

Abstract
Thiol dioxygenases are non-heme mononuclear iron enzymes that catalyze the O2-dependent oxidation of free thiols (-SH) to produce the corresponding sulfinic acid (-SO2–). Regardless of the phylogenic domain, the active site for this enzyme class is typically comprised of two major features: (1) a mononuclear ferrous iron coordinated by three protein-derived histidines and (2) a conserved sequence of outer Fe-coordination-sphere amino acids (Ser-His-Tyr) spatially adjacent to the iron site (∼3 Å). Here, we utilize a promiscuous 3-mercaptopropionic acid dioxygenase cloned from Azotobacter vinelandii (Av MDO) to explore the function of the conserved S-H-Y motif. This enzyme exhibits activity with 3-mercaptopropionic acid (3mpa), L-cysteine (cys), as well as several other thiol-bearing substrates, thus making it an ideal system to study the influence of residues within the highly conserved S-H-Y motif (H157 and Y159) on substrate specificity and reactivity. The values for these residues were determined by pH-dependent steady-state kinetics, and their assignments verified by comparison to H157N and Y159F variants. Complementary electron paramagnetic resonance and Mössbauer studies demonstrate a network of hydrogen bonds connecting H157–Y159 and Fe-bound ligands within the enzymatic Fe site. Crucially, these experiments suggest that the hydroxyl group of Y159 hydrogen bonds to Fe-bound NO and, by extension, Fe-bound oxygen during native catalysis. This interaction alters both the NO binding affinity and rhombicity of the 3mpa-bound iron–nitrosyl site. In addition, Fe coordination of cys is switched from thiolate only to bidentate (thiolate/amine) for the Y159F variant, indicating that perturbations within the S-H-Y proton relay network also influence cys Fe binding denticity.
Graphical Abstract
Enzymes involved in sulfur oxidation and transfer are increasingly being recognized as potential drug targets for the development of antimicrobials, therapies for cancer, and therapies for inflammatory disease.1–4 For instance, patients suffering from neurological disorders such as autism and Down syndrome have significantly lower plasma concentrations of the transsulfuration pathway and methionine cycle products [L-cysteine (cys), homocysteine (hcy), glutathione (gsh), and S-adenosylmethionine (SAM)].5,6 Imbalances in cys metabolism have also been identified in a variety of other neurological disorders (motor neuron disease, Parkinson’s disease, and Alzheimer’s disease).7–9 These observations suggest a potential correlation among impaired sulfur metabolism, oxidative stress, and neurodegenerative disease.5
Cysteine dioxygenase (CDO) and cysteamine (2-aminoethanethiol) dioxygenase (ADO) are the only known mammalian thiol dioxygenases. These enzymes use a single Fe(II) ion within their active site to catalyze the O2-dependent oxidation of sulfur-bearing amino acid derivatives without the need for an external reductant. Among these, mammalian CDO is the best characterized.10–13 This enzyme catalyzes the first committed step in the catabolic dissimilation of cys to produce inorganic sulfate, pyruvate, hypotaurine, and taurine. 14,15 The intracellular cys concentration is the limiting factor in glutathione synthesis; therefore, the activity of CDO directly competes with cellular redox buffering under conditions of low cys availability and oxidative stress.16
Among the structurally characterized enzymes, the thiol dioxygenase active site is comprised of two major features: (1) a mononuclear non-heme iron active site coordinated by three protein-derived histidines and (2) a conserved sequence of outer Fe-coordination-sphere amino acids (Ser-His-Tyr) spatially adjacent to the iron site (∼3 Å). By analogy to the chymotrypsin-like serine proteases, the Ser-His-Tyr (S-H-Y) network has historically been termed the “catalytic triad”.8,17–23 However, this terminology is not strictly correct as synchronized interactions within the S-H-Y proton relay network are not required for catalysis. Several CDO variants that introduce disruptions within the S-H-Y proton relay network have been characterized; while all variants exhibit decreased values relative to that of the wild type (5–20-fold), enzymatic activity is retained in nearly every instance.24–27 Therefore, the S-H-Y motif enhances the catalytic rate and efficiency but is not essential for catalysis.28 This property is inconsistent with the classic definition of a “catalytic triad” in biochemistry.29 We therefore refer to these residues collectively as the S-H-Y motif or proton relay network to avoid misrepresentation.
While this motif has been explored crystallographically,18 the functional relevance of these residues to catalysis remains largely underexplored. As shown in Figure 1A, a notable amendment to point (2) is observed in mammalian CDO. Here, the tyrosine (Y157) present within the S-H-Y motif of Rattus norvegicus CDO (Rn CDO) is covalently cross-linked with a spatially adjacent Cys residue (C93) to yield a C93–Y157 pair.30,31 This post-translational modification is unique to eukaryotic enzymes, whereas bacterial forms utilize an unmodified Tyr residue at this position.
Figure 1.

Structural comparison of mammalian CDO and P. aeruginosa 3MDO active site. (A) The 1.6 Å X-ray diffraction structure of cys-bound R. norvegicus CDO (PDB entry 4IEV).33 For the sake of clarity, the highly conserved S-H-Y motif containing a post-translationally modified C93–Y157 pair is shown below as an inset. Selected atomic distances are designated by dashed lines. (B) The 2.14 Å X-ray diffraction structure of the P. aeruginosa 3MDO active site (PDB entry 4TLF).32 Fe-coordinated solvent ligands designated by 1–3 (blue) and the S-H-Y motif are illustrated as an inset for comparison to CDO. (C) Side-on view of Pa3MDO illustrating the connectivity between Fe-bound solvents and the S-H-Y proton relay network.
Sequence comparison and X-ray diffraction (XRD) studies of annotated bacterial cdo genes reveal two subclasses of bacterial enzymes, which differ in a single outer Fe-coordination-sphere residue (Arg or Gln).17 Figure 1 compares the structure for the substrate-bound mammalian CDO [Protein Data Bank (PDB) entry 4IEV] active site with a “Gln-type” bacterial enzyme cloned from Pseudomonas aeruginosa (PDB entry 4TLF).32,33 In these enzymes, the 3-His facial triad motif is conserved; however, the outer-sphere Arg residue (R60) involved in electrostatic stabilization of the cys substrate is replaced by Gln (Q62). The spatial orientations of the conserved (Ser-His-Tyr) sequence remain invariant. Multiple independent reports demonstrate that the “Gln-type” class of enzymes is more appropriately designated as a 3-mercaptopropionic acid (3mpa) dioxygenase (MDO) rather than a subclass of CDO enzymes.21,22,32,34
The substrate-bound mammalian CDO (Figure 1A) reveals a bidentate coordination of cys to the mononuclear Fe site via a thiolate and neutral amine.25,35,36 Both kinetic and spectroscopic studies demonstrate additional interactions between the cys-bound Fe site and outer-sphere residues (Y157 and R60) within the CDO active site.18,24 These multiple points of interaction are likely responsible for the high substrate specificity exhibited by this enzyme.30 To illustrate, in steady-state reactions using cysteamine (2-aminoethanethiol) (ca) as a substrate in place of cys, a 10000-fold decrease in is reported.30 Therefore, removal of the cys carboxlate has a profound impact on the formation of the CDO ES complex. No catalytic activity whatsoever is observed for thiol substrates lacking an amine functional group.
By contrast, MDO is a promiscuous thiol dioxygenase, capable of accommodating a variety of thiol-bearing substrates over a broad pH range. Indeed, pH (and pD)-dependent steady-state kinetic studies have been reported for this enzyme using 3-mercaptopropionic acid (3mpa), L-cysteine (cys), and cysteamine (2-aminoethanethiol) (ca) as substrates.21,22,32,34 While structures for the substrate-bound MDO are unavailable, a key mechanistic divergence between mammalian CDO and MDO can be inferred from the pH-dependent kinetics of recombinant MDO cloned from Azotobacter vinelandii (Av MDO).
A review of both and data obtained for steady-state reactions with 3mpa, cys, and ca supports the conclusion that interactions between the enzyme and substrate carboxyl represent an important but non-essential interaction for catalysis. To illustrate, reactions of MDO with ca exhibit only a modest decrease in values () relative to those of reactions performed with 3mpa and cys (1.0–1.2 s–1).22 This observation suggests that the rate-limiting chemical steps following formation of the enzyme–substrate (ES) complex are not significantly influenced by the substrate carboxylate group.37 However, the ∼700-fold decrease in obtained in reactions of wild-type Av MDO with 3mpa (72000 ± 9200 M−1 s−1) as compared to that of cys reactions (110 ± 20 M−1 s−1) indicates that the positively charged substrate amino group (present in cys) has a large negative impact on nonchemical events leading up to formation of ES.22 As pointed out by Jameson et al., this can likely be attributed to destabilizing electrostatic interactions between a cationic amino-bearing substrate and the nearby R168.34,38 Because 3mpa lacks an amino functional group, direct comparison of ca and 3mpa data is not appropriate. Instead, a better gauge of the influence of the substrate carboxylate group on catalysis is obtained through comparison of the value determined for cys reactions with that obtained with ca (11 ± 2 M−1 s−1). Here, only a 10-fold difference is observed; therefore, with respect to the reversible nonchemical events leading up to formation of the Av MDO ES complex, introduction of an amino group to the thiol substrate is vastly more impactful than removal of a carboxylate group. In addition, the absence of a substrate carboxylate group has a minimal influence on the rate-limiting chemical steps following formation of the ES complex.
These observations are crucial as both Mm CDO and Av MDO follow an obligate-ordered addition of the substrate prior to oxygen. Thus, direct substrate coordination to the Fe site and subsequent oxygen activation occur only following formation of the catalytically relevant ES complex. For Mm CDO and Av MDO, this obligate-ordered addition also extends to the binding of nitric oxide.22,36 Previous electron paramagnetic resonance (EPR) studies demonstrate formation of iron–nitrosyl species upon addition of nitric oxide to samples of Av MDO preincubated with excess 3mpa, cys, ca, and ethanethiol (et).22 Because both ca and et lack a carboxylate functional group, it is difficult to rationalize how coordination of a substrate carboxylate group to the enzymatic Fe site is a requisite component of the O2-activating ES complex.22 On the basis of these observations, we have argued that binding of the substrate to the Av MDO Fe site occurs via thiolate-only coordination. In our view, this model provides the simplest explanation for all experimental observations.
Jameson and Karplus recently published an AutoDock Vina (ADV) model for 3mpa docking in the Pa3MDO active site, which supports the presence of a catalytically essential salt bridge (∼3.2 Å) between the R168 guanidinium group and 3mpa carboxylate.38 This model was additionally used as a basis to propose a bidentate 3mpa Fe coordination via substrate thiol and carboxylate functional groups. Structurally, this model closely overlays with cys binding to mammalian CDO (shown in Figure 1A), and the proposed salt bridge provides a reasonable explanation for the decreased affinity for amino-bearing substrates. While substitution of a neutral cys amine () for an anionic 3mpa carboxylate () is expected to alter the non-heme iron redox potential, and thus the mechanism of oxygen activation, the influence of this perturbation on the first Fe-coordination sphere was not discussed. However, as noted above, MDO and CDO exhibit vastly different behavior with respect to substrate promiscuity; therefore, arguing in favor of a bidentate 3mpa coordination for the sake of structural continuity with CDO may not be justified. Most critically, the proposed bidentate 3mpa Fe binding model for the MDO fails to address the apparent inconsistencies with the aforementioned kinetic and EPR experiments reported for Av MDO.21,22 Consequently, there is currently no consensus about the nature of 3mpa Fe coordination (thiolate only vs bidentate) within the ES complex.
