

Abstract
Transcription factors regulate hundreds of genes and p53 is no exception. As a stress responsive protein, p53 transactivates an array of downstream targets which define its role in maintaining physiological functions of cells/tissues. Despite decades of studies, our understanding of the p53 in vivo transcriptional program is still incomplete. Here we discuss some of the physiological stressors that activate p53, the pathological and physiological implications of p53 activation and the molecular profiling of the p53 transcriptional program in maintaining tissue homeostasis. We argue that the p53 transcriptional program is spatiotemporally regulated in a tissue-specific manner and define a p53 target signature that faithfully depicts p53 activity. We further emphasize that additional in vivo studies are needed to refine the p53 transactivation profile to harness it for therapeutic purposes.
Subject terms: Cancer, Gene regulation
Facts
The TP53 tumor suppressor encodes a DNA sequence-specific transcription factor that regulates hundreds of genes.
p53 activity is induced in response to multiple physiological stressors.
p53 transcriptional activity is spatio-temporally regulated.
The majority of p53 downstream targets are tissue specific.
Open questions
What are the functions of p53 signature genes in mediating a p53 response in vivo?
Which p53 downstream targets are responsible for the non-cell autonomous response?
Why is expression of some p53 targets restricted to specific cell types in a tissue?
Introduction
The p53 transcription factor
The TP53 tumor suppressor encodes a DNA sequence-specific transcription factor that is a ubiquitously expressed protein but maintained in an inactive, latent form in most cell types. In response to stressors, p53 transcriptionally regulates (up and down) hundreds of downstream targets that eventually determine the fate of individual and surrounding cells.
The 393 amino acid human p53 protein encompasses an amino-terminal transactivation domain (TAD, residues 1–61), a proline rich domain (PRD, residues 61–92), a central DNA binding domain (DBD, residues 94–292), a tetramerization domain (TD, residues 326–353) and a carboxy-terminal regulatory domain (CTD, residues 353–390) [1]. The p53 TAD is further sub-divided into two subdomains; TAD1 (residues 1–40) and TAD2 (residues 41–61) with TAD1 playing a predominant role in p53 transcriptional activity [2]. The p53 protein exists as dimers in normal cells. Under physiological stress, dimers assemble to form the stable functional p53 tetramer (a dimer of dimers) [3–5] which binds a specific DNA responsive element (RE) in the promoters of hundreds of genes. The p53 REs contain two decameric half-site palindromes of the general sequence 5’-RRRCWWGYYY-3’ (R = A/G; W = A/T; Y = C/T), separated by 0–13 base pairs [6]. The deletion of Trp53 in mice leads to normal development (for the most part) indicating it is not an essential gene [7, 8].
The TP53 tumor suppressor is mutated in majority of human tumors with frequencies varying from ~1% in papillary thyroid cancer to >95% in high grade serous ovarian carcinomas [9]. Most of the p53 mutations are missense and with few exceptions are confined to the DBD. Six codons: 175, 245, 248, 249, 273 and 282 were designated “hotspots” because of their increased propensity for mutation [10]. p53 hotspot mutations are further classified as either DNA contact mutations (R248W, R248Q, R273H) or structural mutations (R175H, G245S, R249S, R282H) based on whether the residue has a role in direct DNA contact or in establishment of p53 structure. Missense mutations in the TD also incapacitate p53 transcriptional activity [11].
Given that p53 functions as a gatekeeper for cellular proliferation [12] and its activity determines cell fate, it is tightly regulated in a cell. Mdm2 and a closely related protein Mdm4 are two major negative regulators of p53 activity. In vitro studies have shown that both these proteins bind p53 and mask its TAD [13–15]. Additionally, Mdm2 also promotes p53 degradation through the 26 S proteasome machinery [16–18]. The relationship between Mdm2 and p53 is even more complex as Mdm2 is also a transcriptional target of p53 and thus the two proteins are linked by an autoregulatory loop through which they modulate each other’s levels [19, 20]. Experimental studies have revealed the temporal dynamics of p53 levels following DNA damage. p53 is expressed in a series of discrete pulses, and p53 and Mdm2 exhibit an oscillatory pattern in response to gamma irradiation [21]. Amplification/overexpression of Mdm2/Mdm4 is another mechanism by which p53 activity is dampened in tumors [22, 23]. Importantly, p53 mutations and amplification/overexpression of Mdm2/Mdm4 are mutually exclusive events in tumors further highlighting the close relationship between these partners [23].
Physiological signals that stabilize p53 and activate transcription
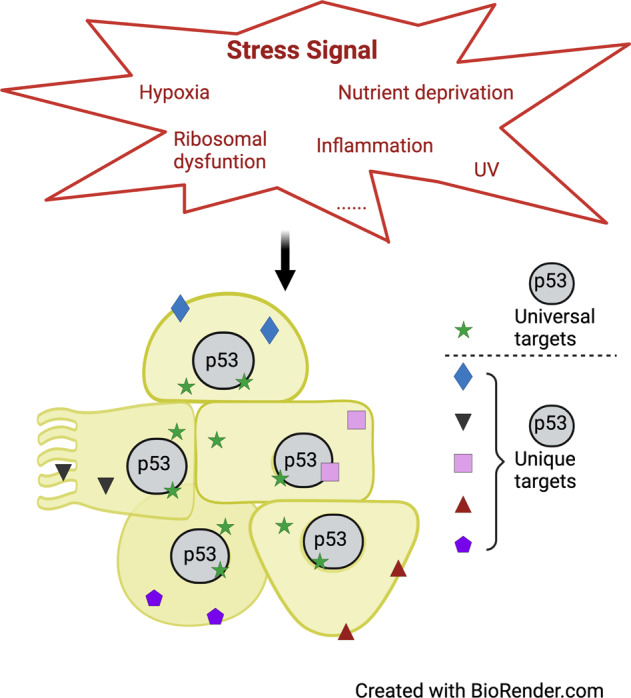
The p53 protein is barely detectable in normal cells and has a very short half-life (~20–30 min) [24, 25]. Multi-decades of research show that p53 is stabilized and activated by a plethora of intrinsic and extrinsic signals that include DNA damage, hypoxia, metabolic dysfunction, perturbations to ribosomal biogenesis, metabolic stress and inflammation amongst other stressors (Fig. 1) [26]. Most of these stressors induce p53 via post-translational modifications at the amino and carboxyl ends of the protein, some of which regulate interaction with Mdm2/4 and oligomerization, respectively. p53-mediated stress response depends on cell type and context as well as the extent, duration, and origin of the stress signal [27]. Growing evidence suggests that the p53 transcriptional response is not limited intracellularly but signals to adjacent cells as well. This section will focus on the physiological signals that regulate p53 activity and the consequences on cell survival.
Fig. 1. A diagram showing major affecters and effectors of p53 transcriptional activity including a universal p53 gene signature.

Top three tissue-specific p53-activated genes from Moyer et al. [73] are shown for each tissue.
Ultraviolet (UV) radiation is an environmental carcinogen that induces DNA damage by causing cyclobutane pyrimidine dimers and 6-4 photoproducts [28]. UV light exposure preferentially activates the ATR protein kinase, which phosphorylates both p53 (at multiple sites including S15 and S37) and its inhibitor MDM2 (S407) [29]. Phosphorylation of p53 removes MDM2 and the phosphorylation of MDM2 reduces MDM2-dependent export of p53 to the cytoplasm; both events lead to p53 accumulation and activation.
