

Abstract
Evolution and change generated an incredible diversity of organisms on this earth. Yet, some processes are so central to life that change is strongly selected against. Synthesis of the eukaryotic messenger RNA is one example. The assemblies that carry out transcription and processing (capping, polyadenylation, and splicing) are so conserved that most genes have recognizable orthologs in yeast and humans. Naturally most would conclude transcription and processing are identical in both sexes. However, this is an assumption. Men and women vastly differ in their physiologies. The incidence of pathologies, symptom presentation, disease outcome, and therapeutic response in each sex vary enormously. Despite the harm ignorance causes women, biological research has been historically carried out without regard to sex. The male mouse was the default mammal. A cultured cell’s sex was considered irrelevant. Attempts to fill this knowledge gap have revealed molecular dissimilarities. For example, the earliest embryonic male and female transcriptomes differ long before fetal sex hormones appear. We used public data to challenge the assumption of sameness by reviewing reports of sex-biased gene expression and gene targeting. We focused on 120 genes encoding non-regulatory proteins involved in mRNA synthesis. Remarkably, genes with recognizable orthologs in yeast and thus LEAST likely to differ, did differ between the sexes. The rapidly growing public databases can be used to compare the expression of any gene in male and female tissues. Appreciating the principles that drive sex differences will enrich our understanding of RNA biology in all humans – men and women.
Graphical/Visual Abstract and Caption

Turning genes on or off is so key that mechanisms are alike between yeast and people. Yet, new molecular approaches are revealing sex differences. Understanding sex differences is needed because most if not all diseases differ in frequency and presentation between men and women.
1. INTRODUCTION
• The centrality of RNA to normal and pathological biology is obvious to readers of WiREs RNA. Ingenious approaches and painstaking experimentation have revealed the nature of the machinery involved in synthesizing mature RNAs. Rigorous studies in vitro and in model organisms and cultured mammalian cells have developed models presented in text books and our classrooms. Remarkably, hundreds of specific proteins are functionally similar between single cell organisms such as yeast and mammals. Such extreme evolutionary homology indicates strong selection for maintaining the accuracy and fidelity of these processes. Direct linkages between RNA synthesis mechanisms revealed in model organisms and specific human diseases have revealed new diagnostic and therapeutic strategies. Thus it is understandable to assume that the stoichiometry of molecular assemblies conserved between yeast and humans would be the same between males and females. However, this is an assumption. In the interest of scientific rigor, we encourage the RNA biology community to use publically available resources to test the assumption that mechanisms involving the core machinery of synthesizing a mature RNA are the same in each sex.
• Why should RNA biologists care about sex differences?
After broad recognition that men and women often differ in disease presentation and therapeutic response, see Box 1, the US National Institutes of Health (NIH) has required including women in NIH-funded clinical research since 1993. The failure to investigate the impact of sex yielded treatments that were ineffective or even harmful to women. Because ignorance of sex differences in pre-clinical data reduced the effectiveness of clinical trials and exacerbates the problem of experimental irreproducibility, the NIH also began requiring investigators to consider sex as a biological variable in 2016 (Clayton & Collins, 2014). One of the most comprehensive reviews of sex differences at all levels from cultured cells to adult non-reproductive organs was prepared by the Institute of Medicine (U.S.). Committee on Understanding the Biology of Sex and Gender Differences (Wizemann & Pardue, 2001), see Box 2. Sadly, two decades following publication, the biological underpinnings of sex differences remain incompletely investigated. Calls for both greater inclusion of females and greater rigor in reporting and analysing sex differences continue (Editorial, 2020; Garcia-Sifuentes & Maney, 2021; Hosman et al., 2022; Ozdemir et al., 2022; Woitowich et al., 2020).
Box 1: Impact of Sex on Human Disease.
Sex and gender influence biochemical, genetic, physiological, physical, and behavioural parameters that alter the incidence, presentation, and course of congenital defects and of diseases, as well as response to therapies (Ozdemir et al., 2022; Rubin, 2022; Westergaard et al., 2019; Wizemann & Pardue, 2001). Modern genomics approaches have identified embryonic gene regulatory differences from the moment of fertilization, long before the embryo synthesizes sex hormones (Deegan et al., 2021). Dissimilarities in immune function; cardiovascular onset and symptoms, bone metabolism, response to toxins and drugs, brain function, pain sensitivity, and cancers have been known for decades (Wizemann & Pardue, 2001). Many studies have documented differences in age of diagnosis (typically lower for men) and disease progression in most, if not all, diseases at the population level (Westergaard et al., 2019). The wide discrepancies have prompted fervent calls for research efforts that include both men and women in clinical studies and that balance and document the sex of cells and animals in lab experiments (Clayton & Collins, 2014; Editorial, 2020; Hosman et al., 2022; Ozdemir et al., 2022; Wizemann & Pardue, 2001; Woitowich et al., 2020). Personalized medicine will fail without consideration of sex in research from the bench to bedside.
Box 2: Mechanisms by which Sex Influences mRNA Abundance.
Sex is biologically defined according to chromosomal complement (XY for males; XX for females in humans) and reproductive organs. The difference in gene dosage on the X or Y chromosomes exert biomedically-relevant changes from fertilization onwards (Deegan et al., 2021; Wizemann & Pardue, 2001). Men, of course, have Y-specific genes such as sex-determining region Y (SRY) which initiates testes development. The random X-inactivation or silencing of most, but not all, genes on one of the two X chromosomes in females or the extra X chromosomes in males with X aneuploidy helps assure similar ratios of X to autosome gene expression as in XY males (Fang et al., 2021). However, a third of the over 1100 X-linked genes are not silenced leaving women with twice the gene dosage as men (Cantone & Fisher, 2017; Fang et al., 2021; Tukiainen et al., 2017). The mosaic expression of maternal or paternal X-linked alleles in females also contributes differences. Sex of the parents controls the expression of over 200 genes by imprinting, whereby a gene is expressed on either the maternal or paternal allele, but not both. Monoallelic expression causes a mutated allele to exhibit a different phenotype based on the parental origin of that allele. Sex hormones such as estrogen and androgen drive the developmental acquisition of female and male features. Furthermore, variation in hormones, for example during puberty, pregnancy, lactation, and menopause, causes sex biases that change over the lifespan. Beyond these biological dissimilarities, gender, a social construct, causes biomedically-relevant environmental experiences. This review focuses on sex, because gender is considerably less well-studied from a molecular and medical perspective.
• Repeating the complex biochemical experiments that underpin the models explaining RNA synthesis mechanisms in both male and female cells is not feasible. However, the growing strength of large, public databases and bioinformatics techniques make a first level assessment of sex differences both rapid and inexpensive. Using publications describing sex differences in global gene expression, we evaluated the RNAs that encode proteins involved in the transcription and processing leading to a mature messenger RNA in tissues from men and women. We limited our analyses to 120 basal machinery proteins whose levels would be reasonably assumed to lack sex bias and to 4 tissues and 2 cultured cell types (Table 2). As described below, differences are apparent in both tissues and in cultured cells. A similar approach can be used to consider sex differences in the expression of any gene.
Table 2. Sex-Biased Genes.
Differential RNA abundance between tissues from males or females was assessed by microarrays or RNA seq as described in all publications except (Lopes-Ramos et al., 2020) which assessed sex bias in the transcription factor network targeting each gene. Table 2 summarizes the authors’ determinations of sex-biased gene expression according to the criteria of each study. The published predictions of sex bias also were compared to the median Transcripts Per Million (TPM) indicated in GTEx Analysis Release v8 (dbGaP Accession phs000424. v8.p2) downloaded April 2022. An asterisk indicates differences between predicted bias and observed raw median differences. Differences with GTEx v8 ratios were most frequent for RNAs with very low abundance or small fold differences. Note, that the approved names are used in all gene expression studies; however, reviews of mechanisms often used aliases. Alternative names can be found here: https://www.genenames.org/.
