

Abstract
Proton MR spectra of the brain, especially those measured at short and intermediate echo times, contain signals from mobile macromolecules (MM). A description of the main MM is provided in this consensus paper. These broad peaks of MM underlie the narrower peaks of metabolites and often complicate their quantification but they also may have potential importance as biomarkers in specific diseases. Thus, separation of broad MM signals from low-molecular-weight metabolites enables accurate determination of metabolite concentrations and is of primary interest in many studies. Other studies attempt to understand the origin of the MM spectrum, to decompose it into individual spectral regions or peaks and to use the components of the MM spectrum as markers of various physiological or pathological conditions in biomedical research or clinical practice. The aim of this consensus paper is to provide an overview and some recommendations on how to handle the MM signals in different types of studies together with a list of open issues in the field which are all summarized at the end of the manuscript.
Keywords: Brain macromolecules, proton magnetic resonance spectroscopy, quantification, parameterization, metabolite quantification, mobile lipids, spectral analysis, fitting
1. Origin of Macromolecule Signals in Proton Spectra
Broad signals underlying the narrower signals of low molecular weight metabolites are observable in 1H MR spectra of the human and animal brain (likely present in other tissues as well) especially at short echo times (TE) and remain detectable at intermediate TE as well (Section 3.2). These signals arise from mobile macromolecules (MM), which display shorter T1 and T2 relaxation times and a lower apparent diffusion coefficient (ADC) compared to metabolites1,2. In the normal brain, MM signals arise mainly from the protons of amino acids within cytosolic proteins3–8, primarily in regions undergoing rapid motions on the time scale of NMR. The ‘mobile’ of MM highlights this fact, although the acronym is interchangeable with ‘macromolecules’ as used by many authors. With onset of disease (e.g., tumors, multiple sclerosis, and stroke) signals from mobile lipids (ML) appear in addition, overlapping with peaks of mobile proteins/peptides, making their separation difficult and thus their sum is mainly reported. Hence, the proteins and lipids detected in vivo with MRS reflect a smaller fraction of the total proteins and lipids of tissue, much of which is bound within membranes, producing extreme line broadening with loss of NMR ‘visibility’.
MM signals upfield of tissue water (~0.5 to 4.5 ppm) correspond to aliphatic (methyl, methylene, and methine) protons, whereas peaks downfield of water (~5.5 to 9.0 ppm) reflect aromatic CH and exchangeable NH protons (amide, amine, and imine). Direct transfer of magnetization between water protons and exchangeable amide or amine protons (tentative assignment based on similarity in chemical shift and exchange rate seen in protein NMR spectra) has been reported using Water Exchange spectroscopy (WEX)9, as well as Chemical Exchange Saturation Transfer (CEST) imaging10. The pattern of aliphatic resonance intensities in WEX spectra resembles brain MM spectra measured in vitro and in vivo, but these resonances have not been assigned specifically to MM nor can exchangeable free amino acids and metabolites identified in the downfield region (e.g., NAA, GSH, ATP, NAD(H)) be excluded. Indirect transfer of label through intramolecular relayed Nuclear Overhauser Effect (NOE) to upfield aliphatic and downfield (possibly aromatic) protons has been reported with these techniques, particularly in CEST imaging11, and may contribute to the appearance of MM spectra.
Post-translational modification of chemical groups in proteins (e.g., methylation, acetylation, glycosylation, sialylation) may contribute to the signals in MM spectra. For example, sharp singlets in the acetyl region (~2.05–2.1 ppm) with relatively longer T2 (similar to the N-acetylaspartate (NAA) acetyl signal) are seen in some fractions of dialyzed brain cytosol4. Many brain proteins are acetylated, including histone and non-histone nuclear proteins, cytoplasmic, mitochondrial proteins and myelin proteins - the primary target of acetylation being lysine (N6ε-acetyl lysine), often with multiple acetyl lysines on a given protein. N-acetylated hexoses of glycoproteins (e.g., N-acetylglucose, -galactose or -neuraminic acid containing oligosaccharides) may contribute signal at 2.05 ppm to brain MM, particularly in necrotic tissue and in cystic tumors12, while methyl protons of fucosylated glycoproteins can contribute at 1.3 ppm13. As glycoproteins are present mainly on the cell surface, these signals originate extracellularly. To the current understanding, MM, such as glycogen or polynucleotides (DNA/RNA), do not contribute to brain MM when isolated and measured in vitro, although potential contributions to the MM spectrum in vivo may exist14.
MM in dialyzed brain cytosol display the same number and pattern of proton signals (relative intensities and chemical shifts) as seen for brain in vivo when metabolites are suppressed via T1- or diffusion-weighting sequences3,4. The same spectral pattern is seen for certain perchloric acid-soluble polypeptides (<40 kD), such as thymosin-β4 and histone-H1 isolated from guinea-pig cerebral cortex6,7,15, as well as microtubule-associated proteins (55–240 kD) isolated from bovine brain16,17. The broad signals from MM in perchloric acid-extracts or dialyzed cytosol disappear upon treatment with strong acid and heat (boiling with 6M HCl for 24 hours), or with proteolytic enzymes, with the appearance of various free amino acids. In contrast, normal brain tissue extracted into chloroform/methanol solutions, which solubilizes all brain lipids (including membrane phospholipids), produces peaks not seen in MM spectra of normal brain. Most significantly, cross-peaks characteristic of fatty acyl chains of lipids are not seen in 2D-Correlated Spectroscopy (COSY) spectra of dialyzed cytosol or whole homogenate of non-diseased brain, ruling out significant lipid contributions to their spectra.
It is well known from protein solution state NMR literature18,19 that sharp signals arise in proton NMR spectra. These signals are generally thought to reflect more mobile regions of polypeptide chains of rapidly tumbling proteins. In contrast, membrane bound proteins investigated by conventional solution NMR yield broad and mostly featureless spectra20. Thus the assignment in vivo of detectable MM to amino acids in freely tumbling cytosolic proteins is consistent with the extensive multiplicity and connectivity in 2D J-RES and COSY spectra of brain MM (discussed below). The closely similar spectral intensity patterns for MM over a large molecular weight range (3.5 to >100 kD), suggest that MM signals are largely non-specific with regard to any particular protein and further support the notion that cytosolic proteins in general contribute to MM spectra. This would explain the highly similar spectral patterns for brain MM during development, across brain regions and species21. MM signals of dialyzed nerve terminal lysates and myelin-enriched fractions from rat brain are qualitatively similar both to MM of dialyzed brain cytosol and to spectra recorded in vivo (unpublished data of K.L. Behar22), suggesting that MM signals may arise from cytosolic proteins/peptides in different cellular compartments, but the distribution is unknown. In principle, altered MM signal intensity, as might be observed with aging or disease, could reflect changes in total protein level or mobility.
1.1. Spectral Characteristics of MM
Broad peaks in MM spectra are composite signals, composed of multiple overlapping and closely spaced multiplets (due to scalar couplings) that originate from different amino acids4,8. Spectral patterns of the same amino acids also differ slightly with respect to their chemical shifts across different proteins23,24. Thus, MM spectra in vivo most likely represent distributions of overlapping multiplets from different amino acids within different proteins25,26, contributing to the apparent broad linewidths of the various peaks (Appendix 1, Table S1).
Chemical shifts, multiplicities and coupling constants of MM signals are consistent with functional groups (methyl, methylene and methine) of various amino acids in polypeptides. Coupling constants of MM signals reflect geminal (two-bond, 2J) and vicinal (three-bond, 3J) scalar couplings. MM signals undergo J-modulation and their appearance changes with TE. The most prominent spin-spin couplings in brain MM are between peaks at 1.70 and 3.0 ppm (M1.70 ↔ M3.00, assigned tentatively to lysineεδ), between the peaks at 0.94 and 2.07 ppm (M0.94 ↔ M2.07, tentatively assigned to branched-chain amino acids, e.g., valineβγ and isoleucineβγ) and ~1.3 ppm to ~4.35 ppm7 (for more details on the nomenclature of the MM components see Table 1).
Table 1:
Description of the main MM peaks. The data presented are mainly extracted from references4,6,8,29,34,39,51,66,70,72
| Recommended nomenclature | Previous nomenclature | ppma | Cross-peaks in 2D spectrab | J, Hzc | Tentative Assignment | Observations |
|---|---|---|---|---|---|---|
| M0.94 | M1 | 0.94 0.88 |
0.94, 2.05 | 7.7(d) | Leucine, isoleucine, valine | -reported in vivo at all magnetic fields -two main peaks4,29,66,72 |
| M1.22 | M2 | 1.22 | 1.22, 4.20 | 6.6(d) | Threonine | -reported in vivo starting at 3 T28,71,81 |
| M1.43 | M3 | 1.43 | 1.43, 1.70 1.43, 4.32 |
7.7(d) | Alanine | -reported in vivo at all magnetic fields |
| M1.70 | M4 | 1.70 | 1.70, 1.43 1.70, 3.00 |
m 7.8(t) |
Lysine, arginine, leucine | -reported in vivo starting at 3 T28,71,81 -scalar coupling between M1.70-M3.00 is important for the detection of GABA |
| M1.81 | - | 1.81 | - | - | To be confirmed in future studies | -reported in vivo in rat brain at 17.2 T29. This peak was identified based on a parameterization of the in vivo acquired MM signal using 32 individual Gaussian functions, therefore its biological relevance needs to be confirmed |
| M1.9 | - | 1.9 | 1.89, 2.03 | - | To be confirmed in future studies | -reported in vivo in rat brain at 9.4 T66 and 17.2 T29. Several peaks were identified based on a parameterization of the in vivo acquired MM signal using 15 or 32 individual Gaussian functions, therefore their biological relevance needs to be confirmed |
| M2.05 | M5 (M5a66) (M5b66) |
2.05 2.00 2.03 |
2.05, 0.94 2.03, 1.89 2.07, 3.22 |
m | Glutamate, glutamine, | -reported in vivo at all magnetic fields |
| M2.07 | - | 2.05, 2.07, 2.11 |
None4,8 | s | To be confirmed in future studies | -peak reported in ref4,8 -peak considered to belong to the MM group M5 in ref4,8 |
| M2.17 | (M5c66) | 2.17 | 2.16, 2.54 2.17, 3.80 |
- | To be confirmed in future studies | -peak reported in ref4,8,29,66,72 and considered to belong to the MM group M5 -reported in vivo starting at 9.4 T in rat brain |
| M2.27 | M6 (M6a)66 (M6b)66 |
2.27 | - | 8.2(m) | Glutamate, glutamine | -reported in vivo starting at 3 T28,71,81 |
| M2.37 | 2.37 | - | - | To be confirmed in future studies | -reported in vivo in rat brain at 9.4 T66, 16.4 T72 and 17.2 T29. Identified based on a parameterization of the in vivo acquired MM signal using 15, 17 or 32 individual Gaussian functions, therefore their biological relevance needs to be confirmed. This resonance might also be caused by the effect of field on strongly coupled methylenes of glutamate/glutamine | |
| M2.50 | (M6c)66 | 2.50 | - | - | To be confirmed in future studies | -reported in vivo in rat brain at 9.4 T66 and 17.2 T29. Identified based on a parameterization of the in vivo acquired MM signal using 15 or 32 individual Gaussian functions, therefore their biological relevance needs to be confirmed. |
| M2.54 | (M6d66) (M739) |
2.54 2.57 |
2.54, 2.16 2.55, 2.74 |
~10,18(dd) | β-methylene protons of aspartyl groups | -reported in vivo starting at 3 T71 and assigned to β-methylene protons of aspartyl groups at 9.4T39 |
| M2.74 | (M839) (M6e66) |
2.74 2.68 |
2.74, 2.55 | ~4,18(dd) | β-methylene protons of aspartyl groups | -reported in vivo starting at 9.4 T and assigned to β-methylene protons of aspartyl groups in ref39 |
| M2.97 | - | 2.97 | - | - | To be confirmed in future studies | -reported in vivo in rat brain at 9.4 T66 and 17.2 T29. Identified based on a parameterization of the in vivo acquired MM signal using 15 or 32 individual Gaussian functions, therefore their biological relevance needs to be confirmed. |
| M3.00 | M7 (M939) (M7a66) (M7b66) |
3.00 3.01 2.96 3.04 |
3.00, 1.70 3.00, 3.31 |
7.6(t) | Lysine, (Cys)2 |
-reported in vivo at all magnetic fields |
| M3.21 | M8a66 (MM851) (M1039) M8b66 |
3.21 3.21 3.21 3.26 |
3.22, 2.07 | - | Valine-Hβ4 αCH protons of protein amino acids39 |
-reported in vivo starting at 3 T28,71,81 -phosphatidyl choline methyl (singlet) may contribute |
| M3.43 – 3.95 | M966 —29 |
3.43 – 3.95 3.54 – 3.95 |
- | To be confirmed in future studies | -reported in vivo in rat brain at 9.4 T66 and 17.2 T29. Identified based on a parameterization of the in vivo MM signal using 15 or 32 individual Gaussian functions, therefore their biological relevance needs to be confirmed. | |
| M3.71 | (M1139) | 3.71 | - | αCH protons of protein amino acids | -reported in vivo starting at 3 T28,71,81 with improved spectral resolution at 7 T | |
| M3.79 | M1239 MM951 |
3.79 3.77 |
3.80, 2.17 3.80, 4.91 |
- | αCH protons of protein amino acids | -reported in vivo starting at 3 T28,71,81 with improved spectral resolution at 7 T |
| M3.87 | M1339 | 3.87 | - | αCH protons of protein amino acids | -reported in vivo starting at 7 T-9.4 T | |
| M3.97 | M1439 | 3.97 | - | αCH protons of protein amino acids | -reported in vivo starting at 3 T28,71,81 with improved spectral resolution at 7 T | |
| M4.05–4.43 | —29 M1066 |
4.05 – 4.42 4.22 – 4.43 |
4.24,1.24 4.32, 1.43 |
Threonine-Hβ4,8 Threonine-Hγ4,8 |
||
| M4.20 | M1539 | 4.20 | - | αCH protons of protein amino acids | -reported in vivo starting at 7 T-9.4 T -at 9.4T and higher several MM peaks were reported in this range, care has to be taken regarding the water suppression in this range |
Small variations in ppm (~0.01–0.04 ppm) can be found in different papers.
MM spectra of healthy brain have shown some variations in peak intensities, for instance between gray matter (GM) and white matter (WM), mainly in humans (Section 6)27,28. With the known (M1.22+M1.43)/M0.94 ratio in a healthy region of brain where ML signals are absent, an increase of this ratio in the diseased brain can be assigned to the contribution of ML, without specific knowledge of the composition of each. Other uses of MM signal ratio combinations, as prior knowledge for individual MM peak intensities estimation, must be employed with caution (Sections 4 and 6). Intensities of several MM signals are highly correlated, as expected due to existing J-couplings and the underlying spectral pattern consisting of multiple resonances from the contributing amino acids. Signals from different amino acids may also be correlated when originating from the same protein/peptide; for example: M0.94 and M3.00 share no J-couplings and are ascribed to different amino acids, yet, both resonances can occur in the same protein (e.g., thymosin-β415). As composite signals reflecting a mix of proteins/peptides of unknown composition and density, the interpretation of variations in MM spectral components are best considered in their totality. The parametrization into individual MM components and their influence on metabolite concentrations has also been evaluated, but further studies are required for the identification of the possible soft constrains and systematic errors (Sections 4 and 6).
1.2. Estimation of MM content
The proton concentration in vivo for the presumed methyl MM signals at 1.22 and 1.43 ppm in rat cortex was estimated by Kauppinen et al.7 (using surface radiofrequency (RF) coil localization and spectral editing) with estimates of ~2 mM and ~4 mM for M1.22 and M1.43, respectively. In line with these results a concentration estimate for M3.00, which shows the least overlap with other resonances, is ~1.7–13 mM4,28,29 as proton density, or 0.8–6.5 mM as lysine residues assuming this peak to represent lysine ε [CH2] only (relaxation effects were taken into account in the calculations). Since lysine constitutes ~6% of total brain protein by weight30–32, and protein is ~10% of tissue weight33, total lysine in protein is ~47 μmol/g brain. Thus, the intensity of M3.00 reflects 2–14% of total lysine, suggesting a large fraction of the total protein is not MRS visible in vivo.
