar programming to the FRβ− resident IMs. This reprogramming includes downregulation of interferon-related genes and monocyte-related genes (e.g., Ly6c2 and Plac8) and upregulation of MHCII-related transcripts. However, some specific gene expression is constrained by origin including higher Il1b in recruited IMs. Thus, although retained recruited IMs largely adopt FRβ− resident IM programming during late inflammation, they may retain potential for an enhanced inflammatory response in the setting of a second insult.
Throughout LPS-induced inflammation, the FRβ+ cluster remains largely restricted to resident IMs. Evaluation of gene regulatory networks with SCENIC revealed activation of signal-dependent transcription factors. Specifically, FRβ+ IMs upregulate genes controlled by the mineralocorticoid receptor (Nr3c2) and thyroid hormone receptor β (Thrb). The former has been implicated in pathologic tissue remodeling in the heart (40), whereas the latter has been implicated in promoting proinflammatory macrophage programming (41). Recruited IMs do not populate the FRβ+ subset after intratracheal LPS; however, it is unknown whether this applies in other inflammatory conditions. For example, FRβ+ IMs have been noted to be more numerous in regions of fibrosis in various forms of human interstitial lung disease (42, 43) and experimental depletion of FRβ+ IMs in mice ameliorated fibrosis after bleomycin (42). Further investigation will be needed to understand the cellular origin of increased FRβ+ ΙΜs that has been observed in these scenarios.
When compared with previous evaluation of IM subsets (2, 9–11, 15), our study has several strengths. First, we used multiple complimentary techniques to evaluate for subsets at homeostasis including unbiased clustering based on surface protein expression by spectral flow cytometry, gene expression with Seurat, and gene regulatory networks with SCENIC. The sequencing data from the current study also benefited from high number of cells, deep sequencing depth, and the multiple replicates. In addition, we evaluated IM subsets and programming during acute inflammation using a lineage tracing model that allowed for quantification and identification of unique monocyte-derived IM clusters.
Our study shares some of the limitations of previous studies, including reliance on lung tissue digestion to liberate IMs before flow cytometry and single-cell RNA sequencing that may alter the basal IM protein expression or transcriptomes. In this context, we note that the “active” FRβ− cluster is defined by expression of inflammatory genes, over half of which have been connected to warm tissue digestion (24). The lack of an analogous “active” cluster within FRβ+ IMs leads us to favor the interpretation that “active” FRβ− are not simply an artifact of tissue processing, but this remains a caveat.
Another limitation relates to the patchy nature of airways-induced inflammation where not all tissue is directly impacted. Important transcriptional changes specific to the resident IMs adjacent to inflammation may have been masked by resident IMs in noninflamed regions. Spatial transcriptomics or administration of inflammatory stimuli to a specific region of the lung could help to eliminate this bias. We relied on CD206 immunostaining to identify IMs in tissue sections. However, not all recruited IMs express CD206 at a high level and some recruited IMs were likely not identified. Quantitative, unbiased histologic evaluation using stereology with a more robust IM marker could reveal the relative contribution of recruited IMs to specific niches. Finally, our scRNA-seq analysis was performed during peak inflammation and early resolution. Whether recruited IMs retain distinct expressions of specific genes at later timepoints is unclear.
Taken as a whole, our study helps to align previous observations regarding lung IM heterogeneity during homeostasis and defines the kinetics and transcriptomic profiles of IM subsets during acute inflammation. In particular, we show that two main subsets of resident IMs exist in the lungs during homeostasis at unique locations and that they are most easily distinguished by the presence or absence of FRβ. During acute lung inflammation, resident IMs are joined by monocytes that are recruited to the lung interstitium where they mature to macrophages, thereby transiently expanding total IM numbers. During early inflammation, recruited IMs have unique transcriptional signatures that suggest they augment inflammation and participate in host defense. As inflammation resolves, IM numbers return to homeostatic levels and the recruited IMs that remain largely adopt the programming of FRβ− resident IMs.
