

Abstract
Immune-mediated thrombotic thrombocytopenic purpura (iTTP) is characterized by a severe ADAMTS13 deficiency due to the presence of anti-ADAMTS13 auto-antibodies, with subsequent accumulation of circulating ultra-large von Willebrand factor (VWF) multimers. The role of endothelial cell activation as a trigger of the disease has been suggested in animal models but remains to be demonstrated in humans. We prospectively obtained plasma from the first plasma exchange of 25 patients during iTTP acute phase. iTTP but not control plasma, induced a rapid VWF release and P-selectin exposure on the surface of dermal human micro-vascular endothelial cell (HMVEC-d), associated with angiopoietin-2 and endothelin-1 secretion, consistent with Weibel-Palade bodies exocytosis. Calcium (Ca2+) blockade significantly decreased VWF release, whereas iTTP plasma induced a rapid and sustained Ca2+ flux in HMVEC-d which correlated in retrospect, with disease severity and survival in 62 iTTP patients. F(ab)’2 fragments purified from the immunoglobulin G fraction of iTTP plasma mainly induced endothelial cell activation with additional minor roles for circulating free heme and nucleosomes, but not for complement. Furthermore, two anti-ADAMTS13 monoclonal antibodies purified from iTTP patients’ B cells, but not serum from hereditary TTP, induced endothelial Ca2+ flux associated with Weibel-Palade bodies exocytosis in vitro, whereas inhibition of endothelial ADAMTS13 expression using small intering RNA, significantly decreased the stimulating effects of iTTP immunoglobulin G. In conclusion, Ca2+-mediated endothelial cell activation constitutes a “second hit” of iTTP, is correlated with the severity of the disease and may constitute a possible therapeutic target.
Introduction
Immune-mediated thrombotic thrombocytopenic purpura (iTTP) is a rare and life-threatening thrombotic microangiopathy (TMA) characterized by a severe thrombocytopenia (<30.109/L) and a mechanical hemolytic anemia. Consequently, ischemic events of variable severity occur, mainly affecting the brain, the heart, and the mesenteric tract. The diagnosis of iTTP relies on the demonstration of ADAMTS13 protease functional deficiency (<10%), due to the presence of anti-ADAMTS13 auto-antibodies.1–3 ADAMTS13 is responsible for the cleavage of ultra-large von Willebrand factor (UL-VWF) into smaller and less thrombotic multimers.4,5 Deficiency of ADAMTS13 activity leads to the accumulation of highly prothrombotic UL-VWF in the patient plasma inducing the formation of multiple platelet-rich thrombi into the microcirculation, consumptive thrombocytopenia, mechanical hemolysis and clinical symptoms.1–3 Despite treatments based on therapeutic plasma exchange (PEX) and immunosuppressive drugs, the mortality rate remains as high as 5-10%.
Animal models demonstrate that in addition to ADAMTS13 deficiency, endothelial UL-VWF exocytosis is necessary to reproduce the disease, suggesting that endothelial cells (EC) may participate in a “second hit” of the disease.6 UL-VWF is the main constituent of endothelial Weibel-Palade bodies (WPB), from which it can rapidly be released upon EC activation.6 In order to obtain a TTP-like disease, ADAMTS13 knockout (KO) mice need to be crossed with mice expressing high intracellular concentrations of VWF, then to be injected with shigatoxin to induce WPB degranulation leading to UL-VWF release, showing that inactivation of the adamts13 gene is not sufficient to induce TTP-like manifestations.7,8,9 In agreement, injections of large concentrations of recombinant VWF can induce TTP in ADAMTS13 KO mice. Moreover, vwf gene deletion results in complete protection in the shigatoxin-induced TTP murine model, demonstrating the absolute requirement of VWF to develop TTP.10,11 Another TTP model consisting in injection of murine anti-ADAMTS13 inhibitory monoclonal antibodies (mAb) into wild-type mice, led to plasma ADAMTS13 deficiency and UL-VWF accumulation without TTP-like symptoms.12 The additional injection of recombinant VWF in this model induces an iTTP-like disease, further demonstrating the essential role of large concentrations of circulating VWF. In a primate model of TTP, injection of a murine anti-human ADAMTS13 inhibitory antibody induced TTP, as demonstrated by the appearance of severe thrombocytopenia, hemolytic anemia, elevated LDH, schistocytes and the occurrence of microthrombi in kidney, heart, brain and spleen.13 However, the primates did not develop end-stage disease, suggesting once again that inhibition of ADAMTS13 alone, may not be sufficient to reproduce a full-spectrum human TTP.
Thus, these experimental models suggest that induction of TTP in animals is a “two-hit” process requiring first, ADAMTS13 protease inactivation and second, an increased VWF release by activated EC. Similar mechanisms however, remain to be demonstrated in humans. In this context, we asked whether iTTP-patient plasma was able to induce WPB exocytosis and tried to identify possible endothelial activators in iTTP-patient plasma. Using plasma prospectively collected from patients during the acute phase of iTTP, we observed induction of UL-VWF release from EC via WPB exocytosis in a Ca2+-dependent pathway. We identified IgG from iTTP patient plasma as the main inducer of endothelial activation and observed that Ca2+-dependent endothelial activation intensity correlated with disease severity.
Methods
Patient characteristics
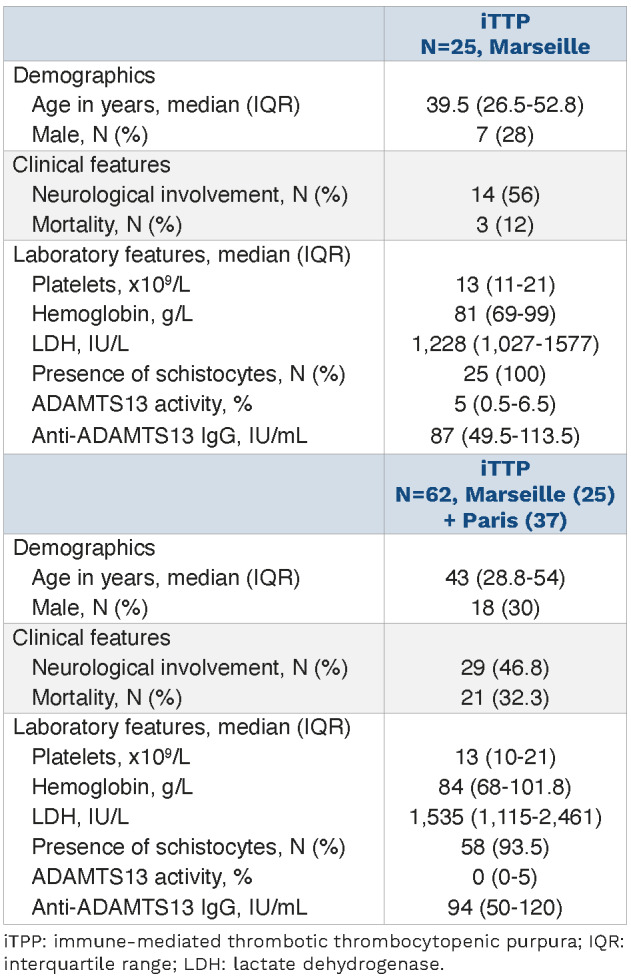
We conducted a prospective study between 2008 and 2011 consisting in a National Clinical Research Project (#2007/23) approved by the Ethical Committee of the “Assistance Publique-Hôpitaux de Marseille”. Informed consent was provided according to the Declaration of Helsinki. Detailed information about the 62 patients can be found in the Online Supplementary Appendix and Table 1.
