

Abstract
A 12-year-old patient with mitochondrial DNA (mtDNA) depletion syndrome due to TK2 gene mutations has been evaluated serially over the last 10 years. We observed progressive muscle atrophy with selective loss of type 2 muscle fibers and, despite severe depletion of mtDNA, normal activities of respiratory chain (RC) complexes and levels of COX II mitochondrial protein in the remaining muscle fibers. These results indicate that compensatory mechanisms account for the slow progression of the disease. Identification of factors that ameliorate mtDNA depletion may reveal new therapeutic targets for these devastating disorders.
Keywords: COX II, Mitochondria DNA depletion, Muscle, Muscle fibers, Respiratory chain, TK2
1. Introduction
MtDNA depletion syndrome (MDS) comprises a group of Mendelian disorders in which mutations in nuclear genes cause quantitative alterations in mtDNA. Mutations in thymidine kinase 2 (TK2) gene cause progressive muscle disease with variable central nervous system involvement [1–3]. Deoxyguanosine kinase (DGUOK) and mtDNA polymerase γ (POLG) gene mutations are associated with encephalopathy and fatal infantile liver disease [4,5]. Deleterious mutation in SUCLA2, the gene encoding the β subunit of the ADP-forming succinyl-CoA synthetase ligase, is associated with encephalomyopathy and mtDNA depletion [6]. Finally, alterations in MPV17, cause infantile hepatocerebral mtDNA depletion and Navajo neurohepatopathy [7,8].
In a patient harboring TK2 mutations (T77M and R161K), in vitro activity of TK2 was markedly reduced [9]. At age 5, he underwent a muscle biopsy, which showed marked mtDNA depletion, but disproportionately modest reductions of the activities of mtDNA-encoded respiratory chain enzyme activities [9]. Here, we re-evaluate this patient who is now 12 years old, and report additional morphological and biochemical data obtained from three muscle biopsies obtained over 10 years.
2. Patient and methods
The clinical features of this 12-year-old patient have been reported [9]. Briefly, this second child of a non-consanguineous couple without family history of neuromuscular disorders was normal until age 19 months when he was noted to have difficulties standing. Physical examination showed weakness of neck flexor and limb–girdle muscles. Serum creatine kinase (CK) was mildly elevated (<400 IU/L, normal value: 30–150 IU/L). Nerve conduction velocities (NCV) were normal and electromyography revealed myogenic abnormalities. The muscle weakness progressed slowly. Currently, he is able to elevate his arms up to the shoulder level against gravity and to walk independently with a waddling gait and mild contractures of both Achilles tendons. He shows no signs of involvement of the central or peripheral nervous systems, liver, kidney, or gastrointestinal tract.
Biochemical analyses were performed in the last muscle biopsy, which was obtained for scientific reasons with the informed consent of his parents, at 12 years of age.
For western blot analyses, monoclonal antibodies against COX II and α-subunit of ATPase V (Molecular Probes, Leiden, The Netherlands) were used and bands were quantitated using densitometry. Southern blot analysis of muscle samples was performed as described [10].
3. Results
Histological studies of quadriceps muscle biopsies obtained at ages 2, 5 and 12 years old revealed progressively decreasing intensities of necrosis, inflammation, and the intra-cytoplasmic neutral lipid accumulation (Fig. 1A); however, within single sections of the first two biopsies, morphological features varied considerably. At age 2, muscle revealed a large number of strongly SDH-positive (ragged-blue) cells, which diminished in subsequent biopsies. At age 5, abundant cytochrome c oxidase (COX) negative fibers with normal SDH staining were seen. In the last biopsy, there were still many COX-negative fibers with normal SDH activity, and the sparse ragged-blue fibers were COX-negative. Over time, the proportion of type I fibers increased as shown by menadione staining (Fig. 1A). The ratios of type I:II fibers increased from 2.02 at age 2 to 3.4 at age 12 (Table 1).
Fig. 1.
