

ABSTRACT
Bacteriophages play central roles in the maintenance and function of most ecosystems by regulating bacterial communities. Yet, our understanding of their diversity remains limited due to the lack of robust bioinformatics standards. Here we present ViroProfiler, an in-silico workflow for analyzing shotgun viral metagenomic data. ViroProfiler can be executed on a local Linux computer or cloud computing environments. It uses the containerization technique to ensure computational reproducibility and facilitate collaborative research. ViroProfiler is freely available at https://github.com/deng-lab/viroprofiler.
KEYWORDS: Virome, microbiome, bacteriophages, bioinformatics, metagenomics
Introduction
Bacteriophages (or phages) are the most abundant biological entities on earth. They play a key role in most ecosystems by regulating bacterial communities. Recent studies suggested that changes in phage composition are associated with several diseases, such as IBD1,2, type 2 diabetes3, malnutrition4, and many more5. Understanding the mechanisms of interactions between phages and their bacterial hosts can provide some insights into the role of these viruses in the environment and the human body.6
The introduction of shotgun metagenomics has significantly improved our understanding of microbial community composition in most ecosystems, including the human body. However, with the introduction of Qiime7 and Mothur8 profiling of bacterial communities has become standardized, no such standard approach is yet available for analyzing the viral community. In addition, compared to metagenomics analyses of the bacterial communities, profiling viruses’ compositions is still highly time-consuming through the current approaches commonly used in the field.
Recently, several tools have been developed to characterize different features of viral contigs after assembly. These tools can be classified into three groups based on their function: 1) tools designed for viral discovery, which include VirSorter29, VIBRANT10, DeepVirFinder11, and VIP12. These tools mainly use homology searches against reference databases or features learned from viral sequences. 2) The second group includes pipelines for virome composition analysis, including VirusSeeker13 MetaVir14, ViromeScan15 and FastViromeExplorer16. 3) The third group includes tools for taxonomy classification or functional annotation, such as VMAGP17 and vConTACT218. However, the function of these tools is mainly limited to identifying a few characterization factors in viral metagenomes. Some of these tools are also highly difficult to install or use for inexperienced users, which makes configuring and integrating them into other tools for generating reproducible data challenging for researchers with limited bioinformatics experience.
Here we present ViroProfiler, a containerized pipeline for viral metagenomic data analysis. ViroProfiler takes advantage of the most recently developed viral metagenomic analysis tools and databases to improve the taxonomy and functional annotation of viruses and their gene products. In addition, ViroProfiler uses containerization to ensure computational reproducibility. ViroProfiler can be executed through a container platform such as Docker and Singularity19 on Linux clusters or cloud computing environments. It can also be installed via the Conda recipe for high-performance computing clusters that don’t support containers.
Results
Overview of the pipeline
Quality control, assembly, and viral discovery
We have included multiple quality control steps for generating an unbiased contig library for downstream analyses in ViroProfiler. These measures ensure to exclude redundancy in the contigs generated, identify prophages and dereplicate highly similar contigs of the same species. This provides a significant advantage to downstream analyses by accurately estimating the relative abundance of viral taxa and metabolic genes in samples. In addition, we included a binning option which enables construction of viral metagenome-assembled genomes (vMAGs) or bins, and provides a more realistic estimation of viral community compositions. After the non-redundant contig library (nrclib) or bins are built, we use VirSorter29, VIBRANT10, DeepVirFinder11 and CheckV20 to detect putative viral sequences. VirSorter2, VIBRANT and CheckV identify viral sequences based on their homology to the reference databases, while DeepVirFinder uses a machine learning model to detect viral sequences. Therefore, it can detect novel viruses not showing homology to the public databases. ViroProfiler provides a scoring system for classifying viral contigs identified by multiple tools in this step (Figure 1).
Figure 1.
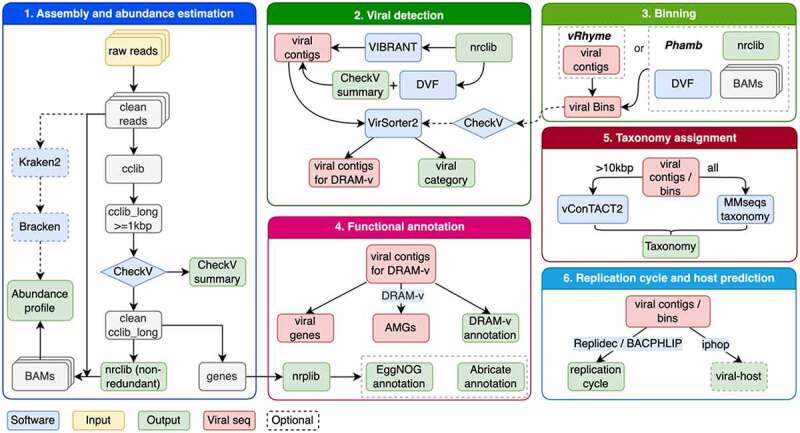
Schematic overview of the ViroProfiler pipeline. Optional steps are indicated with dashed boxes and arrows.