| Approved Gene name | HGNC ID | Location | Approved Gene Symbol | Lung1 | Heart left ventricle1 | Skeletal2 Muscle | Thyroid1 | cultured Fibroblasts3 | newborn (day 1) and adult vascular endothelial cells4 |
|---|---|---|---|---|---|---|---|---|---|
| RNA Pol II TRANSCRIPTION INITIATION (Sainsbury et al., 2015) | |||||||||
| cyclin H | HGNC:1594 | 5q14.3 | CCNH | M>F (Naqvi et al., 2019) | M>F* (Naqvi et al., 2019) | M>F (Naqvi et al., 2019); F>M* (Lopes-Ramos et al., 2020) | |||
| cyclin dependent kinase 7 | HGNC:1778 | 5q13.2 | CDK7 | M>F (Naqvi et al., 2019) | M>F (Naqvi et al., 2019) | F>M (Naqvi et al., 2019); M>F (Lopes-Ramos et al., 2020) | |||
| ERCC excision repair 2, TFIIH core complex helicase subunit | HGNC:3434 | 19q13.32 | ERCC2 | F>M (Naqvi et al., 2019; Oliva et al., 2020) | |||||
| general transcription factor IIA subunit 1 | HGNC:4646 | 14q31.1 | GTF2A1 | M>F (Hartman et al., 2020) | |||||
| general transcription factor IIA subunit 2 | HGNC:4647 | 15q22.2 | GTF2A2 | ||||||
| general transcription factor IIF subunit 1 | HGNC:4652 | 19p13.3 | GTF2F1 | F>M (Naqvi et al., 2019) | F>M (Naqvi et al., 2019) | ||||
| general transcription factor IIF subunit 2 | HGNC:4653 | 13q14.12-q14.13 | GTF2F2 | M>F* (Lopes-Ramos et al., 2020) | |||||
| general transcription factor IIH subunit 2 | HGNC:4656 | 5q13.2 | GTF2H2 | (Hartman, Mokry, et al., 2021) | (Hartman, Mokry, et al., 2021) | ||||
| general transcription factor IIH subunit 5 | HGNC:21157 | 6q25.3 | GTF2H5 | F>M (Naqvi et al., 2019) | |||||
| MNAT1 component of CDK activating kinase | HGNC:7181 | 14q23.1 | MNAT1 | M>F* (Naqvi et al., 2019) | M>F* (Naqvi et al., 2019) | M>F (Lopes-Ramos et al., 2020; Naqvi et al., 2019) | |||
| RNA polymerase II subunit B | HGNC:9188 | 4q12 | POLR2B | F>M (Lopes-Ramos et al., 2020)1 | |||||
| RNA polymerase II subunit C | HGNC:9189 | 16q21 | POLR2C | M>F (Lopes-Ramos et al., 2020) | |||||
| RNA polymerase II, I and III subunit E | HGNC:9192 | 19p13.3 | POLR2E | M>F* (Lopes-Ramos et al., 2020) | M>F (Lopes-Ramos et al., 2020) | ||||
| RNA polymerase II, I and III subunit F | HGNC:9193 | 22q13.1 | POLR2F | M>F (Lopes-Ramos et al., 2020) | |||||
| RNA polymerase II subunit G | HGNC:9194 | 11q12.3 | POLR2G | F>M (Naqvi et al., 2019) | M>F* (Naqvi et al., 2019) | ||||
| RNA polymerase II subunit I | HGNC:9196 | 19q13.12 | POLR2I | M>F (Lopes-Ramos et al., 2020) | |||||
| RNA polymerase II, I and III subunit L | HGNC:9199 | 11p15.5 | POLR2L | M>F (Lopes-Ramos et al., 2020) | |||||
| RNA polymerase II subunit J | HGNC:9197 | 7q22.1 | POLR2J | M>F* (Lopes-Ramos et al., 2020) | M>F (Lopes-Ramos et al., 2020) | ||||
| TATA-box binding protein associated factor 1 | HGNC:11535 | Xq13.1 | TAF1 | F>M* (Lopes-Ramos et al., 2020) | |||||
| TATA-box binding protein associated factor 2 | HGNC:11536 | 8q24.12 | TAF2 | F>M* (Lopes-Ramos et al., 2020) | |||||
| TATA-box binding protein associated factor 3 | HGNC:17303 | 10p14 | TAF3 | F>M (Mayne et al., 2016) | M>F (Naqvi et al., 2019) | ||||
| TATA-box binding protein associated factor 5 | HGNC:11539 | 10q24.33 | TAF5 | F>M (Naqvi et al., 2019) | |||||
| TATA-box binding protein associated factor 7 | HGNC:11541 | 5q31.3 | TAF7 | F>M (Gershoni & Pietrokovski, 2017; Lopes-Ramos et al., 2020) | |||||
| TATA-box binding protein associated factor 9 | HGNC:11542 | 5q13.2 | TAF9 | M>F (Lopes-Ramos et al., 2020) | |||||
| TATA-box binding protein associated factor 10 | HGNC:11543 | 11p15.4 | TAF10 | M>F (Lopes-Ramos et al., 2020) | |||||
| TATA-box binding protein associated factor 11 | HGNC:11544 | 6p21.31 | TAF11 | M>F (Hartman et al., 2020) | |||||
| TATA-box binding protein associated factor 12 | HGNC:11545 | 1p35.3 | TAF12 | M>F (Lopes-Ramos et al., 2020) | |||||
| TATA-box binding protein associated factor 13 | HGNC:11546 | 1p13.3 | TAF13 | M>F (Lopes-Ramos et al., 2020; Naqvi et al., 2019) | |||||
| TATA-box binding protein | HGNC:11588 | 6q27 | TBP | F>M (Lopes-Ramos et al., 2020; Naqvi et al., 2019) | |||||
| POLYADENYLATION (MacDonald, 2019; Tian & Manley, 2017) | |||||||||
| cleavage and polyadenylation specific factor 7 | HGNC:30098 | 11q12.2 | CPSF7 | F>M* (Lopes-Ramos et al., 2020) | |||||
| cleavage stimulation factor subunit 1 | HGNC:2483 | 20q13.2-q13.31 | CSTF1 | F>M (Naqvi et al., 2019) | M>F (Oliva et al., 2020) | ||||
| cleavage stimulation factor subunit 2 | HGNC:2484 | Xq22.1 | CSTF2 | M>F(Naqvi et al., 2019) | M>F (Tukiainen et al., 2017) | ||||
| cleavage stimulation factor subunit 3 | HGNC:2485 | 11p13 | CSTF3 | M>F (Naqvi et al., 2019) | M>F* (Naqvi et al., 2019); F>M (Lopes-Ramos et al., 2020) | M>F (Naqvi et al., 2019); F>M* (Lopes-Ramos et al., 2020) | |||
| RB binding protein 6, ubiquitin ligase | HGNC:9889 | 16p12.1 | RBBP6 | M>F (Naqvi et al., 2019) | |||||
| CAPPING (Galloway & Cowling, 2019,Galloway & Cowling, 2019) | |||||||||
| cap methyltransferase 1 | HGNC:21077 | 6p21.2 | CMTR1 | F>M* (Lopes-Ramos et al., 2020) | |||||
| cap methyltransferase 2 | HGNC:25635 | 16q22.2 | CMTR2 | F>M (Lopes-Ramos et al., 2020) | |||||
| RNA guanylyltransferase and 5'-phosphatase | HGNC:10073 | 6q15 | RNGTT | M>F (Naqvi et al., 2019) | |||||
| RNA guanine-7 methyltransferase | HGNC:10075 | 18p11.21 | RNMT | F>M* (Lopes-Ramos et al., 2020) | |||||
| SPLICING (Bertram et al., 2020; Fica, 2020; Kastner et al., 2019; Verma et al., 2018) | |||||||||
| BR serine/threonine kinase 1 | HGNC:18994 | 19q13.42 | BRSK1 | M>F (Lopes-Ramos et al., 2020) | |||||
| elongation factor Tu GTP binding domain containing 2 | HGNC:30858 | 17q21.31 | EFTUD2 | F>M (Lopes-Ramos et al., 2020; Naqvi et al., 2019) | |||||
| LSM2 homolog, U6 small nuclear RNA and mRNA degradation associated | HGNC:13940 | 6p21.33 | LSM2 | F>M (Oliva et al., 2020) | F>M* (Oliva et al., 2020) | ||||
| LSM3 homolog, U6 small nuclear RNA and mRNA degradation associated | HGNC:17874 | 3p25.1 | LSM3 | M>F (Lopes-Ramos et al., 2020) | |||||
| LSM4 homolog, U6 small nuclear RNA and mRNA degradation associated | HGNC:17259 | 19p13.11 | LSM4 | M>F* (Naqvi et al., 2019) | M>F (Lopes-Ramos et al., 2020) | ||||
| PHD finger protein 5A | HGNC:18000 | 22q13.2 | PHF5A | F>M (Naqvi et al., 2019) | |||||
| pre-mRNA processing factor 3 | HGNC:17348 | 1q21.2 | PRPF3 | F>M (Lopes-Ramos et al., 2020) | F>M (Lopes-Ramos et al., 2020) | ||||
| pre-mRNA processing factor 4 | HGNC:17349 | 9q32 | PRPF4 | F>M (Naqvi et al., 2019) | |||||
| pre-mRNA processing factor 6 | HGNC:15860 | 20q13.33 | PRPF6 | M>F (Lopes-Ramos et al., 2020) | |||||
| pre-mRNA processing factor 8 | HGNC:17340 | 17p13.3 | PRPF8 | F>M (Gershoni & Pietrokovski, 2017; Lopes-Ramos et al., 2020) | F>M* (Lopes-Ramos et al., 2020) | ||||
| pre-mRNA processing factor 31 | HGNC:15446 | 19q13.42 | PRPF31 | F>M* (Naqvi et al., 2019) | F>M* (Naqvi et al., 2019) | ||||
| RNA binding region (RNP1, RRM) containing 3 | HGNC:18666 | 1p21.1 | RNPC3 | F>M* (Lopes-Ramos et al., 2020) | |||||
| spliceosome associated factor 1, recruiter of U4/U6.U5 tri-snRNP | HGNC:10538 | 11q13.1 | SART1 | F>M* (Naqvi et al., 2019) | M>F* (Lopes-Ramos et al., 2020) | M>F (Lopes-Ramos et al., 2020; Naqvi et al., 2019) | |||
| splicing factor 1 | HGNC:12950 | 11q13.1 | SF1 | F>M (Lopes-Ramos et al., 2020) | F>M* (Lopes-Ramos et al., 2020) | ||||
