

ABSTRACT
Ruminococcus gnavus is a prevalent gut microbe reported to occur in higher abundance among individuals with inflammatory bowel disease (IBD). This study reports the isolation and characterization of six bacteriophages (phages) isolated from human fecal material and environmental samples that infect this species. Isolated phages have a siphovirus morphology, with genomes ranging between 36.5 and 37.8 kbp. Genome analysis indicates that the phages have a temperate lifestyle, which was confirmed by their ability to form lysogens on their host bacterial species. In contrast to the finding that phages lyse their host in liquid medium, results from a mouse trial indicate these phages can co-exist with the host bacterium in the gut without causing a significant reduction of R. gnavus. The bacterial counts in the feces of phage-treated mice did not significantly differ in the presence of phage. Furthermore, analysis of publicly available gut virome sequence data indicates a high abundance of these phages among individuals suffering from IBD. This work provides the first insight into how phages interact with R. gnavus in the human gut microbiome.
KEYWORDS: Ruminococcus gnavus, bacteriophage, mouse trial, inflammatory bowel disease
1. Introduction
The human gut microbiome is composed of a dynamic community of numerous microorganisms that engage in different types of symbiotic interactions (mutualism, commensalism, and parasitism) with the metazoan host. Mutualistic microbial symbionts provide a whole array of crucial functions to the human guts, such as digesting complex dietary carbohydrates and synthesizing bioactive molecules (vitamins, amino and fatty acids).1,2 They modulate the human immune system directly (cellular components) or indirectly by producing immunomodulatory substances.3,4 They can also act as a barrier against invading pathogenic bacteria.5 In the healthy human gut, the microbial community exists in a steady state, resulting in long-term stability of its taxonomic and functional composition.6 Insults, such as antibiotic treatments or inflammation, cause the gut microbial community to depart from that steady state, and adopt a new (meta)stable “dysbiotic” state.7 Furthermore, altered gut microbiomes have been observed among people afflicted with inflammatory bowel diseases (IBD), such as Crohn’s disease (CD) and ulcerative colitis (UC).8
Ruminococcus gnavus is a gram-positive anaerobe that is widely prevalent in the gut microbiome of individuals. The most recent literature places the bacterium within the family Lachnospiraceae, 9 and to avoid confusion with other ruminococci classified under family Oscillospiraceae a renaming of R. gnavus as Mediterraneibacter gnavus has been proposed but not validly published.10 The bacterium is 1 of the 57 species that was detected in the feces of more than 50% of 124 European individuals.11 The widespread prevalence of R. gnavus in the gut of individuals can likely be explained by its ability to utilize human mucin as a nutrient source, as observed with R. gnavus strain ATCC 29149, which can produce an intramolecular trans-sialidase encoded by the RgNanH gene producing 2,7-anhydro-Neu5Ac selectively from α2–3-linked sialic acid substrates.12 This makes the enzyme distinct from hydrolytic sialidases found in other mucin-degrading bacteria, such as Bacteroides thetaiotaomicron or Akkermansia muciniphila that produce free sialic acid (Neu5Ac).12
In addition, R. gnavus has evolved to survive and thrive in competitive microbial communities through the production of bacteriocins. For example, R. gnavus E1 was shown to produce the antimicrobial sactipeptide Ruminococcin C in vivo during the colonization of the digestive tract of rats.13 Another aspect of R. gnavus adaptation to the human gut is its involvement in host metabolic pathways, including bile acid metabolism with the production of iso-bile acids.14
R. gnavus is particularly prevalent among individuals with IBD, especially those suffering from CD.15–17 Moreover, blooms of this bacterium among such individuals can be temporal and often correlate with disease flare. The relative abundance of R. gnavus in CD patients can reach a maximum of 69.5% compared to less than 1% in healthy individuals.16,18 Production of capsular polysaccharides (CPS) has been implicated in playing a role in the onset of IBD, where strains of R. gnavus that produce CPS can exist symbiotically with the host immune system. In contrast, strains that do not produce CPS can elicit an inflammatory immune response.19 Another explanation given for the bacterium’s possible role in the generation of inflammation associated with CD is its production of the polysaccharide glucorhamnan that can induce the toll-like receptor 4 (TLR4)-dependent secretion of a pro-inflammatory cytokine tumor necrosis factor-α (TNFα) in dendritic cells.20 The bacterium is also associated with other inflammatory non-IBD conditions, such as spondylarthritis.21
Bacteriophages (phages) are viruses that specifically infect bacteria. Phage numbers are estimated to range between 1:1 and 1:100 compared to bacteria in the human gut.22 They often display a narrow host range, with their infection typically limited to specific strains within a bacterial species. There is growing interest in their development as alternatives to combat bacterial diseases and tools for microbial gut engineering.23,24
Most phages infect their bacterial host through either lytic or lysogenic life cycles. Lytic phages kill their bacterial host through cell lysis, whereas lysogenic phages integrate into the bacterial host genome. Metagenomic studies of the human gut virome show a rich diversity of phages associated with this ecological niche, where phages found within the gut virome are stable over time but highly individual-specific.22,25,26 Individuals with IBD are enriched in temperate phage-related sequences in the gut virome as compared with healthy individuals.27
To our knowledge, phages infecting R. gnavus have not yet been documented. The current study describes the biological characterization and genomic and phylogenetic analyses of six phages related at the genus to subfamily level infecting R. gnavus, obtained from human fecal and environmental samples.
2. Materials and methods
2.1. Bacterial strains and culture conditions
R. gnavus strains were sourced from the Japan Collection of Microorganisms (JCM 6515T = ATCC 29,149T) and the Culture Collection University of Gothenburg (CCUG 52,279, CCUG 51,289, CCUG 54,531, CCUG 43,222, CCUG 49,994, CCUG 57,161, CCUG 57,208, and CCUG 57,137). Upon random screening with 16S rRNA species-specific PCR primers, the R. gnavus strain PS/160 was obtained out of a collection of strict anaerobes isolated from pooled human feces (unpublished data). All R. gnavus strains were propagated using Anaerobe basal broth (ABB, Oxoid – ThermoFisher). All manipulations were carried out at 37°C in strict anaerobic conditions (Type A vinyl anaerobic chamber, Coy Labs).
2.2. Fecal sample collection and the isolation and propagation of phages
Fecal samples used in the present study were randomly selected from a subset of a larger IBD study cohort.28 A total of 33 samples were selected from healthy volunteers and 45 samples from IBD-affected individuals. Manure/slurry tank samples were collected from several locations around a single multi-species farm in Co. Tipperary, Ireland. Human feces and farm samples were split into 1 g aliquots and resuspended in 10 mL of SM buffer (50 mM Tris-HCl pH [7.5], 100 mM NaCl, 8 mM MgSO4). These suspensions were centrifuged, with the resulting supernatants being filtered twice through 0.45 µm pore membrane filters. Then, 2 mL of these filtrates were mixed with 2 mL of 2× ABB soft agar (final agar concentration 0.4% w/v) and 0.5 mL of an overnight culture of R. gnavus JCM 6515T to create an overlay spread on ABB 1.5% w/v agar plates. Plaque formation was assessed after 16–24 h of incubation. Individual plaques were excised and soaked in 100 µL of SM buffer and incubated for 2–3 h with occasional shaking to separate phages from the top agar. The resulting phage preparations were filtered through 0.45 µm pore spin filters. The resulting phage preparation was replated in the same manner as previously described, with this process repeated twice more to produce pure phage stocks.
Two to three rounds of plate propagation were performed to create high-titer phage lysates, followed by two to three rounds of liquid propagation. Briefly, top agar from three plates with near confluent lysis (~104 plaques per plate) was collected in a 15 mL falcon tube, vigorously shaken for 2–3 h, and centrifuged to pellet debris. Obtained supernatants were filtered through 0.45 µm pore membrane filters and used for the subsequent round of propagation. A volume of 0.5–1 mL of phage lysate (or at MOI = 1) was added to 10 mL of exponentially growing host strain R. gnavus JCM 6515T in ABB broth (OD600 = 0.2) and incubated until clearing was observed. Lysates were centrifuged into pellet debris with the supernatant filter sterilized. Phage lysates were stored at 4°C for further experiments.
2.3. The efficiency of lysogeny and host range analysis
For the efficiency of the lysogeny test, 200 µL of high-titer (>109 PFU/mL) phage lysates were spread onto ABB agar plates. A volume of 100 µL of serial dilutions of overnight cultures R. gnavus JCM 6515T were used to inoculate phage-covered and control agar plates. The efficiency of lysogeny was calculated after 48 h of incubation for a percentage of colonies grown on phage-seeded plates relative to the total bacterial number on unseeded/control plates. Ten randomly selected colonies (for each phage) from phage-covered plates were transferred onto ABB agar plates. After three subsequent rounds of streaking, colonies were checked for the presence of prophages using the following primers: phiPS6-F1, R1; phiRg507T2/2-F1, R1; phiRg507T2/3-F1, R1 (Supplementary information 2, Table S1).
The host range of the phages was determined by spotting 10 µL of phage lysates onto a bacterial lawn prepared by the double agar overlay method. Briefly,: 5 mL of molten soft ABB agar (0.4% w/v) was mixed with 0.3 mL of overnight bacterial culture and spread onto ABB agar plates. Phage lysates were spotted after the solidification of the top layer. The formation of the spots was assessed after 16–24 h of incubation.