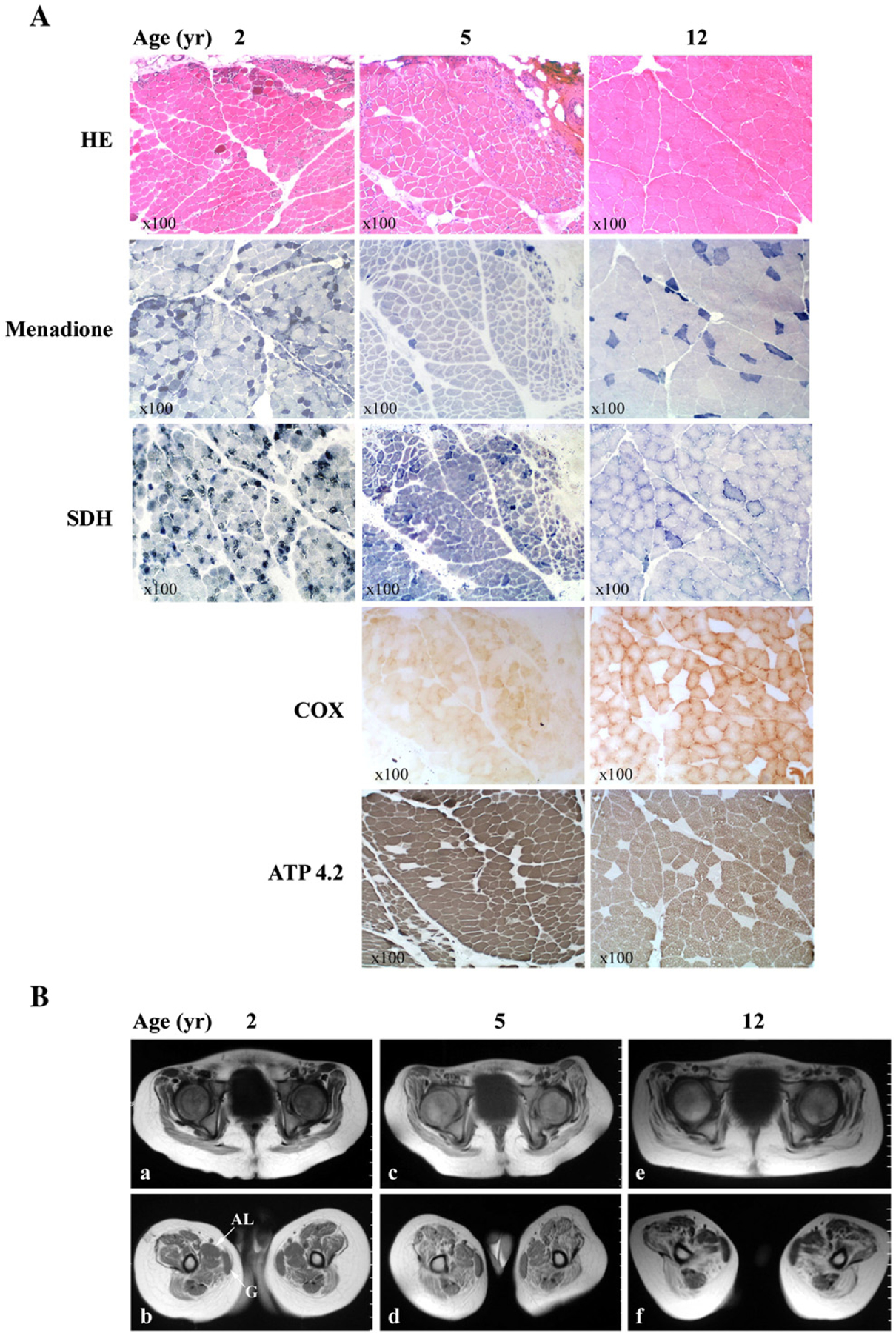
Histological (A) and MRI (B) studies of the patient’s quadriceps muscle samples obtained at 2, 5, and 12 years of age. A) Skeletal muscle stained with hematoxylin–eosin (H&E), menadione, succinate dehydrogenase (SDH), and serial sections taken at 5 and 12 years of age stained with cytochrome c oxidase (COX), and ATPase pH 4.2. B) Axial T1-weighted-TE (TR/TE 500/15) MR images of the patient’s thigh at two different levels at ages 2 (a, b), 5 (c, d) and 12 (e, f) years old, show progressive symmetric and extensive muscle atrophy with fatty infiltration affecting all muscle groups, but gracilis (G) and adductor longus (AL) muscles are relatively spared.