An estimate of the M0.94 signal assuming methyl groups was assessed using ultra-short TE STEAM data acquired from the mouse and human brain. In the human brain (occipital lobe), the MM concentration contributing to M0.94 peak was estimated to be ~ 11.1 μmol CH3/g wet tissue. The same value was assessed from 4 T (TE = 4 ms, TM = 42 ms) and 7 T (TE = 6 ms, TM = 32 ms) spectra using corrections for T1 and T2 relaxation2. A slightly higher concentration of ~15.7 μmol/g was quantified in the mouse hippocampus at 9.4 T (TE = 2 ms, TM = 20 ms). The unsuppressed water resonance was used as an internal reference, assuming 80% brain water content. Of note, the molar concentration of protons (1H) forming the M0.94 signal is 3 times higher than that of the CH3 entities. Furthermore, if M0.94 arises from an equivalent mix of leucine, isoleucine and valine, which together comprise ~16% of human gray matter total protein mass (~70 μmol/g wet wt)31, then M0.94 would reflect ~8–11% [(11–16 μmol/g methyl groups / 2 methyl groups per μmole amino acid)/70 μmol/g wet wt] of their respective concentrations in the total protein. Another study34 performed in humans at 1.5 T (TE = 20 ms) estimated the M1 area (M0.94) at ~40 mmol/kg proton density, which would be equivalent to ~7 mM for combined amino acids if considering a factor of 3 for proton stoichiometry and a factor of 2 for the two methyls per residue of branched-chain amino acids.
Thus, different estimates of MM proton densities suggest that a large fraction of the total protein is not MRS visible in vivo. Further investigations are needed to address the issue of MRS visibility and the extent to which other cellular compartments (e.g., mitochondria and nucleus) might contribute to the in vivo MM spectrum.
1.3. Recommendations on Nomenclature:
In the present manuscript we provide some recommendations on a unified nomenclature of the different MM components, which would be easily expandable to new peaks, MM signals being uniformly described by their resonant frequency in ppm (e.g. M0.94). More details can be found below in Table 1 together with a brief description of each MM signal component.
Furthermore, a clear distinction should be made between MM and ML signals and an underlying baseline35. The ‘baseline’ consists of smoothly varying components and spurious signals arising through imperfections during data acquisition (For details on ‘baseline’, see35).
2. B0 dependence of MM spectrum
2.1. Changes in MM spectral pattern with B0
The apparent “linewidth” of MM components is dictated by four main factors: T2 relaxation, B0 inhomogeneities (ΔB0), multiplicity of J-coupled signals and the overlap of cytosolic protein signals with slightly different chemical shifts. In general, the spectral linewidth under in vivo conditions is determined by T2 relaxation and by microscopic (ΔB0,micro) and residual macroscopic (ΔB0,macro) inhomogeneities of the B0 (i.e., FWHM ~ 1/(πT2) + (γ/2π).ΔB0,micro + (γ/2π).ΔB0,macro)36,37. In addition, the contribution of J-couplings has to be taken into account for the apparent MM signal linewidths since the multiplicity pattern is not directly observable (FWHM >> J). According to relaxation theory (see Section 2.2), the T2 relaxation of MM has a very mild B0 dependence2. However, the line broadening resulting from microscopic ΔB0 increases linearly with B02,36. Even though the ΔB0,macro component can be substantially minimized by successful B0 shimming38, the ΔB0,micro component cannot be eliminated as it originates from intrinsic tissue heterogeneity on a cellular level. Therefore, the effect of ΔB0,micro line broadening should be identical for metabolites and MM. Since MM peaks contain an overlap of multiple J-coupled resonances from different amino acids, and identical contributing amino acids as part of different proteins experience slightly different chemical shifts, an additional increase in the linewidths of MM peaks is expected compared to metabolites. Indeed T2 relaxation plus ΔB0 component alone cannot account for the observed apparent linewidth of MM peaks25.
When assuming high-quality B0 shimming, the apparent M0.94 signal linewidth can be approximated by a simple equation:
| (Eq.1) |
where the term corresponds to a line broadening per tesla (microscopic heterogeneity and chemical shift differences). The contribution of J-coupling was neglected and not included in this simplified formula. The M0.94 signal linewidths (in Hz) assessed from human and animal experimental MRS data follow a linear relationship with B0 from 1.5 T to 16.4 T (Figure 1A). The linewidth was calculated assuming T2 = 32 ms (Section 2.2) and Δν* = 4.73 Hz/T. The M0.94 signal linewidth in ppm (Figure 1B) is determined primarily by T2 relaxation at low B0, while it reduces rapidly with increasing B0 where it becomes nearly B0-independent and approaches the value 2π Δν*/γ. As J-couplings are independent and T2s of MM are nearly independent of the B0 strength, the multiplet widths (in ppm) decrease with B0, which consequently improves the apparent resolution of MM spectra at high B0. In addition, increased B0 transforms complex higher-order spin systems of strongly coupled resonances into first-order multiplets that also may contribute to improve MM spectral resolution at high B0. Such an effect of B0 on strongly coupled MM multiplets can be observed between 3 T and 4 T MM spectra in the region 1.0 – 1.8 ppm (Figure 1C). Only minor improvements in MM spectral resolution can be expected at 7 T or higher B039. Indeed, MM spectra acquired in rat brain at 9.4 T and 14.1 T are very similar (Figure 1C), while highly similar spectral patterns have been observed for the brains of different species (rat, mouse, cat) at 9.4 T (Figure 1C and Section 6).
Figure 1:
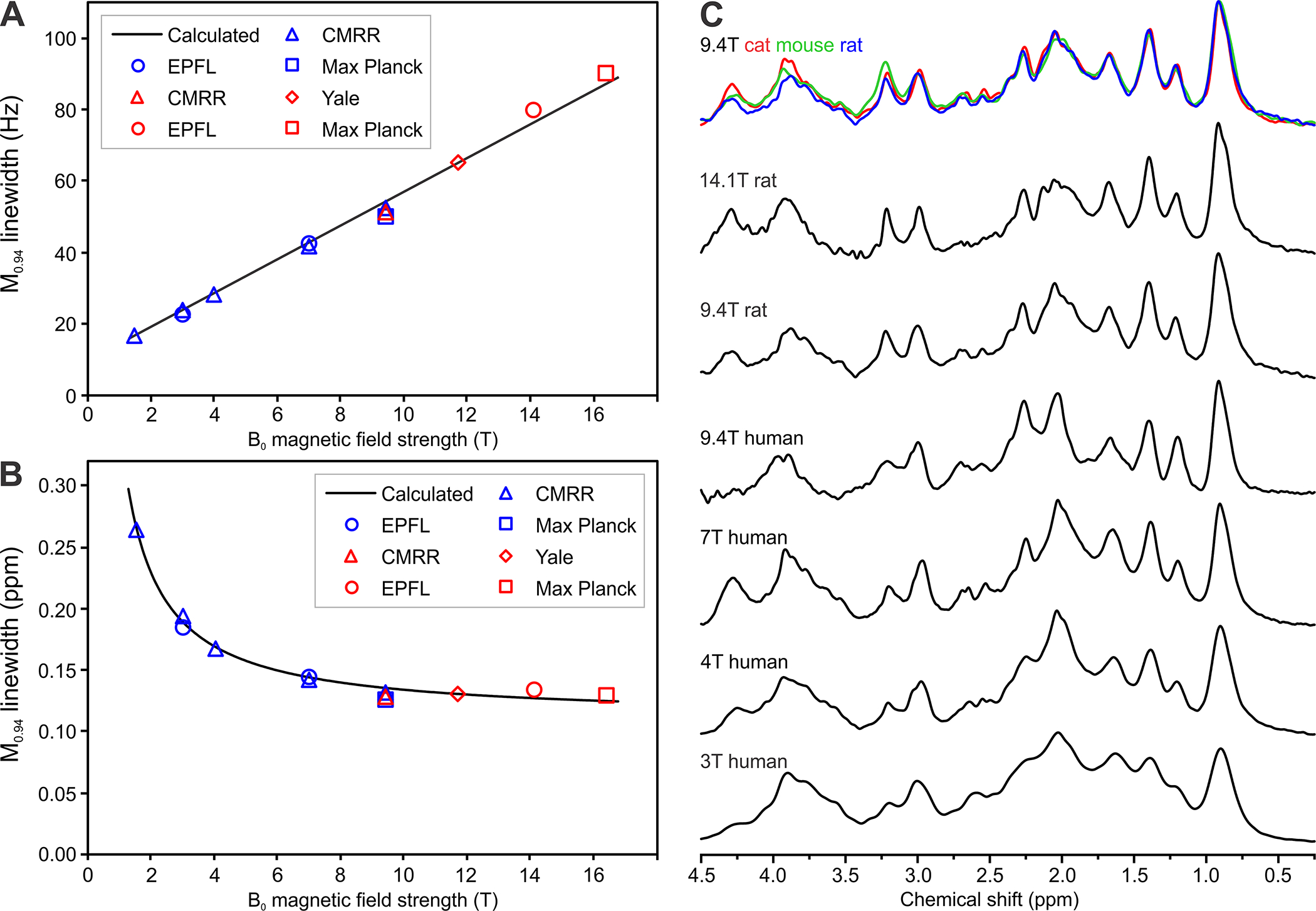
B0 dependence of MM acquired in vivo using 1H MRS.
(A) Dependence of M0.94 signal linewidth on B0 with linewidth expressed in Hz;
(B) Dependence of M0.94 signal linewidth on B0 with linewidth expressed in ppm. Lines calculated for parameters T2 = 32 ms and Δν* = 4.73 Hz/T. Experimental values were assessed using spectra from the CMRR database, spectra provided by co-authors of this paper and spectra from papers27,58,80,81,136. Blue symbols: human MM spectra, red symbols: animal MM spectra.
(C) MM spectra acquired in vivo from the brain of different species at 9.4 T and from human and rat brain at different B0 showing noticeable increased spectroscopic resolution
Spectra are from the following centres: CIBM-EPFL (Centre d’Imagerie Biomedicale, Ecole Polytechnique Federale de Lausanne, Lausanne, Switzerland), CMRR (Center for Magnetic Resonance Research, University of Minnesota, Minneapolis, MN, USA), Max Planck Institute for Biological Cybernetics (Tuebingen, Germany).
Spectra are available online here: https://forum.mrshub.org/t/data-submission-mm-consensus-data-collection/92
2.2. B0 dependence of MM relaxation
MM signals are typically eliminated or isolated from metabolite signals based on differences in T1 and/or T2 relaxation2 (Appendix 1, Tables S2, S3), although differences in molecular diffusion have also been used1,40.
Figure 2 illustrates the B0 dependence of T1 and T2 relaxation for singlet metabolite resonances and MM (Matlab code is provided in Appendix 2). The metabolites include NAA (CH3), tCr (CH3) and tCho (CH3) from a range of publications and B02,29,41,42. Most J-coupled metabolites show shorter T2 than singlets43–47, while T1 for Cr (CH2), glutathione (GSH) and taurine (Tau) (CH2) are noticeably different, falling either below or above this range2,41,48. The MM range includes T1 and T2 values for the M0.94 (M1) to M1.70 (M4) signals2,29 measured in rat brain. Measuring T1 and apparent T2 (J evolution not considered) of MM other than M0.94 to M1.70 is not straightforward due to strong overlap with metabolites, and requires more sophisticated approaches using either double inversion recovery (IR) with optimized combinations of inversion times (TI) and additional elimination of metabolite residuals49,50 or careful elimination of metabolite residuals during post-processing21,51 or combining IR with a diffusion module1,40,52 (Section 3).
Figure 2:
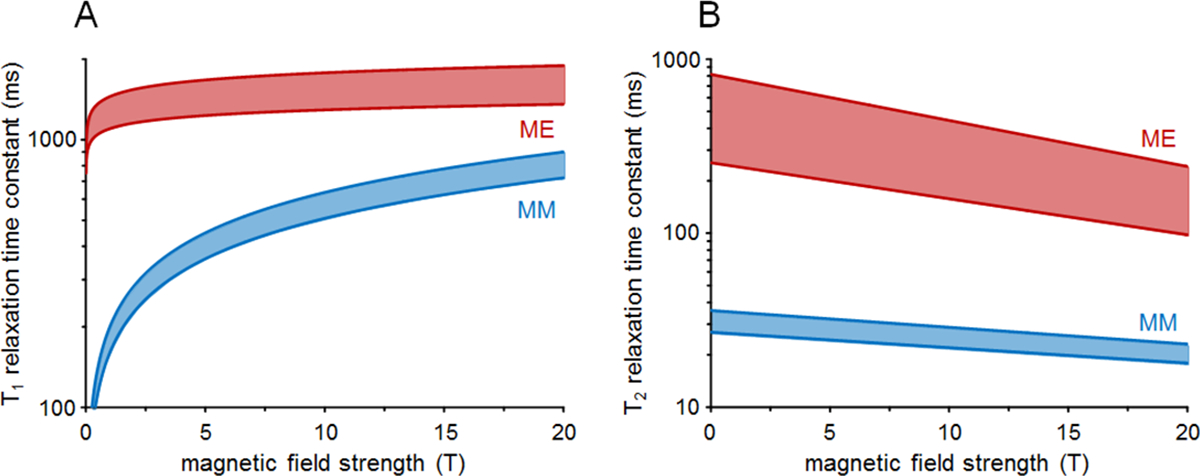
B0 dependence of metabolite (ME) and macromolecule T1 (A) and T2 (B) relaxation. The indicated metabolite ranges include T1 and T2 values for NAA methyl, total creatine methyl and choline methyl signals published in rat2,29,41 and human brain42, whereas the indicated MM ranges include T1 and T2 values for the M0.94 (M1) to M1.70 (M4) signals published in rat brain2,29. Note the logarithmic vertical scale.
Overall, T1 time constants increase and T2 time constants decrease with increasing B0. The slower T1 relaxation of metabolites is in agreement with the Bloembergen-Purcell-Pound (BPP) dipolar relaxation theory53. The T1s of MM increase more strongly with B0, which is also in qualitative agreement with BPP theory for molecules with a longer rotation correlation time2. The apparent T2 time constants of metabolites are shorter than those anticipated by BPP theory. The disagreement can be explained by a loss of phase coherence due to diffusion through microscopic susceptibility gradients42. The T2s of MM has a very mild B0 dependence2. For any value of B0, the T2 of most metabolites is longer than for MM, such that effective suppression of MM can be achieved at longer TEs (see Section 3.2). The main problems of long TE scans are (1) the loss of potentially important MM resonances, (2) the loss of many scalar-coupled metabolite signals; (3) substantial decrease in SNR; and (4) the introduction of T2-weighting, which requires a T2 correction when attempting quantification.
3. Measurement of MM in vivo
MM detection and suppression based on differences in T12,3,8,41 have been reported using single and multiple IR methods (Appendix 1, Table S4). For inversion of magnetization the use of an adiabatic pulse is highly recommended due to broader bandwidth and insensitivity to B1 inhomogeneity.
Figure 3 summarizes the MM signal recovery (A-C) and MM suppression efficiency (D-F) as achieved in metabolite-nulled and MM-nulled MRS. In general, double IR methods (Figures 3B and E) give improved metabolite (Figure 3B) or MM (Figure 3E) suppression over a wider range of T1 times than single IR methods (Figures 3A and D). However, the improved suppression comes at the cost of reduced MM (Figure 3B) or metabolite (Figure 3E) signal recovery and increased T1-weighting. As the difference between MM and metabolite T1 decreases at higher B0, it is harder to suppress one without affecting the recovery of the other. For metabolite-nulled MRS the optimal TIs have only a mild B0 dependence (Figure 3C), whereas for MM-nulled MRS the optimal TIs rapidly increase with B0 (Figure 3F). In all cases, one should be aware of metabolites that are outside the considered T1 range (i.e. tCr methylene2,41, Tau2,41, GSH48) and their residual signals have to be removed by post-processing.