| splicing factor 3a subunit 2 | HGNC:10766 | 19p13.3 | SF3A2 | F>M* (Oliva et al., 2020) | F>M (Naqvi et al., 2019) | F>M* (Lopes-Ramos et al., 2020; Naqvi et al., 2019) | |||
| splicing factor 3a subunit 3 | HGNC:10767 | 1p34.3 | SF3A3 | F>M (Gershoni & Pietrokovski, 2017) | |||||
| splicing factor 3b subunit 2 | HGNC:10769 | 11q13.1 | SF3B2 | F>M (Lopes-Ramos et al., 2020) | F>M* (Lopes-Ramos et al., 2020) | ||||
| splicing factor 3b subunit 3 | HGNC:10770 | 16q22.1 | SF3B3 | M>F (Naqvi et al., 2019) | F>M* (Lopes-Ramos et al., 2020) | ||||
| splicing factor 3b subunit 4 | HGNC:10771 | 1q21.2 | SF3B4 | F>M (Naqvi et al., 2019) | F>M (Lopes-Ramos et al., 2020; Naqvi et al., 2019) | M>F* (Oliva et al., 2020) | |||
| splicing factor 3b subunit 5 | HGNC:21083 | 6q24.2 | SF3B5 | M>F (Lopes-Ramos et al., 2020) | |||||
| splicing factor 3b subunit 6 | HGNC:30096 | 2p23.3 | SF3B6 | F>M (Oliva et al., 2020) | F>M (Oliva et al., 2020) | ||||
| small nuclear ribonucleoprotein U11/U12 subunit 25 | HGNC:14161 | 16p13.3 | SNRNP25 | M>F* (Naqvi et al., 2019); F>M (Oliva et al., 2020) | M>F (Lopes-Ramos et al., 2020; Naqvi et al., 2019) | M>F (Lopes-Ramos et al., 2020) | |||
| small nuclear ribonucleoprotein U11/U12 subunit 35 | HGNC:30852 | 12q24.31 | SNRNP35 | F>M (Naqvi et al., 2019) | M>F* (Naqvi et al., 2019) | F>M* (Naqvi et al., 2019) | |||
| small nuclear ribonucleoprotein U5 subunit 40 | HGNC:30857 | 1p35.2 | SNRNP40 | F>M (Lopes-Ramos et al., 2020) | F>M* (Lopes-Ramos et al., 2020) | ||||
| small nuclear ribonucleoprotein U11/U12 subunit 48 | HGNC:21368 | 6p24.3 | SNRNP48 | F>M (Naqvi et al., 2019) | |||||
| small nuclear ribonucleoprotein U1 subunit 70 | HGNC:11150 | 19q13.33 | SNRNP70 | F>M* (Naqvi et al., 2019) | F>M* (Naqvi et al., 2019) | ||||
| small nuclear ribonucleoprotein U5 subunit 200 | HGNC:30859 | 2q11.2 | SNRNP200 | F>M* (Lopes-Ramos et al., 2020) | |||||
| small nuclear ribonucleoprotein polypeptide C | HGNC:11157 | 6p21.31 | SNRPC | M>F (Lopes-Ramos et al., 2020) | |||||
| small nuclear ribonucleoprotein D1 polypeptide | HGNC:11158 | 18q11.2 | SNRPD1 | F>M (Naqvi et al., 2019) | M>F* (Naqvi et al., 2019) | ||||
| small nuclear ribonucleoprotein D2 polypeptide | HGNC:11159 | 19q13.2-q13.3 | SNRPD2 | M>F (Lopes-Ramos et al., 2020) | |||||
| small nuclear ribonucleoprotein polypeptide E | HGNC:11161 | 1q32.1 | SNRPE | M>F (Hartman et al., 2020) | |||||
| thioredoxin like 4A | HGNC:30551 | 18q23 | TXNL4A | M>F* (Lopes-Ramos et al., 2020; Naqvi et al., 2019) | |||||
| U2 small nuclear RNA auxiliary factor 1 | HGNC:12453 | 21q22.3 | U2AF1 | F>M (Lopes-Ramos et al., 2020) | |||||
| U2 small nuclear RNA auxiliary factor 2 | HGNC:23156 | 19q13.42 | U2AF2 | F>M (Hartman et al., 2020) | |||||
| zinc finger CCHC-type and RNA binding motif containing 1 | HGNC:29620 | 12q12 | ZCRB1 | M>F (Lopes-Ramos et al., 2020) | M>F (Lopes-Ramos et al., 2020) | ||||
| zinc finger matrin-type 5 | HGNC:28046 | 22q12.2 | ZMAT5 | F>M (Naqvi et al., 2019) | F>M* (Naqvi et al., 2019) | F>M* (Naqvi et al., 2019) | |||
| zinc finger CCCH-type, RNA binding motif and serine/arginine rich 2 | HGNC:23019 | Xp22.2 | ZRSR2 | F>M (Kassam et al., 2019; Mayne et al., 2016; Oliva et al., 2020; Tukiainen et al., 2017) | F>M (Kassam et al., 2019; Oliva et al., 2020; Tukiainen et al., 2017) | F>M (Gershoni & Pietrokovski, 2017; Kassam et al., 2019; Lopes-Ramos et al., 2020; Oliva et al., 2020; Tukiainen et al., 2017) | F>M (Gershoni & Pietrokovski, 2017; Kassam et al., 2019; Mayne et al., 2016; Oliva et al., 2020; Tukiainen et al., 2017) | F>M (Gershoni & Pietrokovski, 2017; Kassam et al., 2019; Oliva et al., 2020; Tukiainen et al., 2017) | F>M (Hartman et al., 2020) |
The following genes were not deemed sex-biased in these 6 tissues
Transcription initiation: POLR2H, GTF2B, TAF4, TAF6, TAF8, TAF11, GTF2E1, GTF2E2, GTF2H1, GTF2H3, GTF2H4, ERCC3
Polyadenylation: CPSF1, CPSF2, CPSF3, CPSF4, FIP1L1, WDR33, CSTF2T, NUDT21, CPSF6, SYMPK, PAPOLA, PABPN1
Capping: RAMAC
Splicing: SNRPA, SNRPA1, SNRPB, SF3A1, PPIH, SNU13, DDX23, SNRNP27, SNRPD3, SNRPE, SNRPG, LSM6, LSM7, LSM8, U2AF2
(Gershoni & Pietrokovski, 2017; Hartman, Mokry, et al., 2021; Kassam et al., 2019; Lopes-Ramos et al., 2020; Mayne et al., 2016; Mele et al., 2015; Naqvi et al., 2019; Oliva et al., 2020)
(Gershoni & Pietrokovski, 2017; Hartman, Mokry, et al., 2021; Kassam et al., 2019; Lopes-Ramos et al., 2020; Mele et al., 2015; Naqvi et al., 2019; Oliva et al., 2020)
DOES SEX ALTER THE EXPRESSION OF MY FAVORITE GENE?
• Over the years, we and others have struggled to identify reference genes whose expression is stable between organs, cell types, and physiological conditions including sex. The expression of so called “housekeeping” genes often varies with sex, cell type, or other parameters. One group reviewed published data regarding 50 candidate reference genes to identify good reference genes for cancer studies. The authors concluded that no single gene or even set of genes is entirely stable enough to normalize gene expression in a relatively limited set of 13 cancers (Sharan et al., 2015). As might be expected for a key component of the transcription initiation complex, TATA-box binding protein (TBP) ranked highly in stability. Sharan et al. did not address the potential impact of sex on the variable expression of the candidate reference genes. However, another group tested nine potential reference genes, including TBP, in three types of tissue culture cells exposed to testosterone or estrogen (Fochi et al., 2021). TBP RNA levels were least affected by hormone treatment in cultured cells. Furthermore, Lopes-Ramos et al. predicted that TBP would lack sex bias in 27 of 29 organs (Lopes-Ramos et al., 2020). Naqvi et al. observed a lack of evolutionarily conserved bias in 11 of 12 tested organs (Naqvi et al., 2019). Thus TBP appeared stable between men and women in most tissues. In contrast, in skeletal muscle, Naqvi et al. observed sex-biased differential expression conserved between mammals and Lopes-Ramos et al. observed sex-biased gene regulatory networks. Thus one must be cautious in assuming that sex cannot influence any particular gene, no matter how essential, in all tissues under all physiological situations. Fortunately, the expanding resources of publically available data and analyses can be used to screen the expression of nearly any gene for overt sex bias in many human tissues.
2. USING PUBLIC DATABASES TO ASSESS SEX BIAS IN MANY HUMAN CELL TYPES.
• The Genotype-Tissue Expression (GTEx) Program is a data resource and tissue bank to study the relationship between genetic variants and gene expression in multiple human tissues from hundreds of individuals (https://www.gtexportal.org/home/). The GTEx’s latest dataset (v8) contains RNA-seq data from 54 tissue sites and two cell lines derived from 636 male and 312 female donors. Both protein-coding and non-coding RNA levels are included. Many publications described using the GTEx RNA seq data to assess RNA abundances between the sexes (see Table 1). Table 1 also includes two publications that assesses sex-biased gene expression in microarrays or non-GTEx RNA-seq data. The computational and statistical methodologies in each paper differed greatly and are briefly summarized in Table 1.