Immunofluorescence studies
Confluent human microvascular dermal EC (HMVEC-d) were grown in EC growth basal medium-2 (EBM2) containing 1% fetal bovine serum for 16 hours (h) and activated for 1 hour (h) with either control or iTTP plasma (1/100 in EBM2), washed twice in phosphate buffer saline (PBS), fixed in 1% paraformaldehyde for 10 minutes (min) and labeled with anti-VWF, anti-P-selectin antibodies or with rabbit non-immune serum like described in supplementary methods.
In order to visualize ADAMTS13, HMVEC-d were activated for 20 min with control or iTTP immunoglobulin G (IgG) (30 mg/mL), washed in PBS and labeled as described in the Online Supplementary Appendix. In order to exclude the influence of possible endotoxin contamination of biological samples, all experiments were performed in the presence of 10 mg/mL polymyxin B (Sigma-Aldrich, Saint Louis, USA).
Table 1.
Demographics, clinical and biological data of the 62 immune-mediated thrombotic thrombocytopenic purpura patients included from Marseille and Paris.
Soluble von Willebrand factor, endothelin-1 and angiopoietin-2 measurements
Specific enzyme-linked immunosorbent assays (ELISA) were performed to determine the concentrations of soluble VWF (American Diagnostica, Stanford, USA), endothelin-1 or angiopoietin-2 (R&D system, Minneapolis, USA), according to the manufacturer’s instructions.
Intracellular Ca2+ flux
HMVEC-d were incubated for 1 h with 5 mM fluo-4 AM fluorescent dye (ThermoFisher Scientific, USA) in PBS containing 1% bovine serum albumin (BSA), washed and incubated in PBS 1% BSA for 30 min at 37°C. Cells were stimulated with A23187 (10 µM in EBM2), thrombin (4 IU/mL or 0.5 mM), control or iTTP plasma samples (1/100 in EBM2) for 20 to 1,800 seconds, or with IgG (30 mg/mL in EBM2). Fluorescence analysis used a Cytofluor®, fluorescence multi-well plate reader (PerSeptive-Biosystems, Framingham, USA). Total fluorescence intensity was expressed in arbitrary unit (AU). Intracellular Ca2+ flux was also measured by microscopy (see the Online Supplementary Appendix).
Preparation of IgG-depleted plasma and purification of the IgG fraction
After washing with binding buffer (PBS containing 100 mM Na2HPO4 at pH 7.4), 100 mL of protein A magnetic sepharose beads (GEHealthcare) were incubated for 30 min at 4°C with 300 mL of 1:5 diluted plasma from control or iTTP patients. Beads were retrieved using a magnetic bench and plasma was used for Ca2+ flux induction and IF studies. IgG were eluted using 100 mL of elution buffer (100 mM glycine at pH 12.8) and 5 mL of 1 M TRIS hydrochloride at pH 9.
Human anti-ADAMTS13 monoclonal antibodies
The two anti-human ADAMTS13 mAb TTP73-1 and ELH2-1 were isolated and cloned from B cells of two different iTTP-patients. As previously reported, these two antibodies recognize cryptic epitopes in ADAMTS1314 and were used at 30 mg/mL in experiments.
ADAMTS13 gene silencing
Downregulation of ADAMTS13 mRNA level was achieved using small interfering RNA (siRNA) directed against ADAMTS13 (Ambion, ThermoFisher-Scientific, USA) and Jet Prime kit (Polyplus-transfection, USA) according to the manufacturer’s instructions. As the decrease in ADAMTS13 mRNA level was achieved 24 h after transfection by quantitative real-time polymerase chain reaction (RT-qPCR) and remained stable for 96 h, Ca2+ flux experiments were performed 48 h after transfection.
Statistical analysis
Graphpad Prism v.9.2.0 software (GraphPad-Software Inc., San Diego, USA) was used for statistical analysis. Further details of the methods are available in the Online Supplementary Appendix.
Results
Plasmas from immune-mediated thrombotic thrombocytopenic purpura patients induce von Willebrand factor string formation on the surface of HMVEC-d via Weibel-Palade bodies exocytosis
First, we examined VWF string formation on HMVEC-d by confocal microscopy using anti-VWF labeling on permea-bilized HMVEC-d. After incubation with control plasma samples, anti-VWF labeling showed punctuated fluorescent spots, whereas incubation with iTTP plasma samples induced the appearance of typical UL-VWF strings (Figure 1A). Quantification of the VWF string fluorescence was performed after 30 min of incubation and demonstrated higher VWF fluorescence intensity (FI) when HMVEC-d were stimulated with iTTP than with control plasma samples (FI=6.77 AU/cellx104; interquartile range [IQR], 4.8-8.79 vs. FI=0.53 AU/cellx104; IQR, 0.36-0.87, respectively; P<0.0001; Figure 1B). Kinetic studies showed that VWF strings on the endothelial surface in response to iTTP plasma was observed after 5 min incubation and increased for up to 60 min (Figure 1C) (FI=6.75 AU/cellx104; IQR, 4.81-7.38; FI=8.62 AU/cellx104, IQR, 7.03-14.82; FI=14.5 AU/cellx104; IQR, 10.1-21.68 vs. FI=0.76 AU/cellx104, IQR, 0.71-2.99; P=0.0159; P=0.0079; P=0.0079 respectively at 5, 15 and 60 min vs. 0 min; Figure 1D) Similar data were obtained in various conditions such as dynamic using a flow chamber device (Online Supplementary Figure S1A), macrovascular HUVEC (Online Supplementary Figure S1B), or when using either plasma obtained on citrated tubes or serum, instead of plasma from PEX in order to eliminate a possible Ca2+ overload due to the PEX process (Online Supplementary Figure S1C). In order to exclude possible passive absorption of soluble VWF contained in iTTP plasma on HMVEC-d surface, we used a VWF-affinity column to completely eliminate soluble VWF contained in plasma. Using ELISA, we measured soluble VWF in the supernatant of HMVEC-d stimulated with either VWF-free iTTP or VWF-free control plasma samples for 1 h and observed significantly higher concentrations of endothelial soluble VWF in the former condition (2.13 ng/mL, IQR, 1.76-2.50 vs. 1.05 ng/mL, IQR, 0.98-1.61; P=0.0159; Online Supplementary Figure S1D).