Ribosomopathies, a diverse collection of human genetic disorders that are commonly caused either by haploinsufficiency of ribosomal proteins or due to defects in ribosome biogenesis trigger nucleolar stress that activates p53 [30]. Mutations in RPL5, RPL11, RPS19, RPS24, RPS17, or RPL35A genes disrupt ribosome biogenesis resulting in accumulation of free ribosomal proteins that sequester MDM2 [30, 31]. The consequent stabilization and activation of p53 leads to cell-cycle arrest and apoptosis, which ultimately leads to Diamond-Blackfan anemia [30, 31]. These studies are supported by in vivo studies in mice which show that a number of ribosomal proteins such as L5, L11 and L23, also bind and inhibit Mdm2, thereby stabilizing and activating p53 [32–35]. Ribosomal stress also enhances Mdm2-mediated degradation of Mdm4, thereby releasing p53 from the negative regulation exerted by both proteins [31, 36].
Hypoxia presents as a physiological stress under certain conditions (eg. Hypoventilation, strenuous exercise, cerebral ischemia) [26]. Hypoxic or anoxic conditions stabilize HIF 1α (hypoxia inducible factor) by disrupting its interaction with VHL (von Hippel-Lindau) allowing HIF 1α to bind p53 and promote its stabilization. Additionally, VHL can also interact with p53 directly and promotes p53 phosphorylation and acetylation, leading to its activation [37].
p53 also responds to nutrient availability and manages cell proliferation following metabolic stress. Reduced nutrients or energy levels result in inhibition of the AKT–mTOR pathway and in activation of AMPK (AMP-activated protein kinase), both of which lead to the induction of p53 [38]. Induced p53 in turn activates genes involved in oxidative phosphorylation (OXPHOS) such as GLS2 (glutaminase 2) and SCO2 (synthesis of cytochrome c oxidase 2) and decreases the expression of glucose importers GLUT1 and GLUT4 (glucose transporter 1 and 4) that promote glycolysis [39–44]. Studies in mice show that perturbations in nutrient availability induce p53. Deprivation of serine and glycine amino acids promotes p53 post-translational modification and recruitment at the p21 promoter correlating with cellular accumulation at the G1 stage [44]. Xenograft mice fed a diet lacking serine and glycine display significantly reduced tumor volume and longer survival in a p53-dependent manner. Thus, limited nutrient availability directly correlates with increased p53 transcriptional activity and reduced cell proliferation.
Chronic inflammation has also been linked with p53 activation. p53 is constitutively activated in hepatocytes in patients with chronic liver disease (CLD) [45]. In fact, every causative agent of CLD such as hepatitis B virus X-antigen, the core protein of hepatitis C virus, ethanol exposure and fat accumulation in the liver can activate p53 in hepatocytes [46–48]. A recent study also shows that Mdm2 deletion in hepatocytes activates p53 resulting in apoptosis and a senescence-associated secretory phenotype [45]. p53 activation leads to loss of hepatocytes and subsequent expression of chemokines and humoral factors that expand the hepatic progenitor cell population. These progenitor cells over proliferate, show chromosomal instability and eventually transform. Thus, paradoxically, p53 activation in hepatocytes accelerated the development of liver tumors originating from the expanded pool of the hepatocyte progenitor cells [49].
Thus, numerous physiological signals transiently activate p53 to maintain homeostasis. Constitutive activation of any of these signals would result in increased p53 stability and activity, and pressure cells to dampen its function by either mutating or losing the wild type p53 allele to ensure cell survival. However, lowering the p53 protective shield also exposes the cell to transformation.
Pathological repercussions of increased p53 activity
Given the central role of p53 in regulating diverse cellular responses, inappropriate activation of p53 has pathological consequences which are dependent on the level of p53 activity, stress type and duration and cell/tissue composition. Constitutive induction of p53 effectors can disrupt tissue homeostasis and development. Genetically engineered murine models have been extensively used to understand the physiological consequences of increased p53 activity. Modulating the gene dosage of Mdm2 and Mdm4 in mice has been fundamental to our understanding of p53 physiological functions.
The levels of p53 determine physiological outcomes. Unrestrained activation of p53 in embryogenesis is lethal. Loss of Mdm2 in mice leads to embryonic lethality at pre-implantation due to induction of apoptosis, a phenotype that is rescued by the concomitant deletion of p53 [50, 51]. Similar to Mdm2-null mice, Mdm4 loss in mice also results in p53-dependent embryo lethal phenotypes [52, 53].
In vivo studies using tissue-specific activation of p53 vis-a-vis Mdm2 or Mdm4 deletion and combinations thereof, yield phenotypes that are invariably p53-dependent highlighting the essential role of Mdm2 and Mdm4 in inhibiting p53 activity (reviewed by Eischen and Lozano, 2014). Mdm2 loss always produces a cell lethal event while Mdm4 loss is more nuanced and varies from cell lethality during embryogenesis to a milder reversible phenotype in old mice [54–56].
Murine models with enhanced basal p53 transcriptional activity present a range of phenotypic aberrations wherein tissues with high turnover exhibit major defects [57]. For example, Super p53 (with three copies of p53), Mdm2+/−, and Mdm4+/− mice develop normally, are not tumor prone as expected but succumb to p53-dependent lethality (bone marrow depletion) upon low dose radiation. Mdm2+/−Mdm4+/− double heterozygous mice are developmentally abnormal, and the few progeny that are born die before weaning due to bone marrow ablation. This phenotype is rescued by deletion of a single p53 allele suggesting a stoichiometric relationship between Mdm2, Mdm4, and p53 [58]. Furthermore, Mdm2Puro/∆7-12, Mdm2PND and Mdm25AA mice (hypomorphic Mdm2 alleles with increased p53 activity) exhibit unique p53-dependent pathologies across multiple tissues. These include skin hyperpigmentation, lymphopenia, small testis and decreased number of ovarian follicles resulting in fertility issues (Table 1) [59–62].
Table 1.
p53 induced pathologies in mouse tissues.
| Mouse Genotype | p53 induced pathologies/tissues affected | Rescue/Reversal | Ref. |
|---|---|---|---|
| Mdm2Puro/∆7-12 |
Lymphopoiesis defects Radiation sensitivity (8 Gy WBI) |
NP NP |
[59] [62] |
| Mdm2PND |
Hyperpigmentation in extremities Gonadal atrophy (small testis, follicular defects) Radiation sensitivity (3 Gy WBI) |
Kitl ↓ or p53+/− p53+/− or Puma−/− p53+/− or Puma−/− |
[60] |
| Mdm2P2/P2 |
Radiation sensitivity (6 Gy WBI), BM ablation Radiation resistance (17 Gy SBI), no GI toxicity |
Puma−/− p21−/− |
[75] [78] |
| Mdm25AA | Radiation sensitivity (6 Gy WBI), fertility defects | NP | [61] |
| Super p53 | Radiation resistance (13.4 Gy SBI), no GI toxicity | p21−/− | [79] |
| Mdm2−/−p53ERTAM |
Apoptosis in radiosensitive tissues Inhibition of cell proliferation in apoptosis-resistant tissues |
NP | [67] |
| Mdm4−/−p53ERTAM | Mild/reversible apoptosis and cell proliferation defects in tissues | NP | [55] |
| Mdm2 FM/−;CAG-CreER |
Lethal with severe pathologies in major tissues after four days post Tamoxifen injection Atrophy of splenic white pulp, BM depletion, villus atrophy and crypt apoptosis |
p53−/− | [72] |
| Mdm2 FM/−;CAG-CreER |
Tissue necrosis in pancreas, kidney and intestine 24 hours post Tamoxifen injection No noticeable defects in ovary and heart |
NP | [73] |
NP not performed, BM bone marrow, GI gastrointestinal, WBI whole body irradiation, SBI shielded body irradiation.