Table 1.
Key Points of Publications Assessing Sex-Biased Gene Expression.
| Reference | donor or sample numbers (% male)1 | n samples/tissue males | n samples/tissue females | n tissues | Data source | cutoff for differential expression | primary assessment (Table 2) | total genes analyzed | total sex-biased genes | % genes sex-biased in 1 or more tissues |
|---|---|---|---|---|---|---|---|---|---|---|
| (Mele et al., 2015) | 175 donors (63%) | 14–99 | 11–57 | 43 | GTEx pilot set | False discovery rate (FDR) <0.05 | Any gene with sex bias | 20,110 protein or non-coding genes | 753 (mostly breast) | 0.1– 0.23% per tissue |
| (Mayne et al., 2016) | 2500 samples (68%) | 5–622 | 7–289 | 15 | Compiled microarray data | FDR < 0.05 | Any gene with sex bias | not directly provided | 163–1818 genes | 32% autosomal |
| (Gershoni & Pietrokovski, 2017) | 544 donors (64%) | Number of samples per tissue not specified | NA | 45 | GTEx v6 | p <0.05, fold change ≥ 2 | Any gene with sex bias, excluded mammary gland | 18,670 protein coding genes only | “just over” 6500 | 35% total |
| (Tukiainen et al., 2017) | 449 donors (65%) | 43–228 | 26–133 | 29 | GTEx v6 | FDR < 0.01 | Any X chromosome gene with sex bias, excluded mammary gland | 681 protein-coding or long non-coding genes on the X chromosome | 4% inactivated, 6% variable inactivation, 48% escape genes | |
| (Naqvi et al., 2019) | 740 samples selected (67%) | 17–83 | 9–36 | 12 | GTEx v6p | ≥ 1.05 fold change in 4/5 species | Any gene with sex bias conserved across mammals | 12,939 protein-coding orthologs only | 3161 | 24% total |
| (Kassam et al., 2019) | 617 donors (73%) | 80–491 | 23–125 | 40 | GTEx v7 | Bonferroni corrected significance threshold of PTSSD≤1.58×10−6 | Any gene with sex bias, excluded mammary, pituitary, and minor salivary glands | 23,608 mean protein or non-coding genes per tissue | 131 per tissue | 0.6 % per tissue |
| (Lopes-Ramos et al., 2020) | 548 donors (66%) | 82–290 | 36–162 | 29 | GTEx v6 | FDR <0.05, fold change ≥ 1.5 | Any gene predicted to be targeted by sex-biased gene regulatory network | 30,243 protein or non-coding genes | Overall median 64 differentially expressed genes per tissue (4,181 in breast); overall median 169 differentially targeted per tissue (25,994 in breast) | 0.2 % differentially expressed per tissue; 0.6 % differentially targeted per tissue |
| (Oliva et al., 2020) | 838 donors (61–78%) | 78–469 | 33–237 | 44 | GTEx v82 | Local False Sign Rate (LFSR) ≤ 0.05 | Any gene with sex bias | 35,431 protein or non-coding genes | 473 to 4558 per tissue | 1.3% to 12.9% per tissue |
| (Hartman et al., 2020) | 14 boy-girl newborn twins (50%), 172 adult (75%) | 7/newborn, 129/adult | 7/newborn, 43/adult | 2 | Newborns: RNA seq, Adults: microarray GSE30169 | “intrinsic” p-value < 0.1 at birth and < 0.05 in adults; “acquired” p-value > 0.5 at birth and < 0.05 in adults; any fold change | Any gene with sex bias in HUVEC or HAEC3 | 18630 protein or non-coding genes (same genes queried in RNA seq & microarray) | 2528 at birth in boy/girl twins, 1,798 in adults, 268 in both | 25% of newborn and 14% of adult EC transcriptome |
| (Hartman, Mokry, et al., 2021) | >700 donors (59–69%) | 116–538 | 82–256 | 24 | GTEx v8 | FDR<0.1, fold change ≥ 2 with coexpression in more than 20 permutations of the data | Any gene with sex bias. More than 80 samples of each type and sex | 13,787 protein or non-coding genes | 4062 | 29.5% total |
The papers varied widely in how sample numbers were reported. The numbers, if present, were often buried deeply in supplemental files.
v8 is the latest GTEx Release (dbGaP Accession phs000424.v8.p2) with 54 tissues from 636 male and 312 female donors (85% white). Details are here: https://www.gtexportal.org/home/tissueSummaryPage
HUVEC: human umbilical vein endothelial cells; HAEC: human adult aortic endothelial cells
• We selected 120 proteins involved in mRNA biogenesis from the following major reviews: RNA polymerase II transcription initiation (Sainsbury et al., 2015), capping (Galloway & Cowling, 2019), splicing using both the major and minor spliceosomes (Bertram et al., 2020; Fica, 2020; Kastner et al., 2019; Verma et al., 2018), and polyadenylation (MacDonald, 2019; Tian & Manley, 2017). Other reviews corroborated the involvement of these proteins but were not cited for brevity. We did not assess the noncoding RNAs involved in these processes. Our interpretation of “core” machinery was intentionally broad and we acknowledge that many proteins would reasonably be considered “regulatory”. Either way, a sex-associated difference in cellular concentration may alter the stoichiometry of each complex.
With the caveat that mRNA abundance does not necessarily equal protein levels, we then reviewed the published data regarding the expression of specific mRNAs encoding these proteins in 4 tissues thought to exhibit a lesser to greater extent of sex variation: lung, heart left ventricle, skeletal muscle, and thyroid. We also assessed available data for 2 cultured cell types (fibroblasts and endothelial cells) from males or females. Table 2 summarizes the authors’ determinations of sex-biased gene expression according to the criteria of each study. Because 6 of the 8 papers used earlier GTEx versions, we also compared predictions of sex bias to the raw median Transcripts Per Million (TPM) indicated in the latest GTEx Release (dbGaP Accession phs000424. v8.p2). We note that GTEx v8 has data from 44 different organs common to both sexes and 2 cell types shared between men and women – sex bias may occur in a sample type we did not assess.
SEX BIASES IN RNAS ENCODING BASAL RNA SYNTHESIS MACHINERY (TABLE 2)
• Assessment methodologies.
The 10 studies listed in Table 1 differed greatly in sample choice, methodology, computational models, and statistical criteria for identifying sex bias. Some investigators sought to identify the highest numbers of differentially regulated genes, without regard to fold change. Others were more selective and required a specific fold change cut-off. Sex-biased expression of alternatively processed mRNAs was disregarded because gene models collapsed isoforms to a single gene. Some groups assessed earlier versions of the GTEx data and thus were restricted to samples from fewer donors (Gershoni & Pietrokovski, 2017; Kassam et al., 2019; Lopes-Ramos et al., 2020; Mele et al., 2015; Naqvi et al., 2019; Tukiainen et al., 2017). Eight studies assessed the GTEx abundances primarily for differential expression (Gershoni & Pietrokovski, 2017; Hartman et al., 2020; Kassam et al., 2019; Mayne et al., 2016; Mele et al., 2015; Naqvi et al., 2019; Oliva et al., 2020; Tukiainen et al., 2017). However, several studies excluded certain tissues such as mammary, pituitary, and minor salivary glands specifically because these tissues were found to have many sex-biased genes (Gershoni & Pietrokovski, 2017; Kassam et al., 2019; Tukiainen et al., 2017). The contribution of sex differences in cell type composition was assessed in three papers (Hartman, Mokry, et al., 2021; Lopes-Ramos et al., 2020; Oliva et al., 2020). In contrast to cataloguing differential expression, Lopes-Ramos et al. assessed the targeting of genes by transcription factor networks predicted to regulate genes in a sex-biased manner (Lopes-Ramos et al., 2020). This important study showed that although transcription factors may not themselves be sex-biased, they may activate different sets of genes in males and females. Other important biological parameters such as mapping to the X-chromosome (Kassam et al., 2019; Oliva et al., 2020; Tukiainen et al., 2017), evolutionary conservation of bias (Naqvi et al., 2019), or developmental maintenance of bias over the lifespan (Hartman et al., 2020) were incorporated into other studies.
Perhaps the broadest, most comprehensive net was cast by Oliva et al. using the latest and largest GTEx data set (v8 release, (Oliva et al., 2020)). 35,431 genes including protein-coding, noncoding, and transcribed pseudo genes from all chromosomes including the sex chromosomes were assessed in 44 tissues. Taking into account variation in cell type composition, Oliva et al. deemed 13,294 genes as sex-biased to any degree (local false sign rate, LFSR ≤0.05). 37.5% of all genes exhibited sex-bias in one or more tissues. These genes influence biomedically relevant functions such as drug and hormone response, embryogenesis, reproduction, fat metabolism, cancer, and immune response. Sex specific gene-trait associations were identified only when the results were stratified by sex. We cannot fully understand normal and pathological processes without understanding the cumulative impact of these differences.