Second-sphere residues are frequently employed to modulate the reactivity and or stability of transient Fe–oxo intermediates generated by non-heme oxidase/oxygenase enzymes. For example, within the active site of 2,3-dioxygenase, a second-sphere interaction with a protonated (positively charged) histidine (H200) has a profound influence on the lifetime of transient intermediates as well as the ultimate product formation (intradiol vs extradiol cleavage).39–41 Thus, it is expected that second-sphere interactions also play a pivotal role in regulating thiol dioxygenase reactivity.
With the notable exception of mammalian ADO,42 the S-H-Y motif appears to be conserved among thiol dioxygenases across phylogenic domains. Potentially related, ADO is also the only thiol dioxygenase identified which catalyzes the oxidation of a non-carboxylate bearing thiol-substrate (cysteamine). It is therefore reasonable that this sequence motif serves a critical function in catalysis and/or substrate specificity. The promiscuity of Av MDO, its stability across a broad pH range, and the absence of post-transcriptional modification make this enzyme an ideal system for interrogating the influence of the S-H-Y motif on substrate specificity and catalysis. Comparison of wild-type pH-dependent steady-state kinetics to those of H157N and Y159F variants confirms the assignment of ionizable groups in the pH profile. Supporting pH-dependent kinetic, Mössbauer, and EPR studies performed on wild-type and variant enzymes provide evidence of a hydrogen bonding network connecting H157, Y159, and the Fe-bound ligand (solvent and nitric oxide) within the active site. EPR characterization of the (substrate/NO)-bound enzyme indicates that disruption of this network significantly perturbs the NO binding affinity, coordination geometry, and heterogeneity, as well as the denticity of cys coordination at the Fe site.
MATERIALS AND METHODS
Expression and Purification of Av MDO and Selected Variants.
The isopropyl -d-1-thiogalactopyranoside (IPTG) inducible T7 vector for Av MDO (designated pRP42) was a generous gift from T. Larson (Department of Biochemistry, Virginia Tech, Blacksburg, VA). The pRP42 vector was transformed into chemically competent BL21(DE3) Escherichia coli (Novagen, catalog no. 70236–4) by heat shock (42 °C for 45 s) and grown overnight at 37 °C on a lysogeny broth (LB)43 agar plate in the presence of 100 mg/L ampicillin (Amp). The following day, a single colony was selected for growth in liquid LB (Amp) medium for training on an antibiotic prior to inoculation of a 10 L BF-110 fermentor (New Brunswick Scientific) at 37 °C. The optical density at 600 nm (OD600) was used to monitor cell growth. Induction was initiated by the addition of 1.0 g of IPTG, 78 mg of ferrous ammonium sulfate, and 20 g of casamino acids at an OD600 value of ∼4. At the time of induction, the temperature of the bioreactor was decreased from 37 to 25 °C and agitation was set to maintain an O2 concentration of 20% relative to that of air-saturated media. After 4 h, the cells were harvested and pelleted by centrifugation (Beckman-Coulter Avanti J-E, JA 10.5 rotor) at 18600×g for 15 min. Cell paste was stored at −80 °C prior to purification.
In a typical purification, ∼20 g of frozen cell paste was added to 150 mL of extraction buffer [20 mM HEPES and 50 mM NaCl (pH 8.0)]. Lysosyme, ribonuclease, and deoxyribonuclease were added to the slurry to final concentrations of 10 μg/mL each, and the mixture was stirred slowly on ice for 30 min. The resulting slurry was pulse sonicated (Bronson Digital 250/450) for 15 s on/off at 60% amplitude for a total time of 15 min. The insoluble debris was removed from the cell free extract by centrifugation at 48000×g for 1 h at 4 °C. The supernatant was diluted 1:1 with extraction buffer and then loaded onto a DEAE sepharose fast flow anion exchange column [7 cm (width) × 20 cm (length)] (GE Life Sciences, catalog no. 17070901) pre-equilibrated with 20 mM HEPES and 50 mM NaCl (pH 8.0). The column was washed with 3 column volumes of extraction buffer prior to elution in a linear NaCl gradient (from 50 to 350 mM). Fractions (∼10 mL) were collected overnight and pooled on the basis of enzymatic activity as described elsewhere.22,36 Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) was also used to verify the presence of the recombinant protein (∼23 kDa) in each fraction. Broad range protein molecular weight markers utilized in SDS–PAGE experiments were purchased from Promega (Madison, WI) (catalog no. V8491). The pooled fractions were concentrated to approximately 5–10 mL using an Amicon stir cell equipped with an YM-10 ultrafiltration membrane. Although the expressed Av MDO has a C-terminal His tag, as compared to the DEAE AX method described above, use of immobilized metal affinity chromatography (IMAC) resulted in substantially lower enzymatic activity and yields due to the loss of iron and protein denaturation. Therefore, thrombin protease (Biopharma Laboratories) was added to cleave the C-terminal His tag from Av MDO. In a typical reaction, ∼0.3 molar equivalent of thrombin per Av MDO [based on ultraviolet–visible (UV–vis) absorbance at 280 nm] was added to batches of purified protein for overnight cleavage at 4 °C in HEPES buffer. The remaining thrombin and free (His)6 tag were removed from Av MDO by size exclusion chromatography using a sephacryl S100 column.
All Av MDO variants (H157N and Y159F) were prepared using the QuikChange Lightning Site-Directed Mutagenesis kit (Agilent Technologies). Mutagenic primers (Table S1) were purchased from Integrated DNA Technologies (https://www.idtdna.com), and sequence verification of single-amino acid mutations was performed by Sequetech (Mountain View, CA, http://sequetech.com/). The 57Fe-enriched Av MDO enzyme was prepared for Mössbauer study by growing pRP42-transformed BL21(DE3) cells in M9 media where 57Fe was used as the only iron source during growth and expression of bacteria. All other purification procedures were as described above.
Iron Analysis.
For all batches of Av MDO used in these experiments, spectrophotometric determination of ferrous and ferric iron content was performed using 2,4,6-tripyridyl-s-triazine (TPTZ) according to previously published methods.22,36,44 Briefly, a 200 μL aliquot of ∼100 μM enzyme was hydrolyzed in a microfuge tube by addition of 250 μL of 2 N HCl, 250 μL of 20% (w/v) trichloroacetic acid, and 300 μL of Milli-Q H2O. The sample was heated to 95 °C for 15 min before centrifugation to pellet precipitated protein. Following centrifugation, 750 μL of the sample was added to 300 μL of 1.0 mM TPTZ, 3.0 M acetic acid, and 0.4 mM hydroxyl amine (NH2OH) (pH 6.0). The resulting solution was allowed to equilibrate for 30 min at ambient temperature to ensure complete reduction of all ferric iron to the ferrous oxidation state. The total iron concentration was measured spectrophotometrically at 596 nm ().44 The amount of ferrous iron present in purified Av MDO was determined as described above, except in the absence of a reductant (NH2OH). The amount of ferric iron within samples was determined by difference. For the sake of clarity, the concentrations reported in enzymatic assays reflect the concentration of ferrous iron within samples of Av MDO (FeII-MDO). Protein content was determined by the Bio-Rad protein assay.
Enzyme Assays.
3-Sulfinopropionic acid (3spa) and cysteine sulfinic acid (csa) were assayed using the HPLC method described previously. Instrumental conditions: column, Phenomenex C18 (100 mm × 4.6 mm); mobile phase, 20 mM sodium acetate, 0.6% methanol, and 1% (v/v) heptafluorobutyric acid (pH 2.0). Analytes were detected spectrophotometrically at 218 nm. Reactions (1 mL) were prepared in a buffered solution at the desired pH to obtain a final concentration from 0.1 to 10 mM 3mpa (0.1–60 mM cys). Each reaction was initiated by addition of Av MDO (typically 0.5–1.0 μM) at 20 ± 2 °C. Sample aliquots (250 μL) were removed from the reaction vial at selected time points, and reactions quenched by addition of 10 μL of 1 N HCl (final pH of 2.0). Prior to HPLC analysis, each sample was spin-filtered through a 0.22 μm cellulose acetate membrane (Corning, Spin-X). The product concentration was determined by comparison to calibration curves as described elsewhere.22,30
The rate of dioxygen consumption was determined polarographically using a standard Clark electrode (Hansatech Instruments, Norfolk, England) within a jacketed 2.5 mL cell at 20 ± 2 or 37 ± 2 °C for wild-type or variant enzymes, respectively. The electrode was bathed in a saturated solution of KCl and separated from the buffer using a gas-permeable membrane. The O2 electrode was calibrated as previously described.21,22,30,45 Briefly, an electrode response factor was determined by measuring the voltage deflection upon adding ∼500 units of catalase (Sigma-Aldrich, St. Louis, MO) to a buffered solution containing a known concentration of H2O2 [] at a fixed temperature. The amount of molecular oxygen produced can be calculated on the basis of the known 2:1 stoichiometry of hydrogen peroxide consumed per mole of oxygen produced in catalase reactions. Standardization of initial hydrogen peroxide concentration in solution was performed using the amplex red hydrogen peroxide/peroxidase kit for spectrophotometric determination () of H2O2 (Invitrogen, catalog no. A22188). Once the reaction reached completion, the amplitude of the voltage change was used to determine a response factor for the electrode. For all assay conditions, the sample conditions (temperature, salt concentration, and pH) were carefully matched to those used in enzymatic assays to eliminate any potential influence these physical parameters may have on the electrode response factor. All reactions were initiated by addition of 1.0 μM Av MDO under identical buffer conditions as described for HPLC assays.
The reaction buffers for all pH profile experiments consisted of 20 mM Good’s buffer and 50 mM NaCl. 2-(N-Morpholino)ethanesulfonic acid (MES) was used to buffer reactions over the pH range of 5.5–6.7, and 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES) was used to buffer reactions over the pH range of 6.8–8.4. 2-(Cyclohexylamino)ethanesulfonic acid (CHES) was used to buffer reactions over the pH range of 8.5–10. Reaction temperatures were maintained by a circulating water bath (ThermoFlex 900, Thermo Scientific).
Data Analysis.