Table 1.
Measurement of total, type I and type II fibers in the three muscle biopsies
| Age (years) | 2 | 5 | 12 | |||
|---|---|---|---|---|---|---|
| % | % | % | ||||
| Total fibers | 145.0 | 100 | 190.0 | 100 | 101.0 | 100 |
| Type I | 97.0 | 66.9 | 170.0 | 89.5 | 78.0 | 72.3 |
| Type II | 48.0 | 33.1 | 20.0 | 10.5 | 23.0 | 22.7 |
| Ratio I:II | 2.02 | 8.5 | 3.4 | |||
| Average area (μm) | 1614.43 | 1668.16 | 5150.98 | |||
| Average minor ∅ (μm) | 37.66 | 34.59 | 63.31 | |||
| Fiber ∅ (Mm) from controlsa | 17.5 | 24 | 49.5 |
Mean fiber diameters for normal children biopsied at different ages (from Muscle Biopsy: A modern approach. Dubowith and Brooke 1973 W. B. Saunders Company Ltd. London, 4, 100).
T1-weighted MRI showed progressive and extensive muscle atrophy with fatty infiltration involving all muscle groups symmetrically and bilaterally with partial sparing of gracilis and adductor longus (Fig. 1B). Edema was absent on short tau inversion recovery (STIR) images.
At age 12, muscle showed severe mtDNA depletion (95% reduction) (Fig. 2A). This result was confirmed by real-time PCR (not shown). In contrast, the patient’s muscle showed normal activities of RC complexes (Table 2), and expression of COX II, a mtDNA-encoded protein, was normal compared to control muscles (Fig. 2B). Due to limited available biopsy material, SB and western blot were performed on the sample obtained at age 12, but could not be performed on muscle collected at ages 2 and 5.
Fig. 2.
