

Key Points
-
•
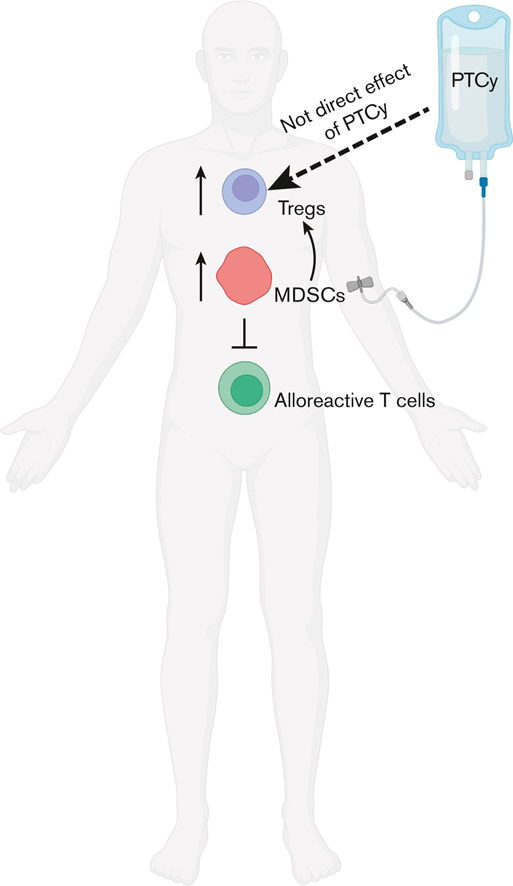
PTCy prevents induction of GVHD from subsequent cell infusion via an indirect effect on Foxp3+ regulatory T cells.
-
•
PTCy modifies the immune environment to expand functional MDSCs, which augment regulatory T-cell recovery.
Visual Abstract
Abstract
Posttransplantation cyclophosphamide (PTCy), given on days +3 and +4, reduces graft-versus-host disease (GVHD) after allogeneic hematopoietic cell transplantation (HCT), but its immunologic underpinnings are not fully understood. In a T-cell–replete, major histocompatibility complex-haploidentical murine HCT model (B6C3F1→B6D2F1), we previously showed that PTCy rapidly induces suppressive mechanisms sufficient to prevent GVHD induction by non-PTCy–exposed donor splenocytes infused on day +5. Here, in PTCy-treated mice, we found that depleting Foxp3+ regulatory T cells (Tregs) in the initial graft but not the day +5 splenocytes did not worsen GVHD, yet depleting Tregs in both cellular compartments led to fatal GVHD induced by the day +5 splenocytes. Hence, Tregs were necessary to control GVHD induced by new donor cells, but PTCy’s impact on Tregs appeared to be indirect. Therefore, we hypothesized that myeloid-derived suppressor cells (MDSCs) play a complementary role. Functionally suppressive granulocytic and monocytic MDSCs were increased in percentages in PTCy-treated mice, and MDSC percentages were increased after administering PTCy to patients undergoing HLA-haploidentical HCT. PTCy increased colony-stimulating factors critical for MDSC development and rapidly promoted the generation of MDSCs from bone marrow precursors. MDSC reduction via anti-Gr1 treatment in murine HCT did not worsen histopathologic GVHD but resulted in decreased Tregs and inferior survival. The clinical implications of these findings, including the potential impact of expanded MDSCs after PTCy on engraftment and cytokine release syndrome, remain to be elucidated. Moreover, the indirect effect that PTCy has on Tregs, which in turn play a necessary role in GVHD prevention by initially transplanted or subsequently infused T cells, requires further investigation.
Introduction
Posttransplantation cyclophosphamide (PTCy) reduces the incidence and severity of graft-versus-host disease (GVHD) after allogeneic hematopoietic cell transplantation (HCT).1 This protective effect thereby allows for the safe use of donors who are not fully HLA-matched, expanding access to the potentially life-saving therapy of HCT to all patients in need.1 However, the mechanisms underlying the effectiveness of PTCy in preventing GVHD have not been well understood.
Recently, we have shown in murine HCT that PTCy prevents GVHD not by eliminating alloreactive T cells but by inducing alloreactive T-cell functional impairment and suppression.2 These data were consistent with those of our prior work showing that CD4+Foxp3+ regulatory T cells (Tregs) are necessary for GVHD prevention by PTCy.3,4 Surprisingly, infusing very high doses of new donor splenocytes did not aggravate GVHD in PTCy-treated mice, even when the new cells were administered as early as 24 hours after PTCy (ie, day +5), suggesting that PTCy rapidly induces highly active suppressive mechanisms2; importantly, the day +5 additional splenocytes were not themselves exposed to PTCy because cyclophosphamide is rapidly cleared within 3 to 4 hours after treatment in mice.5 However, we had not previously characterized the nature of these suppressive mechanisms.
Beyond Tregs, another suppressive cell population that has been shown to play an active role in GVHD control after HCT is that of myeloid-derived suppressor cells (MDSCs).6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 These are a heterogeneous mixture of immature myeloid cells that generally are divided into 2 major subsets (granulocytic MDSCs [G-MDSCs] and monocytic MDSCs [M-MDSCs]),20 mediate suppressive activity via a variety of mechanisms,6,20,21 and recover rapidly after allogeneic HCT.8, 9, 10, 11, 12 MDSCs appear to be increased in mice treated with cyclophosphamide given in a nontransplantation or pretransplantation context.19,22 However, the direct impact of PTCy on MDSCs and the role of MDSCs in PTCy-mediated GVHD prevention have not been previously explored.
In this study, we sought to better understand the nature of the suppressive mechanisms induced by PTCy. We began by investigating the role of Tregs in the prevention of GVHD after new donor splenocyte infusion. We found that Tregs are necessary for this protection, but PTCy appears to have an indirect effect on Tregs. Therefore, we hypothesized that PTCy may be affecting other suppressor cell populations that are active in the early posttransplantation period and thereby contribute to Treg expansion and/or functionality. Here, we focused on the impact of PTCy on MDSCs. We found that phenotypic MDSCs were increased in percentage after PTCy in 3 different murine HCT models and in patients undergoing HLA-haploidentical HCT. These phenotypic murine MDSCs were functionally suppressive of alloreactive T-cell proliferation in mixed lymphocyte cultures. PTCy resulted in increased levels of colony-stimulating factors (CSFs) that drive MDSC generation and quickly promoted the preferential recovery of MDSCs from bone marrow precursors. Depletion of MDSCs did not worsen histopathologic GVHD but did reduce Treg percentages and survival in murine major histocompatibility complex (MHC)-haploidentical HCT.
