

Abstract
Memory CD8 T cells play an important role in the protection against breakthrough infections with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Whether the route of antigen exposure impacts these cells at a functional level is incompletely characterized. Here, we compare the memory CD8 T cell response against a common SARS-CoV-2 epitope after vaccination, infection, or both. CD8 T cells demonstrate comparable functional capacity when restimulated directly ex vivo, independent of the antigenic history. However, analysis of T cell receptor usage shows that vaccination results in a narrower scope than infection alone or in combination with vaccination. Importantly, in an in vivo recall model, memory CD8 T cells from infected individuals show equal proliferation but secrete less tumor necrosis factor (TNF) compared with those from vaccinated people. This difference is negated when infected individuals have also been vaccinated. Our findings shed more light on the differences in susceptibility to re-infection after different routes of SARS-CoV-2 antigen exposure.
Keywords: CD8 T cells, SARS-CoV-2, COVID-19, memory cells, infection, vaccination, influenza, antigen-specific T cells, clonal diversity, transcriptome
Graphical abstract
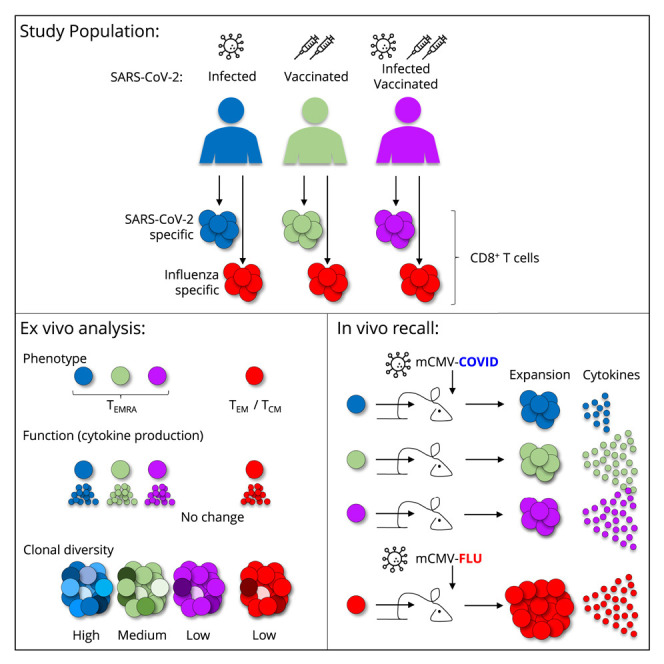
Kavazović et al. compare SARS-CoV-2-specific memory CD8 T cells from people after infection, vaccination, or both. Infection induces a more clonally diverse memory pool than vaccination, but these cells produce less TNF after in vivo recall. Subsequent vaccination negates differences, stressing the importance of boosting irrespective of antigenic history.
Introduction
Protection against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) re-infection is mediated by the concerted action of humoral and cellular immunity. Whereas antibodies bind and neutralize viral particles, T cells recognize and eliminate infected cells. Unfortunately, several viral variants of concern have emerged that are capable of infecting people even though they had generated an antibody response either by prior infection or vaccination.1 , 2 Nevertheless, secondary infection with these strains typically leads to a much milder disease, which has been attributed to the protective effect of memory T cells.3 , 4 Memory T cells against SARS-CoV-2 are long lived and can clear the virus even in individuals with a compromised humoral immune system.5 , 6 However, their functional properties are incompletely characterized, especially with regards to their recall capacity.
Cellular and humoral immunity display key differences in the way they anticipate the re-occurrence of antigen. Antibodies have undergone affinity maturation to target the previously encountered pathogen with very high specificity, whereas cellular immunity contains a much broader scope of clones and associated affinities.7 , 8 Because re-infection is unlikely to occur with the exact same pathogen, the memory pool must anticipate the occurrence of mutations and therefore retain a certain bandwidth within its recognition capacity.7 , 8 For CD8 T cells, this takes the form of increased clonal diversity. We have shown that narrowing the clonal diversity of the memory pool through pharmaceutical or genetic means does not greatly affect its protective effect against the original pathogen but significantly impairs its ability to recognize and eliminate viral mutants.9 , 10 Whether the clonal diversity of the memory CD8 T cell pool against SARS-CoV-2 differs between infected and vaccinated individuals remains understudied.
The functional properties of the CD8 memory T cell pool are impacted by many factors, including the inflammatory environment during priming. A highly inflammatory milieu promotes effector and effector memory differentiation, whereas low inflammation favors formation of central memory cells.11 , 12 The cytokines and co-stimulatory molecules that provide the context of antigen presentation are therefore very important for shaping the identity of the memory pool and are unique for each pathogen. For example, cytomegalovirus infection is associated with more effector/effector memory-type T cells, whereas influenza and respiratory syncytial virus preferentially drive the development of central memory-type T cells.13 Vaccination generates a different inflammatory environment than infection and therefore potentially mediates formation of memory cells with a different functional capacity. Indeed, real-world data over a 52-week follow-up period indicates that the risk of breakthrough infection with SARS-CoV-2 is 3- to 7-fold higher after vaccination than after infection.1 , 2 The underlying molecular mechanism is difficult to investigate, and our current understanding of SARS-CoV-2-specific CD8 T cells is mostly based on the effector memory response that is interrogated directly ex vivo.14 Much less is known about the cells that form secondary effectors upon antigen re-encounter in vivo. Yet, this information is crucial for a better design of new vaccines.
We hypothesized that the antigenic history impacts the scope and functionality of the SARS-CoV-2-specific CD8 memory T cell pool. We directly compared antigen-specific memory CD8 T cells from people with prior infection, vaccination, or both. Whereas the route of antigen exposure had little impact on the phenotype and ex vivo functionality of cells, clonal analysis showed that the memory pool formed after vaccination had a more restricted repertoire than those generated after infection. Using a humanized mouse model, we could show that secondary effectors generated by memory CD8 T cells from infected individuals produced less tumor necrosis factor (TNF) than those from vaccinated people. Importantly, vaccination of people with prior infection widened the scope of the memory pool and increased the ability of secondary effector cells to produce cytokines upon recall in vivo. Vaccination therefore induces a memory CD8 T cell pool that is more potent yet appears less capable of recognizing mutated pathogens due to its narrower scope. Our findings indicate that vaccination of infected individuals levels the differences, emphasizing the importance of booster vaccination.
Results
The route of antigen exposure provokes a unique transcriptional and phenotypic profile in SARS-CoV-2-specific memory CD8 T cells
We collected a cohort of 70 adults that had been preselected for the HLA A∗02 haplotype and divided them in three groups based on SARS-CoV-2 antigenic history (“route” of antigen exposure): prior infection, vaccination, or both. Only people vaccinated with two doses of mRNA vaccines (Pfizer-BioNTech BNT162b2 or Moderna mRNA-1273) were included in the study. On average, people were last exposed to antigen 5–6 months before blood isolation, and groups were largely comparable in composition (Figures S1A and S1B). People were self-declared negative for influenza infection or vaccination at least 1 year before sampling. Peripheral blood mononuclear cells (PBMCs) were collected, and memory CD8 T cells were analyzed using HLA tetramers. We focused our research on CD8 T cells directed against the dominant A∗02 CD8+ T cell epitope of the SARS-CoV-2 spike protein—S269–277 (YLQPRTFLL).15 This epitope had not mutated in any of the major viral strains of concern and was recognized in all individuals carrying the A∗02 haplotype (Figure 1 A and Redd et al.16 and Shomuradova et al.17). In the same individuals, we analyzed CD8 T cells directed against epitope GILGFVFTL of the MP1 protein of influenza, as most individuals of the A∗02 haplotype have memory T cells against this antigen.13 A positive signal for tetramer staining was determined using stringent controls (Figure S1C). The overall frequency of tetramer-specific cells in our study group was comparable to those found previously14 , 18 , 19 , 20 , 21 (Figures 1A and 1B). The lowest frequencies were observed in the infected-only group. The frequency of SARS-CoV-2-specific cells formed after vaccination was not increased by prior infection (Figures 1A and 1B).
