

Abstract
Pulmonary hypertension (PH) is a frequent complication of interstitial lung disease (ILD). Although PH has mostly been described in idiopathic pulmonary fibrosis, it can manifest in association with many other forms of ILD. Associated pathogenetic mechanisms are complex and incompletely understood but there is evidence of disruption of molecular and genetic pathways, with panvascular histopathologic changes, multiple pathophysiologic sequelae, and profound clinical ramifications. While there are some recognized clinical phenotypes such as combined pulmonary fibrosis and emphysema and some possible phenotypes such as connective tissue disease associated with ILD and PH, the identification of further phenotypes of PH in ILD has thus far proven elusive. This statement reviews the current evidence on the pathogenesis, recognized patterns, and useful diagnostic tools to detect phenotypes of PH in ILD. Distinct phenotypes warrant recognition if they are characterized through either a distinct presentation, clinical course, or treatment response. Furthermore, we propose a set of recommendations for future studies that might enable the recognition of new phenotypes.
Keywords: endophenotype, histology, idiopathic pulmonary fibrosis, pathophysiology, pulmonary vascular disease
INTRODUCTION
Pulmonary hypertension (PH) is a common and under‐recognized complication of interstitial lung disease (ILD) that has a great impact on morbidity 1 , 2 and mortality. 3 , 4 , 5 ILD is a heterogeneous group of diseases, 6 and the development of PH in its context can also take many forms; specifically, there are distinct clinical entities under the umbrella of PH associated with ILD (ILD‐PH), with yet more likely to be defined in the future. Crucially, specific phenotypes of ILD‐PH should only be discerned if there are distinct differences in how they present, behave, or respond to treatment (Figure 1). A number of retrospective studies have attempted to define such phenotypes, but a systematic and structured approach has been lacking with most of these descriptions 7 , 8 , 9 which have predominantly been undertaken in idiopathic pulmonary fibrosis (IPF). 10 Therefore, although the association of fibrotic or inflammatory lung diseases with pulmonary vascular abnormalities leads to a high burden of symptoms and poor outcomes, there is still much to elucidate in this broad group of diseases. 5
Figure 1.
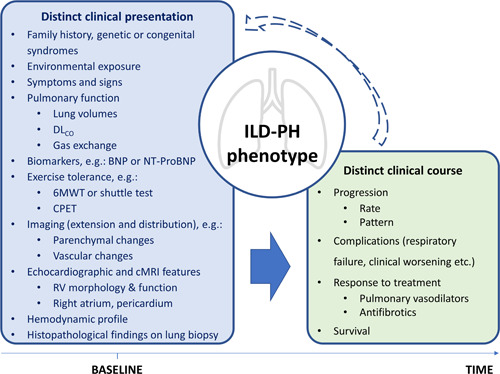
Proposed elements to define phenotypes in PH associated with ILD. These elements are not all necessary to establish a phenotype, but simply constitute traits that might be distinct in phenotypes of ILD‐PH. The arrows indicate how baseline might influence traits observed with progression of disease and vice versa, considering that the diagnosis of PH may be established when both ILD and PH are already advanced in the clinical course. 6MWT, 6‐minute walking test; BNP, brain natriuretic peptide; cMRI, cardiac magnetic resonance imaging; CPET, cardio‐pulmonary exercise test; DLCO, diffusion of carbon monoxide; ILD, interstitial lung disease; NT‐ProBNP, N‐terminal pro‐brain natriuretic peptide; PH, pulmonary hypertension; RV, right ventricle.
The purpose of this work by the IDDI PVRI PH‐Group III workstream is to summarize our current understanding of the pathogenesis and provide evidence and descriptions of different phenotypes in ILD‐PH. It is hoped that this will provide a foundation and framework for descriptions of emerging phenotypes and distinct clinical entities through future observational studies and prospective clinical trials.
PATHOGENESIS
The mechanisms leading to PH and those driving ILD are incompletely understood; how these two processes may combine and to what extent they are linked is an area that requires further research.
In the setting of ILD, PH has traditionally been attributed to fibrotic destruction of the lung parenchyma, leading to loss of pulmonary vascular bed and impaired gas exchange which, due to hypoxic pulmonary vasoconstriction (HPV), contributes to increased pulmonary vascular resistance (PVR). A similar mechanism of parenchymal and blood vessel destruction has historically been invoked for the development of PH in the context of chronic obstructive pulmonary disease (COPD). However, it is now known that some degree of endothelial impairment and arterial remodeling is present in both diseases and this appears to be at least partly independent from hypoxia and destruction of lung parenchyma. Furthermore there is genetic, molecular, and histopathological evidence that pulmonary vascular remodeling is likely caused by different molecular mechanisms in COPD as opposed to IPF. 11 Indeed, even when considering ILD‐PH alone, the pathogenetic mechanisms at play are probably much more complex than previously recognized (Table 1).
Table 1.
Pathogenetic mechanisms of pulmonary hypertension associated with interstitial lung disease.
| Citation | Model | Findings |
|---|---|---|
| Disequilibrium of pro‐ and antiangiogenic pathways | ||
| Ruffenach et al. 12 | Murine model of PH‐PF | ↑ Vascular wall thickness in fibrotic and nonfibrotic areas of PH‐PF patient lungs compared to non‐PH‐PF patients. Role of macrophages and Slug/PIP axis |
| Farkas et al. 25 | Murine model of PF overexpressing TGFβ1 | ↓ Expression of VEGF, increased cell apoptosis, ↓ vascular density. Replacement of VEGF: ↑fibrosis |
| Wu et al. 14 | Human lung tissue and murine bleomycin‐monocrotaline model | ↑ Activation and expression of checkpoint kinases 1/2 in fibroblast and PASMC: proliferative and apoptosis‐resistant endophenotype. Reversible after inhibition |
| Lambers et al. 15 | Murine model of PF | Antifibrotic action of treprostinil. ↓ Recruitment of fibrocyte to sites of vascular remodeling |
| Molecular and genetic abnormalities | ||
| Mura et al. 16 | Human lung tissue: microarray gene expression | NoPH‐PF: proinflammatory gene signature, PH‐PF: pro‐proliferative gene signature |
| Chen et al. 17 | Human lung tissue | ↓ Expression of BMPR2 isoform A in macrophages and PASMC of IPF and IPF‐PH versus normal lung samples. Different miRNA profile |
| Jiang et al. 18 | Murine bleomycin model | ↓ Expression of the BMP9/BMP2/SMAD pathway. Regression of lesions after recombinant BMPR9 |
| Thoré et al. 19 Hernandez‐González et al. 20 | Human PAH families | TBX4 mutation in patients with PAH and ILD, small patella syndrome, and congenital heart disease |
| Eyries et al. 21 | Human PAH families | KDR mutations associated with PAH and ILD |
Abbreviations: BMP2, bone morphogenetic protein 2; BMP9, bone morphogenetic protein 9; BMPR2, bone morphogenetic protein receptor type‐2; ILD, interstitial lung disease; IPF, idiopatic pulmonary fibrosis; IPF‐PH, idiopathic pulmonary fibrosis with pulmonary hypertension; KDR, kinase insert domain receptor gene; NoPH‐PF, pulmonary fibrosis without pulmonary hypertension; PAH, pulmonary arterial hypertension; PASMC, pulmonary arterial smooth muscle cells; PH, pulmonary hypertension; PH‐PF, pulmonary fibrosis with pulmonary hypertension; PIP, prolactin‐induced protein; TBX4, T‐box transcription factor 4; TGF1 β, transforming growth factor β1; VEGF, vascular endothelial growth factor.
Molecular and genetic pathways
Aside from the loss of vessels or HPV, there is increasing evidence of pulmonary vascular disruption including endothelial dysfunction, imbalance between pro‐ and antiangiogenic pathways, and other molecular and genetic abnormalities which may all result in pulmonary vascular remodeling. Recent research into the complex genetic, molecular, and cellular mechanisms underlying pulmonary vasculopathy in ILD might allow the identification of one or more endophenotypes, defined as the linking of genetic and phenotypical traits in a particular subgroup. Potential mechanisms of PH in ILD (Table 1), some important in the pathogenesis of pulmonary arterial hypertension (PAH), are discussed below.
Mura et al. 16 used microarray to compare the gene expression of lung tissue from ILD patients with and without PH, describing specific gene signatures for patients with ILD who did not develop PH (defined by a mean pulmonary artery pressure, mPAP, ≤20 mmHg) and for those who developed severe PH (mPAP ≥40 mmHg). They found that ILD patients without PH mainly had a “proinflammatory” gene signature, while ILD patients with severe PH had a “pro‐proliferative” gene signature. Interestingly, this finding was not confirmed in a study 22 focusing on gene expression from isolated pulmonary distal vasculature, possibly because it included a wide spectrum of PH severity. Another group 12 has shown that in lungs of patients with ILD‐PH, both fibrotic and non‐fibrotic areas exhibited increased thickness of the pulmonary vessel wall, compared with ILD patients without PH. 12 In the same study, which also used a rodent Group 3 PH model of combined bleomycin and monocrotaline inducing both fibrosis and PH respectively, the authors detected increased expression of the transcription factor Slug and its target, the prolactin‐induced protein (PIP); the latter being responsible for inducing pulmonary arterial smooth muscle cell proliferation. 12 Slug was indeed upregulated in macrophages, showing the involvement of cells beyond those strictly pertaining to the lung vascular wall.
Animal models may indeed help understand how the shift from a functionally intact to a diseased pulmonary circulation occurs in ILD patients. 13 , 23 , 24 Farkas et al. 25 used a rodent model of pulmonary fibrosis obtained by overexpressing transforming growth factor β1 (TGFβ1). This model showed decreased expression of vascular endothelial growth factor (VEGF) and of its receptor, leading to endothelial cell apoptosis and lower vascular density. While the replacement of VEGF reduced these phenomena, it also resulted in increased fibrosis. Interestingly, in a bleomycin‐induced mouse model of lung fibrosis, VEGF overexpression attenuated rather than worsened lung fibrosis. 26 This disparate response in different animal models perhaps speaks to the difficulties of identifying the best models to emulate human disease.
Other disruptions of the VEGF pathway, specifically the mutation of the kinase insert domain receptor (KDR) gene, which is a VEGF receptor, have also been described 21 in patients with PAH who also had a low diffusing capacity (DLCO) and mild ILD, thus perhaps providing a link between a specific gene mutation and a clinical phenotype. Intriguingly, family members who were asymptomatic mutation carriers had intermediate reductions in DLCO but no ILD or PAH, while family members without KDR mutation had normal DLCO and normal lung morphology. Thus, it is possible that the development of PH requires a second hit in patients with KDR mutations. In any event, it remains unclear whether the presence of ILD and a severely reduced DLCO associated with KDR mutations results in carriers being predisposed to the development of either PAH or ILD‐PH.
Another study 14 combining human lung tissue from patients with IPF (with and without PH, as evaluated through echocardiography and the size of pulmonary artery on CT scan) and two animal models (bleomycin mouse and bleomycin‐monocrotaline rat models), observed increased DNA damage as well as increased expression and activation of checkpoint kinases 1/2 in lung fibroblasts and pulmonary artery smooth muscle cells, leading to a hyper‐proliferative and apoptosis‐resistant endophenotype. Notably, pharmacological inhibition of the checkpoint kinases 1/2 pathway attenuated both lung fibrosis and pulmonary artery remodeling.
Progress in understanding the genetic architecture of PH is most advanced in heritable and idiopathic PAH. Pathogenic mutations in the bone morphogenetic protein receptor type‐2 (BMPR2) are associated with PAH in families with a history of the condition being found as well as de novo mutations in patients with idiopathic PAH. 27 The expression of BMPR2 is reduced in most forms of clinical and preclinical PH even in the absence of mutations. BMPR2 is a member of the TGF‐β signaling pathway. The current working hypothesis is that reduced BMPR2 activity creates an imbalance in BMP–TGF‐β in favor of TGF‐β, leading to inflammation and angiogenesis. 28 Intriguingly, several studies have linked these genetic anomalies to the development of ILD‐PH. A parallel human‐murine study 17 unveiled reduced expression of BMPR2 in myofibroblasts and pulmonary arterioles from fibrotic lesions of explanted IPF lungs, compared to normal lung samples, as well as increased expression of IL‐6 and phosphorylated STAT3 (p‐STAT3) in the same regions. Both these findings were more prominent in IPF patients with PH than in patients with IPF alone. Furthermore, BMPR2 expression was inversely correlated, and IL‐6 expression directly correlated with mPAP, and microRNAs downregulating BMPR2 were increased differently in IPF patients with and without PH. Blocking IL‐6 and p‐STAT3 reduced expression of those microRNAs, which in the bleomycin murine model allowed expression of BMPR2 and attenuated lung fibrosis and vascular remodeling. Other authors 18 have reported reduced expression of the BMP9/BMP2/SMAD signaling pathway and apoptosis of pulmonary vascular endothelial cells in the same bleomycin murine model, resulting in the development of PH. Interestingly, pulmonary vascular changes were prevented by the administration of recombinant human BMP9 protein. This evidence suggests that restoring the BMPR2‐TGF‐β equilibrium might be an interesting therapeutic strategy in patients with IPF‐PH and possibly in some other forms of ILD‐PH. Importantly, targeting BMP‐TGF‐β signaling by sotatercept (an activin receptor type IIA‐Fc [ActRIIA‐Fc] fusion protein) resulted in a promising reduction in PVR in patients receiving background therapy for PAH, in a Phase 2 study. 28 These findings have since been validated with clinical endpoints in the Phase 3 STELLAR study. 29
Patients bearing mutations of the gene for T‐box transcription factor 4 (TBX4) may develop PAH alongside small patella syndrome (which presents with impaired development of the lower limbs), congenital heart disease and cognitive deficits. The mode of inheritance is autosomal dominant, and PAH typically develops with incomplete penetrance. The disease can manifest either in childhood or in adult life. In two recently published case series, 19 , 20 patients with TBX4 mutations frequently had mild obstructive or restrictive spirometric patterns, DLCO reduction, and pulmonary abnormalities on imaging (emphysema, mosaic distribution, septal lines, micronodules, and ground‐glass opacity), with one patient initially being diagnosed with nonspecific interstitial pneumonia (NSIP) based on the imaging aspect. Some patients manifest combined emphysematous or interstitial abnormalities with plexiform lesions and arterial vasculopathy typical of PAH in the lung tissue.