In the absence of substrate inhibition, initial rate data for product formation and/or O2 consumption were fit to the Michaelis–Menten equation (eq 1) using SigmaPlot version 11.0 (Systat Software Inc., Chicago, IL). From this analysis, both the kinetic parameters ( and ) and the error associated with each value were obtained by nonlinear regression. Under conditions of substrate inhibition (Y159F Av MDO), initial rate data were fit to eq 2 to determine the kinetic parameters ( and ) in addition to the inhibition constant ().
| 1 |
| 2 |
For data sets in which – or plots exhibit limiting non-zero plateaus at low () and high () pH, followed by a transition region and a subsequent decrease beyond , the results were fit to eq 3.21,46 Here, Y is defined as either or , and [H], , and represent the hydrogen ion concentration and the two observable dissociation constants for ionizable groups involved in catalysis, respectively.
| 3 |
For results for which – or profiles decreased only at low or high pH, the data were fit to eq 4.37,47,48 This expression is scaled by a constant scalar quantity (C) that represents the maximum kinetic rate ( or ). The error associated with each parameter (, , , and ) determined from fits to eqs 1 and 2 is reported in Table 1.
Table 1.
Comparison of Steady-State Kinetic Parameters Determined for Wild-Type Av MDO and Selected Variants for 3mpa Reactions
| wild type (20 °C) | H157N (37 °C) | Y159F (37 °C) | |
|---|---|---|---|
| 0.45 ± 0.06 | 0.092 ± 0.003 | 0.081 ± 0.001a | |
| 13 ± 5 | 12 ± 2 | 9 ± 2 | |
| 27000 ± 3000 | 7800 ± 590 | 9000 ± 2700 | |
| coupling (%) | 98 ± 6 (7.5)b | 46 ± 6 (7.5)b | 60 ± 4 (7.5)b |
| 7.8 ± 0.2 | – | – | |
| 9.2 ± 0.1 | 9.6 ± 0.1 | – | |
| 7.4 ± 0.2 | – | – | |
| 9.1 ± 0.1 | 9.5 ± 0.1 | – | |
| fit to eq | 2 | 3 | |
| refs | 21, 22 | this work | this work |
Y159F – and data optimal at the lowest pH values measured (5.5 ± 0.1).
pH value at which the coupling efficiency Y159F log(k /K ) was measured.
| 4 |
Physical Methods.
All UV–visible measurements were taken on an Agilent (Santa Clara, CA) 8453 photodiode array spectrometer using ES Quartz cuvettes (NSG Precision Cells, Farmingdale, NY). X-Band (9 GHz) EPR spectra were recorded on a Bruker (Billerica, MA) EMX Plus spectrometer equipped with a bimodal resonator (Bruker model 4116DM). Low-temperature measurements were taken using an Oxford ESR900 cryostat and an Oxford ITC 503 temperature controller. A modulation frequency of 100 kHz was used for all EPR spectra. All experimental data used for spin quantification were collected under nonsaturating conditions.
Analysis of the EPR spectra utilized the spin Hamiltonian
| 5 |
where D and E are the axial and rhombic zero-field splitting (zfs) parameters, respectively, and g is the g tensor.49 EPR spectra were simulated and quantified using SpinCount written by one of the authors. The simulations were generated upon considering all intensity factors, both theoretical and experimental, to allow for determination of species concentration. The only unknown factor relating the spin concentration to signal intensity was an instrumental factor that is specific to the microwave detection system. This factor was determined by a Cu(EDTA) spin standard prepared from a copper atomic absorption standard solution purchased from Sigma-Aldrich.
Mössbauer spectra were recorded with a spectrometer operating in a constant acceleration mode in a transmission geometry using Janis Research Inc. cryostats that allow for a variation in temperature from 4 to 300 K. Isomer shifts are reported relative to Fe metal at 298 K. Spectral fitting was performed using SpinCount.
EPR/Mössbauer Sample Preparation.
EPR samples of Av MDO were prepared in a glovebox (Coy Laboratory Products Inc., Grass City, MI) with the O2 concentration maintained below 1 ppm. Solutions were degassed on a Schlenk line prior to being transferred into the anaerobic chamber. Analytical grade argon was passed through a copper catalyst (Kontes, Vineland, NJ) to remove trace O2 impurities and then sparged through distilled water to hydrate the gas. Stock solutions of the cys or 3mpa substrate were prepared in anaerobic 20 mM HEPES and 50 mM NaCl, and the pH was adjusted to the desired value prior to addition to the enzyme. Stock NO solutions were prepared by dissolving 5 mg of PROLI-NONOate (Cayman Chemical, 178948–42-0) in a 1.0 mL solution of anaerobic 10 mM sodium hydroxide. Aliquots of degassed stock PROLI-NONOate (20–50 μL) were diluted 1:1 with deoxygenated buffer and allowed to react for 5 min. At pH 7.4, 2 mol of NO is released with each mole of NONOate with a half-life of 6 s at ambient temperature.50,51 Samples of (substrate/NO)-bound enzyme were prepared anaerobically by the addition of 10 molar equivalents of a 22 mM stock NONOate solution to the enzyme preincubated with 10 molar equivalents of the substrate. Samples were allowed to equilibrate on ice for 10 min prior to the sample being transferred into a 4 mm quartz EPR tube (707-SQ-250MM, Wilmad-Lab Glass) with a 250 mL Hamilton gastight syringe equipped with a 6 in. needle. Finally, EPR tubes were frozen slowly in liquid N2 under anaerobic conditions prior to analysis. Note that in some batches of the purified H157N Av MDO variant, partial autoxidation of the enzyme was observed during concentration. Therefore, to ensure full reduction of the H157N (3mpa-bound) enzyme shown in Figure 5, a slight excess (10% relative to iron) of sodium dithionite and methyl viologen was added. This addition had no influence on the observed g-values or zfs parameters.
Figure 5.

X-Band EPR spectra of iron–nitrosyl species produced upon addition of NO to the 3mpa-bound (A) and cys-bound (B) enzyme. (A) Comparison of wild-type (3mpa/NO)-bound Av ES-NO relative to H157N and Y159F variants. For the sake of clarity, all spectra are normalized for Fe concentration. For each spectrum, quantitative simulations (dashed lines) are overlaid on the data for comparison. The isotropic signal shown in the H157N data is an impurity from addition of methyl viologen and dithionite to the sample. (B) EPR spectra for wild-type (cys/NO)-bound Av ES-NO as compared to an equivalent sample prepared with the Y159F variant. The sharp signal in samples prepared with Y159F is attributed to minor dinitrosyl iron species, a common contaminant produced by addition of NO to Fe-containing enzymes.70–73 The black circles (●) designate the more rhombic (cys/NO)-bound confirmation with observed g-values of 4.31, 3.75, and 1.98. Instrumental parameters: microwave frequency, 9.643 GHz; microwave power, 20 μW; modulation frequency, 0.9 mT; temperature, 10 K. The spectroscopic parameters obtained from data in panels A and B are summarized in Tables 3 and 4.
Mössbauer samples were prepared under identical anaerobic conditions and reduced as described for EPR samples. All samples were prepared from a stock solution of 57Fe-enriched enzyme in which the Fe content was verified by the colorimetric assay described above (Iron Analysis). For samples of the enzyme–substrate complex, 250–300 μL aliquots of the 1–3 mM enzyme were mixed with an anaerobic solution containing 10–15 mM substrate (3mpa or cys) titrated to the desired pH at 25 °C. The method used for determining the “apparent pH” for cryogenic samples is presented in the Supporting Information. After each addition, samples were allowed to equilibrate for 15 min on ice prior to being pipetted into Mössbauer cups. Each sample was then frozen slowly (over 1–2 min) by immersion in liquid N2 within the anaerobic chamber.
RESULTS
Steady-State Kinetics of H157N and Y159F Variants of Av MDO.
The wild-type enzyme and selected variants were purified and assayed for iron content and activity as described in Materials and Methods. Expression of this enzyme yields a monomeric protein with a molecular weight of 23 kDa. For each variant (H157N and Y159F), the stoichiometry of iron per protein was determined by TPTZ, Bradford assay, and UV–vis spectroscopy as described previously.36 For the wild-type enzyme, both the iron content (0.83 ± 0.16 mol of Fe per protein) and steady-state kinetic parameters observed at 20 °C were consistent with previously published values21,22 (; , 13 ± 5 μM; , 27000 ± 3000 M–1 s–1). For the sake of clarity, the value of is defined by the maximal velocity normalized for enzyme concentration (). By contrast, both variants have depleted iron content and attenuated specific activity relative to those of the wild-type enzyme. The average ferrous iron contents determined by the TPTZ assay for the H157N and Y159F variants were 0.50 ± 0.04 and 0.63 ± 0.03 mol of Fe per protein, respectively. For wild-type and variant enzymes, >90% of the iron present in samples was observed in the catalytically relevant ferrous oxidation state. To avoid introduction of adventitious iron, which could potentially interfere with kinetic and spectroscopic characterization, reconstitution of purified enzyme variants with iron was not attempted. Instead, concentrations reported in enzymatic assays and spectroscopic experiments reflect the concentration of ferrous iron within samples.22
Oxygen is a co-substrate for dioxygenase reactions; therefore, control experiments were performed to demonstrate that the amount of dissolved oxygen present in buffered 3mpa reactions is sufficient to saturate steady-state kinetics for both the wild type and selected variants. These and others experiments establishing the efficiency at which wild-type and variant enzymes incorporate 1 mol of O2 into the final 3spa product are presented in Figures S1 and S2.
A common method for identifying catalytically relevant proton-dependent ionizations within the (O2/3mpa)-bound enzyme ternary complex is to monitor the influence of pH on . Perturbations within the profile are diagnostic of the irreversible “chemical steps” involved in catalysis. Conversely, information relating to the “non-chemical” reversible steps leading up to the first irreversible process can be obtained from the profile. Beyond a relaxed substrate specificity, Av MDO is also remarkably tolerant to pH changes. This property allows for the measurement of pH-dependent steady-state kinetics for each substrate (3mpa, cys, and ca) over a [H+] range spanning 5 orders of magnitude (5 < pH < 10).21,22 For all of the substrates that were studied, the pH-dependent profile can be rationalized assuming a diprotic enzyme model in which three ionic forms of the enzyme are present [cationic, ; neutral, ; anionic, ].21 Scheme 1 illustrates the kinetic model for the wild-type enzyme resulting in the skewed “bell-shaped” curve in Av MDO–3mpa reactions (Figure 2A). This indicates that electrostatic interaction within the enzymatic active site is the dominant factor driving substrate specificity. This behavior is conserved for both Av MDO and Mm CDO.45
Scheme 1.

Diprotic Kinetic Model for Wild-Type Av MDO-Catalyzed 3mpa Reactions
Figure 2.

Wild-type Av MDO profile for 3mpa reactions (A, ●) as compared to those of H157N (B) and Y159F (C) variants. The profile for Mm CDO-catalyzed cys reactions is shown in panel A (■). The data for both variants were collected at 37 ± 2 °C due to the decreased value of , whereas wild-type data were collected at 20 ± 2 °C. Equations 1 and 2 were used to fit wild-type and H157N variant results, respectively. A standard linear equation was used to fit the pH dependence of data obtained with the Y159F variant. Table 1 summarizes all kinetic parameters obtained from pH-dependent results.
On the basis of (1) the values observed in and data, (2) steady-state results obtained previously for H155A and Y157F Mm CDO variants, and (3) independent EPR and MCD spectroscopic studies with supporting computational modeling,23,24 it was argued that the ionizable groups observed in the profile were associated with the conserved His-157 and Tyr-159 residues within the MDO S-H-Y motif.21 Here, direct assignment of these ionizable residues is made by comparison of data obtained for the active site variants (H157N and Y157F) of Av MDO.