Figure 1.

The route of antigen exposure impacts the transcriptional and phenotypic profile of virus-specific memory cells
PBMCs from 3 groups of people were analyzed directly ex vivo (Inf, people with convalescent COVID-19; Inf/Vacc, people with convalescent COVID-19 who received 2 doses of vaccine; Vacc, nonconvalescent people who received 2 doses of COVID-19 vaccine).
(A) Frequency of antigen-specific (C19- or FLU-tetramer+) cells. Each dot represents one donor (n = 19 – C19-Inf, n = 15 C19-Inf/Vacc, n = 17 C19-Vacc, n = 51 FLU pooled from all 3 groups).
(B) Representative fluorescence-activated cell sorting (FACS) plots of cells stained with HLA A∗02 tetramers loaded with the YLQPRTFLL (C19) epitope of SARS-CoV-2 or GILGFVFTL (FLU) epitope of influenza. Gated is for live CD3+CD8+ cells.
(C) Quantification of TEM, TCM, and TEMRA cell subsets among C19- and FLU-specific CD8+ T cells (n = 18).
(D–F) C19- and FLU-specific CD8+ T cells were sorted and analyzed by RNA sequencing.
(D) Principal-component analysis of virus-specific cells based on all differentially expressed genes (n = 17).
(E) The 200 most differentially expressed genes between C19-tetramer+ cells from the C19-Inf and C19-Vacc groups were subjected to protein network clustering. Shown is the largest node network for each comparison. Inset shows the largest subcluster.
(F) Differential expression of individual genes (n = 17).
(G) Geometric mean fluorescence intensity (GeoMean) of selected markers on C19- and FLU-specific CD8+ T cells by flow cytometry (n = 18). Indicated are means ± SEM.
Statistical significances at ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 using Kruskal-Wallis rank-sum test with Dunn’s post hoc test for multiple comparisons (A) or one-way ANOVA followed with Bonferroni post-testing (C, F, and G).
See also Figure S1.
We next performed a comprehensive phenotypic analysis of the well-defined CD45RA− effector memory (TEM), CD45RA+ effector memory (TEMRA), and central memory (TCM) subsets.22 , 23 SARS-CoV-2-specific CD8 T cells exhibited a similar immunological profile irrespective of antigenic history, with preferential skewing to a TEMRA phenotype. In contrast, influenza-specific cells favored a TEM profile. Moreover, the latter pool contained significantly more TCM cells than SARS-CoV-2-specific cells (Figures 1C and S1D). Subsequently, we sorted antigen-specific cells and performed total transcriptome analysis. Unexpectedly, unbiased clustering grouped cells of infected individuals more closely together with cells of people that were both infected and vaccinated rather than with those that were vaccinated only. Influenza-specific cells clustered separately from the three other groups (Figures 1D and S1E). We next analyzed the 200 most differentially expressed genes between CD8 T cells directed against influenza and SARS-CoV-2 after infection and clustered them using data of known protein-interaction networks. Influenza-specific cells expressed higher amounts of surface markers associated with long-term memory, such as Il7ra, CD8a, and Cd226 (Figures S1E and S1F). We then performed the same network analysis of genes differentially expressed between SARS-CoV-2-specific CD8 T cells of infected or vaccinated individuals. We found that cells from vaccinated individuals had higher expression of intracellular signaling components associated with T cell activation such as Jak3, Itk, and Traf1, whereas they had lower amounts of Stat3 (Figures 1E and 1F). In addition, we observed higher expression of co-stimulatory molecules, such as Cd27 and Tnfrsf6, in SARS-CoV-2-specific cells of vaccinated people. Cells from individuals that were both vaccinated and infected typically showed intermediate expression of these genes (Figure 1F).
To corroborate our findings at a protein level, flow cytometric analysis was performed on virus-specific cells. This confirmed a higher expression of molecules such as CD3ε and CD27 on SARS-CoV-2-specific memory cells of vaccinated people. CD57, a molecule associated with reduced memory potential, was specifically upregulated on SARS-CoV-2-infection-induced cells24 , 25 (Figures 1G and S2A). We did not find induction of other markers typically associated with exhaustion, indicating that these cells are fully functional (Figure S2B). In accordance with their transcriptional profile, antigen-experienced cells from infected/vaccinated individuals mostly showed protein expression of intermediate levels between the other two antigen-experienced groups (Figure 1G).
In summary, the route of antigen exposure has a unique impact on the transcriptional and phenotypic profile of SARS-CoV-2-specific memory cells, even though effects are minor.
The route of antigen exposure does not greatly impact the direct ex vivo cytokine production by antigen-specific cells
To assess the functionality of virus-specific memory CD8+ T cells, we determined their ability to produce cytokines upon direct ex vivo stimulation with single viral peptides. Cytokine responses were observed in all samples (Figure 2 A). Interestingly, we did not find more cells producing cytokines following influenza- than after SARS-CoV-2-peptide stimulation, despite a higher frequency of tetramer+ cells (Figures 1A and 2A). Antigenic history did not greatly impact the ability of the SARS-CoV-2-specific memory pool to produce interferon γ (IFNγ), TNF, and Granzyme B. We only observed an increase in cells able to produce IL-2 or both IFNγ and Granzyme B by memory cells from infected/vaccinated individuals compared with the other two groups (Figure 2A). The ability to produce cytokines did not correlate with the time since last antigen exposure (Figure S2C). Also, no major differences were observed in the polyfunctionality of antigen-specific CD8 T cells (Figure 2B).
Figure 2.

The route of antigen exposure does not greatly impact the direct ex vivo functionality of antigen-specific cells
PBMCs were stimulated with C19 (YLQPRTFLL) or FLU (GILGFVFTL) peptide for 4 h in the presence of brefeldin A and analyzed by flow cytometry.
(A) Representative FACS plots (top row) and quantification of data (bottom row) are shown. Numbers indicate percentages within the quadrant. Cells are gated for live CD3+CD8+ cells. BrefA control, cells stimulated with brefeldin A only (n = 16).
(B) Donut graphs showing polyfunctional analysis of C19- and FLU-specific CD8+ T cells. Relative distribution of single or multiple cytokine-producing cells is shown for IL-2, IFNγ, and TNF (n = 16).
(C and D) PBMCs were stimulated with a peptide pool of immunodominant epitopes of the spike protein of SARS-CoV-2 or of influenza HA for 4 h in the presence of brefeldin A and analyzed by flow cytometry.
(C) Percentage of cells expressing the indicated activation-induced markers (AIMs) (n = 18).
(D) (Top row) Representative FACS plots and (bottom row) quantification of cytokine expression (n = 24). Indicated are means ± SEM.
Statistical significances at ∗p < 0.05 and ∗∗p < 0.01 using one-way ANOVA followed by Bonferroni post-testing (A, C, and D).
See also Figure S2.
Finally, we questioned whether our observations using single epitopes were representative of the general cytokine response of cells directed against viral proteins. Cells were stimulated with peptide pools of the SARS-CoV-2 spike or influenza hemagglutinin (HA) proteins. We did not observe significant changes between groups in the induction of the activation markers CD69, CD137, OX40, CD107a, CD25, or IRF4 (Figures 2C and S2D) nor in their ability to produce cytokines (Figure 2D). In this assay, memory cells from infected/vaccinated individuals did not produce more IL-2, but their increased potential to produce both IFNγ and Granzyme B was retained (Figure 2D).
In summary, the route of antigen exposure does not greatly affect the direct ex vivo ability of memory cells to produce cytokines, though repeated encounters slightly increase their functional capacity.