Finally, recent promising pharmacological data 30 may explain the possible role of the endothelium in triggering epithelial fibrotic changes, based on the direct antifibrotic action of treprostinil, 15 , 31 iloprost, 32 soluble guanylate cyclase modulators, 33 and other vasodilators observed in animal models. However, these findings remain to be further validated in the clinical setting. This might infer that not only does fibrosis drive pulmonary vascular remodeling but also that pulmonary vascular dysfunction might promote the development of fibrosis.
Histopathology
In patients with ILD, histological changes occur at any level of the pulmonary vascular tree, from the main arteries to arterioles, as well as including the capillaries and pulmonary veins. 23 In addition, there are changes involving all layers of the vessel wall thus constituting a “panvasculopathy.” Interestingly, the number of capillaries is increased in non‐fibrotic areas and reduced where fibrosis is more prominent. 34 , 35 This finding is associated with the presence of PH in ILD 36 and has also been observed in systemic sclerosis (SSc)‐related fibrosis with PH. 37 Notably, all the vascular changes, such as hypertrophy and/or hyperplasia of the three arterial wall layers and pulmonary‐bronchial anastomoses, can be found in ILD patients with and without PH but they are generally more pronounced in ILD‐PH patients. 12 , 23 While prominently increased vascular wall thickness may be found in arteries, arterioles, and venules in fibrotic areas, non‐fibrotic areas show a pattern characterized by occlusion of pulmonary venules, muscularization of arterioles, and capillary multiplication 38 (Figure 2). In contrast to these findings, a recent study applied the Heath & Edward's histological grading to pulmonary vascular lesions of explanted end stage fibrotic ILD lungs. The findings included a widespread arterial vasculopathy which was not correlated with the hemodynamic or functional severity of the underlying lung disease 39 ; however, the sites of examination (fibrotic vs. non‐fibrotic areas) were not specified. Interestingly, almost half of the patients had evidence of vascular occlusion with intimal fibrosis and in one‐fifth plexiform lesions were evident; the latter being traditionally considered a hallmark of WHO group 1 PAH pathology. 39 , 40
Figure 2.
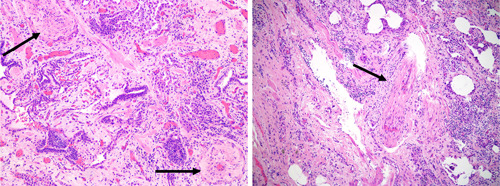
Pulmonary vascular disease histology in interstitial lung disease. On the left panel, a histologic section of explanted lung tissue from a patient with idiopathic pulmonary fibrosis (IPF) who underwent lung transplant, showing marked concentric arterial medial hypertrophy (arrows) (Haematoxylin & Eosin, x100). On the right panel, explanted lung tissue of a patient with IPF without pulmonary hypertension (mean pulmonary artery pressure of 20 mmHg and pulmonary vascular resistance 1.5 wood units, pulmonary arterial wedge pressure 10 mmHg; data obtained 4 months before transplant) but showing signs of venous occlusion. With permission and many thanks to Dr. H. Mani (Inova Fairfax Hospital).
The distinction between progressive vascular rarefaction due to increasing fibrotic disease versus the development of pulmonary vascular changes independent of fibrosis is a key pathogenic factor that is yet to be fully understood. Indeed, while it is commonly held that fibrosis drives the development of vascular changes either directly or indirectly, could the opposite hold true and vascular changes drive the development of fibrosis in a feedback loop?
In summary, the existence of more than one vascular phenotype seems likely given that vascular changes are not necessarily seen in areas of fibrosis and are not related to the severity of the fibrotic lung disease. Furthermore, the sequence of pathogenetic events for interstitial and vascular changes in the lungs is more complex and likely more interlinked than previously thought.
PHENOTYPES OF PH IN ILD
Despite the molecular, genetic, and histological evidence for disruption of the pulmonary vasculature presented above, detection of separate clinical phenotypes has proven more elusive. Whether PH associated with specific forms of ILD should be regarded as distinct phenotypes is open to debate. We have chosen to regard them as such since their pathogenesis and impact from and on the primary disease are likely different. In this section, we summarize a number of entities including combined lung fibrosis and emphysema which constitutes a largely recognized phenotype of ILD‐PH; PH associated with connective tissue and autoimmune diseases, an association that many clinicians consider of special importance; PH in lymphangioleiomyomatosis (LAM); and the largely unrecognized association between post‐tuberculotic lung disease and PH. Finally, we describe tests and tools that might be useful to identify phenotypes of ILD‐PH in future research, as well as previously described subtle patterns and associations which might serve as a blueprint for the identification of future phenotypes.
Recognized and possible phenotypes of ILD‐PH
Combined pulmonary fibrosis and emphysema (CPFE)
CPFE is a tobacco‐related form of ILD characterized by the presence of upper‐lobe emphysema and lower‐lobe fibrosis (most commonly, but not exclusively, IPF), with the minimal extent of emphysema or fibrosis that qualifies for this diagnosis yet to be definitively established. 41 It is a rare syndrome with an unknown prevalence in the general population. However, it has been observed in 0.04%−10% of imaging for lung cancer screening, and is not uncommon among the idiopathic interstitial pneumonias (26%−54%), with a broad range in IPF patients (8%−67%). 41 CPFE is complicated by PH in 30%−50% of cases, 42 which is severe in 68% of patients. 43 There is often relative preservation of airflow and lung volumes as measured by pulmonary function tests, but arterial oxygen and DLCO are severely reduced. 10 There tends to be an inverse correlation between the extent of fibrosis in lung imaging and the DLCO. 44 The presence of severe PH and an FVC < 50% are independent predictors of survival, regardless of the presence of emphysema. 45 Compared to patients with IPF 46 or COPD 47 alone, the severity of PH is increased in CPFE patients, but the likelihood of PH is not increased in CPFE patients compared to IPF patients with a matched extent of disease. 48
There is increasing recognition that many clinical entities exist within the umbrella term of CPFE, including distinct fibrosing ILDs (UIP, fibrotic NSIP, smoking‐related interstitial fibrosis, and desquamative interstitial pneumonia) and varying emphysema types (e.g., “thick‐walled, large cysts variant,” bullae etc.) including their extent and distribution. 41 It is unknown whether these different presentations have an influence on the development or severity of PH; in other words, whether they constitute distinct phenotypes within the broader phenotype of PH associated with CPFE.
Connective tissue and autoimmune disease associated with ILD
PH in the context of autoimmune disease associated with pulmonary fibrosis is a complex clinical entity where an aggressive form of PH 49 , 50 combines with a severe complication of autoimmune disease, that is, pulmonary fibrosis. PH frequently complicates SSc and mixed connective tissue disease and is classified under Group 1 PAH; the most common type of SSc complicated by Group 1 PAH is limited SSc. If SSc, more commonly in the diffuse form, presents with significant pulmonary interstitial involvement, some may categorize it as Group 3 PH. PH can also manifest in interstitial pneumonia with autoimmune features, 51 where a form of ILD is associated with some features but not the full criteria for an autoimmune disease. In patients with SSc presenting with ILD and PH, there is no association between the extent of ILD on imaging and the severity of pulmonary hemodynamics. 52 In one study, patients with pulmonary fibrosis and PH associated with an autoimmune disease had increased survival after the administration of PAH‐targeted therapy, compared with ILD‐PH patients without autoimmune disease. 53 On the contrary, no apparent physiologic differences in response to pulmonary vasodilator treatment were found comparing SSc‐ILD with PH patients and SSc‐PAH 54 patients. This latter study also found that patients with SSc‐ILD associated with PH had worse survival than SSc‐PAH patients, albeit they have better survival than IPF patients with associated PH. 55
The clinical features of PH in the context of autoimmune conditions are influenced by a triad of intertwined pathogenic events in the lung tissue: autoimmunity, fibrosis and/or proliferation, and vasculopathy (Figure 3). The three phenomena are associated with varying degrees of inflammation. Even though ILD‐PH associated with autoimmune disease is not an established phenotype, the argument for differentiating it hinges on the difference in its clinical behavior and prognosis 55 compared to ILD‐PH without autoimmune disease. 53
Figure 3.
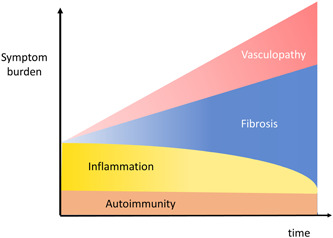
Autoimmune disease, lung fibrosis, and vasculopathy. Autoimmune diseases share with lung fibrosis and pulmonary vascular diseases a variable component of inflammation which, in these entities, plays an important role both in pathogenesis and in symptoms burden. As the disease progresses, fibrosis and pulmonary vasculopathy may affect symptoms more prominently.
Post‐tuberculosis PH
It is estimated that there were 155 million survivors of tuberculosis alive in 2020, many of whom will meet the criteria for post‐tuberculosis lung disease (PTBLD). 56 PTBLD is currently defined as “Evidence of chronic respiratory abnormality, with or without symptoms, attributable at least in part to previous tuberculosis.” 57 Globally, residual respiratory impairment is estimated to occur in one‐fifth of tuberculosis survivors and is more common in low‐income countries. 58 PTBLD may involve the large and small airways as well as parenchyma, and present with either obstructive or restrictive physiology. Chest imaging may demonstrate a spectrum of findings including parenchymal fibrosis, cavities, and destruction, as well as residual pleural disease. 59 PH has been well described after tuberculosis, 60 however the true prevalence is not known, primarily due to lack of access to both echocardiography and right heart catheterizations in settings where PTBLD is most prevalent. 61 One Chinese Registry of 693 patients with Group 3 PH found previous tuberculosis to account for 38.8% of cases. 62 Data also suggests that compared to other causes of Group 3 PH, post‐tuberculosis PH patients may have both higher 1‐year mortality (31.6% vs. 10.0%; p = 0.059) and more frequent readmission rates (78.9% vs. 43.8%; p = 0.009). 63
The mechanisms of post‐tuberculosis PH are not clear, neither is it clear whether it should be considered in Group 3 or Group 5, analogous to other granulomatous diseases such as sarcoidosis. Like other forms of ILD‐PH, the correlation of raised pulmonary artery pressures with extent of radiological parenchymal destruction is not robust. 64 Further, there is evidence of an association between chronic thromboembolic PH and previous tuberculosis, even in settings with relatively low incidence of tuberculosis, 65 highlighting a potentially more complex pathogenesis. Since most post‐tuberculosis PH occurs in low‐ and middle‐income countries, advocacy will be needed to determine its burden, mechanisms, and optimal management. The void of information about post‐tuberculosis PH underscores the huge unmet need with much work needed and strongly encouraged in this area.
Lymphangioleiomyomatosis (LAM)
LAM is a cystic lung disease affecting female patients, where the disruption of the lung parenchyma is caused by the proliferation of smooth muscle cells (called LAM cells) along the lymphatic network of the thorax and abdomen. Among other complications such as pneumothorax, chylous effusions, and benign renal tumors, PH may develop and is classified within Group 3 PH. The prevalence of PH in LAM varies widely from around 8% in general cohorts 66 , 67 , 68 to as high as 45% in pretransplant patients. 69 PH is severe in approximately 20% of cases 68 and is associated with worse dyspnea and exercise tolerance, 66 right heart failure, and haemoptysis. 68 Vascular remodeling and infiltration by LAM cells in the pulmonary artery walls has been described in a small series 68 and is among the possible pathophysiologic mechanisms, together with airflow obstruction and hypoxia. LAM patients with PH have a lower mean FEV1 and DLCO than those who do not develop PH, indicating that PH intervenes at a more advanced stage of the lung disease. The interval between LAM and PH diagnosis is about 9 years on average. Furthermore, within the PH group there is a direct correlation between mPAP or PVR and FEV1, DLCO, and KCO, suggesting that once PH arises, its progression might develop in parallel to the underlying lung disease. The value of mPAP also correlates with the PA/aorta ratio and PA diameter. 70
Clinical tools and tests to identify phenotypes
Symptoms and signs
There is little research describing the clinical presentation of ILD‐PH or change in symptoms heralding the onset of PH in patients with ILD; most data on symptoms and signs emanates from retrospective cohorts. One study 71 found that shortness of breath, fatigue, and swelling were the most frequently reported symptoms, while cough was mentioned when patients were prompted.