2.4. Phage kill-curve assays
For the phage killing curve assays, a culture of R. gnavus JCM 6515T was grown from ABB to OD600 of 0.3 (108 CFU/mL). A volume of 90 µL of cell suspension was dispensed into the wells of flat-bottom 96-well plates. Ten microliters of phage lysate (phiRgPS6, phiRg507T2/2, phiRg507T2/3, and phiRg519T2) or phage mixtures in SM buffer were added to the wells at different MOIs − 1, 0.1, 0.01 and 0.001. Negative control wells were inoculated with SM buffer only. Plates were sealed and immediately transferred to the plate reader. Measurements were taken to monitor OD595 changes for 24 h at 30 min intervals of 5 s shaking before readings. Experiments were performed twice in technical duplicates for each phage or phage mixture. The outcome of these experiments was visualized with the ggplots2 package in R.
2.5. Plaque morphology and electron microscopy
Briefly: 60 mL of high-titer phage lysates (>109 PFU/mL) were spun down in a ultra-centrifuge using an F65L-6×13.5 rotor (ThermoFisher) at 120,000 × g for 3 h. The total phage pellet was resuspended in 5 mL of SM buffer and subsequently purified by a cesium chloride step gradient (5 M/3 M CsCl solutions) with ultra-centrifugation at 105,000 × g for 2.5 h. The phage band was collected by a syringe and subjected to three rounds of buffer exchange by 10-fold dilution with SM buffer and concentrated to the initial volume using Amicon Centrifugal Filter Units MWCO 10 KDa (Millipore). Five microliters of purified phage sample were applied onto Formvar/Carbon 200 Mesh, Cu grids (Electron Microscopy Sciences), negatively stained with 0.5% w/v uranyl acetate and examined by a Tecnai G2 12 BioTWIN transmission electron microscope at UCD Conway Imaging Core Facility (University College Dublin, Dublin, Ireland).
2.6. In vivo mouse study
Ten germ-free C57BL/6 male mice (8 weeks old, bred in-house) were separated into two groups: phage administration group (n = 5) and SM-buffer administration group (control group; n = 5). Each group of mice was housed in two cages, with two and three mice, respectively. Mice were inoculated by oral gavage with 0.2 mL R. gnavus JCM 6515T cells in PBS (2 × 109 CFU) on days 1, 2, and 3 of the experiment. On days 11, 12, and 14, all mice were inoculated by oral gavage with 0.2 mL: of a phage suspension (phiRg507T2/2, phiRg507T2/3, and phiRgPS6) in SM buffer at 1010 PFU for the phage treatment group or 0.2 mL SM buffer for the control group. On days three, seven, 11–23 (once every 2 days), and day 26 mice were placed in new cages and approximately 10 fresh fecal pellets were collected from each cage. Pellets were processed on the same day as collection to ensure the optimal viability of R. gnavus. On day 26, all mice were sacrificed.
To enumerate R. gnavus in mouse feces, feces was resuspended in PBS containing 0.3 mg/mL L-cysteine HCl, 0.3 mg/mL Na-thioglycolate, 1 mg/mL DTT. This underwent a ten-fold serial dilution and was plated onto ABB agar and incubated in anaerobic conditions at 37°C. For phage enumeration, the 10−2 dilution used for bacteria enumeration was filtered-sterilized and used for a ten-fold serial dilution that was subsequently spotted onto an overlay with R. gnavus JCM 6515T as previously described. GraphPad Prism (v8) was used to determine the normality of data using the Shapiro–Wilk test. The unpaired t-test was utilized to compare treatment groups.
On day 23, enumerated R. gnavus isolates were checked for prophages using the following primer pairs: phiRgPS6-F1/R1; phiRg507T2/2-F1/R1; phiRg507T2/3-F1/R1 (Supplementary information 2, Table S1).
2.7. DNA isolation and genome sequencing of Ruminococcus phages
Genomic DNA extraction was conducted as previously described.29 Briefly, phage PEG/NaCl precipitates were collected by centrifugation at 5200 × g for 20 min at 4°C. Pellets were resuspended in 400 µL of SM buffer and extracted by gentle shaking with an equal volume of chloroform followed by centrifugation at 2500 × g for 5 min. The aqueous phase (~360 µL) was aspirated into clean Eppendorf tubes and treated with 8 U of TURBO DNase (ThermoFisher) and 20 U of RNase I (ThermoFisher) in the presence of 1 mM CaCl2 and 5 mM MgCl2 at 37°C for 1 h before inactivating enzymes at 70°C for 10 min. This was followed by a proteinase K (40 µg) treatment in the presence of 0.5% w/v SDS for 20 min at 56°C. Viral particles were then lysed by the addition of 100 µL of Phage Lysis Buffer (4.5 M guanidinium isothiocyanate, 44 mM sodium citrate [pH 7.0], 0.88% w/v sarkosyl and 0.72% v/v 2-mercaptoethanol) at 65°C for 10 min. Lysates were extracted twice with an equal volume of phenol/chloroform/isoamyl alcohol 25:24:1 (ThermoFisher) and subjected to a final round of DNA purification using DNeasy Blood & Tissue Kit (Qiagen). DNA was then quantified using Qubit dsDNA HS Assay Kit and subjected to shotgun library preparation using TruSeq Nano DNA or Nextera XT kits (Illumina), followed by sequencing on the Illumina HiSeq 2500 platform (GATC Biotech AG). Illumina reads were trimmed and filtered using Cutadapt v.2.4 and Trimmomatic v.0.36 as described before,30–32 then assembled with SPAdes v3.10.0.33
2.8. DNA isolation and genome sequencing of R. gnavus
Briefly, genomic DNA extraction was performed on overnight cultures of R. gnavus in ABB using the GenElute kit (Merck), following the manufacturer’s instructions. The Qubit dsDNA HS Assay Kit and Qubit 2.0 Fluorometer (Life Technologies) were used to determine the DNA concentration. The Nextera DNA Flex Library Preparation Kit (Illumina) was used to generate DNA libraries, followed by sequencing on the Illumina NovaSeq platform (Illumina) that was outsourced to AZENTA Life Sciences. Oxford Nanopore sequencing using the Rapid Barcoding Kit (SQK-RBK004; Oxford Nanopore) was performed on the MinION Mk1B platform using MinKNOW software for selected R. gnavus strains. Hybrid assembly of Illumina (trimmed and filtered as above) and Oxford Nanopore reads trimmed and filtered using NanoFilt v2.8.0 was conducted using SPAdes for JCM 6515T, and using Unicycler v0.4.8 for all other strains.34–36
2.9. Bioinformatic analysis
Phage genomes were initially annotated with RASTtk.37 Open reading frames (ORFs) were further investigated using BLASTp using the nr (November 2021) and Phaster (December 2020) databases, Interproscan v5.57–90.0 and HHpred. ARAGORN was used to identify tRNA genes.38–42 Phage genome nucleotide similarity was calculated using VIRIDIC and orthologous genes were discovered by Coregenes 5.0.43,44 Phylogenetic analysis based on the phage proteome was performed with VIPtree and VICTOR.45,46 Phylogenetic trees were edited with iTOL.47 Gegenees v3.1.0 was used to calculate phage proteome homology.48 Pangenome analysis of proteomes was conducted with Proteinortho v6.1.2 [command: proteinortho *.faa -identity = 30 -cov = 70 -singles].49 The resulting.tsv presence-absence table of orthologous groups (OGs) was inspected and visualized using the stats, reshape, gridextra, tidyverse and ggplots2 packages in R. For the presence-absence map of OGs among phages, the proteinothro.tsv file of identified OGs was correlated to protein annotation of analyzed phages. Bacterial genomes were annotated with Prokka v1.14.6, using the Phaster and pVOGs databases for identification of prophage gene products.41,50,51 Pyani v0.2.7 was used for the calculation of average nucleotide identity between bacterial genomes.52 Identified prophages in the genomes of R. gnavus were manually reviewed using the Artemis Comparison Tool (ACT) v18.1.0 or Mauve v10.53,54 Minced v0.4.2 (ctSkennerton/minced) was used for detection of CRISPR spacer sequences and BLASTn (-task BLASTn-short) was used to identify protospacers among phage genomes, possessing no more than 3 misaligned bases.
2.10. Analysis of the prevalence of Ruminococcus phages in the gut microbiome
The human fecal shotgun virome and 16S rRNA amplicon sequencing data previously reported by Norman et al. were accessed through BioProject PRJEB7772.55 Briefly, sequencing reads were processed with Trimmomatic v0.36 for trimming and filtering. Virome reads were aligned to phage genomes using Bowtie v2.1.0 with counts of aligned reads extracted from alignment data with SAMtools v0.1.19 as previously described.31,56–58 For 16S rRNA amplicon reads, counts were generated using BBmap v39.01 on reads aligning with 97% identity [idfilter = 0.97] to 16S rRNA sequences of Ruminococcus gnavus obtained from the Silva 138.1 prokaryotic SSU database.59 Counts of aligned reads were extracted with SAMtools. Boxplots were generated with ggplot2 in R.
2.11. Data availability
Genomes of bacteria and phages described in this study are available at GenBank under accession numbers given in Table 1
Table 1.