Functional annotation and AMG prediction
In the annotation step, the pipeline provides two possible approaches. By default, ViroProfiler uses DRAM-v, the viral mode of DRAM21, an automated pipeline for identifying microbial metabolism. DRAM-v can identify auxiliary metabolic genes (AMGs) in viral sequences and annotating their genomes using multiple publicly available databases. The downside of using DRAM-v for annotation is that it slows down the analyses. Therefore, to overcome this issue, we provide an alternative approach for gene annotation, which relies on searching the EggNOG database22 using eggNOG-mapper23. The latter is helpful if identifying AMGs in viral contigs is out of interest. For the taxonomy assignment, we combine vConTACT218 and MMseqs2 taxonomy24 module searching against NCBI viral RefSeq database. Combining these two methods, we can significantly improve the accuracy of taxonomy assignment to viral sequences from metagenomics data (Figure 1).
Host prediction, and the assessment of replication cycle
The potential hosts of viral sequences are predicted using iPHoP25, a recently developed tool which uses a two-step framework that integrates multiple methods for assigning hosts to different viruses based on their genomic signatures with a < 10% false-discovery rate. In addition, our pipeline allows predicting the replication cycle of viral sequences using BACPHLIP26 and a newly developed in-house software Replidec27, with a combined accuracy of more than 90%. These tools use the genetic signatures of viral sequences, which are associated with three different types of replication cycles in viruses, lytic, lysogenic, and chronic, to predict their replication cycles (Figure 1 and S1).
Visualization and downstream analyses
We developed an R package called vpfkit (short for “ViroProfiler Tookit”) for downstream analyses of ViroProfiler results in R. It contains functions for preprocessing data generated from multiple ViroProfiler steps, and a Shiny APP called ViroProfiler-viewer for visualizing and manipulating results interactively in a web page. ViroProfiler-viewer allows users to filter viral contigs based on their length, quality, and other annotations such as taxonomy, host, and replication type. In addition, a TreeSummarizedExperiment object file can be generated as inputs for downstream analyses in R. Intermediate files from ViroProfiler, such as genome sequences and BAM files, can be used in other software and pipelines, such as MetaPop28 for micro- and macro-viral diversity analyses.
Metagenome analyses and validation of the pipeline
We used a simulated mock dataset29 and an experimental dataset from previous studies to evaluate the performance of ViroProfiler. The mock dataset contains 14 simulated Illumina paired-end sequencing samples, each with 500–1000 viral genomes from the NCBI RefSeq database v69. We analyzed 13 out of the 14 samples using ViroProfiler (sample_12 had no reverse FASTQ file, so it was removed). We compared the viral detection precision and sensitivity of ViroProfiler with Kraken230, and abundance estimation performance with Bracken31.
Specifically, the raw reads from the mock dataset were fed into ViroProfiler for preprocessing, assembly (without binning), annotation, and abundance estimation (”ViroProfiler” in Figure 2). For comparison, Kraken2 and its standard database were used to detect viruses from reads preprocessed by ViroProfiler. Bracken was then used to estimate the abundance of viruses identified by Kraken2 (”BrackenSTD” in Figure 2) and ViroProfiler (”BrackenVPF” in Figure 2), respectively. The taxonomy lineage of viruses was standardized using Taxonkit32 on the NCBI taxonomy database (obtained on 2022-12-15). We compared the performance of these tools in virus identification using precision, sensitivity, and F1 score (harmonic mean of precision and sensitivity) on different taxonomic ranks and abundance thresholds. Our analyses show that ViroProfiler has the best performance (highest F1 score) at the phylum and order levels, especially at lower abundance thresholds, i.e., ViroProfiler can detect low-abundance viruses with high precision and sensitivity. While using Bracken with Kraken2 and its standard database (BrackenSTD) has the highest sensitivity, they showed a lower precision at the phylum and order levels. At the family level, ViroProfiler achieved performance comparable to BrackenSTD, while at the genus and species levels, the sensitivity of ViroProfiler dropped significantly.
Figure 2.
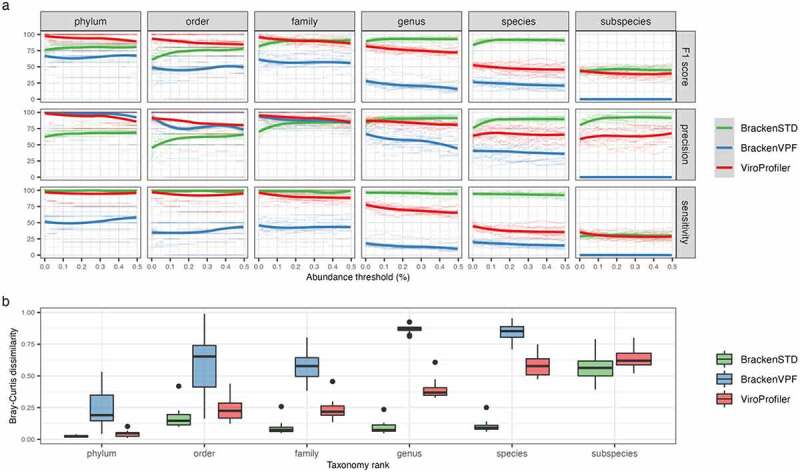
Benchmarking ViroProfiler on mock samples. a) Compares the performance of ViroProfiler with Kraken2 and Bracken in detecting viruses. b) Compares the performance of ViroProfiler and Bracken in providing estimations of viral abundance. BrackenSTD, when Bracken was used with the Kraken2 standard database. BrackenVPF, when Bracken was used with the custom database. Bracken was used for estimating the abundance of identified taxa. Smaller values indicate closer similarity to the true composition profile.