Constitutive activation of p53 has also been associated with aging phenotypes in mice (p53+/m) and humans (MDM2 anti-terminating mutation) [63, 64]. However, failure to observe similar results in Super p53 or hypomorphic Mdm2 mice (Mdm2Puro/∆7-12, Mdm2PND, Mdm25AA) contradict this observation [59–61, 65]. While Super p53 and hypomorphic Mdm2 mice have shorter life spans, they do not exhibit overt aging associated phenotypes. Of note, mice with aging defects express constitutive p53 activity (p53+/m) and also have other genomic aberrations that likely contributed to aging [63]. p53+/m mice are haploinsufficient for 24 genes upstream of p53 [66]. Similarly, the human patient reportedly homozygous for an MDM2 anti-terminating mutation (that renders MDM2 partially dysfunctional) was a result of a consanguineous relationship and was genetically homozygous across a wide spectrum of the genome that likely contributes to progeria [64].
Tissue specificity of p53 transcriptional activity
The p53 response is cell and tissue specific. Inducible murine models and tissue specific Cre-systems have been effectively used to decipher p53 response in a spatio-temporal manner. Conditional restoration of p53ERTAM function in Mdm2-null mice results in irrepressible activation of p53 and death [67]. Classically radiosensitive tissues (thymus, bone marrow etc.) are rapidly ablated while the unbuffered p53 activity triggers profound inhibition of cell proliferation in apoptosis-resistant tissues. Activation of p53 in p53ERTAM Mdm2-null mice results in a marked upregulation of p21 (Cdkn1a) and Puma (Bbc3) in most tissues tested, although the kinetics and extent of induction varies between tissue types. p21 induces cell cycle arrest at the G1/S boundary and Puma induces apoptosis in vivo as determined by classic murine gene knock out studies [68–70]. Of note, other studies with DNA damage-inducing drugs have shown that p53-mediated induction of Puma is not restricted to radiosensitive cells which undergo apoptosis but also occurs in cells that undergo cell cycle arrest and survive [71]. Conditional restoration of p53ERTAM function in Mdm4-null mice similarly revealed a role for Mdm4 in buffering p53 activity in adult normal tissues and their stem cells [55]. Apart from the small intestine, p53 restoration potently induced p21 in all the tissues tested while the proapoptotic gene Puma was induced only in classically radiosensitive tissues (bone marrow, spleen, thymus, and intestinal epithelium). Notably, the effects of transient p53 restoration in the absence of Mdm4 were mild, nonlethal and reversible compared to that of Mdm2 loss.
Another study utilized a conditional Mdm2 allele, Mdm2FM, and a CAG-CreER tamoxifen-inducible recombination system to examine the effects of global Mdm2 loss in adult mice [72]. Increased p53 levels upon Tamoxifen injection (once daily for 3 days) resulted in 100% lethality of Mdm2FM/−;CAG-CreER mice. Most tissues including the kidney, liver, heart, retina and hippocampus exhibit pathological defects correlating with induction of canonical p53 targets p21 and Puma. p21 was activated by approximately 190-fold in the kidney, 40-fold in the heart and 20-fold in cerebellum, cerebrum and eye, but only slightly in the liver and spleen. On the other hand, Puma was elevated approximately 20-fold in the kidney, seven-fold in the eye and heart, three-fold in the cerebrum and two-fold in the cerebellum. In addition, multiple senescence markers, including Cdkn2b (p15), Pml, Ccr6 (Dcr2), p19Arf (Arf/Cdkn2a), Bhlhe40 (Dec1) and p16Ink4a (Cdkn2a), were upregulated to varying degrees in these tissues. These data indicate that the intensity of p53 activity and the fold-induction of downstream targets varies between different tissues.
An acute p53 response was also noted in Mdm2FM/−; CAG-CreER mice just twenty-four hours after a single Tamoxifen injection [73]. Significant increases in p53 protein and associated pathologies was evident in the pancreas, kidney, and intestine as compared to control treated mice. Acinar to ductal metaplasia was noticeable in the pancreas with severe necrotizing pancreatitis along with robust immune cell infiltration. The kidney displayed a two-fold increase in the number of protein casts and dilated tubules. The intestinal crypts, that harbor stem cells and newly differentiated epithelial cells experienced crypt atrophy/drop out. Surprisingly, despite clear evidence of recombination at the Mdm2 locus, the ovary and heart displayed a modest p53 transcriptional response and no observable defects at this time point. The differences in tissue pathologies observed in mice after tamoxifen treatment for 24 h or 3 days again highlight the spatial and temporal variation of the p53 response.
Mdm2 deletion in older Mdm2FM/−; CAG-CreER mice (16–18 month-old) resulted in atrophied spleen and kidney abnormalities, albeit not as extensive as observed in tissues of young mice [72]. The transcriptional activation of p21, Puma and senescence markers was also reduced in older mice when compared to young (2–4-month-old) mice suggesting that p53 activity dampens with age. This was further corroborated by a chromatin immunoprecipitation experiment that revealed dampened p53 binding to the promoters of target genes in older mouse tissues. These data perfectly align with previous report that p53 transcriptional activity and protein stability significantly decline in tissues of old mice [74]. Altogether, these data reinforce the tissue specificity of p53 transcriptional activity. Furthermore, the severity of p53 response in different tissues is dependent on the duration of the p53 activity and the age of mice.
Other models have been used to genetically examine the importance of downstream p53 targets in the physiological response to Mdm2 loss. Enhanced p53 activity in Mdm2P2/P2 mice with a compromised p53-Mdm2 autoregulatory loop makes them exquisitely radiosensitive succumbing to sublethal doses (6 Gy) of ionizing radiation in ∼20 days [75]. The primary cause of this p53-dependent lethality is attributed to bone marrow ablation that is completely reversed by genetic loss of the p53 downstream target Puma but not the loss of the cell cycle arrest and senescent p53 target gene p21. Similarly, in another study, a hypomorphic Mdm2 allele (Mdm2PND) induces p53-hyperactivity phenotypes that include hyperpigmentation of extremities and fertility defects. The skin hyperpigmentation phenotype was driven by p53-mediated Kit-ligand (Kitl) signaling and was not rescued by concomitant deletion of p21 or Puma. The male reproductive phenotypes could be rescued by loss of p53 target Puma but not by deletion of p21 [60]. These illustrative examples of dose- and tissue-specific differences in p53 response in mice underscore the role of distinct downstream targets in manifesting p53-dependent pathologies and warrant a deeper understanding of the p53 response in vivo (Table 1).
Altogether, these studies clearly show that tissues with high turnover potential are more sensitive to p53-dependent apoptosis. This could be attributed to the stem cells in these tissues which are irrevocably damaged by p53 activation. Indeed, LSK (Lin-Sca-Kit + ) stem cells in the bone marrow are highly sensitive to p53 activation [75, 76]. Similarly, wild type bone marrow transplantation in Mdm2P2/P2 mice could protect them from radiation induced bone marrow ablation and death [75].