• Reported sex biases in expression of selected RNA synthesis proteins.
29 of the 43 RNAs encoding proteins involved in transcription initiation were reported to be differentially expressed in one or more of the 6 cell types selected for Table 2 from male and female donors. RNAs encoding 4 of the 5 capping enzymes, 37 of 54 splicing factors, and 5 of 17 polyadenylation factors may be sex-biased according to the criteria of one or more studies. Identifying the 149 observations or predictions of differential gene expression from the 10 reviewed papers (Table 1) in 6 tissue or cell types was time-consuming. However, the median abundance in each sex is freely available on the GTEx portal as shown in Fig. 1. Although the minimally processed data should be interpreted with caution, the posted median values for 106 genes (71%) were consistent with differential expression in that tissue. In other words, a quick look at the GTEx data regarding any specific gene might alleviate fears of substantial or widespread differences between the sexes or reveal interesting and biomedically relevant differences to be investigated further.
Figure 1. Visualization of Consistently Increased ZRSR2 RNA Abundance in Female Tissues.
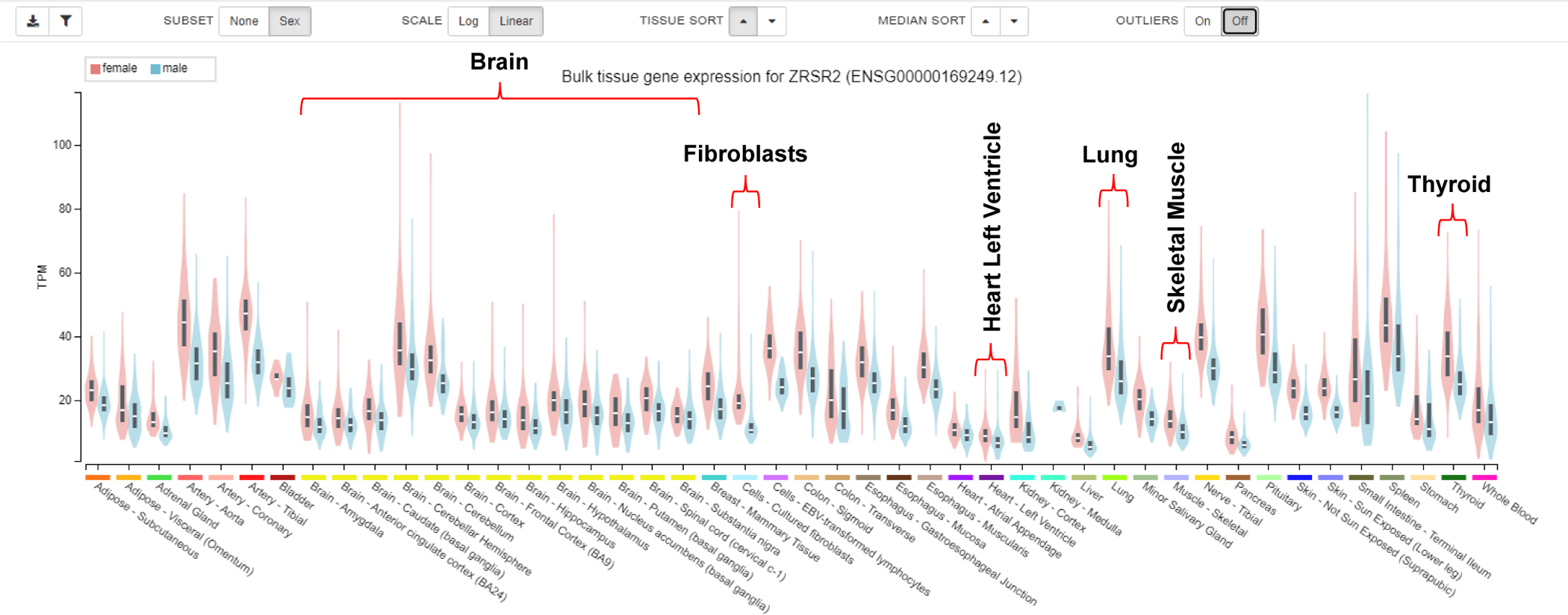
A sample of the bulk gene expression data available from https://www.gtexportal.org/home/ is shown. The transcripts per million (TPM) were calculated from a gene model with isoforms collapsed to a single gene. Data is not otherwise normalized. Box plots show the median and 25th and 75 percentiles. The pink and blue plots illustrate the female and male distributions respectively. Hovering over each plot shows the number of samples and median values on the site. The 5 GTEx tissues from Table 2 are bracketed. The many brain regions included in GTEx are marked because spatially complex RNA metabolism occurs in the brain (Fisher & Feng, 2022; Hilgers, 2022; Landinez-Macias & Urwyler, 2021). Data Souce: GTEx Analysis Release v8 (dbGaP Accession phs000424. v8.p2) downloaded April 2022.
• RNA synthesis genes with the strongest evidence of sex bias.
Replicating the finding of differential expression using different methodologies increases confidence that the differences may be real. Of all 120 genes, the ZRSR2 gene differed the most consistently by far. All tissues from females had significantly higher levels according to all publications except Naqvi et al. and Mele et al. (Mele et al., 2015; Naqvi et al., 2019). Fig. 1 illustrates ubiquitous increased abundance in all female tissues. This was not surprising because this X-linked gene encodes a protein associated with the minor (U12-dependent) spliceosome that is well known to affect diseases differently in men and women (Inoue et al., 2021; Karantanos et al., 2021; Madan et al., 2015; Togami et al., 2022; Verma et al., 2018). ZRSR2 exemplifies how variable X-inactivation influences sex biases (see Box 2).
• RNA Pol II Transcription initiation.
As mentioned above, Naqvi et al. and Lopes-Ramos et al. both predicted that TBP was more abundant in female skeletal muscle (Lopes-Ramos et al., 2020; Naqvi et al., 2019). These two papers were notable in that Naqvi et al. required conservation of sex bias in 4 of 5 different mammals and Lopes-Ramos et al. assessed whether a sex-biased network of transcription factors would target a gene. Lopes-Ramos et al. also measured cell type markers and established that varied proportions of differentiated cell types could not account for sex-biased expression in skeletal muscle. Finally, the median GTEx v8 Transcripts per Million (TPM) values for skeletal muscle were 9.2 for 260 female and 8.1 for 542 male samples. The bottom line is that this core player in transcription initiation may retain its status as a non-sex influenced reference gene in most tissues. However, some aspect of muscle biology may have revealed an unforeseen sex bias. Other transcription initiation genes reported to exhibit sex bias by more than one group include TAF7, TAF13; ERCC2; and MNAT1 (Table 2). Like TBP, the latest GTEx values were consistent with the published predictions for these 4 genes.
• The 5’ and 3’ ends.
None of the genes encoding the 5 capping enzymes or 17 proteins involved in polyadenylation were reported by more than one group to be sex-biased in the 6 selected sample types. Indeed, Naqvi et al. and Lopes-Ramos et al. disagreed on whether CSTF3 was female or male biased in skeletal muscle and thyroid. Although large differences in capping and polyadenylation factors have not been reported, sex may influence the function of some factors at a level other than RNA abundance. CstF-64 encoded by the X-linked gene CSTF2 is profoundly affected by sex because X chromosome inactivation during spermatogenesis blocks participation in polyadenylation (MacDonald, 2019). Instead, τCstF-64, encoded by the autosomal CSTF2t gene, enables the correct polyadenylation of key genes in differentiating male germ cells. CSTF2t also is expressed in many other tissues including brain, but we failed to find consistent reports that the CSTF2t gene was expressed in a sex-biased manner. However, mice lacking the Cstf2t gene showed clear sex-specific differences in behaviour and memory (Harris et al., 2016). Alternate splicing and polyadenylation contribute to a multitude of unique neuronal gene expression patterns (Fisher & Feng, 2022; Hilgers, 2022; Landinez-Macias & Urwyler, 2021). The Cstf2t report highlights the importance of understanding all mechanisms by which sex may impact RNA processing.
• Splicing.
37 proteins involved in the function of the major (U2-dependent) or minor (U12-dependent) spliceosomes are potentially sex-biased in this small set of tissue types. Sex bias was corroborated by 2 or more groups and also apparent in GTEx v8 in the same tissue for PRPF8; TXNL4A; SART1; SNRNP25; and ZRSR2. Numerous cancers and congenital diseases called spliceosomopathies are caused by mutations in genes encoding spliceosome factors (Griffin & Saint-Jeannet, 2020; Yamauchi et al., 2022). These conditions are typically cell- or tissue-specific and presumably reflect variation in composition and regulation of the splicing apparatus. About 9 out of 10 genes are alternately spliced (Wang et al., 2008). An estimated two thirds of human diseases involve splicing (Verma et al., 2018). Even a modest sex bias in a sensitive tissue would influence susceptibility to pathology or response to therapies directed at splicing.