In order to determine whether VWF string formation on HMVEC-d was effectively due to WPB exocytosis, we measured the concentrations of endothelin-1 and of angiopoietin-2, two molecules stored in WPB. Significantly higher concentrations of endothelin-1 (19.02 pg/mL; IQR, 16.88-20.21 vs. 11.55 pg/mL; IQR, 5.79-17.9, respectively; P=0.0102; Figure 1E) and angiopoietin-2 (978 pg/mL; IQR, 619.5-1,200 vs. 614.5 pg/mL; IQR, 445.8-868.3, respectively; P=0.0355; Figure 1F), were present in the supernatants of HMVEC-d when cultured with iTTP compared to control plasma samples. In addition, membrane P-selectin expression on HMVEC-d was observed after stimulation for 10 min with iTTP, but not with control plasma samples (Figure 1G), associated with a 3-time-increased P-selectin FI (FI=290.3 AU/cellx102; IQR, 188.2-596.6 vs. FI=81.23 AU/cellx102; IQR: 61.55-114.3; P<0.0001; Figure 1H). The same data concerning endothelin-1 secretion were observed when HUVEC were used instead of HMVEC-d (40.58 pg/mL; IQR, 25.66-43.52 vs. 19.28 pg/mL; IQR, 16.45-21.56; P=0.0070; Online Supplementary Figure S1E). The endothelin-1 secretion was equivalent when citrated plasma or serum of iTTP-patients were using instead of plasma from PEX (24.65 pg/mL; IQR, 20.39-28.41 and 25.33 pg/mL; IQR, 20.84-32.92 vs. 24.10 pg/mL; IQR, 19.9-24.9; Online Supplementary Figure S1F). Altogether, these data are consistent with the fact that iTTP plasmas obtained after PEX were able to induce WPB exocytosis.
Figure 1.

Induction of Weibel-Palade bodies exocytosis on endothelial cells afer stimulation with immune-mediated thrombotic thrombocytopenic purpura plasma. (A) Confocal microscopy analysis of permeabilized human microvascular endothelial cells from derm (HMVEC-d) incubated with either control plasma (A) or immune-mediated thrombotic thrombocytopenic purpura (iTTP) plasma samples (B) using rabbit anti-von Willebrand Factor (VWF) antibodies and Alexa Fluor 594-conjugated donkey anti-rabbit immunoglobulin G (IgG) (original magnification X63, scale bar=10 mm). (B) Quantification of VWF fluorescence intensity (FI) after HMVEC-d stimulation with either control plasma (Control: n=9) or iTTP plasma samples during the acute phase (iTTP: n=14, ***P<0.001). (C) Kinetics of VWF HMVEC-d exocytosis visualized by fluorescence microscopy after incubation with iTTP plasma samples (original magnification X40, scale bar=10 mm). (D) Quantification of VWF FI over time on HMVEC-d stimulated with iTTP plasma samples (iTTP: n=5, *P<0.05, **P<0.01). (E) Endothelin-1 and (F) angiopoietin-2 concentrations in HMVEC-d supernatants stimulated with either control plasma (n=6) or iTTP plasma samples for 1h (n=25, *P<0.05). (G) P-selectin expression detected by confocal microscopy analysis using anti-rabbit P-selectin antibodies and Alexa Fluor 488-conjugated donkey anti-rabbit IgG (green) in HMVEC-d incubated with control (A) or iTTP plasma samples (B) (original magnification X63, scale bar=10 mm). (H) Percentage of P-selectin FI per cell on HMVEC-d stimulated with control plasma (n=10) (representing 100%) or iTTP plasma samples (n=23, ***P<0.001).
Weibel-Palade bodies exocytosis induced by immune-mediated thrombotic thrombocytopenic purpura plasma is Ca2+-mediated
Two pathways of WPB exocytosis are known: a Ca2+-dependent pathway and a cAMP-dependent pathway. In order to determine whether the Ca2+ signaling cascade was involved in WPB exocytosis induced by iTTP plasma, we used the pharmacological inhibitor MAPTAM. Compared with iTTP plasma samples alone, we observed a significant decrease of VWF FI in the presence of MAPTAM (FI=1.09 AU/Cellx104; IQR, 0.78-1.79 vs. FI= 7.59 AU/Cellx104; IQR, 7.52-8.85; P=0.0079; Figure 2A, B). iTTP plasma-induced endothelin-1 secretion was also reduced by 92% in the presence of MAPTAM (82.1%; IQR, 73-134.8 vs. 8.5%; IQR, 6.9-9.6; P<0.0001; Figure 2C).
We then measured the ability of iTTP plasma to induce Ca2+-flux in EC, using a single-wavelength Ca2+ fluorescent dye. After a 20-second incubation, iTTP plasma samples induced a significantly higher FI level than control plasma samples in HMVEC-d (94 AU; IQR, 78-125.5 vs. 45 AU; IQR, 35.5-76 respectively; P<0.0001; Figure 2D) or in HUVEC (68 AU; IQR, 55.38-114.9 vs. 41.75 AU; IQR, 37.75-54; P=0.0063; Online Supplementary Figure S1G). We also calculated the AUC for Ca2+ flux after 30 min of stimulation and observed that iTTP plasma induced significantly higher Ca2+ mobilization than control plasma samples (1,011 AU; IQR, 472-1,334 vs. 242 AU, IQR, 43.5-388.5; P=0.0002; Figure 2E). The endothelial Ca2+ mobilization was equivalent when using citrated plasma or serum instead of PEX plasma (74.5 AU; IQR, 59.5-94.75 and 66 AU; IQR, 41.75-85 vs. 70 AU; IQR, 48.75-87.5; Online Supplementary Figure S1H).
Immune-mediated thrombotic thrombocytopenic purpura plasma-induced endothelial Ca2+ flux correlates with immune-mediated thrombotic thrombocytopenic purpura severity
In order to establish whether iTTP-induced Ca2+ flux correlated with disease severity, we studied sera from 62 iTTP patients (25 from Marseille added with 37 from Paris) collected during the acute phase, and classified in two different groups depending on survival (Table 1). At 20 seconds, Ca2+ flux appeared significantly higher in cells treated with plasma from non-survivor patients compared with those of survivors (157 AU; IQR, 119.7-262.3 vs. 107 AU; IQR, 70-181, respectively; P=0.0031; Figure 3A). Similarly, after 30 min, area under the curve (AUC) from non-survivors was much higher than those in survivors (6,316 cm2; IQR, 3,066-8,503 vs. 1391 cm2; IQR: 833.5-4,235, respectively; P=0.0002; Figure 3B). In addition, plasma from iTTP patients in remission induced significantly less Ca2+ flux than plasma from the same patients during the acute phase (52.5 AU; IQR: 45-69 vs. 131.5 AU; IQR, 66-177; P=0.0013; Figure 3C) and levels of Ca2+ flux were not different from those induced by control plasma. In agreement, plasma obtained from patients in complete remission did not induce VWF release by HMVEC-d (Figure 3D) which was confirmed by fluorescence quantification (1.19 AUx104; IQR, 0.89-1.99) during remission versus 10.85 AUx104; IQR, 7.59-14.04 during the acute phase; P=0.0286; Figure 3E).