The influence of pH on H157N Av MDO catalysis was measured over the accessible pH range of the enzyme (5.5 < pH < 10). As discussed previously, the rate of 3spa product formation was independent of oxygen concentration for the entire pH regime used in these experiments (Figure S1). The data obtained from the initial rate of H157N variant reactions with 3mpa are shown in Figure 2B. For comparison, the profiles obtained for wild-type Av MDO (■) and Mm CDO (●) are provided in Figure 2A. Each point within these data sets was obtained by fitting the steady-state kinetic results observed at a fixed pH value to the Michaelis–Menten equation (eq 1). The error in each kinetic parameter obtained by nonlinear fitting is indicated graphically by the error bars.
The profile for the wild-type enzyme in 3mpa reactions exhibits a skewed “bell-shaped” curve with two apparent values. Within the acidic branch of this profile (5.5 < pH < 8), both data sets exhibit sigmoidal behavior bracketed by limiting non-zero plateaus at low and high pH. Following a short transition, values decrease beyond with a unitary slope. Over the entire pH range utilized in these experiments, these results can be reasonably fit to eq 3 (solid line). The limiting values of span from to at 20 °C.
By contrast, the data obtained for the H157N variant exhibit only a single ionizable group with a maximum value within the pH-independent region of 0.092 ± 0.003 s−1. Despite the ∼5-fold decrease in observed for the H157N MDO, the observed value (12 ± 2 μM) remains largely invariant over the pH range. Within error, this value is equivalent to that obtained for the wild-type enzyme (13 ± 5 μM). Because the rate of catalysis decreases only at high pH, these results were fit to eq 4 (solid line) to obtain a value of 9.6 ± 0.1. Critically, the absence of in the H157N variant profile (Figure 2B) verifies that this ionizable group is associated with H157. Moreover, relative to the wild-type enzyme , the second midpoint ionization is shifted more basic range (9.6 ± 0.1) for the H157N-catalyzed 3mpa reactions. This perturbation () is well above the statistical error of the assay. A summary of all pH-dependent kinetic results obtained for the wild-type and variant enzymes is provided in Table 1.
Similar pH-dependent steady-state experiments were performed for the Y159F Av MDO variant. Unlike either H157N or the wild-type enzyme, the Y159F variant exhibited substrate inhibition behavior in steady-state kinetic experiments. As illustrated in Figure S3, the extent of this substrate inhibition is pH-dependent. Therefore, rather than the standard Michaelis–Menten equation, Y159F initial rate data were fit to eq 2 to obtain steady-state kinetic parameters ( and ) as well as the inhibition composite constant (). A summary of the kinetic parameters (, , and ) determined at selected pH values and an analysis of these results are provided in Table S2 and Figure S3.
The data obtained for the Y159F variant (triangles) are shown in Figure 2C. Similar to that of the H157N variant, the maximum activity of the Y159F variant is ∼5-fold lower than that of the wild-type enzyme with little perturbation to the observed value obtained (9 ± 2 μM). However, unlike either the wild type or H157N, the Y159F variant exhibits a linear decrease in with an increase in pH. To illustrate, a linear fit (straight line) is overlaid on the Y159F variant results (). Although an increase in pH attenuates enzymatic activity (∼10% decrease in per pH unit), no evidence of either catalytically essential ionization ( or ) is found for the Y159F variant. The linear decrease in activity is likely due to pH-dependent changes in the Fe site redox potential rather than the increased ionic strength from excess −OH as the salt content has little influence on the rate of catalysis.21,22 Regardless, the absence of in this pH profile verifies that Y159 is the catalytically essential ionization associated with (9.2 ± 0.1) for the wild-type enzyme.
As stated previously, these assays provide insight into rate-limiting ionizations within the (3mpa/O2)-bound enzyme ternary complex. While similar analysis can be made from interpretation of the profile to probe protonation events in the reversible “nonchemical” steps leading up to O2 activaiton, these results do not substantially deviate from what is presented above and thus will not be discussed further.
Mössbauer Studies of Resting and Substrate-Bound Av MDO.
Figure 3A shows the spectra collected for samples prepared at pH 6.5 and 9.5. For the resting, free Av MDO enzyme at pH 6.5, the observed zero-field Mössbauer spectrum was fit to two doublets: and from 54% of iron, and and from 46% of iron (Table 2). At pH 9.5, the parameters of both doublets changed slightly, but the relative contributions of each doublet are reversed (, 41%; , 59%). Fitting with a single doublet gave a broad line width of >0.5 mm/s, whereas a fit with two doublets gave a normal line width of <0.4 mm/s. As illustrated in Figure 1C, a Fe-bound solvent (2) ligand is within hydrogen bonding distance (2.67 Å) of the Y159 phenol group. Therefore, ionization of Y159 may affect ligands that are coordinated to iron (i.e., the first Fe-coordination sphere). To study this possibility, samples of Av MDO were prepared over a pH range of 6.5–9.5 and Figure 3 shows the relative amounts of the two species.
Figure 3.

pH-dependent speciation of Mössbauer doublets in Fe(II)-Av MDO. (A) Speciation of Mössbauer doublets ( and ) observed in Fe(II)-Av MDO for samples prepared at pH 6.5 and 9.5. (B) The theoretical lines (red and blue) show the expected behavior assuming a pH-dependent equilibrium with a of 9.1. The apparent pH () at cryogenic temperature is obtained by adding 0.4 pH unit to the pH of the solution at room temperature.
Table 2.
Mössbauer Parameters for Av MDO and Selected Variantsa
| enzyme | ||
|---|---|---|
| wild type | ||
| E (l), pH 6.5 | l.2l | 2.45 |
| E (2), pH 6.5 | l.24 | 2.90 |
| E (l), pH 9.5 | l.23 | 2.63 |
| E (2), pH 9.5 | l.25 | 3.l0 |
| E:3mpa (l) | l.06 | l.79 |
| E:3mpa (2) | l.07 | 2.24 |
| s E: | l.l4 | 3.38 |
| Hl57N | ||
| E (l) | l.2l | 2.34 |
| E (2) | l.2l | 2.8l |
| E:3mpa (l) | l.08 | l.84 |
| E:3mpa (2) | l.05 | 2.26 |
| Y159F | ||
| E | l.2l | 2.5l |
| E:3mpa (l) | l.03 | l.74 |
| E:3mpa (2) | 0.94 | 2.48 |
| s E: |
l.l6 | 3.56 |
All values at pH 8 unless otherwise noted.
The pH of a buffered solution is temperature-dependent and shifts under frozen cryogenic conditions.52–54 Thus, the pH values of the Mössbauer frozen solutions need to be corrected for this shift. The “apparent pH” () was determined under cryogenic conditions by measuring the ionization of metmyoglobin (Mb) both at 37 °C and in frozen solutions. The ferric heme changes from high-spin to low-spin upon deprotonation of the heme-bound aqua ligand. This aqua to alkaline Mb transition can be monitored independently by both UV–vis (37 °C) and cryogenic EPR spectroscopy (10 K).55,56 As illustrated in Figure S4, the value (8.9 ± 0.1) for this transition was measured spectrophotometrically at 37 °C (n = 10). These UV–vis samples were then transferred into EPR tubes and frozen in liquid N2, and the relative amounts of high-spin acid Mb and low-spin alkaline Mb were determined by EPR.52 Under cryogenic conditions, the observed for acid–alkaline Mb is 8.5 ± 0.2, indicating that the apparent pH under cryogenic conditions is +0.4 pH unit more basic relative to measurements made at 37 °C. This value is in good agreement with previous measurements made on similar buffered solutions.52–54
As shown in Figure 3B, using the correction of , the pH-dependent speciation of Mössbauer doublets ( and ) exhibited a pH dependence that agreed with the theoretical titration curve for a single value of 9.1 ± 0.1. Within experimental error, this value matched the obtained for Y159 from fitting log() data versus pH to eq 3. As additional verification that Y159 ionization is responsible for the spectroscopically observed speciation, 57Fe-enriched Mössbauer samples of Av MDO were prepared for both variants (H157N and Y159F) at pH 8.0 and 9.0 (Figure S6). As with the wild-type enzyme, Mössbauer spectra of the H157N variant also exhibited two doublets with parameters close to those of the wild type (Table 2). As with the wild type, the ratio of the two species observed at pH 8.0 changes at pH 9.0, thereby ruling out the possibility that the speciation is attributed to ionization of H157. In contrast, Mössbauer spectra obtained from the Y159F variant at both pH 8 and 9 showed only a single species with no pH dependence (Table 2). Collectively, these results indicate that ionization of Y159 is responsible for the Fe site speciation observed by Mössbauer spectroscopy. This confirms that the tyrosine of the S-H-Y motif directly interacts with a Fe-bound ligand.
Figure 4B shows the change in the Mössbauer spectrum upon addition of 3 molar equivalents of 3mpa per Av MDO at pH 8.0. The Mössbauer parameters of both doublets observed from the free enzyme changed significantly (Table 2; , and ; , and ) with approximately equal relative amounts at pH 8. A minor amount of the enzyme (10%) remained in the unbound form. Like the substrate free enzyme, fitting with a single doublet gave a broad line width of >0.5 mm/s, whereas the fit with two doublets gave a normal line width of <0.4 mm/s. The large decrease in isomer shift is consistent with coordination of the S atom to the Fe site.32,57 We therefore attribute the new species to the binding of 3mpa to the Fe(II) center. The Mössbauer parameters of variants H157N and Y159F displayed a similarly large decrease in the isomer shift in the presence of 3mpa (Table 2) with quadrupole splittings similar to those of the wild type. This suggests that the binding mode of 3mpa and iron coordination of the variants are the same as those of the wild type.
Figure 4.

Mössbauer spectra (4.2 K, 50 mT) for the isolated wild-type Av MDO (A) as compared to the 3mpa-bound (B) and cys-bound (C) enzyme at pH 8. (D) Stoichiometric conversion to the (3mpa/NO)-bound Av MDO is observed upon addition of 10 molar equivalents of NO to the 3mpa-bound enzyme. The isomer shift (δ) and quadrupole splitting () are indicated as . For substrate-bound Av MDO samples, 3mpa and cys were added in 3- and 5-fold molar excesses, respectively. Simulations parameters: , D = 10 cm−1, E/D = 0.01, A = (−25, −21, −23)T, , , .
Figure 4C shows Mössbauer spectra of wild-type Av MDO with a 5-fold molar excess of cys. A new doublet was observed from 30% of the iron (Table 2; , and ) and attributed to the cys-bound enzyme. With 1 equiv of cys, the cys-bound species was only 10% of the iron. Relative to 3mpa, the binding affinity of cys for wild-type Av MDO was significantly lower. As observed for 3mpa binding, the addition of cys to the Fe(II)-Av MDO also caused a decrease in the isomer shift indicative of coordination of thiolate to the Fe site. The large increase in the quadrupole splitting (+0.9 mm/s) for cys-bound relative to 3mpa-bound Av MDO suggests a change in the Fe coordination number and/or geometry.