From a clinical standpoint, experts concur that the appearance of symptoms and signs of right heart failure should prompt screening for PH in ILD patients. 72 However, by this time patients typically have more severe hemodynamic impairment. Therefore more subtle clues, like change in severity or quality of dyspnea, 5 may help raise the index of suspicion for PH in patients who have still not reached this late stage of vascular disease. An increase in oxygen requirements, especially in combination with stable lung volumes, 73 might also alert the physician to intervening pulmonary vascular disease.
It is unknown whether distinct symptoms and signs are more characteristic of any specific phenotype of ILD‐PH, although it is somewhat doubtful that different fibrotic or hemodynamic phenotypes will manifest with a distinct clinical picture. Symptoms have a strong impact on quality of life and on the physical and emotional status of the patient and should therefore play a prominent role in the selection of outcome measures in future trials of PH‐ILD.
Respiratory function
Fibrosis and hemodynamic status exist in a complex interplay. In IPF, the prevalence of PH appears to increase with increasing severity of the IPF; specifically, it has been described in 15% with mild to moderate restriction but increasing to 84% in those with more advanced disease. 3 , 74 , 75 However, the severity of the hemodynamic impairment does not seem to be related to the severity of the underlying fibrosis. 76 This is also confirmed by studies failing to detect a strong correlation between pulmonary function and severity of PH. In a classic study, 7 a retrospective analysis of IPF patients showed a poor correlation between FVC, DLCO, FVC/DLCO, and PH severity. 7 The best predictors of the presence of PH, albeit with low sensitivity and specificity, were DLCO <30% and FVC/DLCO ratio >1.5. Several scores to predict mPAP from pulmonary function tests, 77 sometimes combined with other noninvasive parameters 78 , 79 have been developed; however, these tools still require further external validation in larger cohorts. 80 Data derived from the ARTEMIS‐IPF trial, 74 investigating ambrisentan in mild‐moderate IPF (FVC ≥50% and <5% honeycombing), found that of the 14% patients with PH one‐third had a mPAP ≥35 mmHg, a commonly held threshold for severe PH in lung disease. 10 Data mining of registries have also tried to shed some light on the relationship between lung function and pulmonary hemodynamics. In one such analysis of pretransplant IPF patients, 75 46% had PH, of whom 37% had mPAP <40 mmHg and 9% had mPAP ≥40 mmHg. However, FVC values for those with no PH, moderate PH, and severe PH were very similar (50%, 48%, and 51% of predicted, respectively). 75
The use of inhaled pulmonary vasodilator treatment in ILD‐PH 81 , 82 , 83 highlights the relevance of VA/Q relationships. 84 It is known from classical studies in IPF that VA/Q mismatching accounts for 80% of hypoxemia. 85 While there are theoretic reasons why VA/Q relationships might be worsened with systemically administered vasodilators, this has not borne out as an issue in the few large clinical trials to date. However, it is unknown whether there are specific phenotypes of ILD‐PH patients which due to their pathophysiology develop more VA/Q mismatch than others. If that was the case, these phenotypes would find greater benefit from an inhaled formulation, which should theoretically enable more blood flow to better ventilated areas, 86 as opposed to a systemic vasodilator which may worsen VA/Q mismatch.
The effect of PH on lung volumes is not known; while PAH patients often present with mild restrictive physiology, it is unclear if the presence of PH per se influences lung volumes to any extent in the context of ILD. However, it is notable that there is a very poor correlation between pulmonary pressures and the FVC in patients with ILD. It is possible that evaluating the hemodynamic profile of patients at the time of ILD diagnosis might shed some light on the role of the pulmonary vasculature in the restrictive physiology, while serial hemodynamic assessments would provide even more insight, as demonstrated in small series of IPF patients. 87
Exercise tolerance
Patients with ILD typically experience worsening exercise tolerance and when pulmonary vasculopathy occurs, it impairs exercise even further. In one study of IPF patients awaiting lung transplant, 75 as many as one‐fifth of patients with severe PH (mPAP >40 mmHg) had six‐minute walking test (6MWT) distances of less than 45 m, compared to 14% in the mild‐moderate PH group (mPAP >25 mmHg) and 10% in the group without PH. Furthermore, a persistently high heart rate 1 min into recovery after a 6MWT has prognostic implications in IPF and is somewhat predictive of PH, albeit with low sensitivity (52%) and moderate specificity (74%). 88 Interestingly, in one study 55 of the 6MWT conducted in ILD patients with or without PH, the final oxygen saturation <88% was predictive of worse outcome, while distance was not. In another study of ILD patients with and without PH, the PVR was able to independently predict the distance walked in the 6MWT. 89 The difference in distance walked during the 6MWT might be even more relevant in severe PH (mPAP >35 mmHg), 90 in comparison to patients without PH or those with mild PH. It is very likely that the impairment of exercise tolerance in ILD‐PH patients is multifactorial being that the parenchymal lung disease, the pulmonary vascular dysfunction, comorbidities, and deconditioning all may play varying roles. Larger, ideally prospective studies might be necessary in ILD‐PH to better determine the exact role of all the variables available through the 6MWT in predicting outcomes in these patients. The variables include not only the distance, but also oxygen needs, desaturation nadir, heart rate recovery, and the Borg dyspnea score.
In IPF, ventilatory efficiency during exercise, expressed by VE/VCO2, has been shown 91 , 92 , 93 as the best predictor of PH during cardiopulmonary exercise testing (CPET). This parameter together with the peak oxygen consumption were able to discriminate 94 abnormal response to exercise in ILD patients, which might be a hallmark of early pulmonary vasculopathy. Two small studies by the same group 95 , 96 found that patients with severe PH (mPAP ≥40 mmHg) had lower peak end‐tidal carbon dioxide pressure and higher peak VE/VCO2 on exercise, compared to patients without PH and moderate PH. However, the use of CPET is challenging in larger, late stage studies. Furthermore, in clinical practice the use of CPET is commonly limited to patients not requiring high oxygen supplementation, making the findings less applicable to more hypoxemic patients.
Imaging
Echocardiography remains the cornerstone of screening in all PH Groups. In addition to further risk stratifying for the presence of PH, 97 it allows for an assessment of the possible severity, as well as being an important tool to exclude group 2 PH through the evaluation of the left heart and by ruling out significant valvular disease. 98 Despite its usefulness as a screening and follow‐up tool, echocardiography is burdened by interobserver variability and inaccuracy of measurements of the right ventricular (RV) systolic pressure, potentially leading to the misdiagnosis of PH in patients with lung disease in the absence of a confirmatory right heart catheterization (RHC). 68 , 99 In the latest ESC/ERS guidelines, the ratio of tricuspid annular plane systolic excursion/systolic pulmonary artery pressure, an indirect measure of RV‐pulmonary vascular coupling, has emerged as a useful tool for risk stratification in PAH at baseline. 98 If there is uncoupling in ILD‐PH, this is likely a measure of severity rather than a distinct phenotype. 100 However, it is unknown whether certain phenotypes of ILD‐PH might present more typically with uncoupling or any other features of altered RV function. Indeed, there is no evidence that a particular echocardiographic profile is associated with a specific phenotype of ILD‐PH, although this question has not been specifically investigated.
Experience with cardiac magnetic resonance imaging (cMRI) in ILD‐PH is limited, but two recent studies in ILD cohorts mostly comprised of UIP and fibrotic NSIP patients have provided interesting clues into the features of RV function observed through cMRI. In comparison to healthy controls, both ILD patients with and without PH have increased RV end‐systolic and end‐diastolic indices and reduced RV ejection fractions, with ILD‐PH patients having the lowest values. 101 Unsurprisingly, it was shown that depressed RV function was associated with poorer survival. Furthermore, compared to controls, RV longitudinal strain is progressively impaired in ILD patients without PH, and in ILD‐PH patients compared to those without PH. Differences in RV strain have been shown to have implications for short term outcomes. 102 These data suggests that progressive RV impairment commences even before the development of hemodynamically measured PH and continues to overt RV failure with the establishment of PH. Whether this characteristic may be more prevalent or associated specifically with certain subtypes of ILD, such as UIP and fibrotic NSIP, remains to be investigated.
Screening for PH with CT scans has attractive appeal as it is a cornerstone test in the diagnosis and follow‐up of patients with ILD. Similarly, CT scans can raise suspicion for PH or RV failure in patients with other chronic lung diseases, such as COPD 103 and cystic fibrosis. 104
The quest to identify a simple marker of PH on CT imaging of ILD patients has been elusive. Two studies report pulmonary artery size to be significantly greater in patients with ILD‐PH compared to those without PH. 9 , 89 Another report 105 described pulmonary artery enlargement in CT scans of pulmonary fibrosis patients both with and without PH, deeming this to be an unreliable sign. Indeed, multiple studies have shown a poor correlation between PA measurement per se and PH in the setting of known pulmonary fibrosis. 106 Zisman et al. found no relationship between fibrosis and mPAP on thoracic CT scans of advanced IPF patients, 8 and the same was observed in a mixed cohort of fibrosing idiopathic pneumonias. 89 Nonetheless, an increased PA/aorta ratio, together with other signs of increased pressure in the right ventricle, has been deemed useful in triggering the suspicion of PH in ILD patients based on expert consensus. 72 The incidental finding of an enlarged PA on CT scan has been suggested as a trigger for screening of PH in ILD patients in a recent statement from the Fleischner Society. 107
Despite these prior studies, further research looking for the correlation between imaging and hemodynamic profiles should be undertaken in each specific ILD subgroup. This especially appears to be the case in complex entities such as CPFE. Indeed, a post‐hoc analysis of the RISE‐IIP study, in which riociguat was shown to be harmful in patients with PH associated with idiopathic interstitial pneumonias, 108 demonstrated that the deleterious signal appeared to emanate from those patients with CPFE, and especially from those in whom emphysema predominated. The lesson from this is that clinical trials should consider CT scan imaging at inclusion to control for potential confounding factors. In addition, this highlights the importance of the type and amount of disease burden in the outcome of randomized controlled trials in ILD‐PH. The advent of imaging software that enables better characterization and objective scoring of the parenchymal abnormalities as well as pulmonary vascular volume quantification 109 may shed light on specific morphology phenotypes within the spectrum of ILD‐PH. Artificial intelligence (AI) and in particular machine learning might help the identification of previously unrecognized patterns. 110
While most data about ILD‐PH has emerged from studies of IPF cohorts, the implications of PH complicating other diseases may be equally relevant. In the case of NSIP, the link between the disease and PH is confounded by the high proportion of NSIP cases in patients with underlying connective tissue disease. However, in idiopathic NSIP there is evidence that PH may be present in about one‐third of patients. 111 In patients with chronic hypersensitivity pneumonitis (chronic HP), precapillary PH may also be quite common (44%) 112 and similarly appears to be associated with a worse prognosis. 113 Furthermore, one study of chronic HP patients demonstrated that an increased PA/aorta ratio on CT is associated with worse survival. 114
Hemodynamic profiles
The 6th World Symposium on Pulmonary Hypertension proceedings recommended RHC in ILD patients being evaluated for lung transplantation as well as referral to a PH expert center in cases where there is suspicion of severe PH. In the latter case, enrollment of the patient in a clinical trial, or individualized off‐label treatment might be considered. The 2022 ESC/ERS guidelines 98 have further suggested that RHC be considered when the results might help phenotyping, influence management or when PAH or chronic thromboembolic PH is suspected.
Once it develops, the propensity is for PH in ILD to worsen over time with estimates of mPAP increase in IPF varying widely, from between 1.8 mmHg per year 115 to 3.8 mmHg per month. 87 Any level of PH is associated with reduced survival. 1 , 116 , 117 , 118 In fact, levels of mPAP below the current cut off definition of PH are also associated with reduced exercise capacity and poorer outcomes, 3 , 119 indicating that there is a continuous relationship between an increasing mPAP and mortality. 1 Recently, it has been suggested that the threshold for the diagnosis for PH be lowered to mPAP >20 mmHg with a PVR ≥2 WU, 98 which likely will lead to capturing patients at an earlier stage of pulmonary vascular disease 120 ; this new threshold also has strong physiological basis. 121 , 122 It is unclear whether patients meeting the mPAP threshold of >20 mmHg with and without increased PVR present different phenotypes; however, it is likely that an increased PVR may also be able to discriminate ILD patients with a true vasculopathy. The question is not merely academic, as the identification of a PVR threshold may facilitate the characterization of a hemodynamic profile at major risk of worse outcomes. 123 , 124 In a recent paper, 124 describing a large ILD‐PH cohort from the COMPERA Registry, mPAP did not discriminate worse prognosis while PVR >4 WU was associated with worse survival and >8 WU had the highest discriminatory power. However, there are caveats to this analysis; first, this population was not proportionately representative of less severe cases (median PVR was 7.6 WU, range 6.0−10.6); more than a quarter of patients had CPFE; it is unclear how many patients had documented CT imaging and all patients received vasodilator treatment. Based mostly on this study, the 2022 ESC/ERS PH guidelines suggested using a PVR cut‐off >5 wood units to define severe PH, with the added benefit of homogenizing the severity threshold of Group 3 PH with that of Group 2 PH.