Figure 3:
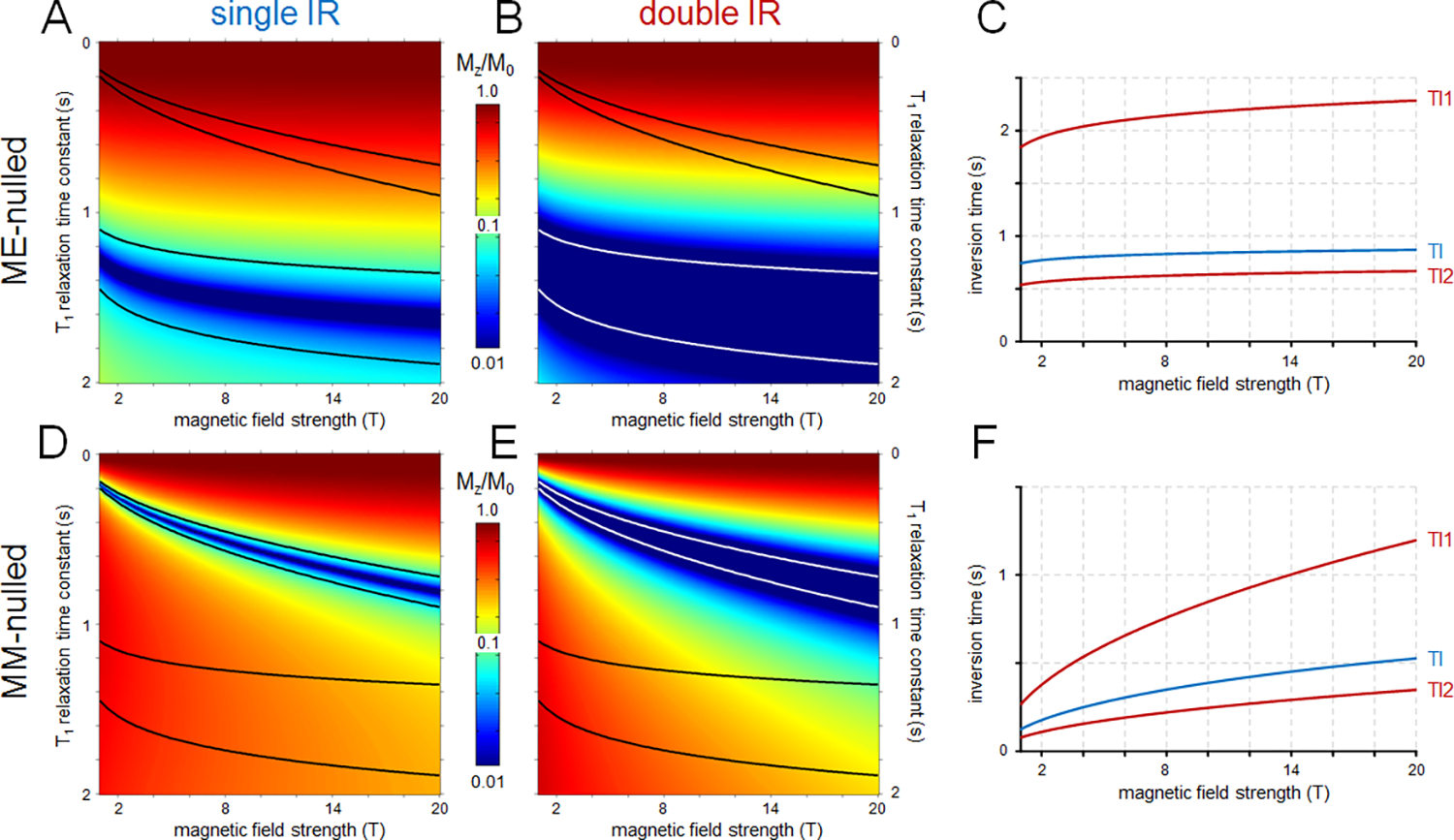
Signal suppression and recovery for (A-C) metabolite-nulled (labelled as ME) and (D-F) MM-nulled MR spectroscopy using (A, D) single inversion recovery (IR, TR=2s) and (B, E) double IR (TR=5s) acquisition strategies as a function of B0 and T1 relaxation time constant published for rat brain. The black and white lines indicate the metabolite and/or MM T1 relaxation ranges. Note the logarithmic vertical scale for all color maps. (C, F) B0 dependence of the optimal inversion recovery times for (C) metabolite-nulled and (F) MM-nulled MRS. The inversion times are optimized to provide the best signal suppression over the T1 ranges indicated in (A, B, D and E). Optimal inversion times for single (TI) and double IR (TI1/TI2) are shown in blue and red, respectively. The Matlab code used to generate these data can be found in Appendix 2.
Diffusion-weighting (DW) combined with IR is another method to measure MM in vivo1,40,52 since MM are expected to have a 10 to 20 times slower diffusion than metabolites40,54. By combining IR with DW (b value of 10 to 11.8 ms/μm2)1,52,55, it was shown in rat brain that a significant attenuation of metabolite residuals can be achieved while the MM signals were almost unaffected1. This eliminates the need for any post-processing1,40,52. However, only few published studies used this method till now, mainly in rodents. The main limitation of this technique is the low SNR (due to the combination of DW at high b-values and IR) which might lead to difficulties in scan-to-scan phasing before averaging. Furthermore, reaching high-enough DW cannot be achieved in some sequences (in particular with short TE spin echo sequences), thus making that approach not a general strategy.
3.1. Removal of residual metabolites
Theoretically, due to faster T1 relaxation of MM compared to metabolites, metabolites are nulled at a specific TI with an almost fully recovered MM. In practice independently of the type of IR method, small residuals of metabolites are still observed in the metabolite-nulled spectrum due to variability in T1 relaxation of metabolites as previously mentioned. These residuals strongly depend on the sequence used and its parameters (TE, TR, TI), on the transmit B1+ inhomogeneity and on B0. Therefore, some studies identified and removed the residuals of the main metabolites such as tCr, NAA, Tau, while others identified additional residuals from tCho, glutamate and glutamine (Glu/Gln) and myo-inositol (Ins) (Appendix 1, Table S4). Residual metabolite signals should be experimentally verified based on: 1) the T1 relaxation times of the metabolites; 2) acquisition of a series of IR spectra using a full range of TI (i.e. 100–1200 ms) where the evolution of the metabolite intensities changes from negative to positive; and 3) acquisition of an IR spectrum with the selected TI but longer TE (around 40 ms) to confirm the residual metabolite signals56–58 (Figures 4A and B). Ideally, MM spectra in vivo should be acquired from reasonably small VOIs using high-quality B0 shimming to optimize the spectral resolution, SNR, water suppression, and minimize baseline distortions or subcutaneous lipid contamination. The contamination of MM spectra by the residual metabolite signals can be efficiently reduced by shortening of TR in the single IR method. The use of a short TR leads to partial saturation of magnetization of metabolites with longer T1, which reduces the sensitivity of metabolite nulling to T1 differences56,58,59. Moreover, TR shortening improves the SNR efficiency for a fixed measuring time.
Figure 4:
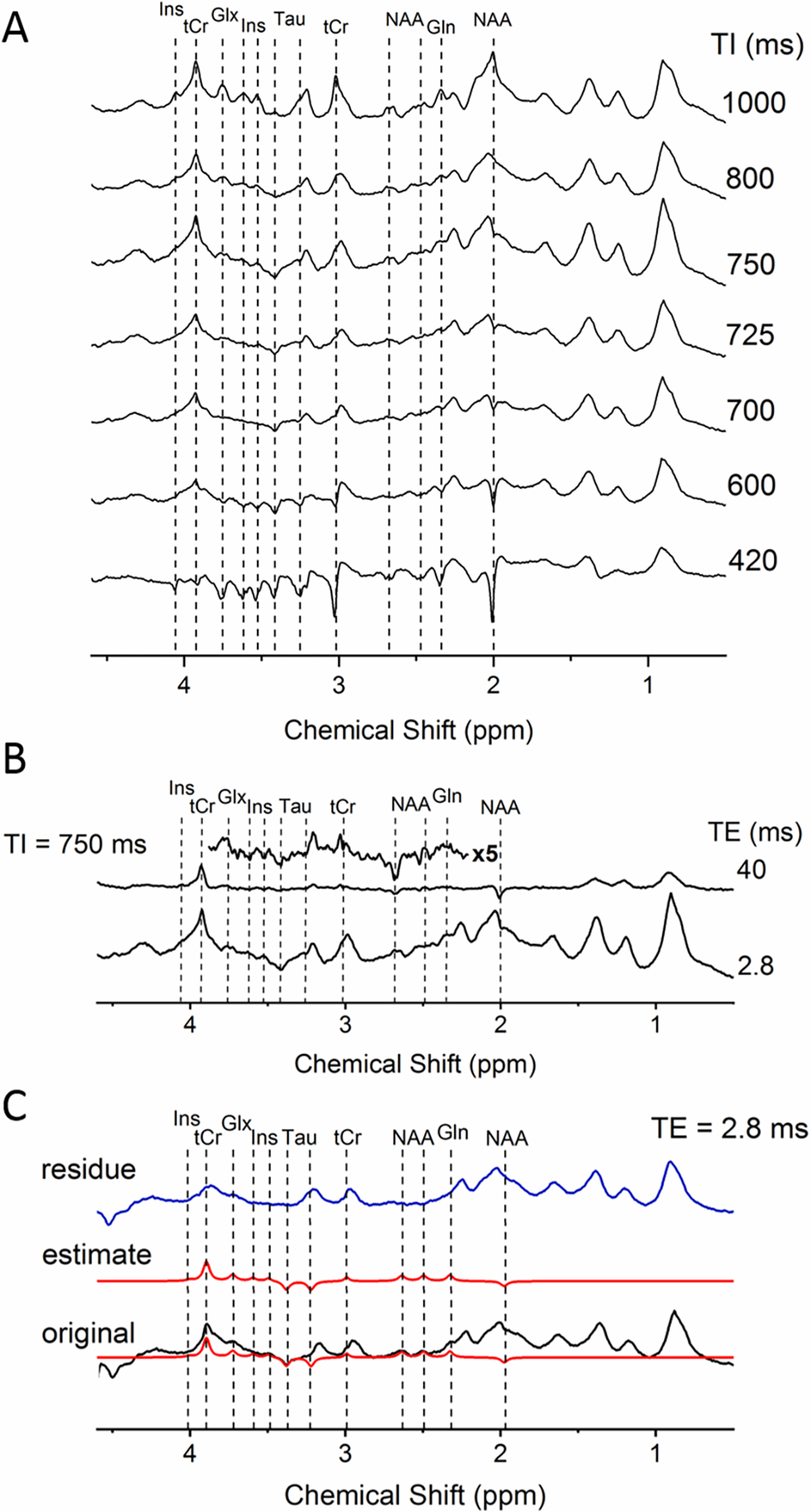
A) A series of IR spectra from rat brain in vivo with TI ranging from 420 to 1000 ms revealing the evolution of metabolite intensities as a function of TI (all the spectra were acquired with TE/TR=2.8/2500 ms at 9.4 T using the SPECIAL sequence in a voxel of 3×3×3mm3 centered in the hippocampus); B) Spectra acquired with a selected TI (750 ms) and TE of 2.8 ms (taken from A) as well as with TE of 40 ms (5x magnified, TE=40 ms spectrum from 2.2–3.8 ppm is shown on the top); C) Original spectra acquired at TI of 750 ms and TE of 2.8 ms (shown in black), estimated fits of the residual metabolites using AMARES (shown in red), and the residue obtained after subtraction of the estimated metabolite signals from the original spectrum (shown in blue). All spectra were acquired in vivo in the rat brain at 9.4 T.
Different approaches/algorithms can be used to eliminate the contribution of metabolite residuals in post-processing. For example, HLSVD (Hankel-Lanczos singular value decomposition) was one of the first algorithms used, but cannot consider the known prior knowledge on the residual metabolites. More recently AMARES 60 (Advanced Method for Accurate, Robust, and Efficient Spectral fitting) was used with constraints on the peak frequency, phase, linewidth, and amplitude to fit the residual metabolites more robustly21 (e.g. in AMARES fitting model prior knowledge is provided only for the residual metabolite peaks to be removed) (Figure 4C). This set of prior knowledge needs to be built by the user. In Figure 4B a spectrum acquired with the TI=750ms was chosen as the one with the least metabolite residuals at 9.4T in the rat brain after acquiring a series of IR spectra (Figure 4A). For the identification of metabolite residuals, in addition to the series of IR spectra where the evolution of the metabolite intensities is changing from negative to positive (Figure 4A, dotted lines), an IR spectrum with TE=40ms was also acquired (TI=750ms, TR=2500 ms). In order to build the set of prior knowledge, special care has to be taken to analyze the behavior of each peak individually at a given TI and TE (the multiplicity of the peak, phase, estimated amplitude based on previously reported relaxation times and linewidth44,61). The following steps and iterations can be performed for fixing the prior knowledge: 1) a flexible prior knowledge and manual inspection to avoid overfitting is used first to remove individually every metabolite residual from the MM spectrum; 2) the obtained results are then used in a second step to construct rigorous prior knowledge of all the metabolite residual peaks combined (still leaving some freedom for the peaks to adjust to different spectra); 3) after removing the peaks the remaining MM can also be fitted to make sure that the final residual is free of any artifacts indicating over- or underestimation of metabolite residuals; 4) if step 3 is validated then the residue from step 2 (the MM spectrum free of residual metabolites, Figure 4C) is saved separately and included in the metabolite basis set. This process requires multiple iterations, but once an adequate set of prior knowledge is built it can be efficiently reused and applied to different spectra with minor adaptations. By using a rigorous prior knowledge and by fixing the phase of each peak, AMARES fits peaks on a non-zero baseline especially when fitting several peaks at the same time. An alternative is to define the MM spectrum by simultaneously fitting a series of IR time spectra where residual metabolite signals are accounted for automatically62,63 or by using a residual metabolite basis set, however very few studies were published to date using these approaches. As such, AMARES or similar/alternative approaches appear to be favorable for the post-processing of MM components21,51,62,63.
3.2. TE dependence of MM
The MM pattern and MM contribution to the overall spectrum depend on the TE and the sequence used (Figure 5). At short TE, MM signals contribute significantly throughout the whole ppm range. At longer TE, the MM contribution relative to metabolites decreases due to shorter T2s. For most non-editing sequences, TE ≥ 150 ms at 3 T and 4 T45,46 and TE ≥ 100 ms at 7 T47 in human brain and 9.4 T in the rat brain are generally sufficient to permit neglecting the MM contribution during quantification (Figure 5). Therefore, the assumptions that MM contribution at TEs around 40–80 ms is negligible, justifying the non-inclusion of MM in the basis set during quantification might not be correct for high SNR spectra. Systematic studies on MM contributions at intermediate and long TE in humans and animals are missing. Because the MM spectral pattern changes with TE due to the J-couplings between different MM resonances4,45 (Section 1), MM spectra should be acquired for each specific TE and sequence.
Figure 5:
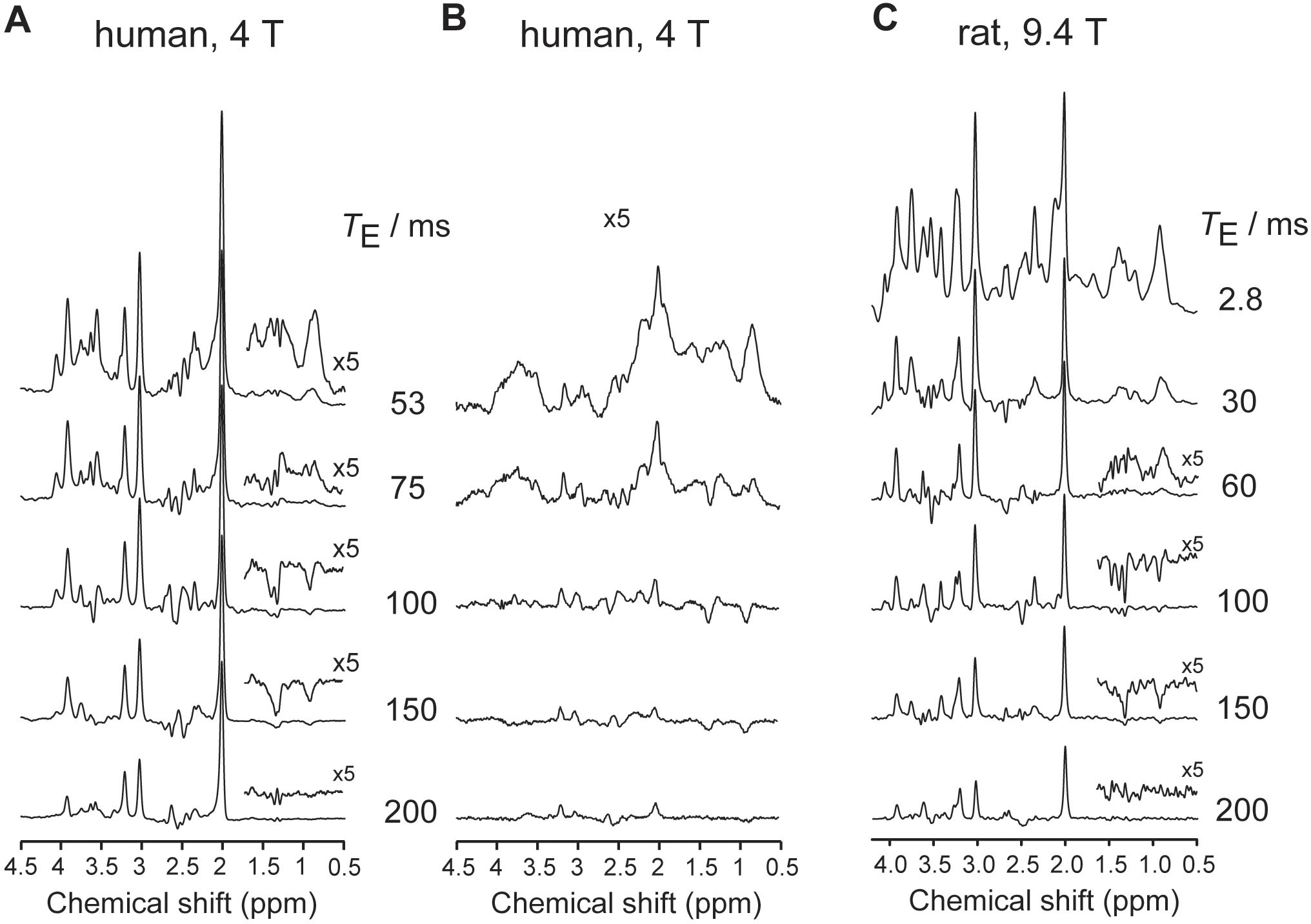
TE dependence of MM. Spectra measured in the human brain in vivo at 4 T at different TEs using (A) LASER sequence and (B) inversion-recovery LASER sequence (occipital lobe, volume-of-interest = 27 mL, TR = 2 s, TI = 0.67 s, 64 averages per TE). Adapted from45 with permission. (C) Spectra measured in the rat brain in vivo at 9.4 T at different TEs using SPECIAL sequence (hippocampus+cortex, volume-of-interest = 27 μL, TR = 4 s, 240 averages per TE).