GenBank accession numbers for genomes of phages and bacteria sequenced in this study.
| Genomes | accession number | |
|---|---|---|
| Ruminococcus phage | phiRg507T2/2 | MT980836 |
| phiRg507T2/3 | MT980837 | |
| phiRg519T2 | MT980838 | |
| phiRgPS6 | MT980839 | |
| phiRgIBDN1 | MT980840 | |
| phiRM10 | MT980841 | |
| R. gnavus bacterial strains | JCM 6515T = ATCC 29149T | CP043051 |
| JCM 6515T (phiRgPS6) clone 1–1 | CP111084 | |
| JCM 6515T (phiRgPS6) clone 2–1 | CP111086 | |
| CCUG 52,279 | JAPRBD000000000 | |
| CCUG 51,289 | JAPRBC000000000 | |
| CCUG 54,531 | JAPRBB000000000 | |
| CCUG 43,222 | JAPRBA000000000 | |
| CCUG 49,994 | JAPRAY000000000 | |
| CCUG 57,161 | JAPRAX000000000 | |
| CCUG 57,208 | JAPRAW000000000 | |
| CCUG 57,137 | JAPRAV000000000 | |
| PS/160 | JAPRAU000000000 | |
2.12.
3. Results
3.1. Isolation of phages against R. gnavus JCM 6515T
Seventy-eight fecal samples from human donors (healthy and those suffering from IBD) and 23 samples of animal and environmental origin (multi-species farm) were subjected to phage screening against R. gnavus JCM 6515T (= ATCC 29,149T). Twelve samples (a pooled fecal sample, stool from eight individuals with IBD, and three farm animal samples) produced plaques. These plaques were observed to be small in size (1–3 mm in diameter) and clear. Ruminococcus phage phiRg507T2/3 was the only isolate to produce plaques surrounded by halos (Supplementary information 1, Figure S1). Plaques from the stool of a healthy individual and those suffering with IBD and the three farm samples were purified through triple passage with isolation of a single plaque on each step. Phages from sample 507T2 yielded two distinct types of plaque morphologies (medium-sized with halos or those pinprick in size without halos), which were purified separately. Ultimately, six phage isolates were selected, which were denoted as phiRgPS6 (pooled fecal sample isolate), phiRg507T2/2, phiRg507T2/3, phiRg519T2, phiRgIBDN1 (IBD isolates), and phiRgRM10 (farm isolate).
3.2. Biological characterization of the phages
Transmission electron microscopy of phiRgPS6, phiRg507T2/2 and phiRg507T2/3 revealed virions with a typical siphovirus morphology with icosahedral heads (55–61 nm in diameter), with long flexible non-contractile tails (195–204 nm) possessing very discreet baseplates (Figure 1a–d and Supplementary information 2, Table S2).
Figure 1.
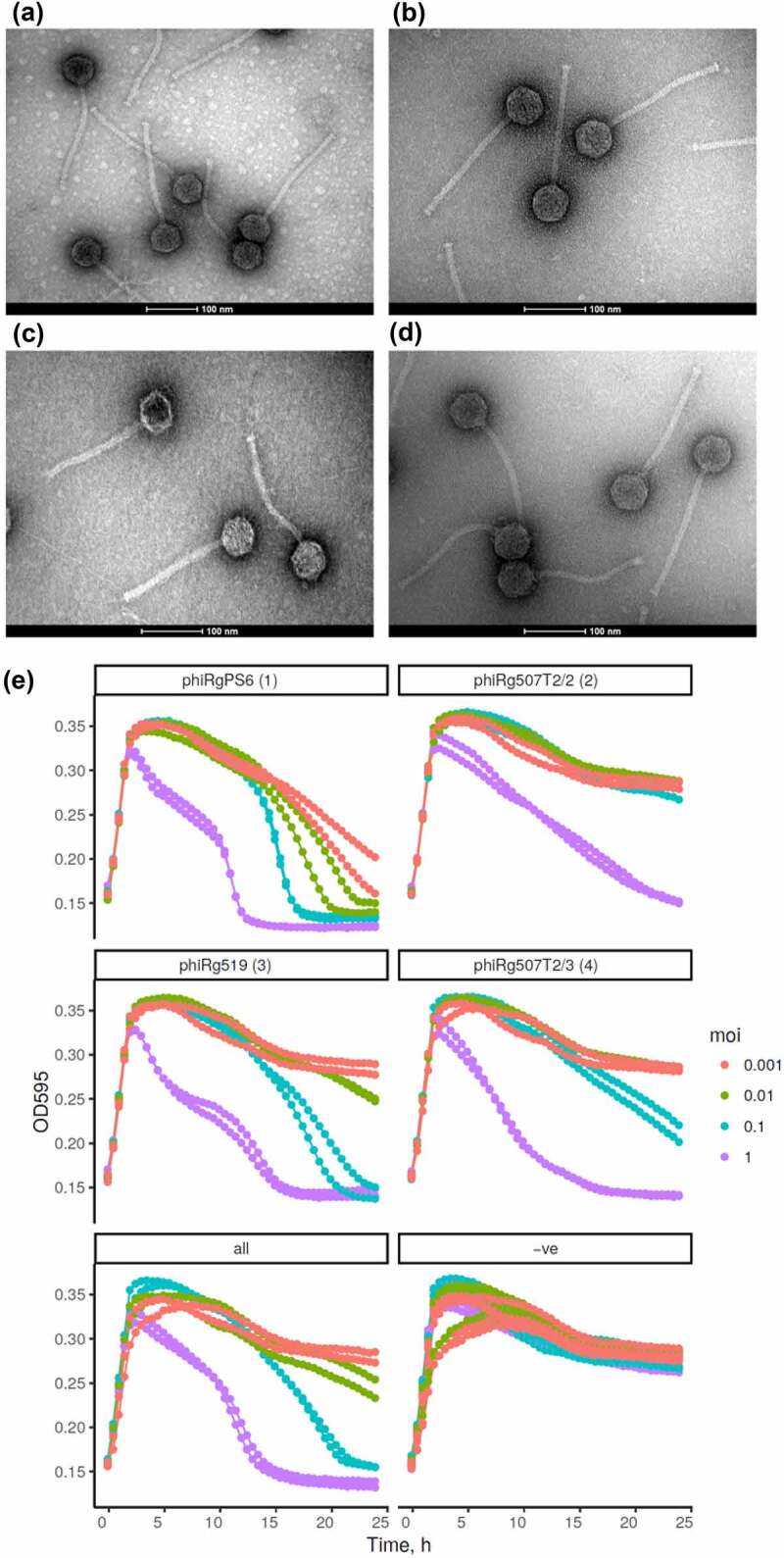
Transmission electron micrographs and phage-host kill curves. The micrographs of Ruminococcus gnavus phages (a) phiRg507t2/2, (b) phiRg507t2/3, and (c) phiRgps6, (d) phiRg519. The scale bar represents 100 nm. (e) Phage kill-curve assays of Ruminococcus gnavus JCM 6515T in response to infection by Ruminococcus phages phiRgps6, phiRg507t2/2, phiRg507t2/3, phiRg519t2 and a phage cocktail of these four phages at varying MOIs from 1×10−3 to 1, compared to a negative control (−ve) of untreated culture of R. gnavus JCM 6515T.
The narrow host range of these phages was confirmed by spotting phage lysate on overlays seeded with eight different strains of R. gnavus obtained from the CCUG collection and strain PS/160, which was isolated during this study. All but two phages had lytic activity limited to a single host strain JCM 6515T. Phages phiRgPS6 and phiRgIBDN1 additionally produced lysis on R. gnavus CCUG 57137 (Supplementary information 2, Table S3).
Kill curve assays were performed to evaluate the lytic activity of phages against R. gnavus in liquid medium. Exponentially growing cultures of JCM 6515T were inoculated with phiRgPS6, phiRg507T2/2, phiRg507T2/3 and phiRg519T2 and a combination of these four phages at four different multiplicities of infection (MOI = 1; 0.1; 0.01; and 0.001). It was found that each phage on its own and a combination of four could cause effective lysis of the bacterial host after 2 h of growth when added at MOI of 1 (Figure 1e). Moreover, bacterial growth remained inhibited for 10–15 h after the initial lysis event. One of the tested phages, phiRgPS6, was able to suppress the host at all tested MOIs.
These phages were also evaluated for their ability to lysogenize their host. Plating of strain JCM 6515T onto agar seeded with a high titer of Ruminococcus phage lysate resulted in isolated colonies that were positive in phage-specific PCR. The efficiency of lysogeny (frequency of formation of lysogens) among the tested colonies was estimated at 35% for phiRgPS6 and 46% for either phage phiRg507T2/2 or phage phiRg507T2/3.
3.3. Genome analysis of Ruminococcus phages and R. gnavus strains
The genomes obtained for the Ruminococcus phages ranged between 36,510 and 37,780 bp (coverage > 1000×) with a GC content of 41–42%. A comparison of nucleotide similarity between genomes showed that all phage isolates were homologous at the nucleotide level. Following the International Committee on Taxonomy of Viruses (ICTV) guidelines,60 a shared nucleotide similarity of less than 95% between phages phiRgPS6, phiRg507T2/2, phiRg507T2/3, phiRg519T2, phiRgIBDN1 and phiRgRM10 allows each to be defined as representing a novel species (Supplementary information 1, Figure S2). Additionally, shared nucleotide between genomes exceeds 55%, with gene synteny conserved between the genomes (Figure 2).
Figure 2.
