

Deep sequencing of 2,860 long-term survivors of pediatric cancer identified accelerated clonal hematopoiesis (CH) and characterized STAT3 as a CH gene specific to Hodgkin lymphoma survivors.
Abstract
We present the first comprehensive investigation of clonal hematopoiesis (CH) in 2,860 long-term survivors of pediatric cancer with a median follow-up time of 23.5 years. Deep sequencing over 39 CH-related genes reveals mutations in 15% of the survivors, significantly higher than the 8.5% in 324 community controls. CH in survivors is associated with exposures to alkylating agents, radiation, and bleomycin. Therapy-related CH shows significant enrichment in STAT3, characterized as a CH gene specific to survivors of Hodgkin lymphoma, and TP53. Single-cell profiling of peripheral blood samples revealed STAT3 mutations predominantly present in T cells and contributed by SBS25, a mutational signature associated with procarbazine exposure. Serial sample tracking reveals that larger clone size is a predictor for future expansion of age-related CH clones, whereas therapy-related CH remains stable decades after treatment. These data depict the distinct dynamics of these CH subtypes and support the need for longitudinal monitoring to determine the potential contribution to late effects.
Significance:
This first comprehensive CH analysis in long-term survivors of pediatric cancer presents the elevated prevalence and therapy exposures/diagnostic spectrum associated with CH. Due to the contrasting dynamics of clonal expansion for age-related versus therapy-related CH, longitudinal monitoring is recommended to ascertain the long-term effects of therapy-induced CH in pediatric cancer survivors.
See related commentary by Collord and Behjati, p. 811.
This article is highlighted in the In This Issue feature, p. 799
INTRODUCTION
Clonal hematopoiesis (CH) refers to the presence of hematopoietic stem cell (HSC) subpopulations that have become genetically distinct from the germline genome via acquisition of somatic mutations. Although CH is an age-related phenomenon in the general population (1), it can also be induced by exogenous factors such as chemotherapy, which can affect both the emergence and evolution of CH clones. In addition to the higher prevalence among people previously treated for malignancy (2, 3), CH is now recognized as a precancerous state of blood cancers (4–7), highlighting the need for monitoring malignant transformation of secondary neoplasms for cancer survivors (1–3).
With the overall improvement of pediatric cancer survival rates, it has become evident that pediatric cancer survivors are at risk of accelerated physiologic aging (8, 9). Despite this, the study of CH in pediatric cancer survivors has been limited by poor representation of this population in studies that focused primarily on survivors of adult-onset cancers (2, 3) or has focused on survivors with limited duration of follow-up or small cohort sizes (10–12). For example, a recent study by Bertrums and colleagues (12) showed that chemotherapy can induce mutations in HSCs directly or indirectly by studying 24 pediatric patients with a median follow-up time of 1.65 years (range, 0.2–10 years). These studies have greatly improved our understanding of the acute landscape of therapy-related CH. However, as the life expectancy of long-term (>5 years) survivors of pediatric cancer can exceed 50 years after treatment (13), the long-term profile of CH in survivors, including the evolutionary trajectory during their lifespan, may inform the design of clinical management of late effects.
To fill this knowledge gap, we analyzed CH in the St. Jude Lifetime Cohort study (SJLIFE), a retrospective cohort with prospective clinical follow-up of pediatric cancer survivors treated at St. Jude Children's Research Hospital since 1962 (14, 15). We performed deep sequencing on DNA extracted from peripheral blood samples obtained from 2,860 SJLIFE survivors (age range, 6.0–66.4 years; median 31.6) to enable comprehensive CH analysis in this relatively young population. Furthermore, we performed statistical modeling to probabilistically assign CH into age- and therapy-related subtypes in order to characterize their distinct dynamics in clonal expansion.
RESULTS
Higher CH Incidence in Pediatric Cancer Survivors
To estimate the prevalence of CH in pediatric cancer survivors, we performed targeted sequencing of 39 hematologic malignancy–associated or cancer-predisposing genes (Supplementary Table S1) using peripheral blood samples from 2,860 SJLIFE survivors and 324 community controls (Fig. 1A). The survivors were followed up over 5.1 to 51.1 years (median 23.5 years) from cancer diagnosis and were ages 6.0 to 66.4 years (median 31.6 years) at the time of sample collection, which was slightly younger than the community controls (18.3–70.2 years, median 34.6; Supplementary Fig. S1). The childhood cancer diagnoses of the survivors included leukemias (35%), lymphomas (19%), central nervous system malignancies (CNS; 11%), and non-CNS solid tumors (35%). Given the relatively young age of our cohort, the median raw sequencing depth was set at 15,987× (1,720× after deduplication) so that we could detect CH clones present at a variant allele frequency (VAF) as low as 0.1%. To reduce the error rate inherent in deep sequencing data, additional analyses, such as computational error suppression (16), outlier detection (17) adjusted for sequencing context (Fig. 1B), and indel realignment (18), were used in variant analysis (Methods). Approximately 40% of the putative variants were subjected to orthogonal validation by digital droplet PCR (ddPCR), whereas the validity of the remaining variants was inferred by modeling the site-specific background error (Fig. 1A; Methods and Supplementary Methods). Altogether, we identified 540 validated CH variants with a median VAF of 0.4% (range, 0.1%–29.5%) for further analysis (Supplementary Table S2).
Figure 1.

Elevated CH in pediatric cancer survivors. A, Study design for CH analysis in survivors. Blood samples from survivors and controls were analyzed by capture sequencing on 39 CH-associated genes. Putative CH variant analysis involves computational error suppression of sequencing reads, outlier analysis of substitutions with the same genomic context, and indel realignment. Approximately 40% of the putative variants (538 total) were successfully assayed by ddPCR, whereas the validity of the remaining variants was inferred by comparing to a background error model. ddPCR, digital droplet PCR; SNV, single-nucleotide variant. B, Outlier analysis based on the sequence context of a CH variant. For each of the 96 genomic triplet context changes, the count and frequency for sequencing context matching the context of the alternative allele as well as the read count were within a sample. Outlier analysis was performed by IsolationForest (17) in both the spaces spanned by the count and coverage and count and frequency. Example shows the GCG>GTC context analysis in an acute lymphoblastic leukemia survivor, in which the DNMT3A R882H mutation was detected as an outlier. cnt., count; cov., coverage. C, Elevated CH prevalence in the survivors (orange) compared with the controls (green) across all five age categories. Significance of difference in the overall prevalence of the two groups was based on Fisher exact test. D, Leukocyte telomere lengths in the CH-positive survivors and controls. Telomere attrition rates were estimated as a regression slope, which was compared by the t test. Bands represent 95% CIs of the least-square regressions.
Higher CH prevalence was found in the survivors compared with the community controls in each age category (Fig. 1C), with a statistically significant increase in the overall prevalence: 15.0% of survivors [95% confidence interval (CI), 13.7%–16.3%] versus 8.6% of controls (95% CI, 5.6–11.7; Fisher exact P = 1.44 × 10−3). When limited to variants with VAF ≥2%, a threshold commonly used for other studies, CH prevalence was 1.99% (57/2,860) and 0.93% (3/324) for survivors and controls, respectively. Notably, at this cutoff, the 1.99% prevalence in our relatively young survivorship cohort (median of 31.6 years) is comparable with the prevalence in a general population ages 50 to 59 years (2.54%, 138/5,441, Fisher exact P = 0.128; ref. 5). As HSCs acquire mutations during cell division, we compared the leukocyte telomere length, a biomarker for cell division, in CH-positive survivors and controls using the accompanying whole-genome sequencing (WGS) dataset (19, 20). The shortening of telomere with age relationship represented by a least-square regression fit showed nearly parallel lines (Fig. 1D). This indicates that the telomere attrition rates, which reflect the rates of underlying HSC division, were very similar between the controls (41.4 base pairs/year) and survivors (45.1; P = 0.413). This suggests that the CH variants were acquired at a similar rate in survivors and controls. Therefore, the increased CH prevalence in the survivors was likely related to cancer and/or treatment-related exposures in childhood rather than an acceleration of cell division.