Methods
Patients
Patients received myeloablative busulfan/fludarabine conditioning followed by T-cell–replete, HLA-haploidentical bone marrow, 50 mg/kg per day PTCy on days +3/+4, and mycophenolate mofetil and sirolimus starting on day +5 (NCT03983850).23 Patients provided informed consent for research studies before specimens were acquired from them. Patient blood samples were subjected to Ficoll density centrifugation to isolate peripheral blood mononuclear cells, which were taken fresh for flow cytometric assessment.
Mice
B6C3F1/Crl, B6D2F1/Crl, B6.SJL-PtprcaPepcb/BoyCrl (Ly5.1), C3H/HeNCrl, and DBA/2NCrl mice were obtained from the Charles River Laboratories. AKR/J mice were obtained from Jackson Laboratories. B6.SJL-PtprcaPepcb/BoyCrl (Ly5.1) females were bred with C3H/HeNCrl males at the National Cancer Institute (NCI) to generate B6C3F1 (CD45.1+CD45.2+) mice, which were used as donors for some experiments. B6C3F1-Foxp3-diphtheria toxin receptor (DTR) mice were bred at the NCI with B6.Foxp3-DTRGFP;luc+ mice as mothers and C3H/HeNCrl mice as fathers.2 Thymectomized B6D2F1/Crl mice were obtained from Charles River Laboratories at 7 to 8 weeks of age after having undergone thymectomy 1 week before shipping. B6D2F1/Crl mice were nonthymectomized unless specifically noted. All donors and recipients were 10 to 12-week-old female littermate controls except for B6C3F1-Foxp3-DTR donor mice, which were 9 to 16-week-old males to allow complete depletion of Foxp3-expressing cells. All mice were maintained in specific pathogen-free conditions, given food and water ad libitum, and treated in accordance with protocols approved by the NCI institutional animal care and use committee.
Murine HCT
HCT was performed as previously described.2 In brief, recipient B6D2F1 mice were irradiated, and 6 to 8 hours later were intravenously administered a graft containing 10 × 106 T-cell–depleted bone marrow cells ± 40 × 106 splenocytes from B6C3F1 (allogeneic) or B6D2F1 (syngeneic) donors. Graft preparation and T-cell depletion of the bone marrow were performed as previously described.2 Mice were monitored daily for survival and euthanized if considered moribund. For survival experiments, blinded evaluations of clinical scores and weights were measured every 3 days as per a standardized scoring rubric.2 Blinded histopathologic assessments were performed as previously described.2
Drug preparation and administration
Cyclophosphamide (Baxter Oncology) and diphtheria toxin (MilliporeSigma) reconstitution and administration were performed as previously described.2 Mice not receiving the drug were always given similar volumes of phosphate-buffered saline (PBS) vehicle intraperitoneally.
MDSC depletion
For in vivo depletion, mice were given intraperitoneal injections of either 200 μg of InVivoMAb anti-mouse Ly6G/Ly6C (Gr1, clone RB6-8C5, Bio X Cell) or InVivoMAb rat immunoglobulin G2b anti-keyhole limpet hemocyanin (isotype control, Bio X Cell) every 4 days for 4 doses starting at day 0, +28, or +150, depending on the experiment. Ex vivo depletion of MDSCs within the allograft was performed by incubating cells with the anti-mouse Ly6G/Ly6C antibody (1:200 dilution) for 30 minutes on ice followed by treatment with guinea pig complement (CedarLane) for 30 minutes at 37°C; for the bone marrow, this treatment was performed along with anti-Thy1.2–mediated T-cell depletion, whereas splenocytes for groups not receiving MDSC-depleted allografts were treated with the complement alone.
Flow cytometry
Up to 3 × 106 viable cells/sample were stained sequentially with the LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (Thermo Fisher), Mouse Fc Block (BD Biosciences, clone 2.4G2), or Human Fc Block (BioLegend) as appropriate, and extracellular antibodies before fixation/permeabilization (eBioscience Foxp3 Staining Kit) and intracellular antibody staining. For compensation purposes, single stains were prepared, and, for some markers such as CD25, CD80, CD115, CD124, F4/80, and PDL1, fluorescence-minus-one controls also were used. Specific monoclonal antibodies used are listed in the supplemental Methods. Data were acquired on a BD Fortessa and analyzed using FCS Express (De Novo Software). Flow cytometric cell sorting was performed on a BD Influx or BD FACSAria II.
Suppression assay
Liver-infiltrating hematopoietic cells were aseptically isolated from PBS-treated or PTCy-treated mice after flushing the liver with sterile PBS, as previously described.2 Viable (LIVE/DEAD-negative) monocytic (CD11b+Ly6G−Ly6Chi) or granulocytic (CD11b+Ly6G+Ly6Clow/int) donor (H2kk+) MDSCs were flow cytometrically isolated on a BD Influx. Flow cytometrically sorted T cells (Thy1.2+) from the spleens of new 10 to 12-week-old B6C3F1 donors were stained with 2.5 μM CellTrace Violet (Thermo Fisher). Stimulator DBA/2 splenocytes were irradiated to 30 Gy and plated at a 3:3:1 ratio of 2 × 105 irradiated DBA/2 splenocyte stimulators to 2 × 105 viable B6C3F1 T-cell responders to 6.7 × 104 M-MDSCs or G-MDSCs in a total volume of 200 μL of cell culture media per well in round-bottom 96-well plates and incubated at 37°C with 5% CO2. On day 5 of culture, the cultured cells were processed for flow cytometric analysis.
Cytokine analysis
Cryopreserved plasma specimens were analyzed in duplicate using a custom 11-plex Mouse Luminex Discovery Assay (Bio-Techne), per the manufacturer’s instructions, with analysis by a Luminex FlexMap3D.
Statistics
The exact log-rank test was used to compare survival distributions. The Wilcoxon rank-sum test was used for weight and clinical score area under the curve comparisons. Weight and clinical score data are shown as the mean ± standard error of the mean. Before t test or one-way analysis of variance, percentages underwent arcsine transformation, and cell counts or cytokine values underwent natural logarithmic transformation. Undetectable cytokine levels were assigned a value of 0.1 before transformation. When analysis of variances were significant, the Holm-Sidak post hoc test was used. Although statistical testing was performed on the transformed data, the nontransformed data are displayed for clarity of understanding. SAS/STAT (13.1-15.1) was used for analyses of survival, weight, and clinical score data. GraphPad Prism (version 9.1) was used for all other statistical analyses and data presentation. All analyses were two-tailed, and P values < .05 are described as statistically significant.
Results
Tregs are necessary to prevent GVHD induction by subsequent donor splenocyte infusion in mice receiving T-cell–replete HCT and PTCy
We previously have shown in our B6C3F1→B6D2F1 murine MHC-haploidentical HCT model that additional (non-PTCy–exposed) donor splenocytes do not exacerbate GVHD when infused as early as 24 hours after PTCy (ie, day +5).2 In this same model, we also had used (B6.Foxp3-DTRGFP × C3H)F1 male donors and thymectomized B6D2F1 recipients to study the impact of Treg depletion on GVHD in PTCy-treated mice,2 confirming our prior findings that Tregs are necessary for GVHD protection by PTCy.3,4 Here, we combined these 2 approaches to better understand the suppressive mechanisms responsible for GVHD prevention after infusion of additional donor splenocytes.