• Myelodysplastic syndromes cause hematopoietic pathologies that increase the risk of leukemia (Karantanos et al., 2021). Relative to women, these blood cancers occur more frequently, present with more aggressive features, and are more likely to cause death in men. Half of patients suffering from myelodysplastic syndromes bear mutations in splicing factors and exhibit aberrant splicing patterns (Inoue et al., 2021; Karantanos et al., 2021; Madan et al., 2015; Togami et al., 2022; Verma et al., 2018). Sex differences in cytidine analog metabolism, stem and progenitor cell compartments, steroid hormone signaling, and X-chromosome inactivation have been hypothesized to explain the striking male bias. A vastly increased incidence in men of mutations in the X-linked ZRSR2 gene support the latter hypothesis (Karantanos et al., 2021; Madan et al., 2015; Togami et al., 2022). As described in Box 2, one allele of most X-chromosome linked genes is inactivated in women, leaving the same functional gene dosage as men. Some genes, including ZRSR2, escape X-inactivation and act more like autosomes. Thus, women express both alleles in all cells and ZRSR2 mRNA abundance consistently exceeds that of men in all tissues (Fig. 1, Table 2 and references therein). Men with mutations in their single X-chromosome lack the protection of a second, unmutated X-chromosome allele. ZRSR2-related cancers fit the classic model of X-linked predisposition to disease. However, men with myelodysplastic syndromes and leukemias also have a higher incidence of U2AF1 and SRSF2 mutations which are encoded on autosomes (Karantanos et al., 2021). The male bias also associated with these splicing factors suggests that some aspect of splicing mechanisms or splicing patterns differs in men and women.
NOTES REGARDING SPECIFIC MECHANISMS AND CONSEQUENCES
• Gene regulatory network (GRN) predictions.
Lopes-Ramos et al. assessed the GTEx v6 data from a GRN perspective (Lopes-Ramos et al., 2020). GRNs model the complex interactions between transcription factors and their target genes. Importantly, two different GRNs may yield similar target gene expression levels in cells under one environmental situation, for example healthy males and females. However, in another situation such as a pathology, the two distinct networks may yield different expression levels in each sex. Lopes-Ramos et al. first tested the hypothesis that sex-biased gene expression would be dominated by hormone-activated receptors (estrogen receptors 1 or 2 or androgen receptor) and responding genes bearing classical sex hormone receptor binding motifs. This hypothesis, whereby genes expressed more highly in females or males would bear estrogen or androgen binding sites respectively, failed. Sex-biased genes were not significantly enriched for receptor binding sites (Kassam et al., 2019; Lopes-Ramos et al., 2020). Because sex hormone receptors were not the sole regulators of sex biases, broader de novo GRN models were developed. Specifically, 8,279 tissue-specific gene regulatory networks were constructed in 29 tissues from transcription factor binding motifs, mRNA abundance patterns, and both direct and indirect protein-protein interactions.
The networks connected to a gene were categorized into male-biased (60% male targeting), female-biased (60% female targeting), or sex-divergent; that is the targeting was split. To allow direct comparison to other publications, Table 2 includes only genes that were targeted by male- or female-biased GRNs according to Lopes-Ramos et al. Sex-biased gene regulation was wide-spread in all tissues. However, only 30% of the differentially targeted genes were also differentially expressed on average (criteria: an absolute change of ≥ 1.5 fold; false discovery rate, FDR < 0.05). Although many of the 120 RNA synthesis genes were considered targeted (Table 2), none met the Lopes-Ramos et al. differential expression cut-off in the six chosen tissues (Lopes-Ramos et al., 2020). However, other groups deemed several factors with sex-biased GRNs to have sex-biased mRNA abundance by other standards such as evolutionary conservation. We agree with the assertion by Lopes-Ramos et al., that sex-biased GRNs that drive similar expression levels in this particular set of healthy males and females may drive sex-biased expression when age, stress, pathology, or other conditions disrupt homeostasis. Latent sex differences in gene regulatory processes controlling RNA factors may contribute to observed differences in disease prognoses and therapeutic responses.
• Evolutionary conservation.
Sexual dimorphism is readily apparent in most if not all animals. Naqvi et al. set out to identify how many orthologous genes were sex-biased in all mammals and how many were uniquely biased by species. Genes were catalogued whose expression differed in 12 shared tissues (adipose, adrenal gland, brain, colon, heart, liver, lung, muscle, pituitary, skin, spleen, and thyroid) from macaque, mouse, rat, or dog males and non-estrous females (Naqvi et al., 2019). RNA seq data from these freshly processed animal tissues were compared to human cadaver data from the Genotype-Tissue Expression Consortium (GTEx, v6p). The GTEx samples were filtered by cause of death, medical history, and pathology notes. When possible, the histology of samples was directly evaluated. This stringent quality control greatly reduced the number of samples relative to all other papers using the GTEx data but presumably reduced variation due to pathologies. To be considered “conserved” sex bias, a relatively low difference of 5% or more in the same sex was required to occur in 4 of the 5 species. We have only included the conserved sex-biased genes in Table 2.
An estimated 23% of the total sex-biased genes in any species were considered to be biased in the common mammalian ancestor (Naqvi et al., 2019). Knowledge regarding the expression of these genes obtained using model organisms is likely to apply to humans. In contrast, more than three fourths of the orthologous genes acquired sex bias after diverging from a common ancestor. Studies of sex bias in model organisms may translate less easily for these genes to humans. Although evolutionary conservation is a high bar, 30 of the 120 genes involved in RNA synthesis exhibited conserved sex-bias in one of the tissues chosen for Table 2. Several of these were also considered to respond to a sex-biased gene regulatory network by Lopes-Ramos et al. (Lopes-Ramos et al., 2020). Typically, gene expression patterns that confer a selective advantage are maintained by redundant regulatory processes. Sex-biased networks may confer such redundancy.
• X-inactivation.
Several groups noted that genes localized to the X-chromosome were observed to be sex-biased somewhat more frequently and to a greater degree. In contrast to 37% of autosomal genes, Oliva et al. found that 47% of X-linked genes were sex-biased (Oliva et al., 2020). The median fold changes of female and male-biased X-linked genes were 1.13 and 1.08, respectively, whereas the fold change for all sex-biased autosomal genes was 1.04. Up to a third of genes located on the X-chromosome may be expressed from both alleles (Cantone & Fisher, 2017; Fang et al., 2021; Tukiainen et al., 2017). Which genes escape from X-inactivation varies greatly between tissues and individuals. Although generally considered to occur randomly during early development and then to remain fixed in somatic cells, X-inactivation is subject to reprogramming and signalling differences (Cantone & Fisher, 2017; Sripathy et al., 2017). For example, antagonism between BMP and TGF-β signalling modifies the expression XIST, the long noncoding RNA that initiates X-chromosome silencing (reviewed in (Shah & Rogers, 2018)). Only 3 of the 120 genes we assessed mapped to the X-chromosome. As described above, ZRSR2 frequently escapes silencing, is expressed at higher levels in female tissues, and contributes to sex bias in blood cancers associated with splicing defects (Inoue et al., 2021; Karantanos et al., 2021; Madan et al., 2015; Togami et al., 2022; Verma et al., 2018). Tukiainen et al. provide a comprehensive list of genes that are reliably silenced, or that escape X-chromosome inactivation to a variable or consistent extent (Tukiainen et al., 2017).
• Influence of parental sex (imprinting).
We focus primarily on cell-autonomous and physiological mechanisms that may cause differences in an individual. However, failing to mention that the maternal and paternal genomes are not expressed identically would be negligent. The expression of 228 protein-coding and non-coding human genes differ according to the sex of the parent who transmitted the gene (Tucci et al., 2019). Only the maternal or paternal allele is expressed for these imprinted genes. None of the genes encoding the 120 selected proteins were imprinted. However, the brain, with its fascinatingly complex RNA biology, is rich in genes whose expression is restricted to one allele (Fisher & Feng, 2022; Hilgers, 2022; Landinez-Macias & Urwyler, 2021; Tucci et al., 2019). For example, the classic reciprocally imprinted Prader-Willi and Angelman neurodevelopmental syndromes involve deletion of an overlapping region. The region includes snoRNAs and the paternally imprinted small nuclear ribonucleoprotein polypeptide N (SNRPN) involved in alternative splicing (Glenn et al., 1993; Lee et al., 2014; Tucci et al., 2019). Those testing mutated or genetically modified RNA metabolism genes in vivo should be aware that alleles inherited from one parent or the other may be expressed differently. As of 2019, 228 human and 260 mouse genes were known to be imprinted (Tucci et al., 2019). However, more sensitive RNA sequencing approaches are discovering new tissue-specific imprinting variation, especially in the brain (Tucci et al., 2019).
• “Intrinsic” vs. acquired sex bias.
An individual’s sex chromosome complement, XX vs. XY, and the subsequent pattern of X-inactivated genes can influence gene expression from fertilization until death. Gene expression differences are detectable from the earliest embryonic stages and may be considered “intrinsic” features of a male or female cell (Deegan et al., 2021; Wizemann & Pardue, 2001). In contrast, the differential impact of male and female gonadal hormones is delayed until gonads develop and begin to drive sexual dimorphism (~12 days in mouse and ~9 weeks in humans). The impact of these hormones also changes drastically throughout the lifespan for example at puberty. In addition, gender identity drives behavioural and environmental effects. Hormonal and gender-associated dissimilarities cause “acquired” differences.