Another group which might represent a distinct phenotype includes those IPF patients with mPAP ≤21 mmHg but with PVR ≥3 WU: in the United Network for Organ Sharing database, they represented 3.6% of IPF patients listed for lung transplantation. Although this group does not meet the current definition of PH, patients had worse survival compared to the IPF group without PH. 120 It is worth noting that comorbidities are common in ILD 75 , 125 , 126 and may be associated or contribute to an increased PVR. Postcapillary PH is found in 5%−20% of patients 7 , 74 , 127 and may coexist with precapillary PH, arguably constituting a different phenotype and possibly having an impact on pulmonary vasodilators efficacy and tolerability. These issues also have important implications for clinical trial design, since the most recent definition of ILD‐PH was not employed in the few clinical trials to date. 81 , 82 , 83
Just as the significance of various hemodynamic groups remains uncertain, the broader question of why some patients with mild IPF develop severe PH 74 and other patients with severe fibrosis never do is yet to be answered. In addition, while the hemodynamic threshold for PH as a predictor for poor survival may be low, the cause of death in ILD‐PH populations remain poorly studied.
DISCUSSION
The first question to address is: what constitutes a phenotype of PH in ILD? There is not one straight answer to this problem. From a practical point of view, one might argue that a phenotype is any distinct entity that has a somewhat unique clinical course, regardless of the underlying ILD or hemodynamic compromise. A recent example of this phenotyping approach is the definition of “progressive pulmonary fibrosis” 128 as an umbrella term for all the fibrotic ILDs that have the propensity for a progressive course. 129 On the other hand, a PH‐COPD pulmonary vascular phenotype 10 , 130 has previously been described 131 , 132 where the common traits are baseline characteristics and not the subsequent, often dismal clinical course. These characteristics include relatively mild airflow obstruction, a severely reduced DLCO, severe hypoxemia, and associated severe PH. Phenotypes may also be defined by a differential response to therapy. These approaches, specifically the definition of a phenotype according to characteristics at diagnosis, clinical course, or treatment response, may also not be mutually exclusive.
In the case of ILD‐PH the disconnect between respiratory function tests, imaging characteristics and hemodynamic profiles suggests that the PH is driven to an extent by factor(s) beyond just the burden or severity of the fibrosis. There could be, for example, a genetic predisposition involving different gene mutations or epigenetics changes, influencing aberrant pathways that contribute to the development of PH. The intrinsic characteristics of the specific ILD in question (IPF, NSIP, or other) may also play a role, as may environmental and even social factors. Cigarette smoking, a common risk factor to some forms of ILD (IPF, 133 CPFE, 41 PLCH, 134 smoking‐related pulmonary fibrosis etc.) has been implicated in vascular remodeling and reduced NO production at the endothelial level, leading to endothelial dysfunction 135 ; but tobacco use might be just one of many intervening environmental agents alongside air pollution, 136 , 137 exposure to chemicals 138 and others. These environmental factors may be further compounded by social and economic factors: higher exposure of low‐income households to chemical substances and air pollution, reduced availability of healthy lifestyle choices such as exercise and salubrious diet, reduced access to medical care among others. All these could contribute to the clinical presentation and subsequent course. Indeed, a complex interaction of all these elements may ultimately be responsible in determining the phenotypic expression of PH in the context of ILD (Figure 4).
Figure 4.
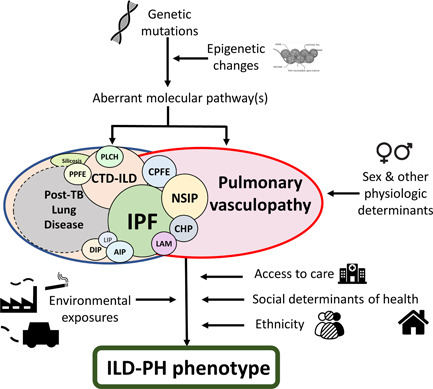
Mechanisms involved in the development of ILD and PH. This figure illustrates some of the potential mechanisms that may lead to the development of PH and ILD in its different forms. The disparate nature of the factors implicated, from prebirth development to exposures throughout life may in part explain the variability in clinical presentation and possibly the existence of different phenotypes. AIP, acute interstitial pneumonia; CHP, chronic hypersensitivity pneumonitis; CPFE, combined pulmonary fibrosis and emphysema; CTD‐ILD, connective tissue disease associated‐ILD; DIP, desquamative interstitial pneumonia; ILD, interstitial lung disease; IPF, idiopathic pulmonary fibrosis; LAM, lymphangioleiomyomatosis; LIP, lymphocytic interstitial pneumonia; NSIP, nonspecific interstitial pneumonia; PH, pulmonary hypertension; PPFE, pleuroparenchymal fibro‐elastosis; TB, tuberculosis.
The nature, evolution, and interaction of these distinct processes may determine the subsequent impact on patients and their outcomes. Indeed, the vascular derangements superimposed on the fibrotic disease process might become the predominant determinant of the patients course. 139 Thus, parenchymal and vascular changes may not necessarily progress in a predictable and colinear fashion; in patients with a profibrotic predisposition there might be more fibrosis progression, while in those with a more “pro‐angiogenetic” genotype 16 the clinical picture of PH might predominate. A recent registry publication 140 has described a group of idiopathic PAH patients with minimal fibrosis on CT scan and no impairment of lung volumes, but very low DLCO and a positive smoking history, presenting very similar outcomes to patients with Group 3 PH. The presence of PH and ILD in this group of patients reflects a “pro‐angiogenetic” phenotype of ILD‐PH that likely exists on a continuum between Group 1 and Group 3 PH. Thus, fibrosis and vasculopathy may exist across a multidimensional spectrum (Figure 5).
Figure 5.
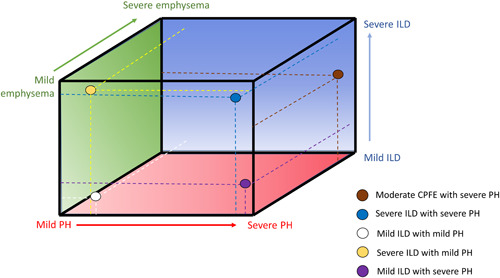
The tri‐dimensional spectrum of ILD, emphysema, and pulmonary vascular disease. The illustration shows the continuum of pathologic changes in the lung which may variably present in ILD‐PH patients. Colored dots are examples of different clinical situations with varying degrees of pathological alterations in the alveolar, interstitial, and vascular compartments and dashed lines indicate where they lay on the tri‐dimensional spectrum. CPFE, combined pulmonary fibrosis and emphysema; ILD, interstitial lung disease; PH, pulmonary hypertension.
The inextricable link between the vasculature and fibrosis in ILD has potential therapeutic implications, as highlighted by the somewhat surreptitious finding of an improved FVC (compared to placebo) in patients treated with sildenafil 141 or inhaled treprostinil 30 in independent double‐blind randomized controlled studies.
One key issue in unraveling the interplay between fibrosis and vascular derangement in ILD patients is the fact that vascular remodeling may precede the development of clinically measurable PH. 39 The implications of pre‐PH vasculopathy are as uncertain as their progression to PH. While the development of PH has a profound impact on outcomes, 120 , 139 it is conceivable that the lung vasculopathy may be a treatment target even before the hemodynamic threshold for PH is reached, as shown by loss of vascular homeostasis even in IPF patients without PH. 17 Given the continuum of the pulmonary vasculopathy, whatever the definition of PH, this is a somewhat arbitrary threshold that might include patients who do not warrant therapy and exclude others who might. Should we disconnect the concept of a distinct hemodynamic threshold with that of treatment in ILD‐PH, favouring instead the notion of a “ILD vasculopathy”? The late stages of the disease course in which PH is usually diagnosed in ILD and the associated poor prognosis, as well as the muted response in those who access treatment later in the disease, 142 support efforts aimed at the earlier detection of a vasculopathy. In this scenario, the lowering of the threshold for the definition of PH in the new ESC/ERS guidelines 98 should be helpful, as is the reintroduction and new definition of exercise PH, which might foster studies earlier in the course of ILD. Until then, the earlier treatment of a vasculopathy in the context of ILD remains speculative and requires further prospective studying. A key point is identifying ILD populations (and subpopulations) that future studies should consider. First and foremost, it is paramount that study populations are identified and described comprehensively and consistently, hence the importance of defining distinct phenotypes. Based on the existing studies, prior shifting definitions have made conclusions sometimes difficult to draw from pooled studies. 41 Thus far most of the previous research in ILD‐PH has focused on IPF and there is very little data on the phenotypical differences of PH in the context of other progressive fibrosing and inflammatory forms of ILD. Previous studies of PH in ARDS and other acute lung inflammatory conditions might provide a roadmap for comparison of inflammatory and fibrosing ILD phenotypes which might shed light onto the relative roles of inflammation and fibrosis on the pulmonary vasculature.
SUMMARY OF RECOMMENDATIONS
The role of registries and consortia. Because of the low prevalence of each diagnostic group within ILD‐PH, data sharing across institutions and countries is integral to providing sufficiently large cohorts to best discern phenotypes. For this reason, the creation of consortia, national and international registries, and other data repositories is strongly encouraged. To achieve this, great efforts need to be made to facilitate the exchange of data, within the appropriate legal framework and ultimately always protecting patients' right to privacy. Similarly, future clinical trials should build in provisions to allow access to patients' data (including CT scan images) beyond the scope of a single study, to maximize the benefit of information that may only be fully realized in combination with other data sets.
Globalization: Greater efforts, including grant proposals and pharmaceutical support to better define and hopefully provide treatments for common, yet underserved phenotypes that are most prevalent in low‐ and middle‐income countries (such as post‐TB PH), are strongly encouraged.
Which analyses should be applied to discern phenotypes? An unbiased approach to determining phenotypes and endophenotypes may be achieved with machine learning and AI in general: practical applications of this approach include the study of genetics, epigenetics, proteomics, metabolomics, and imaging repositories. This may enable cluster analyses to provide evidence of clinical phenotypes, such as it has been applied to PAH, 143 , 144 especially with regard to survival, risk profiling and therapy.
Which specific imaging techniques are advisable? Imaging techniques should include traditional semiquantitative and quantitative measures of the parenchymal lung disease with efforts to better characterize patterns and geographical distribution. Cardiac volumetrics and 4D flow data as well as studies of functional imaging are also needed. Imaging‐based AI creating algorithms through CT and MRI data might better predict which patients have PH. These should ideally be compared with RHC, as well as evaluated for their prognostic accuracy and therapeutic decision making in ILD‐PH. Moreover, for both traditional semiquantitative as well as AI‐based research, multi‐institutional and international data are strongly encouraged to mitigate bias; therefore, the building of international collaborations and consortia is again encouraged to allow for the sharing of data and images.
AUTHOR CONTRIBUTIONS
Lucilla Piccari was involved in the conception and design of the manuscript, conducted the searches and data extraction as well as wrote the first draft of the manuscript. All authors analyzed and interpreted the data, revised the manuscript critically for important intellectual content, approved the final manuscript, and agreed to be accountable for its overall content.
CONFLICTS OF INTEREST STATEMENT
Dr. Lucilla Piccari has received research funding from and served as a speaker for Janssen and Ferrer, advised Janssen, Ferrer and United Therapeutics as well as received support for attending congresses from Janssen, MSD and Ferrer, all of which not related to this manuscript. Prof Katerina Antoniou has a consultant role for Roche, Boehringer‐Ingelheim, GSK, honoraria for lecturing for Roche, Boehringer‐Ingelheim, GSK, Astra‐Zeneca, Chiesi & Menarini. Dr. Paul M. Hassoun serves on a scientific advisory board for Merck, an activity unrelated to the current work. Dr. Sylvia M. Nikkho is an employee of Bayer AG. Dr. Rajan Saggar has a Consulting and Advisory Role for United Therapeutics, Third Pole, Novartis, Acceleron, Aerovate, and Janssen. Dr. Oksana A. Shlobin has consulted for UT, Bayer, Altavant, Aerovate, Jenssen&Jenssen and Merck, and is on the speaker bureau for UT, Bayer, and JJ. Dr. Steven D. Nathan is a consultant for United Therapeutics, Bellerophon, Third Pole, Roche, Boehringer‐Ingelheim, Merck and Daewoong. Dr. Stephen John Wort received honoraria from Janssen, MSD, Bayer and Acceleron for advisory boards; received honoraria from Janssen for educational activity, received unrestricted research grants from Janssen and Bayer, and travel grants, conference registration, and accommodation from Actelion and GSK. The remaining authors declare no conflict of interest.
ETHICS STATEMENT
Not applicable.
ACKNOWLEDGMENTS
The authors wish to thank Dr. Haresh Mani (Pathologist at Inova Fairfax Hospital, US) for providing histologic images of patients with interstitial lung disease. Special thanks to all the members of the Innovative Drug Development Initiative Group 3 Pulmonary Hypertension workstream for their dedication and commitment to the work of the group: Steven H. Abman, Peter Fernandes, Howard M. Lazarus, Horst Olschewski, Mitchell Psotka, Manuel J. Richter, Eric Shen, Norman Stockbridge, and Carmine Dario Vizza. Finally, thank you to the IDDI Leads: Paul Corris, Raymond Benza, Mark Toshner and to the Pulmonary Vascular Research Institute for supporting innovative research in pulmonary vascular disease. All authors agree to be accountable for the overall content of the manuscript.