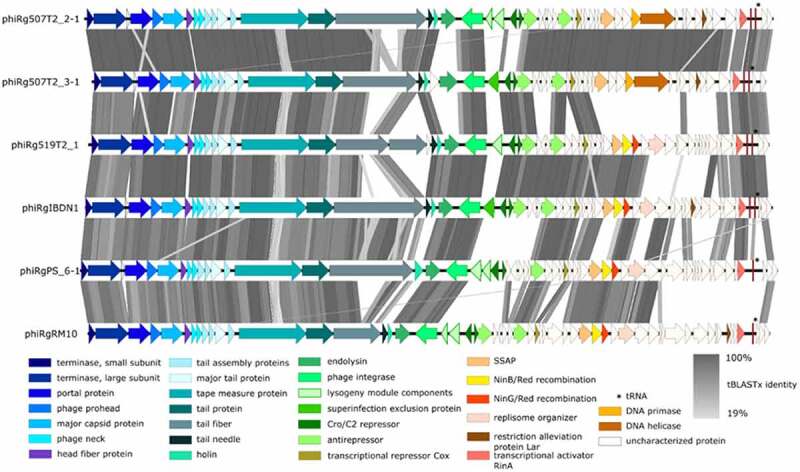
Linear genome map of Ruminococcus phages. Comparison of the genomes of Ruminococcus phages isolated in this study employing BLASTn and visualisation with Easyfig. The genome maps display arrows indicating the locations and orientation of ORFs among different phage genomes. Arrows have been colour-coded describing their predicted roles (see key), and shading between the genome maps indicates the level of identity.
The number of open reading frames (ORFs) found on the genomes of these phages ranged from 54 to 59, with each phage also possessing either one or two tRNA genes (Supplementary information 2, Table S4). To examine the shared proteome of the six phages, we conducted a pangenome analysis of their proteins (identity = 30%, coverage = 70%). These proteins could be placed into 117 orthologous groups (OGs), with the shared proteins (core proteome) among these genomes comprising of 20 OGs (17% of the total). Each phage was found to possess between 2 and 12 unique proteins not shared among other isolates (Supplementary information 2, Figure S3). Only 54% of OGs could be given functional assignments, with overlap seen among numerous OGs annotated with similar functions. Our annotation efforts could place OGs into six major functional categories: lysogeny, virion assembly, host lysis, DNA-related, transcriptional regulation and accessory (Supplementary information 1. Figure S4).
OGs associated with virion structure and assembly were found to be the most conserved among the six phages, with 14 of the 20 OGs universally shared among these phages related to this function. These contained proteins implicated in the formation of the capsid (major capsid & prohead), tail (major tail, tape measure and tail assembly chaperone) and their connection (head-tail connector), as well as those involved in genomic DNA encapsulation (portal protein, large and small terminase). However, proteins implicated to be located in the tail and head fiber structures were found not to be conserved among phages.
All phages share a similar tyrosine integrase (InterPro IPR013762). However, a diverse range of proteins containing the Cro/C1-type HTH domain (IPR001387) is distributed among the phages, being placed among five different OGs. Proteins with this domain are expected to be implicated in the transcriptional repression or activation of prophages entering the lytic life cycle. Seven other OGs involved in transcriptional regulation were also identified.
The greatest diversity was found among the 16 OGs in the DNA replication-related category, with no OGs being shared among all six phages. OGs in this category were annotated as endonuclease, recombinase, single-stranded DNA binding protein, double-strand repair protein, replisome organizer or proteins found to possess a DNA binding or zinc finger domain.
All phages possess a predicted endolysin with an N-acetylmuramoyl-L-alanine amidase activity (IPR002508, IPR002502) and a holin, with proteins of each type placed into two different OGs.
Seven OGs were classified as having an accessory role, with proteins in this category not shared universally among phages. These OGs include gene products implicated in overcoming the host restriction-modification systems, such as DNA methyltransferase (IPR001091) for DNA methylation and the Lar family restriction alleviation protein (PF14354), which provides a countermeasure to restriction enzymes targeting foreign DNA. This group also includes proteins that promote resistance to incoming phage infection, such as superinfection exclusion protein (IPR011434) and siphovirus gp157 (IPR008840).
We sequenced the genomes of the 10 R. gnavus strains utilized in this study to determine if we could identify factors potentially implicated in the resistance of hosts to the Ruminococcus phages. The host strain R. gnavus JCM 6515T was sequenced using Illumina and Oxford Nanopore platforms, generating short and long sequence reads, respectively. This allowed the assembly of a complete circular genome of 3.6 Mbp in size (coverage 88.2×). The remaining nine isolates were sequenced using short reads, resulting in high-quality draft genomes (coverage > 400×) with a low number of contigs (54 [42–102]) and a high N50 (161,630 [131,253–245,696]) (median [range]), with low rates of duplication marker genes (Supplementary information 2, Table S5). The average total length of these 10 assemblies was 3.12 ± 0.19 Mbp (medium ± SD), sharing an average nucleotide identity (ANI) of > 97% with the genome of R. gnavus JCM 6515T.
Of the isolates sequenced, CRISPR arrays were detected among the genomes of CCUG 51289, CCUG 43437 and the host strain JCM 6515T downstream of several Cas proteins associated with a type I-C system. These CRISPR arrays contain 22 ± 5 spacers with a length of 34 ± 1 bp (mean ± SD). A collective total of 68 spacers were identified, with 15 aligning to the Ruminococcus phage genomes with no more than three mismatched bases (Supplementary information 2, Table S6). The three bacterial strains were found to have spacers targeting all six phages. Interestingly, JCM 6515T possesses spacers that perfectly match the target protospacer associated with the genomes of phiRg507T2/2 and phiRg507T2/3 while being sensitive to both phages.
We were also curious to learn if the R. gnavus strains used in this study possess similar prophage elements with lysogenic repressors that could impact infection by the six Ruminococcus phages. Pangenome analysis (identity = 30%, coverage = 70%) with these Rumnoococcus phages and 10 R. gnavus strains did not reveal the presence of prophage elements that shared conserved proteins (large terminase, capsid & portal protein) found among these phages. Furthermore, no gene products were found to be universally shared between phages and these bacterial strains.
3.4. Phylogenetic analysis of Ruminococcus phages
These Ruminococccus phages do not fall into any phage genus currently recognized by the ICTV. Analysis of these phage genomes using BLASTn with the nt database does not identify any cultured phages with genomes that share significant nucleotide homology with those isolated in this study. However, examples of metagenome-assembled genomes (MAGs) of phages originating from the human microbiome could be identified with genus-level nucleotide similarity (Supplementary information 1, Figure S5). This observation indicates that similar Ruminococcus phages are associated with the human microbiome. Additionally, it was possible to identify genomes of R. gnavus with prophage elements that share nucleotide similarity of up to 68% with the Ruminococccus phages (Supplementary information 1, Figure S5).
Analysis with VIPtree allowed the construction of a proteomic phylogram of these phages utilizing 863 genomes. This allowed the placement of these phages in a clade of 14 other related phages possessing genomes ranging from 34 to 52 kbp in size. These related phages possess virions with a siphovirus morphology that infect gram-positive bacterial hosts (Supplementary information 1, Figure S6). Further phylogenetic analysis of this clade using VICTOR to construct a proteomic tree show that the Ruminococcus phages closest evolutionary relationship is that placed within the genus of Svunavirus (Figure 3a). This observation was further confirmed by Gegenees (tBLASTx), with proteomes of the Ruminococcus phages sharing the greatest level of homology (19–20% identity) among inspected phages (Figure 3b). However, the connection between the Ruminococcus phages and this phage genus is distant due to the phylogenetic distance indicated in the VICTOR phylogram and low protein homology in Gegenees analysis.
Figure 3.
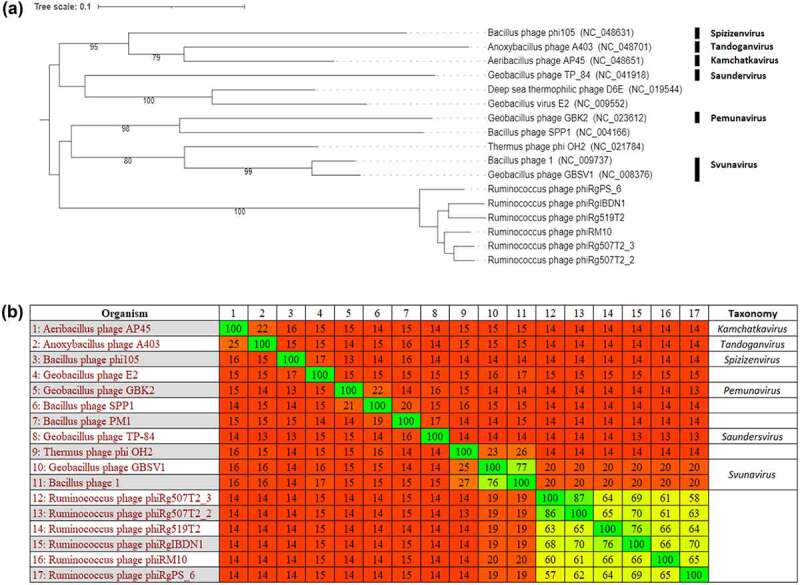
Phylogenetic analysis of Ruminococcus phages. (A) VICTOR-generated phylogenomic Genome-BLAST Distance Phylogeny (GBDP) tree inferred using the formula D4 and yielding average support of 67%. (B) a tBLASTx heatmap generated using Gegenees with accurate parameters – fragment length: 200 bp; and step size: 100 bp; threshold: 5%. The phylogram and heatmap includes this study’s Ruminococcus phages and those that share an evolutionary connection. The genus (if allocated) of phages in these analyses is illustrated.