The purified IgG fraction from immune-mediated thrombotic thrombocytopenic purpura plasma induced Ca2+-dependent Weibel-Palade bodies exocytosis
In order to identify the soluble mediators contained in iTTP plasma involved in WPB exocytosis, we studied several candidates. As iTTP is an autoimmune disease, we first investigated the role of circulating IgG. After depletion of the IgG fraction from iTTP plasma, we observed a significant decrease of VWF FI release by HMVEC-d (Figure 4A). In agreement, the Ca2+ flux was reduced by 47% in HMVEC-d cells treated with IgG-depleted plasma compared with complete plasma (127.5 AU; IQR, 83.5-188 vs. 68 AU; IQR, 45.5-113, respectively; P=0.0003; Figure 4B). Conversely, we tested the ability of the purified IgG fraction from iTTP or control plasma to activate HMVEC-d. VWF exocytosis was observed in HMVEC-d cultured with iTTP IgG, but not with control IgG (Figure 4C). Using ELISA, we measured soluble VWF released in the supernatants of HMVEC-d and observed significantly increased VWF concentrations in HMVEC-d stimulated by iTTP IgG compared with control IgG (3.42 ng/mL; IQR, 2.68-4.18 vs. 2.72 ng/mL; IQR, 2.08-3.03, respectively; P=0.0273; Figure 4D). In agreement, we observed a significantly higher Ca2+ flux in HMVEC-d stimulated with iTTP IgG than with control IgG fractions (77.25 AU; IQR, 54.88-109.8 vs. 46.75 AU; IQR, 41-54, respectively; P=0.0011; Figure 4E). These results were confirmed by directly measuring Ca2+ flux in living cells stimulated with either iTTP or control IgG, using microscopy (Online Supplementary Figure S2A, B). In order to determine which fraction of the immunoglobulin was involved in EC activation, we purified the F(ab’)2 fragments from the IgG fraction, and observed that F(ab’)2 fragments induced both VWF string formation (Figure 4F) and a twice higher endothelial Ca2+ flux compared to the complete iTTP IgG fraction (130.8; IQR, 86.11-197.2 vs. 265.3; IQR, 206.6-315.3; P=0.002; Figure 4G).
Figure 2.

Weibel-Palade bodies exocytosis induced by immune-mediated thrombotic thrombocytopenic purpura plasma is Ca2+-mediated. (A) Von Willebrand Factor (VWF) secretion detected by fluorescence microscopy, using rabbit anti-VWF antibodies and Alexa Fluor 594-conjugated donkey anti-rabbit immunoglobulin G (IgG) after incubation of permeabilized human microvascular endothelial cells from derm (HMVEC-d) with immune-mediated thrombotic thrombocytopenic purpura (iTTP) plasma samples in the absence (A, B) or in the presence of the Ca2+ chelator MAPTAM (C, D) (original magnification X40, scale bar=30 mm). (B) VWF fluorescence intensity (FI) quantification by cell comparing HMVEC-d stimulated with iTTP plasma samples in absence or presence of MAPTAM (iTTP: n=5, **P<0.01). (C) Endothelin-1 concentration measured in the supernatants of HMVEC-d incubated with iTTP plasma samples (n=10) in the absence (representing 100%) or in the presence of MAPTAM (***P<0.001). (D) Fluorescence intensity (AU) of the Ca2+ flux in HMVEC-d incubated for 20 seconds with either control plasma samples (n=13), iTTP plasma samples (n=25, ***P<0.001) or A23187 Ca2+ ionophore (n=3). (E) Area under the curve (AUC) (cm2) from Ca2+ flux curves after 30 minutes of HMVEC-d incubation with either control plasma (n=13), iTTP plasma samples (n=25, ***P<0.001) or A23187 Ca2+ ionophore (n=3).
Immune-mediated thrombotic thrombocytopenic purpura IgG induced Weibel-Palade bodies degranulation partly via ADAMTS13 recognition on endothelial cells
In order to identify the endothelial antigen target of iTTP-IgG, we concentrated on ADAMTS13, the main protein involved in iTTP pathogenesis. Since it was impossible to create an ADAMTS13 affinity column in order to eliminate anti-ADAMTS13 auto-antibodies contained in iTTP-purified IgG, we used different procedures. First, we compared the ability of hereditary TTP (hTTP) serum samples which do not contain anti-ADAMTS13 auto-antibodies to induce Weibel-Palade bodies degranulation, with iTTP serum samples. Compared to that induced by iTTP serum samples, we observed that VWF release induced by hTTP serum was 65% lower (6.61 ng/mL; IQR, 4.45-12.84 vs. 17.27 ng/mL; IQR, 11.09-24.88; P=0.0317; Figure 5A) and that the Ca2+ flux induced by hTTP serum samples appeared significantly reduced (113.5 AU; IQR, 112.7-118.4 vs. 126.7 AU; IQR, 123.3-131.7; P=0.0079; Figure 5B). After we verified that extracellular expression of endothelial ADAMTS13 was not modified when EC were stimulated with IgG from iTTP-patients or control plasma samples (Online Supplementary Figure S3A) (90.4%; IQR, 62.78-116.2 vs. 116.3 %; IQR, 73.37-144.7; P=0.3357; Online Supplementary Figure S3B), we inhibited endothelial ADAMTS13 expression using specific siRNA (70% of inhibition, 1; IQR, 1-1 vs. 0.29; IQR, 0.16-0.41; P=0.0313; Figure 5C,) and observed that inhibition of ADAMTS13 expression was associated with a significant reduction of the iTTP IgG-induced Ca2+ flux (33.44 AU; IQR, 25.88-35.22 vs. 47.15 AU; IQR, 31.42-73.9; P=0.0084; Figure 5D). We further tested the ability of two anti-ADAMTS13 antibodies, TTP73-1 or ELH2-1, previously isolated and cloned from the B cells of two iTTP-patients,14 to induce WPB exocytosis in HMVEC-d. Both TTP73-1 and ELH2-1 weakly induced VWF tethering on HMVEC-d membranes (Figure 5E). When cultured in the presence of TTP73-1 but not of or ELH2-1, higher soluble VWF concentrations were measured in HMVEC-d supernatants compared to stimulation with control IgG (3.7 ng/mL; IQR, 3.3-4.3 for TTP73-1 and 1.7 ng/mL; IQR, 1.6-2 for ELH2-1 vs. 0.8 ng/mL; IQR, 0.7-0.99; P=0.0357 and P=0.0571 respectively; Figure 5F), as well as higher endothelin-1 secretion (14.1 pg/mL; IQR, 11.8-15.4 for TTP73-1 and 11.5 pg/mL; IQR, 10.8-14.4 for ELH2-1 vs. 6.4 pg/mL; IQR, 4.2-11.5; P=0.0317 and P=0.0952 respectively; Figure 5G). In addition, an increased but nonsignificant Ca2+ flux was also induced by TTP73-1 and ELH2-1, compared to control IgG (15.01 AU, IQR, 11.55-19.31 and 14.91 AU; IQR, 6.26-18.39, respectively, vs. 3.2 AU, IQR, 0.59-5.83; P=0.10; Figure 5H).