4. Mathematical modeling of MM for metabolite and MM quantification
4.1. Quantification/parameterization of MM
Quantitative comparison of MM content or MM composition between cohorts of subjects or different brain locations is facilitated by modeling the experimental MM in terms of interpretable MR signals. Signal integration of the raw measured MM spectrum or after post-processing64 is also a possibility, though is less flexible and accurate. Thus, most often the MM spectrum is parameterized into a number of Lorentzian, Gaussian or Voigt lines representing easily interpretable MR signal entities. However, parameterization of the MM into regular MR signal components is non-trivial since the number and nature of contributing chemical entities is a priori unknown65. The best chemical information for parameterization still dates to the pioneering work of Behar et al.4 where ex-vivo NMR showed signals at 14 frequencies, with the seven main peak groups labeled as M1 (M0.94) through M7 (M3.00) (see above). However, this original signal model was often not used directly for parameterization, mostly because the appearance of the MM is B0-dependent, not all peaks are easy to identify and the nomenclature was arbitrary. Most researchers have thus devised heuristic models based on the visual appearance of their own MM spectra or closely matching previous data. Though, very often the original labeling of peak groups as M1 (M0.94) through (M7) (M3.00) and later up to M10 (M4.20) has been maintained in many reports. The actual models used between 4 and 32 Gaussian or Lorentzian lines28,29,39,51,66–72(Appendix 1, Table S4). It is likely that future improvements in SNR and spectral resolution will warrant more complex mathematical models to model the MM profile accurately (Table 1).
Alternative parameterizations without predefined choice of a number of interpretable component peaks have been suggested as well. To that end, MM spectra have been either described point-by-point in the frequency domain (from saturation recovery data)34 or as a sum of overlapping densely and equally spaced Voigt lines29,62,63 which can be grouped into interpretable features with common characteristics in hindsight. Both approaches have the advantage to be model-free, but are a mathematical representation rather than a physical or physiological model. Thus they are well suited to represent single MM spectra or a set of interrelated MM spectra recorded at specific acquisition parameters, but do not yield models that are generalizable. None of the above approaches can fully represent J-coupling modulations with TE in case of editing experiments or 2D J-resolved spectra.
As previously mentioned the MM pattern is also influenced by the sequence used and its parameters (i.e TE, TR). Hence MM have also been parameterized in terms of relaxation times. An effective T2 (T2eff) that includes both relaxation as well as J-evolution effects has mostly been determined from metabolite-nulled scans with different TEs (Appendix 1, Table S3), and T1 has been derived from scans with multiple inversion or saturation recovery periods (Appendix 1, Table S2). One approach tried to include the entire set of TE and IR series into one spectral fit model to simultaneously quantify metabolites and macromolecules62,63,73. Another recent approach used measured T1 and T2 times of all MM resonances to derive a MM model that can be adapted to any sequence and scan parameters from experimentally acquired MM spectra obtained by one specific sequence74. Both approaches are still under development.
4.2. Consideration of MM signal during quantification of metabolites
For a reliable quantification of metabolites from brain 1H MR spectra containing MM contributions, the MM spectrum has to be subtracted before spectral fitting75,76 or the MM spectrum or its components have to be included in the basis set used for linear combination model fitting 51,57,67,71,77–81. The second option is more common.
A widely used approach in estimating MM models is to suppress the metabolite signals using an IR sequence to determine a single spectrum - containing only MM signals at fixed relative amplitudes (Section 3). This MM model spectrum is subsequently included in the basis set used by the fitting algorithm, incorporating prior knowledge of the MM signals and therefore improving fitting stability. Whilst the IR sequence reduces the SNR, the reduced overlap between metabolites and MM signals improves model accuracy over the use of a purely mathematically estimated MM spectrum57,71,78,80–82. To create a single MM basis spectrum that includes all MM resonances, it may be sufficient to average the metabolite-nulled MM spectra acquired in vivo from several healthy subjects assuming the residual metabolite signals have been removed. A further approach involves averaging the parameterized MM signals after fitting the MM spectrum acquired in vivo (see below).
Alternatively, a parameterization into independent MM signals provides a higher level of analysis flexibility and yields noise-free MM models when compared to the direct use of experimentally acquired MM data in the fitting process. To that end, the experimental MM spectra can be modeled as either a sum of splines62,83 or a combination of broad symmetric resonances with Lorentzians, Gaussian or Voigt lines each with characteristic frequency and lineshape parameters51,67,71,77,80. Certain groups of subjects can be assumed to have identical MM profiles and therefore each individually modelled MM signal can then be combined at fixed proportions, which reduces the degrees of freedom in the fit and makes it more robust. Conversely, it may be known that a specific MM moiety is a disease biomarker. Then, a MM fitting model with the freedom to quantify this specific signal separately is useful. Alternatively, individually modelled MM signals can be included in the metabolite basis set and quantified together with metabolites67,77. These components can also be combined into one or more signals and used with metabolite spectra for analysis29,51,84. However, the increased number of fitted parameters without constraints may lead to overfitting51. In such a case, the fitted amplitudes of MM signals may lose their biochemical meaning. Additional studies are required to evaluate the best prior knowledge or soft constraints to be used in the fitting process to avoid over-parameterization.
An alternative approach for estimating the MM signals exploits the short T2* relaxation of MM from the first time domain points of the MRS signal85,86. The Subtract-QUEST86 algorithm was used to compare this approach versus experimentally obtained spectra of MM in the quantification of in vivo 1H MRS data82,87. Significant differences in the calculated concentrations were obtained when using the short T2* relaxation estimation of the MM56,82,87.
In conclusion, accurate quantification of metabolites in short and intermediate TE 1H MR spectra require an equally accurate assessment of MM as demonstrated over a range of field strengths56,57,71,78–82,87. While MM measured at a low B0 of 1.5–3 T appear smoother8,81, the complexity of MM spectra increases at higher fields29,56–58,78,80,87,88 requiring additional scrutiny in their modeling. Differences in metabolite concentrations can be seen when comparing the mathematically generated MM models using different algorithms71,81,82 with the MM measured in vivo. The smooth approximation of spline or another type of mathematical fitting for MM does not completely reproduce the in vivo spectral pattern at higher B056,58,78,80,88. Therefore, experimentally measured MM are recommended for all B0.
A detailed description of different MRS quantification algorithms to handle MM quantification is provided in Appendix 3 and in a recent book chapter83.
5. MM-coediting in spectral editing (GABA+)
MRS of GABA in vivo, the major inhibitory neurotransmitter, is technically challenging because of the presence of overlapping resonances from metabolites, such as creatine, glutamate/glutamine, GSH, homocarnosine, and NAA. For this reason, spectral editing is employed for GABA detection89 including J-difference editing90,91 and doubly selective multiple quantum filtering method89,92. These methods exploit the J-coupling of GABA β and γ methylene protons resonating at 1.9 and 3.0 ppm. However, because of finite bandwidth of the frequency-selective editing pulse set at 1.9 ppm, the MM resonance at 1.7 ppm, which is coupled to the MM resonance at 3.0 ppm, is also partially inverted3,4,8 (Figure 6), contributing to the net signal measured in the difference spectrum at 3.0 ppm. GABA+MM (or GABA+) values are widely reported in the literature93–95. In doubly selective, multiple quantum filtering methods, MM signals at 3.0 ppm are also partially co-edited by the double-band selective pulse, but the MM signal contribution is smaller than for MEGA-PRESS because of the increased frequency selectivity of the double-banded selective pulse for refocusing both coupled partners92,96.
Figure 6.
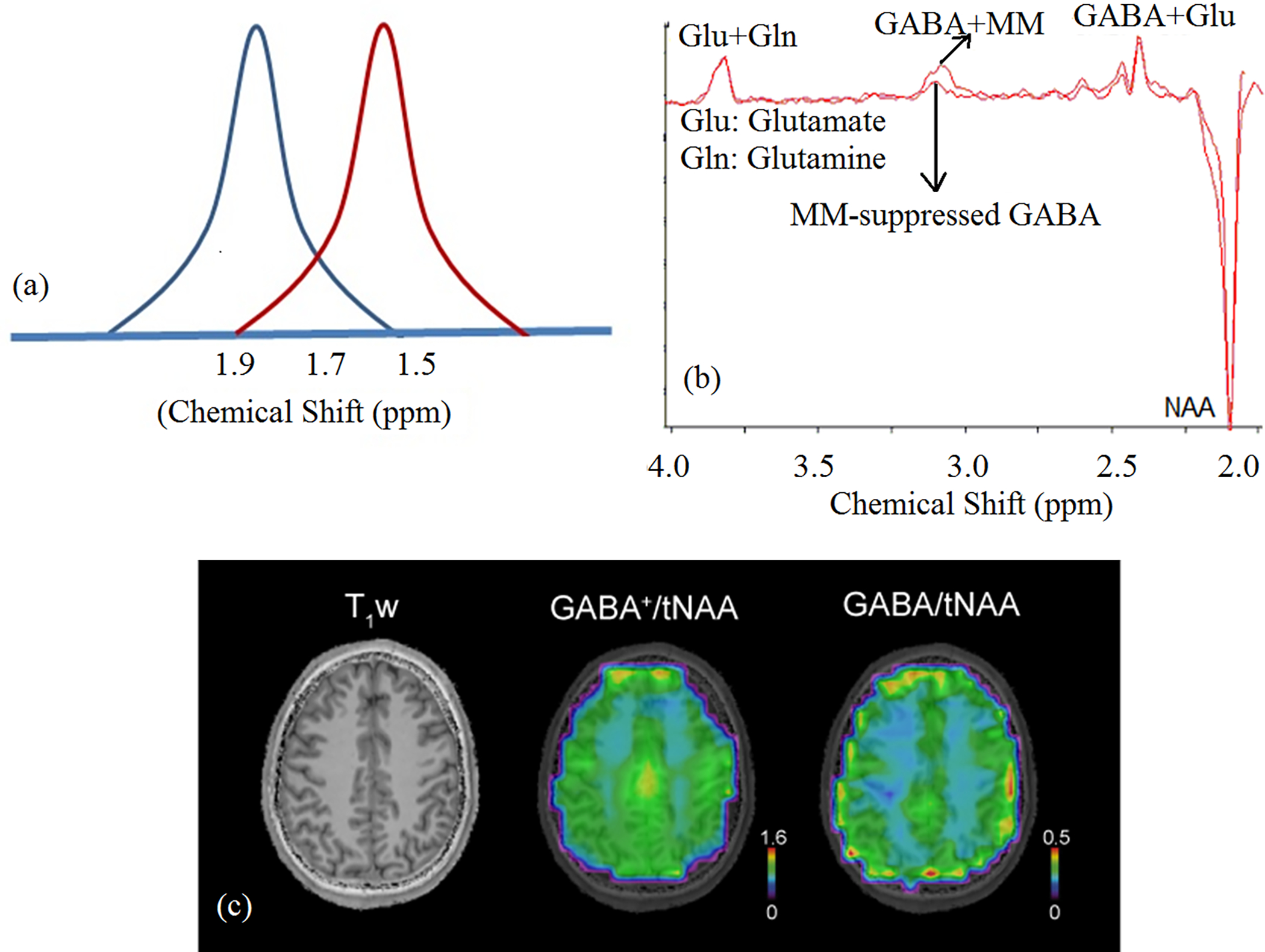
A) Schematic representation of MM coediting. Gaussian pulse (blue) set at 1.9 ppm partially excites 1.7 ppm MM resonance to result in MM coediting. In symmetric pulsing, the ON and OFF resonance pulses are set at 1.9 (blue) and 1.5 ppm (red) respectively, resulting in MM-suppressed GABA signal104. B) Single-subject MM-coedited GABA (GABA+MM) and MM-suppressed GABA spectra using symmetric pulsing with MEGA-LASER sequence at 7 T. (Adapted from reference137 with permission); C) T1-weighted MRI, metabolic maps of GABA+/tNAA (i.e. GABA+MM3.00) and GABA/tNAA (7 T, nominal voxel volume ~1.4 ml, GABA measured using IR MM-nulling100). The GM/WM contrast increased 2.15-fold in GABA/NAA compared to GABA+/NAA, as also shown in a previous study using MQ GABA editing138. This may be attributed to a reduced dilution effect of MM contribution that has less contrast between GM and WM than GABA and to an elevated abundance of the underlying MM component (M3.00) in WM, but further investigation is needed. (Note: tNAA measured with EDIT-OFF IR-ON was used for normalization in both cases). MM-suppressed MEGA-edited GABA measurement139 has shown similar GM/WM difference of GABA as MQ GABA editing138.
The measurement of GABA+ may be acceptable under certain conditions (e.g., healthy controls or no MM changes expected), but when studying the change of GABA levels in certain disease conditions and different age groups, it is essential to account for the MM contributions97. Quantification and comparison of GABA levels among different groups can be affected by possible differences in MM contamination. For example, studies conducted at 1.5 T, 4.1 T and 7 T27,34,68,98,99 reported significantly higher MM levels in GM than in WM, whereas no difference was observed in another study conducted at 3 T and 7 T28. Another study involving GABA editing 7 T100 found M3.00 to be higher in WM than in GM. Any dependence of MM level on tissue composition will lead to differences in the calculated GABA level. Thus, variations in voxel positioning and inter-subject variability in GM/WM content will lead to increased variance in the measured GABA. In addition, because the ~ 0.93 and 1.35 ppm co-edited MM resonances are not related to the 3.0–1.7 ppm coupling8,91, it is not possible at present to calculate the M3.00 contribution to the edited GABA+ signal in vivo by reference to other (non-overlapping) MM resonances.
The larger the bandwidth of the frequency-selective editing pulse at 1.9 ppm the higher will be the excitation of MM1.70 and its coupled component, M3.00. However, for fixed bandwidth of the inversion pulse the editing selectivity (of GABA over MM3.00) improves with increasing field strength. Conversely, a higher bandwidth will reduce the susceptibility to misadjustment of the editing pulse frequency caused by motion and field drift101, which alters MM coediting91. The relative contributions of GABA and MM to the edited GABA+ resonance also depend on the timing pattern of the editing pulse102.
Improving selectivity of the editing pulse by performing single quantum editing with a numerically optimized pulse has been used to reduce MM coediting103. Additional strategies could be employed using multiple quantum filtering to reduce MM coediting by adjusting the frequency separation between the two frequency selection bands and by repeating the double-band selective pulse92,96. MM contamination in MEGA-PRESS can be minimized by (i) performing an additional metabolite nulling scan91 using IR; (ii) applying frequency-selective pulses symmetrically with respect to the 1.7 ppm MM resonance (i.e., at 1.9 and 1.5 ppm, respectively, Figure 6)104, or by using a longer TE than the proposed 68 ms (1/2J)105.
When MM co-editing cannot be avoided, the variance of possible MM co-editing should be mitigated by standardization of editing pulses102 and real-time updating of the editing pulse frequency106,107, or avoiding measurements after high gradient duty cycle sequences including functional and diffusion MRI with echo-planar sequences. A recent multi-site study has shown excellent stability and reproducibility of GABA+ measurements compared to MM-suppressed GABA measurements mostly due to site-to-site misadjustments in editing frequency108. However, every effort should be made to acquire MM-suppressed GABA for a more unambiguous quantification.
6. Brain regional dependence of MM
Due to the spectroscopic overlap and low SNR of MM in vivo, a precise characterization of their regional differences is yet to be established. Single voxel spectroscopy (SVS) has previously shown spatial/tissue differences in MM27,34,109. Recent magnetic resonance spectroscopic imaging (MRSI) studies51,68,110 have provided an improved spatial coverage and indicate that these differences may be larger than expected51,68. Thus, a careful characterization may be important for MRS quantification of metabolites in both healthy and diseased brain21,67,109,111–115.