SARS-CoV-2 infection causes a broader T cell receptor (TCR)-V repertoire than vaccination
We have previously demonstrated that a memory CD8 T cell pool of increased clonal diversity has better protective effects against mutated pathogens.9 , 10 Real-world data indicate that the protection against breakthrough infection provided by immunological memory formed after infection is better than after vaccination.1 , 2 We therefore hypothesized that the clonal diversity of the memory CD8 T cell pool is larger after infection than after vaccination. To elucidate this, we performed sequence analysis of the TCR-α and -β chains of sorted, SARS-CoV-2-, or influenza-specific CD8 T cells. We observed clonal expansion in all individuals, as most reads were recovered more than twice (Figures 3A and S3A). Stringent clonal selection had taken place, with the 20% most frequent TCR chains (quintet 1) together making up more than 50% of total reads in almost all individuals, with no major differences between groups (Figure 3A). Similarly, we observed a comparable dominance of the five most frequently recovered TCR chains, which comprised more than 25% of all reads in most people.
Figure 3.

SARS-CoV-2 infection causes a broader TCR repertoire than vaccination
C19- and FLU-specific CD8+ T cells were sorted and TCR-α and -β chains were analyzed by RNA sequencing.
(A) Donut graph of the distribution of the TCR-α (green) and -β (orange) chains of a representative individual for each group. Inner ring shows reads recovered once (blue), twice (yellow), or >2 times (orange/green). Middle ring shows relative contribution of TCR chains segregated in quintets and ranked based on their relative frequency within the total pool. Outer ring shows the contribution of the 5 most frequent TCR chains to the total pool.
(B) Average length of the VDJ region (n = 17).
(C) Average contribution of each TCR chain to the total pool (n = 17).
(D) Contribution of the 15 most dominant TCR-β and -α chains to the total antigen-specific response (n = 17). Dashed lines indicate SEM.
(E) Representative TCR-Vβ usage of four individuals. The ten most frequent TCR chains for each TCR-Vβ are color coded, and chains recovered with lower frequency are gray.
Indicated are means ± SEM and statistical significances at ∗∗p < 0.05 and ∗∗∗p < 0.01 by one-way ANOVA followed by Bonferroni post-testing (B), Kruskal-Wallis rank-sum test with Dunn’s post hoc test for multiple comparisons (C), and unpaired Mann-Whitney test (D).
See also Figure S3.
We did observe that the average length of the VDJ region of both the TCR-α and -β chain differed between groups, with the longest reads found in SARS-CoV-2-infected individuals and the shortest in those people that had only been vaccinated (Figure 3B). In addition, the antigenic history affected the relative contribution of reads recovered with low frequency. On average, vaccination resulted in clones with a larger frequency (Figure S3B). This corresponded with a reduced contribution of low-frequency clones, most notably for the TCR-β chain (Figures 3C and 3D). Interestingly, vaccination of infected individuals further reduced the average frequency of specific TCR chains (Figure 3C), suggesting that vaccination recruits new low-frequency clones into the memory pool.
We questioned whether changes in the distribution of T cell receptor chains also translate into a different skewing of TCR subunit usage. Memory cells directed against influenza showed a strong preference for the TCR-Vα 27 and TCR-Vβ 19 genes (Figures 3F and S3D). As previously reported,14 SARS-CoV-2-specific cells favored usage of the TCR-Vα 12-1 and TCR-Vβ 7-9 genes. Strikingly, whereas we did not find major differences between the groups at the level of individual reads (Figure 3A), skewing toward the use of a specific TCR-V gene was much higher in vaccinated people than in infected people (Figures 3E, S3C, and S3D). Cells from infected/vaccinated people showed a level of diversity that more closely resembled that of infected individuals, especially with regards to TCR-Vβ usage (Figures 3E and S3D).
Thus, compared with infection, vaccination against SARS-CoV-2 leads to a lower frequency of TCR chains used with low abundance within the memory CD8 T cell pool, whereas the prominence of the most dominant chains is not greatly affected.
Vaccination-induced memory cells produce more TNF after in vivo recall
In vitro restimulation provides valuable information about the direct functionality of cells but does not recapitulate the in vivo recall capacity of CD8 memory T cells. Current animal models using hACE2TG mice have investigated the ability of murine CD8 T cells to respond to SARS-CoV-2,26 but how human memory cells perform in vivo is unknown. We therefore set up a mouse model to interrogate the recall capacity of human cells in vivo in response to a real viral infection (Figure 4 A). We generated two strains of murine cytomegalovirus that express either the YLQPRTFLL epitope of SARS-CoV-2 (mCMV-COVID) or GILGFVFTL of influenza (mCMV-FLU), inserted at the C terminus of the viral immediate-early (IE2) protein. This system allows for a direct comparison of antigen-specific T cells directed against two different pathogens, as it negates differences in peptide processing and antigenic load while avoiding the impact of humoral immunity. As a control, we used a virus expressing the SIINFEKL epitope of ovalbumin (mCMV-N4).27 , 28 Immunodeficient NSG mice expressing a human HLA A∗02 transgene were transferred intraperitoneally (i.p.) with human PBMCs. Next, animals were infected with recombinant mCMV and injected with a single shot of human interleukin-2 (IL-2). Infection with mCMV-N4 did not result in an expansion of donor CD8 T cells, indicating that neither the murine pathogen nor human IL-2 alone leads to bystander activation of human cells (Figure S4A).
Figure 4.

Vaccination-induced memory cells have increased functional potential after in vivo recall
PBMCs were analyzed pretransfer for the frequency of tetramer+ cells. Cells were divided into two equal fractions and transferred to NSG-HLA:A∗02 transgenic mice. On the same day, animals were infected i.p. with 2 × 105 PFU mCMV-COVID or mCMV-FLU. The next day, mice were injected i.p. with human IL-2 (100 ng/mouse). 7 days post-infection, donor cells in peritoneal exudate cells (PECs) were analyzed (n = 25).
(A) Experimental setup.
(B) Line graph showing expansion of cells of the same donor as fold increase over pretransfer, comparing C19- with FLU-specific CD8+ T cells.
(C) Representative FACS plots of CD8+ T cell stained with HLA-A∗02 tetramers pretransfer or 7 days post-infection. Non-HLA-A∗02 cells are included as negative control. Numbers indicate percentages. Gated is for live hCD45+hCD8+ cells.
(D) Fold increase of antigen-specific cells segregated by C19 groups.
(E) Correlation between time after last antigen exposure and the expansion of virus-specific cells after mCMV-C19 infection.
(F–G) On day 7 post-infection, PECs were re-stimulated in vitro with PMA/IONO for 4 h in the presence of Brefeldin A and analyzed by flow cytometry. Shown are (F) cytokine production of live hCD45+hCD8+tetramer+ cells and (G) representative FACS plots gated for hCD45+hCD8+tetramer+ cells. Indicated are means ± SEM.
p values were calculated using paired Student t test (B) and Kruskal-Wallis rank-sum test with Dunn’s post hoc test for multiple comparisons (F). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
See also Figure S4.
To assess the expansion of antigen-specific cells in vivo, PBMCs from individuals were divided into two equal fractions and transferred i.p. to separate NSG-HLA A∗02TG mice, which were subsequently infected with mCMV-COVID or mCMV-FLU. On day 7 post-infection, the peritoneal exudate cells (PECs) were isolated. The frequencies of C19- and influenza-specific CD8 T cells were determined by tetramer+ staining and compared with those before transfer (Figures 4B and 4C). Both viruses induced a marked expansion of virus-specific cells (Figure 4B). However, the relative increase was significantly larger for influenza-specific cells (22.7-fold ± 29.8) compared with SARS-CoV-2-specific cells (8-fold ± 6.9) from the same person. The recall capacity of SARS-CoV-2-specific cells was not greatly impacted by the antigenic history nor by time since last antigen exposure (Figures 4D and 4E).
To test the functional capacity of antigen-specific secondary effector cells formed in vivo, they were restimulated in vitro, and cytokine production was quantified within tetramer+ populations (Figure S4B). Influenza-specific cells produced the most IFNγ and TNF and displayed a larger frequency of polyfunctional cells (Figures 4F and 4G). Importantly, SARS-CoV-2-specific secondary effector CD8 T cells formed by memory cells from infected people produced significantly less TNF compared with the vaccinated group. This difference was not observed in individuals that were both infected and vaccinated (Figures 4F and 4G). The ability to produce IFNγ was not impacted by the antigenic history.