Figure 3.

In vitro endothelial Ca2+ flux intensity induced by immune-mediated thrombotic thrombocytopenic purpura plasma is associated with clinical severity and prognosis. (A) Fluorescence intensity (AU) of the Ca2+ flux measured at 20 seconds in permeabilized human microvascular endothelial cells from derm (HMVEC-d) stimulated with immune-mediated thrombotic thrombocytopenic purpura (iTTP) plasma samples of survivors (n=41) or non-survivors patients (n=21, **P<0.01). (B) Area under the curve (AUC) from Ca2+ flux curves after 30 minutes of HMVEC-d stimulated with iTTP plasma samples of survivors (n=41) or non-survivors (n=21, ***P<0.001). (C) Fluorescence intensity (AU) of the Ca2+ flux in HMVEC-d incubated for 20 seconds with plasma samples from iTTP patients in acute phase (iTTP-AP) or from same patients in remission (iTTP-R) (n=11, **P<0.01). Horizontal dotted line represents median Ca2+ mobilization induced in HMVEC-d by control plasma. (D) Von Willebrand Factor (VWF) secretion and tethering detected by fluorescence microscopy using rabbit anti-VWF antibodies and Alexa Fluor 594-conjugated donkey anti-rabbit immunoglobulin (red) after 1-hour-incubation of HMVEC-d with iTTP plasma samples collected in acute phase (A) or during remission (B). Cell nuclei are labeled in blue using DAPI (original magnification X40, scale bar=10 mm). (E) VWF fluorescence quantification after the HMVEC-d stimulation with iTTP plasma from patients in acute phase (iTTP-AP: n=4), or in remission (iTTP-R: n=4, *P<0.05).
Free heme and nucleosomes, but not complement, play minor roles in immune-mediated thrombotic thrombocytopenic purpura plasma-induced Weibel-Palade bodies exocytosis
The concentrations of free heme were significantly increased in iTTP compared to control plasma samples (11.2 mM; IQR, 6.98-20.35 vs. 9 mM; IQR, 8.2-9.6; P=0.0369; Figure 6A), but returned to control levels after remission (0.63 mM; IQR, 0.44-1.03 vs. 11.2 mM; IQR, 6.98-20.35; P<0.0001; Figure 6A). Free heme concentrations however, were equivalent between survivor and non-survivor patients (10.96 mM; IQR, 8.2-14.07 vs. 12.96 mM; IQR, 2.55-43.56; P=0.6368; Online Supplementary Figure S4A). Inhibition of heme by addition of large concentrations of hemopexin, weakly reduced iTTP plasma-induced VWF exocytosis (Figure 6B), endothelin-1 release (69.86%; IQR, 27.59-136.1 vs. 79.86%; IQR, 54.79-162.6; P<0.0494; Figure 6C) and iTTP-induced Ca2+ flux (30% inhibition, 133.3 AU; IQR, 89.25-242.8 vs. 107.5 AU; IQR, 61.88-208.3; P=0.0063; Figure 6D). iTTP plasma also contained significant enrichment in nucleosomes concentrations compared to control plasma samples (6.34-fold increase; IQR, 3.37-12.14 vs. 1.15-fold increase; IQR; 0.12-1.78; P<0.0001; Figure 6E) which was no longer observed after remission (0.81-fold increase; IQR, 0.53-1.52 vs. 6.34-fold increase; IQR, 3.37-12.14 in acute phase; P<0.0001; Figure 6E), in accordance with previous studies.15 Nucleosomes enrichment however, was not different between survivors and non-survivors (3.57-fold increase; IQR, 1.76-5.24 vs. 3.51-fold increase; IQR, 1.24-7.49; P=0.9295; Online Supplementary Figure S4B). Nucleosomes signalization is dependent of the phosphatidylserine activation and can be inhibited by Annexin V. Addition of Annexin V to iTTP plasma weakly but significantly reduced VWF exocytosis (Figure 6F) and endothelin-1 release by HMVEC-d (10% inhibition, 72.62%; IQR, 64.16-126.1 vs. 80.11%; IQR; 67.52-149.6; P=0.0052; Figure 6G), as well as Ca2+ flux induced by iTTP plasma (13%-reduction, 216 AU; IQR, 102.8-262.8 vs. 246.5 AU; IQR, 128-304.5; P=0.0011; Figure 6H).
Figure 4.

Immunoglobulin G contained in immune-mediated thrombotic thrombocytopenic purpura plasma are mainly involved in permeabilized human microvascular endothelial cells from derm activation. (A) Von Willebrand Factor (VWF) secretion detected by fluorescence microscopy after permeabilized human microvascular endothelial cells from derm (HMVEC-d) incubation with complete immune-mediated thrombotic thrombocytopenic purpura (iTTP) (A) or immunoglobulin (IgG)-depleted (B) plasma samples (original magnification X20, scale bar=30 mm). (B) Fluorescence intensity (AU) of the Ca2+ flux induction in HMVEC-d after a 20-second incubation with complete iTTP or IgG-depleted plasma samples (n=15, ***P<0.001). (C) VWF secretion detected by fluorescence microscopy after HMVEC-d incubation with IgG purified from control (A) or iTTP plasma samples (b) (original magnification X40, scale bar=30 mm). (D) VWF quantification by enzyme-linked immunosorbant assay in the supernatants of cells treated with IgG purified from control (n=7) or iTTP plasma samples (n=16, *P<0.05). (E) Fluorescence intensity (AU) of Ca2+ flux induction in HMVEC-d after 20 seconds incubation with IgG from control (n=6) or iTTP plasma samples (n=16, **P<0.01). (F) VWF secretion detected by fluorescence microscopy after HMVEC-d incubation with IgG (a) from iTTP plasma samples or F(ab)’2 fragments (b) purified from these IgG (original magnification X63, scale bar=20 mm). (G) Fluorescence intensity (AU) of Ca2+ flux induction in HMVEC-d after a 20-second of incubation with medium alone (EBM2), A23187 calcium (Ca2+) ionophore, IgG (IgG) from iTTP plasma samples, or F(ab)’2 fragments purified from these IgG (n=10, **P<0.01).
Figure 5.