APPENDIX
Single-Cell Computational Methods
Cell Ranger was used to align and count transcripts for each individual 10X sample (using Cell Ranger version 3.1.0) using the mm10 reference genome, and Cell Ranger aggr was performed on all samples for visualization in the Loupe Browser (UMAP embeddings and clusters from Seurat were imported into the visualization). Seurat (version 4.0.0) was then used to filter the cells, integrate all samples, find UMAP embeddings for the cells, and calculate clusters. Cells with greater than 10% mitochondrial content, fewer than 500 expressed genes, or fewer than 1,000 transcripts were removed. The transcript counts for each sample were normalized and the top 2,000 were selected based on the “versus” method of FindVariableFeatures. Reciprocal principal component analysis was used to integrate all samples, using 2,000 features (nfeatures = 2,000), three cell anchors (k.anchor = 3), and the top 30 dimensions of the underlying data (dims = 1:30). The UMAP embeddings for the integrated data were found using the RunUMAP function with the top 70 principal components of the scaled, integrated data. Clustering was performed using the standard Seurat Louvain method (dims = 1:70) with a resolution parameter of 1.2. Cluster marker genes were computed using Seurat's FindAllMarkers function, using an adjusted P value of 0.05 and a minimum average log2 fold-change of 0.4.
IM subsetting of the cells involved removing unwanted clusters and rerunning principal component analysis, then recalculating the UMAP embeddings and clusters as above but with different parameters. For the day 0 integration, the UMAP embedding used the top 30 principal components; the clustering with two groups used a resolution parameter of 0.2, whereas the four-group version used a resolution parameter of 0.3. For the days 0, 3, and 6 integration, the UMAP embedding used the top thirty principal components and a resolution parameter of 0.19.
Heat maps with dendrograms were created using the clustermap function of the Seaborn Python library, v. 0.11.2 (44). Gene expression for each group was the mean of the log-normalized, standardized transcript counts. For Fig. 3D, this standardization used all cells in the day 0 IM subset. For Figs. 5F and 6, E and H, the expression was standardized over all cells in the IM subset of days 0, 3, and 6 after LPS. Gene Ontology (GO) term enrichment was calculated using Fisher's exact test on DEGs with adjusted P values less than 0.05 and average log2 fold-changes greater than 0.5. Annotations of mouse genes to GO biological process terms were provided by the Mouse Genome Informatics group (45), downloaded November 2021. Regulon specificity scores were calculated using the SCENIC method (see below). Pearson’s correlation between groups was calculated using the mean of the standardized, log-normalized expression for the top 2,000 genes by coefficient of variation. Hierarchical clustering of all heat maps used the Ward method.
The expression dot plots were created using the scatterplot function of the Seaborn Python library. Gene expression for each group was the mean of the log-normalized, standardized transcript counts. For Figs. 2, D and E, and 3C, the expression was standardized over all cells in the day 0 IM subset. For Fig. 5C, the expression was standardized over all cells in the IM subset of days 0, 3, and 6 after LPS. The percent expression was the percentage of cells with at least one count of the given transcript.
SCENIC (specifically, pySCENIC version 0.11.2) was used to perform regulon analysis. The Jaspar database (46) was used to define the potential transcription factors. The mouse cis-acting motif annotations file was used (https://resources.aertslab.org/cistarget/motif2tf/motifs-v9-nr.mgi-m0.001-o0.0.tbl), along with the 500 bp upstream, 100 bp downstream ranking database (https://resources.aertslab.org/cistarget/databases/mus_musculus/mm10/refseq_r80/mc9nr/gene_based/mm10__refseq-r80__500bp_up_and_100bp_down_tss.mc9nr.genes_vs_motifs.rankings.feather). Enrichment of regulons within clusters was performed using the pySCENIC regulon_specific_scores function.
DATA AVAILABILITY
The transcriptional data are publicly accessible in the Gene Expression Omnibus (GSE218884). All other data will be made avilable upon reasonable request.
SUPPLEMENTAL DATA
Supplemental Tables S1–S10 and Supplemental Figs. S1–S8: https://doi.org/10.6084/m9.figshare.20315406.v1.