Thus, whereas the proliferative capacity is not affected by the route of antigen exposure, we show that vaccination leads to a superior ability of secondary effectors to produce TNF compared to infection.
Discussion
Infection and vaccination both induce potent immunological memory against SARS-CoV-2, but its real-world protective effect is not equal, especially against breakthrough infection with new strains.1 , 2 We here characterized the phenotype and function of memory CD8 T cells directed against a common viral epitope in the context of different histories of antigen exposure. We find that infection induces fewer antigen-specific cells with a different phenotypic and transcriptional profile than vaccination. Whereas this did not translate to differences in functionality when directly analyzed ex vivo, it did cause a reduced capacity to produce TNF after recall in vivo. Nevertheless, its increased TCR-chain diversity potentially allows it to better recognize epitopes carrying point mutations. Importantly, vaccination after prior infection appeared to correct the functional disadvantage of CD8 T cell memory while mostly retaining TCR-chain diversity.
The phenotypic and functional properties of memory cells are determined by many factors, including the nature and route of antigen exposure as well as the inflammatory environment during their formation.12 , 29 , 30 , 31 , 32 , 33 , 34 Not surprisingly, we found several differences in the properties of antigen-specific memory formed after infection and vaccination. Compared with vaccination, SARS-CoV-2 infection induced memory with higher surface levels of CD57 and lower CD27, which is associated with an effector memory rather than a central memory profile.22 , 23 Indeed, whereas direct ex vivo functionality was comparable, TNF production after in vivo recall was lower by infection-induced cells, suggesting a different memory potential. This may have great clinical impact since deficiency of TNF was shown to greatly exacerbate pathology following respiratory infection.35 Importantly, people with rheumatoid arthritis who were treated with anti-TNF as part of their normal therapy were shown to be more susceptible to SARS-CoV-2 infection.36 Thus, a reduced capacity to produce TNF may predispose people to more severe infection upon viral re-encounter.
Interestingly, IFNγ production of secondary effectors was not different between groups. In chronic infection, exhausted CD8 T cells are known to lose their ability to produce TNF before IFNγ.37 , 38 Both SARS-CoV-2 infection and vaccination were shown to induce a subset of exhausted cells,14 , 39 , 40 though this was only investigated directly ex vivo and not in recall models. We did not observe increased expression of key exhaustion markers41 in cells from infected individuals before recall. However, we did find lower expression of signal transduction molecules such as Traf1, Jak3, and Itk. This latter molecule was shown to drive nuclear factor κB (NF-κB)-mediated cytokine release by T cells.42 Whether reduced Itk-mediated signaling is the underlying cause of the more exhausted profile in secondary effector cells of infected individuals remains to be determined. Importantly, vaccination after initial infection promoted both the direct ex vivo effector function and the functional recall capacity of these cells. This may explain why vaccination after infection provides the best protection against re-infection.1
Our study corroborates previous findings that both SARS-CoV-2 infection and vaccination favor acquisition of a TEMRA phenotype,20 , 43 , 44 whereas influenza infection promotes TEM/TCM differentiation.21 TCM cells are thought to have increased recall capacity over effector-type memory cells,22 , 23 which is in accordance with our observation that influenza-specific cells expanded more potently than SARS-CoV-2-specific cells in vivo. Alternatively, differences in the properties of the cognate antigen derived from influenza and SARS-CoV-2 may underly the observed differences.
The antigenic history also impacted the TCR-chain composition of the memory CD8 T cell pool. Our observations are in line with previous reports, which indicate that infection and vaccination lead to a roughly equal contribution of dominant clones.14 , 43 , 45 However, a focus on only the most prominent clones underevaluates the total protective scope of antigen-specific memory. The generation of a broad pool of low-frequency memory cells is of clinical importance because they are typically of lower specificity for the original antigen and are therefore more likely to recognize mutated epitopes.7 Indeed, animals with a genetic deficiency in key genes controlling memory cell diversity have increased susceptibility to infection with mutated pathogens.9 , 10 , 46 Moreover, low-frequency memory clones can make a dominant contribution to the secondary effector response upon breakthrough infection with SARS-CoV-2.45 Our findings indicate that infection leads to a larger number of subdominant clones than vaccination. In addition, we find that infection-induced memory is less restrictive in its TCR-V gene usage, allowing for a greater potential to recognize mutated pathogens.14 , 47 This observation provides an explanation for why prior infection is associated with better protection against breakthrough infection than vaccination.
Our findings help in the development next-generation vaccines and indicate that the induction of a broader memory CD8 T cell repertoire may include the strengths from both routes of antigen exposure, which is likely to be beneficial for the protection against breakthrough infection.
Limitations of the study
This study has several limitations. First, we did not perform single-cell RNA sequencing analysis of antigen-specific cells. Therefore, we cannot draw definitive conclusions with regards to the clonal diversity within groups, as we do not know which TCR-α and -β pairs are present. However, our findings are in line with studies that did do single-cell RNA sequencing,14 making it likely that clonal diversity is indeed higher after infection than after vaccination. Second, we have investigated the ability of human memory cells to respond to a secondary viral challenge in mice. We therefore cannot exclude that differences in memory cell behavior are at least partially the result of the xenogeneic environment. Finally, our study was limited to one HLA A∗02-restricted SARS-CoV-2 epitope. Whereas this haplotype is highly prevalent in some human populations and has been shown to be representative of the general T cell response against SARS-CoV-2,5 , 14 we cannot exclude that other viral epitopes will induce memory CD8 T cells with different properties.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti human CD3 monoclonal antibody (clone: OKT3), Alexa Fluor™ 700 | eBioscience | Cat. # 56-0037-42 |
| Anti human CD8a monoclonal antibody (clone: OKT8) APC-eFluor™ 780 | eBioscience | Cat. # 47-0086-42 |
| Fixable Viability Dye eFluor 506 | eBioscience | Cat. # 65-0866-14 |
| Anti human CD45RA monoclonal antibody (clone: HI100) Super Bright™ 645 | eBioscience | Cat. # 64-0458-42 |
| Anti human CD45RO monoclonal antibody (clone: UCHL1) PE-eFluor™ 610 | eBioscience | Cat. # 61-0457-42 |
| Anti human CD27 monoclonal antibody (clone: O323) Super Bright™ 780 | eBioscience | Cat. # 78-0279-42 |
| Anti human CD57 monoclonal antibody (clone: QA17A04) Brilliant Violet 711™ |
BioLegend | Cat. # 393328 |
| Anti human IFNγ monoclonal antibody (clone: 4S.B3) FITC | eBioscience | Cat. # 11-7319-82 |
| Anti human TNFα monoclonal antibody (clone: MAb11) PE-Cyanine7 | eBioscience | Cat. # 25-7349-82 |