Hartman et al. cleverly used new-born boy-girl twins to tease apart intrinsic vs. acquired sex differences in endothelial cells (Hartman et al., 2020). Because both twins experience 9 months of a similar environment, an assumption was made that the human umbilical vein endothelial cells (HUVEC) transcriptome from these twins would largely reflect cell intrinsic sex differences. In contrast, the transcriptome of human adult aortic endothelial cells (HAEC from heart transplant donors) would have acquired sex differences from a lifetime of added hormonal and gender-associated influences. The RNA synthesis genes, GTF2A1, TAF11, SNRPE, U2AF2, and ZRSR2, were differentially expressed both at birth and in adulthood and thus considered intrinsically sex-biased in endothelial cells (Table 2). Vascular endothelial cell biology profoundly influences coronary artery health. Many of the intrinsically sex-biased vascular endothelial genes also have been associated with coronary artery disease (Hartman et al., 2020). This overlap accentuates the importance of addressing the causes of transcriptome sex differences.
• Sex bias in diseased tissues – a cardiovascular example.
We have focused on data primarily from presumed healthy tissues. The same group that performed the endothelial cell twin study also characterized differentially expressed genes and inferred gene regulatory networks in atherosclerotic aortic tissue (Hartman, Owsiany, et al., 2021). Using a unique collection of age-matched samples from 160 women and 160 men from the STARNET (Stockholm-Tartu Atherosclerosis Reverse Network Engineering Task) project, major differences in gene expression and network connectivity were identified in these atherosclerotic samples. This differential expression likely drives the observed sex dimorphism of this disease (Bjorkegren & Lusis, 2022). Among the 120 genes involved in the basal RNA synthesis machinery, TAF2, PRPF3, SNRNP40, SNRPD3, and ZRSR2 were all deemed more abundant in atherosclerotic tissue from females relative to males (Hartman, Owsiany, et al., 2021). Women have been vastly underrepresented in previous cardiovascular disease studies. Balanced studies are critical for correcting erroneous or incomplete conclusions regarding the biology of cardiovascular disease in women.
CONCLUSIONS: CAVEATS AND FUTURE DIRECTIONS
• Making an mRNA molecule is fundamental. Vast variation in transcription and processing mechanisms between men and women must be selected against. But just like body morphology shows sexual dimorphism in homologous parts, the RNA synthesis machinery may vary. Differences are likely small, just as the male and female skulls diverge in ways that only a trained anatomist might recognize. The studies assessed here used diverse bioinformatics workflows and statistical criteria, different wet lab processes (RNAseq vs. microarray), included or excluded non-coding RNAs and/or the sex chromosomes. Shared tissues were excluded in some studies simply because the number of sex-biased genes was considered excessive. Organs contain multiple cell types with distinct transcriptomes. With a few exceptions, the sample sets were flawed. More than half of the GTEx donors were male. Two thirds were greater than 50 years of age. 85% were white. Any description of sex-biased expression of alternatively processed isoforms with distinct functions and regulatory modules was lacking. Gene annotation lists are imperfect in that female-biased genes had fewer HUGO Gene Nomenclature Committee symbol annotations than male-biased genes (Hartman, Owsiany, et al., 2021). Despite these pervasive failings reflecting past inattention to sex as a key biological variable, several studies corroborated findings of sex-bias in vital proteins involved in RNA synthesis.
• Differences for components of the basal synthesis machinery may be small, but the cumulative effects may influence both health and disease. Differences in the proteins and noncoding RNAs that regulate the machinery are likely to exert far greater effects. Examining sex specific differences in RNA abundance with these easily used and expanding public data bases should become routine. Although not perfectly representative, the latest GTEx dataset (v8) contains DNA data from 838 post-mortem donors and 17,382 RNA-seq samples from 54 tissue sites and two cell lines and makes stratification by sex straight-forward. Open access, international projects such as the Human Cell Atlas consortium aim to map every cell type in the human body. Although initial analyses were not stratified by sex ((Liu & Zhang, 2022) and refs. therein), it is anticipated that future studies will address this omission. The STARNET collection was designed with particular care to represent both sexes of patients with or without coronary artery disease (Bjorkegren & Lusis, 2022; Hartman, Owsiany, et al., 2021). RNA-seq data is available for 7 tissues: blood, free internal mammary artery, atherosclerotic aortic root, subcutaneous fat, visceral abdominal fat, skeletal muscle, and liver from 1,300 diseased patients and about 400 donors without heart disease. With resources such as these, scientists can begin to gauge the universality of the mechanisms that govern RNA biology.
Acknowledgments
We thank Hebah Ahmed and Maansi Chalasani for thorough review and helpful comments.
Funding Information
Funding was provided by grants from the National Heart, Lung, and Blood Institute (R01HL114751) and National Institutes of Aging (R56AG050762) to MBR.
Footnotes
Diane E. Garsetti, no conflicts of interest
Khushboo Sahay, no conflicts of interest
Yue Wang, no conflicts of interest
Melissa B. Rogers, no conflicts of interest
Contributor Information
Diane E. Garsetti, Rutgers - New Jersey Medical School (NJMS), Department of Microbiology, Biochemistry, and Molecular Genetics
Khushboo Sahay, Rutgers - NJMS, Department of Microbiology, Biochemistry, and Molecular Genetics.
Yue Wang, Rutgers - NJMS, Department of Microbiology, Biochemistry, and Molecular Genetics.
Melissa B. Rogers, Rutgers - NJMS, Department of Microbiology, Biochemistry, and Molecular Genetics.
References
- Bertram K, El Ayoubi L, Dybkov O, Agafonov DE, Will CL, Hartmuth K, . . . Luhrmann R. (2020). Structural Insights into the Roles of Metazoan-Specific Splicing Factors in the Human Step 1 Spliceosome. Mol Cell, 80(1), 127–139 e126. doi: 10.1016/j.molcel.2020.09.012 [DOI] [PubMed] [Google Scholar]
- Bjorkegren JLM, & Lusis AJ (2022). Atherosclerosis: Recent developments. Cell, 185(10), 1630–1645. doi: 10.1016/j.cell.2022.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantone I, & Fisher AG (2017). Human X chromosome inactivation and reactivation: implications for cell reprogramming and disease. Philos Trans R Soc Lond B Biol Sci, 372(1733). doi: 10.1098/rstb.2016.0358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton JA, & Collins FS (2014). Policy: NIH to balance sex in cell and animal studies. Nature, 509(7500), 282–283. doi: 10.1038/509282a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deegan DF, Nigam P, & Engel N (2021). Sexual Dimorphism of the Heart: Genetics, Epigenetics, and Development. Front Cardiovasc Med, 8, 668252. doi: 10.3389/fcvm.2021.668252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Editorial. (2020). Accounting for sex and gender makes for better science. Nature, 588(7837), 196. doi: 10.1038/d41586-020-03459-y [DOI] [PubMed] [Google Scholar]
- Fang H, Deng X, & Disteche CM (2021). X-factors in human disease: impact of gene content and dosage regulation. Hum Mol Genet, 30(R2), R285–R295. doi: 10.1093/hmg/ddab221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fica SM (2020). Cryo-EM snapshots of the human spliceosome reveal structural adaptions for splicing regulation. Curr Opin Struct Biol, 65, 139–148. doi: 10.1016/j.sbi.2020.06.018 [DOI] [PubMed] [Google Scholar]
- Fisher E, & Feng J (2022). RNA splicing regulators play critical roles in neurogenesis. Wiley Interdiscip Rev RNA, e1728. doi: 10.1002/wrna.1728 [DOI] [PubMed] [Google Scholar]
- Fochi S, Orlandi E, Ceccuzzi L, Rodolfo M, Vergani E, Turco A, . . . Gomez-Lira M. (2021). Identification of suitable mRNAs and microRNAs as reference genes for expression analyses in skin cells under sex hormone exposure. Gene, 769, 145336. doi: 10.1016/j.gene.2020.145336 [DOI] [PubMed] [Google Scholar]
- Galloway A, & Cowling VH (2019). mRNA cap regulation in mammalian cell function and fate. Biochim Biophys Acta Gene Regul Mech, 1862(3), 270–279. doi: 10.1016/j.bbagrm.2018.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Sifuentes Y, & Maney DL (2021). Reporting and misreporting of sex differences in the biological sciences. Elife, 10. doi: 10.7554/eLife.70817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershoni M, & Pietrokovski S (2017). The landscape of sex-differential transcriptome and its consequent selection in human adults. BMC Biol, 15(1), 7. doi: 10.1186/s12915-017-0352-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenn CC, Porter KA, Jong MT, Nicholls RD, & Driscoll DJ (1993). Functional imprinting and epigenetic modification of the human SNRPN gene. Hum Mol Genet, 2(12), 2001–2005. doi: 10.1093/hmg/2.12.2001 [DOI] [PubMed] [Google Scholar]