This was expected, as in contrast to ViroProfiler, which uses lowest common ancestor (LCA) of all genes in viral contigs for taxonomy assignment, Kraken2 relies on LCA of exact k-mer matches of partial genomes, which increases sensitivity when the viral sequences have representatives in the Kraken2 reference database. Since Kraken2 standard database and the mock dataset are highly similar, we created a custom database that only included viral contigs annotated by ViroProfiler to evaluate the performance of Kraken 2 when these two are less alike. Our results showed that BrackenVPF had the lowest sensitivity in all taxonomic ranks. Even at the phylum level, where ViroProfiler had>95% sensitivity and precision, BrackenVPF had only~50% sensitivity (BrackenVPF in Figure 2a). In addition, we compared the performance of BrackenSTD and BrackenVPF with ViroProfiler in estimating the viral abundances using the mock dataset. We compared the abundance profile generated by ViroProfiler, BrackenSTD, and BrackenVPF with the true composition profile from the original study using Bray-Curtis dissimilarity (Figure 2b). ViroProfiler and BrackenSTD showed similar performance at the phylum and order levels, while Kraken2 and Bracken with the standard database (BrackenSTD) performed better at the family, genus, and species levels. However, when Kraken2 and Bracken were used with the custom database (BrackenVPF), it showed the lowest performance in all taxonomic ranks.
Altogether, our analyses show that ViroProfiler can accurately classify viruses at phylum, order, and family levels. In addition, Viroprofiler provides a database-independent approach for viral classification, contrary to Kraken2. This is especially useful for metagenomic studies, as metagenomes usually include viruses with no homology to the reference database.
To evaluate the performance of ViroProfiler on real datasets, we randomly selected and analyzed 20 out of 266 samples from a previous study of viral community composition in fecal samples from ulcerative colitis (UC) patients and healthy individuals2. Using ViroProfiler, we significantly improved the viral discovery rate by identifying 761 viral contigs compared to 183 contigs assembled by the authors. We also observe differences in phage community composition identified by the earlier study compared to the ViroProfiler findings. For example, contrary to the initial analyses, we observed a higher proportion of Podoviridae in samples from healthy individuals than in UC patients (34.6% vs 12.3%). In addition, we did not observe significant differences in diversity scores, as seen in the initial analyses. Moreover, through ViroProfiler, we used DRAM-v, which with a higher accuracy, to strictly identify AMGs in viral contigs, contrary to the initial study that relied on the general functional capacity of the viral contigs, which could be misleading2. Finally, ViroProfiler assigned a host to each viral contig, showing that UC patients carry fewer phages that infect Bacteroidia than healthy individuals (Figure 3).
Figure 3.
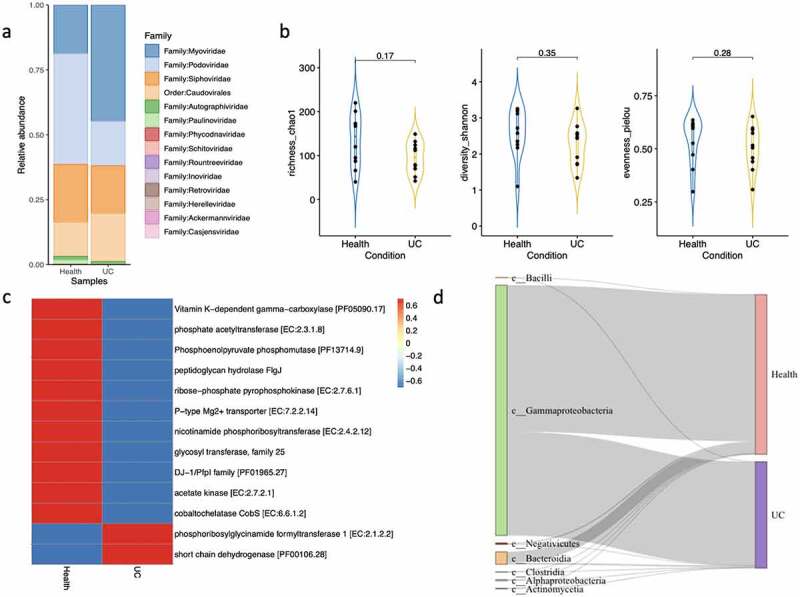
a) Relative abundance of viral contigs generated by ViroProfiler; b) Violin plots show different diversity indexes; c) Heatmap of AMGs predicted in viral contigs from healthy and UC samples; d) Sankey plot of host prediction for different viral contigs.
Computational requirements
ViroProfiler can be installed on most operating systems that support Conda and containerization techniques. However, it is recommended to run the pipeline on a High-Performance Computing (HPC) system. The minimum hard disk requirement for the databases and container images is~80GB. However, additional storage space is required if users want to run optional modules such as EggNOG annotation and PHAMB binning. A detailed storage space requirement for each module is available in supplementary table 1.
Our benchmarking analysis on 13 mock datasets using Helmholtz Munich’s Scientific Computing HPC cluster (1 to 20 CPUs and 1 to 120 GB of RAM for each process) was finished in 12 hours. Host prediction was the most time-consuming and took 10 hours to complete. However, most analyses can be run in parallel; therefore, using more computational resources will decrease the running time. The execution times and the computational resources used for each step are provided in supplementary figure S1 and supplementary file 1, respectively.