Of note, increased p53 activity is not necessarily deleterious in all tissues. Murine studies also indicate that p53 activity plays a protective role in gastrointestinal (GI) tissue. p53 wild type mice subsist better than p53-null mice after high dose radiation to the intestine [77]. Similarly, enhanced acute p53 activity in irradiated Mdm2P2/P2 mice protects them from GI failure as compared to wild type mice [78]. Intestinal cells residing in the +4 and higher positions, but not crypt cells, exhibit decreased apoptosis, increased p21 expression, and hyper-proliferation to re-establish intestinal integrity of irradiated Mdm2P2/P2 mice. This effect could also be recapitulated in wild type mice upon pharmacological augmentation of p53 activity with an Mdm2 inhibitor. In contrast, deletion of p21 in Mdm2P2/P2 mice renders them radiosensitive implicating its role in gut radioprotection. Similar results were observed upon ionizing radiation (IR) exposure of Super p53 mice which carry an extra copy of p53 (Table 1) [79]. Thus, different p53-downstream targets are involved in the manifestation of tissue-specific pathologies and play a role in determining the paradoxical radiation response in hematopoetic and gastrointestinal tissues.
Molecular profiling of the p53 transcriptional program
Despite all these studies, our understanding of the p53 physiological transcriptional program remains inadequate. Multiple RNA-seq, microarray and other in silico studies were carried out to identify global p53 targets scattered across the genome. These have been further refined with the addition of chromatin immunoprecipitation-sequencing (ChIP-seq) studies to validate the direct transcriptional targets of p53.
An unbiased analysis of chromatin occupancy by p53 using ChIP-seq on MCF7 cells (a human breast tumor cell line) treated with either 5FU (5-fluorouracil), Nutlin3a (an Mdm2 inhibitor) or RITA (compound that reactivates p53) was carried out by Nikulenkov et al. [80]. Overlaying the combined ChIP-seq data with microarray expression data from Nutlin3a treated cells identified 320 differentially expressed genes that included 280 novel and 40 previously known p53 targets. Of these, 254 genes were upregulated in the Nutlin3a treatment group. In another study, Menendez et al. compared either doxorubicin vs untreated, or Nutlin3a vs DMSO treated U2OS cells (TP53 wild type osteosarcoma cells) to identify p53 targets [81]. Overlaying the results with p53 ChIP analyses revealed 275 genes that were both bound by p53 and also differentially expressed after doxorubicin or Nutlin3a treatment. To identify direct p53 targets, Allen et al. utilized Global Run-On sequencing (GRO-seq) analyses on isogenic cell lines (HCT116 colorectal cancer cells) with or without TP53 after short term treatment with Nutlin-3a [82]. This methodology identified 198 gene loci whose transcription was significantly induced in Nutlin-3a treated p53 wild type cells. Comparison of the above three studies wherein cancer cell lines were treated with Nutlin3a identified an overlap of only 43 genes that were commonly upregulated (Fig. 2A). p53 canonical targets BAX, CDKN1A, FAS, and MDM2 were included in this list. Thus, in conclusion, these studies highlight the commonality and also the uniqueness of the p53 transcriptional program across multiple cell types (breast, bone, colon). Nonetheless, as noted earlier, these studies were carried out in different cancer cell lines which have major chromosomal rearrangements and epigenetic changes that differ from normal precancerous cells. Moreover, isogenic cell line controls with p53 loss was lacking in the first two studies. Some of the drugs used in these studies to activate p53 also induce p53-independent pathways and transcriptional programs. Other confounding factors including differences in methodology, time point of analysis post damage, and different criterion for target selection have likely obfuscated a true physiological response.
Fig. 2. Molecular profiling of the p53 transcription factor.

A Comparison of upregulated genes from the Allen et al., Menendez et al. and Nikulenkov et al. data sets that identify common p53 targets in cancer cell lines treated with Nutlin 3a. B Comparison of Li et al., and Kenzelmann Broz et al. data sets from mouse embryonic stem (mES) cells and mouse embryonic fibroblasts (MEFs) to identify common p53 targets. C Identification of 7 common genes activated by p53 gene in murine tissues. Rederived from Moyer et al. [73]. D Comparison of Moyer et al., and Tanikawa et al. data sets to identify common p53 targets. http://www.interactivenn.net/ was used to generate the Venn diagrams [101]. Some shared common genes are listed. Numbers denote either the total number of genes expressed (in parentheses) or the number of unique/shared genes.
p53 transcriptional studies have also been carried out in undifferentiated and differentiated normal cells. A study using ChIP-seq and microarray profiling on mouse embryonic stem cells (mES cells) treated with the DNA-damaging agent adriamycin identified 3697 genes as direct p53 targets of which 2070 were activated while 1627 were repressed following treatment [83]. This is an approximately 10 fold increase in the number of genes activated by p53 compared to cancer cell lines. Similarly, Kenzelmann Broz et al. performed RNA-seq and ChIP-seq on p53 wild type and p53-null mouse embryo fibroblasts (MEFs) that were treated with the DNA-damaging agent doxorubicin to identify upregulated gene promoters bound by p53 [84]. The study identified 432 upregulated genes that contained p53 binding sites. Comparison of the above two studies revealed 245 common genes that are p53-dependent and upregulated in both studies (Fig. 2B). Differences in the upregulated genes from these murine cell-based studies reveal that p53 activity is cell state and DNA damage dependent.
To elucidate the p53 transcriptional landscape in vivo, Tanikawa et al. treated p53−/− and p53+/+ mice with 10 Gy IR and harvested tissues 24 h later [85]. RNA-seq analysis revealed that a total of 3551 genes were induced in 24 tissue types collected. Superimposing these data with ChIP-seq data from Kenzelmann Broz et al. [84] identified 741 genes bound by p53. Further in-depth analysis revealed that 37 genes were induced in more than 7 tissues. Thus, while many genes are commonly activated by p53, the majority are tissue specific. To identify p53 global targets, our lab took a slightly different approach and compared p53 upregulated genes in conditional Mdm2FM/−; CAG-CreER and Mdm2FM/− mice (control) following Tamoxifen-mediated recombination and deletion of Mdm2 [73]. We performed RNA sequencing of pancreas, kidney, heart, ovary and intestine tissues to elucidate the transcriptional programs activated by p53 and overlaid tissue-specific RNA-sequencing data with existing p53 ChIP-sequencing data [84]. A total of 414 p53-specific upregulated and 27 downregulated genes were identified in these tissues. Further analyses yielded a common p53 transcriptional signature that consisted of only seven genes that included Mdm2 (the conditional Mdm2 allele retains the p53 binding sites in the promoter), and other genes involved in known p53 functions such as cell cycle arrest (Ccng1, Gtse1, Psrc1), apoptosis (Eda2r), and DNA repair (Polκ, Zfp365) (Fig. 2C). Surprisingly, p21, a canonical target gene considered the gold standard in assessing p53 activity, was not significantly upregulated in the heart and ovary after Mdm2 deletion and not included in the list. Nonetheless, p21 was upregulated in 3/5 tissues (pancreas, kidney, and intestine) and was also upregulated in more than 7 tissues in Tanikawa et al. study. Comparison of these studies revealed that except for PolK, the other six gene targets were common to both set of studies (Fig. 2D). Of note, comparison of mES and MEFs RNA-seq data (Fig. 2B) also showed upregulation of the six of seven genes from Moyer et al. with the exception of Gtse1. A compilation of these data suggests that the seven genes and p21 define an eight gene signature that can be informatively used to identify p53 transcriptional status. The human counterparts of these genes also carry p53 responsive elements. Many of the genes identified have not been studied in much detail and further studies of their functions will provide much needed insights into the p53 transcriptional program. Thus, these studies highlight the large repertoire of direct p53 targets and the most of p53 these transcribed genes are tissue specific.