Associations of CH Status with Therapeutic Variables
To investigate the origin of elevated CH in pediatric cancer survivors, we analyzed the association of CH with demographic variables and cancer treatment exposures. Although no association was found with sex and race/ethnicity, the prevalence of CH was correlated with age in both survivors and controls as expected (Supplementary Table S3). We next sought to evaluate the association of specific therapies with CH, as cancer treatments typically involve multiple therapeutic agents and modalities. In multivariable logistic regression analysis, after adjusting for age at sample acquisition and age at diagnosis, CH was associated with alkylating agents, bleomycin, and estimated radiotherapy (RT) dose to active bone marrow (Fig. 2A). Specifically, exposures to alkylating agents and RT were independently associated with CH in a dose-dependent manner, which was not observed for bleomycin (Fig. 2B). As treatment differs by cancer type, we analyzed the association of CH prevalence by diagnosis (Fig. 2C). After adjusting for age, survivors of Hodgkin lymphoma (HL), soft-tissue sarcoma, germ cell tumor, rhabdomyosarcoma, neuroblastoma, non-HL, or acute lymphoblastic leukemia were more likely to develop CH compared with the controls; these diagnoses were collectively represented as Dx*, while those with insignificant odds ratio were labeled Dx (Fig. 2C). We also found that a significantly higher proportion of survivors with Dx* had been exposed to alkylating agents (all dose tertiles; details in Methods) than those of Dx (Fig. 2D; 67.0% vs. 35.9%, proportion test P = 1.83 × 10−57). Bleomycin was administered to only a small number of cases (n = 147), with the second tertile exhibiting a significantly higher proportion in cancer diagnoses significantly associated with CH risk (2.47% vs. 0.20%, P = 7.18 × 10−6). The third tertile of bleomycin dose was primarily among survivors of osteosarcoma, a cancer type that barely missed the cutoff for Dx* (Fig. 2C), which led to the statistically insignificant finding in this category (Supplementary Fig. S2). There was no difference in the distribution of RT third tertile (18.3% vs. 17.2%, P = 0.477). However, in a subset analysis of survivors treated without chemotherapy (n = 475; Supplementary Fig. S3), in whom a potential effect-masking by chemotherapy was uncovered, we similarly observed a significantly higher proportion of the third tertile (30.9% vs. 16.1%, P = 2.09 × 10−5).
Figure 2.

Associations of CH prevalence with clinical variables. A, Odds ratios and 95% CIs for CH and cancer therapies. Multivariable logistic analysis adjusted for other therapy doses, age at blood sampling, and age at diagnosis. Cumulative doses were used for chemotherapy. For RT, maximum region-specific dose was used after adjusting for the active bone marrow distribution at the diagnosis age. Variables were scaled by standard deviation. Bars represent 95% CIs. *, P < 0.05; **, P < 0.01; ***, P < 0.001. B, Odds ratio between significant therapies in A and CH over the dose tertile. Odds ratios were similarly adjusted as in A. Bands represent 95% CIs. *, P < 0.05; **, P < 0.01; ***, P < 0.001. C, Age-adjusted odds ratios of CH relative to the control were estimated for diagnoses with 50 survivors or more. Uncommon diagnoses with <50 cases were combined as “Others.” Diagnoses in blue indicate a significant odds ratio and are collectively denoted as Dx*. Bars represent 95% CIs. *, q (Benjamini–Hochberg corrected P) < 0.05; **, q < 0.01; ***, q < 0.001. D, Comparison of therapy intensity in CH-associated cancer types (Dx*, blue) with those not associated with CH (Dx, black). The frequency of therapy intensities found significant in B was compared between the two groups by proportion test. The analysis on the bottom right was restricted to survivors who had not received chemotherapy.
Although exposures to alkylating agents, bleomycin, and RT were associated with elevated CH prevalence in the survivors, there may exist additional contributing factors. To address this hypothesis, we performed a mediation analysis to quantify how much of the CH association with cancer type could be explained by these three therapies (21). Exposure to the statistically significant tertiles of alkylating agents, bleomycin, or RT was defined as a dichotomous mediator in the analysis. After adjusting for ages at diagnosis and at sample collection, the therapy mediator explained 74% of the association between CH and cancer diagnosis, confirming that the CH development in our cohort was largely contributed by these therapies.
CH Association with Germline Pathogenic Mutations in Survivors
In addition to the therapy variables, germline mutations may have also played a modifier role in CH development. We first analyzed the CH association with 112 germline pathogenic mutations identified in the SJLIFE cohort, which were previously classified based on the American College of Medical Genetics and Genomics (ACMG) guidelines in 60 cancer predisposition genes (CPG) known to be associated with autosomal dominant cancer predisposition syndromes with moderate to high penetrance (ref. 20; Supplementary Table S4). The CH prevalence was 8.9% in carriers of CPG mutations versus 15.3% in noncarriers, respectively (Supplementary Table S4). The reduced prevalence in the carriers, albeit statistically insignificant (Fisher exact P = 0.078), likely reflected the fact that more than 50% of such mutations occurred in survivors of cancer types with the lowest CH incidence due to less intensive therapy (Fig. 2C)—that is, retinoblastoma (n = 35), Wilms tumor (n = 9), or CNS malignancies (n = 24).
We also studied the effect of deficiency in DNA damage repair (DDR) on CH prevalence (Supplementary Table S4) using germline pathogenic mutations in 127 DDR genes selected by our prior study (22). The overall CH prevalence, 17.9% in carriers versus 14.9% in noncarriers (Fisher exact P = 0.417), was not affected by germline mutation status. However, when stratifying the survivors by therapy exposure, nonirradiated survivors (n = 1,359) had a significantly higher CH prevalence in mutation carriers (21.2%) than in noncarriers (11.7%, Fisher exact P = 0.0496), indicating that RT might have masked the pathogenic effect of mutations in irradiated cases (P = 0.730).
Molecular Characteristics of Age- versus Therapy-Related CH Subtypes
We proposed an additive model for CH clones detected in survivors based on the association of therapy with CH prevalence (Fig. 2) and a cell division rate comparable between survivors and community controls in this cohort (Fig. 1D). Under this model, CH in survivors is an admixture of therapy- and age-related clones. Age-related CH clones in the survivors are generated at a rate similar to those in the community controls over time (Supplementary Fig. S4). As the vast majority of CH-positive survivors (85% or 365/430) harbored only a single CH event, we classified the clones into age- or therapy-related categories using two steps. First, the probability of developing age-related CH in a given age category was estimated by logistic regression using the control data. Second, the probability of developing CH in a given age category and by therapy exposure was estimated in the survivor cohort. In the second regression, the age effect estimated by the first regression was included as an offset term so that the age impact on a survivor and a control will be the same at a given age category as in the proposed model (Supplementary Fig. S4; Methods). Using these two models, we computed probabilities of developing CH for each survivor, given their age and treatment exposures, and when the estimated contribution from therapy was higher than that of age, the CH of the sample was designated as “inferred therapy-related” (iTR) and otherwise as “inferred age-related” (iAR). Of the 430 CH-positive survivors, this approach found 214 samples as iAR, which accounted for 7.5% in the survivor cohort, comparable with 8.6% CH prevalence in the control (Fisher exact P = 0.439). Prevalence of iAR in the survivors increased with age, matching the course of the community controls (Fig. 3A).
Figure 3.