In the first set of experiments, the initial allograft was Foxp3-DTR–expressing, whereas the splenocytes infused on day +5 were wild-type (WT), allowing for depletion only of Tregs contained in the initial allograft. In groups receiving T-cell–replete HCT and PTCy, mice depleted of allograft Tregs but receiving the additional WT splenocyte infusion had similar outcomes compared with those of mice receiving the additional WT splenocyte infusion without Treg depletion and also had improved clinical scores and weights compared with those of mice depleted of graft Tregs but not receiving the additional WT splenocytes (Figure 1), suggesting that the newly infused splenocytes were able to compensate for Treg depletion of the initial allograft.
Figure 1.

Infusion of additional PTCy-nonexposed splenocytes on day +5 does not induce GVHD and even protects against GVHD induced by Treg depletion of the initial day 0 allograft in mice treated with T-cell–replete HCT and PTCy. (A) Schematic of the experimental design. On day 0, 10- to 12-week-old thymectomized female recipient B6D2F1 mice received TBI of 10.5 Gy in a single fraction, and 6 to 8 hours later received transplantation with 10 × 106 TCD BM cells ± 40 × 106 splenocytes (Splen) from 9- to 16-week-old male (B6.Foxp3-DTRGFP × C3H)F1 donors or 10- to 12-week-old WT female B6C3F1 donors. PBS vehicle or 25 mg/kg per day PTCy was administered intraperitoneally on days +3 and +4. DT 25 μg/kg per day was given intraperitoneally on days 0, +1, +6, and +7. Vehicle control or 120 million WT B6C3F1 splenocytes were infused on day +5. Thus, the initial Foxp3-DTR graft, but not the additional WT splenocyte infusion, was susceptible to Treg depletion. (B) A day +5 splenocyte infusion led to rapid GVHD induction and universal fatality when given to mice that received transplantation only with TCD BM allografts. (C) Mice also receiving T-cell–replete splenocyte allografts and PTCy did not have aggravation of GVHD when given a day +5 WT splenocyte infusion, even when Tregs in the initial allograft (but not the day +5 splenocyte infusion) were depleted via administration of DT. This contrasted with worse GVHD seen in Treg-depleted mice not receiving the day +5 WT splenocyte infusion. The combined results are shown from 2 independent experiments with n = 4 to 5 mice per group per experiment. Survival outcomes were compared using the exact log-rank test, and AUC comparisons of weights and clinical scores were performed using Wilcoxon rank-sum test. AUC, area under the curve; BM, bone marrow; DT, diphtheria toxin; NS, not significantly different; TBI, total body irradiation; TCD, T-cell–depleted.
However, in experiments wherein Tregs were depleted both in the initial graft and in the day +5 splenocytes, universally fatal GVHD ensued (Figure 2), which was significantly worse than that seen in Treg-depleted mice not receiving additional splenocyte infusion. These results suggested that Tregs are necessary to prevent GVHD after additional splenocyte infusion for mice receiving T-cell–replete HCT and PTCy, but a direct effect of PTCy on Tregs is not required. Based on these results and our past data showing that Tregs play an increasingly important role in PTCy-mediated GVHD prevention as time progresses after transplantation,2 we hypothesized that other suppressive cell populations may be playing a critical role in facilitating Treg expansion and controlling GVHD early after PTCy.
Figure 2.

Foxp3+Tregs are necessary to prevent GVHD induction after an additional splenocyte infusion on day +5 in mice treated with T-cell–replete HCT and PTCy. (A) Schematic of the experimental design. Thymectomized B6D2F1 mice underwent transplantation as in Figure 1 except that the day +5 splenocyte infusion was 40 × 106 splenocytes from (B6.Foxp3-DTRGFP × C3H)F1 male donors, and DT was administered intraperitoneally on days 0, +1, +5, +6, +11, and +12 to allow complete depletion of Tregs both in the initial Foxp3-DTR graft and in the day +5 Foxp3-DTR splenocytes. (B) Day +5 splenocyte infusion again led to rapid GVHD induction and fatality when given to mice receiving T-cell–depleted BM allografts only. (C) Mice also receiving T-cell–replete splenocytes and PTCy were protected from GVHD induction by day +5 splenocytes, except in groups also having Tregs depleted (both in the initial allograft and the day +5 splenocyte infusion), in which case severe and universally fatal GVHD ensued. The Treg-depleted plus day +5 splenocyte-infused group also had significantly worse survival and weights than those of mice depleted of Tregs that did not receive a day +5 splenocyte infusion, showing that the day +5 splenocytes exacerbated GVHD in this context beyond that simply induced by depleting Tregs in the initial graft. Thus, Tregs, either from the initial allograft and/or the day +5 splenocytes, are necessary to protect PTCy-treated recipients against GVHD induced by a new splenocyte infusion. Combined results are shown from 2 independent experiments with n = 5 mice per group per experiment except for certain control groups: DTR BM/WT Splen day 0, PTCy, DT (total n = 5), DTR BM/DTR Splen day 0 (total n = 4), and WT BM/WT Splen day 0 (total n = 6). Survival outcomes were compared using the exact log-rank test, and AUC comparisons of weights and clinical scores were performed using Wilcoxon rank-sum test. AUC, area under the curve; BM, bone marrow; DT, diphtheria toxin.
MDSC percentages are increased in mice and patients treated with PTCy
We previously found that the percentages of donor leukocytes that are CD11b+CD3−NK1.1−B220− cells are increased in PTCy-treated mice.2 Given that both MDSC populations are CD11b+ and that prior studies suggested a role for MDSCs in GVHD prevention,6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18 we examined PTCy’s influence on MDSCs in our B6C3F1→B6D2F1 HCT model. The older classification for identifying MDSCs delineated CD11b+Gr1high cells as G-MDSCs and CD11b+Gr1low cells as M-MDSCs. Using this classification, we found at day +21 that there seemed to be a dose effect of PTCy on MDSCs, particularly G-MDSCs, but there was no substantial benefit for a dose above the optimal PTCy dose in this model of 25 mg/kg per day on days +3 or +4 (supplemental Figure 1).2
To further examine the dynamics of MDSC recovery after PTCy in our B6C3F1→B6D2F1 HCT model, we conducted experiments wherein we assessed these 2 MDSC populations in the allograft, at posttransplantation day +3 (before PTCy), and at posttransplantation days +7 and +21 (in mice receiving PBS vehicle or 25 mg/kg per day PTCy on days +3 or +4) (Figure 3A-B). To delineate MDSC populations more definitively, we adopted the newer MDSC classification (G-MDSCs: CD11b+Ly6G+Ly6Cint/low and M-MDSCs: CD11b+Ly6G−Ly6Chigh).20 We also used CD45.1+CD45.2+ B6C3F1 donors to best differentiate donor-derived MDSCs from any residual recipient-derived MDSCs and not exclude myeloid cells with low MHC class I expression.