A discussion of the universally induced p53 gene targets is warranted. As noted above, Mdm2 as a canonical target of p53 encodes the major p53 inhibitor and likely fine tunes the p53 response. Mdm2 is the only p53 target identified in all studies discussed above. Ccng1, (cyclin G1) is a canonical p53 target gene bearing a p53 response element in the first intron of the gene. Ccng1-deficient mice are developmentally normal and viable [86, 87]. Paradoxically, Ccng1-null mice are highly radiosensitive and exhibit a reduced incidence of hepatic tumors upon exposure to hepatocarcinogens followed by partial hepatectomy [86]. These data imply increased p53 tumor suppressor activity. Perhaps, in response to damage, Ccng1 loss feeds back to activate p53. Another common p53 target, Gtse1 (G2 and S phase expressed protein) encodes a microtubule associated protein that accumulates in the nucleus after DNA damage. Gtse1 binds p53 and shuttles it out of the nucleus [88]. Gtse1 overexpression delays the G2/M phase of the cell cycle. In line with these data, knockdown of Gtse1 in cell lines increases p53 protein levels and activity that synergizes with DNA damage to further increase the levels and activity of p53 [88]. Gtse1 directly interacts with the C-terminal regulatory domain of p53 and negatively impacts p53 activity. Thus, both Ccng1 and Gtse1 seem to function like Mdm2 (although not near as potent) in that they are upregulated by p53 and in turn inhibit p53 activity. Perhaps the effect of these proteins on cell cycle transition contributes to this feedback inhibition. Also, as these experiments were performed at a single time point and thus, the dynamic nature of p53 regulation is not evident in these studies. The in vivo relevance of these interactions remains unknown. Another p53 target, Psrc1 (Proline/serine-rich coiled-coil protein 1) encodes a microtubule-associated protein that controls chromosome condensation and segregation by regulating the mitotic spindle. Depletion of Psrc1 perturbs chromosome condensation and alignment at the metaphase equator [89]. Thus, three universally expressed p53 targets encode proteins that control various aspects of the cell cycle. It is possible that the expression of these genes is restricted to certain cell types in a tissue and/or also dependent on the cell cycle stage of the particular cell. Nonetheless, their important role in p53 regulation of the cell cycle remains indisputable.
Eda2r (also known as XEDAR) is a transmembrane receptor that belongs to the TNF-receptor super family of proteins known to induce apoptosis. The Eda2r gene was previously identified as a p53 target [90, 91]. Eda2r overexpression increases caspase 3 activity and causes anoikis, a form of cell death that occurs in anchorage dependent cells. Reduced expression of EDA2R is observed in mutant p53 breast, colorectal and lung cancers [90] and in pancreatic ductal adenocarcinoma and glioblastoma multiforme (unpublished data). Of note, Eda2r deletion in mice results in viable fertile mice that live a normal life span [92]. Other reports suggest that EDA2R is significantly upregulated in wild type TP53 cancer types [93]. However, for unknown reasons, EDA2R upregulation is not sufficient to initiate cell death in these tumors. Clearly a more in-depth characterization of Eda2r is needed to clarify its biological role in officiating p53 functions.
The p53 target Polκ encodes DNA polymerase κ, a traditionally error-prone polymerase that is overexpressed in some tumors [94]. The main cellular strategy to tolerate DNA damage during replication is to synthesize DNA past the damage. The up regulation of Polκ by p53 in vivo suggests a cellular survival mechanism by which p53 pushes through DNA damage but with accumulation of errors. Polκ-null mice are viable and fertile but have a shortened life span for unknown reasons [95]. Polκ-null mouse embryo fibroblasts (MEFs) are sensitive to UV radiation and die more quickly than control MEFs and are also hypersensitive to killing by benzo[a]pyrene [95, 96]. Many tissues from Polκ-null mice also have a higher spontaneous mutation frequency (mostly G:C to A:T base pair changes) beginning at 9 months as compared to normal controls [97]. Thus, in a toxic stressful environment, Polκ is required acutely for replication and cell survival. The long-term consequences of constant/repetitive damage are unknown but may lead to transformation. The last p53 transcriptional target included the universal p53 signature list is Zfp365 which encodes a zinc-finger protein that promotes stability of fragile sites and telomeres [98]. Thus, both Polκ and Zfp365 are taking stock of the integrity of the genome of the cell.
While the compilation of these studies has defined a p53 signature over space and time that could be used to measure/validate p53 activation, the most fascinating aspect of these studies is the broad nature of the p53 response and the number of tissue specific genes regulated by p53 (Figs. 1 and 2). Whether any of these genes is a cell-specific essential mediator of the p53 response needs to be examined as such evidence would increase the repertoire of p53 reactivation targets.
The non-cell autonomous nature of the p53 response
Physiologically, there is evidence for a non-cell autonomous response to p53 activation [26, 99]. For example, deletion of Mdm2 in the intestine (using Villin Cre-mediated recombination) results in a dramatic increase in p53-dependent apoptosis in differentiated cells [100]. These cellular abnormalities trigger activation of canonical Wnt and EGFR-Ras/MAPK pathways which increase proliferation of stem cells that retain Mdm2 and promote survival of the organism. This implies that cell-cell communication occurs to ensure functional integrity of the organ. In addition, the acinar to ductal metaplasia observed in the pancreas 24 h after ubiquitous Mdm2 depletion, is also non-cell autonomous as the phenotype disappears with acinar-specific deletion of Mdm2 [73]. While these phenotypes are p53-dependent, whether they are due to p53 transcriptional regulation of target genes is not known. Nonetheless, many genes activated by p53 in vivo encode proteins that promote cell signaling. For example, Eda2r (discussed above) is a cell surface receptor, while Gdf15, Hgf, Jag2, Lif, Ltbp2, Ngf, Pdgfc, and Tnfsf9 (identified in the Moyer et al. study) encode cytokines and growth factors. More studies are needed to understand the roles of these genes and factors in non-cell autonomous response to p53 activation.
Conclusions and future directions
Thus, numerous physiological signals activate the p53 tumor suppressor and it in turn regulates a transcriptional program that is dependent on context and age. In vitro and in vivo studies have cataloged the global p53 transcriptional program and identified a universal 8-gene signature of p53 transcriptional activity that could be used to assess p53 activation in general. However, as p53 activity and resulting pathologies vary enormously amongst tissues, identifying the tissue-specific nature of the p53 response is needed. The p53 response in a non-cell autonomous manner suggests communication between adjacent neighboring cells is also an important function of p53 yet remains poorly understood. Moreover, the spatio-temporal nature of p53 activity should be further investigated by analyses of the transcriptional program at different times post stress and as a function of aging to determine if dampened p53 activity is gradual or punctuated. Single cell RNA sequencing and other techniques may be utilized to further extend our understanding of p53 function at a cellular level within the physiological complexity of an organ.
One caveat of some of the studies published thus far is the supra-physiological levels of unleashed p53 activity. Careful use of drugs/stressors at physiologically relevant dosages to specifically induce p53-dependent pathways and transcriptional programs will improve the quality of the data. Finally, besides analyzing mRNA levels of p53 targets, high quality proteomic analysis of the proteins expressed by these genes is needed. This may aid in understanding of p53 stabilization/activation which is very heterogenous in tissues.
Overall, the fact that p53 activates cell cycle inhibitors that arrest cell proliferation, other genes that induce apoptosis, and Mdm2 which encodes the major p53 inhibitor leading to cell survival supports a dynamic view of p53 surveillance and transcriptional activation. Systematic in vivo studies characterizing the p53 transcriptional profile will help refine the actionable repertoire of p53 transactivation targets for therapeutic purposes.