EPR Studies of Substrate-Bound Iron–Nitrosyl Species Produced in Wild-Type MDO and Selected S-H-Y Motif Variants.
The Mössbauer experiments described above verify that pH-dependent ionization of Y159 within the Av MDO S-H-Y motif influences the Fe site in the absence of a substrate. An obvious extension is to determine how this interaction influences the (substrate/O2)-bound Av MDO ternary complex. Given the short lifetime of the O2-bound complex, nitric oxide was substituted for molecular oxygen for characterization by EPR spectroscopy as described previously.36
Addition of NO to a substrate-bound mononuclear non-heme iron site typically yields an species characterized by an axial EPR spectrum . The spin manifold can be described by an antiferromagnetic coupling between a high-spin and a bound NO− anion .58–60 In these experiments, samples of the (3mpa/NO)-bound enzyme were prepared for the H157N and Y159F variants of Av MDO for comparison to the wild-type enzyme. Addition of NO to samples containing a wild-type Av MDO pre-equilibrated with 10 molar equivalents 3mpa produced the EPR spectra shown in Figure 5A. The substrate-bound site (Av ES-NO) has observed g- values of 4.06, 3.96, and 2.01, consistent with a nearly axial () spin state. The axial zero-field splitting term for this complex was measured previously by fitting the variable-temperature EPR data to a Boltzmann population curve.36 Within error, the magnitude of the D value determined here was consistent with those reported for multiple iron–nitrosyl complexes.61 Mössbauer data (shown in Figure 4D) collected for equivalent samples of 57Fe-enriched Av MDO verify stoichiometric conversion of the 3mpa-bound enzyme to the (Av ES-NO) iron–nitrosyl complex.
As with the wild-type enzyme, equivalent samples of Av ES-NO prepared from H157N and Y159F variants also exhibit an iron–nitrosyl species upon addition of NO to the substrate-bound enzyme. However, both exhibit larger rhombic distortion (E/D values of 0.012 and 0.016, respectively). Analytical simulations for each species (dashed lines) were performed to determine the concentration of Av ES-NO produced for each enzyme. Relative to the starting ferrous iron concentration, the wild-type enzyme yields nearly stoichiometric conversion to the iron–nitrosyl species. By contrast, the yields for H157N (65%) and Y159F (10%) were significantly lower. These observations verify that disruption of the S-H-Y proton relay network alters both the NO binding affinity and the geometry at the (3mpa/NO)-bound ternary complex.
Unlike the mammalian cysteine dioxygenase (Mm CDO), coordination of cys to the Fe site of Av MDO is believed to occur via thiol-only coordination.36 Computational studies have suggested that the bidentate (thiol/amine) cys denticity was responsible for the unusual spin state reported for (cys/NO)-bound Mm CDO.36,62–64 In effect, the additional coordination of the neutral amine provides sufficient ligand field to overcome the pairing energy, yielding a low-spin Fe site. Thus, the (cys/NO)-bound Mm CDO active site can be considered a NO radical bound to a low-spin ferrous site.36,63 Up to this point, the Mm ES-NO complex was the only example of an species in a non-heme iron enzyme.
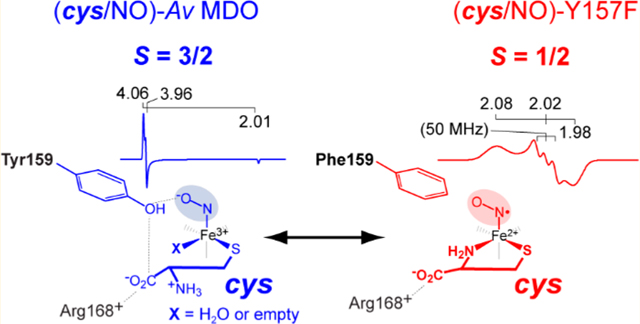
Unlike those of Mm ES-NO, the EPR spectra shown in Figure 5B for the wild-type (cys/NO)-bound ternary complex are consistent with an electronic ground state. The Mössbauer experiments presented above confirm cys-thiolate binding at the Fe site, but the iron–nitrosyl complex observed by EPR was inconsistent with the additional coordination of the cys amine group. While the cys carboxylate group could potentially form a second Fe ligand for bidentate coordination, pH-dependent steady-state assays indicated that the substrate carboxylate was not required for catalysis. We therefore conclude that substrate (3mpa and cys) binding occurs solely through coordination of thiolate to the active site Fe center.
As described previously, two sets of g-values (4.06, 3.96, and 2.00 and 4.31, 3.75, and 1.98) observed for the (cys/NO)-bound enzyme indicated two distinct conformers differing in rhombicity (E/D value). As indicated in Table 3, analytical EPR simulations of each species ( and 0.055) indicated relative concentrations of 0.7 and 0.3, respectively. Collectively, these accounted for 44% of the total iron present. The nearly axial species has the same E/D value (0.008) as the (3mpa/NO)-bound ternary complex, whereas the second signal exhibited a significantly higher rhombicity indicating a more distorted geometry.22
Table 3.
EPR Simulation Parameters for Substrate-Bound Av ES-NO Speciesa
| wild type | H157N | Y159F | |
|---|---|---|---|
| 3mpa-bound | |||
| D (cm−1) | 10 ± 2 | 10 ± 2 | 10 ± 2 |
| E/D | 0.0080 | 0.0124 | 0.016 |
| <0.001 | 0.002 | 0.005 | |
| 0.9 | 0.9 | 0.9 | |
| % Fe | 100 | 65 | 10 |
| wild type | Y159F | ||
| cys-bound | |||
| D (cm−1) | 10 ± 2 | 10 ± 2 | 10 ± 2 |
| E/D | 0.008 | 0.055 | 0.020 |
| <0.001 | 0.005 | ||
| 0.9 | 0.9 | 0.9 | |
| % Fe | 31 | 13 | 10 |
By contrast, the EPR spectrum obtained for cys-bound Av ES-NO prepared from the Y159F variant exhibited three distinct sets of observable g-values. The first signal has observed g-values of 4.14, 3.90, and 2.00 indicating the iron-nitrosyl complex with a slightly higher rhombicity relative to that of the wild-type (cys/NO)-bound ternary complex. Similar to samples prepared with 3mpa (Figure 5A), the (cys/NO)-bound Y159F variant shown in Figure 5B exhibits greater rhombicity and spectral line width compared to those of similar samples prepared with the wild-type enzyme. Of note, the second, more rhombically distorted species (designated by ● in Figure 5B) was absent in samples of Av ES-NO prepared from the Y159F variant.
Two additional signals were observed in the g ∼ 2 region indicative of the formation of two distinct spin species. The sharp signal with g-values of 2.04, 2.04, and 2.01 shown in gray was attributed to a known dinitrosyl iron (DNIC) complex frequently observed during NO addition experiments.65–67 This signal accounted for <30% of the total Fe within the sample and will not be discussed further. The contribution of DNIC was subtracted from the spectra, and the resulting signal (Figure 5B, colored red) exhibited g-values of 2.08, 2.02, and 1.98. A triplet hyperfine splitting A of 50 MHz was observed at the central g value of 2.02. The spectroscopic parameters observed for this iron–nitrosyl site (Table 4) were close to those of the low-spin iron–nitrosyl species observed in samples of Mm ES-NO and CDO model complexes.36,63 Figure 6 shows a simulation for this iron–nitrosyl species observed in the difference spectra. The spin concentration of this species corresponded to 52% of the total iron in the sample. The presence of the minor contribution (10%) from the nearly axial species suggests an equilibrium between monoand bidentate coordination. This change in substrate Fe-coordination denticity implies that Y159 affects substrate binding in the wild-type enzyme, possibly through hydrogen bonding interactions with the Fe-bound axial solvent. This argument was strengthened by the observation that a nearly equivalent cys-bound Av ES-NO signal (g = 2.07, 2.01, 1.98) was produced in the wild-type enzyme at pH values near or above the of Y159 (Figure S4). However, the spin concentration of this species indicated it is in the minority (<8% of total iron) in comparison to the 52% yield of the Y159 variant. A summary of the EPR parameters used to simulate the (substrate/NO)-bound Av ES-NO is provided in Table 3 and Table 4 .
Table 4.
EPR Parameters for Selected Low-Spin Iron–Nitrosyl Species Produced in Enzymatic Sites and Model Complexes
| g-values | A-values (MHz) | ||||||
|---|---|---|---|---|---|---|---|
|
|
|
||||||
| A2 (14N) | A2 (14N) | A2 (14N) | ref | ||||
| wild-type Mm ES-NO | 2.071 | 2.022 | 1.976 | 27 | 60 | 28 | 36 |
| Y157F Av ES-NO | 2.080 | 2.017 | 1.984 | 34 | 50 | 24 | this work |
| wild-type Av ES-NO (pH 9.4) | 2.071 | 2.014 | 1.981 | 35 | 53 | 39 | this work |
| 2.047 | 2.007 | 1.962 | 40 | 59 | 40 | 63 | |
| 2.072 | 2.021 | 1.992 | 47 | 62 | 50 | 59 | |
| 2.040 | 2.036 | 2.015 | - | - | - | this work | |
Figure 6.

X-Band EPR spectra of the (cys/NO)-bound Av ES-NO complex produced in the Y159F variant. Instrumental parameters: microwave frequency, 9.643 GHz; microwave power, 20 μW; modulation frequency, 0.9 mT; temperature, 10 K. For comparison, the spectroscopic parameters obtained for analogous iron–nitrosyls produced in Mm CDO and model complexes are provided in Table 4.
DISCUSSION
Both Av MDO and Mm CDO exhibit diprotic pH-dependent enzyme kinetics (Figure 2A) consistent with the presence of three ionic enzyme forms [Scheme 1; cationic, ; neutral, ; anionic, ].21 In this work, H157 and Y159 are unambiguously assigned as the ionizable residues observed in data obtained for Av MDO in reactions with 3mpa, cys, and ca. A key observation from these studies is that variants within the S-H-Y motif (H157N and Y159F) exhibit decreased values relative to that of the wild-type enzyme without significant perturbation to the 3mpa value. This suggests that the 3mpa affinity is not significantly influenced by the S-H-Y motif but instead a nonreversible chemical step following formation of the ES complex.
These pH-dependent kinetic studies also reveal that H157 and Y159 are in direct hydrogen bonding contact with each other. To illustrate, in reactions of 3mpa with wild-type Av MDO, data demonstrate that Y159 exhibits a value of 9.2 ± 0.1. This value becomes more basic (9.6 ± 0.1) in H157N-catalyzed 3mpa reactions. The perturbation is well above the statistical error of the assay, and thus, the Gibbs free energy for ionization of the Y159 phenolic group is decreased by 2.4 kJ mol−1 by hydrogen bonding with H157 relative to asparagine. Furthermore, while the observed for the H157N variant is attenuated relative to that of the wild-type enzyme, retention of this proton relay network is clearly essential for catalysis as the non-hydrogen bonding H155A variant of Mm CDO exhibits only trace enzymatic activity and is vastly uncoupled.24 Finally, the absence of the H157 ionization () in data obtained from Y159Fcatalyzed 3mpa reactions suggests that the influence of H157 ionization is propagated through an interaction between the ES complex and Y159. Thus, in the absence of Y159, ionization of H157 does not significantly alter a rate-limiting chemical step during catalysis.