ADAMTS13 is involved in immune-mediated thrombotic thrombocytopenic purpura IgG-induced Weibel-Palade bodies degranulation. (A) Von Willebrand Factor (VWF) quantification by enzyme-linked immunosorbant assay in the supernatants of cells treated with immune-mediated thrombotic thrombocytopenic purpura (iTTP) serum (n=5) or hereditary TTP serum samples (hTTP, n=5, *P<0.05). (B) Fluorescence intensity (AU) of Ca2+ flux induction in permeabilized human microvascular endothelial cells from derm (HMVEC-d) after a 20-second incubation with serum from iTTP patients (n=5) or from hTTP patients (n=5, **P<0.01). (C) ADAMTS-13 mRNA relative quantification in HMVEC-d cultured in the presence of control small interfering RNA (siRNA) (Ct si) or siRNA targeting ADAMTS13 (ADAMTS13 Si) (n=6, *P<0.05). (D) Fluorescence intensity (AU) of Ca2+ flux induction in HMVEC-d transfected with control or anti-ADAMTS13 siRNA, then stimulated for 20 seconds with IgG from iTTP plasma samples (n=15, **P<0.01). (E) VWF expression detected by fluorescence microscopy in HMVEC-d incubated with IgG from control patient samples (a) TTP73-1 monoclonal antibody (mAb) (B) or ELH2-1 mAb (C) (original magnification X40, scale bar=30 mm). (F) VWF or (G) endothelin-1 concentrations in supernatants of HMVEC-d incubated with IgG from control patient samples (Control) (n=3), TTP73-1 or ELH2-1 mAb (n=5, *P<0.05). (H) Fluorescence intensity (AU) of Ca2+ flux measured at 20 seconds in HMVEC-d stimulated in a similar fashion as in (G) (n=3, ns: not significant).
Figure 6.

Heme and nucleosomes participate of the immune-mediated thrombotic thrombocytopenic purpura patient’s plasma induced Ca2+-mediated endothelial activation. (A) Free heme concentration (mM) measured in control (n=13), immune-mediated thrombotic thrombocytopenic purpura (iTTP) patients in acute phase (iTTP-AP, n=62) or iTTP patients in remission (iTTP-R, n=14) plasma samples (*P<0.05, ***P<0.001). (B) Von Willebrand Factor (VWF) expression detected by fluorescence microscopy after permeabilized human microvascular endothelial cells from derm (HMVEC-d) incubation with heme or iTTP plasma samples in the absence (A, B) or in the presence (C, D) of hemopexin (+Hx) (original magnification X40, scale bar=30 mm). (C) Endothelin-1 concentrations in the supernatants of HMVEC-d incubated with iTTP plasma samples in the absence (representing 100%) or in the presence of hemopexin (n=14, *P<0.05). (D) Fluorescence intensity (AU) of the Ca2+ flux induction in HMVEC-d after a 20-second incubation with iTTP plasma samples alone or after preincubation with hemopexin (n=18, **P<0.01). (E) Total nucleosomes in control (n=13), iTTP patients in acute phase (iTTP-AP, n=62) or iTTP patients in remission (iTTP-R, n=14) plasma samples (***P<0.001). (F) VWF secretion detected by fluorescence microscopy after incubation of HMVEC-d with iTTP plasma samples in the absence (A) or in the presence of Annexin V (B) (original magnification X40, scale bar=30 mm). (G) Endothelin-1 concentrations in the supernatants of HMVEC-d incubated with iTTP plasma samples in the absence (representing 100%) or in the presence of Annexin V (n=16, **P<0.01). (H) Fluorescence intensity (AU) of the Ca2+ flux induction in HMVEC-d after a 20-second incubation with either iTTP plasma samples alone, or in the presence of Annexin V (n=17, **P<0.01).
In order to investigate the role of complement on iTTP plasma-induced endothelial activation, we performed the same experiments with heated plasma or in the presence of eculizumab. None of these treatments significantly influenced endothelial VWF (Online Supplementary Figure S5A) or endothelin-1 secretion after 1 hour of stimulation (29.45 pg/mL; IQR, 23.61-41.76 and 20.85 pg/mL; IQR, 16.01-27.10 vs. 23.27 pg/mL; IQR, 19.29-30.39, not significant; Online Supplementary Figure S5B), as well as endothelial Ca2+ flux intensity after 20 seconds (111.8 AU; IQR, 74.85-148.8 and 100.2 AU; IQR, 79.43-287.4 vs. 99.41 AU; IQR, 72.49-210.7, not significant; Online Supplementary Figure S5C). Since iTTP plasma is likely to contain thrombin, we reproduced the same experiments in the presence of hirudin and observed no significant modification (187.5 AU; IQR, 82.75-240 vs. 180 AU; IQR, 89.75-262.5, not significant; Online Supplementary Figure S5D). We also used the general serine protease inhibitor PPACK on the iTTP plasma induced VWF release and Ca2+ flux increase and observed no significant difference (6.298 ng/mL; IQR, 5.859-6.924 vs. 6.345 ng/mL; IQR, 6.095-8.615, not significant; Online Supplementary Figure S5E, and 149.3 AU; IQR, 134.2-154.8 vs. 144.4 AU; IQR, 138.5-155.9, not significant; Online Supplementary Figure S5F, respectively).
Discussion
In the present study, we observed for the first time that plasma or serum from iTTP patients in acute phase, but not in remission, were able to induce VWF secretion via WPB exocytosis by a mechanism involving a Ca2+ flux. We ruled out a possible passive adhesion of soluble VWF contained in iTTP plasma on the EC surface, since VWF tethering remained present on the EC membrane even after complete depletion of VWF contained in iTTP plasma and concluded that VWF secretion was an active mechanism likely due to WPB exocytosis. WPB exocytosis was indeed confirmed by the fact that iTTP plasma not only induced VWF and P-selectin membrane exposure, but also en-dothelin-1 and angiopoietin-2 secretion by EC, all components of the WPB. WPB exocytosis is known to involve two different signaling pathways. One is rapid and Ca2+-mediated, whereas the other is linked to adenylate cyclase activation.6 Both the WPB exocytosis and the rapid and sustained intracellular Ca2+ flux induced by iTTP plasma were inhibited by the Ca2+ chelator MAPTAM, demonstrating a Ca2+-mediated signaling pathway. The possible Ca2+ overload of iTTP plasma due to the PEX process was excluded by the observation that the serum of the iTTP-patients had similar effects as plasma and that plasma of non-TTP patients treated with PEX, was not able to activate EC.