Acknowledgements
We thank Jovanka Gencel-Augusto, Shunbin Xiong and Xiaojie Yu for suggestions on this manuscript.
Author contributions
CS and VP analyzed the data. VP and GL conceived the idea, wrote the manuscript and prepared the figures.
Funding
VP is partially supported by R50-CA251703. Mdm2 studies are funded by NCI grant CA47296 to GL.
Competing interests
The authors declare no competing interests.
Footnotes
Edited by G. Melino
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Joerger AC, Fersht AR. Structural biology of the tumor suppressor p53. Annu Rev Biochem. 2008;77:557–82. doi: 10.1146/annurev.biochem.77.060806.091238. [DOI] [PubMed] [Google Scholar]
- 2.Sullivan KD, Galbraith MD, Andrysik Z, Espinosa JM. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 2018;25:133–43. doi: 10.1038/cdd.2017.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clore GM, Gronenborn AM. Determining the structures of large proteins and protein complexes by NMR. Trends Biotechnol. 1998;16:22–34. doi: 10.1016/S0167-7799(97)01135-9. [DOI] [PubMed] [Google Scholar]
- 4.Lee W, Harvey TS, Yin Y, Yau P, Litchfield D, Arrowsmith CH. Solution structure of the tetrameric minimum transforming domain of p53. Nat Struct Biol. 1994;1:877–90. doi: 10.1038/nsb1294-877. [DOI] [PubMed] [Google Scholar]
- 5.Jeffrey PD, Gorina S, Pavletich NP. Crystal structure of the tetramerization domain of the p53 tumor suppressor at 1.7 angstroms. Science. 1995;267:1498–502. doi: 10.1126/science.7878469. [DOI] [PubMed] [Google Scholar]
- 6.Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9:402–12. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- 7.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr., Butel JS, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–21. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 8.Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/S0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 9.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–9. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hainaut P, Pfeifer GP. Somatic TP53 mutations in the era of genome sequencing. Cold Spring Harb Perspect Med. 2016;6:a026179. doi: 10.1101/cshperspect.a026179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gencel-Augusto J, Lozano G. p53 tetramerization: at the center of the dominant-negative effect of mutant p53. Genes Dev. 2020;34:1128–46. doi: 10.1101/gad.340976.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zilfou JT, Lowe SW. Tumor suppressive functions of p53. Cold Spring Harb Perspect Biol. 2009;1:a001883. doi: 10.1101/cshperspect.a001883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–45. doi: 10.1016/0092-8674(92)90644-R. [DOI] [PubMed] [Google Scholar]
- 14.Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature. 1993;362:857–60. doi: 10.1038/362857a0. [DOI] [PubMed] [Google Scholar]
- 15.Shvarts A, Steegenga WT, Riteco N, van Laar T, Dekker P, Bazuine M, et al. MDMX: a novel p53-binding protein with some functional properties of MDM2. EMBO J. 1996;15:5349–57. doi: 10.1002/j.1460-2075.1996.tb00919.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 17.Marine JC, Lozano G. Mdm2-mediated ubiquitylation: p53 and beyond. Cell Death Differ. 2010;17:93–102. doi: 10.1038/cdd.2009.68. [DOI] [PubMed] [Google Scholar]
- 18.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–9. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 19.Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993;7:1126–32. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- 20.Barak Y, Juven T, Haffner R, Oren M. mdm2 expression is induced by wild type p53 activity. EMBO J. 1993;12:461–8. doi: 10.1002/j.1460-2075.1993.tb05678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geva-Zatorsky N, Rosenfeld N, Itzkovitz S, Milo R, Sigal A, Dekel E, et al. Oscillations and variability in the p53 system. Mol Syst Biol. 2006;2:2006 0033. doi: 10.1038/msb4100068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oliner JD, Saiki AY, Caenepeel S. The role of MDM2 amplification and overexpression in tumorigenesis. Cold Spring Harb Perspect Med. 2016;6:a026336. doi: 10.1101/cshperspect.a026336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wasylishen AR, Lozano G. Attenuating the p53 pathway in human cancers: many means to the same end. Cold Spring Harb Perspect Med. 2016;6:a026211. doi: 10.1101/cshperspect.a026211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pant V, Xiong S, Iwakuma T, Quintas-Cardama A, Lozano G. Heterodimerization of Mdm2 and Mdm4 is critical for regulating p53 activity during embryogenesis but dispensable for p53 and Mdm2 stability. Proc Natl Acad Sci USA. 2011;108:11995–2000. doi: 10.1073/pnas.1102241108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Itahana K, Mao H, Jin A, Itahana Y, Clegg HV, Lindstrom MS, et al. Targeted inactivation of Mdm2 RING finger E3 ubiquitin ligase activity in the mouse reveals mechanistic insights into p53 regulation. Cancer Cell. 2007;12:355–66. doi: 10.1016/j.ccr.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 26.Boutelle AM, Attardi LD. p53 and tumor suppression: it takes a network. Trends Cell Biol. 2021;31:298–310. doi: 10.1016/j.tcb.2020.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–31. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 28.Latonen L, Taya Y, Laiho M. UV-radiation induces dose-dependent regulation of p53 response and modulates p53-HDM2 interaction in human fibroblasts. Oncogene. 2001;20:6784–93. doi: 10.1038/sj.onc.1204883. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y, Tavana O, Gu W. p53 modifications: exquisite decorations of the powerful guardian. J Mol Cell Biol. 2019;11:564–77. doi: 10.1093/jmcb/mjz060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Narla A, Ebert BL. Ribosomopathies: human disorders of ribosome dysfunction. Blood. 2010;115:3196–205. doi: 10.1182/blood-2009-10-178129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pant V, Quintas-Cardama A, Lozano G. The p53 pathway in hematopoiesis: lessons from mouse models, implications for humans. Blood. 2012;120:5118–27. doi: 10.1182/blood-2012-05-356014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lohrum MA, Ludwig RL, Kubbutat MH, Hanlon M, Vousden KH. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell. 2003;3:577–87. doi: 10.1016/S1535-6108(03)00134-X. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Y, Wolf GW, Bhat K, Jin A, Allio T, Burkhart WA, et al. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol Cell Biol. 2003;23:8902–12. doi: 10.1128/MCB.23.23.8902-8912.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dai MS, Lu H. Inhibition of MDM2-mediated p53 ubiquitination and degradation by ribosomal protein L5. J Biol Chem. 2004;279:44475–82. doi: 10.1074/jbc.M403722200. [DOI] [PubMed] [Google Scholar]
- 35.Dai MS, Zeng SX, Jin Y, Sun XX, David L, Lu H. Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol Cell Biol. 2004;24:7654–68. doi: 10.1128/MCB.24.17.7654-7668.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gilkes DM, Chen L, Chen J. MDMX regulation of p53 response to ribosomal stress. EMBO J. 2006;25:5614–25. doi: 10.1038/sj.emboj.7601424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roe JS, Youn HD. The positive regulation of p53 by the tumor suppressor VHL. Cell Cycle. 2006;5:2054–6. doi: 10.4161/cc.5.18.3247. [DOI] [PubMed] [Google Scholar]