On the basis of these observations, further inferences can be made with regard to the results reported previously for the wild-type enzyme utilizing cysteamine (ca) as a substrate. In this report, ionization of H157 () was observed in ca reactions, but the second ionization () associated with Y159 was absent. Because the protonation of H157 was observed in data obtained for ca reactions, it can be inferred that Y159 remains in contact with the ES complex. Otherwise, similar to the Y159F-catalyzed 3mpa reactions, ionization of H157 would not be present in the profile. Instead, the Y159 value likely becomes more basic and outside the pH regime assayed . This observation supports the conclusion that Y159 directly interacts with the substrate (3mpa or cys) carboxylate group, and disruption of this interaction increased the Y159 value. These observations highlight the critical importance of interactions of the substrate with Y159 of the S-H-Y motif, a property both Av MDO and Mm CDO appear to share.24 As an interesting side note, unlike Av MDO, the Pseudomonas Pa3MDO exhibits no activity in reactions with cysteamine.32 This observation suggests that despite their high degree of sequence homology, there may be distinct geometric or coordination differences with respect to how these enzymes bind the substrate.
As shown above, the Mössbauer spectra of wild-type Av MDO show two species that are related by the ionization of Y159 (Table 2). This observation suggests that the hydrogen bonding network connecting H157 to Y159 is further extended to an Fe-bound ligand. Previous Mössbauer data reported for Pa3MDO at pH 8.0 exhibit parameters ( and ) similar to those observed for Av MDO.32 However, the Pa3MDO spectra were reported as a single doublet with a line width much greater than is typical of a single Fe(II) species; consequently, the reported Mössbauer parameters are the average of those stated here for Av MDO.
Upon addition of 3mpa, both species of Av MDO are affected as indicated by the change in the parameters of both doublets (Table 2). In the presence of 3mpa or cys, the large decrease in isomer shift relative to the substrate free enzyme is consistent with direct S atom coordination of the substrate to the Fe(II) site. However, there is a large difference in the value of for the two substrates: cys (3.38 mm/s) versus 3mpa (1.79 and 2.24 mm/s). Similar behavior was reported previously for cys-bound Pa3MDO.32
Recently, Goldberg and Jameson et al. reported the structural and spectroscopic comparison of the mammalian C93G variant of CDO with a series of five- and six-coordinatae model complexes with an equivalent Fe-coordination sphere.68 In that report, it was observed that the quadrupole splitting for six-coordinate model complexes were significantly higher (, 3.14–3.55 mm/s) relative to those of the five-coordinate Fe sites (, 1.97–2.28 mm/s). Given the near equivalence in Fe-coordination sphere and active site geometry, the observations reported from the combined enzyme/model study of mammalian CDO can be used as a basis for assignment of the coordination number at the 3mpa- and cys-bound Av MDO Fe site. Regardless of pH, both doublets observed for the 3mpa-bound enzyme exhibited a (1.79 and 2.24 mm/s) more consistent with those reported for the five-coordinate model complexes. Conversely, the larger of the cys-bound wild-type enzyme (3.38 mm/s) suggests that the enzyme Fe site is six-coordinate. This trend is conserved among the 3mpa- and cys-bound H157N and Y159F variants, indicating that disruption of the S-H-Y motif does not alter the coordination number at the substrate-bound Fe site. From this analysis, we can conclude that the coordination number is not equivalent for the 3mpa- and cys-bound enzymes.
EPR characterization of the 3mpa- and cys-bound Av ES-NO species produced in the wild-type enzyme as compared to H157N and Y159F variants complements our previous studies in which multiple thiol-bearing substrates (3mpa, cys, et, and ca) were used to investigate molecular interaction and Fe site coordination within the Av MDO active site.22 The amount of (3mpa/NO)-bound iron–nitrosyl species produced in samples prepared from H157N and Y159F Av MDO is significantly smaller, and these variants also exhibit greater rhombic distortion. The decreased NO affinity for S-H-Y motif variants suggests that hydrogen bonding to Y159 is important for stabilization of the Fe-bound NO. The O atom of Y159 is 2.67 Å from the solvent molecule (2), which is the presumed O2 binding site and thus could form a hydrogen bond to the Fe-bound NO. Perturbation of this hydrogen bond (for H157N) or the loss of the hydrogen bond (for Y159F) likely results in a lower NO affinity. Qualitatively, this argument is supported by the increased value observed for both H157N and Y159F variants with respect to molecular oxygen as well as their decreased (O2/3mpa) coupling efficiencies (Figures S1 and S2). The strong Fe–NO bond causes the electronic symmetry of most complexes to be nearly axial and changes in E/D reflect alterations in the Fe–NO bond angle. Therefore, relative to the wild-type enzyme, the increased E/D for the (substrate/NO)-bound variants is consistent with hydrogen bonding interactions between Y159 and the Fe-bound NO which distorts the Fe-NO bond angle.
These observations are consistent with reports for Mm CDO in which it has been demonstrated that interactions between the phenol group of the C93–Y157 pair and the cys carboxyl group influence the enzymatic activity, coupling efficiency, and geometry of the substrate-bound active site.24 Thus, both Av MDO and Mm CDO appear to utilize direct interactions between the Fe-bound substrate carboxylate group and terminal Tyr reside hydroxyl group within the S-H-Y motif to regulate the structure of the ES complex, thereby optimizing enzymatic activity.
Typically, addition of nitric oxide to a substrate-bound mononuclear non-heme iron center results in the formation of an species. Mm CDO is an exception to this because (cys/NO)-bound Mm CDO produces a low-spin species.36 While unusual among non-heme iron enzymes, synthetic complexes have been reported in the literature. Wieghardt et al. have characterized several iron–nitrosyl model complexes with amine and thiolate coordination59,69 and found that the ground state of these complexes can be described as a ligand-centered NO• radical coordinated to a low-spin ferrous iron.59,60 Goldberg et al. reported a pair of synthetic complexes specifically designed to model the substrate-bound active site of CDO. The NO-bound complexes also exhibit a similar electronic configuration. In addition, detailed spectroscopic (EPR, Mössbauer, and S atom K-edge XAS) and computational density functional theory (DFT) modeling are all consistent with the assignment of this species as a low-spin ferrous iron with a ligand-centered radical.63 Computational studies reported by Solomon et al. suggest that this atypical low-spin configuration is a consequence of the bidentate cys coordination to the Fe site.62
Until now, Mm ES-NO was the only example of an species in a non-heme iron enzyme. However, as demonstrated in Figure 5B, a low-spin species with similar g-values and hyperfine splitting is observed from cys-bound Av ES-NO prepared from the Y159F variant of Av MDO. This change in substrate Fe-coordination denticity implies that Y159 regulates substrate binding in the wild-type enzyme, potentially through hydrogen bonding interactions with one or more Fe-bound solvent molecules as demonstrated by pH-dependent Mössbauer experiments. This argument is strengthened by the observation that a similar bidentate cys-bound () Av ES-NO EPR signal can be produced in the wild-type enzyme at pH values near or above the of Y159 (Figure S4). Of note, perturbations in both g- and A-values observed in samples of the low-spin iron–nitrosyl signal produced from the cys-bound wild-type enzyme relative to the Y159F Av MDO variant (Table 4) suggest the presence of hydrogen bonding between the Y159 hydroxyl group and Fe-bound NO. This conclusion is supported by the increased rhombicity and heterogeneity observed for EPR signals observed for H157N and Y159F variants relative to the wild-type enzyme.
In summary, the experiments presented here verify the presence of a catalytically enhancing proton relay network connecting conserved residues (H157 and Y159) within the S-H-Y motif of Av MDO to a Fe-bound ligand. The changes relative to the wild type in both the NO affinity and the rhombicity of the substrate-bound iron–nitrosyl species suggest that Y159 forms a hydrogen bond to the Fe-bound NO and, by extension, may also form a hydrogen bond with Fe-bound oxygen.
Mössbauer data for 3mpa-bound Av MDO are suggestive of a five-coordinate site. Given the high degree of structural similarity between CDO and MDO illustrated in Figure 1C, it is reasonable to assume that displacement of solvent water (1) by substrate–thiolate coordination occurs at the same position (trans to H91 of the 3-His facial triad). This leaves solvent 2 2.67 Å from the Y159 phenol group as the most likely point of interaction with Y159 as 3 is 0.7 Å more distant. As an interesting point of observation, solvent 2 also exhibits the lowest thermal B factor among all other Fe-bound solvents (PDB entry 4TLF), suggesting a relatively tight, or structurally constrained, Fe coordination.
DFT computational modeling and X-ray crystallographic data for (cys/NO)-bound mammalian CDO suggest that the site occupied by solvent 2, trans to H89, represents the most likely oxygen binding site.28,36 As stated previously, a bidentate model for 3mpa Fe coordination has been proposed in which solvent 3 is displaced by the 3mpa carboxylate group trans to H142.38 However, for reasons outlined in the introductory section, this model is inconsistent with our interpretation of experimental results, namely, kinetic and EPR spectroscopic studies performed on Av MDO using multiple thiol-bearing substrates (3mpa, cys, ca, and et).21,22 There are also technical concerns to consider (discussed in the Supporting Information) when evaluating the reliability of an ADV model generated from a heavily edited (solvent-deleted and 3mpabound) posed starting structure. Therefore, in the absence of corroborating experimental data, we exclude the possibility of bidentate 3mpa coordination and instead suggest retention of a solvent water within the ES complex. Because solvent 2 is the putative oxygen binding site, this water is likely lost upon 3mpa Fe coordination to accommodate oxygen binding. We therefore propose retention of solvent 3, which bridges Y159 and R168, to yield a (3mpa/H2O)-bound Fe site.
With respect to cys binding, the Mössbauer value (3.38 mm/s) of the cys-bound enzyme is suggestive of a six-coordiante complex. Thus, following binding of cys thiolate at the 3-His-coordinated Fe site, two Fe sites remain available. While the active site could certainly accommodate the bidentate coordination of the neutral cys amine, this is ruled out as addition of NO to the cys-bound enzyme does not produce a low-spin iron–nitrosyl species analogous to the six-coordinate (cys/NO)-bound Mm CDO.28,36,62–64 Similarly, coordination of the substrate carboxylate has been ruled out on the basis of the aforementioned kinetic and EPR data.21,22 Therefore, the remaining Fe sites are likely coordinated by solvent. As indicated in Scheme 2, addition of nitric oxide is expected to displace at least one solvent water leaving one bound solvent or an open coordination site (designated X).
Scheme 2.