Piccari L, Allwood B, Antoniou K, Chung JH, Hassoun PM, Nikkho SM, Saggar R, Shlobin OA, Vitulo P, Nathan SD, Wort SJ. Pathogenesis, clinical features, and phenotypes of pulmonary hypertension associated with interstitial lung disease: a consensus statement from the Pulmonary Vascular Research Institute's Innovative Drug Development Initiative ‐ Group 3 Pulmonary Hypertension. Pulm Circ. 2023;13:e12213. 10.1002/pul2.12213
Steven D. Nathan and Stephen John Wort contributed equally to this work, therefore they are both last author.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable as no new data generated.
REFERENCES
- 1. Hayes D, Black SM, Tobias JD, Kirkby S, Mansour HM, Whitson BA. Influence of pulmonary hypertension on patients with idiopathic pulmonary fibrosis awaiting lung transplantation. Ann Thorac Surg. 2016;101:246–52. [DOI] [PubMed] [Google Scholar]
- 2. Judge EP, Fabre A, Adamali HI, Egan JJ. Acute exacerbations and pulmonary hypertension in advanced idiopathic pulmonary fibrosis. Eur Respir J. 2012;40:93–100. [DOI] [PubMed] [Google Scholar]
- 3. Kimura M, Taniguchi H, Kondoh Y, Kimura T, Kataoka K, Nishiyama O, Aso H, Sakamoto K, Hasegawa Y. Pulmonary hypertension as a prognostic indicator at the initial evaluation in idiopathic pulmonary fibrosis. Respiration. 2013;85:456–63. [DOI] [PubMed] [Google Scholar]
- 4. Hoeper MM, Behr J, Held M, Grunig E, Vizza CD, Vonk‐Noordegraaf A, Lange TJ, Claussen M, Grohé C, Klose H, Olsson KM, Zelniker T, Neurohr C, Distler O, Wirtz H, Opitz C, Huscher D, Pittrow D, Gibbs JSR. Pulmonary hypertension in patients with chronic fibrosing idiopathic interstitial pneumonias. PLoS One. 2015;10:e0141911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nikkho SM, Richter MJ, Shen E, Abman SH, Antoniou K, Chung J, Fernandes P, Hassoun P, Lazarus HM, Olschewski H, Piccari L, Psotka M, Saggar R, Shlobin OA, Stockbridge N, Vitulo P, Vizza CD, Wort SJ, Nathan SD. Clinical significance of pulmonary hypertension in interstitial lung disease: a consensus statement from the Pulmonary Vascular Research Institute's innovative drug development initiative—group 3 pulmonary hypertension. Pulm Circ. 2022;12(3):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Johannson KA, Chaudhuri N, Adegunsoye A, Wolters PJ. Treatment of fibrotic interstitial lung disease: current approaches and future directions. Lancet. 2021;6736:1–11. [DOI] [PubMed] [Google Scholar]
- 7. Nathan SD, Shlobin OA, Ahmad S, Urbanek S, Barnett SD. Pulmonary hypertension and pulmonary function testing in idiopathic pulmonary fibrosis. Chest. 2007;131:657–63. [DOI] [PubMed] [Google Scholar]
- 8. Zisman DA, Karlamangla AS, Ross DJ, Keane MP, Belperio JA, Saggar R, Lynch JP, Ardehali A, Goldin J. High‐resolution chest CT findings do not predict the presence of pulmonary hypertension in advanced idiopathic pulmonary fibrosis. Chest. 2007;132:773–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Alhamad EH, Al‐Boukai AA, Al‐Kassimi FA, Alfaleh HF, Alshamiri MQ, Alzeer AH, Al‐Otair HA, Ibrahim GF, Shaik SA. Prediction of pulmonary hypertension in patients with or without interstitial lung disease: reliability of CT findings. Radiology. 2011;260:875–83. [DOI] [PubMed] [Google Scholar]
- 10. Nathan SD, Barbera JA, Gaine SP, Harari S, Martinez FJ, Olschewski H, Olsson KM, Peacock AJ, Pepke‐Zaba J, Provencher S, Weissmann N, Seeger W. Pulmonary hypertension in chronic lung disease and hypoxia. Eur Respir J. 2019;53:1801914. 10.1183/13993003.01914-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hoffmann J, Wilhelm J, Marsh LM, Ghanim B, Klepetko W, Kovacs G, Olschewski H, Olschewski A, Kwapiszewska G. Distinct differences in gene expression patterns in pulmonary arteries of patients with chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis with pulmonary hypertension. Am J Respir Crit Care Med. 2014;190:98–111. [DOI] [PubMed] [Google Scholar]
- 12. Ruffenach G, Umar S, Vaillancourt M, Hong J, Cao N, Sarji S, Moazeni S, Cunningham CM, Ardehali A, Reddy ST, Saggar R, Fishbein G, Eghbali M. Histological hallmarks and role of Slug/PIP axis in pulmonary hypertension secondary to pulmonary fibrosis. EMBO Mol Med. 2019;11:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Farkas L, Gauldie J, Voelkel NF, Kolb M. Pulmonary hypertension and idiopathic pulmonary fibrosis: a tale of angiogenesis, apoptosis, and growth factors. Am J Respir Cell Mol Biol. 2011;45:1–15. [DOI] [PubMed] [Google Scholar]
- 14. Wu WH, Bonnet S, Shimauchi T, Toro V, Grobs Y, Romanet C, Bourgeois A, Vitry G, Omura J, Tremblay E, Nadeau V, Orcholski M, Breuils‐Bonnet S, Martineau S, Ferraro P, Potus F, Paulin R, Provencher S, Boucherat O. Potential for inhibition of checkpoint kinases 1/2 in pulmonary fibrosis and secondary pulmonary hypertension. Thorax. 2021;77(3):247–58. [DOI] [PubMed] [Google Scholar]
- 15. Lambers C, Roth M, Jaksch P, Muraközy G, Tamm M, Klepetko W, Ghanim B, Zhao F. Treprostinil inhibits proliferation and extracellular matrix deposition by fibroblasts through cAMP activation. Sci Rep. 2018;8:1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mura M, Anraku M, Yun Z, McRae K, Liu M, Waddell TK, Singer LG, Granton JT, Keshavjee S, de Perrot M. Gene expression profiling in the lungs of patients with pulmonary hypertension associated with pulmonary fibrosis. Chest. 2012;141:661–73. 10.1378/chest.11-0449 [DOI] [PubMed] [Google Scholar]
- 17. Chen N, Collum SD, Luo F, Weng T, Le TT, M Hernandez A, Philip K, Molina JG, Garcia‐Morales LJ, Cao Y, Ko TC, Amione‐Guerra J, Al‐Jabbari O, Bunge RR, Youker K, Bruckner BA, Hamid R, Davies J, Sinha N, Karmouty‐Quintana H. Macrophage bone morphogenic protein receptor 2 depletion in idiopathic pulmonary fibrosis and group III pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2016;311:238–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jiang Q, Liu C, Liu S, Lu W, Li Y, Luo X, Ma R, Zhang C, Chen H, Chen Y, Zhang Z, Hong C, Guo W, Wang T, Yang K, Wang J. Dysregulation of BMP9/BMPR2/SMAD signalling pathway contributes to pulmonary fibrosis and pulmonary hypertension induced by bleomycin in rats. Br J Pharmacol. 2021;178:203–16. [DOI] [PubMed] [Google Scholar]
- 19. Thoré P, Girerd B, Jaïs X, Savale L, Ghigna MR, Eyries M, Levy M, Ovaert C, Servettaz A, Guillaumot A, Dauphin C, Chabanne C, Boiffard E, Cottin V, Perros F, Simonneau G, Sitbon O, Soubrier F, Bonnet D, Remy‐Jardin M, Chaouat A, Humbert M, Montani D. Phenotype and outcome of pulmonary arterial hypertension patients carrying a TBX4 mutation. Eur Respir J. 2020;55:1902340. 10.1183/13993003.02340-2019 [DOI] [PubMed] [Google Scholar]
- 20. Hernandez‐Gonzalez I, Tenorio J, Palomino‐Doza J, Martinez Meñaca A, Morales Ruiz R, Lago‐Docampo M, Valverde Gomez M, Gomez Roman J, Enguita Valls AB, Perez‐Olivares C, Valverde D, Gil Carbonell J, Garrido‐Lestache Rodríguez‐Monte E, del Cerro MJ, Lapunzina P, Escribano‐Subias P. Clinical heterogeneity of pulmonary arterial hypertension associated with variants in TBX4. PLoS One. 2020;15:e0232216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eyries M, Montani D, Girerd B, Favrolt N, Riou M, Faivre L, Manaud G, Perros F, Gräf S, Morrell NW, Humbert M, Soubrier F. Familial pulmonary arterial hypertension by KDR heterozygous loss of function. Eur Respir J. 2020;55:1902165. [DOI] [PubMed] [Google Scholar]
- 22. Patel NM, Kawut SM, Jelic S, Arcasoy SM, Lederer DJ, Borczuk AC. Pulmonary arteriole gene expression signature in idiopathic pulmonary fibrosis. Eur Respir J. 2013;41:1324–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ruffenach G, Hong J, Vaillancourt M, Medzikovic L, Eghbali M. Pulmonary hypertension secondary to pulmonary fibrosis: clinical data, histopathology and molecular insights. Respir Res. 2020;21:303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Keane MP, Arenberg DA, Lynch JP, Whyte RI, Iannettoni MD, Burdick MD, Wilke CA, Morris SB, Glass MC, DiGiovine B, Kunkel SL, Strieter RM. The CXC chemokines, IL‐8 and IP‐10, regulate angiogenic activity in idiopathic pulmonary fibrosis. J Immunol. 1997;159:1437–43. [PubMed] [Google Scholar]
- 25. Farkas L, Farkas D, Ask K, Möller A, Gauldie J, Margetts P, Inman M, Kolb M. VEGF ameliorates pulmonary hypertension through inhibition of endothelial apoptosis in experimental lung fibrosis in rats. J Clin Invest. 2009;119:1298–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Murray LA, Habiel DM, Hohmann M, Camelo A, Shang H, Zhou Y, Coelho AL, Peng X, Gulati M, Crestani B, Sleeman MA, Mustelin T, Moore MW, Ryu C, Osafo‐Addo AD, Elias JA, Lee CG, Hu B, Herazo‐Maya JD, Knight DA, Hogaboam CM, Herzog EL. Antifibrotic role of vascular endothelial growth factor in pulmonary fibrosis. JCI Insight. 2017;2(16):e92192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lane KB, Machado RD, Pauciulo MB, Thomson JR, Phillips 3rd JA, Loyd JE, Nichols WC, Trembath RC, International PPH Consortium . Heterozygous germline mutations in BMPR2, encoding a TGF‐beta receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;55:81–4. [DOI] [PubMed] [Google Scholar]
- 28. Newman JH. Molecular rescue in pulmonary arterial hypertension. N Engl J Med. 2021;384:1271–72. [DOI] [PubMed] [Google Scholar]
- 29. Hoeper MM, Badesch DB, Ghofrani HA, Gibbs JSR, Gomberg‐Maitland M, McLaughlin VV, Preston IR, Souza R, Waxman AB, Grünig E, Kopeć G, Meyer G, Olsson KM, Rosenkranz S, Xu Y, Miller B, Fowler M, Butler J, Koglin J, de Oliveira Pena J, Humbert M, STELLAR Trial Investigators. Phase 3 Trial of Sotatercept for Treatment of Pulmonary Arterial Hypertension. N Engl J Med. 2023. 10.1056/NEJMoa2213558. [DOI] [Google Scholar]
- 30. Nathan SD, Waxman A, Rajagopal S, Case A, Johri S, DuBrock H, De La Zerda DJ, Sahay S, King C, Melendres‐Groves L, Smith P, Shen E, Edwards LD, Nelsen A, Tapson VF. Inhaled treprostinil and forced vital capacity in patients with interstitial lung disease and associated pulmonary hypertension: a post‐hoc analysis of the INCREASE study. Lancet Respir Med. 2021;2600:1–9. [DOI] [PubMed] [Google Scholar]
- 31. Nikitopoulou I, Manitsopoulos N, Kotanidou A, Tian X, Petrovic A, Magkou C, Ninou I, Aidinis V, Schermuly RT, Kosanovic D, Orfanos SE. Orotracheal treprostinil administration attenuates bleomycin‐induced lung injury, vascular remodeling, and fibrosis in mice. Pulm Circ. 2019;9:1–14. 10.1177/2045894019881954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhu Y, Liu Y, Zhou W, Xiang R, Jiang L, Huang K, Xiao Y, Guo Z, Gao J. A prostacyclin analogue, iloprost, protects from bleomycin‐induced pulmonary fibrosis in mice. Respir Res. 2010;11:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sandner P, Stasch JP. Anti‐fibrotic effects of soluble guanylate cyclase stimulators and activators: a review of the preclinical evidence. Respir Med. 2017;122:S1–9. [DOI] [PubMed] [Google Scholar]
- 34. Ebina M, Shimizukawa M, Shibata N, Kimura Y, Suzuki T, Endo M, Sasano H, Kondo T, Nukiwa T. Heterogeneous increase in CD34‐positive alveolar capillaries in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2004;169:1203–08. [DOI] [PubMed] [Google Scholar]