Molecular features of inferred age- versus therapy-related CH. A, Comparable prevalence of iAR CH in the survivors (blue) and CH in the controls (green). Fisher exact test showed no significant difference between the two groups (P = 0.439). B, Mutation spectra of single-nucleotide variants (SNV) in the control (green), iAR (blue), and iTR (purple). Spectrum frequency was compared by the χ2 test followed by post hoc χ2. Only significant comparisons are shown. Post hoc P value was adjusted by the Šidák method. C, Distribution of mutation count per sample. The distribution in each group was compared by the Kruskal–Wallis test. D, CH mutation frequency in the top 10 genes identified in the survivors and controls. The CH mutations in survivors were further stratified into iAR and iTR groups, and the top five genes were compared with the control by a two-sided proportion test. *, q (Benjamini–Hochberg corrected P) < 0.05 and ***, q < 0.001, with red indicating a proportion higher than control and blue if lower. E,STAT3 mutations identified in this study (top, number in the circle represents occurrence) and the COSMIC database (v97) for hematologic and lymphoid tumors (bottom, y-axis shows occurrence). F, Cell phenotype and STAT3 Y640F genotype in blood samples from three survivors profiled by Mission Bio's Tapestri assay on single-cell amplicon-based DNA and protein sequencing. Cells were mapped by uniform manifold approximation and projection (UMAP) by protein antibody data and colored by their corresponding cell types. Those harboring Y640F mutation are labeled in red. DP, double-positive; NK, natural killer. G, Mutation burden and signature analysis for SJHL018702 by scWGS. The peripheral blood sample was sorted for viable cells followed by multiple displacement amplification to generate scWGS data in STAT3 Y640F–mutant and wild-type (wt) cells (left). Mutation burden by somatic SNVs identified from scWGS using bulk WGS as a control is shown as a bar plot with Y640F-mutant cells marked by a red star (right). Mutational signatures of Y640F-mutant cells and wt cells are shown at the bottom.
To provide independent validation for this model, we first compared the mutation spectra of iAR and iTR, which reflects the underlying mutagenesis process and subsequent selection. As shown in Fig. 3B, the spectra from the control and iAR were nearly identical, exhibiting dominance of the cytosine (C)>thymine (T) transition in 63.0% and 65.8% of the substitutions, respectively, consistent with expected age-related processes (5). By contrast, the iTR spectrum was different from iAR in that the C>T transition accounted for only 45.7% of substitutions (post hoc P = 2.05 × 10−3). Although samples with four CH mutations were observed only in iTR, the distribution of CH mutation burden did not differ across the control, iAR, and iTR (Kruskal–Wallis P = 0.454; Fig. 3C). Next, we examined the mutation frequency in DNMT3A, KRAS, TP53, TET2, and STAT3, the five most frequently mutated genes in both survivors and controls (Fig. 3D). These five genes had similar mutation frequencies in control and iAR samples of the survivors, consistent with the iAR classification. By contrast, the frequencies in iTR showed considerable differences—significantly higher frequency was found for STAT3 (17.9% in iTR vs. 6.25% in controls, FDR q = 2.61 × 10−6) and TP53 (17.5% vs. 12.5%, q = 0.033), and a lower rate was found for DNMT3A (19.0% vs. 28.1%, q = 2.48 × 10−5). Although elevated TP53 mutation frequency in therapy-related CH is consistent with previous studies that reported TP53 as a therapy-related CH gene (2, 3, 23), STAT3 has not been characterized as related to therapy.
Therapy-Related STAT3 Mutations in Survivors of HL
Identification of STAT3 as a frequently mutated CH gene is a new finding in our pediatric cancer survivorship cohort. Notably, all STAT3 mutations were in the Src homology 2 domain (Fig. 3E), which contains mutation hotspots in hematologic and lymphoid cancers. The most frequent hotspot mutation, Y640F, was also the predominant mutation in our cohort present in 36 cases. Interestingly, STAT3 mutations were significantly enriched in survivors of HL (9.3% in HL survivors vs. 1.4% in survivors of pediatric cancers other than HL, Fisher exact P = 9.21 × 10−14; Supplementary Table S5). This suggests that STAT3 is an HL-specific CH gene, which was further supported by the lack of enrichment of STAT3 mutations in survivors of cancers other than HL (1.4% in other cancers vs. 0.6% in controls, P = 0.426).
For three survivors with STAT3 Y640F, we sought to identify the mutant cell phenotype by the Mission Bio Tapestri assay, which jointly profiles the genotype and phenotype in the same single cell using oligo-conjugated antibodies (Methods). Although STAT3 Y640F was detected in all blood cell types, it was highly enriched in T cells across all three cases (Fig. 3F; Supplementary Fig. S5). Among the subtypes of T cells, the CD8+ populations consistently harbored this mutation (Supplementary Fig. S5). This suggests that the mutant T cells may have accelerated proliferation, as STAT3 Y640F has previously been identified as a gain-of-function driver mutation in T-cell large granular lymphocyte leukemia, a lymphoproliferative disorder characterized by CD3+ CD8+ T-cell expansion (24). The broad spectrum of mutation-positive cell types coupled with its predominance in T cells argues against the possibility that Y640F arises from residual HL, as classic HL is of B-cell origin (25, 26).
To evaluate whether mutagenic processes related to therapy might explain the association between STAT3 mutation and HL, we performed single-cell WGS (scWGS) analysis on mutant and wild-type cells in HL survivors (Methods) and were able to analyze four cells for each genotype from SJHL018702, an iTR case whose blood sample collected 22 years after the initial HL diagnosis was used for this purpose (Fig. 3G). Somatic single-nucleotide variants (SNV) were identified by using the bulk WGS data as the matching control (Methods). Compared with the expected mutation burden of 1,000 to 2,000 for a normal peripheral blood cell aged 37.7 years (27), all cells had elevated mutation burden. The mutation burden of STAT3 Y640F–mutant cells was 3 to 4 times higher than those of the wild-type cells (Fig. 3G, middle). Mutation signature analysis of these cells only extracted COSMIC SBS25, a postchemotherapy signature specific to HL cell lines (28) or normal tissues of HL survivors (29, 30), at a cosine similarity of 0.91. Multiple signatures, including COSMIC SBS1, 5, 11, and 25 with the relative contribution of 2.4%, 24.5%, 16.1%, and 57.5%, respectively, at a cosine similarity of 0.94, were extracted for the wild-type cells (Fig. 3G, right). SBS25 preferentially converts T to adenine (A) when C or guanine (G) precedes. Therefore, the probability that Y640F (GTA>GAA) was induced by SBS25 was estimated to be 0.985 to 1.0 in the mutant cells.
The near 100% probability of Y640F being induced by SBS25 based on mutation signature analysis of scWGS data prompted us to examine the sequence context of all STAT3 mutations in our cohort. Indeed, N647I (GTT>GAT), the only other STAT3 mutation (besides Y640F) enriched in HL (Supplementary Table S5), also matches the predominant pattern of SBS25. Recently, Santarsieri and colleagues (30) found that procarbazine, an alkylating drug used in HL treatment, is likely responsible for the SBS25 signature. In our cohort, 192 of 356 HL survivors, including SJHL018702 (Fig. 3G), were exposed to procarbazine. We found that prior exposure to procarbazine was associated with CH-positive HL survivors harboring mutations of G/C[T>A]N context—that is, the pattern that matches SBS25 (Supplementary Table S6; Fisher exact P = 5.55 × 10−6)—but not those with other mutation contexts (P = 0.893), suggesting the specific contribution of procarbazine to SBS25.