Our objective was then to identify the main endothelial activators contained in iTTP plasma/serum and we showed that the IgG fraction purified from iTTP plasma was able to induce a Ca2+ flux and subsequent WPB exocytosis associated with VWF secretion, whereas IgG depletion resulted in an almost 60% reduction of EC activation by iTTP plasma. Noteworthy, was the fact that IgG stimulating effects were reproduced by the F(ab’)2 fractions and were thus linked to a putative antigen recognition on EC. Since iTTP is known to be caused by acquired antibodies directed against ADAMTS13, we investigated the role of these antibodies in EC activation. We did not succeed in directly purifying anti-ADAMTS13 Ab from the iTTP IgG fraction, thus we used different ways to determine whether ADAMTS13 played a role in endothelial activation by purified IgG. First, we compared the ability of serum from iTTP with those of hTTP to induce endothelial activation. Hereditary TTP are due to ADAMTS13 genetic deficiency and do not contain anti-ADAMTS13 auto antibodies. We observed a significant reduction (almost 60%) of endothelial activation using hTTP serum samples. Second, we observed that efficient inhibition of ADAMTS13 expression in HMVEC-d using siRNA was associated with a significant reduction of EC degranulation induced by iTTP IgG fraction. Third, we used two previously described human anti-ADAMTS13 mAb, cloned from iTTP isolated B cells,14 and observed that both of them weakly induced Ca2+ flux in EC as well as VWF and endothelin-1 secretion, confirming that anti-ADAMTS13 IgG were at least in part, responsible for iTTP-induced Ca2+-mediated EC activation. In most of the patients, anti-ADAMTS13 antibodies behave as inhibitors of the ADAMTS13 protease, but in 10-20% of iTTP-patients, these antibodies demonstrate other mechanisms of interaction with ADAMTS13, such as increased clearance,16,17 conformational changes,14,18 or increased metalloprotease activity19 without modifying ADAMTS13 quantification in ELISA.20,21 Our data suggest that in addition to these various interactions, anti-ADAMTS13 antibodies may also induce endothelial activation and VWF exocytosis, the well-known second hit of the disease.8 This observation may appear contradictory with animal models suggesting that anti-ADAMTS13 antibodies do not induce VWF release. Indeed, injection of an inhibiting anti-human ADAMTS13 mAb into baboons did not increase circulating VWF concentrations, but induced histological lesions of TTP, therefore suggesting that at least local VWF occured.13 Also in humans the sole presence of anti-ADAMT13 auto antibodies seems insufficient to induce the disease, which appears frequently related to precipitating events such as pregnancy, surgery or infections.22 These apparently contradictory results may have several explanations. First that our observations in vitro may not have an important significance in vivo. However, despite some heterogeneity, our results show a significant association of Ca2+-induced endothelial activation in vitro with TTP prognosis and survival. Second, that not all anti-ADAMTS13 auto-antibodies have the capacity to induce WPB degranulation or that iTTP IgG may recognize additional endothelial targets to induce this effect. This is consistent with the fact that in our experiments, inhibition of ADAMTS13 expression did not completely suppress the effects of iTTP IgG and that anti-ADAMTS13 mAb weakly activated EC in vitro. Other anti-EC antibodies, as suggested by previous reports in iTTP and other autoimmune diseases, may participate in endothelial activation.23 Moreover, in support of the role of anti-ADAMTS13 in endothelial VWF release, it may be noticeable that during hTTP, even very low circulating concentrations of ADAMTS13 may not be associated with disease bouts, and that serum of hTTP which do not contain anti-ADAMTS13 auto-antibodies did not induce a strong endothelial activation in our experiments.
We also observed high free heme concentrations in iTTP plasma, in the order of magnitude of those observed in sickle cell disease and HUS patients.24,25 Heme-induced VWF exocytosis and participated in iTTP-induced Ca2+-mediated EC activation since VWF exocytosis was decreased by 30% by hemopexin. Free heme binds to TLR4 on EC and has been shown to induce WPB exocytosis.26,27 Interestingly, TLR4, also the receptor for LPS, may mediate Ca2+ flux and endothelial permeability, as shown in lung EC.28 Moreover, hemoglobin resulting from intravascular hemolysis has been shown to inactivate ADAMTS13.29 Furthermore, in accordance with previous reports showing that histones could induced WPB exocytosis,30 we observed elevated concentrations of nucleosomes in iTTP plasma samples which weakly participated in iTTP-induced Ca2+-mediated EC. DNA/histone complexes known to contribute to thrombosis have been reported elevated in the plasma of TMA patients15 and have been shown to induce TTP in a zebrafish model, underscoring their potential importance in iTTP pathogenesis.31 Despite previous evidence showing the amplifying role of complement in endothelial activation,32 our experiments did not show a role for complement in this very initial endothelial activation during iTTP.
To date, except for markers of tissue ischemic injury, no real prognostic factor has been reported in iTTP, although anti-ADAMTS13 antibodies of various isotypes, including IgA, may be associated with more severe forms.33,34 In our study, the amount of in vitro endothelial Ca2+ flux induced by 62 iTTP plasma or serum samples appeared to be associated with disease severity and death, whereas neither free heme, nor nucleosome concentrations correlated with iTTP severity. This result may indicate that several plasmatic components including anti-ADAMTS13 IgG, free heme, nucleosomes and possibly others converge to induce Ca2+-mediated EC activation. We have previously observed in a prospective study that circulating EC counts correlated with disease severity.35 Thus, iTTP early prognosis and death may be related to the intensity of endothelial activation/injury. Moreover, the identification of Ca2+ as the main cellular messenger of iTTP-induced WPB degranulation may possibly help to design a new therapeutic strategy targeting endothelial activation.
Supplementary Material
Acknowledgments
Members of the french ENDO-13 study group include D. Bourdessoule, CHU de Limoges; K. Clabault, CHU de Rouen; C. Daubin, CHU de Caen and G. Choukroun, CHU Amiens, France.
Funding Statement
Funding: This study was supported by research funding from the French ministry of Health (Projet National de Recherche Clinique 2007, #2007/23) to GK.