Proposed (cys/NO) Coordination for Wild-Type Av MDO and the Y159F Variant
By contrast, addition of NO to samples of the Y159F Av MDO variant precomplexed with cys yields a similar low-spin iron–nitrosyl EPR signal that has been reported for six-coordinate (cys/NO)-bound Mm CDO. Therefore, bidentate coordination of cys via thiolate and neutral amine can be supported in the absence of the Y159 hydroxyl group or, to a lesser extent, following deprotonation of Y159-OH at elevated pH. A possible explanation for this observation is related to the apparent network of hydrogen bonds connecting Y159 and the Fe-bound solvents shown in Figure 1C. Solvent 2 is the closest water to the Y159 phenol group (2.67 Å), whereas 3 appears to bridge Y159 and R168 (each separated by ∼3.4 Å). This network of hydrogen bonds extends from the S-H-Y motif network to Fe-bound solvents and appears to orchestrate substrate coordination at the Fe site. Disruption of this proton relay network, by ionization of Y159 or removal (as in the case of the Y159F variant), may open up additional Fe sites for coordination or influence the positioning of R168 within the active site. In either case, it would appear that regulation of cys-coordination denticity at the Av MDO Fe site is more nuanced than previously appreciated.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the Department of Chemistry and Biochemistry at The University of Texas at Arlington Shimadzu Center for Advanced Analytical Chemistry for the use of HPLC and LC-MS/MS instrumentation.
Funding
This work was supported by National Institute of Health (National Institute of General Medical Sciences) Grants R15 GM117511-01 (B.S.P.) and R01 GM077387 (M.P.H.).
ABBREVIATIONS
- CDO
cysteine dioxygenase
- MDO
3-mercaptopropionic acid dioxygenase
- ADO
cysteamine dioxygenase
- 3mpa
3-mercaptopropionic acid
- cys
L-cysteine
- ca
cysteamine (2-aminoethanethiol)
- et
ethanethiol
- 3spa
3-sulfinopropionic acid
- csa
cysteine sulfinic acid
- Mm
Mus musculus
- Rn
Rattus norvegicus
- Av
Azotobacter vinelandii
- Pa
Pseudomonas aeruginosa
- HPLC
high-performance liquid chromatography
- EPR
electron paramagnetic resonance
- LC-MS
liquid chromatography–mass spectrometry
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.9b00674.
Oxygen saturation kinetics and coupling efficiencies for wild-type Av MDO and its H157N and Y159F variants, Dixon–Webb analysis of pH-dependent Y159F variant substrate inhibition, calibration of the apparent pH at cryogenic temperatures, X-band EPR data showing pH-dependent formation of the low-spin iron–nitrosyl complex in wild-type Av MDO, and Mössbauer spectra of H157N and Y159F Av MDO variants at pH 8.0 and 9.0 (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Reddie KG, and Carroll KS (2008) Expanding the functional diversity of proteins through cysteine oxidation. Curr. Opin. Chem. Biol 12, 746–754. [DOI] [PubMed] [Google Scholar]
- (2).Winyard PG, Moody CJ, and Jacob C. (2005) Oxidative activation of antioxidant defence. Trends Biochem. Sci 30, 453–461. [DOI] [PubMed] [Google Scholar]
- (3).Trachootham D, Alexandre J, and Huang P. (2009) Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat. Rev. Drug Discovery 8, 579–591. [DOI] [PubMed] [Google Scholar]
- (4).Bhave DP, Muse WB III, and Carroll KS (2007) Drug Targets in Mycobacterial Sulfur Metabolism. Infect. Disord.: Drug Targets 7, 140–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Deth R, Muratore C, Benzecry J, Power-Charnitsky V-A, and Waly M. (2008) How environmental and genetic factors combine to cause autism: A redox/methylation hypothesis. Neuro-Toxicology 29, 190–201. [DOI] [PubMed] [Google Scholar]
- (6).James SJ, Cutler P, Melnyk S, Jernigan S, Janak L, Gaylor DW, and Neubrander JA (2004) Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am. J. Clin. Nutr 80, 1611–1617. [DOI] [PubMed] [Google Scholar]
- (7).Gordon C, Emery P, Bradley H, and Waring H. (1992) Abnormal sulphur oxidation in systemic lupus erythematosus. Lancet 339, 25–26. [DOI] [PubMed] [Google Scholar]
- (8).Dominy JE Jr., Simmons CR, Karplus PA, Gehring AM, and Stipanuk MH (2006) Identification and Characterization of Bacterial Cysteine Dioxygenases: a New Route of Cysteine Degradation for Eubacteria. J. Bacteriol 188, 5561–5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Heafield MT, Fearn S, Steventon GB, Waring RH, Williams AC, and Sturman SG (1990) Plasma cysteine and sulfate levels in patients with motor neurone, Parkinson’s and Alzheimer’s disease. Neurosci. Lett 110, 216–220. [DOI] [PubMed] [Google Scholar]
- (10).Stipanuk MH (2004) Sulfur Amino Acid Metabolism: Pathways for Production and Removal of Homocysteine and Cysteine. Annu. Rev. Nutr 24, 539–577. [DOI] [PubMed] [Google Scholar]
- (11).Ewetz L, and Sorbo B. (1966) Characteristics of the Cysteinesulfinate-Forming Enzyme System in Rat Liver. Biochim. Biophys. Acta, Enzymol. Biol. Oxid 128, 296–305. [DOI] [PubMed] [Google Scholar]
- (12).Sorbo B, and Ewetz L. (1965) The Enzymatic Oxidation of Cysteine to Cysteinesulfinate in Rat Liver. Biochem. Biophys. Res. Commun 18, 359–363. [DOI] [PubMed] [Google Scholar]
- (13).Lombardini JB, Singer TP, and Boyer PD (1969) Cysteine Oxygenase. J. Biol. Chem 244, 1172–1175. [PubMed] [Google Scholar]
- (14).Stipanuk M, Simmons C, Andrew Karplus P, and Dominy J. (2011) Thiol dioxygenases: unique families of cupin proteins. Amino Acids 41, 91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Vitvitsky V, Garg SK, and Banerjee R. (2011) Taurine Biosynthesis by Neurons and Astrocytes. J. Biol. Chem 286, 32002–32010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Kabil O, and Banerjee R. (2010) Redox Biochemistry of Hydrogen Sulfide. J. Biol. Chem 285, 21903–21907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Driggers CM, Hartman SJ, and Karplus PA (2015) Structures of Arg- and Gln-type bacterial cysteine dioxygenase homologs. Protein Sci. 24, 154–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Driggers CM, Kean KM, Hirschberger LL, Cooley RB, Stipanuk MH, and Karplus PA (2016) Structure-Based Insights into the Role of the Cys–Tyr Crosslink and Inhibitor Recognition by Mammalian Cysteine Dioxygenase. J. Mol. Biol 428, 3999–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Simmons CR, Liu Q, Huang Q, Hao Q, Begley TP, Karplus PA, and Stipanuk MH (2006) Crystal Structure of Mammalian Cysteine Dioxygenase: A NOVEL MONONUCLEAR IRON CENTER FOR CYSTEINE THIOL OXIDATION. J. Biol. Chem 281, 18723–18733. [DOI] [PubMed] [Google Scholar]
- (20).Davies CG, Fellner M, Tchesnokov EP, Wilbanks SM, and Jameson GNL (2014) The Cys-Tyr Cross-Link of Cysteine Dioxygenase Changes the Optimal pH of the Reaction without a Structural Change. Biochemistry 53, 7961–7968. [DOI] [PubMed] [Google Scholar]
- (21).Crowell JK, Sardar S, Hossain MS, Foss FW Jr, and Pierce BS (2016) Non-chemical proton-dependent steps prior to O2-activation limit Azotobacter vinelandii 3-mercaptopropionic acid dioxygenase (MDO) catalysis. Arch. Biochem. Biophys 604, 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Pierce BS, Subedi BP, Sardar S, and Crowell JK (2015) The “Gln-Type” Thiol Dioxygenase from Azotobacter vinelandii Is a 3-Mercaptopropionic Acid Dioxygenase. Biochemistry 54, 7477–7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Blaesi EJ, Fox BG, and Brunold TC (2015) Spectroscopic and Computational Investigation of the H155A Variant of Cysteine Dioxygenase: Geometric and Electronic Consequences of a Third-Sphere Amino Acid Substitution. Biochemistry 54, 2874–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Li W, Blaesi EJ, Pecore MD, Crowell JK, and Pierce BS (2013) Second-sphere interactions between the C93-Y157 cross-link and the substrate-bound Fe site influence the O2-coupling efficiency in mouse cysteine dioxygenase. Biochemistry 52, 9104–9119. [DOI] [PubMed] [Google Scholar]
- (25).Ye S, Wu X. a., Wei L, Tang D, Sun P, Bartlam M, and Rao Z. (2007) An Insight into the Mechanism of Human Cysteine Dioxygenase: Key Roles of the Thioether-Bonded Tyrosine-Cysteine Cofactor. J. Biol. Chem 282, 3391–3402. [DOI] [PubMed] [Google Scholar]
- (26).Dominy JE, Hwang J, Guo S, Hirschberger LL, Zhang S, and Stipanuk MH (2008) Synthesis of Amino Acid Cofactor in Cysteine Dioxygenase Is Regulated by Substrate and Represents a Novel Post-translational Regulation of Activity. J. Biol. Chem 283, 12188–12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Njeri CW, and Ellis HR (2014) Shifting redox states of the iron center partitions CDO between crosslink formation or cysteine oxidation. Arch. Biochem. Biophys 558, 61–69. [DOI] [PubMed] [Google Scholar]
- (28).Li J, Koto T, Davis I, and Liu A. (2019) Probing the Cys-Tyr Cofactor Biogenesis in Cysteine Dioxygenase by the Genetic Incorporation of Fluorotyrosine. Biochemistry 58, 2218–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Dodson G, and Wlodawer A. (1998) Catalytic triads and their relatives. Trends Biochem. Sci 23, 347–352. [DOI] [PubMed] [Google Scholar]
- (30).Li W, and Pierce BS (2015) Steady-state substrate specificity and O2-coupling efficiency of mouse cysteine dioxygenase. Arch. Biochem. Biophys 565, 49–56. [DOI] [PubMed] [Google Scholar]
- (31).Easson LH, and Stedman E. (1933) Studies on the relationship between chemical constitution and physiological action: Molecular dissymmetry and physiological activity. Biochem. J 27, 1257–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Tchesnokov EP, Fellner M, Siakkou E, Kleffmann T, Martin LW, Aloi S, Lamont IL, Wilbanks SM, and Jameson GNL (2015) The cysteine dioxygenase homologue from Pseudomonas aeruginosa is a 3-mercaptopropionate dioxygenase. J. Biol. Chem 290, 24424–24437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Driggers CM, Cooley RB, Sankaran B, Hirschberger LL, Stipanuk MH, and Karplus PA (2013) Cysteine Dioxygenase Structures from pH4 to 9: Consistent Cys-Persulfenate Formation at Intermediate pH and a Cys-Bound Enzyme at Higher pH. J. Mol. Biol 425, 3121–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Fellner M, Aloi S, Tchesnokov EP, Wilbanks SM, and Jameson GNL (2016) Substrate and pH-Dependent Kinetic Profile of 3-Mercaptopropionate Dioxygenase from Pseudomonas aeruginosa. Biochemistry 55, 1362–1371. [DOI] [PubMed] [Google Scholar]