Dynamic Characteristics of Age- versus Therapy-Related CH Clones
To examine the clonal expansion pattern, we plotted VAF, a surrogate measure for clone size, for iAR over age and for iTR over follow-up time, respectively (Fig. 4A). We tested if the pattern differed across clone sizes by performing quantile regression for the lower 25th (Q25th), median (Q50th), and upper 25th (Q75th) quartiles of VAF values, which respectively correspond to smaller, medium, and larger clones. Although iTR clones, regardless of their size quantiles, remained stable over follow-up, there was a modest but significant increase of the Q25th quartile in iAR over age. The iAR pattern was replicated when we analyzed two independent age-related CH datasets from previous studies using the same approach (refs. 2, 31; Supplementary Fig. S6). Encouraged by this reproducible finding, we further analyzed the correlation between clone size and growth direction using serial samples (Fig. 4B). VAF change during the two time points (median time interval, 4.13 years; range, 0.23–7.9) was plotted to show the growth direction (i.e., expanding and nonexpanding) of an iAR (n = 46) or iTR (n = 55) clone (Supplementary Fig. S7; Supplementary Table S7); the majority of CH clones did not expand in this analysis (76% of iAR and 64% of iTR, Fisher exact P = 0.179; inset in Fig. 4B). Among the iAR clones, there was a tendency for the nonexpanding clones to be smaller than the expanding clones at the initial time point (nonexpanding and expanding clone median VAF, 0.22% vs. 1.25%; Mann–Whitney P = 6.28 × 10−4; Fig. 4B), suggesting the trend at Q25th was due to the loss of smaller clones. We speculate that this modest Q25th trend in iAR could be a prelude to the precipitous decline in diversity of HSCs in the elderly (>70 years old) reported recently by Mitchell and colleagues (32). By contrast, such a tendency was not detected in iTR clones (0.33 vs. 0.48%, P = 0.183), reminiscent of the stable VAF over follow-up times across the survivorship cohort (Fig. 4A). We then tested whether these patterns could arise by performing a simulation using the branching HSC division model, which was developed to explain age-related CH dynamics (33). Indeed, simulated CH clone trajectories recapitulated the association between VAF and the growth pattern of iAR (Supplementary Fig. S8). To evaluate whether the contrasting pattern in iTR could be attributed to the early onset of therapy-related clones, we performed a second simulation using a modified model that generated CH trajectories driven by the same dynamics but initiated in the range of childhood cancer treatment (i.e., 0–20 years old; Methods). However, this modified simulation produced a pattern similar to iAR, refuting the hypothesis that the timing of mutational acquisition led to the lack of an association between clone expansion and VAF in iTR.
Figure 4.

Dynamics of inferred age- versus therapy-related CH. A, VAF distribution along age for iAR and along follow-up for iTR. Longitudinal trend was estimated by quantile regression on VAF quartiles: Q25th (lower 25%), Q50th (median), and Q75th (upper 25%). Estimated slopes are shown. **, P < 0.01. B, Association between clone size and subsequent expansion revealed by serial analysis. For each serial sample pair, clone fate was classified as expanding or nonexpanding based on the statistical test. The proportion of the expanding fate did not differ between iAR and iTR (inset, Fisher exact P = 0.179). Log-transformed VAFs at the earlier time point were compared by the t test between expanding and nonexpanding fates. Horizontal lines in the violin represent the VAF (log scale) observed at the initial time point.
The median follow-up time of the serial samples profiled in this study was 33.1 years (range, 14.0–48.4 years), and this long follow-up time allowed us to evaluate the long-term effect of cytotoxic therapies on the growth rate of CH clones in iTR and iAR clones. There was no significant difference in the growth rate of iAR and iTR clones harboring the most frequently mutated genes, including TP53 (Supplementary Fig. S9, top). This indicates that there is a minimal long-term effect of cytotoxic therapies on accelerating the growth of clones harboring mutations, including TP53, a DNA damage response gene—a different effect was shown previously in samples obtained with a short follow-up time (median was 0.79 years; ref. 3). Interestingly, iTR clones with TP53 mutations were significantly larger than their iAR counterparts (Supplementary Fig. S9, bottom), which can be attributed to an early clonal expansion under the selective pressure of therapy, consistent with the observation by Bolton and colleagues (3) using samples with short follow-up time. Altogether, these findings suggest that the selective pressure of cancer therapy on clonal expansion may not be permanent and can be attenuated over decades.
DISCUSSION
In this first comprehensive analysis on CH of long-term survivors of pediatric cancer, we detected a higher CH prevalence in the survivor cohort compared with the community controls (Fig. 1C). The results are consistent with findings from recent studies that reported the effect of cancer therapy on CH in cohorts comprised mostly of adult cancer patients (2, 3). More importantly, the long-term follow-up (median 24 years) of the SJLIFE cohort enabled us to confirm that elevated CH is a chronic condition that persists for decades, which has potential clinical relevance to inform clinical trials of early interventions in childhood cancer survivors. Our study analyzed only survivors who were alive at the cohort enrollment and thus able to provide a blood sample for sequencing. A review of all >5-year survivors at St. Jude found that only 0.68% succumbed to secondary leukemias/myelodysplastic syndrome before they could be recruited to SJLIFE. Given the very low frequency of such events, our CH analysis provides a general representation of long-term survivors of pediatric cancer.
Although radiotherapy was found related to CH development by Bolton and colleagues (3) as well as this study, the association with chemotherapeutic agents can be affected by different cancer diagnoses examined in different cohorts. For example, platinum-based agents were associated with CH in the mostly adult cohort (3) but not in our pediatric cancer survivors. Notably, leukemias and lymphomas, which account for 54% (1,557/2,860) of the cases in our cohort, were rarely (1.1% or 17/1,557) treated with platinum. After excluding these hematologic malignancies, multivariable logistic analysis (Fig. 2A) showed a marginal, albeit barely insignificant, association between CH and platinum (odds ratio: 1.14; 95% CI, 0.996–1.31; P = 0.057). By contrast, platinum-based agents were the most frequently used drug in the study by Bolton and colleagues (3) with 44% of the patients being exposed (5,236 exposed vs. 6,695 unexposed), rendering sufficient statistical power to detect such an association.
HSC turnover rates of survivors versus controls were comparable, as indicated by their near-identical telomere attrition rate (Fig. 1D). This provides a rationale for separating age- versus therapy-related events in the survivor cohort. A striking molecular finding in therapy-related CH is the identification of STAT3 as a CH gene in HL survivors (Supplementary Table S5). Single-cell profiling showed that STAT3 mutations are predominantly present in T cells and are therefore unlikely to arise from residual disease, as classic HL is of B-cell origin (25, 26). The sequence context of the two enriched STAT3 mutations in HL, Y640F and N647I, also matches the predominant pattern of the COSMIC mutation signature SBS25. The causality of SBS25 to STAT3 Y640F was further supported, with an estimated probably of 0.985 to 1.0, by scWGS data generated from an HL survivor exposed to procarbazine. Recently, Santarsieri and colleagues (30) found that procarbazine, an alkylating drug used in HL treatment, is likely responsible for SBS25 and recommended that procarbazine be replaced with dacarbazine because dacarbazine is as effective without incurring an excess mutation burden (30). Our findings from scWGS profiling as well as correlative analysis of procarbazine and CH mutation context in the HL survivors (Supplementary Table S6) provide further support for their recommendation.
Although clone size estimated by VAF can predict the subsequent growth of age-related CH clones (Fig. 4B; Supplementary Fig. S7), therapy-related clones appear to lack this feature, at least within the VAF range analyzed in this study (first and third quartiles, 0.2% and 0.7%). This difference in clone dynamics may be attributable to nonmutational factors in addition to the higher mutation burden suggested by the targeted sequencing of bulk samples (Fig. 3C) and scWGS (Fig. 3G). Because of their young age at cancer diagnosis, the survivors analyzed in this study received cancer therapy when their hematopoietic and immune systems were still undergoing development (median 7.1 years old at diagnosis; range, 0.0–23.6), which might have modulated normal development (34), especially for survivors with CH classified as iTR. Although mutated cells with relatively low fitness are routinely eliminated by immune system surveillance in healthy individuals (35, 36), the altered microenvironments in the survivors might have imposed a weaker selection on CH clones, thereby tolerating clones with various adaptation potentials, including those behaving erratically.