- 38.Humpton TJ, Vousden KH. Regulation of cellular metabolism and hypoxia by p53. Cold Spring Harb Perspect Med. 2016;6:a026146. doi: 10.1101/cshperspect.a026146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004;64:2627–33. doi: 10.1158/0008-5472.CAN-03-0846. [DOI] [PubMed] [Google Scholar]
- 40.Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–93. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 41.Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer. 2009;9:691–700. doi: 10.1038/nrc2715. [DOI] [PubMed] [Google Scholar]
- 42.Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci USA. 2010;107:7455–60. doi: 10.1073/pnas.1001006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scherz-Shouval R, Weidberg H, Gonen C, Wilder S, Elazar Z, Oren M. p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc Natl Acad Sci USA. 2010;107:18511–6. doi: 10.1073/pnas.1006124107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maddocks OD, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb E, et al. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature. 2013;493:542–6. doi: 10.1038/nature11743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Makino Y, Hikita H, Fukumoto K, Sung JH, Sakano Y, Murai K, et al. Constitutive activation of the tumor suppressor p53 in hepatocytes paradoxically promotes non-cell autonomous liver carcinogenesis. Cancer Res. 2022;82:2860–73. doi: 10.1158/0008-5472.CAN-21-4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kao CF, Chen SY, Chen JY, Wu, Lee YH. Modulation of p53 transcription regulatory activity and post-translational modification by hepatitis C virus core protein. Oncogene. 2004;23:2472–83. doi: 10.1038/sj.onc.1207368. [DOI] [PubMed] [Google Scholar]
- 47.Derdak Z, Lang CH, Villegas KA, Tong M, Mark NM, de la Monte SM, et al. Activation of p53 enhances apoptosis and insulin resistance in a rat model of alcoholic liver disease. J Hepatol. 2011;54:164–72. doi: 10.1016/j.jhep.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yahagi N, Shimano H, Matsuzaka T, Sekiya M, Najima Y, Okazaki S, et al. p53 involvement in the pathogenesis of fatty liver disease. J Biol Chem. 2004;279:20571–5. doi: 10.1074/jbc.M400884200. [DOI] [PubMed] [Google Scholar]
- 49.Barton MC, Lozano G. p53 activation paradoxically causes liver cancer. Cancer Res. 2022;82:2824–5. doi: 10.1158/0008-5472.CAN-22-2065. [DOI] [PubMed] [Google Scholar]
- 50.Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378:206–8. doi: 10.1038/378206a0. [DOI] [PubMed] [Google Scholar]
- 51.Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378:203–6. doi: 10.1038/378203a0. [DOI] [PubMed] [Google Scholar]
- 52.Parant J, Chavez-Reyes A, Little NA, Yan W, Reinke V, Jochemsen AG, et al. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet. 2001;29:92–5. doi: 10.1038/ng714. [DOI] [PubMed] [Google Scholar]
- 53.Migliorini D, Lazzerini Denchi E, Danovi D, Jochemsen A, Capillo M, Gobbi A, et al. Mdm4 (Mdmx) regulates p53-induced growth arrest and neuronal cell death during early embryonic mouse development. Mol Cell Biol. 2002;22:5527–38. doi: 10.1128/MCB.22.15.5527-5538.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xiong S, Van Pelt CS, Elizondo-Fraire AC, Fernandez-Garcia B, Lozano G. Loss of Mdm4 results in p53-dependent dilated cardiomyopathy. Circulation. 2007;115:2925–30. doi: 10.1161/CIRCULATIONAHA.107.689901. [DOI] [PubMed] [Google Scholar]
- 55.Garcia D, Warr MR, Martins CP, Brown Swigart L, Passegue E, Evan GI. Validation of MdmX as a therapeutic target for reactivating p53 in tumors. Genes Dev. 2011;25:1746–57. doi: 10.1101/gad.16722111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Francoz S, Froment P, Bogaerts S, De Clercq S, Maetens M, Doumont G, et al. Mdm4 and Mdm2 cooperate to inhibit p53 activity in proliferating and quiescent cells in vivo. Proc Natl Acad Sci USA. 2006;103:3232–7. doi: 10.1073/pnas.0508476103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eischen CM, Lozano G. The Mdm network and its regulation of p53 activities: a rheostat of cancer risk. Hum Mutat. 2014;35:728–37. doi: 10.1002/humu.22524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Terzian T, Wang Y, Van Pelt CS, Box NF, Travis EL, Lozano G. Haploinsufficiency of Mdm2 and Mdm4 in tumorigenesis and development. Mol Cell Biol. 2007;27:5479–85. doi: 10.1128/MCB.00555-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mendrysa SM, O’Leary KA, McElwee MK, Michalowski J, Eisenman RN, Powell DA, et al. Tumor suppression and normal aging in mice with constitutively high p53 activity. Genes Dev. 2006;20:16–21. doi: 10.1101/gad.1378506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pant V, Xiong S, Chau G, Tsai K, Shetty G, Lozano G. Distinct downstream targets manifest p53-dependent pathologies in mice. Oncogene. 2016;35:5713–21. doi: 10.1038/onc.2016.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pant V, Aryal NK, Xiong S, Chau GP, Fowlkes NW, Lozano G. Alterations of the Mdm2 C-terminus differentially impact its function in vivo. Cancer Res. 2022;82:1313–20. doi: 10.1158/0008-5472.CAN-21-2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mendrysa SM, McElwee MK, Michalowski J, O’Leary KA, Young KM, Perry ME. mdm2 Is critical for inhibition of p53 during lymphopoiesis and the response to ionizing irradiation. Mol Cell Biol. 2003;23:462–72. doi: 10.1128/MCB.23.2.462-473.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, et al. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002;415:45–53. doi: 10.1038/415045a. [DOI] [PubMed] [Google Scholar]
- 64.Lessel D, Wu D, Trujillo C, Ramezani T, Lessel I, Alwasiyah MK, et al. Dysfunction of the MDM2/p53 axis is linked to premature aging. J Clin Invest. 2017;127:3598–608. doi: 10.1172/JCI92171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu D, Prives C. Relevance of the p53-MDM2 axis to aging. Cell Death Differ. 2018;25:169–79. doi: 10.1038/cdd.2017.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Donehower LA. Using mice to examine p53 functions in cancer, aging, and longevity. Cold Spring Harb Perspect Biol. 2009;1:a001081. doi: 10.1101/cshperspect.a001081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ringshausen I, O’Shea CC, Finch AJ, Swigart LB, Evan GI. Mdm2 is critically and continuously required to suppress lethal p53 activity in vivo. Cancer Cell. 2006;10:501–14. doi: 10.1016/j.ccr.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 68.Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82:675–84. doi: 10.1016/0092-8674(95)90039-X. [DOI] [PubMed] [Google Scholar]