In MRSI, spatial mapping of MM using IR-based metabolite nulling is complicated by low SNR and spatial B1-inhomogeneities. Free induction decay (FID)-MRSI is therefore particularly suited for mapping MM due to the complete absence of any TE (i.e., no J-coupling and T2-related signal loss) resulting in the best possible sensitivity for these ultra-short T2 MM components2,8. Direct spectral fitting of individual MM as part of the basis set rather than analysis of the metabolite nulled spectrum was therefore proposed51. This requires a strict control of the fitting parameters. In particular, when the different MM components are mapped individually, one must be extremely careful about over-parameterization51,66,67. Another post-processing approach of extracting the MM contribution from FID-MRSI was proposed by Lam et al.110.
Mapping of individual MM by using them as part of the basis set has provided insights into the spectroscopic differences between GM and WM51. The MM components in the ranges from 0.5–2.3 ppm and 3.6–4.0 ppm, as measured by FID-MRSI at 7 T, tend to be higher in GM compared to WM (the MM/NAA ratios were 15% to 40% higher in GM than in WM)51, which is in agreement with the results of previous SVS studies27,34. In contrast, the MM peaks at 3.0 and 3.2 ppm do not follow this trend, being higher in WM than in GM34,51 (Figure 7). The observation that MM contribution in this frequency range in WM is higher than in GM was recently replicated using MEGA-edited MRSI at 3 T and 7 T100,116,117 and was also consistent with results from other “MM-sensitive” techniques (e.g., T1rho)118. This has important implications for undesired co-edited components of MM in spectral editing at ~3 ppm (e.g., GABA+MM).
Figure 7:
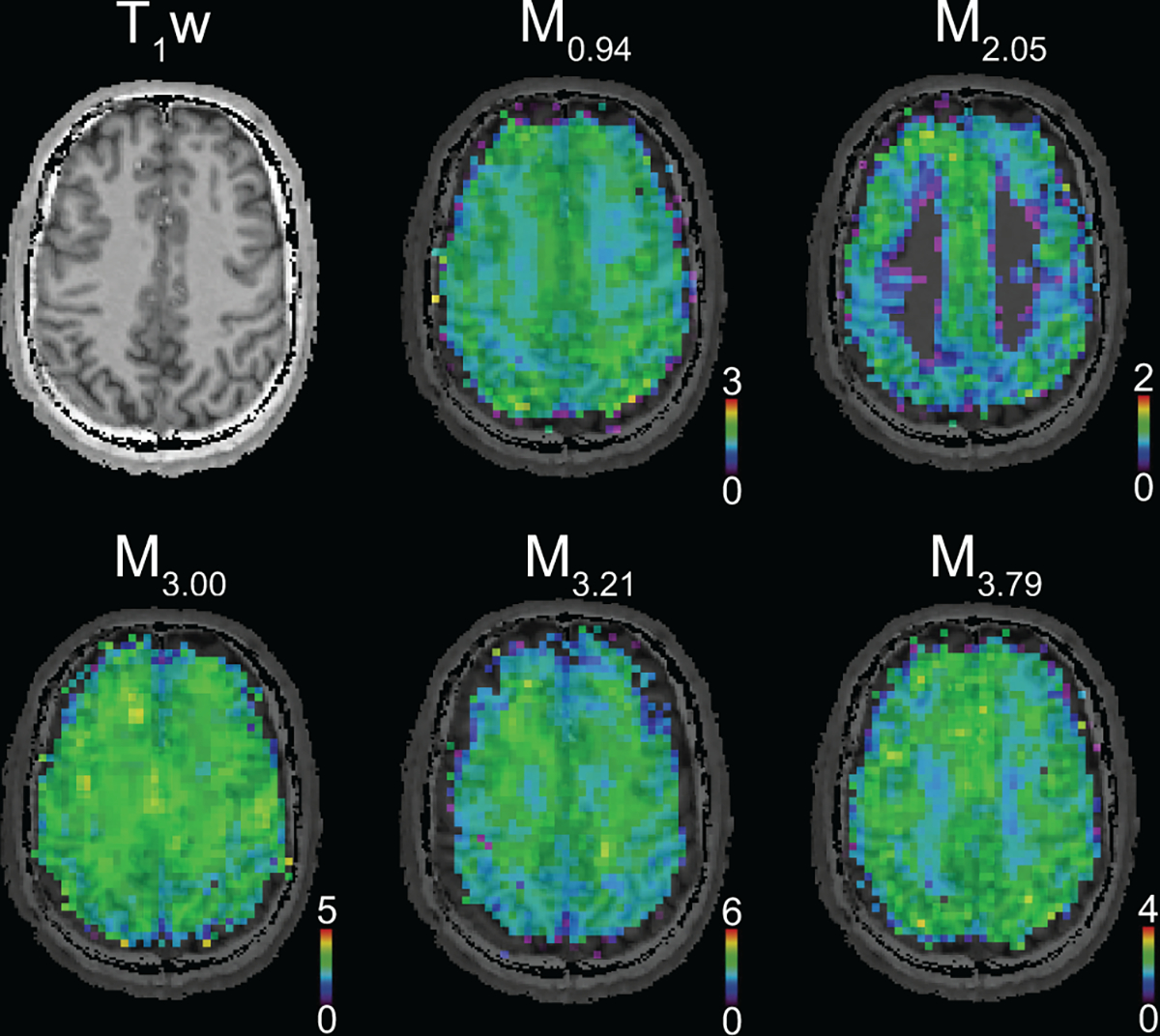
A) T1-weighted MRI and metabolic maps of MM components obtained from a healthy human brain using simultaneous quantification of metabolites and MM from FID-MRSI data. (acquired at 7 T, nominal voxel volume ~0.32 ml51). MM components show regional differences in healthy brain and their signal intensities are typically higher in GM than in WM.
The regional differences may be linked to amino acids inside cytosolic proteins3–8 that contribute to the MM signals (Appendix 1, Table S1). Some of the regional and tissue differences that are revealed by MM mapping could be explained by different T1 relaxation of individual MM resonances2,49,119. Overall, the reliability of mapping MM components varies significantly between different spectral ranges:
0.94 ppm – the M0.94 peak is large and does not overlap with metabolites, making the mapping of this component straightforward. If lipids are present in the spectrum due to disease or outer-volume contamination, the M0.94 peak may be affected.
1.2 – 1.7 ppm (M1.22-M1.70) – Lipids present in the spectrum due to disease or outer-volume contamination may impede reliable mapping despite a lack of overlap with abundant metabolites.
2 – 4 ppm (M2.05-M3.97) – these MM overlap strongly with metabolites. Hence, correlations with metabolite concentrations should be checked carefully.
Although preclinical studies become attractive due to a large number of disease models, little attention has been given to preclinical MM. In animal models, the typical assumption that no substantial differences exist between regions and species has been evaluated in rodents in hippocampus, cortex and striatum21,120. This is mainly due to the fact that rodent brains contain mostly GM and only minor variability of MM in rats and mice has been observed21,120. No significant differences in metabolite concentrations were found when different MM spectra were used for the metabolite quantification of a given brain region21,120. However, care has to be taken when removing residual metabolites, since this procedure can lead to a slight variability in the shape of the MM120.
7. Age dependence of MM
In the rodent brain, the MM content has been shown to increase during postnatal development with no changes to the macromolecular pattern121. The MM content was quantified from spectra in three brain regions, cortex, striatum, and hippocampus, using LCModel and corrected for age-dependent changes in brain water content. At the time of writing, there are no published reports on the MM content in aging rodent brain.
In human brain, the MM content and pattern have been shown to differ with age34,64. Higher MM content was observed in middle-aged (25 – 55 years) compared to young (< 25 years) subjects in centrum semiovale34 and in older (67 – 88 years) compared to young (19–31 years) adults in the occipital and posterior cingulate cortex64 (Figure 8). The greatest MM pattern differences associated with age occurred around 1.7 ppm. In the occipital cortex and the posterior cingulate cortex, the largest differences in intensities were observed for the MM resonances around 1.7 ppm and 2.0 ppm64. These age-associated differences in MM pattern and content could not be explained by differences in the tissue content (lower gray matter content in older adults). The differences in MM pattern require use of age-specific MM spectra for quantification. However, because the patterns were the same in two brain regions, there is no indication that region-specific MM spectra are needed when studying these brain regions.
Figure 8:
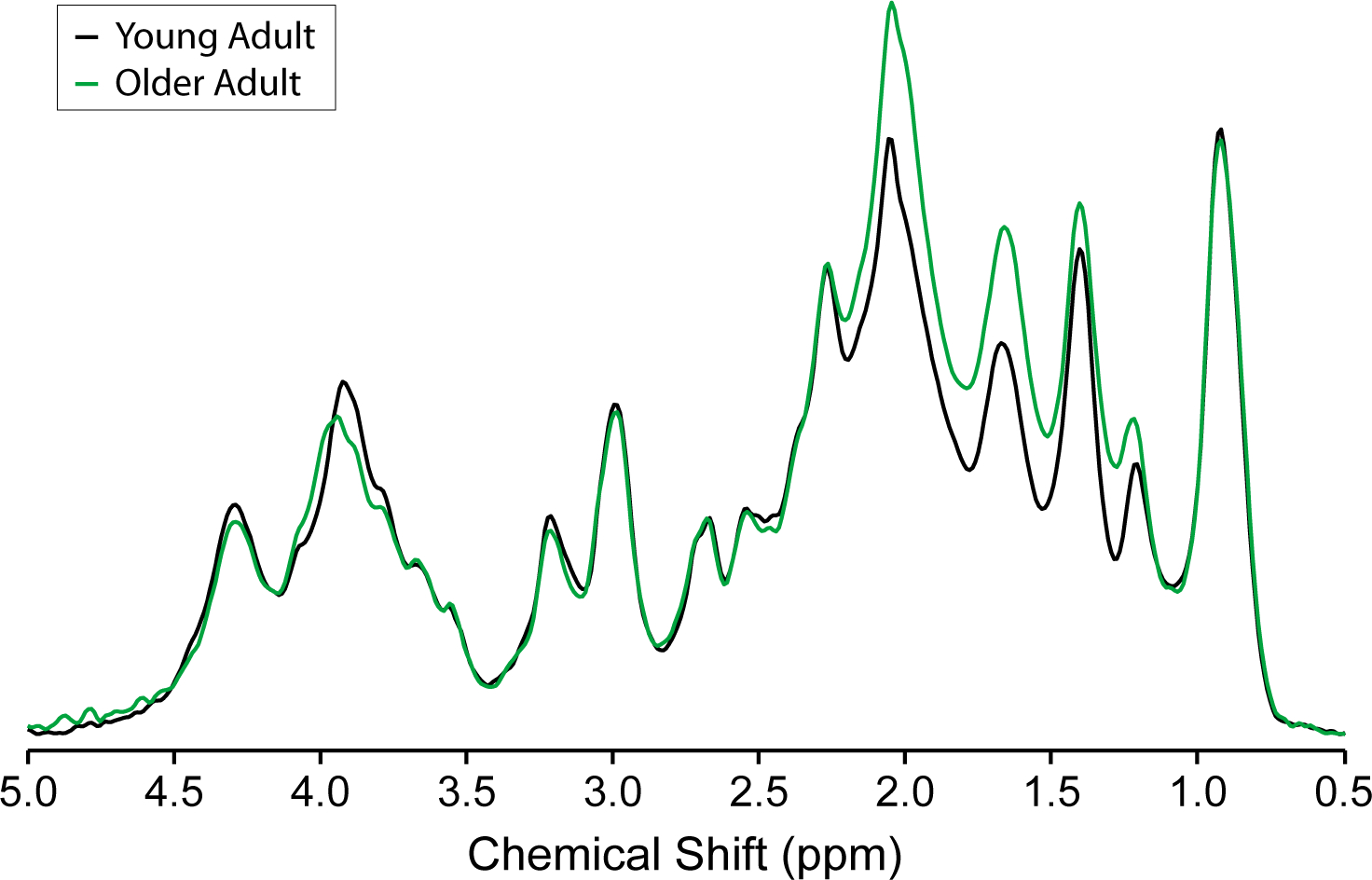
Age-associated MM differences. Average metabolite-nulled macromolecular spectra measured from four young adults (26 ± 4 years) and three older adults (73 ± 3 years) normalized to water reference and taking into consideration GM, WM and cerebrospinal fluid content, as well as T2 of water in different compartments. Clear age-associated differences in MM pattern are apparent, as the spectra overlap completely at 0.9 ppm, but diverge at several other chemical shifts. A content difference is also apparent, as the spectra are normalized, and the MM spectrum for young adults lies below the MM spectrum for older adults to a greater extent in the 1–2.3 ppm range than it lies above the MM spectrum for older adults in the 3–4.5 ppm range. Adapted from reference64 with permission. 7 T, single inversion recovery technique combined with STEAM, TR = 2 s, TE = 8 ms, TM = 32 ms, TI = 0.68 s, 8-mL volume of interest in the occipital cortex, 1664 averages for the young adults, 960 averages for the older adults.
8. Disease dependence of MM and ML
The clinical literature is extremely limited on the application of MM contributions using MRS with most reports on the MM of healthy people62,79,81. Even though in preclinical studies there are pulse sequences available for MM measurements in vivo, to date, few studies on MM in disease have been reported.
Research publications reporting on the measurement of MM in human brain pathologies have included brain tumors, multiple sclerosis (MS), and stroke. With onset of disease, signals from ML appear that overlap with peaks of MM, thus mainly changes in ML+MM were reported and more precisely in the region 0.9 to 1.9 ppm due to the easier accessibility. These ML consist mainly of neutral triglyceride and cholesterol esters in the form of cytoplasmic lipid droplets, rather than membrane lipids, although mobile components of membrane phospholipid (choline methyl, 3.2 ppm) might contribute. ML are not subcutaneous lipid signals arising from imperfect localization (considered as artifacts in MR spectra), thus these terms should not be used interchangeably. Proton signals of mobile lipids have been assigned to methyl –CH3 (0.9 ppm) and methylene –(CH2)n– (1.3 ppm), allylic (2.05 ppm), α-acyl (2.3 ppm), bis-allylic (2.8 ppm) methylene and vinylic (5.4 ppm) methine, which overlap the MM signals122. Mobile lipids can be distinguished qualitatively from mobile proteins/peptides by the higher proton density of methylene (1.3 ppm) over terminal methyl (0.9 ppm) groups, expressed as the CH2/CH3 ratio. The relative intensities of the 1.3 ppm and 0.9 ppm lipid peaks may reflect differences in chain lengths, correlation times (and visibility) of the methylene protons along the chain length, as well as presence of cholesterol esters representing different classes of lipid or proteolipid involved.
There are only a few reports of MM+ML in 1H MRS of MS. One study of acute MS reported changes in ML+MM at 1.3 and 0.9 ppm123. In another study114, acute and chronic MS lesions were compared with healthy brain tissue, finding an increase in MM+ML in the acute (but not chronic) lesions at 0.9 and 1.3 ppm and no changes at 2.1 and 3.0 ppm. The changes seen in the acute MS lesions were suggested to arise from both ML and MM, the latter comprising proteolipid protein from myelin fragments.
There is substantial literature reporting the potential use of 1H MRS to classify brain tumors. In low grade glioma, ML is low, whereas in high grade gliomas ML can dominate the spectrum, depending on the level of necrosis. Different types of brain tumors have been discriminated based on the spectrum profile, including MM+ML, and classifiers built for computer aided diagnostics124,125 allowing the prediction of the tumor type and grade. In a study on the relationship between distance to the malignant glioma core and spectral pattern, ML+MM were an important factor for demarcation of the solid brain tumor126. Short and long TE spectroscopic patterns of normal appearing white matter, meningioma, metastases, low grade astrocytoma, anaplastic astrocytoma, and glioblastoma were (visually) compared127, while the complete MM+ML signal of different types of human brain tumors has also been investigated67. Moreover, the apparent T2 of the ML component at 1.3 ppm was shown to be different between glioblastoma and brain metastases in patients128. A preclinical study performed in a mouse glioblastoma model showed MM+ML and MM changes21, being partially consistent with in vivo and ex vivo HRMAS results from humans129. Besides a large variation of MM+ML at 1.3 ppm in the tumor region induced by glioma-initiating cells, changes also appeared at 2.8 ppm (similar chemical shift to polyunsaturated fatty acids) and 3.6–3.7 ppm21. The origin of these signals remains to be characterized more thoroughly.
In stroke, only a few studies have been performed, showing changes mainly in signals from ML113,130–132, which in subacute stroke patients may represent ML in macrophages or other cells113,130,131. A preclinical study performed in the rat hippocampus after mild and moderate traumatic brain injury revealed a substantial change at 1.3 ppm133. To demonstrate the potential of 1H MRS analysis including the ML and MM contributions in the clinical setting, a clinical case showing the longitudinal effects of a transient, but severe systemic hypoxia on the ML, MM and metabolites in the human brain is described in Appendix 1 using the spectrIm MM model presented in Appendix 3.