Our study has several limitations. First, CH characterization was based on deep sequencing of a panel of 39 genes (Supplementary Table S1), and missing newly identified CH genes is an inherent problem with this approach. For example, our gene panel does not include PPM1D and CHEK2, which were recognized as prominent driver genes in therapy-related CH in recent studies (3, 23). To assess the impact of missing data, we performed targeted sequencing on PPM1D, as the mutation frequency is twice as high as CHEK2's (3). By including only samples available for PPM1D analysis (2,185 survivors and 311 controls; Supplementary Fig. S10A and S10B), there is a slight increase of CH prevalence in survivors by 0.3% (from the original 15.0%–15.3%), which does not have a major impact on the main findings presented in this study. Second, CH variants in noncoding regions or in nondriver genes, which are not assayed by gene panels, can be important markers for defining the CH landscape. A recent study by Mitchell and colleagues (32) performed WGS on single cell–derived colonies of HSCs and found that the majority of expanding HSC subpopulations are not characterized by known drivers. We do recognize, however, that with the current technology, unbiased approaches such as high-coverage (e.g., 100×) WGS or scWGS as used by Mitchell and colleagues (32) are not scalable to profiling a larger cohort such as SJLIFE. Future studies may consider leveraging the strengths of both approaches: The insights gained from a global view of the entire cohort, enabled by panel sequencing, can be complemented by evolutional trajectory mapped by unbiased genome-wide sequencing in selected cases.
In this study, we profiled the blood samples collected at patients’ recent hospital follow-up visits to maximize the sensitivity in CH detection. Despite this, the relatively young ages of the survivors in our cohort (median 31.6 years; range, 6.0–66.4) may not have unveiled the full scope of adverse health conditions that will require continued follow-up efforts. Furthermore, our study has unveiled a growth profile of CH clones different from samples collected from our long-term follow-up cohort from that of samples collected closer to the time of cancer diagnosis and treatment, as in the previous study by Bolton and colleagues (3). These data support the need for longitudinal surveillance of CH in pediatric cancer survivors to evaluate its chronicity and association with clone size, a key feature used for risk stratification in other populations (1).
METHODS
Study Population
Participants were enrolled in the SJLIFE study, a retrospective cohort with prospective clinical follow-up of childhood cancer survivors treated at St. Jude Children's Research Hospital (37). This study was conducted in accordance with the Declaration of Helsinki and approved by the St. Jude Children's Research Hospital institutional review board. Consent for participants under 18 years of age was provided by a parent or legal guardian. All participants ages 14 years and older provided written informed consent; individuals ages between 8 and 13 years provided verbal assent. Eligibility criteria and sample quality control (QC) for sequencing analysis were described previously (ref. 20; Supplementary Methods S1). The current study included pediatric cancer patients who survived at least 5 years since diagnosis. A community control group (SJLIFE controls) consisting of 324 individuals without a history of pediatric cancer with frequency-matched demographic information (i.e., age, sex, and race/ethnicity) was included for comparison purposes. For individuals with blood samples collected at multiple time points, the most recent samples were used for the initial discovery phase of targeted sequencing. Demographic information and key clinical variables used for this study are presented in Supplementary Table S8.
CH Variant Detection
Peripheral blood samples were processed for DNA library construction via hybrid capture–based enrichment over the coding regions of 39 genes implicated in hematologic malignancy or cancer predisposition (Supplementary Table S1). Libraries were sequenced on a NovaSeq 6000 to a target depth of 10,000, and reads were mapped to the GRCh37 genome by BWA (Supplementary Methods S2) followed by running CleanDeepSeq (16) to remove reads with a high error rate. Prior benchmark analysis based on dilution experiment showed that error suppression by CleanDeepSeq enables the detection of variants at VAF of 0.05% to 0.1% (16) and showed high concordance with unique molecular identifier (UMI)–based VAF in our recent testing on a publicly available UMI dataset (38). CH variants with VAF >0.1% were identified in the following four steps: (i) Putative somatic CH SNVs were detected as outliers of alternative allele count and alternative allele fraction distribution of trinucleotide context substitutions (e.g., GCG>GTG) within each sample (Supplementary Methods S3.1). Putative somatic indels were detected by Bambino (39), which was modified for indel detection (40), with realignment (ref. 18; details are described in Supplementary Methods S3.2). This approach has higher sensitivity in detecting rare variants than conventional approaches used for somatic variant detection (Supplementary Methods S3.3). (ii) Nonprotein coding or synonymous SNVs as well as nonprotein-coding indels were filtered. (iii) Orthogonal validation by ddPCR was performed for variants in ∼40% of the genes. Given the novelty of STAT3 CH variants, we analyzed all STAT3 variants by ddPCR provided there were sufficient DNA samples for the assay. Details are described in Supplementary Methods S4 and Supplementary Table S9. (iv) For variants not selected for ddPCR or having a failed assay, their validity was inferred by building a binomial model based on read counts of the validated and notvalidated readout of ddPCR assay (Supplementary Methods S5). Also, amplicon-based sequencing on PPM1D was performed targeting exons 5 and 6 where somatic CH mutations are primarily found (2) and detailed in Supplementary Methods S6. The number of variant calls processed at each filtering step is shown with the CH variant detection flowchart (Supplementary Methods S7).
Germline Genetic Data Source
Carrier status of germline mutations (20, 22) and telomere length estimation (19) were extracted from previously published data. For analyses related to CPG mutations, we used only bona fide pathogenic variants on 60 genes that have been associated with autosomal dominant cancer predisposition syndromes (SJCPG60; refs. 20, 41); thus, variants classified as “likely pathogenic” were not included in this analysis. Variants annotated as mosaic in Wang and colleagues (20) were excluded, as they could be potential CH variants. Analyses related to mutations in DDR genes were based on Qin and colleagues (22), which analyzed 127 DDR genes from six pathways: homologous recombination, nonhomologous end joining, nucleotide excision repair, mismatch repair, base excision repair, and Fanconi anemia. We only included mutations classified as “Pathogenic” by ClinVar (version 2022-10-1), similar to CPG.
Treatment Exposure Analysis
Treatment details were extracted from medical records. Chemotherapy variables were expressed as cumulative dose received per body surface area. To estimate the RT doses relevant to CH, we estimated the maximum region-specific doses abstracted from RT oncology records to the age-specific body distribution of active bone marrow (42). For survivors who had received autologous transplants (n = 70), we modified the therapy doses by excluding therapies undertaken between stem cell harvest and transplantation. For those who received allogenic (n = 1) or syngeneic (n = 1) transplantation, we set the therapy exposures to zero assuming complete replacement by the donor cells. The treatment data in Supplementary Table S8 were based on the modified values. To facilitate the comparison of various magnitudes, all cumulative doses were scaled by standard deviation. When the therapy dose was divided into tertiles, the exposed population was divided into three parts at the 1/3 and 2/3 quantiles of the ordered dose distribution so that the first, second, and third tertiles represented low, medium, and high levels of exposure, respectively. In mediation analysis, pediatric cancer diagnoses were binned into CH-enriched and not-enriched groups (Fig. 2C) represented as Dx* (including HL, soft-tissue sarcoma, germ cell tumor, rhabdomyosarcoma, neuroblastoma, non-HL, acute lymphoblastic leukemia) and Dx (including acute myeloid leukemia, osteosarcoma, Ewing sarcoma family of tumors, retinoblastoma, CNS malignancies, Wilms tumor, and others), respectively. This binary diagnosis was examined by defining the CH status as outcome and the exposure to alkylating agents, bleomycin, or RT with a significant dose (Fig. 2B) as a binary mediator. Logistic regression adjusted for age at sample collection and age at diagnosis was used to assess the mediation.
Modeling Age- and Therapy-Related CH
Using the control data, the probability, 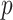 , to develop age-related CH was estimated at a given age category binned as in Fig. 1. Age categories [0, 18) and [18, 30) were combined because the controls were >18 years old.
, to develop age-related CH was estimated at a given age category binned as in Fig. 1. Age categories [0, 18) and [18, 30) were combined because the controls were >18 years old.
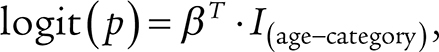 |
where 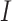 are indicators for each age category.
are indicators for each age category.