| Anti human IL-2 monoclonal antibody (clone: MQ1-17H12) APC | eBioscience | Cat. # 17-7029-82 |
| Anti human Granzyme B monoclonal antibody (clone: N4TL33) PE | eBioscience | Cat. # 12-8896-42 |
| Fixable Viability Dye eFluor™ 780 | eBioscience | Cat. # 65-0865-14 |
| Anti human KIR2D monoclonal antibody (clone: NKVFS1) FITC | Miltenyi Biotec | Cat. # 130-092-687 |
| Anti human CD8a monoclonal antibody (clone: RPA-T8) PerCP-Cyanine5.5 | eBioscience | Cat. # 45-0088-42 |
| Anti human CD45 monoclonal antibody (clone: HI30) PE-Cyanine7 | eBioscience | Cat. # 25-0459-42 |
| Anti human HLA-A2 monoclonal antibody (clone: BB7.2) PE-Cyanine7 | eBioscience | Cat. # 25-9876-42 |
| Anti human HLA-A3 monoclonal antibody (clone: GAP.A3) APC | eBioscience | Cat. # 17-5754-42 |
| Anti human CD3 monoclonal antibody (clone: OKT3) FITC | eBioscience | Cat. # 11-0037-42 |
| Anti human CD4 monoclonal antibody (clone: OKT4) PerCP-Cyanine5.5 | eBioscience | Cat. # 45-0048-42 |
| Anti human CD96 (TACTILE) monoclonal antibody (clone: NK92.39) PE-Cyanine7 | BioLegend | Cat. # 338415 |
| Anti human CD197 (CCR7) monoclonal antibody (clone: G043H7) Brilliant Violet 421™ | BioLegend | Cat. # 353207 |
| Anti human CD7 monoclonal antibody (clone: REA1244) PerCP-Vio® 700 | Miltenyi Biotec | Cat. # 130-124-937 |
| Anti mouse CD45.1 monoclonal antibody (clone: A20) FITC | eBioscience | Cat. # 11-0453-82 |
| Anti human IRF4 monoclonal antibody (clone:REA201) PE-Vio770 | Miltenyi Biotec | Cat. # 130-100-909 |
| Anti human CD134 (OX40) monoclonal antibody (clone: ACT35) PE | Miltenyi Biotec | Cat. # 130-116-488 |
| Anti human CD25 monoclonal antibody (clone: REA570) APC-Vio770 | Miltenyi Biotec | Cat. # 130-123-469 |
| Anti human CD137 monoclonal antibody (clone: 4B4-1) APC | BioLegend | Cat. # 309810 |
| Anti human CD107a monoclonal antibody (clone: REA792) FITC | Miltenyi Biotec | Cat. # 130-111-620 |
| Bacteria and virus strains | ||
| WT MCMV (MW97.01) | In house produced | Wagner et al.,48 |
| mCMV-N4 | In house produced | Lemmermann et al., 201028 |
| mCMV-YLQPRTFLL | In house produced | N/A |
| mCMV- GILGFVFTL | In house produced | N/A |
| GS1783 bacteria | In house produced | Tischer et al., 201049 |
| Biological samples | ||
| PBMCs | Collected in this study | N/A |
| Chemicals peptides and recombinant proteins | ||
| Brefeldin A | eBioscience | Cat. # 00-4506-51 |
| Histopaque 1077 | Sigma Aldrich | Cat. # 10771 |
| Phorbol myristate acetate (PMA) | InvivoGen | Cat. # tlrl-pma |
| Ionomycin | InvivoGen | Cat. # inh-ion |
| YLQPRTFLL peptide | JPT Peptide Technologies | N/A |
| GILGFVFTL peptide | JPT Peptide Technologies | N/A |
| PepTivator Influenza A (H1N1) HA | Miltenyi Biotec | 130-099-803 |
| PepTivator SARS-CoV-2 Prot_S | Miltenyi Biotec | 130-126-701 |
| Critical commercial assays | ||
| Intracellular Fixation & Permeabilization Buffer Set | eBioscience | Cat. # 88-8824-00 |
| Super Bright Complete Staining Buffer | eBioscience | Cat. # SB-4401-75 |
| Deposited data | ||
| RNA sequencing data | GEO | GEO: GSE202262 |
| Experimental Models: Organisms | ||
| Mouse: NSG-HLA:A∗02 transgenic | The Jackson Laboratory | Strain code: 9617 |
| Oligonucleotides | ||
| ie2_FLU_fwd (BAC mutagenesis) CTT TCT CTT GAC CAG AGA CCT GGT GAC CGT CAG GAA GAA GAT TCA GCC TCT GAC TAA GGG GAT TTT AGG ATT TGT GTT CAC GCT CTG AGA GGA CAC GAG GAC GCA TCG TGG CCG GAT CTC | Metabion GmbH | N/A |
| ie2_FLU_rev (BAC mutagenesis) CCT CTT TAT TTA TTG ATT AAA AAC CAT GAC ATA CCT CGT GTC CTC TCA GAG CGT GAA CAC AAA TCC TAA AAT CCC CTT AGT CAG AGG CTG AAT CTT CTT CCG TGA CCA CGT CGT GGA ATG C | Metabion GmbH | N/A |
| ie2_COVID-19_fwd (BAC mutagenesis) CTT TCT CTT GAC CAG AGA CCT GGT GAC CGT CAG GAA GAA GAT TCA GTG CAG CTT ATT ATG TTA TCT TCA ACC TAG GAC TTT TCT ATT ATG AGA GGA CAC GAG GAC GCA TCG TGG CCG GAT CTC | Metabion GmbH | N/A |
| CCT CTT TAT TTA TTG ATT AAA AAC CAT GAC ATA CCT CGT GTC CTC TCA TAA TAG AAA AGT CCT AGG TTG AAG ATA ACA TAA TAA GCT GCA CTG AAT CTT CTT CCG TGA CCA CGT CGT GGA ATG C | Metabion GmbH | N/A |
| Recombinant DNA | ||
| mCMV BAC plasmid pSM3fr | In house produced | Tischer et al., 201049 |
| Software and algorithms | ||
| Flow Jo | FLOWJO, LLC (Tree Star) | v10.8.1 |
| GraphPad Prism | GraphPad Software | v8.3.0 |
| Cytoscape | Cytoscape.org | v3.9.1 |
| STRING | EMBL | V11.5 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Felix M. Wensveen (Felix.Wensveen@medri.uniri.hr).
Materials availability
All models generated in this paper will be shared freely upon request to the lead contact.
Experimental models and subject details
Study subjects
Prior to initiation of the study, the Clinical Hospital Rijeka Ethics committee approved this research under license 003-05/20-1/84. Appropriate informed consent forms were signed by all people. We conducted the research in accordance and agreement with the International Conference on Harmonization guidelines on Good Clinical Practice and with the Declaration of Helsinki. Adult donors (≥18 years old) were recruited at the Clinical Hospital Rijeka and only HLA-A∗02+ people (dominant haplotype in the Croatian population (Grubic et al. Int J Immunogenet. 2014) were included in the study (70 individuals). Peripheral blood mononuclear cells (PBMCs) were isolated from peripheral blood (PB) by density gradient centrifugation using Histopaque 1077 Density Gradient Medium (Sigma-Aldrich, St. Louis, USA). After isolation, the cells were cryopreserved in fetal bovine serum (Corning) with 10% dimethyl sulfoxide (Sigma-Aldrich, St. Louis, USA) until use. In the present study, we categorized people into three groups: Infected (C19-Inf, n=22) – people with convalescent COVID-19 6 months’ post COVID-19 infection; Infected/Vacc (C19-Inf/Vacc, n=24) – people with convalescent COVID-19 1 month after 2nd dose of COVID-19 vaccine; Vaccinated (C19-Vacc, n=24) – non-convalescent people 1 month after 2nd dose of COVID-19 vaccine. Only people vaccinated with 2 doses of mRNA vaccines (Pfizer-BioNTech BNT162b2 or Moderna mRNA-1273 COVID-19 vaccine) were included in the study.
Mice
Mice were strictly age- and sex-matched within experiments and were held in the local animal facility in Rijeka, Croatia, under specific pathogen-free conditions and handled in accordance with institutional, national and/or EU guidelines. NSG-HLA:A∗02 transgenic mice (line 14570) were purchased from The Jackson Laboratory. All animal experiments were done with approval from the University of Rijeka Medical Faculty Ethics Committee and Croatian Ministry of Agriculture, Veterinary and Food Safety Directorate under license UP/I-322-01/20-01/17.
Method details
HLA class I tetramer staining and multi-parameter flow cytometry
First, subject samples were tested for HLA haplotype by flow cytometry and only people with an HLA-A∗02 haplotype were included in this study. CD8+ T cells were enriched from PBMCs using magnetic-activated cell sorting CD8 microbeads (Miltenyi Biotec, Auburn, CA, USA) according to the manufacturer’s instructions. Enriched CD8+ T cells were stained with APC-conjugated HLA class I tetramer (SARS-CoV-2 (C19) – epitope YLQPRTFLL on Spike protein) and PE-conjugated HLA class I tetramer (Influenza (FLU) – epitope GILGFVFTL on MP1 protein) (Tetramer Shop, Denmark) for 15 min at 37°C in dark, washed, and then stained with fluorochrome-conjugated antibodies for specific surface markers for 30 min at 4°C in dark. This allowed us to directly compare within the same person C19- and FLU- specific CD8+ T cells. Multi-parameter flow cytometry was performed using a BD FACSAria IIu (BD Biosciences) and analyzed using FlowJo software version 10.8.1 (Tree Star). All antibodies are summarized in Table S1. (key resources table).