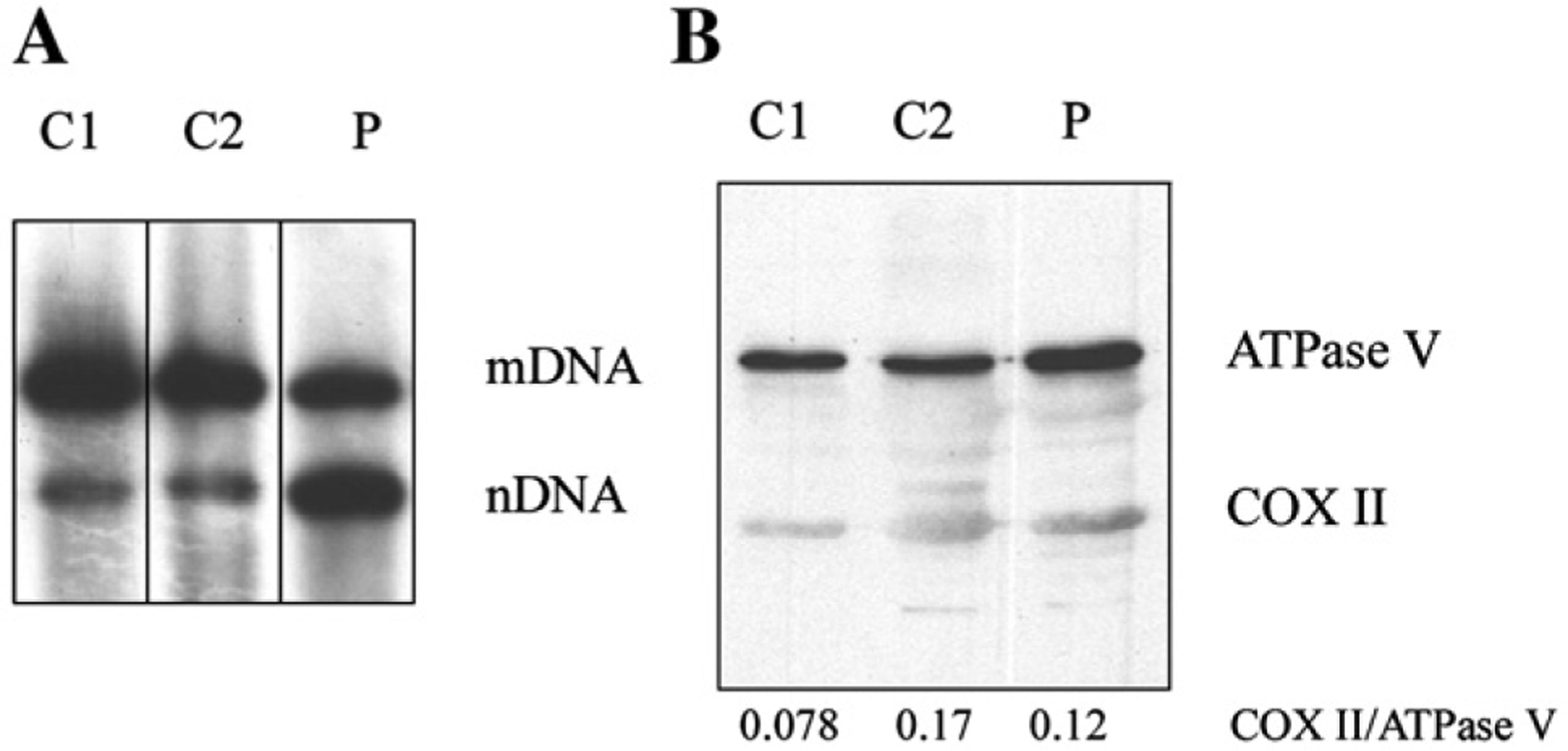
A) Southern blot analysis of total DNA obtained from the last muscle biopsy (age 12) of the patient (P) and two age-matched control muscle samples (C1 and C2), showed severe depletion of mtDNA. Probes for mtDNA and the nuclear DNA (nDNA) encoded 18 S rRNA genes have been used. B) Western blot analysis showed normal protein levels of COX II at age 12. Ratios of the mtDNA-encoded COX II relative to the nDNA-encoded ATPase V are indicated.
Table 2.
Values of respiratory chain enzyme activities
| nmol/min mg | mU/U CS | |||
|---|---|---|---|---|
| Patient | Ref. Value | Patient | Ref. Value | |
| C I+III | 22.93 | 12–56 | 221.3 | 107–560 |
| SDH | 23.44 | 10–25 | 226.2 | 57–239 |
| C II+III | 14.54 | 7–24 | 140.3 | 75–149 |
| C III | 72.54 | 55–259 | 700 | 610–1760 |
| C IV | 97.34 | 59–170 | 939.4 | 590–1300 |
| CS | 103.62 | 71–200 |
4. Discussion
Here, we describe progression of mitochondrial alterations in muscle of a patient with myopathic MDS and atypically slow progression and long survival. Despite very low levels of mtDNA in the last muscle biopsy, RC activities, and expression of mtDNA-encoded COX II protein were normal. Similar to two other patients with late-onset myopathic MDS with long survival, our patient had partial mtDNA depletion confined to muscle [3,10,11]. To account for this mild phenotype, we performed analyses of the histological and biochemical features of his muscle biopsies collected over between ages 2 and 12.