GRANTS
This work was supported by the National Institutes of Health and National Heart, Lung, and Blood Institute. Grant numbers R01HL149741, R01HL130938, R35HL140039, R00HL141658, F32HL156289, and T32HL007085.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
P.K.M., E.F.R., P.M.H., W.J.J., and A.L.M. conceived and designed research; P.K.M., S.A.M., T.-H.T., and A.L.M. performed experiments; P.K.M., K.C.A., T.-H.T., and A.L.M. analyzed data; P.K.M., K.C.A., E.M.K., K.J.M., P.M.H., W.J.J., and A.L.M. interpreted results of experiments; P.K.M. and K.C.A. prepared figures; P.K.M. drafted manuscript; P.K.M., K.C.A., S.A.M., E.M.K., K.J.M., E.F.R., P.M.H., W.J.J., and A.L.M. edited and revised manuscript; P.K.M., K.C.A., S.A.M., T.-H.T., E.M.K., K.J.M., E.F.R., P.M.H., W.J.J., and A.L.M. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Jazalle McClendon for assistance.
REFERENCES
- 1. Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol 14: 392–404, 2014. doi: 10.1038/nri3671. [DOI] [PubMed] [Google Scholar]
- 2. Schyns J, Bai Q, Ruscitti C, Radermecker C, De Schepper S, Chakarov S, Farnir F, Pirottin D, Ginhoux F, Boeckxstaens G, Bureau F, Marichal T. Non-classical tissue monocytes and two functionally distinct populations of interstitial macrophages populate the mouse lung. Nat Commun 10, 2019. doi: 10.1038/s41467-019-11843-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Janssen WJ, Barthel L, Muldrow A, Oberley-Deegan RE, Kearns MT, Jakubzick C, Henson PM. Fas determines differential fates of resident and recruited macrophages during resolution of acute lung injury. Am J Respir Crit Care Med 184: 547–560, 2011. doi: 10.1164/rccm.201011-1891OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sabatel C, Radermecker C, Fievez L, Paulissen G, Chakarov S, Fernandes C, Olivier S, Toussaint M, Pirottin D, Xiao X, Quatresooz P, Sirard J-C, Cataldo D, Gillet L, Bouabe H, Desmet CJ, Ginhoux F, Marichal T, Bureau F. Exposure to bacterial CpG DNA protects from airway allergic inflammation by expanding regulatory lung interstitial macrophages. Immunity 46: 457–473, 2017. doi: 10.1016/j.immuni.2017.02.016. [DOI] [PubMed] [Google Scholar]
- 5. Mould KJ, Jackson ND, Henson PM, Seibold M, Janssen WJ. Single cell RNA sequencing identifies unique inflammatory airspace macrophage subsets. JCI Insight 4: 126556, 2019. doi: 10.1172/jci.insight.126556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mould KJ, Barthel L, Mohning MP, Thomas SM, McCubbrey AL, Danhorn T, Leach SM, Fingerlin TE, O'Connor BP, Reisz JA, D'Alessandro A, Bratton DL, Jakubzick CV, Janssen WJ. Cell origin dictates programming of resident versus recruited macrophages during acute lung injury. Am J Respir Cell Mol Biol 57: 294–306, 2017. doi: 10.1165/rcmb.2017-0061OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, et al.. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med 214: 2387–2404, 2017. doi: 10.1084/jem.20162152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gibbings SL, Thomas SM, Atif SM, McCubbrey AL, Desch AN, Danhorn T, Leach SM, Bratton DL, Henson PM, Janssen WJ, Jakubzick CV. Three unique interstitial macrophages in the murine lung at steady state. Am J Respir Cell Mol Biol 57: 66–76, 2017. doi: 10.1165/rcmb.2016-0361OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chakarov S, Lim HY, Tan L, Lim SY, See P, Lum J, Zhang X-M, Foo S, Nakamizo S, Duan K, Kong WT, Gentek R, Balachander A, Carbajo D, Bleriot C, Malleret B, Tam JKC, Baig S, Shabeer M, Toh S-A, Schlitzer A, Larbi A, Marichal T, Malissen B, Chen J, Poidinger M, Kabashima K, Bajenoff M, Ng LG, Angeli V, Ginhoux F. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science 363, 2019. doi: 10.1126/science.aau0964. [DOI] [PubMed] [Google Scholar]