RNA sequencing and TCR analysis
PBMCs were stained with APC (C19)- and PE (FLU)- conjugated HLA class I tetramers (Tetramershop), antibodies directed against CD8 and CD3, and viability dye. Live FLU+CD8+CD3+ and C19+CD8+CD3+ T cells were sorted in RLT buffer using a BD FACSAria IIu (BD Biosciences). Total (bulk) RNA was isolated using QIAGEN RNeasy micro kit according to manufacturer’s protocols. Samples were sent to GENEWIZ for further processing. RNA library preparations and sequencing reactions were conducted at Azenta US, Inc (South Plainfield, NJ, USA). Ultra-low input RNA sequencing library was prepared by using SMART-Seq HT kit for full-length cDNA synthesis and amplification (Takara, San Jose, CA, USA), and Illumina Nextera XT (Illumina, San Diego, CA, USA) library was used for sequencing library preparation. Briefly, cDNA was fragmented and an adaptor was added using Transposase, followed by limited-cycle PCR to enrich and add index to the cDNA fragments. The sequencing library was validated on the Agilent TapeStation (Agilent Technologies, Palo Alto, CA, USA). The sequencing libraries were multiplexed and clustered on a flowcell. After clustering, the flowcel was loaded on an Illumina HiSeq instrument according to manufacturer’s instructions. The samples were sequenced using a 2x150 Paired End (PE) configuration. Image analysis and base calling were conducted by the HiSeq Control Software (HCS). Raw sequence data (.bcl files) generated from Illumina HiSeq was converted into fastq files and de-multiplexed using Illumina's bcl2fastq 2.17 software. One mis-match was allowed for index sequence identification. Transcriptome data was analyzed using the R2 Genomics and Visualization platform (http://r2.amc.nl). To generate protein interaction maps, differentially expressed genes were analyzed using the STRING platform version 11.5 (https://string-db.org), with a confidence (score) cutoff of 0.4. Visualization of networks was done using cytoscape version 3.9.1. to illustrate largest subnetworks. For TCR analysis, Bulk RNA sequencing reads were aligned to reference V, D, J and C genes of T cell receptors with MiXCR version 2.1.11 (Illumina), which assembled the clonotypes and extracted the α and β chains of the CDR3 regions. The MiXCR clonotype output was further imported to vdjtools (https://vdjtools-doc.readthedocs.io/en/master/#) where the non-functional (non-coding) clonotypes were filtered and individual clonotypes were normalized by dividing their read counts by the total read count of the sample clonotypes. The resulted TRB CDR3 sequence-based clonotypes were used for downstream statistical analysis. Reads retrieved only once or twice were excluded from the analysis to prevent background of sequencing errors.
In vitro intracellular cytokine staining (ICS) assay
To measure cytokine production directly ex vivo, PBMCs were cultured in RPMI 1640 medium (PAN-Biotech), supplemented with 10% FCS (PAN-Biotech) and 2-ME (Sigma-Aldrich). A total of 1x106 PBMCs per well in a U-bottom 96-well plate (Cellstar) were stimulated with C19 (YLQPRTFLL) or FLU (GILGFVFTL) peptide (JPT Peptide Technologies, Germany) or with a peptide pool of SARS-CoV-2 spike (PepTivator SARS-CoV-2 Prot_S, Miltenyi) or influenza HA (PepTivator Influenza A HA, Miltenyi) peptides in a final concentration of 1 μg/mL for 4h in the presence of Brefeldin A (eBioscience). After incubation, PBMCs were collected and washed before performing surface staining (FVD, CD8). Fixation and permeabilization of cells for intracellular staining (IFNγ, TNF, IL-2 and Granzyme B) were done with the intracellular fixation and permeabilization set (eBioscience).
Generation of recombinant viruses
mCMV-COVID (YLQPRTFLL) and mCMV-FLU (GILGFVFTL) were generated by en-passant BAC mutagenesis of the mCMV BAC plasmid pSM3fr as follows: a kanR cassette flanked by homologous viral sequences was amplified from plasmid pori6K-RIT using the oligonucleotide pairs ie2_FLU_fwd / ie2_FLU_rev or ie2_COVID-19_fwd / ie2_COVID-19_rev (see key resources table). The resulting products were transformed into GS1783 bacteria carrying pSM3fr and the peptide coding sequence was integrated at the C-terminus of the viral ie2 gene by Red recombination. Subsequently the BACs pmCMV-FLU and pmCMV-COVID were purified and the successful integration of the peptide-coding sequence mutagenesis was confirmed by sequencing (Eurofins Genomics). The recombinant virus mCMV-FLU and mCMV-COVID were reconstituted in and high-titer cell culture derived virus stocks were prepared by standard protocol.
In vivo NSG experiments
PBMCs were divided into two equal fractions and transferred to NSG-HLA:A∗02 transgenic mice. Fraction of PBMCs was stained prior the transfer to determine the percentage of antigen-specific cells. On the same day mice were infected intraperitoneal (i.p.) with 2 x 106 PFU - mCMV-COVID or mCMV-FLU. Next day mice were injected i.p. with 100ng/mouse of human IL-2 (PeproTech). Mice were sacrificed 7 days post infection and peritoneal exudate cells (PEC) were isolated. For the analysis of the expansion cells were labeled with APC (C19)- and PE (FLU)- conjugated HLA class I tetramers, washed and stained with FVD to determine live cells, anti-mouse CD45.1 to exclude mouse cells and anti-human CD8 and anti-human CD45 to determine human PBMCs. To measure cytokine production, cells were stimulated with phorbol myristate acetate (PMA, InvivoGen) and Ionomycin (InvivoGen) for 4h in the presence of Brefeldin A (eBioscience). After incubation, cells were stained with HLA-A∗02 C19 or FLU tetramer, following were surface staining (FVD, anti-human CD8, anti-human CD45). Fixation and permeabilization of cells for intracellular staining (IFNγ and TNF) was done with the intracellular fixation and permeabilization set (eBioscience). Flow cytometry analysis was done on a FACSVerse (BDBiosciences) and analyzed with FlowJo software (Tree Star).
Quantification and statistical analysis
Statistical analysis
Data are presented as mean ± SEM or as individual values. Statistical significance was determined by paired Student’s t test, one-way ANOVA with Bonferroni's post-test correction for multiple comparisons or Kruskal-Wallis rank-sum test with Dunn’s post hoc test for multiple comparisons using Graph Pad Prism 8. ∗p < 0.05, ∗∗p < 0.01 and ∗∗∗p < 0.001.
Acknowledgments
We thank all participants of the study. We thank Sali Slavić Stupac, Ante Miše, and Michaela Gašparević for technical support. We thank Prof. Stipan Jonjić for providing mice. This work was supported by grants of the University of Rijeka (18-152-1301 to F.M.W.) and the Croatian Science Foundation (IP-2016-06-8027, IP-2020-02-7928, and IP-CORONA-2020-04-2045 to F.M.W.).
Author contributions
I.K. designed and performed experiments and co-wrote the article. C.D., J.K., and M.B. performed bioinformatic RNA sequencing and TCR analysis. D.G. performed experiments and generated figures. N.A.L. generated new viral strains. M.R.F., I.B., and D.C.G. included study subjects and collected material. F.M.W. designed experiments, co-wrote the article, and supervised the project. All authors critically revised the manuscript for important intellectual content.
Declaration of interests
The authors declare no competing interests.
Published: April 4, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112395.