- 35. Renzoni EA, Walsh DA, Salmon M, Wells AU, Sestini P, Nicholson AG, Veeraraghavan S, Bishop AE, Romanska HM, Pantelidis P, Black CM, du Bois RM. Interstitial vascularity in fibrosing alveolitis. Am J Respir Crit Care Med. 2003;167:438–43. [DOI] [PubMed] [Google Scholar]
- 36. Kim KH, Maldonado F, Ryu JH, Eiken PW, Hartman TE, Bartholmai BJ, Decker PA, Yi ES. Iron deposition and increased alveolar septal capillary density in nonfibrotic lung tissue are associated with pulmonary hypertension in idiopathic pulmonary fibrosis. Respir Res. 2010;11:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Seki A, Anklesaria Z, Saggar R, Dodson MW, Schwab K, Liu MC, Charan Ashana D, Miller WD, Vangala S, DerHovanessian A, Channick R, Shaikh F, Belperio JA, Weigt SS, Lynch JP, Ross DJ, Sullivan L, Khanna D, Shapiro SS, Sager J, Gargani L, Stanziola A, Bossone E, Schraufnagel DE, Fishbein G, Xu H, Fishbein MC, Wallace WD, Saggar R. Capillary proliferation in systemic‐sclerosis‐related pulmonary fibrosis: association with pulmonary hypertension. ACR Open Rheumatol. 2019;1:26–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Colombat M, Mal H, Groussard O, Capron F, Thabut G, Jebrak G, Brugière O, Dauriat G, Castier Y, Lesèche G, Fournier M. Pulmonary vascular lesions in end‐stage idiopathic pulmonary fibrosis: histopathologic study on lung explant specimens and correlations with pulmonary hemodynamics. Hum Pathol. 2007;38:60–5. [DOI] [PubMed] [Google Scholar]
- 39. Dotan Y, Stewart J, Gangemi A, Wang H, Aneja A, Chakraborty B, Dass C, Zhao H, Marchetti N, D'Alonzo G, Cordova FC, Criner G, Mamary AJ. Pulmonary vasculopathy in explanted lungs from patients with interstitial lung disease undergoing lung transplantation. BMJ Open Respir Res. 2020;7(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Humbert M, Guignabert C, Bonnet S, Dorfmüller P, Klinger JR, Nicolls MR, Olschewski AJ, Pullamsetti SS, Schermuly RT, Stenmark KR, Rabinovitch M. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J. 2018;53(1):1801887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cottin V, Selman M, Inoue Y, Wong AW, Corte TJ, Flaherty KR, Han MK, Jacob J, Johannson KA, Kitaichi M, Lee JS, Agusti A, Antoniou KM, Bianchi P, Caro F, Florenzano M, Galvin L, Iwasawa T, Martinez FJ, Morgan RL, Myers JL, Nicholson AG, Occhipinti M, Poletti V, Salisbury ML, Sin DD, Sverzellati N, Tonia T, Valenzuela C, Ryerson CJ, Wells AU. Syndrome of combined pulmonary fibrosis and emphysema: an official ATS/ERS/JRS/ALAT research statement. Am J Respir Crit Care Med. 2022;206:e7–e41. 10.1164/rccm.202206-1041ST [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cottin V, Nunes H, Brillet PY, Delaval P, Devouassoux G, Tillie‐Leblond I, Israel‐Biet D, Court‐Fortune I, Valeyre D, Cordier JF, Groupe d'Etude et de Recherche sur les Maladies Orphelines Pulmonaires (GERM O P) . Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity. Eur Respir J. 2005;26:586–93. [DOI] [PubMed] [Google Scholar]
- 43. Cottin V, Le Pavec J, Prevot G, Mal H, Humbert M, Simonneau G, Cordier JF. Pulmonary hypertension in patients with combined pulmonary fibrosis and emphysema syndrome. Eur Respir J. 2010;35:105–11. [DOI] [PubMed] [Google Scholar]
- 44. Matsuoka S, Yamashiro T, Matsushita S, Kotoku A, Fujikawa A, Yagihashi K, Nakajima Y. Quantitative CT evaluation in patients with combined pulmonary fibrosis and emphysema. Academic Radiol. 2015;22:626–31. [DOI] [PubMed] [Google Scholar]
- 45. Mejía M, Carrillo G, Rojas‐Serrano J, Estrada A, Suárez T, Alonso D, Barrientos E, Gaxiola M, Navarro C, Selman M. Idiopathic pulmonary fibrosis and emphysema. Chest. 2009;136:10–5. [DOI] [PubMed] [Google Scholar]
- 46. Ryerson CJ, Hartman T, Elicker BM, Ley B, Lee JS, Abbritti M, Jones KD, King TE, Ryu J, Collard HR. Clinical features and outcomes in combined pulmonary fibrosis and emphysema in idiopathic pulmonary fibrosis. Chest. 2013;144:234–40. [DOI] [PubMed] [Google Scholar]
- 47. Tomioka H, Mamesaya N, Yamashita S, Kida Y, Kaneko M, Sakai H. Combined pulmonary fibrosis and emphysema: effect of pulmonary rehabilitation in comparison with chronic obstructive pulmonary disease. BMJ Open Respir Res. 2016;3:e000099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jacob J, Bartholmai BJ, Rajagopalan S, Karwoski R, Nair A, Walsh SLF, Barnett J, Cross G, Judge EP, Kokosi M, Renzoni E, Maher TM, Wells AU. Likelihood of pulmonary hypertension in patients with idiopathic pulmonary fibrosis and emphysema. Respirology. 2018;23:593–9. [DOI] [PubMed] [Google Scholar]
- 49. Gall H, Felix JF, Schneck FK, Milger K, Sommer N, Voswinckel R, Franco OH, Hofman A, Schermuly RT, Weissmann N, Grimminger F, Seeger W, Ghofrani HA. The giessen pulmonary hypertension registry: survival in pulmonary hypertension subgroups. J Heart Lung Transplant. 2017;36:957–67. [DOI] [PubMed] [Google Scholar]
- 50. Hurdman J, Condliffe R, Elliot CA, Davies C, Hill C, Wild JM, Capener D, Sephton P, Hamilton N, Armstrong IJ, Billings C, Lawrie A, Sabroe I, Akil M, O'Toole L, Kiely DG. ASPIRE registry: assessing the spectrum of pulmonary hypertension identified at a REferral centre. Eur Respir J. 2012;39:945–55. [DOI] [PubMed] [Google Scholar]
- 51. Fischer A, Antoniou KM, Brown KK, Cadranel J, Corte TJ, du Bois RM, Lee JS, Leslie KO, Lynch DA, Matteson EL, Mosca M, Noth I, Richeldi L, Strek ME, Swigris JJ, Wells AU, West SG, Collard HR, Cottin V. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J. 2015;46:976–87. [DOI] [PubMed] [Google Scholar]
- 52. Fischer A, Swigris JJ, Bolster MB, Chung L, Csuka ME, Domsic R, Frech T, Hinchcliff M, Hsu V, Hummers LK, Gomberg‐Maitland M, Mathai SC, Simms R, Steen VD. Pulmonary hypertension and interstitial lung disease within PHAROS: impact of extent of fibrosis and pulmonary physiology on cardiac haemodynamic parameters. Clin Exp Rheumatol. 2014;32(6 Suppl 86):S–109-14. [PubMed] [Google Scholar]
- 53. Saggar R, Giri PC, Deng C, Johnson D, McCloy MK, Liang L, Shaikh F, Hong J, Channick RN, Shapiro SS, Lynch JP, Belperio JA, Weigt SS, Ramsey AL, Ross DJ, Sayah DM, Shino MY, Derhovanessian A, Sherman AE, Saggar R. Significance of autoimmune disease in severe pulmonary hypertension complicating extensive pulmonary fibrosis: a prospective cohort study. Pulm Circ. 2021;11:1–12. 10.1177/20458940211011329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chauvelot L, Gamondes D, Berthiller J, Nieves A, Renard S, Catella‐Chatron J, Ahmad K, Bertoletti L, Camara B, Gomez E, Launay D, Montani D, Mornex JF, Prévot G, Sanchez O, Schott AM, Subtil F, Traclet J, Turquier S, Zeghmar S, Habib G, Reynaud‐Gaubert M, Humbert M, Cottin V. Hemodynamic response to treatment and outcomes in pulmonary hypertension associated with interstitial lung disease versus pulmonary arterial hypertension in systemic sclerosis: data from a study identifying prognostic factors in pulmonary hypertension ass. Arthritis Rheum. 2021;73:295–304. [DOI] [PubMed] [Google Scholar]
- 55. Alhamad EH, Cal JG, Alrajhi NN, Alharbi WM. Predictors of mortality in patients with interstitial lung disease‐associated pulmonary hypertension. J Clin Med. 2020;9:3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dodd PJ, Yuen CM, Jayasooriya SM, van der Zalm MM, Seddon JA. Quantifying the global number of tuberculosis survivors: a modelling study. Lancet Infect Dis. 2021;21:984–92. [DOI] [PubMed] [Google Scholar]
- 57. Allwood BW, Van Der Zalm MM, Amaral AFS, Byrne A, Datta S, Egere U, Evans CA, Evans D, Gray DM, Hoddinott G, Ivanova O, Jones R, Makanda G, Marx FM, Meghji J, Mpagama S, Pasipanodya JG, Rachow A, Schoeman I, Shaw J, Stek C, van Kampen S, von Delft D, Walker NF, Wallis RS, Mortimer K. Post‐tuberculosis lung health: perspectives from the first international symposium. Int J Tuberc Lung Dis. 2020;24:820–8. [DOI] [PubMed] [Google Scholar]
- 58. Alene KA, Wangdi K, Colquhoun S, Chani K, Islam T, Rahevar K, Morishita F, Byrne A, Clark J, Viney K. Tuberculosis related disability: a systematic review and meta‐analysis. BMC Med. 2021;19:203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Allwood BW, Byrne A, Meghji J, Rachow A, van der Zalm MM, Schoch OD. Post‐tuberculosis lung disease: clinical review of an under‐recognised global challenge. Respiration. 2021;100:751–63. [DOI] [PubMed] [Google Scholar]
- 60. Akkara SA, Shah AD, Adalja M, Akkara AG, Rathi A, Shah DN. Pulmonary tuberculosis: the day after. Int J Tuberc Lung Dis. 2013;17:810–3. [DOI] [PubMed] [Google Scholar]
- 61. Walsh KF, Lui JK. Post‐tuberculosis pulmonary hypertension: a case of global disparity in health care. Lancet Global Health. 2022;10:e476. [DOI] [PubMed] [Google Scholar]
- 62. Chen Y, Liu C, Lu W, Li M, Hadadi C, Wang EW, Yang K, Lai N, Huang J, Li S, Zhong N, Zhang N, Wang J. Clinical characteristics and risk factors of pulmonary hypertension associated with chronic respiratory diseases: a retrospective study. J Thorac Dis. 2016;8:350–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Park SY, Lee CY, Kim C, Jang SH, Park YB, Park S, Hwang YI, Lee MG, Jung KS, Kim DG. One‐year prognosis and the role of brain natriuretic peptide levels in patients with chronic cor pulmonale. J Korean Med Sci. 2015;30:442–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bhattacharyya P, Saha D, Bhattacherjee P, Das S, Bhattacharyya P, Dey R. Tuberculosis associated pulmonary hypertension: the revelation of a clinical observation. Lung India. 2016;33:135–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Park SY, Lee SM, Shin JW, Choi BW, Kim H, Lee JS, Lee SD, Park SS, Moon HS, Park YB. Epidemiology of chronic thromboembolic pulmonary hypertension in Korea: results from the Korean registry. Korean J Intern Med. 2016;31:305–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Freitas CSG, Baldi BG, Jardim C, Araujo MS, Sobral JB, Heiden GI, Kairalla RA, Souza R, Carvalho CRR. Pulmonary hypertension in lymphangioleiomyomatosis: prevalence, severity and the role of carbon monoxide diffusion capacity as a screening method. Orphanet J Rare Dis. 2017;12:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Taveira‐dasilva AM, Hathaway OM, Sachdev V, Shizukuda Y, Birdsall CW, Moss J. Pulmonary artery pressure in lymphangioleiomyomatosis. Chest. 2007;132:1573–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Cottin V, Harari S, Humbert M, Mal H, Dorfmüller P, Jaïs X, Reynaud‐Gaubert M, Prevot G, Lazor R, Taillé C, Lacronique J, Zeghmar S, Simonneau G, Cordier JF. Pulmonary hypertension in lymphangioleiomyomatosis: characteristics in 20 patients. Eur Respir J. 2012;40:630–40. [DOI] [PubMed] [Google Scholar]
- 69. Reynaud‐Gaubert M, Mornex JF, Mal H, Treilhaud M, Dromer C, Quétant S, Leroy‐Ladurie F, Guillemain R, Philit F, Dauriat G, Grenet D, Stern M. Lung transplantation for lymphangioleiomyomatosis: the French experience. Transplantation. 2008;86:515–20. [DOI] [PubMed] [Google Scholar]