- 10. Ural BB, Yeung ST, Damani-Yokota P, Devlin JC, de Vries M, Vera-Licona P, Samji T, Sawai CM, Jang G, Perez OA, Pham Q, Maher L, Loke P, Dittmann M, Reizis B, Khanna KM. Identification of a nerve-associated, lung-resident interstitial macrophage subset with distinct localization and immunoregulatory properties. Sci Immunol 5: eaax8756, 2020. doi: 10.1126/sciimmunol.aax8756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dick SA, Wong A, Hamidzada H, Nejat S, Nechanitzky R, Vohra S, Mueller B, Zaman R, Kantores C, Aronoff L, Momen A, Nechanitzky D, Li WY, Ramachandran P, Crome SQ, Becher B, Cybulsky MI, Billia F, Keshavjee S, Mital S, Robbins CS, Mak TW, Epelman S. Three tissue resident macrophage subsets coexist across organs with conserved origins and life cycles. Sci Immunol 7: eabf7777, 2022. doi: 10.1126/sciimmunol.abf7777. [DOI] [PubMed] [Google Scholar]
- 12. Bain CC, MacDonald AS. The impact of the lung environment on macrophage development, activation and function: diversity in the face of adversity. Mucosal Immunol 15: 223–234, 2022. doi: 10.1038/s41385-021-00480-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McCubbrey AL, Allison KC, Lee-Sherick AB, Jakubzick CV, Janssen WJ. Promoter specificity and efficacy in conditional and inducible transgenic targeting of lung macrophages. Front Immunol 8: 1618, 2017. doi: 10.3389/fimmu.2017.01618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McCubbrey AL, Barthel L, Mohning MP, Redente EF, Mould KJ, Thomas SM, Leach SM, Danhorn T, Gibbings SL, Jakubzick CV, Henson PM, Janssen WJ. Deletion of c-FLIP from CD11bhi macrophages prevents development of bleomycin-induced lung fibrosis. Am J Respir Cell Mol Biol 58: 66–78, 2018. doi: 10.1165/rcmb.2017-0154OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gibbings SL, Goyal R, Desch AN, Leach SM, Prabagar M, Atif SM, Bratton DL, Janssen W, Jakubzick CV. Transcriptome analysis highlights the conserved difference between embryonic and postnatal-derived alveolar macrophages. Blood 126: 1357–1366, 2015. doi: 10.1182/blood-2015-01-624809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 33: 495–502, 2015. doi: 10.1038/nbt.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine J-C, Geurts P, Aerts J, van den Oord J, Atak ZK, Wouters J, Aerts S. SCENIC: Single-cell regulatory network inference and clustering. Nat Methods 14: 1083–1086, 2017. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Etzerodt A, Maniecki MB, Møller K, Møller HJ, Moestrup SK. Tumor necrosis factor α-converting enzyme (TACE/ADAM17) mediates ectodomain shedding of the scavenger receptor CD163. J Leukoc Biol 88: 1201–1205, 2010. doi: 10.1189/jlb.0410235. [DOI] [PubMed] [Google Scholar]
- 19. Kneidl J, Löffler B, Erat MC, Kalinka J, Peters G, Roth J, Barczyk K. Soluble CD163 promotes recognition, phagocytosis and killing of Staphylococcus aureus via binding of specific fibronectin peptides. Cell Microbiol 14: 914–936, 2012. doi: 10.1111/j.1462-5822.2012.01766.x. [DOI] [PubMed] [Google Scholar]
- 20. Yona S, Kim K-W, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, Hume DA, Perlman H, Malissen B, Zelzer E, Jung S. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38: 79–91, 2013. [Erratum in Immunity 38: 1073–1079, 2013]. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Joshi N, Watanabe S, Verma R, Jablonski RP, Chen C-I, Cheresh P, Markov NS, Reyfman PA, McQuattie-Pimentel AC, Sichizya L, Lu Z, Piseaux R, Kirchenbuechler D, Flozak AS, Gottardi CJ, Cuda CM, Perlman H, Jain M, Kamp DW, Budinger GRS, Misharin AV. A spatially restricted fibrotic niche in pulmonary fibrosis is sustained by M-CSF/M-CSFR signaling in monocyte-derived alveolar macrophages. Eur Respir J 55: 1900646, 2020. doi: 10.1183/13993003.00646-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589, 2010. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, Garner H, Trouillet C, de Bruijn MF, Geissmann F, Rodewald H-R. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518: 547–551, 2015. doi: 10.1038/nature13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Denisenko E, Guo BB, Jones M, Hou R, de Kock L, Lassmann T, Poppe D, Clément O, Simmons RK, Lister R, Forrest ARR. Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus RNA-seq workflows. Genome Biol 21: 130, 2020. doi: 10.1186/s13059-020-02048-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guth AM, Janssen WJ, Bosio CM, Crouch EC, Henson PM, Dow SW. Lung environment determines unique phenotype of alveolar macrophages. Am J Physiol Lung Cell Mol Physiol 296: L936–L946, 2009. doi: 10.1152/ajplung.90625.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dahlgren MW, Molofsky AB. Adventitial cuffs: regional hubs for tissue immunity. Trends Immunol 40: 877–887, 2019. doi: 10.1016/j.it.2019.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med 354: 610–621, 2006. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- 28. Puttur F, Denney L, Gregory LG, Vuononvirta J, Oliver R, Entwistle LJ, Walker SA, Headley MB, McGhee EJ, Pease JE, Krummel MF, Carlin LM, Lloyd CM. Pulmonary environmental cues drive group 2 innate lymphoid cell dynamics in mice and humans. Sci Immunol 4: eaav7638, 2019. doi: 10.1126/sciimmunol.aav7638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sokol CL, Camire RB, Jones MC, Luster AD. The chemokine receptor CCR8 promotes the migration of dendritic cells into the lymph node parenchyma to initiate the allergic immune response. Immunity 49: 449–463.e6, 2018. doi: 10.1016/j.immuni.2018.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hung CF, Rohani MG, Lee S-S, Chen P, Schnapp LM. Role of IGF-1 pathway in lung fibroblast activation. Respir Res 14: 102, 2013. doi: 10.1186/1465-9921-14-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhou X, Franklin RA, Adler M, Jacox JB, Bailis W, Shyer JA, Flavell RA, Mayo A, Alon U, Medzhitov R. Circuit design features of a stable two-cell system. Cell 172: 744–757.e17, 2018. doi: 10.1016/j.cell.2018.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cahill TJ, Sun X, Ravaud C, Villa del Campo C, Klaourakis K, Lupu I-E, Lord AM, Browne C, Jacobsen SEW, Greaves DR, Jackson DG, Cowley SA, James W, Choudhury RP, Vieira JM, Riley PR. Tissue-resident macrophages regulate lymphatic vessel growth and patterning in the developing heart. Dev Camb Engl 148: dev194563, 2021. doi: 10.1242/dev.194563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lim HY, Lim SY, Tan CK, Thiam CH, Goh CC, Carbajo D, Chew SHS, See P, Chakarov S, Wang XN, Lim LH, Johnson LA, Lum J, Fong CY, Bongso A, Biswas A, Goh C, Evrard M, Yeo KP, Basu R, Wang JK, Tan Y, Jain R, Tikoo S, Choong C, Weninger W, Poidinger M, Stanley RE, Collin M, Tan NS, , et al. Hyaluronan receptor LYVE-1-expressing macrophages maintain arterial tone through hyaluronan-mediated regulation of smooth muscle cell collagen. Immunity 49: 326–341.e7, 2018. doi: 10.1016/j.immuni.2018.06.008. [DOI] [PubMed] [Google Scholar]