Figure 3.

Phenotypic MDSCs are relatively expanded after PTCy in mice and in patients. (A-D) Ten- to 12-week-old female nonthymectomized recipient mice underwent irradiation (10.5 Gy) and transplantation with TCD BM cells and splenocytes from 10- to 12-week-old female donors. Cell doses were 10 × 106 TCD BM cells and 40 × 106 splenocytes for the B6C3F1→B6D2F1 and C3H→AKR models and 5 × 106 TCD BM cells and 50 × 106 splenocytes for the C3H→B6D2F1 model.2 PBS vehicle or 25 mg/kg per day PTCy was administered intraperitoneally on days +3 and +4. (A-B) Data are shown for the B6C3F1→B6D2F1 MHC-haploidentical HCT model using CD45.1+CD45.2+ B6C3F1 donors and WT B6D2F1 (CD45.1−CD45.2+) recipients. Donor cells were gated based on CD45.1 vs CD45.2 expression. (A) Donor MDSC content in the allograft and at day +3 (before PTCy treatment). (B) PTCy increased the percentages of donor G-MDSCs (CD11b+Ly6G+Ly6Cint/low) and M-MDSCs (CD11b+Ly6G−Ly6Chigh) in various tissues at days +7 and +21. (C) M-MDSCs also were increased at day +6 in the C3H→B6D2F1 MHC-disparate HCT model. Donor cells were gated based on H2kk-positivity. (D) Both G-MDSCs and M-MDSCs were increased at day +7 in the C3H→AKR MHC-matched HCT model. (E) This increase in M-MDSCs also was seen in early posttransplantation samples from patients (n = 5) treated with T-cell–replete, HLA-haploidentical BM transplantation using PTCy. G-MDSCs in patients were not reported because differentiating true G-MDSCs from normal neutrophils contaminating the buffy coat layer of Ficolled blood may not be reliable. For panels A-D, combined results from 2 independent experiments are shown with n = 4 per group per experiment. ∗P < .05, ∗∗P < .01, ∗∗∗P < .001, ∗∗∗∗P < .0001, and NS = not significantly different on unpaired t test.
We found that there were low levels of donor G-MDSCs and M-MDSCs at day +3, but these percentages increased substantially by days +7 and +21 (Figure 3A-B). Furthermore, across hematopoietic (lymph nodes, spleen) and GVHD-target (liver) tissues, the percentages of both donor G-MDSCs and M-MDSCs were increased in PTCy-treated mice compared with PBS-treated mice, particularly at day +7 (Figure 3A-B). We also studied the effect of PTCy on MDSC recovery in 2 other murine HCT models (C3H→B6D2F1 [MHC-disparate], day +6; C3H→AKR [MHC-matched], day +7), again finding that MDSCs, particularly M-MDSCs in these models, were increased in percentages in PTCy-treated mice (Figure 3C-D). This increase in M-MDSCs was also observed early after transplantation in patients treated with HLA-haploidentical HCT and PTCy (Figure 3E).
Expanded MDSCs are functionally suppressive
MDSCs obtained from mice that underwent transplantation in our B6C3F1→B6D2F1 MHC-haploidentical HCT model were functionally suppressive of proliferation of CD8+ and conventional (Foxp3−) CD4+ T cells in response to allogeneic stimulation in vitro (Figure 4). There was no major difference in the suppressive capacity of G-MDSCs retrieved from PTCy-treated mice compared with those from PBS-treated mice, although M-MDSCs from PTCy-treated mice appeared to have somewhat heightened suppressive capability. There were also significant differences between PTCy-treated and PBS-treated mice in other phenotypic markers associated with MDSCs (supplemental Figures 2 and 3).
Figure 4.

MDSCs recovered after PTCy are functionally suppressive of alloreactive T-cell proliferation. Mice underwent transplantation in the B6C3F1→B6D2F1 MHC-haploidentical HCT model as in Figure 3A-B except that WT B6C3F1 mice were used as donors. Mice were given PBS vehicle or 25 mg/kg per day PTCy on days +3 and +4. At day +7, livers were flushed from the heart through the portal vein, and liver-infiltrating cells were isolated. Donor G-MDSCs and M-MDSCs were flow cytometrically sorted from these liver-infiltrating cells, whereas T cells were flow cytometrically sorted from the spleens of new B6C3F1 donors. The T cells were stimulated in vitro with irradiated (30 Gy) host-parental (DBA/2) splenocytes at 2 × 105 responders and 2 × 105 stimulators per well with or without 6.7 × 104 G-MDSCs or M-MDSCs per well (3:3:1 ratio of responders to stimulators to MDSCs). Both G-MDSCs and M-MDSCs suppressed the proliferation of CD8+ and/or CD4+Foxp3− conventional T cells. The combined results from 2 independent experiments are shown with n = 4 to 5 per group per experiment. ∗P < .05, ∗∗P < .01, and ∗∗∗P < .001 on one-way analysis of variance (ANOVA) with the Holm-Sidak post hoc test using the T cells alone group as the control.
PTCy increases levels of CSFs that drive MDSC expansion and promotes the differentiation of MDSCs from bone marrow precursors
Increased levels of MDSCs after PTCy may be due to the preferential survival of MDSCs or increased differentiation of myeloid cells to become MDSCs. Given the very low levels of MDSCs at day +3 (day of PTCy administration) (Figure 3A) and the short lifespan of MDSCs,24 we hypothesized that PTCy may be promoting MDSC differentiation. We first examined plasma levels of cytokines important for MDSC growth/expansion and/or activation.25,26 Despite the effects on MDSCs being more robust in GVHD-target and secondary lymphoid organs than in the blood (Figure 3C-D), we found that plasma levels of granulocyte-macrophage (GM), granulocyte (G), and/or macrophage (M) CSF were increased in PTCy-treated mice across the 3 HCT models (Figure 5A-C). In contrast, cytokines involved in MDSC activation were variably affected (Figure 5A-C), likely reflecting the multifaceted impact of PTCy on T cells as well as MDSCs.2,26,27
Figure 5.