References
- 1.Kremer Hovinga JA, Coppo P, Lämmle B, Moake JL, Miyata T, Vanhoorelbeke K. Thrombotic thrombocytopenic purpura. Nat Rev Div Primers. 2017;3:17020. [DOI] [PubMed] [Google Scholar]
- 2.Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood. 2017;129(21):2836-2846. [DOI] [PubMed] [Google Scholar]
- 3.Scully M, Cataland S, Coppo P, et al. Consensus on the standardization of terminology in thrombotic thrombocytopenic purpura and related thrombotic microangiopathies. J Thromb Haemost. 2017;15(2):312-322. [DOI] [PubMed] [Google Scholar]
- 4.Tsai HM, Lian EC. Antibodies to von Willebrand factor-cleaving protease in acute thrombotic thrombocytopenic purpura. N Engl J Med. 1998;339(22):1585-1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Furlan M, Robles R, Galbusera M, et al. von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura and the hemolytic-uremic syndrome. N Engl J Med. 1998;339(22):1578-1584. [DOI] [PubMed] [Google Scholar]
- 6.Rondaij MG, Bierings R, Kragt A, van Mourik JA, Voorberg J. Dynamics and plasticity of Weibel-Palade bodies in endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26(5):1002-1007. [DOI] [PubMed] [Google Scholar]
- 7.Banno F, Kokame K, Okuda T, et al. Complete deficiency in ADAMTS13 is prothrombotic, but it alone is not sufficient to cause thrombotic thrombocytopenic purpura. Blood. 2006;107(8):3161-3166. [DOI] [PubMed] [Google Scholar]
- 8.Motto DG, Chauhan AK, Zhu G, et al. Shigatoxin triggers thrombotic thrombocytopenic purpura in genetically susceptible ADAMTS13-deficient mice. J Clin Invest. 2005;115(10):2752-2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nolasco LH, Turner NA, Bernardo A, et al. Hemolytic uremic syndrome-associated Shiga toxins promote endothelial-cell secretion and impair ADAMTS13 cleavage of unusually large von Willebrand factor multimers. Blood. 2005;106(13):4199-4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schiviz A, Wuersch K, Piskernik C, et al. A new mouse model mimicking thrombotic thrombocytopenic purpura: correction of symptoms by recombinant human ADAMTS13. Blood. 2012;119(25):6128-6135. [DOI] [PubMed] [Google Scholar]
- 11.Chauhan AK, Walsh MT, Zhu G, Ginsburg D, Wagner DD, Motto DG. The combined roles of ADAMTS13 and VWF in murine models of TTP, endotoxemia, and thrombosis. Blood 2008;111(7):3452-3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deforche L, Tersteeg C, Roose E, et al. Generation of anti-murine ADAMTS13 antibodies and their application in a mouse model for acquired thrombotic thrombocytopenic purpura. PLoS One. 2016;11(8):e0160388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feys HB, Roodt J, Vandeputte N, et al. Thrombotic thrombocytopenic purpura directly linked with ADAMTS13 inhibition in the baboon (Papio ursinus). Blood. 2010;116(12):2005-2010. [DOI] [PubMed] [Google Scholar]
- 14.Roose E, Vidarsson G, Kangro K, et al. Anti-ADAMTS13 Autoantibodies against cryptic epitopes in immune-mediated thrombotic thrombocytopenic purpura. Thromb Haemost. 2018;118(10):1729-1742. [DOI] [PubMed] [Google Scholar]
- 15.Fuchs TA, Kremer Hovinga JA, Schatzberg D, Wagner DD, Lämmle B. Circulating DNA and myeloperoxidase indicate disease activity in patients with thrombotic microangiopathies. Blood. 2012;120(6):1157-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shelat SG, Smith P, Ai J, Zheng XL. Inhibitory autoantibodies against ADAMTS-13 in patients with thrombotic thrombocytopenic purpura bind ADAMTS-13 protease and may accelerate its clearance in vivo. J Thromb Haemost. 2006;4(8):1707-1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thomas MR, de Groot R, Scully MA, Crawley JTB. Pathogenicity of anti-ADAMTS13 autoantibodies in acquired thrombotic thrombocytopenic purpura. EBioMedicine. 2015;2(8):942-952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roose E, Schelpe A-S, Tellier E, et al. Open ADAMTS13, induced by antibodies, is a biomarker for subclinical immune-mediated thrombotic thrombocytopenic purpura. Blood. 2020;136(3):353-361. [DOI] [PubMed] [Google Scholar]
- 19.Schelpe A-S, Petri A, Roose E, et al. Antibodies that conformationally activate ADAMTS13 allosterically enhance metalloprotease domain function. Blood Adv. 2020;4(6):1072-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dekimpe C, Roose E, Tersteeg C, et al. Anti-ADAMTS13 autoantibodies in immune-mediated thrombotic thrombocytopenic purpura do not hamper ELISA-based quantification of ADAMTS13 antigen. J Thromb Haemost. 2020;18(4):985-990. [DOI] [PubMed] [Google Scholar]
- 21.Dekimpe C, Roose E, Kangro K, et al. Determination of anti-ADAMTS-13 autoantibody titers in ELISA: influence of ADAMTS-13 presentation and autoantibody detection. J Thromb Haemost. 2021;19(9):2248-2255. [DOI] [PubMed] [Google Scholar]
- 22.Morgand M, Buffet M, Busson M, et al. High prevalence of infectious events in thrombotic thrombocytopenic purpura and genetic relationship with toll-like receptor 9 polymorphisms: experience of the French Thrombotic Microangiopathies Reference Center. Transfusion. 2014;54(2):389-397. [DOI] [PubMed] [Google Scholar]
- 23.Praprotnik S, Blank M, Levy Y, et al. Anti-endothelial cell antibodies from patients with thrombotic thrombocytopenic purpura specifically activate small vessel endothelial cells. Int Immunol. 2001;13(2):203-210. [DOI] [PubMed] [Google Scholar]
- 24.Frimat M, Tabarin F, Dimitrov JD, et al. Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome. Blood. 2013;122(2):282-292. [DOI] [PubMed] [Google Scholar]
- 25.Reiter CD, Wang X, Tanus-Santos JE, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med. 2002;8(12):1383-1389. [DOI] [PubMed] [Google Scholar]
- 26.Belcher JD, Chen C, Nguyen J, et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vasoocclusion in murine sickle cell disease. Blood. 2014;123(3):377-390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Figueiredo RT, Fernandez PL, Mourao-Sa DS, et al. Characterization of heme as activator of Toll-like receptor 4. J Biol Chem. 2007;282(28):20221-20229. [DOI] [PubMed] [Google Scholar]
- 28.Tauseef M, Knezevic N, Chava KR, et al. TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxin-induced lung vascular permeability and inflammation. J Exp Med. 2012;209(11):1953-1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Studt J-D, Kremer Hovinga JA, Antoine G, et al. Fatal congenital thrombotic thrombocytopenic purpura with apparent ADAMTS13 inhibitor: in vitro inhibition of ADAMTS13 activity by hemoglobin. Blood. 2005;105(2):542-544. [DOI] [PubMed] [Google Scholar]
- 30.Michels A, Albánez S, Mewburn J, et al. Histones link inflammation and thrombosis through the induction of Weibel-Palade body exocytosis. J Thromb Haemost. 2016;14(11):2274-2286. [DOI] [PubMed] [Google Scholar]
- 31.Zheng L, Abdelgawwad MS, Zhang D, et al. Histone-induced thrombotic thrombocytopenic purpura in adamts13-/- zebrafish depends on von Willebrand factor. Haematologica. 2020;105(4):1107-1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ruiz-Torres MP, Casiraghi F, Galbusera M, et al. Complement activation: the missing link between ADAMTS-13 deficiency and microvascular thrombosis of thrombotic microangiopathies. Thromb Haemost. 2005;93(3):443-452. [DOI] [PubMed] [Google Scholar]
- 33.Ferrari S, Scheiflinger F, Rieger M, et al. Prognostic value of anti-ADAMTS 13 antibody features (Ig isotype, titer, and inhibitory effect) in a cohort of 35 adult French patients undergoing a first episode of thrombotic microangiopathy with undetectable ADAMTS 13 activity. Blood. 2007;109(7):2815-2822. [DOI] [PubMed] [Google Scholar]
- 34.Ferrari S, Mudde GC, Rieger M, Veyradier A, Kremer Hovinga JA, Scheiflinger F. IgG subclass distribution of anti-ADAMTS13 antibodies in patients with acquired thrombotic thrombocytopenic purpura. J Thromb Haemost. 2009;7(10):1703-1710. [DOI] [PubMed] [Google Scholar]
- 35.Widemann A, Pasero C, Arnaud L, et al. Circulating endothelial cells and progenitors as prognostic factors during autoimmune thrombotic thrombocytopenic purpura: results of a prospective multicenter French study. J Thromb Haemost. 2014;12(10):1601-1609. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.