- Griffin C, & Saint-Jeannet JP (2020). Spliceosomopathies: Diseases and mechanisms. Dev Dyn, 249(9), 1038–1046. doi: 10.1002/dvdy.214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JC, Martinez JM, Grozdanov PN, Bergeson SE, Grammas P, & MacDonald CC (2016). The Cstf2t Polyadenylation Gene Plays a Sex-Specific Role in Learning Behaviors in Mice. PLoS One, 11(11), e0165976. doi: 10.1371/journal.pone.0165976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman RJG, Kapteijn DMC, Haitjema S, Bekker MN, Mokry M, Pasterkamp G, . . . den Ruijter HM. (2020). Intrinsic transcriptomic sex differences in human endothelial cells at birth and in adults are associated with coronary artery disease targets. Sci Rep, 10(1), 12367. doi: 10.1038/s41598-020-69451-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman RJG, Mokry M, Pasterkamp G, & den Ruijter HM (2021). Sex-dependent gene co-expression in the human body. Sci Rep, 11(1), 18758. doi: 10.1038/s41598-021-98059-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman RJG, Owsiany K, Ma L, Koplev S, Hao K, Slenders L, . . . den Ruijter HM. (2021). Sex-Stratified Gene Regulatory Networks Reveal Female Key Driver Genes of Atherosclerosis Involved in Smooth Muscle Cell Phenotype Switching. Circulation, 143(7), 713–726. doi: 10.1161/CIRCULATIONAHA.120.051231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgers V (2022). Regulation of neuronal RNA signatures by ELAV/Hu proteins. Wiley Interdiscip Rev RNA, e1733. doi: 10.1002/wrna.1733 [DOI] [PubMed] [Google Scholar]
- Hosman FL, Engels S, den Ruijter HM, & Exalto LG (2022). Call to Action for Enhanced Equity: Racial/Ethnic Diversity and Sex Differences in Stroke Symptoms. Front Cardiovasc Med, 9, 874239. doi: 10.3389/fcvm.2022.874239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue D, Polaski JT, Taylor J, Castel P, Chen S, Kobayashi S, . . . Abdel-Wahab O. (2021). Minor intron retention drives clonal hematopoietic disorders and diverse cancer predisposition. Nat Genet, 53(5), 707–718. doi: 10.1038/s41588-021-00828-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karantanos T, Jain T, Moliterno AR, Jones RJ, & DeZern AE (2021). Sex-Related Differences in Chronic Myeloid Neoplasms: From the Clinical Observation to the Underlying Biology. Int J Mol Sci, 22(5). doi: 10.3390/ijms22052595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassam I, Wu Y, Yang J, Visscher PM, & McRae AF (2019). Tissue-specific sex differences in human gene expression. Hum Mol Genet, 28(17), 2976–2986. doi: 10.1093/hmg/ddz090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastner B, Will CL, Stark H, & Luhrmann R (2019). Structural Insights into Nuclear pre-mRNA Splicing in Higher Eukaryotes. Cold Spring Harb Perspect Biol, 11(11). doi: 10.1101/cshperspect.a032417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landinez-Macias M, & Urwyler O (2021). The Fine Art of Writing a Message: RNA Metabolism in the Shaping and Remodeling of the Nervous System. Front Mol Neurosci, 14, 755686. doi: 10.3389/fnmol.2021.755686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MS, Lin YS, Deng YF, Hsu WT, Shen CC, Cheng YH, . . . Li C. (2014). Modulation of alternative splicing by expression of small nuclear ribonucleoprotein polypeptide N. FEBS J, 281(23), 5194–5207. doi: 10.1111/febs.13059 [DOI] [PubMed] [Google Scholar]
- Liu Z, & Zhang Z (2022). Mapping cell types across human tissues. Science, 376(6594), 695–696. doi: 10.1126/science.abq2116 [DOI] [PubMed] [Google Scholar]
- Lopes-Ramos CM, Chen CY, Kuijjer ML, Paulson JN, Sonawane AR, Fagny M, . . . DeMeo DL. (2020). Sex Differences in Gene Expression and Regulatory Networks across 29 Human Tissues. Cell Rep, 31(12), 107795. doi: 10.1016/j.celrep.2020.107795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald CC (2019). Tissue-specific mechanisms of alternative polyadenylation: Testis, brain, and beyond (2018 update). Wiley Interdiscip Rev RNA, 10(4), e1526. doi: 10.1002/wrna.1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan V, Kanojia D, Li J, Okamoto R, Sato-Otsubo A, Kohlmann A, . . . Koeffler HP. (2015). Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat Commun, 6, 6042. doi: 10.1038/ncomms7042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayne BT, Bianco-Miotto T, Buckberry S, Breen J, Clifton V, Shoubridge C, & Roberts CT (2016). Large Scale Gene Expression Meta-Analysis Reveals Tissue-Specific, Sex-Biased Gene Expression in Humans. Front Genet, 7, 183. doi: 10.3389/fgene.2016.00183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mele M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M, . . . Guigo R. (2015). Human genomics. The human transcriptome across tissues and individuals. Science, 348(6235), 660–665. doi: 10.1126/science.aaa0355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naqvi S, Godfrey AK, Hughes JF, Goodheart ML, Mitchell RN, & Page DC (2019). Conservation, acquisition, and functional impact of sex-biased gene expression in mammals. Science, 365(6450). doi: 10.1126/science.aaw7317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva M, Munoz-Aguirre M, Kim-Hellmuth S, Wucher V, Gewirtz ADH, Cotter DJ, . . . Stranger BE. (2020). The impact of sex on gene expression across human tissues. Science, 369(6509). doi: 10.1126/science.aba3066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdemir BC, Gerard CL, & Espinosa da Silva C (2022). Sex and Gender Differences in Anticancer Treatment Toxicity: A Call for Revisiting Drug Dosing in Oncology. Endocrinology, 163(6). doi: 10.1210/endocr/bqac058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin JB (2022). The spectrum of sex differences in cancer. Trends Cancer, 8(4), 303–315. doi: 10.1016/j.trecan.2022.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainsbury S, Bernecky C, & Cramer P (2015). Structural basis of transcription initiation by RNA polymerase II. Nat Rev Mol Cell Biol, 16(3), 129–143. doi: 10.1038/nrm3952 [DOI] [PubMed] [Google Scholar]
- Shah TA, & Rogers MB (2018). Unanswered Questions Regarding Sex and BMP/TGF-beta Signaling. J Dev Biol, 6(2). doi: 10.3390/jdb6020014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharan RN, Vaiphei ST, Nongrum S, Keppen J, & Ksoo M (2015). Consensus reference gene(s) for gene expression studies in human cancers: end of the tunnel visible? Cell Oncol (Dordr), 38(6), 419–431. doi: 10.1007/s13402-015-0244-6 [DOI] [PubMed] [Google Scholar]
- Sripathy S, Leko V, Adrianse RL, Loe T, Foss EJ, Dalrymple E, . . . Bedalov A. (2017). Screen for reactivation of MeCP2 on the inactive X chromosome identifies the BMP/TGF-beta superfamily as a regulator of XIST expression. Proc Natl Acad Sci U S A, 114(7), 1619–1624. doi: 10.1073/pnas.1621356114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B, & Manley JL (2017). Alternative polyadenylation of mRNA precursors. Nat Rev Mol Cell Biol, 18(1), 18–30. doi: 10.1038/nrm.2016.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togami K, Chung SS, Madan V, Booth CAG, Kenyon CM, Cabal-Hierro L, . . . Lane AA. (2022). Sex-Biased ZRSR2 Mutations in Myeloid Malignancies Impair Plasmacytoid Dendritic Cell Activation and Apoptosis. Cancer Discov, 12(2), 522–541. doi: 10.1158/2159-8290.CD-20-1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucci V, Isles AR, Kelsey G, Ferguson-Smith AC, & Erice Imprinting G (2019). Genomic Imprinting and Physiological Processes in Mammals. Cell, 176(5), 952–965. doi: 10.1016/j.cell.2019.01.043 [DOI] [PubMed] [Google Scholar]
- Tukiainen T, Villani AC, Yen A, Rivas MA, Marshall JL, Satija R, . . . MacArthur DG. (2017). Landscape of X chromosome inactivation across human tissues. Nature, 550(7675), 244–248. doi: 10.1038/nature24265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma B, Akinyi MV, Norppa AJ, & Frilander MJ (2018). Minor spliceosome and disease. Semin Cell Dev Biol, 79, 103–112. doi: 10.1016/j.semcdb.2017.09.036 [DOI] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, . . . Burge CB. (2008). Alternative isoform regulation in human tissue transcriptomes. Nature, 456(7221), 470–476. doi: 10.1038/nature07509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westergaard D, Moseley P, Sorup FKH, Baldi P, & Brunak S (2019). Population-wide analysis of differences in disease progression patterns in men and women. Nat Commun, 10(1), 666. doi: 10.1038/s41467-019-08475-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wizemann TM, & Pardue ML (Eds.). (2001). Exploring the Biological Contributions to Human Health: Does Sex Matter? Washington (DC). [PubMed] [Google Scholar]
- Woitowich NC, Beery A, & Woodruff T (2020). A 10-year follow-up study of sex inclusion in the biological sciences. Elife, 9. doi: 10.7554/eLife.56344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi H, Nishimura K, & Yoshimi A (2022). Aberrant RNA splicing and therapeutic opportunities in cancers. Cancer Sci, 113(2), 373–381. doi: 10.1111/cas.15213 [DOI] [PMC free article] [PubMed] [Google Scholar]