- 69.Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4:321–8. doi: 10.1016/S1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- 70.Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–8. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- 71.Michalak EM, Villunger A, Adams JM, Strasser A. In several cell types tumour suppressor p53 induces apoptosis largely via Puma but Noxa can contribute. Cell Death Differ. 2008;15:1019–29. doi: 10.1038/cdd.2008.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang Y, Xiong S, Li Q, Hu S, Tashakori M, Van Pelt C, et al. Tissue-specific and age-dependent effects of global Mdm2 loss. J Pathol. 2014;233:380–91. doi: 10.1002/path.4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Moyer SM, Wasylishen AR, Qi Y, Fowlkes N, Su X, Lozano G. p53 drives a transcriptional program that elicits a non-cell-autonomous response and alters cell state in vivo. Proc Natl Acad Sci USA. 2020;117:23663–73. doi: 10.1073/pnas.2008474117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Feng Z, Hu W, Teresky AK, Hernando E, Cordon-Cardo C, Levine AJ. Declining p53 function in the aging process: a possible mechanism for the increased tumor incidence in older populations. Proc Natl Acad Sci USA. 2007;104:16633–8. doi: 10.1073/pnas.0708043104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pant V, Xiong S, Jackson JG, Post SM, Abbas HA, Quintas-Cardama A, et al. The p53-Mdm2 feedback loop protects against DNA damage by inhibiting p53 activity but is dispensable for p53 stability, development, and longevity. Genes Dev. 2013;27:1857–67. doi: 10.1101/gad.227249.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang YV, Leblanc M, Fox N, Mao JH, Tinkum KL, Krummel K, et al. Fine-tuning p53 activity through C-terminal modification significantly contributes to HSC homeostasis and mouse radiosensitivity. Genes Dev. 2011;25:1426–38. doi: 10.1101/gad.2024411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Komarova EA, Kondratov RV, Wang K, Christov K, Golovkina TV, Goldblum JR, et al. Dual effect of p53 on radiation sensitivity in vivo: p53 promotes hematopoietic injury, but protects from gastro-intestinal syndrome in mice. Oncogene. 2004;23:3265–71. doi: 10.1038/sj.onc.1207494. [DOI] [PubMed] [Google Scholar]
- 78.Pant V, Xiong S, Wasylishen AR, Larsson CA, Aryal NK, Chau G, et al. Transient enhancement of p53 activity protects from radiation-induced gastrointestinal toxicity. Proc Natl Acad Sci USA. 2019;116:17429–37. doi: 10.1073/pnas.1909550116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kirsch DG, Santiago PM, di Tomaso E, Sullivan JM, Hou WS, Dayton T, et al. p53 controls radiation-induced gastrointestinal syndrome in mice independent of apoptosis. Science. 2010;327:593–6. doi: 10.1126/science.1166202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nikulenkov F, Spinnler C, Li H, Tonelli C, Shi Y, Turunen M, et al. Insights into p53 transcriptional function via genome-wide chromatin occupancy and gene expression analysis. Cell Death Differ. 2012;19:1992–2002. doi: 10.1038/cdd.2012.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Menendez D, Nguyen TA, Freudenberg JM, Mathew VJ, Anderson CW, Jothi R, et al. Diverse stresses dramatically alter genome-wide p53 binding and transactivation landscape in human cancer cells. Nucleic Acids Res. 2013;41:7286–301. doi: 10.1093/nar/gkt504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Allen MA, Andrysik Z, Dengler VL, Mellert HS, Guarnieri A, Freeman JA, et al. Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. Elife. 2014;3:e02200. doi: 10.7554/eLife.02200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li M, He Y, Dubois W, Wu X, Shi J, Huang J. Distinct regulatory mechanisms and functions for p53-activated and p53-repressed DNA damage response genes in embryonic stem cells. Mol Cell. 2012;46:30–42. doi: 10.1016/j.molcel.2012.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kenzelmann Broz D, Spano Mello S, Bieging KT, Jiang D, Dusek RL, Brady CA, et al. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013;27:1016–31. doi: 10.1101/gad.212282.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tanikawa C, Zhang YZ, Yamamoto R, Tsuda Y, Tanaka M, Funauchi Y, et al. The transcriptional landscape of p53 signalling pathway. EBioMedicine. 2017;20:109–19. doi: 10.1016/j.ebiom.2017.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jensen MR, Factor VM, Fantozzi A, Helin K, Huh CG, Thorgeirsson SS. Reduced hepatic tumor incidence in cyclin G1-deficient mice. Hepatology. 2003;37:862–70. doi: 10.1053/jhep.2003.50137. [DOI] [PubMed] [Google Scholar]
- 87.Ohno S, Ikeda JI, Naito Y, Okuzaki D, Sasakura T, Fukushima K, et al. Comprehensive phenotypic analysis of knockout mice deficient in cyclin G1 and cyclin G2. Sci Rep. 2016;6:39091. doi: 10.1038/srep39091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Monte M, Benetti R, Buscemi G, Sandy P, Del Sal G, Schneider C. The cell cycle-regulated protein human GTSE-1 controls DNA damage-induced apoptosis by affecting p53 function. J Biol Chem. 2003;278:30356–64. doi: 10.1074/jbc.M302902200. [DOI] [PubMed] [Google Scholar]
- 89.Zhang L, Shao H, Zhu T, Xia P, Wang Z, Liu L, et al. DDA3 associates with microtubule plus ends and orchestrates microtubule dynamics and directional cell migration. Sci Rep. 2013;3:1681. doi: 10.1038/srep01681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tanikawa C, Furukawa Y, Yoshida N, Arakawa H, Nakamura Y, Matsuda K. XEDAR as a putative colorectal tumor suppressor that mediates p53-regulated anoikis pathway. Oncogene. 2009;28:3081–92. doi: 10.1038/onc.2009.154. [DOI] [PubMed] [Google Scholar]
- 91.Brosh R, Sarig R, Natan EB, Molchadsky A, Madar S, Bornstein C, et al. p53-dependent transcriptional regulation of EDA2R and its involvement in chemotherapy-induced hair loss. FEBS Lett. 2010;584:2473–7. doi: 10.1016/j.febslet.2010.04.058. [DOI] [PubMed] [Google Scholar]
- 92.Newton K, French DM, Yan M, Frantz GD, Dixit VM. Myodegeneration in EDA-A2 transgenic mice is prevented by XEDAR deficiency. Mol Cell Biol. 2004;24:1608–13. doi: 10.1128/MCB.24.4.1608-1613.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Donehower LA, Soussi T, Korkut A, Liu Y, Schultz A, Cardenas M, et al. Integrated analysis of TP53 gene and pathway alterations in the cancer genome atlas. Cell Rep. 2019;28:3010. doi: 10.1016/j.celrep.2019.08.061. [DOI] [PubMed] [Google Scholar]
- 94.Lange SS, Takata K, Wood RD. DNA polymerases and cancer. Nat Rev Cancer. 2011;11:96–110. doi: 10.1038/nrc2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schenten D, Gerlach VL, Guo C, Velasco-Miguel S, Hladik CL, White CL, et al. DNA polymerase kappa deficiency does not affect somatic hypermutation in mice. Eur J Immunol. 2002;32:3152–60. doi: 10.1002/1521-4141(200211)32:11<3152::AID-IMMU3152>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 96.Ogi T, Shinkai Y, Tanaka K, Ohmori H. Polkappa protects mammalian cells against the lethal and mutagenic effects of benzo[a]pyrene. Proc Natl Acad Sci USA. 2002;99:15548–53. doi: 10.1073/pnas.222377899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Stancel JN, McDaniel LD, Velasco S, Richardson J, Guo C, Friedberg EC. Polk mutant mice have a spontaneous mutator phenotype. DNA Repair (Amst) 2009;8:1355–62. doi: 10.1016/j.dnarep.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhang Y, Shin SJ, Liu D, Ivanova E, Foerster F, Ying H, et al. ZNF365 promotes stability of fragile sites and telomeres. Cancer Disco. 2013;3:798–811. doi: 10.1158/2159-8290.CD-12-0536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 2014;14:359–70. doi: 10.1038/nrc3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Valentin-Vega YA, Okano H, Lozano G. The intestinal epithelium compensates for p53-mediated cell death and guarantees organismal survival. Cell Death Differ. 2008;15:1772–81. doi: 10.1038/cdd.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Heberle H, Meirelles GV, da Silva FR, Telles GP, Minghim R. InteractiVenn: a web-based tool for the analysis of sets through Venn diagrams. BMC Bioinforma. 2015;16:169. doi: 10.1186/s12859-015-0611-3. [DOI] [PMC free article] [PubMed] [Google Scholar]