The morphological findings of our patient’s first muscle biopsy at age 2 are very similar to those of infants with the typical myopathic MDS. The similarities are probably related to the comparable ages and stages of the diseases of the patients at the time of their biopsies. However, unlike the typical rapid progressive myopathy and infantile death of most patients with TK2 mutations, our patient’s disease progressed slowly and allowed investigation of the evolution of the muscle changes. At age 5, necrosis and lipid accumulation were less prominent and by age 12, the remaining muscle fibers appeared normal with minimal endomysial fibrosis. One of the novel observations of this paper is the demonstration of simultaneous progressive loss of muscle mass and improvements of mitochondrial functions in the surviving muscle fibers. Thus, two processes may be occurring in the myopathy caused by TK2 mutations. In all cases, muscle initially develops variations in fiber size, vacuolation, increased lipid content, necrosis, and marked interstitial fibrosis with significant loss of muscle mass. At this point, as observed in most cases of myopathic MDS, the pathological alteration may rapid progress and can cause death in infancy. Alternatively, as demonstrated in our patient with prolonged survival, muscle atrophy may progress slowly with fatty infiltration (Fig. 1B), preservation of normal appearing muscle fibers, a selective loss of type II, and survival of type I fibers.
Molecular alterations could account for the prolonged progression of the myopathy in a subset of patients with TK2 mutations. The most straightforward explanation for the slow evolution of weakness would be mild deficiency of TK2 compared to typical patients. Biochemical characterization of each of the two mutant forms of TK2 protein in our patient, T77M and R161K showed drastic reductions in the catalytic rates, measured as increased Km and decreased Vmax, nevertheless, the heterodimeric T77/R161K enzyme could have higher activity than either homodimers [9]. This hypothetical mild TK2 deficiency is not entirely satisfactory in our patient because his biopsy at age 2 was similar to those of patients with the typical severe form of myopathic MDS. Moreover, measurement of TK2 activity in the patient’s fibroblasts revealed severe TK2 deficiency (7% residual activity relative to control) [9]. Alternatively, molecular adaptations could compensate for the mtDNA depletion. Indirect evidence from another patient with MDS supports this hypothesis; Barthélémy et al. described an 8-year-old boy who had an isolated myopathic form of MDS with severe mtDNA depletion (95% reduction of mtDNA) and normal activities of the RC enzymes. Steady-state levels of mitochondrial transcripts were also normal and indicating a transcriptional compensatory mechanism [11]. On re-evaluation of this patient at age 14, Vilà et al. identified pathogenic mutations in his TK2 gene [3]. Analysis of skeletal muscle biopsy revealed severe atrophy, diminished activities of respiratory enzymes, but normal levels of mtDNA. Restoration of normal mtDNA levels in muscle was attributed to atrophy or loss of myofibers with severe mtDNA depletion with selective survival of non-atrophic fibers harboring normal levels of mtDNA. These findings suggested that upregulation of mitochondrial transcription and mtDNA levels in surviving myofibers can ameliorate the clinical phenotype of patients with MDS [3].
The discordance between low amounts of mtDNA and normal levels of mitochondrial transcripts, proteins and respiratory chain enzyme activities is not unique to these two patients. In another patient with MDS due to TK2 deficiency, Durham et al. measured the minimum amount of wild-type mtDNA molecules required to maintain COX activity in skeletal muscle, which showed a mosaic of variable COX staining [12]. As in our patient, the authors found that some muscle retained residual COX activity despite a very low content of mtDNA molecules. They attributed this incongruity to either compensatory increases in transcription, as originally was suggested by Barthelemy et al. [11], or functional complementation by RC subunits synthesized in adjacent segments of the same muscle fiber containing a greater number of mtDNA molecules [12].
Compensatory molecular mechanisms probably exist in other forms of mtDNA depletion. In asymptomatic HIV-infected individuals treated with stavudine plus didanosine, peripheral blood monocytes showed mtDNA depletion, but retained normal levels of COX activity and COX II expression, indicating that transcriptional or post-transcriptional upregulation can compensate for drug-induced mtDNA depletion [13]. Similarly, in cultured Drosophila melanogaster cells with severe depletion of mtDNA, normal levels of mitochondrial transcripts were maintained [14]. Over expression of mitochondrial transcription factors could stabilize the few residual mtDNA molecules, and also increase their transcription rates, ensuring normal levels of mtDNA transcripts and protein synthesis.