Discussion
Viral communities are central to the maintenance of most ecosystems, including the human body. The introduction of shotgun metagenomics has provided opportunities to study these communities. Yet, analyses of generated data require applying multiple bioinformatic tools and need relevant programming skills. We believe ViroProfiler, a containerized pipeline for virome data analysis, can address these issues. ViroProfiler combines stand-alone analytical tools and databases with a workflow management system which enables flexible and reproducible analyses of virome data in an interactive environment while significantly shortening the processing time.
We benchmarked ViroProfiler using mock datasets and compared its performance to the existing tools for classifying viruses. ViroProfiler showed high accuracy in classifying viruses at taxonomic ranks higher than genus. Moreover, it can detect viral replication cycles, predict hosts, and identify AMGs in viral sequences. We also used ViroProfiler for analyzing previously published experimental viral metagenome data as part of our validation step. We then compared our results with the original analyses, which showed significant improvement in multiple profiling steps, including viral discovery, taxonomy assignment, functional annotation, host and replication cycle predictions. This was achieved while less than ten percent of the published data were analyzed.
In conclusion, we believe that ViroProfiler can substantially improve the quality of data analyses in virome research and pave the ground for more standardized characterization of the viral communities from complex ecosystems. However, ViroProfiler is specifically designed for classifying viruses in samples with isolated viruses. Therefore, excessive environmental contaminations, usually found in metagenome sequences, could increase the running time of the pipeline and result in lower precision. Yet, this is a general issue with virome studies, and it is recommended to isolate the viral fractions before sequencing for an accurate estimation of viruses in the samples.
Methods
The pipeline
ViroProfiler integrates state-of-the-art bioinformatic tools via Conda environments and containerization techniques for processing viral metagenomic sequences in a nf-core32 based Nextflow33 pipeline (Figure 1). It executes series of standard viral metagenomics analysis subsequently or separately if part of the analysis has been done elsewhere. The installation process is described in detail at https://github.com/deng-lab/viroprofiler. For ensuring reproducible analyses, a specific version of the pipeline can always be run by using the version parameter in the command line (-r <version>). In addition, each container used in the workflow is tagged by the accompanying tool version, pre-build and stored on Docker Hub (https://hub.docker.com/u/denglab). The benefit of containers is that users don’t need to install multiple software that may cause conflict. Each container contains one or more sub-workflows that is versioned, and Nextflow will automatically download and manage the containers used in each step. Core modules of ViroProfiler and integrated tools are listed in Table 1.
Table 1.
Core modules and integrated tools of ViroProfiler.
| Software | Module | License | Reference |
|---|---|---|---|
| metaSPAdes | Assembly | NA | 35 |
| vRhyme | Binning | GPL v3 | 36 |
| Phamb | Binning | MIT | 37 |
| CheckV | Virus detection and QC | BSD | 20 |
| VirSorter2 | Virus detection | GPL v2 | 9 |
| DeepVirFinder | Virus detection | USC-RL v1.0 | 11 |
| VIBRANT | Virus detection and gene annotation | GPL v3 | 10 |
| DRAM | Functional annotation | GPL v3 | 21 |
| eggnog-mapper | Functional annotation | GPL v3 | 22,23 |
| abricate | Functional annotation | GPL v2 | https://github.com/tseemann/abricate |
| MMseqs2 | Taxonomy assignment | GPL v3 | 24 |
| vConTACT2 | Taxonomy assignment | GPL v3 | 18 |
| Bacphlip | Replication cycle prediction | MIT | 26 |
| Replidec | Replication cycle prediction | MIT | 27 |
| iPHoP | Host prediction | GPL v3 | 25 |
| CoverM | Abundance estimation | GPL v3 | https://github.com/wwood/CoverM |
| Kraken2 | Virus detection | MIT | 30 |
| Bracken | Abundance estimation | GPL v3 | 31 |
Quality control
The quality control of raw sequencing reads is performed using fastp37. The high-quality reads are generated by following five consecutive steps: 1) trimming adapters, 2) removing low-quality reads and 3) trimming the low-quality bases (Q < 20) at the end of reads, 4) removing the trimmed reads with length<30bp, and 5) if decontamination option is enabled, reads that show homology to mammalian host genomes will be removed38. This is specifically beneficial for identification of AMGs as the previous studies20 have shown that the removal of host contamination substantially improves the accuracy of AMG identification and interpretation of viral-encoded functions.
Genome assembly and dereplication
Each sample was individually assembled using metaSPAdes34. The assembled contigs were then merged into a multi-FASTA file and contigs shorter than a threshold (ex. 1kbp) were excluded from the further analyses. This step generated the long “complete contig library” (cclib_long). The quality of cclib_long was then evaluated using CheckV20, which were assessed for their quality, completeness, and potential contamination. The host flanking region were also removed from the final contigs. To remove redundancy in the contig library, we dereplicated the cclib_long by clustering contigs following the MIUViG guidelines (95% ANI – Average Nucleotide Identity and 85% AF – Aligned Fraction)39 using custom python script anical.py and aniclust.py from CheckV. This step generated a non-redundant contig library (nrclib) for downstream analyses.