Supplemental information
Data and code availability
-
•
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Gazit S., Shlezinger R., Perez G., Lotan R., Peretz A., Ben-Tov A., Herzel E., Alapi H., Cohen D., Muhsen K., et al. SARS-CoV-2 naturally acquired immunity vs. Vaccine-induced immunity, reinfections versus breakthrough infections: a retrospective cohort study. Clin. Infect. Dis. 2022;75:e545–e551. doi: 10.1093/cid/ciac262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dhumal S., Patil A., More A., Kamtalwar S., Joshi A., Gokarn A., Mirgh S., Thatikonda P., Bhat P., Murthy V., et al. SARS-CoV-2 reinfection after previous infection and vaccine breakthrough infection through the second wave of pandemic in India: an observational study. Int. J. Infect. Dis. 2022;118:95–103. doi: 10.1016/j.ijid.2022.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mallajosyula V., Ganjavi C., Chakraborty S., McSween A.M., Pavlovitch-Bedzyk A.J., Wilhelmy J., Nau A., Manohar M., Nadeau K.C., Davis M.M. CD8(+) T cells specific for conserved coronavirus epitopes correlate with milder disease in COVID-19 patients. Sci. Immunol. 2021;6:eabg5669. doi: 10.1126/sciimmunol.abg5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peng Y., Felce S.L., Dong D., Penkava F., Mentzer A.J., Yao X., Liu G., Yin Z., Chen J.L., Lu Y., et al. An immunodominant NP105-113-B∗07:02 cytotoxic T cell response controls viral replication and is associated with less severe COVID-19 disease. Nat. Immunol. 2022;23:50–61. doi: 10.1038/s41590-021-01084-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dan J.M., Mateus J., Kato Y., Hastie K.M., Yu E.D., Faliti C.E., Grifoni A., Ramirez S.I., Haupt S., Frazier A., et al. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science. 2021;371:eabf4063. doi: 10.1126/science.abf4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bange E.M., Han N.A., Wileyto P., Kim J.Y., Gouma S., Robinson J., Greenplate A.R., Hwee M.A., Porterfield F., Owoyemi O., et al. CD8(+) T cells contribute to survival in patients with COVID-19 and hematologic cancer. Nat. Med. 2021;27:1280–1289. doi: 10.1038/s41591-021-01386-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kavazović I., Polić B., Wensveen F.M. Cheating the hunger games; mechanisms controlling clonal diversity of CD8 effector and memory populations. Front. Immunol. 2018;9:2831. doi: 10.3389/fimmu.2018.02831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wensveen F.M., van Gisbergen K.P., Eldering E. The fourth dimension in immunological space: how the struggle for nutrients selects high-affinity lymphocytes. Immunol. Rev. 2012;249:84–103. doi: 10.1111/j.1600-065X.2012.01156.x. [DOI] [PubMed] [Google Scholar]
- 9.Kavazović I., Han H., Balzaretti G., Slinger E., Lemmermann N.A.W., Ten Brinke A., Merkler D., Koster J., Bryceson Y.T., de Vries N., et al. Eomes broadens the scope of CD8 T-cell memory by inhibiting apoptosis in cells of low affinity. PLoS Biol. 2020;18:e3000648. doi: 10.1371/journal.pbio.3000648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Gisbergen K.P., Klarenbeek P.L., Kragten N.A.M., Unger P.P.A., Nieuwenhuis M.B.B., Wensveen F.M., ten Brinke A., Tak P.P., Eldering E., Nolte M.A., van Lier R.A.W. The costimulatory molecule CD27 maintains clonally diverse CD8(+) T cell responses of low antigen affinity to protect against viral variants. Immunity. 2011;35:97–108. doi: 10.1016/j.immuni.2011.04.020. [DOI] [PubMed] [Google Scholar]
- 11.Badovinac V.P., Porter B.B., Harty J.T. CD8+ T cell contraction is controlled by early inflammation. Nat. Immunol. 2004;5:809–817. doi: 10.1038/ni1098. [DOI] [PubMed] [Google Scholar]
- 12.Pipkin M.E., Sacks J.A., Cruz-Guilloty F., Lichtenheld M.G., Bevan M.J., Rao A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity. 2010;32:79–90. doi: 10.1016/j.immuni.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Aalderen M.C., Remmerswaal E.B.M., Verstegen N.J.M., Hombrink P., ten Brinke A., Pircher H., Kootstra N.A., ten Berge I.J.M., van Lier R.A.W. Infection history determines the differentiation state of human CD8+ T cells. J. Virol. 2015;89:5110–5123. doi: 10.1128/JVI.03478-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Minervina A.A., Pogorelyy M.V., Kirk A.M., Crawford J.C., Allen E.K., Chou C.H., Mettelman R.C., Allison K.J., Lin C.Y., Brice D.C., et al. SARS-CoV-2 antigen exposure history shapes phenotypes and specificity of memory CD8 T cells. medRxiv. 2022 doi: 10.1101/2021.07.12.21260227. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Habel J.R., Nguyen T.H.O., van de Sandt C.E., Juno J.A., Chaurasia P., Wragg K., Koutsakos M., Hensen L., Jia X., Chua B., et al. Suboptimal SARS-CoV-2-specific CD8(+) T cell response associated with the prominent HLA-A∗02:01 phenotype. Proc. Natl. Acad. Sci. USA. 2020;117:24384–24391. doi: 10.1073/pnas.2015486117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Redd A.D., Nardin A., Kared H., Bloch E.M., Abel B., Pekosz A., Laeyendecker O., Fehlings M., Quinn T.C., Tobian A.A.R. Minimal crossover between mutations associated with omicron variant of SARS-CoV-2 and CD8(+) T-cell epitopes identified in COVID-19 convalescent individuals. mBio. 2022;13:e0361721. doi: 10.1128/mbio.03617-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shomuradova A.S., Vagida M.S., Sheetikov S.A., Zornikova K.V., Kiryukhin D., Titov A., Peshkova I.O., Khmelevskaya A., Dianov D.V., Malasheva M., et al. SARS-CoV-2 epitopes are recognized by a public and diverse repertoire of human T cell receptors. Immunity. 2020;53:1245–1257.e5. doi: 10.1016/j.immuni.2020.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peng Y., Mentzer A.J., Liu G., Yao X., Yin Z., Dong D., Dejnirattisai W., Rostron T., Supasa P., Liu C., et al. Broad and strong memory CD4(+) and CD8(+) T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat. Immunol. 2020;21:1336–1345. doi: 10.1038/s41590-020-0782-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferretti A.P., Kula T., Wang Y., Nguyen D.M.V., Weinheimer A., Dunlap G.S., Xu Q., Nabilsi N., Perullo C.R., Cristofaro A.W., et al. Unbiased screens show CD8(+) T cells of COVID-19 patients recognize shared epitopes in SARS-CoV-2 that largely reside outside the spike protein. Immunity. 2020;53:1095–1107.e3. doi: 10.1016/j.immuni.2020.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rha M.S., Jeong H.W., Ko J.H., Choi S.J., Seo I.H., Lee J.S., Sa M., Kim A.R., Joo E.J., Ahn J.Y., et al. PD-1-Expressing SARS-CoV-2-specific CD8(+) T cells are not exhausted, but functional in patients with COVID-19. Immunity. 2021;54:44–52.e3. doi: 10.1016/j.immuni.2020.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kared H., Redd A.D., Bloch E.M., Bonny T.S., Sumatoh H., Kairi F., Carbajo D., Abel B., Newell E.W., Bettinotti M.P., et al. SARS-CoV-2-specific CD8+ T cell responses in convalescent COVID-19 individuals. J. Clin. Invest. 2021;131:e145476. doi: 10.1172/JCI145476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamann D., Baars P.A., Rep M.H., Hooibrink B., Kerkhof-Garde S.R., Klein M.R., van Lier R.A. Phenotypic and functional separation of memory and effector human CD8+ T cells. J. Exp. Med. 1997;186:1407–1418. doi: 10.1084/jem.186.9.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sallusto F., Lenig D., Förster R., Lipp M., Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 24.Brenchley J.M., Karandikar N.J., Betts M.R., Ambrozak D.R., Hill B.J., Crotty L.E., Casazza J.P., Kuruppu J., Migueles S.A., Connors M., et al. Expression of CD57 defines replicative senescence and antigen-induced apoptotic death of CD8+ T cells. Blood. 2003;101:2711–2720. doi: 10.1182/blood-2002-07-2103. [DOI] [PubMed] [Google Scholar]