We are investigating which factors might allow survival of a subpopulation of muscle fibers during the evolution of myopathy due to TK2 gene mutations. Identification of the compensatory mechanisms for mtDNA depletion may reveal new therapeutic targets for these devastating disorders.
Acknowledgements
We thank Antoni L. Andreu for helpful discussions and David Lligé for technical assistance. This work has been supported by Spanis Fondo de Investigación Sanitaria (FIS PI040415) (MRV) the FUNDISMUN Foundation and a grant from the Muscular Dystrophy Association (MH).
References
- [1].Saada A, Shaag A, Mandel H, Nevo Y, Eriksson S, Elpeleg O. Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nat Genet 2001;29:342–4. [DOI] [PubMed] [Google Scholar]
- [2].Mancuso M, Salviati L, Sacconi S, Otaegui D, et al. Mitochondrial DNA depletion: mutations in thymidine kinase gene with myopathy and SMA. Neurology 2002;59:1197–202. [DOI] [PubMed] [Google Scholar]
- [3].Vilà MR, Segovia-Silvestre T, Gámez J, Marina A, et al. Reversion of mtDNA depletion in a patient with TK2 deficiency. Neurology 2003;60:1203–5. [DOI] [PubMed] [Google Scholar]
- [4].Mandel H, Szargel R, Labay V, Elpeleg O, et al. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat Genet 2001;29:337–41. [DOI] [PubMed] [Google Scholar]
- [5].Naviaux RK, Nguyen KV. POLG mutations associated with Alpers’ syndrome and mitochondrial DNA depletion. Ann Neurol 2004;55: 706–12. [DOI] [PubMed] [Google Scholar]
- [6].Elpeleg O, Miller C, Hershkovitz E, Bitner-Glindzicz M, et al. Deficiency of the ADP-forming succinyl-CoA synthase activity is associated with encephalomyopathy and mitochondrial DNA depletion. Am J Hum Genet 2005;76:1081–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Spinazzola A, Viscomi C, Fernandez-Vizarra E, et al. MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat Genet 2006; 38:570–5. [DOI] [PubMed] [Google Scholar]
- [8].Karadimas CL, Vu TH, Holve SA, et al. Navajo neurohepatopathy is caused by a mutation in the MPV17 gene. Am J Hum Genet 2006;79: 544–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wang L, Limongelli A, Vilà MR, Carrara F, et al. Molecular insight into mitochondrial DNA depletion syndrome in two patients with novel mutations in the deoxyguanosine kinase and thymidine kinase 2 genes. Mol Genet Metab 2005;84:75–82. [DOI] [PubMed] [Google Scholar]
- [10].Vu TH, Tanji K, Valsamis H, DiMauro S, Bonilla E. Mitochondrial DNA depletion in a patient with long survival. Neurology 1998;51: 1190–3. [DOI] [PubMed] [Google Scholar]
- [11].Barthélémy C, Ogier de Baulny H, Diaz J, Cheval MA, et al. Late-onset mitochondrial DNA depletion: DNA copy number, multiple deletions, and compensation. Ann Neurol 2001;49:607–17. [PubMed] [Google Scholar]
- [12].Durham SE, Bonilla E, Samuels DC, DiMauro S, Chinnery PF. Mitochondrial DNA copy number threshold in mtDNA depletion myopathy. Neurology 2005;65:453–5. [DOI] [PubMed] [Google Scholar]
- [13].Miró O, Lopez S, Rodriguez de la Concepción M, Martinez E, et al. Upregulatory mechanisms compensate for mitochondrial DNA depletion in asymptomatic individuals receiving stavudine plus didanosine. J Acquir Immune Defic Syndr 2004;37:1550–5. [DOI] [PubMed] [Google Scholar]
- [14].Seidel-Rogol BL, Shadel GS. Modulation of mitochondrial transcription in response to mtDNA depletion and repletion in HeLa cells. Nucleic Acids Res 2002;30:1929–34. [DOI] [PMC free article] [PubMed] [Google Scholar]