Non radiological ex vivo clinical applications of the metabolic and biopsy data have been done using HRMAS spectroscopy and differences in metabolites, MM and ML between various brain tumors have been measured134.
The number of studies evaluating MM or ML+MM changes in disease is still limited, but they provide substantial evidence that MM+ML changes are relevant and should be taken into account in the quantification step. Suggestions on how to handle them would be: 1) to use a MM spectrum acquired in vivo in its totality, if feasible; 2) if it is known or visible in the 1H MR spectrum that a specific MM moiety is changing, then a MM fitting model with the freedom to measure this signal separately is required (i.e. add a separate simulated MM or lipid component as already done in patients with adrenoleukodystrophy135); 3) or to use a parameterized MM spectrum with well-defined soft constraints to avoid over-parameterization (i.e. fix ratios of all MM peaks in the parameterized MM spectrum, except for a small number of specific ones). In this context, future studies should focus on evaluating MM changes in additional pathologies together with a precise identification of the origins of these MM peaks and the underlying mechanisms.
9. Dissemination
In order to streamline and standardize the analysis of MM contribution to 1H MRS spectra without duplicating effort, we recommend sharing of MM models with the MRS community. Dissemination of sequence- and field-strength-specific MM models can be accomplished through sharing either parameterization of MM resonances or complete experimentally measured MM basis functions for linear-combination analysis, along with settings and control files, as well as a complete documentation of how these data were acquired. (see Table 2, recommendations 14+15). Preferably, MM data should be collected in a centralized public repository, and made available free of charge or license. We encourage the use of the MRSHub (https://www.mrshub.org), a resource designed by the recently established Committee for MRS Data and Code Sharing, a standing committee under the auspices of the ISMRM MRS Study Group. The MRSHub features resources for dissemination of analysis software as well as data, and also includes a discussion forum to address open issues in the field of MM in an open and collaborative fashion (https://forum.mrshub.org/t/data-submission-mm-consensus-data-collection/92).
Table 2:
Current recommendations and open issues in the field to improve the general knowledge about MM
| Recommendations | |
| 1. | If the objective of a study is to determine metabolite concentrations and/or metabolite ratios, then the MM contribution has to be removed or included in the basis set used for quantification, especially for TE’s below 80 ms. |
| 2. | We recommend to use the unified nomenclature of the MM components (Table 1), which can be easily expanded to new peaks. |
| 3. | The MM spectrum is different from a spectral baseline. These terms should not be used interchangeably. |
| 4. | Since the shape, amplitudes and relaxation times of different MM contributions vary with B0, we recommend to measure the MM spectra for metabolite quantification specifically for each B0 field strength and each acquisition sequence used. |
| 5. | We recommend to acquire MM spectra in vivo using adiabatic RF pulse(s) and with the highest possible spatial and spectral resolution (i.e. high quality B0 shimming), high SNR, efficient water suppression and no baseline distortions, and to avoid subcutaneous lipid contamination. |
| 6. | We recommend to identify properly and eliminate residual peaks of metabolites from any MM spectrum measured in vivo (Section 3.1). Some residual peaks of metabolites are always present independent of the sequence used to measure them. AMARES or similar approaches with the ability to consider prior knowledge for the residual metabolite peaks, are recommended for post-processing of the MM spectrum. |
| 7. | Both single and double inversion methods of MM measurement are recommended since they provide good MM signal recovery, while the double inversion methods provide improved metabolite suppression (e.g. for mapping in the presence of B1+ - inhomogeneities) at the expense of stronger T1-weighting (e.g. reduced MM signal recovery). |
| 8. | If the goal of the study is to estimate individual MM peaks, care has to be taken to avoid over-parameterization of the fitting, for instance via including some soft constraints on the relative amplitudes, frequencies and linewidth of the different MM components. One possible indication for overfitting is the presence of strong correlation between a MM component and an overlapping metabolite peak. |
| 9. | In human brain, the MM content and pattern change in older adult subjects (≥ 60 years), therefore age-specific MM spectra are required for metabolite estimation (i.e. a spectrum per 5 years after the age of 60). |
| 10. | It should be kept in mind that the MM content of some individual MM peaks varies across brain regions in humans. This should be particularly considered for low abundant metabolites, such as GABA+ in spectral editing, where the contribution of MM to GABA+ seem to be higher in WM than in GM. |
| 11. | The MM content and spectral pattern does not seem to differ in hippocampus, cortex and striatum in healthy rodents (rats and mice). Thus, fitting metabolite concentrations assuming the constant shape of the MM spectrum can be a practical approach. Therefore, we recommend to use one single MM spectrum and to provide a clear description of this MM spectrum when publishing the data. |
| 12. | The MM and ML content and pattern change in disease in both humans and rodents. We recommend characterizing the MM and ML contribution in each specific disease by measuring the MM or MM+ML spectrum in vivo. |
| 13. | The choice of the approach to handle the MM highly depends on the circumstances:1) a single MM basis spectrum when metabolite concentrations are of primary interest; 2) individual MM components if MM measures are desired as disease biomarkers. |
| 14. | To avoid duplicated effort, we recommend sharing of the various MM models through a list of parameters or data points. |
| 15. | We recommend that each publication contains a clear description of how MM were handled during the metabolite quantification step and also a brief description of the parameters used for acquisition, voxel position and size, quality of the shimming reported as water linewidth, the names of metabolite residuals eliminated and the type of post-processing used. |
| Open issues in the field of MM | |
| 1. | Identification of the biological background of individual MM peaks, especially those yet unknown MM peaks observed at ultra-high B0. Improvement of the assignment of the individual MM peaks to particular amino acids and further investigate the origin of the MM signal with respect to contributions from structured versus unstructured cytosolic proteins, membrane bound proteins and large protein complexes in order to better understand any spatial/tissue differences. Investigation of the contribution of other types of macromolecules, such as sugars or DNA/RNA. Performing additional ex vivo validation studies. |
| 2. | Measurement of T1 and T2 relaxation times of different MM peaks (M1.81 to M4.20) at different B0 in humans and rodents. |
| 3. | Analyze the possible contribution of signals of metabolites with short T1 relaxation times, such as GSH, in the measured metabolite-nulled MM spectrum48. |
| 4. | Performing additional studies on how MM vary with TE using different types of acquisition sequences (i.e. LASER, STEAM, PRESS, SPECIAL) and determine a threshold of TE above which the MM contribution is insignificant and does not need to be considered in the quantification step |
| 5. | Improvement of IR or DIR methods for MM measurement in vivo. Development of alternative methods for MM mapping, which will not be based on IR-nulling or spectral quantification. |
| 6. | Identification of possible soft constrains and systematic errors in fitting individual MM peaks. |
| 7. | Confirmation of the observed regional differences in MM spectra in humans. Measurement of MM in additional brain regions in rodents (i.e. cerebellum, thalamus) to confirm the lack of brain regional changes in MM in rodent brain. |
| 8. | Performing additional studies on quantification of individual MM peaks in normal versus diseased brain tissue. Rapid changes (in hours/days) in the 0.9–1.8 ppm range are observed in response to (systemic) hypoxia, and slow changes in relative amplitudes are observed on the timescale of several weeks. Prediction of patient outcome seems possible, and further systematic investigations are needed. Continuation of the studies on ML, MM+ML and MM only changes in pathologies and performing a precise identification of the origins of these MM peaks and the mechanisms behind. Determination of the most suitable approach on how to handle the MM contributions in pathologies. |
| 9. | Evaluate whether the age-associated MM pattern and content differences observed in the occipital cortex and posterior cingulate cortex are more widely spread throughout the brain. Evaluate whether changes with age occur in GM, WM or in both tissue types. Identify the causes of these differences. Assess the effect of age-associated differences in the MM spectral pattern on quantification of non-edited and edited spectra. Acquire data from neo-natal through to young adult subjects to fully characterize the age-dependence of the MM contribution to human/rodent brain spectra. |
10. Concluding remarks and recommendations
This manuscript is written as an experts’ consensus recommendation and aims to summarize the present knowledge in the field of MM contribution in brain 1H MRS measurements. At the time of writing the authors, experts in the MM field, agreed on several recommendations and provided a list with future studies needed to improve the general knowledge about MM. The recommendations and problems to be addressed in the future are summarized in Table 2.
Supplementary Material
Acknowledgements:
These experts’ consensus recommendations were arrived at from senior members of the authorship, who are all longtime experts in MRS methodology in general and this special topic in particular. The trainee members of the authorship are also well familiar with this topic and have fulfilled the requirements for authorship by contributing data, literature search, and/or assisting in drafting, discussing, and revising the manuscript.
A further group of MRS experts, all of them with years of expertise in the field of MRS methodology, and/or macromolecules handling in 1H MRS brain spectra was collected to support the recommendations as the “Experts Collaborators Group on Contribution of macromolecules to brain 1H MR spectra”. The members of the group are listed in Appendix 1, Supplementary Table S5.
The authors are grateful for the input of Dr J Valette in Section 3, Prof. R. Bartha and Dr. S. Provencher in Appendix 3, and Dr Georg Oeltzschner in dissemination section and Appendix 3.
Financial information:
The preparation of this manuscript was supported by the Swiss National Science Foundation (320030-175984, RK; 310030_173222/1, CC); Horizon 2020/CDS-QUAMRI Grant number: 634541 (AH, TB, and SMM); SYNAPLAST Grant number: 679927 (AH and AW); US National Institutes of Health (P41-P41EB015909, MP; R01 MH109159, KLB; P41-EB027061, P30-NS076408 MM and IT); Austrian Science Fund (FWF)(P 30701-B27, WB); Ministry of Education, Youth and Sports of the Czech Republic (CZ.02.1.01/0.0/0.0/16_013/0001775, ZS and JS)
Abbreviations:
- ADC
apparent diffusion coefficient
- AMARES
advanced method for accurate, robust, and efficient spectral fitting
- BPP
Bloembergen-Purcell-Pound
- CEST
chemical exchange saturation transfer
- COSY
correlated spectroscopy
- DIR
double inversion recover
- DW
diffusion-weighting
- FID
free induction decay
- FWHM
full width at half maximum
- GM
gray matter
- HLSVD
Hankel-Lanczos singular value decomposition
- HRMAS
high-resolution magic angle spinning
- IR
inversion recovery
- J-RES
J-resolved spectroscopy
- LASER
localization by adiabatic selective refocusing
- MEGA-PRESS
Mescher-Garwood point-resolved spectroscopy
- ML
mobile lipids
- MM
mobile macromolecules
- MQ
multiple quantum
- MR
magnetic resonance
- MRI
magnetic resonance imaging
- MRS
magnetic resonance spectroscopy
- MRSI
magnetic resonance spectroscopic imaging
- MS
multiple sclerosis
- NOE
nuclear Overhauser effect
- PRESS
point-resolved spectroscopy
- QUEST
quantitation based on semi-parametric quantum estimation
- RF
radiofrequency
- SNR
signal-to-noise ratio
- SPECIAL
spin-echo full-intensity acquired localized spectroscopy
- STEAM
stimulated echo acquisition mode
- SVS
single voxel spectroscopy
- TE
echo time
- TI
inversion time
- TM
mixing time, middle time
- TR
repetition time
- VOI
volume of interes
- WEX
water exchange
- WM
white matter
Contributor Information
Cristina Cudalbu, Center for Biomedical Imaging, Ecole Polytechnique Fédérale de Lausanne, Lausanne, Switzerland.
Kevin L. Behar, Magnetic Resonance Research Center and Department of Psychiatry, Yale University, New Haven, CT USA
Pallab K. Bhattacharyya, Imaging Institute, Cleveland Clinic Foundation, Cleveland, OH, USA
Wolfgang Bogner, High Field MR Centre, Department of Biomedical Imaging and Image-Guided Therapy, Medical University of Vienna; Christian Doppler Laboratory for Clinical Molecular MR Imaging, Vienna, Austria.
Tamas Borbath, Max Planck Institut für Biologische Kybernetik, Baden-Württemberg, Germany and Faculty of Science, Eberhard-Karls Universität Tübingen, Baden-Württemberg, Germany.
Robin A. de Graaf, Department of Radiology and Biomedical Imaging, Yale University, New Haven, CT, USA
Rolf Gruetter, Laboratory for functional and metabolic imaging, Ecole Polytechnique Fédérale de Lausanne, Lausanne, Switzerland.
Anke Henning, Advanced Imaging Research Center, University of Texas Southwestern Medical Center, Dallas, TX, United States and Max Planck Institute for Biological Cybernetics, Tuebingen, Germany.
Christoph Juchem, Departments of Biomedical Engineering and Radiology, Columbia University, New York, NY, USA.
Roland Kreis, Departments of Radiology and Biomedical Research, University of Bern, Bern, Switzerland.
Phil Lee, Department of Radiology, Hoglund Brain Imaging Center, University of Kansas Medical Center, Kansas City, KS, USA.
Hongxia Lei, Center for Biomedical Imaging, Ecole Polytechnique Fédérale de Lausanne, Lausanne, Switzerland.
Małgorzata Marjańska, Center for Magnetic Resonance Research and Department of Radiology, University of Minnesota, Minneapolis, MN, USA.
Ralf Mekle, Center for Stroke Research Berlin (CSB), Charité Universitätsmedizin Berlin, Berlin, Germany.
Saipavitra Murali-Manohar, Max Planck Institut für Biologische Kybernetik, Baden-Württemberg, Germany and Faculty of Science, Eberhard-Karls Universität Tübingen, Baden-Württemberg, Germany.
Michal Považan, Russell H. Morgan Department of Radiology and Radiological Science, The Johns Hopkins University School of Medicine, Baltimore, MD, USA.
Veronika Rackayová, Laboratory of Functional and Metabolic Imaging, Ecole Polytechnique Fédérale de Lausanne (EPFL), Lausanne, Switzerland; Center for Biomedical Imaging, Ecole Polytechnique Fédérale de Lausanne, Lausanne, Switzerland.
Dunja Simicic, Laboratory of Functional and Metabolic Imaging, Ecole Polytechnique Fédérale de Lausanne (EPFL), Lausanne, Switzerland; Center for Biomedical Imaging, Ecole Polytechnique Fédérale de Lausanne, Lausanne, Switzerland.
Johannes Slotboom, University Institute of Diagnostic and Interventional Neuroradiology, University Hospital Bern and Inselspital, 3010 Bern, Switzerland.
Brian J Soher, Center for Advanced MR Development, Department of Radiology, Duke University Medical Center, Durham, NC, USA.
Zenon Starčuk, Jr., The Czech Academy of Sciences, Institute of Scientific Instruments, Brno, Czech Republic
Jana Starčuková, The Czech Academy of Sciences, Institute of Scientific Instruments, Brno, Czech Republic.
Ivan Tkáč, Center for Magnetic Resonance Research and Department of Radiology, University of Minnesota, Minneapolis, MN, USA.
Stephen Williams, School of Health Sciences (Emeritus), University of Manchester, Manchester, UK..
Martin Wilson, Centre for Human Brain Health and School of Psychology, University of Birmingham, Birmingham, UK..
Andrew Martin Wright, Max Planck Institut für Biologische Kybernetik, Baden-Württemberg, Germany and IMPRS for Cognitive and Systems Neuroscience, Eberhard-Karls Universität Tübingen, Baden-Württemberg, Germany.
Lijing Xin, Center for Biomedical Imaging, Ecole Polytechnique Fédérale de Lausanne, Lausanne, Switzerland.
Vladimír Mlynárik, High Field MR Centre, Department of Biomedical Imaging and Image-Guided Therapy, Medical University of Vienna; Christian Doppler Laboratory for Clinical Molecular MR Imaging, Vienna, Austria.