Under the proposed model (Supplementary Fig. S4), a survivor and a control in the same age bin would have a similar chance to develop age-related CH. Therefore, the survivor's probability, 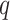 , to develop any CH, which may be age- or therapy-related, was estimated by fixing the age effect as an offset term:
, to develop any CH, which may be age- or therapy-related, was estimated by fixing the age effect as an offset term:
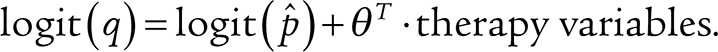 |
The therapy variables included the dose tertiles for alkylating agents, RT, and bleomycin, which were found significantly associated with CH in the multivariable logistic analysis with alkylating agents, RT, bleomycin, platinum, anthracyclines, vinca alkaloids, methotrexate, dactinomycin, epipodophyllotoxins, age at sample collection, and age at diagnosis. The age at diagnosis was also included to capture potential age effects specific to the survivors. Assuming that the age- and therapy-related etiologies were mutually exclusive, CH clones were inferred as therapy-related (iTR) if they belonged to a survivor whose estimated probability for therapy-related CH was higher than the probability for age-related CH:
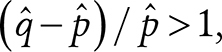 |
otherwise, inferred as age-related (iAR).
Joint Analysis on Cell Phenotyping and STAT3 Y640F Mutation Status by Tapestri
The blood samples from three survivors were analyzed; for SJHL018072, a serial sample collected 3 years after the one used for panel sequencing was used due to material exhaustion. Cryopreserved survivor samples were washed with FACS buffer and quantified using a Luna-FL Dual Fluorescence cell counter. Cells (0.5 × 106–4.0 × 106 viable cells) were then resuspended in cell staining buffer (#420201, BioLegend) in the concentration of 25,000 cells/μL and incubated with TruStain FcX, and 1 × Tapestri blocking buffer (Mission Bio) for 15 minutes on ice. Then TotalSeq-D Human Heme Oncology Cocktail v1.0 (#399906, BioLegend), which contains the pool of 45 oligo-conjugated antibodies, was added and incubated for 30 minutes on ice. Cells were then washed three times with prechilled cell staining buffer (#420201, BioLegend) followed by resuspension of the cells in the Tapestri cell suspension buffer (Mission Bio). We followed the Tapestri Single-cell DNA+Protein sequencing v2 (custom panel) user guide to process the samples for the single-cell amplicon-based DNA and protein sequencing. In brief, after counting the resuspended cells on an automated cell counter (Luna Biosystems), resuspended cells (2,500–3,500 cells/μL) were encapsulated using a Tapestri microfluidics cartridge and lysed. A forward primer mix (30 μmol/L each) for the antibody tags was added before barcoding. Barcoded samples were then subjected to targeted PCR amplification of a custom amplicon covering the STAT3 Y640F locus. DNA PCR products were then isolated from individual droplets and purified with Ampure XP beads as per the user's guide. The DNA PCR products were then used as a PCR template for library generation with additional 4 more PCR cycles and repurified using Ampure XP beads. Protein PCR products were incubated with Tapestri pullout oligo (5 μmol/L) at 96°C for 5 minutes followed by incubation on ice for 5 minutes. Protein PCR products were then purified using streptavidin beads (Mission Bio) and were used as a PCR template for the incorporation of i5/i7 Illumina indices followed by purification using Ampure XP beads. All libraries, both DNA and protein, were quantified using an Agilent Bioanalyzer and pooled for sequencing on an Illumina NextSeq 550 or NovaSeq 6000. The resulting FASTQ files for single-cell DNA and protein libraries were uploaded via the Tapestri portal for analysis by Tapestri's pipeline. This pipeline determines the genotype by analyzing the amplified DNA segment and the phenotype by reading the oligo-tag conjugated to the antibody in the same cell that is indexed by i5/i7 Illumina sequences. The analysis of results contains the 2D coordinate in the cluster space for each cell as well as meta information for the genotype and phenotype, which was visualized in Fig. 3F. The same data were used to summarize cellular fraction of STAT3-mutant cells in each cell type as shown in Supplementary Fig. S5.
Single-Cell Whole-Genome Amplification and STAT3 Y640F Genotyping
Thawed survivor blood samples (the same as those used for the Tapestri assay), which had been cryopreserved, were washed and resuspended in a culture medium. The resuspended cells were stained with Calcein and DAPI for viable cell sorting. Calcein-positive/DAPI-negative cells were sorted into 96-well DNA loBind Semi Skirted plates containing 4 μL of cell storage buffer (Qiagen, #150370). The whole-genome amplification reactions were assembled in a pre-PCR workstation, which was decontaminated with ultraviolet irradiation before every experiment. Multiple displacement amplification (MDA) was carried out according to the Repli-g Advanced DNA Single Cell Kit (Qiagen, #150365). The amplified whole-genome amplification yield was in the range of 25 to 40 μg and was stored at −20°C until further processing STAT3 genotyping and sequencing library preparation. Aliquots from the amplified DNA material were diluted to 25 ng/μL, and the targeted region was amplified by PCR using Q5 Hot Start High-Fidelity 2 × Master Mix (New England BioLabs, #M0494L). Two primer sets were used to generate the amplicons. The first set included the forward primer GGAAAGAAAAAATGGGCAG and the reverse AAATCAACAACTACCTGGG. The second set was TTAAGTCTTTTCCCCTTCG for forward and TCAACAACTACCTGGGTC for reverse, respectively. The PCR fragments were cleaned up using ExoSAP-IT PCR product cleanup reagent (Thermo Fisher Scientific, #78202). The chromatogram from Sanger sequencing was visually inspected for the STAT3 Y640F mutation.
scWGS and Variant Identification
For amplified DNA materials from STAT3 Y640F or wild-type cells, Illumina-compatible whole-genomic DNA libraries were constructed using the Kapa HyperPrep Kit (Roche, #07962363001) and IDT for Illumina TruSeq DNA UD Indexes (Illumina, #20023784). Library QC was performed using a Bioanalyzer High Sensitivity DNA Analysis chip (Agilent, #5067-4626) and MiSeq Nano kit (Illumina, cat. #15036717). The scWGS libraries were denatured with PhiX spike-in, and sequencing was performed on a NovaSeq 6000 using S2 or S4 flow cells and standard workflow to generate 150-cycle paired-end reads. The reads were mapped to the GRCh37 reference genome by BWA. The mapped datasets were required to cover >70% of the genome at a depth of 10 reads or more.
GATK HaplotypeCaller (4.0.2.1) was applied at the default setting to the single-cell datasets and the bulk WGS dataset (20) from the same survivor. GATK's VariantFiltration was used to filter the raw calls: –filter-expression “QD < 2.0” –filter-expression “FS > 60.0” –filter-expression “MQ < 40.0” –filter-expression “MQRankSum < -12.5” –filter-expression “ReadPosRankSum < -8.0” –filter-expression “SOR > 3.0” –filter-expression “QUAL < 30” -window 10. MDA reaction tends to yield a nonuniform amplification and thus could provide a biased genotype representation due to allele dropout. To select datasets with relatively uniform amplification, we performed a QC check that requires 10× coverage in >70% of the human genome in scWGS, and >90% genotype concordance of heterozygous SNPs in bulk WGS sample in regions with >10× coverage in both bulk WGS and scWGS. For scWGS data that passed QC, SNVs private to scWGS (i.e., absent in bulk WGS) were considered putative somatic SNVs. Of the scWGS data generated from three HL survivors, only those from SJHL018072 passed the QC check (93.7%–95.7%), whereas the other two (SJHL017906 and SJHL019340) failed (59.1–87.2%) despite repeated effort for all available DNA materials. From the QC-failed case, SNVs unique to scWGS were also collected for the purpose of building a panel of artifactual SNVs caused by MDA. The final mutation set for SJHL018072 was prepared by filtering with the SNVs recurring in the other two cases, which may represent MDA/sequencing/artifacts, as suggested by the presence of SBS57, an error signature.