PTCy results in increased levels of CSFs and the preferential differentiation of G-MDSCs from bone marrow precursors. Plasma levels of cytokines involved in MDSC growth/expansion and/or differentiation were assessed in (A) MHC-haploidentical (B6C3F1→B6D2F1, day +7), (B) MHC-disparate (C3H→B6D2F1, day +6), and (C) MHC-matched (C3H→AKR, day +7) HCT models,2 showing consistent increases in M-CSF and variable impact on GM-CSF and G-CSF. VEGF was consistently increased, whereas TNF-α and IL-10 were consistently decreased after PTCy. Effects on other cytokines were more model-dependent. For G-CSF in the C3H→B6D2F1 model, a single value was above the extrapolatable range and was graphed at 100 000. (D) Schematic of an experimental design in which mice underwent transplantation in the B6C3F1→B6D2F1 MHC-haploidentical HCT model as in Figure 4 but also on day +3 received 1.5 × 106 new bone marrow cells that had been flow cytometrically sorted to remove cells that were Ly6G+, Ly6C+, Th1.2+, CD19+, or NK1.1+. (E-F) Seven days after the infusion of these lineage-negative bone marrow cells (ie, posttransplantation day +10), flow cytometry was performed on spleens and bone marrow, showing increased percentages of G-MDSCs after PTCy both in (E) cells derived from the lineage-negative bone marrow given on day +3 and (F) cells derived from the initial allograft given on day 0. The lack of difference in M-MDSCs may be due to a variable effect between these 2 cell types or due to the lack of increase in M-MDSCs in these tissue types and model as shown in Figure 3B at day +7. For all parts, combined results of 2 independent experiments with n = 3 to 4 mice per group per experiment are shown. ∗P < .05, ∗∗P < .01, ∗∗∗P < .001, ∗∗∗∗P < .0001, and NS = not significantly different on unpaired t test. CSF, colony-stimulating factor; G, granulocyte; GM, granulocyte-macrophage; IL, interleukin; M, macrophage; ND, not detected; TNF, tumor necrosis factor; VEGF, vascular endothelial growth factor.
To show more definitively that PTCy was promoting MDSC development from myeloid progenitors, we flow cytometrically sorted Ly6C−Ly6G−Thy1.2−CD19−NK1.1− bone marrow progenitor cells from new B6C3F1 donors and infused these cells on posttransplantation day +3 to mice that previously underwent transplantation using our B6C3F1→B6D2F1 HCT model (Figure 5D). At 7 days after administration, G-MDSCs composed a large percentage of cells derived from these bone marrow progenitors (Figure 5E); percentages of these G-MDSCs had significantly increased in PTCy-treated mice compared with those in vehicle-treated mice (Figure 5E), similar to that observed in G-MDSCs from the original allograft infused on day 0 (Figure 5F), confirming that PTCy augments the differentiation of MDSCs from bone marrow precursors.
Prolonged depletion of Gr1+ cells at early but not late posttransplantation time points worsens outcomes in PTCy-treated mice after allogeneic HCT
Next, we examined the role of MDSCs in GVHD prevention after PTCy by depleting MDSCs. Ex vivo incubation of the donor bone marrow and splenocytes with an anti-Gr1 antibody resulted in substantial reductions of G-MDSCs, but not M-MDSCs, without affecting other immune cell subsets (supplemental Figure 4A). Nevertheless, this treatment had no effect on survival or clinical GVHD in mice that received transplantation with our B6C3F1→B6D2F1 HCT model (supplemental Figure 4B). This lack of effect was likely driven by the rapid recovery of G-MDSCs by day +7 to levels at or above those of mice receiving nondepleted allografts (supplemental Figure 4C). Consequently, this only transient depletion did not alter the effects of PTCy on Tregs or alloreactive T-cell proliferation and differentiation (supplemental Figure 4D-E).2,28,29
To produce more robust and longer-lasting MDSC depletion, we serially administered the anti-Gr1 antibody to transplanted mice. Initial studies of post-HCT depletion showed that MDSCs began to recover by the fifth day after anti-Gr1 injection, so we administered anti-Gr1 antibody (or anti-keyhole limpet isotype control antibody) every 4 days from day 0 to +12 (Figure 6A; supplemental Figure 5), resulting in persistent reduction of both G-MDSCs and M-MDSCs (supplemental Figure 5). Anti-Gr1 treatment had no major impact on outcomes in syngeneic HCT (Figure 6B), whereas anti-Gr1 depletion immediately after transplantation led to significantly inferior survival, weights, and clinical scores in PTCy-treated mice undergoing allogeneic HCT (Figure 6C). In contrast, Gr1+ cell depletion at days +28 or +150 only had a transient effect on weights but no impact on survival or clinical scores after allogeneic HCT using PTCy (supplemental Figure 6).
Figure 6.

Gr1+cell depletion immediately after transplantation worsens survival and clinical outcomes in mice undergoing MHC-haploidentical HCT with PTCy. (A) Schematic of the experimental design. On day 0, 10- to 12-week-old recipient female WT B6D2F1 mice underwent irradiation (10.5 Gy) and transplantation with 10 × 106 TCD BM cells ± 40 × 106 splenocytes (Splen) from 10- to 12-week-old female WT B6C3F1 donors. PBS vehicle or 25 mg/kg per day PTCy was administered intraperitoneally on days +3 and +4. Anti-Gr1 (200 μg/mouse per day) or an isotype control antibody (rat IgG2b anti-keyhole limpet hemocyanin) was given intraperitoneally on days 0, +4, +8, and +12. (B-C) Anti-Gr1 depletion had no appreciable impact in (B) syngeneic HCT, (C) but led to inferior survival, weights, and clinical scores in mice treated with allogeneic MHC-haploidentical HCT using PTCy. The combined results from 2 independent experiments are shown with n = 5 mice per group per experiment. Survival outcomes were compared using the exact log-rank test, and AUC comparisons of weights and clinical scores were performed using Wilcoxon rank-sum test.
Gr1+ cell depletion slightly reduces Treg percentages but does not worsen histopathologic GVHD
Given the rapid fatality of PTCy-treated mice treated with anti-Gr1 immediately after transplantation, we studied the effect of anti-Gr1 treatment on histopathologic GVHD and Treg recovery at days +6 (PBS-treated) or +10 (PTCy-treated). MDSC depletion via anti-Gr1 resulted in a reduction in Treg percentages (Figure 7A). However, anti-Gr1 treatment did not increase histopathologic GVHD (Figure 7B; supplemental Tables 1 and 2), but the rapid fatality in mice treated with anti-Gr1 early after transplantation, seen in Figure 6, appeared related, at least in part, to infection (Figure 7C).
Figure 7.