References
- 1.Kunz N, Cudalbu C, Mlynarik V, Hüppi PS, Sizonenko SV., Gruetter R. Diffusion-weighted spectroscopy: A novel approach to determine macromolecule resonances in short-echo time 1H-MRS. Magn Reson Med. 2010;64(4):939–946. [DOI] [PubMed] [Google Scholar]
- 2.Graaf RA de Brown PB, Mcintyre S Nixon TW, Behar KL Rothman DL. High Magnetic Field Water and Metabolite Proton T 1 and T 2 Relaxation in Rat Brain In Vivo. Magn Reson Med. 2006;56(2):386–94. [DOI] [PubMed] [Google Scholar]
- 3.Behar KL, Ogino T. Assignment of resonances in the 1H spectrum of rat brain by two-dimensional shift correlated and J-resolved NMR spectroscopy. Magn Reson Med. 1991;17(2):285–303. [DOI] [PubMed] [Google Scholar]
- 4.Behar KL, Ogino T. Characterization of macromolecule resonances in the 1H NMR spectrum of rat brain. Magn Reson Med. 1993;30(1):38–44. [DOI] [PubMed] [Google Scholar]
- 5.Arus C, Yen-Chung C, Barany M. Proton magnetic resonance spectra of excised rat brain. Assignments of resonances. Physiol Chem Phys Med NMR. 1985;17(1):23–33. [PubMed] [Google Scholar]
- 6.Kauppinen RA, Kokko H, Williams SR. Detection of Mobile Proteins by Proton Nuclear Magnetic Resonance Spectroscopy in the Guinea Pig Brain Ex Vivo and Their Partial Purification. J Neurochem. 1992;58(3):967–74. [DOI] [PubMed] [Google Scholar]
- 7.Kauppinen RA, Niskanen T, Hakumäki J, Williams SR. Quantitative analysis of 1H NMR detected proteins in the rat cerebral cortex in vivo and in vitro. NMR Biomed. 1993;6(4):242–7. [DOI] [PubMed] [Google Scholar]
- 8.Behar KL, Rothman DL, Spencer DD, Petroff OAC. Analysis of macromolecule resonances in1H NMR spectra of human brain. Magn Reson Med. 1994;32(3):294–302. [DOI] [PubMed] [Google Scholar]
- 9.Mori S, van Zijl PCM, Johnson MON, Berg JM. Water Exchange Filter (WEX Filter) for Nuclear Magnetic Resonance Studies of Macromolecules. J Am Chem Soc. 1994;116(26):11982–11984. [Google Scholar]
- 10.van Zijl PCM, Yadav NN. Chemical exchange saturation transfer (CEST): what is in a name and what isn’t? Magn Reson Med. 2011;65(4):927–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heo HY, Jones CK, Hua J, et al. Whole-brain amide proton transfer (APT) and nuclear overhauser enhancement (NOE) imaging in glioma patients using low-power steady-state pulsed chemical exchange saturation transfer (CEST) imaging at 7T. J Magn Reson Imaging. 2016;44(1):41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Candiota AP, Majós C, Bassols A, et al. Assignment of the 2.03 ppm resonance in in vivo 1H MRS of human brain tumour cystic fluid: contribution of macromolecules. Magn Reson Mater Physics, Biol Med. 2004;17(1):36–46. [DOI] [PubMed] [Google Scholar]
- 13.Mountford C, Quadrelli S, Lin A, Ramadan S. Six fucose-α(1–2) sugars and α-fucose assigned in the human brain using in vivo two-dimensional MRS. NMR Biomed. 2015;28(3):291–6. [DOI] [PubMed] [Google Scholar]
- 14.Soares AF, Gruetter R, Lei H. Technical and experimental features of Magnetic Resonance Spectroscopy of brain glycogen metabolism. Anal Biochem. 2017;529:117–126. [DOI] [PubMed] [Google Scholar]
- 15.Kauppinen RA, Nissinen T, Kärkkäinen AM, et al. Detection of thymosin β4 in situ in a guinea pig cerebral cortex preparation using 1H NMR spectroscopy. J Biol Chem. 1992;15(267(14)):9905–10. [PubMed] [Google Scholar]
- 16.Woody RW, Roberts GCK, Clark DC, Bayley PM. 1 H NMR evidence for flexibility in microtubule-associated proteins and microtubule protein oligomers. FEBS Lett. 1982;141(2):181–184. [DOI] [PubMed] [Google Scholar]
- 17.Woody RW, Clark DC, Roberts GCK, Martin SR, Bayley PM. Molecular Flexibility in Microtubule Proteins: Proton Nuclear Magnetic Resonance Characterization. Biochemistry. 1983;22(9):2186–2192. [DOI] [PubMed] [Google Scholar]
- 18.Wüthrich K The way to NMR structures of proteins. Nat Struct Biol. 2001;8:923–925. [DOI] [PubMed] [Google Scholar]
- 19.Wüthrich K Protein structure determination in solution by NMR spectroscopy. J Biol Chem. 1990;265(36):22059–62. [PubMed] [Google Scholar]
- 20.Marassi FM, Opella SJ. NMR structural studies of membrane proteins. Curr Opin Struct Biol. 1998;8(5):640–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Craveiro M, Clément-Schatlo V, Marino D, Gruetter R, Cudalbu C. In vivo brain macromolecule signals in healthy and glioblastoma mouse models: 1H magnetic resonance spectroscopy, post-processing and metabolite quantification at 14.1 T. J Neurochem. 2014;129(5):806–815. [DOI] [PubMed] [Google Scholar]
- 22.Behar KL. Dealing with macromolecules. In: ISMRM, Morning Categorical Course “Spectroscopy: The Brain and Beyond”.; 2004. [Google Scholar]
- 23.Mielke SP, Krishnan VV. Characterization of protein secondary structure from NMR chemical shifts. Prog Nucl Magn Reson Spectrosc. 2009;54(3–4):141–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Dios AC, Pearson JG, Oldfield E. Secondary and tertiary structural effects on protein NMR chemical shifts: An ab initio approach. Science (80-). 1993;260(5113):1491–6. [DOI] [PubMed] [Google Scholar]
- 25.Borbath T, Murali-Manohar S, Wright AM, Henning A. T2 Relaxation Times of Macromolecules in Human Brain Spectra at 9.4 T. 27th Annu Meet Exhib Int Soc Magn Reson Med (ISMRM 2019), Montréal, QC, Canada. 2019. [Google Scholar]
- 26.Borbáth T, Murali-Manohar S, Henning A. Towards a Fitting Model of Macromolecular Spectra: Amino Acids. In: ISMRM.; 2019:1068. [Google Scholar]
- 27.Schaller B, Xin L, Gruetter R. Is the macromolecule signal tissue-specific in healthy human brain? a 1H MRS study at 7 tesla in the occipital lobe. Magn Reson Med. 2014;72(4):934–940. [DOI] [PubMed] [Google Scholar]
- 28.Snoussi K, Gillen JS, Horska A, et al. Comparison of brain gray and white matter macromolecule resonances at 3 and 7 Tesla. Magn Reson Med. 2015;74(3):607–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lopez-Kolkovsky AL, Mériaux S, Boumezbeur F. Metabolite and Macromolecule T1 and T2 Relaxation Times in the Rat Brain in vivo at 17.2T. Magn Reson Med. 2016;75(2):503–514. [DOI] [PubMed] [Google Scholar]
- 30.Beach EF, Munks B, Robinson A. The Amino Acid Composition of Animal Tissue Protein. J Biol Chem. 1943;148:431–439. [Google Scholar]
- 31.Robinson N, Williams CB. Amino acids in human brain. Clin Chim Acta. 1965;12:311–317. [Google Scholar]
- 32.Smith MH. The amino acid composition of proteins. J Theor Biol. 1966;13:261–282. [Google Scholar]
- 33.Clouet DH, Gaitonde MK. THE CHANGES WITH AGE IN THE PROTEIN COMPOSITION OF THE RAT BRAIN. J Neurochem. 1956;1(2):126–133. [DOI] [PubMed] [Google Scholar]
- 34.Hofmann L, Slotboom J, Boesch C, Kreis R. Characterization of the macromolecule baseline in localized 1H-MR spectra of human brain. Magn Reson Med. 2001;46(5):855–863. [DOI] [PubMed] [Google Scholar]
- 35.Kreis R, Boer V, Choi I-Y, et al. Terminology for the characterization of in vivo MR spectroscopy methods and MR spectra: Background and experts’ consensus recommendations. NMR Biomed. 2019;submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tkác I, Andersen P, Adriany G, Merkle H, Ugurbil K, Gruetter R. In vivo 1H NMR spectroscopy of the human brain at 7 T. Magn Reson Med. 2001;46(3):451–6. [DOI] [PubMed] [Google Scholar]
- 37.Juchem C, de Graaf RA. B0 magnetic field homogeneity and shimming for in vivo magnetic resonance spectroscopy. Anal Biochem. 2017;529:17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Juchem C, Boer VO, Cudalbu C, et al. B0 Shimming for In Vivo MR Spectroscopy: Experts’ consensus recommendations. NMR Biomed. [DOI] [PubMed] [Google Scholar]
- 39.Giapitzakis IA, Avdievich N, Henning A. Characterization of macromolecular baseline of human brain using metabolite cycled semi-LASER at 9.4T. Magn Reson Med. 2018;80(2):462–473. [DOI] [PubMed] [Google Scholar]
- 40.Döring A, Adalid V, Boesch C, Kreis R. On the exploitation of slow macromolecular diffusion for baseline estimation in MR spectroscopy using 2D simultaneous fitting. In: Joint 26th Meeting of ISMRM and 35th Meeting of the ESMRMB, Paris (F).; 2018:1315. [Google Scholar]
- 41.Cudalbu C, Mlynárik V, Xin L, Gruetter R. Comparison of T1 Relaxation Times of the Neurochemical Profile in Rat Brain at 9.4 Tesla and 14.1 Tesla. Magn Reson Med. 2009;62(4):862–867. [DOI] [PubMed] [Google Scholar]
- 42.Michaeli S, Garwood M, Zhu X, et al. Proton T 2 Relaxation Study of Water, N-acetylaspartate, and Creatine in Human Brain Using Hahn and Carr-Purcell Spin Echoes at 4T and 7T. 2002;633:629–633. [DOI] [PubMed] [Google Scholar]
- 43.Wyss PO, Bianchini C, Scheidegger M, et al. In vivo estimation of transverse relaxation time constant (T2) of 17 human brain metabolites at 3T. Magn Reson Med. 2018;80(2):452–461. [DOI] [PubMed] [Google Scholar]
- 44.Xin L, Gambarota G, Cudalbu C, Mlynárik V, Gruetter R. Single spin-echo T 2 relaxation times of cerebral metabolites at 14.1 T in the in vivo rat brain. Magn Reson Mater Physics, Biol Med. 2013;26(6):549–554. [DOI] [PubMed] [Google Scholar]
- 45.Deelchand DK, Henry P-G, Ugurbil K, Marjanska M. Measurement of Transverse Relaxation Times of J-Coupled Metabolites in the Human Visual Cortex at 4 T. Magn Reson Med. 2012;67:891–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deelchand DK, Auerbach EJ, Kobayashi N, Marjanska M. Transverse Relaxation Time Constants of the Five Major Metabolites in Human Brain Measured In Vivo Using LASER and PRESS at 3 T. Magn Reson Med. 2018;79:1260–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marjańska M, Auerbach EJ, Valabrègue R, Moortele P Van De, Adriany G, Garwood M. Localized 1 H NMR spectroscopy in different regions of human brain in vivo at 7T : T 2 relaxation times and concentrations of cerebral metabolites. NMR Biomed. 2012;25:332–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choi IY, Lee P. Doubly selective multiple quantum chemical shift imaging and T1relaxation time measurement of glutathione (GSH) in the human brain in vivo. NMR Biomed. 2013;26(1):28–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murali-Manohar S, Wright AM, Borbath T, Henning A. Longitudinal Relaxation times of Macromolecular Resonances at 9.4 T in Human Brain. 27th Annu Meet Exhib Int Soc Magn Reson Med (ISMRM 2019), Montréal, QC, Canada. 2019. [Google Scholar]
- 50.Murali-Manohar S, Borbath T, Wright AM, Soher B, Mekle R, Henning A. T2 relaxation times of macromolecules and metabolites in the human brain at 9.4 T. Magn Reson Med. 2020:Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 51.Považan M, Strasser B, Hangel G, et al. Simultaneous mapping of metabolites and individual macromolecular components via ultra-short acquisition delay 1 H MRSI in the brain at 7T. Magn Reson Med. 2017;79(3):1231–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ligneul C, Palombo M, Valette J. Metabolite diffusion up to very high b in the mouse brain in vivo: Revisiting the potential correlation between relaxation and diffusion properties. Magn Reson Med. 2017;77(4):1390–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bloembergen N, Purcell EM, Pound RV. Relaxation effects in nuclear magnetic resonance absorption. Phys Rev. 1948;73(7):679–712. [Google Scholar]
- 54.Pfeuffer J, Tkáč I, Gruetter R. Extracellular-intracellular distribution of glucose and lactate in the rat brain assessed noninvasively by diffusion-weighted 1H nuclear magnetic resonance spectroscopy in vivo. J Cereb Blood Flow Metab. 2000;20(4):736–746. [DOI] [PubMed] [Google Scholar]
- 55.Ligneul C, Palombo M, Hernández-Garzón E, et al. Diffusion-weighted magnetic resonance spectroscopy enables cell-specific monitoring of astrocyte reactivity in vivo. Neuroimage. 2019;191:457–469. [DOI] [PubMed] [Google Scholar]
- 56.Cudalbu C, Mlynarik V, Gruetter R. Handling macromolecule signals in the quantification of the neurochemical profile. J Alzheimers Dis. 2012;31 Suppl 3:S101–15. [DOI] [PubMed] [Google Scholar]
- 57.Cudalbu C, Mlynrik V, Xin L, Gruetter R. Quantification of in vivo short echo-time proton magnetic resonance spectra at 14.1 T using two different approaches of modelling the macromolecule spectrum. Meas Sci Technol. 2009;20:104034 (7pp). [Google Scholar]
- 58.Mlynárik V, Cudalbu C, Xin L, Gruetter R. 1H NMR spectroscopy of rat brain in vivo at 14.1 Tesla: Improvements in quantification of the neurochemical profile. J Magn Reson. 2008;194(2):163–168. [DOI] [PubMed] [Google Scholar]
- 59.Marjanska M, Deelchand DK, Hodges JS, et al. Altered macromolecular pattern and content in the aging human brain. NMR Biomed. 2018;(31:e3865):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vanhamme L, Van Huffel S. AMARES: Advanced Method for Accurate, Robust and Efficient Spectral fitting of MRS data with use of prior knowledge. 1997;43(129):1–2. [DOI] [PubMed] [Google Scholar]
- 61.Govindaraju V, Young K, Maudsley AA. Proton NMR chemical shifts and coupling constants for brain metabolites. NMR Biomed. 2000;13:129–153. [DOI] [PubMed] [Google Scholar]
- 62.Chong DGQ, Kreis R, Bolliger CS, Boesch C, Slotboom J. Two-dimensional linear-combination model fitting of magnetic resonance spectra to define the macromolecule baseline using FiTAID, a Fitting Tool for Arrays of Interrelated Datasets. Magn Reson Mater Physics, Biol Med. 2011;24(3):147–164. [DOI] [PubMed] [Google Scholar]
- 63.Kreis R, Slotboom J, Hofmann L, Boesch C. Integrated data acquisition and processing to determine metabolite contents, relaxation times, and macromolecule baseline in single examinations of individual subjects. Magn Reson Med. 2005;54:761–8. [DOI] [PubMed] [Google Scholar]
- 64.Marjańska M, Deelchand DK, Hodges JS, et al. Altered macromolecular pattern and content in the aging human brain. NMR Biomed. 2018;31(2):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bhogal AA, Schür RR, Houtepen LC, et al. 1H–MRS processing parameters affect metabolite quantification: The urgent need for uniform and transparent standardization. NMR Biomed. 2017;30(11):1–9. [DOI] [PubMed] [Google Scholar]
- 66.Lee HH, Kim H. Parameterization of spectral baseline directly from short echo time full spectra in 1H-MRS. Magn Reson Med. 2017;78(3):836–847. [DOI] [PubMed] [Google Scholar]
- 67.Seeger U, Klose U, Mader I, Grodd W, Na T. Parameterized Evaluation of Macromolecules and Lipids in Proton MR Spectroscopy of Brain Diseases. 2003;28:19–28. [DOI] [PubMed] [Google Scholar]
- 68.Považan M, Hangel G, Strasser B, et al. Mapping of brain macromolecules and their use for spectral processing of 1H-MRSI data with an ultra-short acquisition delay at 7T. Neuroimage. 2015;121:126–135. [DOI] [PubMed] [Google Scholar]
- 69.Pfeuffer J, Juchem C, Merkle H, Nauerth A, Logothetis NK. High-field localized 1H NMR spectroscopy in the anesthetized and in the awake monkey. Magn Reson Imaging. 2004;22(10):1361–1372. [DOI] [PubMed] [Google Scholar]
- 70.Otazo R, Mueller B, Ugurbil K, Wald L, Posse S. Signal-to-Noise Ratio and Spectral Linewidth Improvements Between 1.5 and 7 Tesla in Proton Echo-Planar Spectroscopic Imaging. 2006;56:1200–1210. [DOI] [PubMed] [Google Scholar]