Mutational Signature Analysis
We ran signature profiler (https://github.com/AlexandrovLab/SigProfilerSingleSample) to query against COSMIC signatures (v3.1, GRCh37) with the following options: ref = “GRCh37” and exome = False. Given the elevated mutation burden in STAT3 Y640F–mutant cells compared with the wild-type cells, we performed the analysis by combining all SNVs from the mutant cells to generate a profile for the mutant cells and combining all SNVs from the wild-type cells to generate a profile for the wild-type cells. The signatures from these two categories are shown in Fig. 3G.
Serial Sample Analysis
Eighty-four survivors had serial blood samples collected from multiple SJLIFE visits. These samples were analyzed for CH variants as described in the section “CH Variant Detection.” For the pair of time points in each serial sample, the growth direction of a CH clone was determined as expanding or nonexpanding by comparing the two VAFs with a binomial test or taking the concordance of the ddPCR concentration estimates for samples in which the readings were available at both time points.
Simulation Study
The stochastic clone dynamics model proposed by Watson and colleagues (33) was used to simulate the age-related CH dynamics. In addition to the original model, a modified model was prepared to study trajectories with early onsets as in CH-positive survivors. This model elevates the mutation rate [original rate by Watson and colleagues (33): 3 × 10−6/cell] by 5 to 10 times during a random time window starting at age 0 to 15 with a span of 1 to 5 years. Trajectories starting within the hypermutative windows were analyzed. To simulate the serial sample analysis, clones were subjected to a two time-point sampling if VAF stayed >0.1% (the detection limit of this study). The two VAFs were compared to label the clone as expanding or nonexpanding.
Computations
Statistical analyses, simulation, and visualization were performed in Python. Comparisons were considered significant if two-sided P values were < 0.05.
Data Availability
The BAM files for panel sequencing and scWGS are available on St. Jude Cloud under accession SJC-DC-1020 (https://platform.stjude.cloud/data/cohorts?dataset_accession=SJC-DS-1020).
Supplementary Material
Figure S1: Age distribution of the cohort. Figure S2: The distribution of bleomycin 3rd tertile. Figure S3: Distribution of cancer types treated without alkylating agents, platinum, anthracyclines, bleomycin, dactinomycin, epipodophyllotoxins, methotrexate, vinca alkaloids. Figure S4: Conceptual illustration for age- and therapy-related clonal hematopoiesis. Figure S5: Distribution of cell phenotypes harboring STAT3 Y640F mutation. Figure S6: Regression analysis on public dataset to reproduce the clone expansion pattern in main Figure 4A. Figure S7: Longitudinal view of the serial samples. Figure S8: Simulation analysis to reproduce the pattern in main Figure 4B. Figure S9: Comparison of clone growth rates and clone sizes between age- and therapy-related CH. Figure S10: Additional sequencing for PPM1D. Table S1: Gene panel. Table S3: Comparison between CH and sex, race and age. Table S4: Comparison between CH status and pathogenic germline mutations. Separately analyzed for cancer predisposition genes and DNA damage repair genes. Table S5: Enrichment of STAT3 mutations in Hodgkin lymphoma survivors. Table S6: Association between procarbazine exposure and mutation pattern in Hodgkin lymphoma survivors.
Table S2: List of CH mutations identified. Table S7: Mutations analyzed in serial analysis. Table S8: Clinical features of the survivors. Table S9: List of primers used in ddPCR validation.
Acknowledgments
This study was supported by a Cancer Center Support Grant (P30 CA21765) to St. Jude Children's Research Hospital, R01 CA216345 (MPIs: Y. Yasui and J. Zhang), R01 CA216391 (PI: J. Zhang), U01 CA195547 1 (MPIs: M.M. Hudson and K.K. Ness), and the American Lebanese Syrian Associated Charities. We thank Mr. David Rosenfield and team for supporting the analysis of panel sequencing and scWGS data and Mr. Michael Edmonson for his help on proofreading the manuscript. We are grateful to Ms. Delaram Rahbarinia, Mr. Michael Macias, and Mr. Clay McLeod for uploading the sequencing datasets to St. Jude Cloud. We also thank Ms. Jessica Baedke, Ms. Kyla Shelton, Ms. Huiqi Wang, and Dr. Emily Finch for their effort in collecting the clinical information. We thank Dr. Louis Staudt for discussions related to STAT3 mutation in HL. Finally, we acknowledge the very insightful comments from the two reviewers, which helped us to substantially improve the quality of this study.
Footnotes
Note: Supplementary data for this article are available at Cancer Discovery Online (http://cancerdiscovery.aacrjournals.org/).
Authors’ Disclosures
K.K. Ness reports grants from the NIH during the conduct of the study. M.M. Hudson reports grants from the NCI during the conduct of the study. No disclosures were reported by the other authors.
Authors’ Contributions
K. Hagiwara: Conceptualization, data curation, software, formal analysis, investigation, visualization, methodology, writing–original draft, writing–review and editing. S. Natarajan: Data curation, formal analysis, validation, investigation. Z. Wang: Conceptualization, resources, data curation, formal analysis, investigation, methodology. H. Zubair: Data curation, validation, investigation, methodology. H.L. Mulder: Supervision, validation, investigation, methodology. L. Dong: Validation, methodology. E.M. Plyler: Methodology. P. Thimmaiah: Validation, methodology. X. Ma: Software, methodology. K.K. Ness: Conceptualization, resources, data curation, funding acquisition, writing–review and editing. Z. Li: Resources. D.A. Mulrooney: Resources, data curation, investigation, writing–review and editing. C.L. Wilson: Resources, data curation, investigation, writing–review and editing. Y. Yasui: Conceptualization, resources, data curation, formal analysis, supervision, funding acquisition, investigation, methodology, writing–review and editing. M.M. Hudson: Conceptualization, resources, supervision, funding acquisition, project administration, writing–review and editing. J. Easton: Data curation, supervision, validation, investigation, methodology, writing–review and editing. L.L. Robison: Conceptualization, resources, data curation, supervision, project administration, writing–review and editing. J. Zhang: Conceptualization, resources, data curation, formal analysis, supervision, funding acquisition, project administration, writing–review and editing.