3.5. Phage–host interaction in a murine in vivo model
A 28-day mouse trial was conducted to assess the dynamics of the six Ruminococcus phages and the potential infection of their host within the murine GIT (experimental design presented in Supplementary 1, Figure S7). Germ-free mice were gavaged with R. gnavus JCM 6515T at the onset of the trial (days 1, 2 & 3) in two equal groups of mice (n = 5). On days 11, 12 and 14, each group of mice were gavaged with either SM buffer or a Ruminococcus phage mixture (phiRg507T2/2, phiRg507T2/3, and phiRgPS6). Post-treatment, mice from both groups followed a similar trend of body weight gain till the end of the trial (Supplementary information 1, Figure S8). Post-gavage of R. gnavus (from days 3 to 23), the bacterium could be consistently enumerated in mouse feces of both treatment groups, with counts ranging between 3 × 105 and 4 × 1010 CFU/g (Figure 4a). No significant difference in bacterial numbers could be detected post phage administration in the phage treatment group. Furthermore, viable phage could be detected on the two inspected time points (days 16 & 21) post-gavage among the phage-treated group, with counts ranging from 1.4 × 107 to 1 × 1010 PFU/g, with MOI values ranging between 1 × 10−2 to 1 × 104 (Figure 4b). These findings indicate that phages were indeed able to reach the gut of mice and replicate on the R. gnavus host without causing a significant reduction of the host population density.
Figure 4.
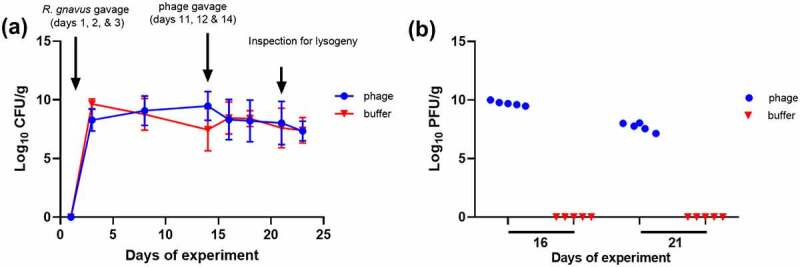
A mouse trial to investigate the interaction of R. gnavus JCM 6515T and a Ruminococcus phage mixture. (a) CFU/g counts of R. gnavus and (b) PFU/g counts of Ruminococcus phages in mice faeces from those treated (n = 5) and untreated (n = 5) with phage. Time points at which mice were gavaged with host bacterium and phages and when R. gnavus was inspected for phage lysogeny are illustrated.
On day 23, a PCR targeting Ruminococcus phages was conducted on bacterial isolates (triple streaked to ensure purity) from the phage-treated group. This investigation found that 35 out of 72 (48.6%) colonies tested positive for the presence of prophages phiRgPS6 and phiRg507T2/2 or phiRg507T2/3, indicating their lysogenization by these phages.
3.6. Lysogeny of the Ruminococcus phages
We were interested in further studying the lysogenization of R. gnavus JCM 6515T by the phages isolated in this study to understand how this interaction may impact the ecology of such phages and R. gnavus in the gut.
Liquid cultures of R. gnavus JCM 6515T lysogenized by phages phiRgPS6 (clones 1–1 and 2–1), phiRg507T2/2 (clone 11) and phiRg507T2/3 (clones 21–1 and 22–1) were inspected for the production of active phage virions resulting from subpopulations of cells where prophages have entered the lytic cycle. Filtered supernatants were spotted onto an overlay seeded with a non-lysognised JCM 6515T where it was possible to detect zones of lysis indicating the presence of viable phage. Indicating that R. gnavus lysogenized by these phages possess a sub-population of cells that undergo spontaneous lysis, releasing viable phage in the gut.
We proceeded to determine the range of superinfection immunity conferred to R. gnavus JCM 6515T by lysogenization. The sensitivity of R. gnavus JCM 6515T lysogenized by either phage phiRgPS6, phiRg507T2/2 or phiRg507T2/3 was investigated by spot assay using lysates of the six Ruminococcus phages (Table 2). Phages could not plaque the cultures of JCM 6525T lysogenized with the same phage. Additionally, a crossover of resistance was observed among isolates of JCM 6515T lysogenized by phiRg507T2/2 and phiRg507T2/3, where lysates of either phage could not plaque on these isolates. Additionally, phage phiRg519T2 was found to be unable to plaque on JCM 6525T lysogenized by phiRg507T2/3.
Table 2.
Sensitivity of R. gnavus JCM 6515T with prophage of Ruminococcus phages against lysates of the same phages.
| Phage | JCM 6515T lysogenised by (isolate) |
||
|---|---|---|---|
| phiRgPS6 (1–1) | phiRg507T2/2 (11) | phiRg507T2/3 (21–1) | |
| phiRgPS6 | - | + | + |
| phiRg507T2/2 | + | - | + |
| phiRg507T2/3 | + | - | - |
| phiRg519T2 | + | + | - |
Results recorded as −, no plaque formation; +, plaque formation.
We were also curious to learn how these prophages integrate into the genome of their host. To determine this, cultures of R. gnavus JCM 6515T lysogenized by phage phiRgPS6 (clones 1–1 and 2–1) were subjected to short and long read-based sequencing to obtain complete circular genomes (coverage > 100×). Alignment of these genomes to the native wild type JCM 6515T shows that the genome of phage phiRgPS6 had integrated into that of the host. For clone 1–1, an intergenic region between CDSs FXV78_RS15645 and FXV78_RS15640 (position: 3,137,358–3,137,378 bp) was identified as the attB site for this phage (Figure 5a). This sequence was identified to be 21 bp in length with the same sequence associated with the attP site of phage phiRgPS6 (position: 9,654–19,674 bp, sequence: 5’ – tcttaagcatttttaagattt − 3’). Furthermore, this sequence was found to be preserved with the attL and attR for the prophage form of phiRgPS6.
Figure 5.
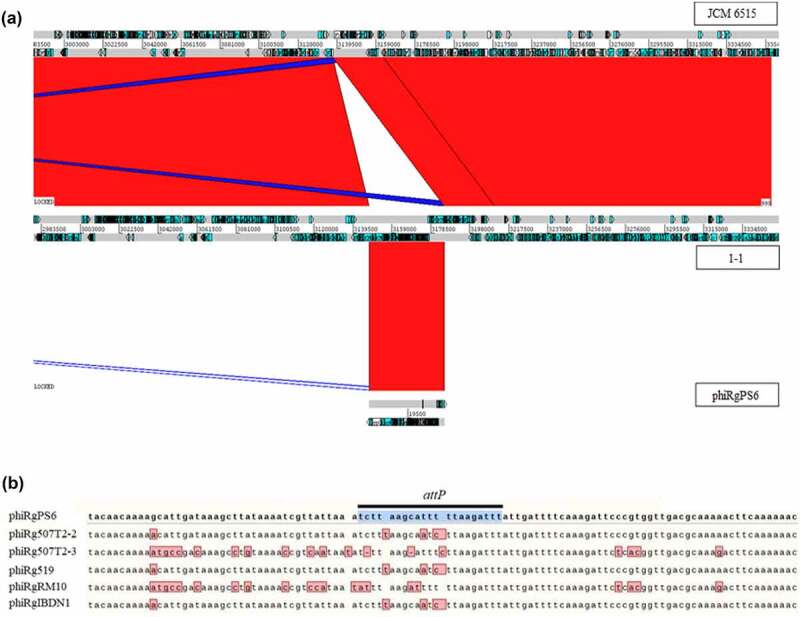
Lysogenetisation of R. gnavus JCM 6515T by Ruminococcus phage phiRgps6. (a) Genome alignment visualised with the Artemis Comparison Tool (ACT) of the wild-type (WT) R. gnavus JCM 6515T versus a clone 1–1 lysogenised by Ruminococcus phage phiRgps6, as well as the genome of the phage phiRgps6. Locations of direct homology (as determined with BLASTn) between aligned genomes are indicated with areas of red. (b) Genome alignment of the attP of Ruminococcus phage phiRgps6 with the other five Ruminococcus phages in this study, illustrating sequence diversity of potential attP sites among their genomes.
A conjugation element is located at the boundary of the prophage integration site (FXV78_RS15645 – FXV78_RS15800) for JCM 6515T clone 2–1, with a similar element found upstream (FXV78_RS12270 – FXV78_RS12425). At the boundaries of these elements, there appears to be the possibility of a genomic inversion that is 668,564 bp in size, as found in clone 2–1 (Supplementary information 1, Figure S9). This observation may relate to the presence of integrase genes situated at the boundary of these conjugation elements.
PCR was conducted on JCM 6515T clones 1–1 and 2–1 utilizing primers targeting the prophage and host genome to confirm the observations made in genomic sequence analysis Supplementary information 2, Table S1. These PCRs resulted in amplicons of the predicted size (Supplementary information 1, Figure S10) that were subsequently sequenced to confirm the location of the prophage.
We also examined publicly available genomes of R. gnavus, found to contain prophages sharing similarity at the nucleotide level with the phages of this study. Analysis of the boundary points of these elements allowed identification of those with a predicted attB site located at a similar intergenic region (relative to the genome of JCM 6515T) as with R. gnavus JCM 6515T with a prophage of phiRgPS6 (Supplementary information 2, Table S7). It also appears that related prophages can target attB sites associated with tRNA genes (Supplementary information, Table S7). Additionally, the alignment of the attP site of phage phiRgPS6 to the other five Ruminococcus phages shows sequence diversity at this locus (Figure 5b).