- 71.Birch R, Peet AC, Dehghani H, Wilson M. Influence of Macromolecule Baseline on 1 H MR Spectroscopic Imaging Reproducibility. Magn Reson Med. 2017;77:34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hong S-T, Balla DZ, Shajan G, Choi C, Uğurbil K, Pohmann R. Enhanced Neurochemical Profile of the Rat Brain using In Vivo 1H NMR spectroscopy at 16.4T. Magn Reson Med. 2011;65(1):28–34. [DOI] [PubMed] [Google Scholar]
- 73.Hoefemann M, Bolliger C, van derVeen JW, Kreis R. About the need for a comprehensive description of the macromolecular baseline signal for MR fingerprinting and multidimensional fitting of MR spectra. In: ISMRM.; 2019:1069. [Google Scholar]
- 74.Wright AM, Murali-Manohar S, Henning A. Relaxation corrected and Sequence-dependent Macromolecule Baseline Model. In: ISMRM.; 2019:2247. [Google Scholar]
- 75.Kassem MNE, Bartha R. Quantitative proton short-echo-time LASER spectroscopy of normal human white matter and hippocampus at 4 Tesla incorporating macromolecule subtraction. Magn Reson Med. 2003;49(5):918–27. [DOI] [PubMed] [Google Scholar]
- 76.Penner J, Bartha R. Semi-LASER 1H MR spectroscopy at 7 Tesla in human brain: Metabolite quantification incorporating subject-specific macromolecule removal. Magn Reson Med. 2015;74(1):4–12. [DOI] [PubMed] [Google Scholar]
- 77.Bartha R, Drost DJ, Williamson PC. Factors affecting the quantification of short echo in-vivo1H MR spectra: Prior knowledge, peak elimination, and filtering. NMR Biomed. 1999;12(4):205–216. [DOI] [PubMed] [Google Scholar]
- 78.Pfeuffer J, Tkac I, Provencher SW, Gruetter R. Towards an In Vivo Neurochemical Profile : Quantification of 18 Metabolites in Short-Echo-Time 1H NMR Spectra of the Rat Brain. J Magn Reson. 1999;141:104–120. [DOI] [PubMed] [Google Scholar]
- 79.Hofmann L, Slotboom J, Jung B, Maloca P, Boesch C, Kreis R. Quantitative 1H-magnetic resonance spectroscopy of human brain: Influence of composition and parameterization of the basis set in linear combination model-fitting. Magn Reson Med. 2002;48(3):440–453. [DOI] [PubMed] [Google Scholar]
- 80.Giapitzakis IA, Borbath T, Murali-Manohar S, Avdievich N, Henning A. Investigation of the influence of macromolecules and spline baseline in the fitting model of human brain spectra at 9.4T. Magn Reson Med. 2018;(June 2018):746–758. [DOI] [PubMed] [Google Scholar]
- 81.Schaller B, Xin L, Cudalbu C, Gruetter R. Quantification of the neurochemical profile using simulated macromolecule resonances at 3 T. NMR Biomed. 2013;26(5):593–599. [DOI] [PubMed] [Google Scholar]
- 82.Gottschalk M, Lamalle L, Segebarth C. Short-TE localised 1 H MRS of the human brain at 3 T : quantification of the metabolite signals using two approaches to account for macromolecular signal contributions. 2008;(21):507–517. [DOI] [PubMed] [Google Scholar]
- 83.Henning A Advanced spectral quantification: Parameter handling, nonparametric pattern modeling, and multidimensional fitting. eMagRes. 2016:981–994. [Google Scholar]
- 84.Provencher SW. Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magn Reson Med. 1993;30(6):672–679. [DOI] [PubMed] [Google Scholar]
- 85.Coenradie Y, Beer R De, Ormondt D Van, Lyon B. Background-signal Parameterization in In Vivo MR Spectroscopy. Time.:6–8. [Google Scholar]
- 86.Ratiney H, Sdika M, Coenradie Y, Cavassila S, van Ormondt D, Graveron-Demilly D. Time-domain semi-parametric estimation based on a metabolite basis set. NMR Biomed. 2005;18(1):1–13. [DOI] [PubMed] [Google Scholar]
- 87.Cudalbu C, Beuf O, Cavassila S. In vivo short echo time localized 1H MRS of the rat brain at 7 T: Influence of two strategies of background-accommodation on the metabolite concentration estimation using QUEST. J Signal Process Syst. 2009;55(1–3). [Google Scholar]
- 88.Cudalbu C, Mlynárik V, Xin L, Gruetter R. Comparison of two approaches to model the macromolecule spectrum for the quantification of short TE 1H MRS spectra. In: IST 2008 - IEEE Workshop on Imaging Systems and Techniques Proceedings.; 2008. [Google Scholar]
- 89.Choi I-Y, Andronesi O, Barker P, et al. Spectral editing in 1H magnetic resonance spectroscopy: Experts’ consensus recommendations. NMR Biomed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rothman DL, Petroff OA, Behar KL, Mattson RH. Localized 1H NMR measurements of gamma-aminobutyric acid in human brain in vivo. Proc Natl Acad Sci. 1993;90(12):5662–5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Terpstra M, Ugurbil K, Gruetter R. Direct in vivo measurement of human cerebral GABA concentration using MEGA-editing at 7 Tesla. Magn Reson Med. 2002;47(5):1009–1012. [DOI] [PubMed] [Google Scholar]
- 92.Choi IY, Lee SP, Merkle H, Shen J. Single-shot two-echo technique for simultaneous measurement of GABA and creatine in the human brain in vivo. Magn Reson Med. 2004;51(6):1115–21. [DOI] [PubMed] [Google Scholar]
- 93.Donahue MJ, Near J, Blicher JU, Jezzard P. Baseline GABA concentration and fMRI response. Neuroimage. 2010;53:392–8. [DOI] [PubMed] [Google Scholar]
- 94.Aguila MER, Lagopoulos J, Leaver AM, et al. Elevated levels of GABA+ in migraine detected using 1H-MRS. NMR Biomed. 2015;28:890–7. [DOI] [PubMed] [Google Scholar]
- 95.O’Gorman RL, Michels L, Edden RA, Murdoch JB, Martin E. In vivo detection of GABA and glutamate with MEGA-PRESS: Reproducibility and gender effects. J Magn Reson Imaging. 2011;33:1262–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Choi IY, Lee SP, Shen J. In vivo single-shot three-dimensionally localized multiple quantum spectroscopy of GABA in the human brain with improved spectral selectivity. J Magn Reson. 2005;172(1):9–16. [DOI] [PubMed] [Google Scholar]
- 97.Bhattacharyya PK. Macromolecule contamination in GABA editing using MEGA-PRESS should be properly accounted for. Neuroimage. 2014;84:1111–1112. [DOI] [PubMed] [Google Scholar]
- 98.McLean MA, Barker GJ. Concentrations and magnetization transfer ratios of metabolites in gray and white matter. Magn Reson Med. 2006;56(6):1365–1370. [DOI] [PubMed] [Google Scholar]
- 99.Pan JW, Mason GF, Pohost GM, Hetherington HP. Spectroscopic imaging of human brain glutamate by water-suppressed J-refocused coherence transfer at 4.1 T. Magn Reson Med. 1996;36(1):7–12. [DOI] [PubMed] [Google Scholar]
- 100.Moser P, Hingerl L, Strasser B, et al. Whole-slice mapping of GABA and GABA + at 7T via adiabatic MEGA-editing, real-time instability correction, and concentric circle readout. Neuroimage. 2019;184(April 2018):475–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Harris AD, Glaubitz B, Near J, et al. Impact of frequency drift on gamma-aminobutyric acid-edited MR spectroscopy. Magn Reson Med. 2014;72:941–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Harris AD, Puts NAJ, Wijtenburg SA, et al. Normalizing data from GABA-edited MEGA-PRESS implementations at 3 Tesla. Magn Reson Imaging. 2017;42:8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hetherington HP, Newcomer BR, Pan JW. Measurements of human cerebral GABA at 4.1 T using numerically optimized editing pulses. Magn Reson Med. 1998;39:6–10. [DOI] [PubMed] [Google Scholar]
- 104.Henry PG, Dautry C, Hantraye P, Bloch G. Brain gaba editing without macromolecule contamination. Magn Reson Med. 2001;45:517–20. [DOI] [PubMed] [Google Scholar]
- 105.Edden RAE, Puts NAJ, Barker PB. Macromolecule-suppressed GABA-edited magnetic resonance spectroscopy at 3T. Magn Reson Med. 2012;68:657–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Henry PG, Van De Moortele PF, Giacomini E, Nauerth A, Bloch G. Field-frequency locked in vivo proton MRS on a whole-body spectrometer. Magn Reson Med. 1999;42:636–42. [DOI] [PubMed] [Google Scholar]
- 107.Bogner W, Gagoski B, Hess AT, et al. 3D GABA imaging with real-time motion correction, shim update and reacquisition of adiabatic spiral MRSI. Neuroimage. 2014;103:290–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mikkelsen M, Barker PB, Bhattacharyya PK, et al. Big GABA : Edited MR spectroscopy at 24 research sites. Neuroimage. 2017;159:32–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mader I, Seeger U, Karitzky J, Erb M, Schick F, Klose U. Proton magnetic resonance spectroscopy with metabolite nulling reveals regional differences of macromolecules in normal human brain. J Magn Reson Imaging. 2002;16(5):538–46. [DOI] [PubMed] [Google Scholar]
- 110.Lam F, Li Y, Clifford B, Liang ZP. Macromolecule mapping of the brain using ultrashort-TE acquisition and reference-based metabolite removal. Magn Reson Med. 2018;79(5):2460–2469. [DOI] [PubMed] [Google Scholar]
- 111.Sibbitt WL, Haseler LJ, Griffey RR, Friedman SD, Brooks WM. Neurometabolism of active neuropsychiatric lupus determined with proton MR spectroscopy. Am J Neuroradiol. 1997;18(7):1271–1277. [PMC free article] [PubMed] [Google Scholar]
- 112.Saunders DE, Howe F a, van den Boogaart A, Griffiths JR, Brown MM. Discrimination of metabolite from lipid and macromolecule resonances in cerebral infarction in humans using short echo proton spectroscopy. J Magn Reson Imaging. 1997;7(6):1116–21. [DOI] [PubMed] [Google Scholar]
- 113.Graham GD, Hwang J-H, Rothman DL, Prichard JW. Spectroscopic Assessment of Alterations in Macromolecule and Small-Molecule Metabolites in Human Brain After Stroke. Stroke. 2001;32(12):2797–2802. [DOI] [PubMed] [Google Scholar]
- 114.Mader I, Seeger U, Weissert R, et al. Proton MR spectroscopy with metabolite-nulling reveals elevated macromolecules in acute multiple sclerosis. Brain. 2001;124(5):953–961. [DOI] [PubMed] [Google Scholar]
- 115.Howe FA, Opstad KS. 1H MR spectroscopy of brain tumours and masses. NMR Biomed. 2003;16(3):123–31. [DOI] [PubMed] [Google Scholar]
- 116.Povazan M, Hnilicova P, Hangel G, et al. Detection of MM using metabolite-nulled MEGA-LASER at 3T – A possible effect on GABA+ signal. Proc Intl Soc Mag Reson Med. 2017;25. [Google Scholar]
- 117.Hnilicová P, Považan M, Strasser B, et al. Spatial variability and reproducibility of GABA-edited MEGA-LASER 3D-MRSI in the brain at 3 T. NMR Biomed. 2016;29(11):1656–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Andronesi OC, Bhat H, Reuter M, Mukherjee S, Caravan P, Rosen BR. Whole brain mapping of water pools and molecular dynamics with rotating frame MR relaxation using gradient modulated low-power adiabatic pulses. Neuroimage. 2014;89:92–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Geades N, Wismans C, Damen M, et al. Evidence for regional and spectral differences of macromolecule signals in human brain using a crusher coil at 7 Tesla. In: Proc. Intl. Soc. Mag. Reson. Med. 24 (2016).; 2016. [Google Scholar]
- 120.Xin L, Mlynarik V, Lei H, Gruetter R. Influence of regional macromolecule baseline on the quantification of neurochemical profile in rat brain. In: Proc. Intl. Soc. Mag. Reson. Med.; 2010:5. [Google Scholar]
- 121.Tkac I, Rao R, Georgieff MK, Gruetter R. Developmental and regional changes in the neurochemical profile of the rat brain determined by in vivo 1H NMR spectroscopy. Magn Reson Med. 2003;50(1):24–32. [DOI] [PubMed] [Google Scholar]
- 122.Schmitz JE, Kettunen MI, Hu DE, Brindle KM. 1H MRS-visible lipids accumulate during apoptosis of lymphoma cells in vitro and in vivo. Magn Reson Med. 2005;54:43–50. [DOI] [PubMed] [Google Scholar]
- 123.Wolinsky JS, Narayana PA, Fenstermacher MJ. Proton magnetic resonance spectroscopy in multiple sclerosis. Neurology. 1990;40(11):1764–9. [DOI] [PubMed] [Google Scholar]
- 124.García-Gómez JM, Luts J, Julià-Sapé M, et al. Multiproject–multicenter evaluation of automatic brain tumor classification by magnetic resonance spectroscopy. Magn Reson Mater Physics, Biol Med. 2009;22(1):5–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Durmo F, Rydelius A, Cuellar Baena S, et al. Multivoxel 1H-MR Spectroscopy Biometrics for Preoprerative Differentiation Between Brain Tumors. Tomogr (Ann Arbor, Mich). 2018;4(4):172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Pedrosa de Barros N, Meier R, Pletscher M, et al. On the relation between MR spectroscopy features and the distance to MRI-visible solid tumor in GBM patients. Magn Reson Med. 2018;80(6):2339–2355. [DOI] [PubMed] [Google Scholar]
- 127.Howe FA, Barton SJ, Cudlip SA, et al. Metabolic profiles of human brain tumors using quantitative in vivo 1H magnetic resonance spectroscopy. Magn Reson Med. 2003;49(2):223–232. [DOI] [PubMed] [Google Scholar]
- 128.Opstad KS, Griffiths JR, Bell BA, Howe FA. Apparent T 2 Relaxation Times of Lipid and Macromolecules : A Study of High-Grade Tumor Spectra. 2008;184:178–184. [DOI] [PubMed] [Google Scholar]
- 129.Opstad KS, Wright AJ, Bell BA, Griffiths JR, Howe FA. Correlations between in vivo 1H MRS and ex vivo 1H HRMAS metabolite measurements in adult human gliomas. J Magn Reson Imaging. 2010;31(2):289–97. [DOI] [PubMed] [Google Scholar]
- 130.Oz G, Alger JR, Barker PB, et al. The MRS Consensus Group. Clinical proton MR spectroscopy in central nervous system disorders. Radiology. 2014;270(3):658–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hwang JH, Graham GD, Behar KL, Alger JR, Prichard JW, Rothman DL. Short echo time proton magnetic resonance spectroscopic imaging of macromolecule and metabolite signal intensities in the human brain. Magn Reson Med. 1996;35(5):633–9. [DOI] [PubMed] [Google Scholar]
- 132.Petroff OAC, Graham GD, Blamire AM, et al. Spectroscopic imaging of stroke in humans: Histopathology correlates of spectral changes. Neurology. 1992;42(7):1349–54. [DOI] [PubMed] [Google Scholar]
- 133.Singh K, Trivedi R, Verma A, et al. Altered metabolites of the rat hippocampus after mild and moderate traumatic brain injury – a combined in vivo and in vitro 1H–MRS study. NMR Biomed. 2017;30:e3764. [DOI] [PubMed] [Google Scholar]
- 134.Opstad KS, Bell BA, Griffiths JR, Howe FA. An investigation of human brain tumour lipids by high-resolution magic angle spinning 1 H MRS and histological analysis. NMR Biomed. 2008;21(7):677–685. [DOI] [PubMed] [Google Scholar]
- 135.Oz G, Tkac I, LR C, et al. Assessment of adrenoleukodystrophy lesions by high field MRS in non-sedated pediatric patients. Neurology. 2005;64(3):434–441. [DOI] [PubMed] [Google Scholar]
- 136.Tkáč I, Öz G, Adriany G, Uǧurbil K, Gruetter R. In vivo 1H NMR spectroscopy of the human brain at high magnetic fields: Metabolite quantification at 4T vs. 7T. Magn Reson Med. 2009;62(4):868–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bhattacharyya PK, Lowe KJ. Macromolecule-suppressed GABA acquisition at 7T with commonly available Gaussian editing pulses. In: ISMRM.; 2018:26:1285. [Google Scholar]
- 138.Choi IY, Lee SP, Merkle H, Shen J. In vivo detection of gray and white matter differences in GABA concentration in the human brain. Neuroimage. 2006;33(1):85–93. [DOI] [PubMed] [Google Scholar]
- 139.Bhattacharyya PK, Phillips MD, Stone LA, Lowe MJ. In vivo magnetic resonance spectroscopy measurement of gray-matter and white-matter gamma-aminobutyric acid concentration in sensorimotor cortex using a motion-controlled MEGA point-resolved spectroscopy sequence. Magn Reson Imaging. 2011;29(3):374–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.