- (35).McCoy JG, Bailey LJ, Bitto E, Bingman CA, Aceti DJ, Fox BG, and Phillips GN Jr. (2006) Structure and mechanism of mouse cysteine dioxygenase. Proc. Natl. Acad. Sci. U. S. A 103, 3084–3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Pierce BS, Gardner JD, Bailey LJ, Brunold TC, and Fox BG (2007) Characterization of the nitrosyl adduct of substrate-bound mouse cysteine dioxygenase by electron paramagnetic resonance: electronic structure of the active site and mechanistic implications. Biochemistry 46, 8569–8578. [DOI] [PubMed] [Google Scholar]
- (37).Cook PF, and Cleland WW (2007) Enzyme Kinetics and Mechanisms, Garland Science, New York. [Google Scholar]
- (38).Aloi S, Davies CG, Karplus PA, Wilbanks SM, and Jameson GNL (2019) Substrate Specificity in Thiol Dioxygenases. Biochemistry 58, 2398–2407. [DOI] [PubMed] [Google Scholar]
- (39).Hayden J, Farquhar E, Que L, Lipscomb J, and Hendrich M. (2013) NO binding to Mn-substituted homoprotocatechuate 2,3-dioxygenase: relationship to O2 reactivity. JBIC, J. Biol. Inorg. Chem 18, 717–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Mbughuni MM, Chakrabarti M, Hayden JA, Bominaar EL, Hendrich MP, Münck E, and Lipscomb JD (2010) Trapping and spectroscopic characterization of an FeIII-superoxo intermediate from a nonheme mononuclear iron-containing enzyme. Proc. Natl. Acad. Sci. U. S. A 107, 16788–16793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Fielding AJ, Lipscomb JD, and Que L. (2012) Characterization of an O2 Adduct of an Active Cobalt-Substituted Extradiol-Cleaving Catechol Dioxygenase. J. Am. Chem. Soc 134, 796–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Wang Y, Griffith WP, Li J, Koto T, Wherritt DJ, Fritz E, and Liu A. (2018) Cofactor Biogenesis in Cysteamine Dioxygenase: C-F Bond Cleavage with Genetically Incorporated Unnatural Tyrosine. Angew. Chem., Int. Ed 57, 8149–8153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Notomista E, Lahm A, Di Donato A, and Tramontano A. (2003) Evolution of Bacterial and Archaeal Multicomponent Monooxygenases. J. Mol. Evol 56, 435–445. [DOI] [PubMed] [Google Scholar]
- (44).Collins PF, Diehl H, and Smith GF (1959) 2,4,6-Tripyridyl-s-triazine as Reagent for Iron. Determination of Iron in Limestone, Silicates, and Refractories. Anal. Chem 31, 1862–1867. [Google Scholar]
- (45).Crowell JK, Li W, and Pierce BS (2014) Oxidative uncoupling in cysteine dioxygenase is gated by a proton-sensitive intermediate. Biochemistry 53, 7541–7548. [DOI] [PubMed] [Google Scholar]
- (46).Emanuele JJ, and Fitzpatrick PF (1995) Mechanistic studies of the flavoprotein tryptophan 2-monooxygenase. 1. Kinetic mechanism. Biochemistry 34, 3710–3715. [DOI] [PubMed] [Google Scholar]
- (47).Denu JM, and Fitzpatrick PF (1994) pH and kinetic isotope effects on the oxidative half-reaction of D-amino-acid oxidase. J. Biol. Chem 269, 15054–15059. [PubMed] [Google Scholar]
- (48).Cleland WW (1982) [22] The use of pH studies to determine chemical mechanisms of enzyme-catalyzed reactions. In Methods in Enzymology (Daniel LP, Ed.) pp 390–405, Academic Press. [DOI] [PubMed] [Google Scholar]
- (49).Abragam A, and Bleaney B. (1970) Electron Paramagnetic Resonance of Transition Ions. International Series of Monographs on Physics, Oxford University Press. [Google Scholar]
- (50).Caranto JD, Weitz A, Hendrich MP, and Kurtz DM Jr. (2014) The nitric oxide reductase mechanism of a flavo-diiron protein: identification of active-site intermediates and products. J. Am. Chem. Soc 136, 7981–7992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Saavedra JE, Southan GJ, Davies KM, Lundell A, Markou C, Hanson SR, Adrie C, Hurford WE, Zapol WM, and Keefer LK (1996) Localizing Antithrombotic and Vasodilatory Activity with a Novel, Ultrafast Nitric Oxide Donor. J. Med. Chem 39, 4361–4365. [DOI] [PubMed] [Google Scholar]
- (52).Orii Y, and Morita M. (1977) Measurement of the pH of Frozen Buffer Solutions by Using pH Indicators. J. Biochem 81, 163–168. [DOI] [PubMed] [Google Scholar]
- (53).Sieracki NA, Hwang HJ, Lee MK, Garner DK, and Lu Y. (2008) A temperature independent pH (TIP) buffer for biomedical biophysical applications at low temperatures. Chem. Commun. (Cambridge, U. K.), 823–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Williams-Smith DL, Bray RC, Barber MJ, Tsopanakis AD, and Vincent SP (1977) Changes in apparent pH on freezing aqueous buffer solutions and their relevance to biochemical electron-paramagnetic-resonance spectroscopy. Biochem. J 167, 593–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Svistunenko DA, Sharpe MA, Nicholls P, Blenkinsop C, Davies NA, Dunne J, Wilson MT, and Cooper CE (2000) The pH dependence of naturally occurring low-spin forms of methaemoglobin and metmyoglobin: an EPR study. Biochem. J 351, 595–605. [PMC free article] [PubMed] [Google Scholar]
- (56).George P, and Hanania G. (1952) The ionization of acidic metmyoglobin. Biochemi J. 52, 517–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Chen VJ, Orville AM, Harpel MR, Frolik CA, Surerus KK, Münck E, and Lipscomb JD (1989) Spectroscopic studies of isopenicillin N synthase. A mononuclear nonheme Fe2+ oxidase with metal coordination sites for small molecules and substrate. J. Biol. Chem 264, 21677–21681. [PubMed] [Google Scholar]
- (58).Wanat A, Schneppensieper T, Stochel G, van Eldik R, Bill E, and Wieghardt K. (2002) Kinetics, Mechanism, and Spectroscopy of the Reversible Binding of Nitric Oxide to Aquated Iron(II). An Undergraduate Text Book Reaction Revisited. Inorg. Chem 41, 4–10. [DOI] [PubMed] [Google Scholar]
- (59).Li M, Bonnet D, Bill E, Neese F, Weyhermüller T, Blum N, Sellmann D, and Wieghardt K. (2002) Tuning the Electronic Structure of Octahedral Iron Complexes [FeL(X)] (L = 1-Alkyl-4,7-bis(4-tert-butyl-2-mercaptobenzyl)-1,4,7-triazacyclononane, X = Cl, CH3O, CN, NO). The S = 1/2 - S = 3/2 Spin Equilibrium of [FeLPr(NO)]. Inorg. Chem 41, 3444–3456. [DOI] [PubMed] [Google Scholar]
- (60).Sellmann D, Blum N, Heinemann FW, and Hess BA (2001) Synthesis, Reactivity, and Structure of Strictly Homologous 18 and 19 Valence Electron Iron Nitrosyl Complexes. Chem. - Eur. J 7, 1874–1880. [DOI] [PubMed] [Google Scholar]
- (61).Brown CD, Neidig ML, Neibergall MB, Lipscomb JD, and Solomon EI (2007) VTVH-MCD and DFT Studies of Thiolate Bonding to {FeNO}7/{FeO2}8 Complexes of Isopenicillin N Synthase: Substrate Determination of Oxidase versus Oxygenase Activity in Nonheme Fe Enzymes. J. Am. Chem. Soc 129, 7427–7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Diebold AR, Neidig ML, Moran GR, Straganz GD, and Solomon EI (2010) The Three-His Triad in Dke1: Comparisons to the Classical Facial Triad. Biochemistry 49, 6945–6952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).McQuilken AC, Ha Y, Sutherlin KD, Siegler MA, Hodgson KO, Hedman B, Solomon EI, Jameson GNL, and Goldberg DP (2013) Preparation of Non-heme {FeNO}7 Models of Cysteine Dioxygenase: Sulfur versus Nitrogen Ligation and Photorelease of Nitric Oxide. J. Am. Chem. Soc 135, 14024–14027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Straganz GD, Diebold AR, Egger S, Nidetzky B, and Solomon EI (2010) Kinetic and CD/MCD Spectroscopic Studies of the Atypical, Three-His-Ligated, Non-Heme Fe2+ Center in Diketone Dioxygenase: The Role of Hydrophilic Outer Shell Residues in Catalysis. Biochemistry 49, 996–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Ye S, and Neese F. (2010) The Unusual Electronic Structure of Dinitrosyl Iron Complexes. J. Am. Chem. Soc 132, 3646–3647. [DOI] [PubMed] [Google Scholar]
- (66).Butler AR, and Megson IL (2002) Non-Heme Iron Nitrosyls in Biology. Chem. Rev 102, 1155–1166. [DOI] [PubMed] [Google Scholar]
- (67).Reginato N, McCrory CTC, Pervitsky D, and Li L. (1999) Synthesis, X-ray Crystal Structure, and Solution Behavior of Fe(NO)2(1-MeIm)2: Implications for Nitrosyl Non-Heme-Iron Complexes with g = 2.03. J. Am. Chem. Soc 121, 10217–10218. [Google Scholar]
- (68).Gordon JB, McGale JP, Prendergast JR, Shirani-Sarmazeh Z, Siegler MA, Jameson GNL, and Goldberg DP (2018) Structures, Spectroscopic Properties, and Dioxygen Reactivity of 5- and 6-Coordinate Nonheme Iron(II) Complexes: A Combined Enzyme/Model Study of Thiol Dioxygenases. J. Am. Chem. Soc 140, 14807–14822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Surawatanawong P, Sproules S, Neese F, and Wieghardt K. (2011) Electronic Structures and Spectroscopy of the Electron Transfer Series [Fe(NO)L2]z (z = 1+, 0, 1−, 2−,3−; L = Dithiolene). Inorg. Chem 50, 12064–12074. [DOI] [PubMed] [Google Scholar]
- (70).Reginato N, McCrory CTC, Pervitsky D, and Li L. (1999) Synthesis, X-ray Crystal Structure, and Solution Behavior of Fe(NO)2(1-MeIm)2: Implications for Nitrosyl Non-heme-Iron Complexes with g = 2.03. J. Am. Chem. Soc 121, 10217–10218. [Google Scholar]
- (71).Lu S, Libby E, Saleh L, Xing G, Bollinger JMJ, and Moenne-Loccoz P. (2004) Characterization of NO adducts of the diiron center in protein R2 of Escherichia coli ribonucleotide reductase and site-directed variants; implications for the O2 activation mechanism. JBIC, J. Biol. Inorg. Chem 9, 818–827. [DOI] [PubMed] [Google Scholar]
- (72).Butler AR, and Megson IL (2002) Non-Heme Iron Nitrosyls in Biology. Chem. Rev 102, 1155–1165. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.