3.7. Prevalence of Ruminococcus phages in the human gut virome
As previously discussed, the abundance of R. gnavus has been reported to be higher in individuals suffering from IBD than in healthy individuals, especially among those with CD.15–17 We were interested in determining if a similar trend could be observed for Ruminococcus phages. To determine if that was the case, we reexamined sequence data from the study of IBD and healthy gut microbiomes and viromes reported by Norman et al. 55 The 16S rRNA dataset from this study is composed of 45 people suffering from CD, 70 people suffering from UC and 32 healthy individuals. Enumeration of reads that aligned (97% identity) to representative 16S rRNA genes of R. gnavus shows an increased abundance of the bacterium among individuals with CD compared to the healthy controls (Figure 6a). The virome dataset of the Norman et al. study is composed of 37 individuals suffering from CD, 42 individuals with UC, and 64 healthy individuals. We could detect sequence reads aligning to the genomes of the Ruminococcus phage among 9% of the healthy individuals, 54% of the individuals with CD and 42% of the individuals with UC. Furthermore, the highest number of aligned reads were detected among viromes of individuals suffering from CD and UC (Figure 6b).
Figure 6.
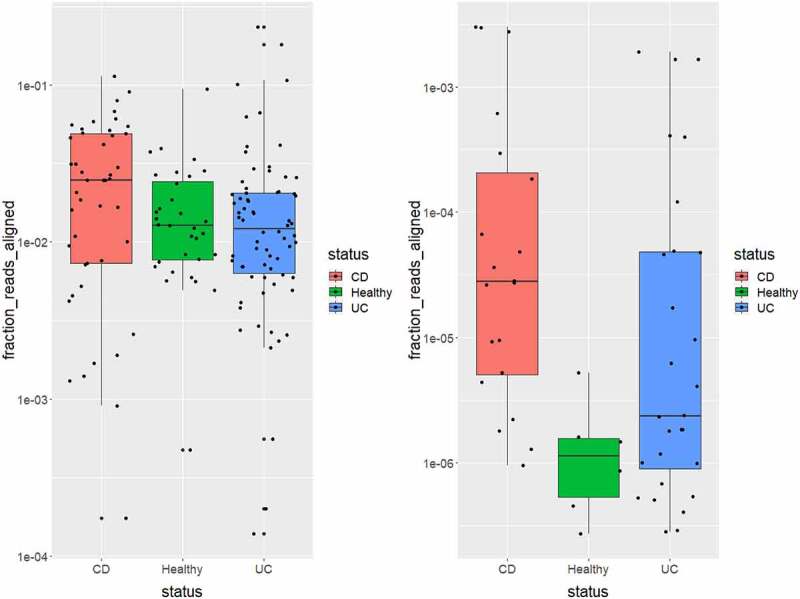
Prevalence of Ruminococcus gnavus and Ruminococcus phages in examined human faecal 16S rRNA and viriome sequence data. (a) the fraction of reads aligning to R. gnavus 16 rRNA gene from faecal 16S rRNA data and (b) the fraction of reads aligned from faecal viromes of individuals with Crohn’s disease (CD), ulcerative colitis (UC) and those deemed healthy with the genomes of the six Ruminococcus phages isolated in this study.
4. Discussion
To date, little is known about phages that infect R. gnavus and how their infection may affect the behavior of this bacterium in the human gut. In this study, we report the isolation and characterization of six novel phages that infect the type strain of R. gnavus JCM 6515T. The phages have a siphovirus morphology, with their host range limited to no more than two out of a panel of 10 R. gnavus strains, with phages possessing a temperate lifestyle. The phage genomes range between 36.5 and 37.8 kb in size, with genes encoding hallmark proteins implicated in the lysogenic life cycle, such as integrase and putative repressor proteins containing the Cro/C1-type HTH domain (IPR001387).
We demonstrated the lysogenization of R. gnavus strain JCM 6515T by Ruminococcus phage phiRgPS6, where the attB site for this phage is located within an intergenic region on the boundary of a conjugation element. Analysis of public databases allowed the identification of R. gnavus genomes with prophage elements that share significant nucleotide similarity (up to 68%) with these newly isolated Ruminococcus phages. Furthermore, examining these prophage elements allowed the identification of strains with a predicted attB site situated at a similar intergenic region as found for phage phiRgPS6. We also identified similar prophage elements with a predicted attB site associated with a tRNA gene. The latter observation has been made among other species of gut bacteria.61 The alignment of the known attP site of phage phiRgPS6 with the five other Ruminococus phages demonstrated a diversity of the nucleotide sequence within this region among the six phages. This diversity may indicate that the six phages target different attB sites at different loci that can occur within an R. gnavus genome. However, it may also ensure such phages can overcome the emerging sequence diversity of their target attB site that is likely to occur among different strains of their host bacterium.
We also show that when R. gnavus JCM 6515T is lysogenized by phages phiRgPS6, phiRg507T2/2 and phiRg507T2/3, it becomes immune to lytic superinfection by the same phage. It is possible to observe cross-immunity with cultures of JCM 6515T lysogenized with different prophages. For example, JCM 6525T lysogenized with phiRg507T2/2 was resistant to lysis by phiRg507T2/3. The observed superinfection immunity is expected to be a consequence of prophage repressor proteins simultaneously maintaining a prophage in the lysogenic cycle and interfering with lytic infection by a similar temperate phage.62 However, this finding did not explain the lack of plaque formation with the Runminococcus phages among the additional nine R. gnavus strains used in host range analysis, as these bacterial strains were found not to possess prophage elements with homology to the phages examined in this study.
CRISPR-Cas is an adaptive immune system found among archaea and bacteria that acts against foreign invading DNA. These systems store foreign DNA sequences from past exposures as “spacers” that form part of the CRISPR arrays. Transcribed spacers (short crRNAs) form a complex with Cas proteins that cleave foreign DNA possessing a corresponding protospacer sequence.63 We examined the presence of CRISPR arrays among these R. gnavus genomes, where they were detected in strains CCUG 51289, CCUG 43437 and JCM 6515T downstream of Cas proteins related to a type I-C system. However, this generated more questions than possible answers for strain JCM 6515T regarding its sensitivity to Ruminococcus phage infection. All three strains possess spacers targeting protospacers with no more than three base mismatches found among the six Ruminococcus phages. Additionally, JCM 6515T had spacers that perfectly matched protospacers found among phages phiRg507T2/2 and phiRg507T2/3. Their lack of effect against infection by these phages may relate to a mismatch of the PAM sequence associated with protospacers or may suggest that these phages may possess proteins with anti-CRISPR activity.64,65 Other avenues that remain to be explored for phage resistance among R. gnavus strains include the action of restriction-modification systems, the presence of abortive infection systems and cell surface modifications that could impede phage absorption.66
In a mouse trial utilizing germ-free mice mono-colonized with R. gnavus JCM 6515T over 27 days, no significant reduction of this bacterium was detected in mouse feces after gavage with a mixture of phages (phiRg507T2/2, phiRg507T2/3 & phiRgPS6), 11 days post gavage with the host bacterium. Additionally, phages could be detected in the feces of mice (days 16 & 21), suggesting phage and the bacterial host could exist in the mouse gut in a manner that did not significantly impact host numbers. There are two mechanisms through which these phages could interact with the host bacterium that could explain this observation. First, phages can infect the bacterium through the lytic life cycle but do so in a manner where an insufficient number of cells are infected or at a speed that does not outpace the growth of the host bacterium (below the inundation threshold).67 Second, phages could lysogenize the host, and a subpopulation could release viable phages. The latter option is supported by a phage-specific PCR that confirmed that 48.6% (35/72) colonies of R. gnavus JCM 6515T isolated from feces (day 23 of the trial) were lysogenized by phage. Furthermore, liquid cultures of R. gnavus JCM 6515T lysogenized by phage phiRgPS6 spontaneously release viable phage. However, the observed outcome of the mouse trial may likely result from Ruminococus phages interacting with R. gnavus through a combination of both mechanisms.
We also show that Ruminococcus phages, like those isolated in this study, are more prevalent and abundant in the gut of individuals with IBD, as determined by the alignment of publicly available gut virome sequence reads from 143 individuals. The prevalence and abundance of these phages among individuals with IBD, especially CD, correlate with the high prevalence and abundance of R. gnavus typically found among such individuals.15,17,68
Our results indicate that there is more likely a positive rather than a negative correlation between the high prevalence and abundance of R. gnavus within the gut of those with IBD and the type of Ruminococcus phages isolated in this study. This conclusion begets some interesting questions that merit further investigation. For example, if a high number of such phages can occur alongside a high number of R. gnavus, does this result in increased lysis events of the host bacterium within the gut? If so, can the products of R. gnavus lysis play a role in stimulating the human immune system that induces inflammation associated with IBD? Indeed, lytic phage infection has been shown to release cellular components (Pathogen Associated Molecular Patterns) that can elicit an immune response.69 Follow-on work that may help answer these questions could include in vitro assays based on immune system cells to examine pro-inflammatory signals.19 Or animal trials using interleukin-10-deficient mice that are inoculated with R. gnavus with the phages of this study versus the bacterium alone, allowing the detection of an altered immune response.70
Supplementary Material
Funding Statement
This research was conducted with the financial support of Science Foundation Ireland (SFI) under Grant Number SFI/12/RC/2273 (Colin Hill & Paul Ross), a Science Foundation Ireland’s Spokes Programme which is co-funded under the European Regional Development Fund under Grant Number SFI/14/SP APC/B3032, and a research grant from Janssen Biotech, Inc (Colin Hill & Paul Ross). Andrey Shkoporov was supported by a Wellcome Trust Research Career Development Fellowship [220646/Z/20/Z], and the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement No 101001684). This research was funded in whole, or in part, by the Wellcome Trust [220646/Z/20/Z]. For the purpose of open access, the authors have applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission.