- 70. Baldi BG, Fernandes CJCS, Heiden GI, Freitas CSG, Sobral JB, Kairalla RA, Carvalho CRR, Souza R. Association between pulmonary artery to aorta diameter ratio with pulmonary hypertension and outcomes in diffuse cystic lung diseases. Medicine. 2021;100:e26483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. DuBrock HM, Nathan SD, Reeve BB, Kolaitis NA, Mathai SC, Classi PM, Nelsen AC, Olayinka‐Amao B, Norcross LN, Martin SA. Pulmonary hypertension due to interstitial lung disease or chronic obstructive pulmonary disease: a patient experience study of symptoms and their impact on quality of life. Pulm Circ. 2021;11:1–9. 10.1177/20458940211005641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rahaghi FF, Kolaitis NA, Adegunsoye A, de Andrade JA, Flaherty KR, Lancaster LH, Lee JS, Levine DJ, Preston IR, Safdar Z, Saggar R, Sahay S, Scholand MB, Shlobin OA, Zisman DA, Nathan SD. Screening strategies for pulmonary hypertension in patients with interstitial lung disease. Chest. 2022;162:145–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Behr J, Nathan SD. Pulmonary hypertension in interstitial lung disease: screening, diagnosis and treatment. Curr Opin Pulm Med. 2021;27:396–404. 10.1097/mcp.0000000000000790 [DOI] [PubMed] [Google Scholar]
- 74. Raghu G, Nathan SD, Behr J, Brown KK, Egan JJ, Kawut SM, Flaherty KR, Martinez FJ, Wells AU, Shao L, Zhou H, Henig N, Szwarcberg J, Gillies H, Montgomery AB, O'Riordan TG. Pulmonary hypertension in idiopathic pulmonary fibrosis with mild‐to‐moderate restriction. Eur Respir J. 2015;46:1370–77. [DOI] [PubMed] [Google Scholar]
- 75. Shorr AF, Wainright JL, Cors CS, Lettieri CJ, Nathan SD. Pulmonary hypertension in patients with pulmonary fibrosis awaiting lung transplant. Eur Respir J. 2007;30:715–21. [DOI] [PubMed] [Google Scholar]
- 76. Fartoukh M, Humbert M, Capron F, Maître S, Parent F, Le Gall C, Sitbon O, Hervé P, Duroux P, Simonneau G. Severe pulmonary hypertension in histiocytosis X. Am J Respir Crit Care Med. 2000;161:216–23. [DOI] [PubMed] [Google Scholar]
- 77. Zisman DA, Ross DJ, Belperio JA, Saggar R, Lynch JP, Ardehali A, Karlamangla AS. Prediction of pulmonary hypertension in idiopathic pulmonary fibrosis. Respir Med. 2007;101:2153–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sonti R, Gersten RA, Barnett S, Brown AW, Nathan SD. Multimodal noninvasive prediction of pulmonary hypertension in IPF. Clin Respiratory J. 2019;13:567–73. [DOI] [PubMed] [Google Scholar]
- 79. Parikh R, Konstantinidis I, O' Sullivan DM, Farber HW. Pulmonary hypertension in patients with interstitial lung disease: a tool for early detection. Pulm Circ. 2022;12(4):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zisman DA, Karlamangla AS, Kawut SM, Shlobin OA, Saggar R, Ross DJ, Schwarz MI, Belperio JA, Ardehali A, Lynch JP, Nathan SD. Validation of a method to screen for pulmonary hypertension in advanced idiopathic pulmonary fibrosis. Chest. 2008;133:640–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Nathan SD, Flaherty KR, Glassberg MK, Raghu G, Swigris J, Alvarez R, Ettinger N, Loyd J, Fernandes P, Gillies H, Kim B, Shah P, Lancaster L. A randomized, double‐blind, placebo‐controlled study of pulsed, inhaled nitric oxide in subjects at risk of pulmonary hypertension associated with pulmonary fibrosis. Chest. 2020;158:637–45. [DOI] [PubMed] [Google Scholar]
- 82. Waxman A, Restrepo‐Jaramillo R, Thenappan T, Ravichandran A, Engel P, Bajwa A, Allen R, Feldman J, Argula R, Smith P, Rollins K, Deng C, Peterson L, Bell H, Tapson V, Nathan SD. Inhaled treprostinil in pulmonary hypertension due to interstitial lung disease. N Engl J Med. 2021;384:325–34. [DOI] [PubMed] [Google Scholar]
- 83. King CS, Flaherty KR, Glassberg MK, Lancaster L, Raghu G, Swigris JJ, Argula RG, Dudenhofer RA, Ettinger NA, Feldman J, Johri S, Fernandes P, Parsley E, Shah PS, Nathan SD. A Phase‐2 exploratory randomized controlled trial of INOpulse in patients with fibrotic interstitial lung disease requiring oxygen. Ann Am Thorac Soc. 2022;19:594–602. [DOI] [PubMed] [Google Scholar]
- 84. Behr J. Inhaled treprostinil in pulmonary hypertension in the context of interstitial lung disease: a success, finally. Am J Respir Crit Care Med. 2022;205:144–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Agusti A, Barberà JA. Contribution of multiple inert gas elimination technique to pulmonary Medicine. 2. Chronic pulmonary diseases: chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Thorax. 1994;49:924–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Olschewski H, Ardeschir Ghofrani H, Walmrath D, Schermuly R, Temmesfeld‐Wollbrück B, Grimminger F, Seeger W. Inhaled prostacyclin and iloprost in severe pulmonary hypertension secondary to lung fibrosis. Am J Respir Crit Care Med. 1999;160:600–7. [DOI] [PubMed] [Google Scholar]
- 87. Nathan SD, Shlobin OA, Ahmad S, Koch J, Barnett SD, Ad N, Burton N, Leslie K. Serial development of pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Respiration. 2008;76:288–94. [DOI] [PubMed] [Google Scholar]
- 88. Swigris JJ, Olson AL, Shlobin OA, Ahmad S, Brown KK, Nathan SD. Heart rate recovery after six‐minute walk test predicts pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Respirology. 2011;16:439–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Yoo DK, Zompatori M, Barrile A, Rossi G, D'Amato D, Sergiacomi G, Rogliani P, Mura M. Associated pulmonary hypertension is an independent contributor to exercise intolerance in chronic fibrosing interstitial pneumonias. Respiration. 2018;96:543–51. [DOI] [PubMed] [Google Scholar]
- 90. Leuchte HH, Neurohr C, Baumgartner R, Holzapfel M, Giehrl W, Vogeser M, Behr J. Brain natriuretic peptide and exercise capacity in lung fibrosis and pulmonary hypertension. Am J Respir Crit Care Med. 2004;170:360–5. [DOI] [PubMed] [Google Scholar]
- 91. Gläser S, Obst A, Koch B, Henkel B, Grieger A, Felix SB, Halank M, Bruch L, Bollmann T, Warnke C, Schäper C, Ewert R. Pulmonary hypertension in patients with idiopathic pulmonary fibrosis—the predictive value of exercise capacity and gas exchange efficiency. PLoS One. 2013;8:e65643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Boutou AK, Pitsiou GG, Trigonis I, Papakosta D, Kontou PK, Chavouzis N, Nakou C, Argyropoulou P, Wasserman K, Stanopoulos I. Exercise capacity in idiopathic pulmonary fibrosis: the effect of pulmonary hypertension. Respirology. 2011;16:451–8. [DOI] [PubMed] [Google Scholar]
- 93. Gille T, Laveneziana P. Cardiopulmonary exercise testing in interstitial lung diseases and the value of ventilatory efficiency. Eur Respir Rev. 2021;30:200355. 10.1183/16000617.0355-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Degani‐Costa LH, Levarge B, Digumarthy SR, Eisman AS, Harris RS, Lewis GD. Pulmonary vascular response patterns during exercise in interstitial lung disease. Eur Respir J. 2015;46:738–49. [DOI] [PubMed] [Google Scholar]
- 95. Armstrong HF, Thirapatarapong W, Dussault NE, Bartels MN. Distinguishing pulmonary hypertension in interstitial lung disease by ventilation and perfusion defects measured by cardiopulmonary exercise testing. Respiration. 2013;86:407–13. [DOI] [PubMed] [Google Scholar]
- 96. Armstrong HF, Schulze PC, Bacchetta M, Thirapatarapong W, Bartels MN. Impact of pulmonary hypertension on exercise performance in patients with interstitial lung disease undergoing evaluation for lung transplantation. Respirology. 2014;19:675–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Bax S, Bredy C, Kempny A, Dimopoulos K, Devaraj A, Walsh S, Jacob J, Nair A, Kokosi M, Keir G, Kouranos V, George PM, McCabe C, Wilde M, Wells A, Li W, Wort SJ, Price LC. A stepwise composite echocardiographic score predicts severe pulmonary hypertension in patients with interstitial lung disease. ERJ Open Res. 2018;4:00124‐2017. 10.1183/23120541.00124-2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger RMF, Brida M, Carlsen J, Coats AJS, Escribano‐Subias P, Ferrari P, Ferreira DS, Ghofrani HA, Giannakoulas G, Kiely DG, Mayer E, Meszaros G, Nagavci B, Olsson KM, Pepke‐Zaba J, Quint JK, Rådegran G, Simonneau G, Sitbon O, Tonia T, Toshner M, Vachiery JL, Vonk Noordegraaf A, Delcroix M, Rosenkranz S. 2022 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2022;61:2200879. 10.1183/13993003.00879-2022 [DOI] [PubMed] [Google Scholar]
- 99. Arcasoy SM, Christie JD, Ferrari VA, Sutton MSJ, Zisman DA, Blumenthal NP, Pochettino A, Kotloff RM. Echocardiographic assessment of pulmonary hypertension in patients with advanced lung disease. Am J Respir Crit Care Med. 2003;167:735–40. [DOI] [PubMed] [Google Scholar]
- 100. Tello K, Ghofrani HA, Heinze C, Krueger K, Naeije R, Raubach C, Seeger W, Sommer N, Gall H, Richter MJ. A simple echocardiographic estimate of right ventricular‐arterial coupling to assess severity and outcome in pulmonary hypertension on chronic lung disease. Eur Respir J. 2019;54:1802435. [DOI] [PubMed] [Google Scholar]
- 101. Kato S, Sekine A, Kusakawa Y, Ogura T, Futaki M, Iwasawa T, Kirigaya H, Gyotoku D, Iinuma N, Iguchi K, Nakachi T, Fukui K, Kimura K, Umemura S. Prognostic value of cardiovascular magnetic resonance derived right ventricular function in patients with interstitial lung disease. J Cardiovasc Magn Reson. 2015;17:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kamide H, Kato S, Hayakawa K, Fukui K, Kitamura H, Ogura T, Iwasawa T, Kimura K, Tamura K, Utsunomiya D. Impairment of right ventricular strain evaluated by cardiovascular magnetic resonance feature tracking in patients with interstitial lung disease. Int J Cardiovasc Imaging. 2021;37:1073–83. [DOI] [PubMed] [Google Scholar]
- 103. Wells JM, Iyer AS, Rahaghi FN, Bhatt SP, Gupta H, Denney TS, Lloyd SG, Dell'Italia LJ, Nath H, Estepar RSJ, Washko GR, Dransfield MT. Pulmonary artery enlargement is associated with right ventricular dysfunction and loss of blood volume in small pulmonary vessels in chronic obstructive pulmonary disease. Circulation: Cardiovasc Imaging. 2015;8(4):e002546. 10.1161/CIRCIMAGING.114.002546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Zouk AN, Gulati S, Xing D, Wille KM, Rowe SM, Wells JM. Pulmonary artery enlargement is associated with pulmonary hypertension and decreased survival in severe cystic fibrosis: a cohort study. PLoS One. 2020;15:e0229173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Devaraj A, Wells AU, Meister MG, Corte TJ, Hansell DM. The effect of diffuse pulmonary fibrosis on the reliability of CT signs of pulmonary hypertension. Radiology. 2008;249:1042–49. [DOI] [PubMed] [Google Scholar]
- 106. Tan RT, Kuzo R, Goodman LR, Siegel R, Haasler GR, Presberg KW. Utility of CT scan evaluation for predicting pulmonary hypertension in patients with parenchymal lung disease. Chest. 1998;113:1250–56. [DOI] [PubMed] [Google Scholar]
- 107. Remy‐Jardin M, Ryerson CJ, Schiebler ML, Leung ANC, Wild JM, Hoeper MM, Alderson PO, Goodman LR, Mayo J, Haramati LB, Ohno Y, Thistlethwaite P, van Beek EJR, Knight SL, Lynch DA, Rubin GD, Humbert M. Imaging of pulmonary hypertension in adults: a position paper from the Fleischner Society. Eur Respir J. 2021;57:2004455. 10.1183/13993003.04455-2020 [DOI] [PubMed] [Google Scholar]
- 108. Nathan SD, Cottin V, Behr J, Hoeper MM, Martinez FJ, Corte TJ, Keogh AM, Leuchte H, Mogulkoc N, Ulrich S, Wuyts WA, Yao Z, Ley‐Zaporozhan J, Müller‐Lisse UG, Scholle FD, Brüggenwerth G, Busse D, Nikkho S, Wells AU. Impact of lung morphology on clinical outcomes with riociguat in patients with pulmonary hypertension and idiopathic interstitial pneumonia: a post hoc subgroup analysis of the RISE‐IIP study. J Heart Lung Transplant. 2021;40:494–503. 10.1016/j.healun.2021.02.006 [DOI] [PubMed] [Google Scholar]