Gr1+cell depletion immediately after transplantation reduces Treg percentages but does not worsen histopathologic GVHD. Mice underwent transplantation as in Figure 6 and then were taken for flow cytometric and histopathologic assessment at day +6 (PBS-treated) or day +10 (PTCy-treated). Anti-Gr1 (200 μg per mouse per day) or an isotype control antibody was given intraperitoneally on days 0 and +4 for mice evaluated at day +6 or on days 0, +4, and +8 for mice evaluated at day +10. (A) Gr1+ depletion resulted in a reduction in the percentages of Tregs in the blood and/or liver. (B) Gr1+ depletion did not significantly increase histopathologic GVHD after either PBS or PTCy treatment. (C) Histopathology from some mice treated with allogeneic HCT and Gr1 depletion showed bacterial infection in the gastrointestinal tract. (Left) Colon with focal submucosal bacterial colonies with associated inflammatory cell infiltrate, H&E, original magnification ×100. (Right) Liver with bacterial colonies within parenchyma with associated acute inflammatory cell infiltrate, H&E, original magnification ×200. The combined results from 2 independent experiments are shown for panels A-B with n = 4 per group per experiment except for the allo-PTCy anti-Gr1 group (n = 4 total because of excess deaths before day +10).
Discussion
Our prior studies have shown that PTCy prevents GVHD through its effects on both alloreactive conventional and regulatory T cells27; PTCy induces alloreactive conventional T-cell functional impairment and the survival and rapid reconstitution of Tregs that further control GVHD.2, 3, 4,27 Importantly, the influence of Treg depletion was not seen within the first few weeks after transplantation,2 suggesting that other mechanisms may be more dominant in the early posttransplantation period. Moreover, our prior work revealed that potent suppressive mechanisms sufficient to prevent GVHD induction by new splenocyte infusions are established as early as 24 hours after PTCy treatment.2 Yet, we had not previously explored whether this effect was due to Tregs or other suppressor cell populations.
Here, we found that Tregs are required for this protection against GVHD induction by new donor T cells given to PTCy-treated mice. Tregs in the additional splenocyte infusion are sufficient for this protection, but because these Tregs were not themselves exposed to PTCy, it appears that the effect of PTCy on Tregs is indirect. This finding suggests that another cellular or immunologic change directly induced by PTCy may be responsible for the Treg-mediated suppression in this context.
MDSCs, whether endogenous,8, 9, 10,12,14,30,31 in vivo–modified,8,13,15, 16, 17,19,32 or ex vivo–expanded,6,7,18,33,34 have shown the potential to inhibit conventional T-cell functionality, expand Tregs, and reduce GVHD severity. Early studies suggested that cyclophosphamide treatment of adult mice that did not undergo transplantation induced several different suppressor cell populations, one of which was CD11b-expressing.22 Induction of MDSCs by cyclophosphamide may occur when this drug is given as part of the conditioning for transplantation19 as well as after transplantation, as we have shown here. In our current study, we found that functional MDSCs were significantly increased in percentages early after PTCy, in part driven by increased levels of CSFs important for MDSC growth and differentiation and by the consequent enhanced differentiation of MDSCs from bone marrow precursors after PTCy. Different CSFs were increased after PTCy in the various HCT models, but these corresponded to the MDSC types that were increased after PTCy in each respective model. Furthermore, the administration of an anti-Gr1 antibody early after transplantation worsened survival, weights, and clinical scores in PTCy-treated mice, but only in the context of allogeneic HCT, which was consistent with MDSC-mediated suppression of GVHD. However, histopathologic examination of anti-Gr1–treated mice revealed no evidence of aggravated GVHD but did show microabscesses and bacterial tissue invasion.
Therefore, it is unclear whether MDSCs are important for GVHD protection in PTCy-treated mice, or whether these mice may have been dying of infection due to the depletion of Gr1-expressing neutrophils in the setting of mucosal disruption secondary to radiation and GVHD rather than exacerbation of histopathologic GVHD secondary to MDSC depletion. If the mortality in allogeneic HCT was due to infection and not GVHD, the lack of negative effect of Gr1-depleting antibody treatment in mice receiving syngeneic HCT may be due to reduced tissue disruption given the absence of concomitant GVHD at the time of neutrophil depletion, and thus decreased opportunity for bacterial or fungal translocation.
Nonetheless, we cannot fully rule out that MDSCs may play a role in preventing GVHD after PTCy. Prior studies have shown clinical effects of lower-dose MDSC infusions in ameliorating GVHD that did not translate to histopathologic improvement.7 In addition, increased MDSCs after PTCy in our models were particularly prominent within the liver, a GVHD target organ. Furthermore, we observed high, rapid mortality of PTCy-treated mice given anti-Gr1, so only the least affected mice that were able to survive to day +10 were assessed, potentially creating a bias toward lower GVHD histopathologic scores in that group. Indeed, many of those surviving mice still had a substantial amount of M-MDSCs present at day +10 (supplemental Figure 5). Perhaps the mice that died had more significant GVHD, and the ones that survived only did so because of the higher MDSC content. Conversely, PTCy promotes the continual generation of new MDSCs, leading to incomplete depletion with intermittent antibody treatment and thereby potentially confounding the intended effects of anti-Gr1 depletion. Moreover, the lack of clinical or immunologic effect of ex vivo anti-Gr1 depletion was likely due to the quite transient and incomplete (only G-MDSCs affected) depletion.
Even if MDSCs do not play a role in GVHD prevention by PTCy, we are unable with this experimental system to assess whether MDSCs are important in preventing GVHD following new donor splenocyte infusion after PTCy; the high mortality rate seen in mice receiving anti-Gr1 depletion without splenocyte infusion would obscure any effect. This high mortality and the only transient effect of ex vivo MDSC depletion also limit the ability to test the impact of expanded MDSCs after PTCy on beneficial lymphocyte responses, particularly those directed against infection or malignancy.
Interestingly, PTCy did not provide protection against GVHD induced by new donor splenocytes when given on day +5 to mice receiving only T-cell–depleted bone marrow transplants on day 0 (supplemental Figure 7). These results suggest either that there is another important subset, whether MDSCs or others, present in the day 0 transplanted splenocytes that mediates a protective effect or that there may be an important interplay between the donor alloreactive T-cell responses and suppressive effects mediated by non–T-cell subsets from either the splenocytes or the bone marrow graft. Indeed, the immunologic cytokine and proteomic milieu modulate the suppressive activity of MDSCs,6,8,33,35 MDSCs must be present during T-cell priming to prevent GVHD,7 and antigen-specific T-cell responses may enhance the immunosuppressive activity of MDSCs.26 PTCy appears to directly and indirectly affect both T cells and MDSCs, and the consequent impact on the inflammatory cytokine milieu might be expected to both positively and negatively affect MDSCs by promoting MDSC expansion (increased CSFs) but limiting MDSC activation (eg, decreased tumor necrosis factor-α and interleukin-10); even so, our suppression assays suggest that MDSCs in PTCy-treated mice have similar or even enhanced suppressive capability as do MDSCs in vehicle-treated mice.