Viral contig binning
Due to the limitation of assemblers, we usually get fragmented contigs of a viral genome. To overcome this limitation, ViroProfiler uses binning approach that relies on Phamb36 and vRhyme35 to identify contigs that belong to the same genome and classify them as a bin, or viral metagenome-assembled genome (vMAG). Phamb is a recently developed tool for binning phage genomes that relies on DeepVirFinder for viral contig discovery and a deep-learning algorithm for contig binning40. It requires>50,000 contigs as input, which sometimes can not be met. In that case, users can choose vRhyme for the binning step, which uses multi-sample coverage effect size comparisons between scaffolds, protein redundancy scoring mechanism, and machine learning model to detect bins. Viral quality, completeness and contamination ratio of bins were then assessed using CheckV. Binning is set as an optional step in ViroProfiler because the risk of false positive and the fact that contigs in a bin is connected randomly, which might not represent the actual viral genomes.
Viral contig identification
ViroProfiler integrates five different tools for identification of viral sequences: 1) VirSorter29, 2) MMseqs2 taxonomy assignment24 based on NCBI viral RefSeq, 3) CheckV20, 4) DeepVirFinder11 and 5) VIBRANT10. Briefly, contigs or bins are identified as viruses when they satisfy one of the following criteria: 1) identified as viruses in category 1, 2, 4, or 5 by VioSorter2 with default parameters (–virome mode); 2) classified as viruses by Mmseqs2 taxonomy module; 3) classified as complete, high-quality, medium-quality and low-quality by CheckV; 4) have a score>0.9 and p-value<0.01 in the DeepVirFinder prediction; 5) identified as viruses by VIBRANT. Viral detection tools were selected based on their approach to identifying viral sequences. VirSorter2, VIBRANT, MMseqs taxonomy module, and CheckV identify viral sequences based on the homology of proteins in contigs to reference databases, which is more reliable than non-homology-based tools like DeepVirFinder. However, DeepVirFinder employs a machine-learning model trained on viral genomic signatures to distinguish viral sequences from non-viral sequences. Therefore, it can detect novel viruses with no homology to the reference databases. While homology-based tools like VirSorter2 and VIBRANT tend to have lower false positive rates on longer contigs (e.g.>3 kbp), non-homology-based tools like DeepVirFinder have shown higher sensitivity, making them more suitable for analyzing short contigs (e.g.<3 kbp) and detecting novel viruses41–43.
ViroProfiler provides a confidence classification to the contigs or bins identified as viruses using the following criteria, 1) “high confident” is assigned if they are classified by VIBRANT, or as category 1,2 by VirSorter2, or as viruses by mmseqs2 taxonomy module, or have “Complete”, “High-quality”, “Medium-quality” annotation in CheckV; 2) “low confident” are rest contigs that predicted as viral sequences by DeepVirFinder, and “unclassified” by MMseqs2 taxonomy module or have “Low quality” annotation in CheckV.
Gene prediction and protein function annotation
To keep as many potential genes as possible, contigs in cclib_long are fed into Prodigal44 for predicting protein-coding genes and translating them to proteins. To remove redundancy and improve annotation speed in downstream analysis, proteins are clustered using MMseqs245 using thresholds of minimum identity (0.7 by default) and coverage (0.9 by default). These thresholds can be modified in the params.yml config file before running the pipeline. Representative proteins of these clusters are used to make the non-redundant protein library (nrplib), which is assigned a computationally predicted function and gene ontology using eggNOG-mapper23 searching against the EggNOG database22. This step will not be necessary in case prediction of AMGs is planned as DRAM-v also provides functional annotations. Functional annotations of viral contigs are annotated using DRAM-v, which searches viral genes against multiple databases, such as KEGG46, PFAM47, VOGDB (https://vogdb.org/) and NCBI viral RefSeq48. DRAM-v also detects auxiliary metabolic genes (AMGs) in viral genomes. In addition, antimicrobial resistance and virulence genes can be identified using Abricate (https://github.com/tseemann/abricate) to search genes against CARD49, ResFinder50 and VFDB51, 52 databases.
Taxonomy assignment
Taxonomy assignment of viral contigs is performed using a combination of viral genome clustering and voting-based classification approaches. Briefly, for viral contigs longer than 10 kbp, their protein sequences are fed into vConTACT253 for virus clustering and taxonomy annotation. Since vConTACT2 does not report taxonomy names at the species and subspecies level, we combine vConTACT2 clustering with the MMseqs2 taxonomy module24 using the NCBI viral RefSeq as references. MMseqs2 assigns taxonomy to viral sequences by comparing their proteins to reference databases and determining taxonomy using the lowest common ancestor. MMseqs2 was selected as it is fast and sensitive24. We combine the MMseqs2 results with viral clusters (VCs) generated by VConTACT2. When VCs contain multiple contigs with different taxonomies, we use LCA to assign the final taxonomy. However, users could manually check these VCs and determine taxonomy based on their domain knowledge. To be consistent with taxonomy assignment, names and lineages are standardized using taxonkit32 and an in-house python script.
Host and replication cycle prediction
We used iPHoP to predict virus-host ranges25, which integrates multiple methods to provide host predictions. This makes its predictions highly reliable compared to other tools available for host prediction. However, iPHoP has a big database (~200GB), thus we set host prediction as an optional step. Users can skip this step if they are not interested in the host predictions. The virus replication cycle is predicted using BACPHLIP26 and Replidec27.