References
- 1. Köhnke T, Majeti R. Clonal hematopoiesis: from mechanisms to clinical intervention. Cancer Discov 2021;11:2987–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Coombs CC, Zehir A, Devlin SM, Kishtagari A, Syed A, Jonsson P, et al. Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell 2017;21:374–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bolton KL, Ptashkin RN, Gao T, Braunstein L, Devlin SM, Kelly D, et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat Genet 2020;52:1219–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 2014;20:1472–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371:2488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014;371:2477–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Desai P, Mencia-Trinchant N, Savenkov O, Simon MS, Cheang G, Lee S, et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat Med 2018;24:1015–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ness KK, Kirkland JL, Gramatges MM, Wang Z, Kundu M, McCastlain K, et al. Premature physiologic aging as a paradigm for understanding increased risk of adverse health across the lifespan of survivors of childhood cancer. J Clin Oncol 2018;36:2206–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Robison LL, Hudson MM. Survivors of childhood and adolescent cancer: life-long risks and responsibilities. Nat Rev Cancer 2014;14:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Collord G, Park N, Podestà M, Dagnino M, Cilloni D, Jones D, et al. Clonal haematopoiesis is not prevalent in survivors of childhood cancer. Br J Haematol 2018;181:537–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Coorens THH, Collord G, Lu W, Mitchell E, Ijaz J, Roberts T, et al. Clonal hematopoiesis and therapy-related myeloid neoplasms following neuroblastoma treatment. Blood 2021;137:2992–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bertrums EJM, Rosendahl Huber AKM, de Kanter JK, Brandsma AM, van Leeuwen AJCN, Verheul M, et al. Elevated mutational age in blood of children treated for cancer contributes to therapy-related myeloid neoplasms. Cancer Discov 2022;12:1860–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yeh JM, Ward ZJ, Chaudhry A, Liu Q, Yasui Y, Armstrong GT, et al. Life expectancy of adult survivors of childhood cancer over 3 decades. JAMA Oncol 2020;6:350–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hudson MM, Ness KK, Gurney JG, Mulrooney DA, Chemaitilly W, Krull KR, et al. Clinical ascertainment of health outcomes among adults treated for childhood cancer. JAMA 2013;309:2371–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hudson MM, Ehrhardt MJ, Bhakta N, Baassiri M, Eissa H, Chemaitilly W, et al. Approach for classification and severity grading of long-term and late-onset health events among childhood cancer survivors in the St. Jude lifetime cohort. Cancer Epidemiol Biomarkers Prev 2017;26:666–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ma X, Shao Y, Tian L, Flasch DA, Mulder HL, Edmonson MN, et al. Analysis of error profiles in deep next-generation sequencing data. Genome Biol 2019;20:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu FT, Ting KM, Zhou ZH. Isolation forest. In: 2008 Eighth IEEE International Conference on Data Mining; 2008Dec 15–19; Pisa, Italy. New York: IEEE. p. 413–22. [Google Scholar]
- 18. Hagiwara K, Edmonson MN, Wheeler DA, Zhang J. indelPost: harmonizing ambiguities in simple and complex indel alignments. Bioinformatics 2022;38:549–51. [DOI] [PubMed] [Google Scholar]
- 19. Song N, Li Z, Qin N, Howell CR, Wilson CL, Easton J, et al. Shortened leukocyte telomere length associates with an increased prevalence of chronic health conditions among survivors of childhood cancer: a report from the St. Jude Lifetime Cohort. Clin Cancer Res 2020;26:2362–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang Z, Wilson CL, Easton J, Thrasher A, Mulder H, Liu Q, et al. Genetic risk for subsequent neoplasms among long-term survivors of childhood cancer. J Clin Oncol 2018;36:2078–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. VanderWeele TJ. Mediation analysis: a practitioner's guide. Annu Rev Public Health 2016;37:17–32. [DOI] [PubMed] [Google Scholar]
- 22. Qin N, Wang Z, Liu Q, Song N, Wilson CL, Ehrhardt MJ, et al. Pathogenic germline mutations in DNA repair genes in combination with cancer treatment exposures and risk of subsequent neoplasms among long-term survivors of childhood cancer. J Clin Oncol 2020;38:2728–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pich O, Reyes-Salazar I, Gonzalez-Perez A, Lopez-Bigas N. Discovering the drivers of clonal hematopoiesis. Nat Commun 2022;13:4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Koskela HL, Eldfors S, Ellonen P, van Adrichem AJ, Kuusanmäki H, Andersson EI, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med 2012;366:1905–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Küppers R, Rajewsky K, Zhao M, Simons G, Laumann R, Fischer R, et al. Hodgkin disease: Hodgkin and Reed-Sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B cells at various stages of development. Proc Natl Acad Sci U S A 1994;91:10962–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Marafioti T, Hummel M, Anagnostopoulos I, Foss HD, Falini B, Delsol G, et al. Origin of nodular lymphocyte-predominant Hodgkin's disease from a clonal expansion of highly mutated germinal-center B cells. N Engl J Med 1997;337:453–8. [DOI] [PubMed] [Google Scholar]
- 27. Machado HE, Mitchell E, Øbro NF, Kübler K, Davies M, Leongamornlert D, et al. Diverse mutational landscapes in human lymphocytes. Nature 2022;608:724–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Petljak M, Alexandrov LB, Brammeld JS, Price S, Wedge DC, Grossmann S, et al. Characterizing mutational signatures in human cancer cell lines reveals episodic APOBEC mutagenesis. Cell 2019;176:1282–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee-Six H, Olafsson S, Ellis P, Osborne RJ, Sanders MA, Moore L, et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature 2019;574:532–7. [DOI] [PubMed] [Google Scholar]
- 30. Santarsieri A, Mitchell E, Sturgess K, Brice P, Menne TF, Osborne W, et al. Replacing procarbazine with dacarbazine in escalated BEACOPP dramatically reduces the post treatment haematopoietic stem and progenitor cell mutational burden in Hodgkin lymphoma patients with no apparent loss of clinical efficacy. Blood 2022;140( Suppl 1):1761–4. Abstract nr 624. [Google Scholar]
- 31. Acuna-Hidalgo R, Sengul H, Steehouwer M, van de Vorst M, Vermeulen SH, Kiemeney LALM, et al. Ultra-sensitive sequencing identifies high prevalence of clonal hematopoiesis-associated mutations throughout adult life. Am J Hum Genet 2017;101:50–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mitchell E, Spencer Chapman M, Williams N, Dawson KJ, Mende N, Calderbank EF, et al. Clonal dynamics of haematopoiesis across the human lifespan. Nature 2022;606:343–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Watson CJ, Papula AL, Poon GYP, Wong WH, Young AL, Druley TE, et al. The evolutionary dynamics and fitness landscape of clonal hematopoiesis. Science 2020;367:1449–54. [DOI] [PubMed] [Google Scholar]
- 34. Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell 2015;28:690–714. [DOI] [PubMed] [Google Scholar]
- 35. Caiado F, Pietras EM, Manz MG. Inflammation as a regulator of hematopoietic stem cell function in disease, aging, and clonal selection. J Exp Med 2021;218:e20201541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Laconi E, Marongiu F, DeGregori J. Cancer as a disease of old age: changing mutational and microenvironmental landscapes. Br J Cancer 2020;122:943–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Howell CR, Bjornard KL, Ness KK, Alberts N, Armstrong GT, Bhakta N, et al. Cohort profile: the St. Jude Lifetime Cohort study (SJLIFE) for paediatric cancer survivors. Int J Epidemiol 2021;50:39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Young AL, Challen GA, Birmann BM, Druley TE. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat Commun 2016;7:12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Edmonson MN, Zhang J, Yan C, Finney RP, Meerzaman DM, Buetow KH. Bambino: a variant detector and alignment viewer for next-generation sequencing data in the SAM/BAM format. Bioinformatics 2011;27:865–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hagiwara K, Ding L, Edmonson MN, Rice SV, Newman S, Easton J, et al. RNAIndel: discovering somatic coding indels from tumor RNA-Seq data. Bioinformatics 2020;36:1382–90. Available from: 10.1093/bioinformatics/btz753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med 2015;373:2336–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cristy M. Active bone marrow distribution as a function of age in humans. Phys Med Biol 1981;26:389–400. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Age distribution of the cohort. Figure S2: The distribution of bleomycin 3rd tertile. Figure S3: Distribution of cancer types treated without alkylating agents, platinum, anthracyclines, bleomycin, dactinomycin, epipodophyllotoxins, methotrexate, vinca alkaloids. Figure S4: Conceptual illustration for age- and therapy-related clonal hematopoiesis. Figure S5: Distribution of cell phenotypes harboring STAT3 Y640F mutation. Figure S6: Regression analysis on public dataset to reproduce the clone expansion pattern in main Figure 4A. Figure S7: Longitudinal view of the serial samples. Figure S8: Simulation analysis to reproduce the pattern in main Figure 4B. Figure S9: Comparison of clone growth rates and clone sizes between age- and therapy-related CH. Figure S10: Additional sequencing for PPM1D. Table S1: Gene panel. Table S3: Comparison between CH and sex, race and age. Table S4: Comparison between CH status and pathogenic germline mutations. Separately analyzed for cancer predisposition genes and DNA damage repair genes. Table S5: Enrichment of STAT3 mutations in Hodgkin lymphoma survivors. Table S6: Association between procarbazine exposure and mutation pattern in Hodgkin lymphoma survivors.
Table S2: List of CH mutations identified. Table S7: Mutations analyzed in serial analysis. Table S8: Clinical features of the survivors. Table S9: List of primers used in ddPCR validation.
Data Availability Statement
The BAM files for panel sequencing and scWGS are available on St. Jude Cloud under accession SJC-DC-1020 (https://platform.stjude.cloud/data/cohorts?dataset_accession=SJC-DS-1020).