- 25.Verma K., Ogonek J., Varanasi P.R., Luther S., Bünting I., Thomay K., Behrens Y.L., Mischak-Weissinger E., Hambach L. Human CD8+ CD57- TEMRA cells: too young to be called "old". PLoS One. 2017;12:e0177405. doi: 10.1371/journal.pone.0177405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Winkler E.S., Bailey A.L., Kafai N.M., Nair S., McCune B.T., Yu J., Fox J.M., Chen R.E., Earnest J.T., Keeler S.P., et al. SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nat. Immunol. 2020;21:1327–1335. doi: 10.1038/s41590-020-0778-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lemmermann N.A.W., Kropp K.A., Seckert C.K., Grzimek N.K.A., Reddehase M.J. Reverse genetics modification of cytomegalovirus antigenicity and immunogenicity by CD8 T-cell epitope deletion and insertion. J. Biomed. Biotechnol. 2011;2011:812742. doi: 10.1155/2011/812742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lemmermann N.A.W., Gergely K., Böhm V., Deegen P., Däubner T., Reddehase M.J. Immune evasion proteins of murine cytomegalovirus preferentially affect cell surface display of recently generated peptide presentation complexes. J. Virol. 2010;84:1221–1236. doi: 10.1128/JVI.02087-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shen C.H., Talay O., Mahajan V.S., Leskov I.B., Eisen H.N., Chen J. Antigen-bearing dendritic cells regulate the diverse pattern of memory CD8 T-cell development in different tissues. Proc. Natl. Acad. Sci. USA. 2010;107:22587–22592. doi: 10.1073/pnas.1016350108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.West E.E., Youngblood B., Tan W.G., Jin H.T., Araki K., Alexe G., Konieczny B.T., Calpe S., Freeman G.J., Terhorst C., et al. Tight regulation of memory CD8(+) T cells limits their effectiveness during sustained high viral load. Immunity. 2011;35:285–298. doi: 10.1016/j.immuni.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buchholz V.R., Flossdorf M., Hensel I., Kretschmer L., Weissbrich B., Gräf P., Verschoor A., Schiemann M., Höfer T., Busch D.H. Disparate individual fates compose robust CD8+ T cell immunity. Science. 2013;340:630–635. doi: 10.1126/science.1235454. [DOI] [PubMed] [Google Scholar]
- 32.Kedzierska K., Venturi V., Field K., Davenport M.P., Turner S.J., Doherty P.C. Early establishment of diverse T cell receptor profiles for influenza-specific CD8(+)CD62L(hi) memory T cells. Proc. Natl. Acad. Sci. USA. 2006;103:9184–9189. doi: 10.1073/pnas.0603289103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Bree G.J., van Leeuwen E.M.M., Out T.A., Jansen H.M., Jonkers R.E., van Lier R.A.W. Selective accumulation of differentiated CD8+ T cells specific for respiratory viruses in the human lung. J. Exp. Med. 2005;202:1433–1442. doi: 10.1084/jem.20051365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Badovinac V.P., Harty J.T. Manipulating the rate of memory CD8+ T cell generation after acute infection. J. Immunol. 2007;179:53–63. doi: 10.4049/jimmunol.179.1.53. [DOI] [PubMed] [Google Scholar]
- 35.Tuazon Kels M.J., Ng E., Al Rumaih Z., Pandey P., Ruuls S.R., Korner H., Newsome T.P., Chaudhri G., Karupiah G. TNF deficiency dysregulates inflammatory cytokine production, leading to lung pathology and death during respiratory poxvirus infection. Proc. Natl. Acad. Sci. USA. 2020;117:15935–15946. doi: 10.1073/pnas.2004615117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keewan E., Beg S., Naser S.A. Anti-TNF-alpha agents modulate SARS-CoV-2 receptors and increase the risk of infection through notch-1 signaling. Front. Immunol. 2021;12:641295. doi: 10.3389/fimmu.2021.641295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wherry E.J., Blattman J.N., Murali-Krishna K., van der Most R., Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 2003;77:4911–4927. doi: 10.1128/jvi.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mackerness K.J., Cox M.A., Lilly L.M., Weaver C.T., Harrington L.E., Zajac A.J. Pronounced virus-dependent activation drives exhaustion but sustains IFN-gamma transcript levels. J. Immunol. 2010;185:3643–3651. doi: 10.4049/jimmunol.1000841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kusnadi A., Ramírez-Suástegui C., Fajardo V., Chee S.J., Meckiff B.J., Simon H., Pelosi E., Seumois G., Ay F., Vijayanand P., Ottensmeier C.H. Severely ill COVID-19 patients display impaired exhaustion features in SARS-CoV-2-reactive CD8(+) T cells. Sci. Immunol. 2021;6:eabe4782. doi: 10.1126/sciimmunol.abe4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Diao B., Wang C., Tan Y., Chen X., Liu Y., Ning L., Chen L., Li M., Liu Y., Wang G., et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19) Front. Immunol. 2020;11:827. doi: 10.3389/fimmu.2020.00827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blank C.U., Haining W.N., Held W., Hogan P.G., Kallies A., Lugli E., Lynn R.C., Philip M., Rao A., Restifo N.P., et al. Defining 'T cell exhaustion'. Nat. Rev. Immunol. 2019;19:665–674. doi: 10.1038/s41577-019-0221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gallagher M.P., Conley J.M., Vangala P., Garber M., Reboldi A., Berg L.J. Hierarchy of signaling thresholds downstream of the T cell receptor and the Tec kinase ITK. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2025825118. e2025825118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adamo S., Michler J., Zurbuchen Y., Cervia C., Taeschler P., Raeber M.E., Baghai Sain S., Nilsson J., Moor A.E., Boyman O. Signature of long-lived memory CD8(+) T cells in acute SARS-CoV-2 infection. Nature. 2022;602:148–155. doi: 10.1038/s41586-021-04280-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao Y., Cai C., Wullimann D., Niessl J., Rivera-Ballesteros O., Chen P., Lange J., Cuapio A., Blennow O., Hansson L., et al. Immunodeficiency syndromes differentially impact the functional profile of SARS-CoV-2-specific T cells elicited by mRNA vaccination. Immunity. 2022;55:1732–1746.e5. doi: 10.1016/j.immuni.2022.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sureshchandra S., Lewis S.A., Doratt B.M., Jankeel A., Coimbra Ibraim I., Messaoudi I. Single-cell profiling of T and B cell repertoires following SARS-CoV-2 mRNA vaccine. JCI Insight. 2021;6:e153201. doi: 10.1172/jci.insight.153201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wensveen F.M., Klarenbeek P.L., van Gisbergen K.P.J.M., Pascutti M.F., Derks I.A.M., van Schaik B.D.C., Ten Brinke A., de Vries N., Cekinovic D., Jonjic S., et al. Pro-apoptotic protein Noxa regulates memory T cell population size and protects against lethal immunopathology. J. Immunol. 2013;190:1180–1191. doi: 10.4049/jimmunol.1202304. [DOI] [PubMed] [Google Scholar]
- 47.Rowntree L.C., Petersen J., Juno J.A., Chaurasia P., Wragg K., Koutsakos M., Hensen L., Wheatley A.K., Kent S.J., Rossjohn J., et al. SARS-CoV-2-specific CD8(+) T-cell responses and TCR signatures in the context of a prominent HLA-A∗24:02 allomorph. Immunol. Cell Biol. 2021;99:990–1000. doi: 10.1111/imcb.12482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wagner M., Jonjic S., Koszinowski U.H., Messerle M. Systematic excision of vector sequences from the BAC-cloned herpesvirus genome during virus reconstitution. J. Virol. 1999;73:7056–7060. doi: 10.1128/jvi.73.8.7056-7060.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tischer B.K., Smith G.A., Osterrieder N. En passant mutagenesis: A two step markerless red recombination system. Methods Mol. Biol. 2010;634:421–430. doi: 10.1007/978-1-60761-652-8_30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.