Viral abundance estimation
ViroProfiler provides two approaches for viral abundance estimation. The first approach uses Bracken to estimate the abundance of each taxonomic category from the Kraken2 classification results. This provides accurate estimates of viral sequences with representatives in the Kraken2 reference database. However, Kraken2 fails to identify novel viruses with no homology to the databases. Therefore, the second approach estimates viral abundance based on mapping clean reads to ViroProfiler assembled viral contigs. Briefly, clean reads are mapped to contigs in nrclib using bowtie254 to create BAM files for each sample. Next, CoverM (https://github.com/wwood/CoverM) is used to remove spurious read mappings at less than 90% identity in BAM files and then calculate the number of reads (−m count), trimmed mean of coverage (-m trimmed_mean) and covered fraction (-m covered_fraction) of each contig across all samples. In the downstream analyses, the abundance of a viral contig in a sample is usually set to zero if reads from that contig cover less than a threshold percentage (ex. 50%) in the sample. This refinement of the abundance table can be generated in ViroProfiler-viewer in an interactive way. Finally, if the abundance of genes is of interest, featureCounts55 is used to calculate number of reads mapped to each protein-coding gene. Altogether, these two approaches can accurately estimate viral abundance regardless of their homology to reference databases.
Supplementary Material
Funding Statement
This work was funded by the German Research Foundation (D.F.G. Emmy Noether program, Project No. 273124240, SFB 1371, Project No. 395357507), Marie Sklodowska-Curie Actions Innovation Training Networks grant agreement no. 955974 (VIROINF), and the European Research Council Starting grant (ERC StG 803077) awarded to L.D. J.R was supported by the China Scholarship Council (CSC).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Author contributions
J.R. developed the software. M.K.M. and J.R. drafted the manuscript. J.R and X.P. performed the analyses. J.X. wrote the documentation. M.K.M. and L.D. conceived and supervised the project. All authors reviewed and approved the manuscript.
Data and software availability
ViroProfiler is available at https://github.com/deng-lab/viroprofiler. The development version of the pipeline will be updated once the dependent software are updated. The stable version will be updated yearly. The R package vpfkit is available at https://github.com/deng-lab/vpfkit. All data and reproducible analysis scripts used in this study are available as an R package at https://github.com/deng-lab/vpfpaper.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19490976.2023.2192522.
References
- 1.Clooney AG, Sutton TDS, Shkoporov AN, Holohan RK, Daly KM, O’regan O, Ryan FJ, Draper LA, Plevy SE, Ross RP, et al. Whole-virome analysis sheds light on viral dark matter in inflammatory bowel disease. Cell Host & Microbe. 2019;26:764–778.e5. doi: 10.1016/j.chom.2019.10.009. [DOI] [PubMed] [Google Scholar]
- 2.Zuo T, X-J L, Zhang Y, Cheung CP, Lam S, Zhang F, Tang W, Ching JYL, Zhao R, Chan PKS, et al. Gut mucosal virome alterations in ulcerative colitis. Gut. 2019;68:1169–11. doi: 10.1136/gutjnl-2018-318131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ma Y, You X, Mai G, Tokuyasu T, Liu C.. A human gut phage catalog correlates the gut phageome with type 2 diabetes. Microbiome. 2018;6:24. doi: 10.1186/s40168-018-0410-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mirzaei MK, Khan MAA, Ghosh P, Taranu ZE, Taguer M, Ru J, Chowdhury R, Kabir MM, Deng L, Mondal D, et al. Bacteriophages isolated from stunted children can regulate gut bacterial communities in an age-specific manner. Cell Host & Microbe. 2020;27:199–212.e5. doi: 10.1016/j.chom.2020.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma T, Ru J, Xue J, Schulz S, Mirzaei MK, Janssen K-P, Quante M, Deng L. Differences in gut virome related to Barrett esophagus and esophageal adenocarcinoma. Microorganisms. 2021;9:1701. doi: 10.3390/microorganisms9081701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Noble WS, Lewitter F. A quick guide to organizing computational biology projects. PLoS Comput Biol. 2009;5:e1000424. doi: 10.1371/journal.pcbi.1000424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo J, Bolduc B, Zayed AA, Varsani A, Dominguez-Huerta G, Delmont TO, Pratama AA, Gazitúa MC, Vik D, Sullivan MB, et al. VirSorter2: a multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome. 2021;9:37. doi: 10.1186/s40168-020-00990-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kieft K, Zhou Z, Anantharaman K. VIBRANT: automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome. 2020;8:90. doi: 10.1186/s40168-020-00867-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ren J, Song K, Deng C, Ahlgren NA, Fuhrman JA, Li Y, Xie X, Poplin R, Sun F. Identifying viruses from metagenomic data using deep learning. Quantitative Biology. 2020;8:64–77. doi: 10.1007/s40484-019-0187-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Y, Wang H, Nie K, Zhang C, Zhang Y, Wang J, Niu P, Ma X. VIP: an integrated pipeline for metagenomics of virus identification and discovery. Sci Rep. 2016;6. doi: 10.1038/srep23774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao G, Wu G, Lim ES, Droit L, Krishnamurthy S, Barouch DH, Virgin HW, Wang D. VirusSeeker, a computational pipeline for virus discovery and virome composition analysis. Virology. 2017;503:21–30. doi: 10.1016/j.virol.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roux S, Faubladier M, Mahul A, Paulhe N, Bernard A, Debroas D, Enault F. Metavir: a web server dedicated to virome analysis. Bioinformatics. 2011;27:3074–3075. doi: 10.1093/bioinformatics/btr519. [DOI] [PubMed] [Google Scholar]