- 109. Rahaghi FN, Nardelli P, Harder E, Singh I, Sánchez‐Ferrero GV, Ross JC, San José Estépar R, Ash SY, Hunsaker AR, Maron BA, Leopold JA, Waxman AB, San José Estépar R, Washko GR. Quantification of arterial and venous morphologic markers in pulmonary arterial hypertension using CT imaging. Chest. 2021;160:2220–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Dwivedi K, Sharkey M, Condliffe R, Uthoff JM, Alabed S, Metherall P, Lu H, Wild JM, Hoffman EA, Swift AJ, Kiely DG. Pulmonary hypertension in association with lung disease: quantitative CT and artificial intelligence to the rescue? State‐of‐the‐art review. Diagnostics. 2021;11:679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. King CS, Brown AW, Shlobin OA, Weir N, Libre M, Franco‐Palacios D, Ahmad S, Nathan SD. Prevalence and impact of WHO group 3 pulmonary hypertension in advanced idiopathic nonspecific interstitial pneumonia. Eur Respir J. 2018;52:1800545. 10.1183/13993003.00545-2018 [DOI] [PubMed] [Google Scholar]
- 112. Oliveira RKF, Pereira CAC, Ramos RP, Ferreira EVM, Messina CMS, Kuranishi LT, Gimenez A, Campos O, Silva CMC, Ota‐Arakaki JS. A haemodynamic study of pulmonary hypertension in chronic hypersensitivity pneumonitis. Eur Respir J. 2014;44:415–24. [DOI] [PubMed] [Google Scholar]
- 113. Oliveira RKF, Ota‐Arakaki JS, Gomes PS, Gimenez A, Messina CMS, Ramos RP, Ferreira EVM, Systrom DM, Pereira CAC. Pulmonary haemodynamics and mortality in chronic hypersensitivity pneumonitis. Eur Respir J. 2018;51:1800430. [DOI] [PubMed] [Google Scholar]
- 114. Chung JH, Montner SM, Adegunsoye A, Oldham JM, Husain AN, Vij R, Noth I, Strek ME. CT findings associated with survival in chronic hypersensitivity pneumonitis. Eur Radiol. 2017;27:5127–35. [DOI] [PubMed] [Google Scholar]
- 115. Teramachi R, Taniguchi H, Kondoh Y, Ando M, Kimura T, Kataoka K, Suzuki A, Furukawa T, Sakamoto K, Hasegawa Y. Progression of mean pulmonary arterial pressure in idiopathic pulmonary fibrosis with mild to moderate restriction. Respirology. 2017;22:986–90. [DOI] [PubMed] [Google Scholar]
- 116. Nadrous HF, Pellikka PA, Krowka MJ, Swanson KL, Chaowalit N, Decker PA, Ryu JH. Pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Chest. 2005;128:2393–99. [DOI] [PubMed] [Google Scholar]
- 117. Lettieri CJ, Nathan SD, Barnett SD, Ahmad S, Shorr AF. Prevalence and outcomes of pulmonary arterial hypertension in advanced idiopathic pulmonary fibrosis. Chest. 2006;129:746–52. [DOI] [PubMed] [Google Scholar]
- 118. Piccari L, Wort SJ, Meloni F, Rizzo M, Price LC, Martino L, Salvaterra E, Scelsi L, López Meseguer M, Blanco I, Callari A, Pérez González V, Tuzzolino F, McCabe C, Rodríguez Chiaradía DA, Vitulo P, REHAR Registry Investigators . The effect of borderline pulmonary hypertension on survival in chronic lung disease. Respiration. 2022;101(8):717–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Oliveira RKF, Waxman AB, Hoover PJ, Dellaripa PF, Systrom DM. Pulmonary vascular and right ventricular burden during exercise in interstitial lung disease. Chest. 2020;158:350–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Nathan SD, Barnett SD, King CS, Provencher S, Barbera JA, Pastre J, Shlobin OA, Seeger W. Impact of the new definition for pulmonary hypertension in patients with lung disease: an analysis of the united network for organ sharing database. Pulm Circ. 2021;11:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Kovacs G, Olschewski A, Berghold A, Olschewski H. Pulmonary vascular resistances during exercise in normal subjects: a systematic review. Eur Respir J. 2012;39:319–28. [DOI] [PubMed] [Google Scholar]
- 122. Kovacs G, Berghold A, Scheidl S, Olschewski H. Pulmonary arterial pressure during rest and exercise in healthy subjects: a systematic review. Eur Respir J. 2009;34:888–94. [DOI] [PubMed] [Google Scholar]
- 123. Corte TJ, Wort SJ, Gatzoulis MA, Macdonald P, Hansell DM, Wells AU. Pulmonary vascular resistance predicts early mortality in patients with diffuse fibrotic lung disease and suspected pulmonary hypertension. Thorax. 2009;64:883–8. [DOI] [PubMed] [Google Scholar]
- 124. Olsson KM, Hoeper MM, Pausch C, Grünig E, Huscher D, Pittrow D, Rosenkranz S, Gall H. Pulmonary vascular resistance predicts mortality in patients with pulmonary hypertension associated with interstitial lung disease: results from the COMPERA registry. Eur Respir J. 2021;58:2101483. [DOI] [PubMed] [Google Scholar]
- 125. Kizer JR, Zisman DA, Blumenthal NP, Kotloff RM, Kimmel SE, Strieter RM, Arcasoy SM, Ferrari VA, Hansen‐Flaschen J. Association between pulmonary fibrosis and coronary artery disease. Arch Intern Med. 2004;164:551–6. [DOI] [PubMed] [Google Scholar]
- 126. Nathan SD, Barnett SD, Urban BA, Nowalk C, Moran BR, Burton N. Pulmonary embolism in idiopathic pulmonary fibrosis transplant recipients. Chest. 2003;123:1758–63. [DOI] [PubMed] [Google Scholar]
- 127. Teramachi R, Taniguchi H, Kondoh Y, Kimura T, Kataoka K, Yokoyama T, Furukawa T, Yagi M, Sakamoto K, Hashimoto N, Hasegawa Y. Impact of post‐capillary pulmonary hypertension on mortality in interstitial lung disease. Respir Investig. 2021;59:342–9. [DOI] [PubMed] [Google Scholar]
- 128. Raghu G, Remy‐jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, Kreuter M, Lynch DA, Maher TM, Martinez FJ, Molina‐Molina M, Myers JL, Nicholson AG, Ryerson CJ, Strek ME, Troy LK, Wijsenbeek M, Mammen MJ, Hossain T, Bissell BD, Herman DD, Hon SM, Kheir F, Khor YH, Macrea M, Antoniou KM, Bouros D, Buendia‐Roldan I, Caro F, Crestani B, Ho L, Morisset J, Olson AL, Podolanczuk A, Poletti V, Selman M, Ewing T, Jones S, Knight SL, Ghazipura M, Wilson KC. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults, an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2022;205:e18–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Hambly N, Farooqi MM, Dvorkin‐Gheva A, Donohoe K, Garlick K, Scallan C, Chong SG, MacIsaac S, Assayag D, Johannson KA, Fell CD, Marcoux V, Manganas H, Morisset J, Comes A, Fisher JH, Shapera S, Gershon AS, To T, Wong AW, Sadatsafavi M, Wilcox PG, Halayko AJ, Khalil N, Cox G, Richeldi L, Ryerson CJ, Kolb M. Prevalence and characteristics of progressive fibrosing interstitial lung disease in a prospective registry. Eur Respir J. 2022;60:2102571. [DOI] [PubMed] [Google Scholar]
- 130. Kovacs G, Agusti A, Barberà JA, Celli B, Criner G, Humbert M, Sin DD, Voelkel N, Olschewski H. Pulmonary vascular involvement in chronic obstructive pulmonary disease. Is there a pulmonary vascular phenotype? Am J Respir Crit Care Med. 2018;198:1000–11. [DOI] [PubMed] [Google Scholar]
- 131. Chaouat A, Bugnet AS, Kadaoui N, Schott R, Enache I, Ducoloné A, Ehrhart M, Kessler R, Weitzenblum E. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;172:189–94. [DOI] [PubMed] [Google Scholar]
- 132. Thabut G, Dauriat G, Stern JB, Logeart D, Levy A, Marrash‐Chahla R, Mal H. Pulmonary hemodynamics in advanced COPD candidates for lung volume reduction surgery or lung transplantation. Chest. 2005;127:1531–36. [DOI] [PubMed] [Google Scholar]
- 133. Bae W, Lee CH, Lee J, Kim YW, Han K, Choi SM. Impact of smoking on the development of idiopathic pulmonary fibrosis: results from a nationwide population‐based cohort study. Thorax. 2022;77:470–6. [DOI] [PubMed] [Google Scholar]
- 134. Tazi A, De Margerie C, Naccache JM, Fry S, Dominique S, Jouneau S, Lorillon G, Bugnet E, Chiron R, Wallaert B, Valeyre D, Chevret S. The natural history of adult pulmonary Langerhans cell histiocytosis: a prospective multicentre study. Orphanet J Rare Dis. 2015;10:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Blanco I, Piccari L, Barberà JA. Pulmonary vasculature in COPD: the silent component. Respirology. 2016;21:984–94. [DOI] [PubMed] [Google Scholar]
- 136. Aguilar PM, Carrera LG, Segura CC, Sánchez MIT, Peña MFV, Hernán GB, Rodríguez IE, Zapata RMR, Lucas EZD, Álvarez PDA, Bueno EV, Sánchez CP, Walther RÁS. Relationship between air pollution levels in Madrid and the natural history of idiopathic pulmonary fibrosis: severity and mortality. J Int Med Res. 2021;49:030006052110290. 10.1177/03000605211029058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Johannson KA, Vittinghoff E, Lee K, Balmes JR, Ji W, Kaplan GG, Kim DS, Collard HR. Acute exacerbation of idiopathic pulmonary fibrosis associated with air pollution exposure. Eur Respir J. 2014;43:1124–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Dumas O, Despreaux T, Perros F, Lau E, Andujar P, Humbert M, Montani D, Descatha A. Respiratory effects of trichloroethylene. Respir Med. 2018;134:47–53. [DOI] [PubMed] [Google Scholar]
- 139. King CS, Nathan SD. Pulmonary hypertension due to interstitial lung disease. Curr Opin Pulm Med. 2019;25:459–67. [DOI] [PubMed] [Google Scholar]
- 140. Hoeper MM, Dwivedi K, Pausch C, Lewis RA, Olsson KM, Huscher D, Pittrow D, Grünig E, Staehler G, Vizza CD, Gall H, Distler O, Opitz C, Gibbs JSR, Delcroix M, Park DH, Ghofrani HA, Ewert R, Kaemmerer H, Kabitz HJ, Skowasch D, Behr J, Milger K, Lange TJ, Wilkens H, Seyfarth HJ, Held M, Dumitrescu D, Tsangaris I, Vonk‐Noordegraaf A, Ulrich S, Klose H, Claussen M, Eisenmann S, Schmidt KH, Swift AJ, Thompson AAR, Elliot CA, Rosenkranz S, Condliffe R, Kiely DG, Halank M. Phenotyping of idiopathic pulmonary arterial hypertension: a registry analysis. Lancet Respir Med. 2022;10(10):937–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Kolb M, Raghu G, Wells AU, Behr J, Richeldi L, Schinzel B, Quaresma M, Stowasser S, Martinez FJ. Nintedanib plus sildenafil in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2018;379:1722–31. 10.1056/NEJMoa1811737 [DOI] [PubMed] [Google Scholar]
- 142. Waxman AB, Jaramillo RR, Thenappan T, Engel P, Bajwa AA, Ravichandran AK, Feldman JP, Hajari Case A, Tapson VF, Smith P, Deng C, Shen E, Nathan SD. Long‐term effects of inhaled treprostinil in patients with pulmonary hypertension due to interstitial lung disease: the INCREASE study open‐label extension. Am J Respir Crit Care Med. 2022;205:A5671. [Google Scholar]
- 143. Launay D, Montani D, Hassoun PM, Cottin V, Le Pavec J, Clerson P, Sitbon O, Jaïs X, Savale L, Weatherald J, Sobanski V, Mathai SC, Shafiq M, Cordier JF, Hachulla E, Simonneau G, Humbert M. Clinical phenotypes and survival of pre‐capillary pulmonary hypertension in systemic sclerosis. PLoS One. 2018;13:e0197112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Hoeper MM, Pausch C, Grünig E, Klose H, Staehler G, Huscher D, Pittrow D, Olsson KM, Vizza CD, Gall H, Benjamin N, Distler O, Opitz C, Gibbs JSR, Delcroix M, Ghofrani HA, Rosenkranz S, Ewert R, Kaemmerer H, Lange TJ, Kabitz HJ, Skowasch D, Skride A, Jureviciene E, Paleviciute E, Miliauskas S, Claussen M, Behr J, Milger K, Halank M, Wilkens H, Wirtz H, Pfeuffer‐Jovic E, Harbaum L, Scholtz W, Dumitrescu D, Bruch L, Coghlan G, Neurohr C, Tsangaris I, Gorenflo M, Scelsi L, Vonk‐Noordegraaf A, Ulrich S, Held M. Idiopathic pulmonary arterial hypertension phenotypes determined by cluster analysis from the COMPERA registry. J Heart Lung Transplant. 2020;39:1435–44. 10.1016/j.healun.2020.09.011 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable as no new data generated.