- 34. Choi J, Park J-E, Tsagkogeorga G, Yanagita M, Koo B-K, Han N, Lee J-H. Inflammatory signals induce AT2 cell-derived damage-associated transient progenitors that mediate alveolar regeneration. Cell Stem Cell 27: 366–382.e7, 2020. doi: 10.1016/j.stem.2020.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Katsura H, Kobayashi Y, Tata PR, Hogan BLM. IL-1 and TNFα contribute to the inflammatory niche to enhance alveolar regeneration. Stem Cell Reports 12: 657–666, 2019. doi: 10.1016/j.stemcr.2019.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. House IG, Savas P, Lai J, Chen AXY, Oliver AJ, Teo ZL, Todd KL, Henderson MA, Giuffrida L, Petley EV, Sek K, Mardiana S, Gide TN, Quek C, Scolyer RA, Long GV, Wilmott JS, Loi S, Darcy PK, Beavis PA. Macrophage-derived CXCL9 and CXCL10 are required for antitumor immune responses following immune checkpoint blockade. Clin Cancer Res 26: 487–504, 2020. doi: 10.1158/1078-0432.CCR-19-1868. [DOI] [PubMed] [Google Scholar]
- 37. Bronger H, Singer J, Windmüller C, Reuning U, Zech D, Delbridge C, Dorn J, Kiechle M, Schmalfeldt B, Schmitt M, Avril S. CXCL9 and CXCL10 predict survival and are regulated by cyclooxygenase inhibition in advanced serous ovarian cancer. Br J Cancer 115: 553–563, 2016. doi: 10.1038/bjc.2016.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Romagnani P, Lasagni L, Annunziato F, Serio M, Romagnani S. CXC chemokines: the regulatory link between inflammation and angiogenesis. Trends Immunol 25: 201–209, 2004. doi: 10.1016/j.it.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 39. Landsman L, Jung S. Lung macrophages serve as obligatory intermediate between blood monocytes and alveolar macrophages. J Immunol 179: 3488–3494, 2007. doi: 10.4049/jimmunol.179.6.3488. [DOI] [PubMed] [Google Scholar]
- 40. Fraccarollo D, Thomas S, Scholz C-J, Hilfiker-Kleiner D, Galuppo P, Bauersachs J. Macrophage mineralocorticoid receptor is a pleiotropic modulator of myocardial infarct healing. Hypertension 73: 102–111, 2019. doi: 10.1161/HYPERTENSIONAHA.118.12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Perrotta C, Buldorini M, Assi E, Cazzato D, De Palma C, Clementi E, Cervia D. The thyroid hormone triiodothyronine controls macrophage maturation and functions: protective role during inflammation. Am J Pathol 184: 230–247, 2014. doi: 10.1016/j.ajpath.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 42. Nagai T, Tanaka M, Hasui K, Shirahama H, Kitajima S, Yonezawa S, Xu B, Matsuyama T. Effect of an immunotoxin to folate receptor β on bleomycin-induced experimental pulmonary fibrosis. Clin Exp Immunol 161: 348–356, 2010. doi: 10.1111/j.1365-2249.2010.04182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schniering J, Benešová M, Brunner M, Haller S, Cohrs S, Frauenfelder T, Vrugt B, Feghali-Bostwick C, Schibli R, Distler O, Müller C, Maurer B. 18F-AzaFol for detection of folate receptor-β positive macrophages in experimental interstitial lung disease—a proof-of-concept study. Front Immunol 10: 2724, 2019. doi: 10.3389/fimmu.2019.02724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Waskom ML. seaborn: statistical data visualization. J Open Source Softw 6: 3021, 2021. doi: 10.21105/joss.03021. [DOI] [Google Scholar]
- 45. Bult CJ, Blake JA, Smith CL, Kadin JA, Richardson JE; the Mouse Genome Database Group. Mouse Genome Database (MGD) 2019. Nucleic Acids Res 47: D801–D806, 2019. doi: 10.1093/nar/gky1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Castro-Mondragon JA, Riudavets-Puig R, Rauluseviciute I, Berhanu Lemma R, Turchi L, Blanc-Mathieu R, Lucas J, Boddie P, Khan A, Manosalva Pérez N, Fornes O, Leung TY, Aguirre A, Hammal F, Schmelter D, Baranasic D, Ballester B, Sandelin A, Lenhard B, Vandepoele K, Wasserman WW, Parcy F, Mathelier A. JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res 50: D165–D173, 2022. doi: 10.1093/nar/gkab1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Tables S1–S10 and Supplemental Figs. S1–S8: https://doi.org/10.6084/m9.figshare.20315406.v1.
Data Availability Statement
The transcriptional data are publicly accessible in the Gene Expression Omnibus (GSE218884). All other data will be made avilable upon reasonable request.