Disclosure statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Authors’ contributions
C.B. and E.V.K. performed bioinformatics, analyzed results and wrote the manuscript; E.V.K. and A.N.S. conducted wet lab work; L.S. performed genomic sequencing of R. gnavus strains; C.M.H. designed and performed the mouse trail; B.G. & L.A.D. managed the project; R.P.R., A.N.S. and C.H. secured funding and supervised the project. All authors reviewed and edited the manuscript.
Consent for publication
The manuscript does not contain any individual personal data in any form.
Ethics approval and Consent to participate
The studies involving human participants were reviewed and approved by the Cork Research Ethics Committee.
Availability of data and materials
The genomes of bacteria used are freely available on the NCBI website, accession numbers are provided in Table 1.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19490976.2023.2194794.
References
- 1.Morowitz MJ, Carlisle EM, Alverdy JC.. Contributions of intestinal bacteria to nutrition and metabolism in the critically Ill. Surg Clin North Am. 2011;91(4):771. doi: 10.1016/j.suc.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhattacharya T, Ghosh TS, Mande SS, Aziz RK. Global profiling of carbohydrate active enzymes in human gut microbiome. Plos One. 2015;10(11):e0142038. doi: 10.1371/journal.pone.0142038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hill DA, Artis D. Intestinal bacteria and the regulation of immune cell homeostasis. Annu Rev Immunol. 2010;28(1):623–19. doi: 10.1146/annurev-immunol-030409-101330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brestoff JR, Artis D. Commensal bacteria at the interface of host metabolism and the immune system. Nat Immunol. 2013;14:676. doi: 10.1038/ni.2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buffie CG, Pamer EG. Microbiota-mediated colonization resistance against intestinal pathogens. Nat Rev Immunol. 2013;13(11):790–801. doi: 10.1038/nri3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL, Clemente JC, Knight R, Heath AC, Leibel RL, et al. The long-term stability of the human gut microbiota. Science (80-). 2013;341(6141):341. doi: 10.1126/science.1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shaw LP, Bassam H, Barnes CP, Walker AS, Klein N, Balloux F. Modelling microbiome recovery after antibiotics using a stability landscape framework. Isme J. 2019;13:1845. doi: 10.1038/s41396-019-0392-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tamboli CP, Neut C, Desreumaux P, Colombel JF. Dysbiosis in inflammatory bowel disease. Gut. 2004;53:1. doi: 10.1136/gut.53.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sorbara MT, Littmann ER, Fontana E, Moody TU, Kohout CE, Gjonbalaj M, Eaton V, Seok R, Leiner IM, Pamer EG. Functional and genomic variation between human-derived isolates of Lachnospiraceae reveals inter- and intra-species diversity. Cell Host & Microbe. 2020;28:134. doi: 10.1016/j.chom.2020.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Togo AH, Diop A, Bittar F, Maraninchi M, Valero R, Armstrong N, Dubourg G, Labas N, Richez M, Delerce J, et al. Description of mediterraneibacter massiliensis, gen. nov., sp. nov., a new genus isolated from the gut microbiota of an obese patient and reclassification of Ruminococcus faecis, Ruminococcus lactaris, Ruminococcus torques, Antonie Van Leeuwenhoek. Int J Gen Mol Microbiol. 2018;111:2107–2128. doi: 10.1007/s10482-018-1104-y. [DOI] [PubMed] [Google Scholar]
- 11.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464(7285):59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tailford LE, Owen CD, Walshaw J, Crost EH, Hardy-Goddard J, Le Gall G, de Vos Wm, Taylor GL, Juge N, de Vos WM. Discovery of intramolecular trans-sialidases in human gut microbiota suggests novel mechanisms of mucosal adaptation. Nat Commun. 2015;6:7624. doi: 10.1038/ncomms8624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crost EH, Ajandouz EH, Villard C, Geraert PA, Puigserver A, Fons M. Ruminococcin C, a new anti-Clostridium perfringens bacteriocin produced in the gut by the commensal bacterium Ruminococcus gnavus E1. Biochimie. 2011;93:1487–1494. doi: 10.1016/j.biochi.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 14.Devlin AS, Fischbach MA. A biosynthetic pathway for a prominent class of microbiota-derived bile acids. Nat Chem Biol. 2015;11:685. doi: 10.1038/nchembio.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Png CW, Lindén SK, Gilshenan KS, Zoetendal EG, McSweeney CS, Sly LI, McGuckin MA, Florin THJ. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am J Gastroenterol. 2010;105:2420–2428. doi: 10.1038/ajg.2010.281. [DOI] [PubMed] [Google Scholar]
- 16.Nishino K, Nishida A, Inoue R, Kawada Y, Ohno M, Sakai S, Inatomi O, Bamba S, Sugimoto M, Kawahara M, et al. Analysis of endoscopic brush samples identified mucosa-associated dysbiosis in inflammatory bowel disease. J Gastroenterol. 2018;53:95–106. doi: 10.1007/s00535-017-1384-4. [DOI] [PubMed] [Google Scholar]
- 17.Hall AB, Yassour M, Sauk J, Garner A, Jiang X, Arthur T, Lagoudas GK, Vatanen T, Fornelos N, Wilson R, et al. A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med. 2017;9. doi: 10.1186/s13073-017-0490-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Willing BP, Dicksved J, Halfvarson J, Andersson AF, Lucio M, Zheng Z, Järnerot G, Tysk C, Jansson JK, Engstrand L. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology. 2010;139:1844–1854.e1. doi: 10.1053/j.gastro.2010.08.049. [DOI] [PubMed] [Google Scholar]
- 19.Henke MT, Brown EM, Cassilly CD, Vlamakis H, Xavier RJ, Clardy J. Capsular polysaccharide correlates with immune response to the human gut microbe Ruminococcus gnavus. Proc Natl Acad Sci U S A. 2021;118:1–7. doi: 10.1073/pnas.2007595118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henke MT, Kenny DJ, Cassilly CD, Vlamakis H, Xavier RJ, Clardy J. Ruminococcus gnavus, a member of the human gut microbiome associated with Crohn’s disease, produces an inflammatory polysaccharide. Proc Natl Acad Sci U S A. 2019;116:12672–12677. doi: 10.1073/pnas.1904099116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Breban M, Tap J, Leboime A, Said-Nahal R, Langella P, Chiocchia G, Furet JP, Sokol H. Faecal microbiota study reveals specific dysbiosis in spondyloarthritis. Ann Rheum Dis. 2017;76:1614–1622. doi: 10.1136/annrheumdis-2016-211064. [DOI] [PubMed] [Google Scholar]
- 22.Shkoporov AN, Hill C. Bacteriophages of the human gut: the “Known unknown” of the microbiome. Cell Host & Microbe. 2019;25:195–209. doi: 10.1016/j.chom.2019.01.017. [DOI] [PubMed] [Google Scholar]
- 23.Albright MBN, Louca S, Winkler DE, Feeser KL, Haig SJ, Whiteson KL, Emerson JB, Dunbar J. Solutions in microbiome engineering: prioritizing barriers to organism establishment. Isme J. 2021;1616(2):331–338. doi: 10.1038/s41396-021-01088-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kwiatek M, Parasion S, Nakonieczna A. Therapeutic bacteriophages as a rescue treatment for drug-resistant infections – an in vivo studies overview. J Appl Microbiol. 2020;128:985–1002. doi: 10.1111/jam.14535. [DOI] [PubMed] [Google Scholar]
- 25.Shkoporov A, Clooney A, Sutton T, Ryan F, Daly K, Nolan J, McDonnell S, Khokhlova E, Draper L, Forde A, et al. The human gut virome is highly diverse, stable, and individual specific. Cell Host & Microbe. 2019;26:527–541.e5. doi: 10.1016/j.chom.2019.09.009. [DOI] [PubMed] [Google Scholar]
- 26.Shkoporov AN, Turkington CJ, Hill C. Mutualistic interplay between bacteriophages and bacteria in the human gut. Nat Rev Microbiol. 2022;20(12):737–749. doi: 10.1038/s41579-022-00755-4. [DOI] [PubMed] [Google Scholar]
- 27.Clooney AG, Sutton TDS, Shkoporov AN, Holohan RK, Daly KM, O’regan O, Ryan FJ, Draper LA, Plevy SE, Ross RP, et al. Whole-virome analysis sheds light on viral dark matter in inflammatory bowel disease. Cell Host & Microbe. 2019;26:764–778.e5. doi: 10.1016/j.chom.2019.10.009. [DOI] [PubMed] [Google Scholar]
- 28.Stockdale SR, Harrington RS, Shkoporov AN, Ev K, Daly KM, McDonnell SA, O’reagan O, Nolan JA, Sheehan D, Lavelle A, et al. Metagenomic assembled plasmids of the human microbiome vary across disease cohorts. Sci Rep. 2022;12. doi: 10.1038/s41598-022-13313-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shkoporov AN, Ryan FJ, Draper LA, Forde A, Stockdale SR, Daly KM, McDonnell SA, Nolan JA, Sutton TDS, Dalmasso M, et al. Reproducible protocols for metagenomic analysis of human faecal phageomes. Microbiome. 2018;6:68. doi: 10.1186/s40168-018-0446-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shkoporov AN, Stockdale SR, Lavelle A, Kondova I, Heuston C, Upadrasta A, Khokhlova EV, van der Kamp I, Ouwerling B, Draper LA, et al. Viral biogeography of the mammalian gut and parenchymal organs. Nat Microbiol. 2022;7(8):1301–1311. doi: 10.1038/s41564-022-01178-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bolger AMM, Lohse M, Usadel B. Genome analysis Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 33.Bankevich A, Nurk S, Antipov D, Gurevich AAA, Dvorkin M, Kulikov ASS, Lesin VMM, Nikolenko SII, Pham S, Prjibelski ADD, et al. Spades: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kolmogorov M, Yuan J, Lin Y, Pevzner PA. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol. 2019;37:540–546. doi: 10.1038/s41587-019-0072-8. [DOI] [PubMed] [Google Scholar]