- 15.Rampelli S, Soverini M, Turroni S, Quercia S, Biagi E, Brigidi P, Candela M. ViromeScan: a new tool for metagenomic viral community profiling. BMC Genomics. 2016;17:165. doi: 10.1186/s12864-016-2446-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tithi SS, Aylward FO, Jensen RV, Zhang L. FastViromeExplorer: a pipeline for virus and phage identification and abundance profiling in metagenomics data. PeerJ. 2018;6:e4227. doi: 10.7717/peerj.4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lorenzi HA, Hoover J, Inman J, Safford T, Murphy S, Kagan L, Williamson SJ. The Viral MetaGenome Annotation Pipeline (VMGAP): an automated tool for the functional annotation of viral metagenomic shotgun sequencing data. Stand Genomic Sci. 2011;4:418–429. doi: 10.4056/sigs.1694706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bin Jang H, Bolduc B, Zablocki O, Kuhn JH, Roux S, Adriaenssens EM, Brister JR, Kropinski AM, Krupovic M, Lavigne R, et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat Biotechnol. 2019;37:632–639. doi: 10.1038/s41587-019-0100-8. [DOI] [PubMed] [Google Scholar]
- 19.Kurtzer GM, Sochat V, Mw B, Gursoy A. Singularity: scientific containers for mobility of compute. PLoS One. 2017;12:e0177459. doi: 10.1371/journal.pone.0177459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nayfach S, Camargo AP, Schulz F, Eloe-Fadrosh E, Roux S, Kyrpides NC . CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat Biotechnol. 2021;39(5): 578–585. doi: 10.1038/s41587-020-00774-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shaffer M, Borton MA, McGivern BB, Zayed AA, La Rosa SL, Solden LM, Liu P, Narrowe AB, Rodríguez-Ramos J, Bolduc B, et al. DRAM for distilling microbial metabolism to automate the curation of microbiome function. Nucleic Acids Res. 2020;48:8883–8900. doi: 10.1093/nar/gkaa621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huerta-Cepas J, Szklarczyk D, Heller D, Hernández-Plaza A, Forslund SK, Cook H, Mende DR, Letunic I, Rattei T, Jensen LJ, et al. Ggnog 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2018;47:D309–14. doi: 10.1093/nar/gky1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cantalapiedra CP, Hernández-Plaza A, Letunic I, Bork P, Huerta-Cepas J, Tamura K. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol Biol Evol. 2021;38:5825–5829. doi: 10.1093/molbev/msab293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mirdita M, Steinegger M, Breitwieser F, Söding J, Levy Karin E, Kelso J. Fast and sensitive taxonomic assignment to metagenomic contigs. Bioinformatics. 2021;37:3029–3031. doi: 10.1093/bioinformatics/btab184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roux S, Camargo AP, Coutinho FH, Dabdoub SM, Dutilh BE, Nayfach S, Tritt A . iPHoP: an integrated machine-learning framework to maximize host prediction for metagenome-assembled virus genomes. bioRxiv. 2022. doi: 10.1101/2022.07.28.501908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hockenberry AJ, Co W. BACPHLIP: predicting bacteriophage lifestyle from conserved protein domains. PeerJ. 2021;9:e11396. doi: 10.7717/peerj.11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peng X, Ru J, Mirzaei MK, Deng L. Replidec – use I Bayes classifier to identify virus lifecycle from metagenomics data. bioRxiv. 2022. doi: 10.1101/2022.07.18.500415. [DOI] [Google Scholar]
- 28.Gregory AC, Gerhardt K, Zhong Z-P, Bolduc B, Temperton B, Konstantinidis KT, Sullivan MB. MetaPop: a pipeline for macro- and microdiversity analyses and visualization of microbial and viral metagenome-derived populations. Microbiome. 2022;10:49. doi: 10.1186/s40168-022-01231-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roux S, Emerson JB, Eloe-Fadrosh EA, Sullivan MB. Benchmarking viromics: an in silico evaluation of metagenome-enabled estimates of viral community composition and diversity. PeerJ. 2017;5:e3817. doi: 10.7717/peerj.3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wood DE, Lu J, Langmead B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019;20:257. doi: 10.1186/s13059-019-1891-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu J, Breitwieser FP, Thielen P, Salzberg SL. Bracken: estimating species abundance in metagenomics data. PeerJ Computer Science. 2017;3:e104. doi: 10.7717/peerj-cs.104. [DOI] [Google Scholar]
- 32.Shen W, Ren H. TaxonKit: a practical and efficient NCBI taxonomy toolkit. Journal of Genetics and Genomics. 2021;48:844–850. doi: 10.1016/j.jgg.2021.03.006. [DOI] [PubMed] [Google Scholar]
- 33.Ewels PA, Peltzer A, Fillinger S, Patel H, Alneberg J, Wilm A, Garcia MU, Di Tommaso P, Nahnsen S. The nf-core framework for community-curated bioinformatics pipelines. Nat Biotechnol. 2020;38:276–278. doi: 10.1038/s41587-020-0439-x. [DOI] [PubMed] [Google Scholar]