How PTCy promotes MDSC differentiation from myeloid precursors is unknown. The enhanced levels of CSFs likely play a role, but why these levels are elevated in PTCy-treated mice is unclear. Plasma levels of these factors generally are tightly controlled and are cleared by myeloid cells.36 It is possible that the initial reduction in myeloid numbers related to the cytotoxic effects of cyclophosphamide may lead to the surge in these levels and consequently drive the recovery of myeloid cells, including the preferential recovery of MDSCs. Such a hypothesis would be consistent with the rapid recovery of G-MDSCs from bone marrow precursors (Figure 5E) and after ex vivo depletion of MDSCs within the allografts (supplemental Figure 4).
Overall, these findings are important for understanding more deeply the mechanistic biology of PTCy, including differentiating immune effects that are mechanistic vs those that may only be epiphenomenal. Nevertheless, even epiphenomenal changes may have critical clinical implications, including the potential that elevated MDSC levels may reduce global inflammation or modify the risk profile when other cellular therapies are administered, as is frequently necessary clinically to address mixed/falling chimerism, infectious complications, or malignancy relapse. Indeed, a recent report suggests that early MDSCs may play a role in engraftment in patients with sickle cell disease undergoing HLA-haploidentical HCT using PTCy.37 Thus, the clinical implications of expanded MDSCs after PTCy remain to be elucidated. Furthermore, we have shown that PTCy directly promotes differentiation of MDSCs from myeloid precursors, expanding our understanding of the hematologic and immunologic impact of this ubiquitously used chemotherapeutic. Finally, these studies provide important novel insight into the role of Tregs in PTCy’s ability to prevent GVHD induction by new donor T-cell infusions; Tregs are required for GVHD prevention in this context, but a direct effect of PTCy on Tregs is not required. Therefore, we are continuing to explore the role of other immunosuppressive non–T-cell subsets that may interact with Tregs and work to prevent GVHD after PTCy.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Acknowledgments
The authors thank Devorah Gallardo, Veena Kapoor, Nga Voong Hawk, and William Telford for their technical assistance. The authors thank Jon Inglefield and Yanyu Wang of the Clinical Support Laboratory of the Frederick National Laboratory for Cancer Research for performing the cytokine analyses. The visual abstract was created using BioRender.com.
This work was supported by the Intramural Research Program of the National Cancer Institute of the National Institutes of Health and by the Lasker Foundation.
Authorship
Contribution: C.G.K. designed the study; R.E.F. and M.T.P. contributed to the study design; R.E.F., N.S.N., M.T.P., N.V., S.M.K., S.K.M., X.L., A.d.P., L.P.W., and C.G.K. performed experiments; R.E.F., N.S.N., N.V., S.M.K., and C.G.K. analyzed the data; M.A.E. performed blinded assessments of histopathology; D.J.V. and H.C.-W. designed and performed the statistical analyses; all authors interpreted data; R.E.F., N.S.N., N.V., and C.G.K. designed and created the tables and figures; R.E.F. and C.G.K. wrote the manuscript; and all authors revised the manuscript.
Footnotes
Data are available on request from the corresponding author, Christopher G. Kanakry (christopher.kanakry@nih.gov).
The full-text version of this article contains a data supplement.
Supplementary Material
References
- 1.Kanakry CG, Fuchs EJ, Luznik L. Modern approaches to HLA-haploidentical blood or marrow transplantation. Nat Rev Clin Oncol. 2016;13(1):10–24. doi: 10.1038/nrclinonc.2015.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wachsmuth LP, Patterson MT, Eckhaus MA, Venzon DJ, Gress RE, Kanakry CG. Post-transplantation cyclophosphamide prevents graft-versus-host disease by inducing alloreactive T cell dysfunction and suppression. J Clin Invest. 2019;129(6):2357–2373. doi: 10.1172/JCI124218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ganguly S, Ross DB, Panoskaltsis-Mortari A, et al. Donor CD4+ Foxp3+ regulatory T cells are necessary for posttransplantation cyclophosphamide-mediated protection against GVHD in mice. Blood. 2014;124(13):2131–2141. doi: 10.1182/blood-2013-10-525873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kanakry CG, Ganguly S, Zahurak M, et al. Aldehyde dehydrogenase expression drives human regulatory T cell resistance to posttransplantation cyclophosphamide. Sci Transl Med. 2013;5(211):1–12. doi: 10.1126/scitranslmed.3006960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Said R, Abdel-Rehim M, Sadeghia B, Al-Hashemi S, Hassan M. Cyclophosphamide pharmacokinetics in mice: a comparison between retro orbital sampling versus serial tail vein bleeding. Open Pharmacol J. 2007;1:30–35. [Google Scholar]
- 6.Highfill SL, Rodriguez PC, Zhou Q, et al. Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1-dependent mechanism that is up-regulated by interleukin-13. Blood. 2010;116(25):5738–5747. doi: 10.1182/blood-2010-06-287839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Messmann JJ, Reisser T, Leithauser F, Lutz MB, Debatin KM, Strauss G. In vitro-generated MDSCs prevent murine GVHD by inducing type 2 T cells without disabling antitumor cytotoxicity. Blood. 2015;126(9):1138–1148. doi: 10.1182/blood-2015-01-624163. [DOI] [PubMed] [Google Scholar]
- 8.Wang D, Yu Y, Haarberg K, et al. Dynamic change and impact of myeloid-derived suppressor cells in allogeneic bone marrow transplantation in mice. Biol Blood Marrow Transplant. 2013;19(5):692–702. doi: 10.1016/j.bbmt.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Billiau AD, Fevery S, Rutgeerts O, Landuyt W, Waer M. Transient expansion of Mac1+Ly6-G+Ly6-C+ early myeloid cells with suppressor activity in spleens of murine radiation marrow chimeras: possible implications for the graft-versus-host and graft-versus-leukemia reactivity of donor lymphocyte infusions. Blood. 2003;102(2):740–748. doi: 10.1182/blood-2002-06-1833. [DOI] [PubMed] [Google Scholar]
- 10.Luyckx A, Schouppe E, Rutgeerts O, et al. Subset characterization of myeloid-derived suppressor cells arising during induction of BM chimerism in mice. Bone Marrow Transplant. 2012;47(7):985–992. doi: 10.1038/bmt.2011.207. [DOI] [PubMed] [Google Scholar]
- 11.Demosthenous C, Sakellari I, Douka V, Papayanni PG, Anagnostopoulos A, Gavriilaki E. The role of myeloid-derived suppressor cells (MDSCs) in graft-versus-host disease (GVHD) J Clin Med. 2021;10(10):1–16. doi: 10.3390/jcm10102050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guan Q, Blankstein AR, Anjos K, et al. Functional myeloid-derived suppressor cell subsets recover rapidly after allogeneic hematopoietic stem/progenitor cell transplantation. Biol Blood Marrow Transplant. 2015;21(7):1205–1214. doi: 10.1016/j.bbmt.2015.04.015. [DOI] [PubMed] [Google Scholar]