- 35.Wick RR, Judd LM, Gorrie CL, Holt KE, Phillippy AM. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol. 2017;13:e1005595. doi: 10.1371/journal.pcbi.1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Coster W, D’hert S, Schultz DT, Cruts M, Van Broeckhoven C, Berger B. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. 2018;34:2666–2669. doi: 10.1093/bioinformatics/bty149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brettin T, Davis JJ, Disz T, Edwards RA, Gerdes S, Olsen GJ, Olson R, Overbeek R, Parrello B, Pusch GD, et al. Rasttk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci Rep. 2015;5:8365. doi: 10.1038/srep08365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Söding J, Biegert A, Lupas AN, Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:W244–8. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones P, Binns D, H-YHYY C, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinform. 2009;10:1–9. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, Wishart DS. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016;44:W16–21. doi: 10.1093/nar/gkw387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Laslett D, Canback B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004;32:11–16. doi: 10.1093/nar/gkh152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moraru C, Varsani A, Kropinski AM. VIRIDIC—a novel tool to calculate the intergenomic similarities of prokaryote-infecting viruses. Viruses. 2020;12:1268. doi: 10.3390/v12111268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Turner D, Reynolds D, Seto D, Mahadevan P. CoreGenes3.5: a webserver for the determination of core genes from sets of viral and small bacterial genomes. BMC Res Notes. 2013;6:140. doi: 10.1186/1756-0500-6-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nishimura Y, Yoshida T, Kuronishi M, Uehara H, Ogata H, Goto S, Valencia A. ViPTree: the viral proteomic tree server. Bioinformatics. 2017;33:2379–2380. doi: 10.1093/bioinformatics/btx157. [DOI] [PubMed] [Google Scholar]
- 46.Meier-Kolthoff JP, Göker M, Kelso J. VICTOR: genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics. 2017;33:3396–3404. doi: 10.1093/bioinformatics/btx440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Letunic I, Bork P. Interactive Tree of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2019;47:W256–259. doi: 10.1093/nar/gkz239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ågren J, Sundström A, Håfström T, Segerman B, Ahmed N. Gegenees: fragmented alignment of multiple genomes for determining phylogenomic distances and genetic signatures unique for specified target groups. Plos One. 2012;7:e39107. doi: 10.1371/journal.pone.0039107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lechner M, Findeiß S, Steiner L, Marz M, Pf S, Sj P. Proteinortho: detection of (Co-)orthologs in large-scale analysis. BMC Bioinform. 2011;12:124. doi: 10.1186/1471-2105-12-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grazziotin AL, Koonin EV, Kristensen DM. Prokaryotic virus orthologous groups (pVogs): a resource for comparative genomics and protein family annotation. Nucleic Acids Res. 2017;45:D491–8. doi: 10.1093/nar/gkw975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 52.Pritchard L, Glover RH, Humphris S, Elphinstone JG, Toth IK. Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal Methods. 2016;8(1):12–24. doi: 10.1039/C5AY02550H. [DOI] [Google Scholar]
- 53.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 54.Darling AE, Mau B, Perna NT, Stajich JE. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. Plos One. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Norman JM, Handley SA, Baldridge MT, Droit L, Liu CY, Keller BC, Kambal A, Monaco CL, Zhao G, Fleshner P, et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell. 2015;160:447–460. doi: 10.1016/j.cell.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shkoporov AN, Khokhlova EV, Fitzgerald CB, Stockdale SR, Draper LA, Ross RP, Hill C. Φcrass001 represents the most abundant bacteriophage family in the human gut and infects Bacteroides intestinalis. Nat Commun. 2018;91(9):1–8. doi: 10.1038/s41467-018-07225-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:41. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Turner D, Kropinski AM, Adriaenssens EM. A roadmap for genome-based phage taxonomy. Viruses. 2021;13:506. doi: 10.3390/v13030506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Buttimer C, Bottacini F, Shkoporov AN, Draper LA, Ross P, Hill C. Selective isolation of eggerthella lenta from human faeces and characterisation of the species prophage diversity. Microorganisms. 2022;10:195. doi: 10.3390/microorganisms10010195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bondy-Denomy J, Qian J, Westra ER, Buckling A, Guttman DS, Davidson AR, Maxwell KL. Prophages mediate defense against phage infection through diverse mechanisms. Isme J. 2016;10(12):2854–2866. doi: 10.1038/ismej.2016.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bhaya D, Davison M, Barrangou R. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu Rev Genet. 2011;45:273–297. doi: 10.1146/annurev-genet-110410-132430. [DOI] [PubMed] [Google Scholar]
- 64.Hynes AP, Rousseau GM, Agudelo D, Goulet A, Amigues B, Loehr J, Romero DA, Fremaux C, Horvath P, Doyon Y, et al. Widespread anti-CRISPR proteins in virulent bacteriophages inhibit a range of Cas9 proteins. Nat Commun. 2018. 91;9:1–10. doi: 10.1038/s41467-018-05092-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Makarova KS, Haft DH, Barrangou R, Brouns SJJ, Charpentier E, Horvath P, Moineau S, Mojica FJM, Wolf YI, Yakunin AF, et al. Evolution and classification of the CRISPR–Cas systems. Nat Rev Microbiol. 2011;9:467–477. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Labrie SJ, Samson JE, Moineau S. Bacteriophage resistance mechanisms. Nat Rev Microbiol. 2010;8:317–327. doi: 10.1038/nrmicro2315. [DOI] [PubMed] [Google Scholar]
- 67.Danis-Wlodarczyk K, Dąbrowska K, Abedon ST. Phage therapy: the pharmacology of antibacterial viruses. In: Coffey A, Buttimer C, editors. Bacterial viruses exploitation for biocontrol and therapeutics. Caister Academic Press; 2020. [Google Scholar]
- 68.Nishino K, Nishida A, Inoue R, Kawada Y, Ohno M, Sakai S, Inatomi O, Bamba S, Sugimoto M, Kawahara M, et al. Analysis of endoscopic brush samples identified mucosa-associated dysbiosis in inflammatory bowel disease. 2018;53:95–106. doi: 10.1007/s00535-017-1384-4 [DOI] [PubMed] [Google Scholar]
- 69.Tetz G, Tetz V. Bacteriophages as new human viral pathogens. Microorg. 2018;6:54. doi: 10.3390/microorganisms6020054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Eun CS, Mishima Y, Wohlgemuth S, Liu B, Bower M, Carroll IM, Sartor RB, McCormick BA. Induction of bacterial antigen-specific colitis by a simplified human microbiota consortium in gnotobiotic interleukin-10 −/− mice. Infect Immun. 2014;82:2239–2246. doi: 10.1128/IAI.01513-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genomes of bacteria and phages described in this study are available at GenBank under accession numbers given in Table 1
Table 1.
GenBank accession numbers for genomes of phages and bacteria sequenced in this study.
| Genomes | accession number | |
|---|---|---|
| Ruminococcus phage | phiRg507T2/2 | MT980836 |
| phiRg507T2/3 | MT980837 | |
| phiRg519T2 | MT980838 | |
| phiRgPS6 | MT980839 | |
| phiRgIBDN1 | MT980840 | |
| phiRM10 | MT980841 | |
| R. gnavus bacterial strains | JCM 6515T = ATCC 29149T | CP043051 |
| JCM 6515T (phiRgPS6) clone 1–1 | CP111084 | |
| JCM 6515T (phiRgPS6) clone 2–1 | CP111086 | |
| CCUG 52,279 | JAPRBD000000000 | |
| CCUG 51,289 | JAPRBC000000000 | |
| CCUG 54,531 | JAPRBB000000000 | |
| CCUG 43,222 | JAPRBA000000000 | |
| CCUG 49,994 | JAPRAY000000000 | |
| CCUG 57,161 | JAPRAX000000000 | |
| CCUG 57,208 | JAPRAW000000000 | |
| CCUG 57,137 | JAPRAV000000000 | |
| PS/160 | JAPRAU000000000 | |
The genomes of bacteria used are freely available on the NCBI website, accession numbers are provided in Table 1.