- 34.Di Tommaso P, Chatzou M, Floden EW, Barja PP, Palumbo E, Notredame C. Nextflow enables reproducible computational workflows. Nat Biotechnol. 2017;35:316–319. doi: 10.1038/nbt.3820. [DOI] [PubMed] [Google Scholar]
- 35.Nurk S, Meleshko D, Korobeynikov A, Pa P. metaSpades: a new versatile metagenomic assembler. Genome Res. 2017;27:824–834. doi: 10.1101/gr.213959.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kieft K, Adams A, Salamzade R, Kalan L, Anantharaman K. vRhyme enables binning of viral genomes from metagenomes. Nucleic Acids Res. 2022;50:e83. doi: 10.1093/nar/gkac341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johansen J, Plichta DR, Nissen JN, Jespersen ML, Shah SA, Deng L, Stokholm J, Bisgaard H, Nielsen DS, Sørensen SJ, et al. Genome binning of viral entities from bulk metagenomics data. Nat Commun. 2022;13:965. doi: 10.1038/s41467-022-28581-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen S, Zhou Y, Chen Y, Gu J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–90. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gregory AC, Zablocki O, Zayed AA, Howell A, Bolduc B, Sullivan MB. The gut virome database reveals age-dependent patterns of virome diversity in the human gut. Cell Host & Microbe. 2020;28(5):724–740.e8. doi: 10.1016/j.chom.2020.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roux S, Adriaenssens EM, Dutilh BE, Koonin EV, Kropinski AM, Krupovic M, Kuhn JH, Lavigne R, Brister JR, Varsani A, et al. Minimum Information about an Uncultivated Virus Genome (MIUViG). Nat Biotechnol. 2019;37:29–37. doi: 10.1038/nbt.4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nissen JN, Johansen J, Allesøe RL, Sønderby CK, Armenteros JJA, Grønbech CH, Jensen LJ, Nielsen HB, Petersen TN, Winther O, et al. Improved metagenome binning and assembly using deep variational autoencoders. Nat Biotechnol. 2021;39:1–6. doi: 10.1038/s41587-020-00777-4. [DOI] [PubMed] [Google Scholar]
- 42.Schackart KE, Graham JB, Ponsero AJ, Hurwitz BL. Evaluation of computational phage detection tools for metagenomic datasets. Front Microbiol. 2023;14. doi: 10.3389/fmicb.2023.1078760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pratama AA, Bolduc B, Zayed AA, Zhong Z-P, Guo J, Vik DR, Gazitúa MC, Wainaina JM, Roux S, Sullivan MB. Expanding standards in viromics: in silico evaluation of dsDNA viral genome identification, classification, and auxiliary metabolic gene curation. PeerJ. 2021;9:e11447. doi: 10.7717/peerj.11447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Glickman C, Hendrix J, Strong M. Simulation study and comparative evaluation of viral contiguous sequence identification tools. BMC Bioinform. 2021;22:329. doi: 10.1186/s12859-021-04242-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hyatt D, Chen G-L, LoCascio PF, Land ML, Larimer FW, Lj H. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Steinegger M, Söding J. Mmseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat Biotechnol. 2017;35:1026–1028. doi: 10.1038/nbt.3988. [DOI] [PubMed] [Google Scholar]
- 47.Kanehisa M, Goto SK. Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mistry J, Chuguransky S, Williams L, Qureshi M, Salazar GA, Sonnhammer ELL, Tosatto SCE, Paladin L, Raj S, Richardson LJ, et al. Pfam: the protein families database in 2021. Nucleic Acids Res. 2021;49:D412–9. doi: 10.1093/nar/gkaa913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li W, O’neill KR, Haft DH, DiCuccio M, Chetvernin V, Badretdin A, Coulouris G, Chitsaz F, Derbyshire MK, Durkin AS, et al. RefSeq: expanding the prokaryotic genome annotation pipeline reach with protein family model curation. Nucleic Acids Res. 2021;49:D1020–8. doi: 10.1093/nar/gkaa1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alcock BP, Raphenya AR, Lau TTY, Tsang KK, Bouchard M, Edalatmand A, Huynh W, Nguyen AL, Cheng AA, Liu S, et al. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020;48:D517–25. doi: 10.1093/nar/gkz935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Florensa AF, Kaas RS, Clausen PTLC, Aytan-Aktug D, Aarestrup FMY. ResFinder an open online resource for identification of antimicrobial resistance genes in next-generation sequencing data and prediction of phenotypes from genotypes. Microbial Genomics. 2022;8:000748. doi: 10.1099/mgen.0.000748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu B, Zheng D, Jin Q, Chen L, Yang J. VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019;47:D687–92. doi: 10.1093/nar/gky1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bolduc B, Jang HB, Doulcier G, You Z-Q, Roux S, Mb S. vContact: an iVirus tool to classify double-stranded DNA viruses that infect archaea and bacteria. PeerJ. 2017;5:e3243. doi: 10.7717/peerj.3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012. Apr;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ViroProfiler is available at https://github.com/deng-lab/viroprofiler. The development version of the pipeline will be updated once the dependent software are updated. The stable version will be updated yearly. The R package vpfkit is available at https://github.com/deng-lab/vpfkit. All data and reproducible analysis scripts used in this study are available as an R package at https://github.com/deng-lab/vpfpaper.