- 13.Joo YD, Lee SM, Lee SW, et al. Granulocyte colony-stimulating factor-induced immature myeloid cells inhibit acute graft-versus-host disease lethality through an indoleamine dioxygenase-independent mechanism. Immunology. 2009;128(1 suppl):e632–640. doi: 10.1111/j.1365-2567.2009.03048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang J, Chen HM, Ma G, et al. The mechanistic study behind suppression of GVHD while retaining GVL activities by myeloid-derived suppressor cells. Leukemia. 2019;33(8):2078–2089. doi: 10.1038/s41375-019-0394-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang K, Lv M, Chang YJ, et al. Early myeloid-derived suppressor cells (HLA-DR−/lowCD33+CD16−) expanded by granulocyte colony-stimulating factor prevent acute graft-versus-host disease (GVHD) in humanized mouse and might contribute to lower GVHD in patients post allo-HSCT. J Hematol Oncol. 2019;12(31):1–16. doi: 10.1186/s13045-019-0710-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morecki S, Gelfand Y, Yacovlev E, Eizik O, Shabat Y, Slavin S. CpG-induced myeloid CD11b+Gr-1+ cells efficiently suppress T cell-mediated immunoreactivity and graft-versus-host disease in a murine model of allogeneic cell therapy. Biol Blood Marrow Transplant. 2008;14(9):973–984. doi: 10.1016/j.bbmt.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 17.Yin J, Li L, Wang C, Zhang Y. Increased Galectin-9 expression, a prognostic biomarker of aGVHD, regulates the immune response through the Galectin-9 induced MDSC pathway after allogeneic hematopoietic stem cell transplantation. Int Immunopharm. 2020;88:1–11. doi: 10.1016/j.intimp.2020.106929. [DOI] [PubMed] [Google Scholar]
- 18.Park MJ, Baek JA, Kim SY, et al. Myeloid-derived suppressor cells therapy enhance immunoregulatory properties in acute graft versus host disease with combination of regulatory T cells. J Transl Med. 2020;18(483):1–14. doi: 10.1186/s12967-020-02657-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shuyu E, Seth A, Vogel P, Sommers M, Ong T, Pillai AB. Bidirectional immune tolerance in nonmyeloablative MHC-mismatched BMT for murine beta-thalassemia. Blood. 2017;129(22):3017–3030. doi: 10.1182/blood-2016-03-704387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bronte V, Brandau S, Chen SH, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7 doi: 10.1038/ncomms12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baumann T, Dunkel A, Schmid C, et al. Regulatory myeloid cells paralyze T cells through cell-cell transfer of the metabolite methylglyoxal. Nat Immunol. 2020;21(5):555–566. doi: 10.1038/s41590-020-0666-9. [DOI] [PubMed] [Google Scholar]
- 22.Brooks-Kaiser JB L, Hoskin D. Heterogeneity of splenic natural suppressor cells induced in mice by treatment with cyclophosphamide. Immunopharmacology. 1990;25:117–129. doi: 10.1016/0162-3109(93)90015-i. [DOI] [PubMed] [Google Scholar]
- 23.McAdams MJ, Hyder M, Dimitrova D, et al. Phase I/II study of reduced dosing of post-transplantation cyclophosphamide (PTCy) after HLA-haploidentical bone marrow transplantation. Blood. 2021;138(suppl 1):101. [Google Scholar]
- 24.Ostrand-Rosenberg S, Fenselau C. Myeloid-derived suppressor cells: immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J Immunol. 2018;200(2):422–431. doi: 10.4049/jimmunol.1701019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Millrud CR, Bergenfelz C, Leandersson K. On the origin of myeloid-derived suppressor cells. Oncotarget. 2017;8(2):3649–3665. doi: 10.18632/oncotarget.12278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagaraj S, Youn JI, Gabrilovich DI. Reciprocal relationship between myeloid-derived suppressor cells and T cells. J Immunol. 2013;191(1):17–23. doi: 10.4049/jimmunol.1300654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nunes NS, Kanakry CG. Mechanisms of graft-versus-host disease prevention by post-transplantation cyclophosphamide: an evolving understanding. Front Immunol. 2019;10:1–15.2668. doi: 10.3389/fimmu.2019.02668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hadjis AD, Nunes NS, Khan SM, et al. Post-transplantation cyclophosphamide uniquely restrains alloreactive CD4(+) T-cell proliferation and differentiation after murine MHC-haploidentical hematopoietic cell transplantation. Front Immunol. 2022;13 doi: 10.3389/fimmu.2022.796349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wachsmuth LP, Patterson MT, Eckhaus MA, Venzon DJ, Kanakry CG. Optimized timing of post-transplantation cyclophosphamide in MHC-haploidentical murine hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2020;26(2):230–241. doi: 10.1016/j.bbmt.2019.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jensen KP, Hongo DA, Ji X, et al. Development of immunosuppressive myeloid cells to induce tolerance in solid organ and hematopoietic cell transplant recipients. Blood Adv. 2021;5(17):3290–3302. doi: 10.1182/bloodadvances.2020003669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hongo D, Tang X, Baker J, Engleman EG, Strober S. Requirement for interactions of natural killer T cells and myeloid-derived suppressor cells for transplantation tolerance. Am J Transplant. 2014;14(11):2467–2477. doi: 10.1111/ajt.12914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mielcarek M, Martin PJ, Torok-Storb B. Suppression of alloantigen-induced T-cell proliferation by CD14+ cells derived from granulocyte colony-stimulating factor-mobilized peripheral blood mononuclear cells. Blood. 1997;89(5):1629–1634. [PubMed] [Google Scholar]
- 33.Koehn BH, Apostolova P, Haverkamp JM, et al. GVHD-associated, inflammasome-mediated loss of function in adoptively transferred myeloid-derived suppressor cells. Blood. 2015;126(13):1621–1628. doi: 10.1182/blood-2015-03-634691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koehn BH, Blazar BR. Role of myeloid-derived suppressor cells in allogeneic hematopoietic cell transplantation. J Leukoc Biol. 2017;102(2):335–341. doi: 10.1189/jlb.5MR1116-464R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koehn BH, Saha A, McDonald-Hyman C, et al. Danger-associated extracellular ATP counters MDSC therapeutic efficacy in acute GVHD. Blood. 2019;134(19):1670–1682. doi: 10.1182/blood.2019001950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Metcalf D. The colony-stimulating factors and cancer. Nat Rev Cancer. 2010;10(6):425–434. doi: 10.1038/nrc2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bhat DK, Olkhanud PB, Gangaplara A, et al. Early myeloid derived suppressor cells (eMDSCs) are associated with high donor myeloid chimerism following haploidentical HSCT for sickle cell disease. Front Immunol. 2021;12 doi: 10.3389/fimmu.2021.757279. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.