

Abstract
The mechanisms of many diseases, including central nervous system disorders, are regulated by circadian rhythms. The development of brain disorders such as depression, autism, and stroke is strongly associated with circadian cycles. Previous studies have shown that cerebral infarct volume is smaller at night (active phase) than during the day (inactive phase) in ischemic stroke rodent models. However, the underlying mechanisms remain unclear. Increasing evidence suggests that glutamate systems and autophagy play important roles in the pathogenesis of stroke. Here, we report that GluA1 expression was decreased and autophagic activity was increased in active-phase male mouse models of stroke compared with the inactive-phase models. In the active-phase model, induction of autophagy decreased the infarct volume, whereas inhibition of autophagy increased the infarct volume. Meanwhile, GluA1 expression was decreased following activation of autophagy and increased following inhibition of autophagy. We used Tat-GluA1 to uncouple p62, an autophagic adapter, from GluA1 and found that this blocked the degradation of GluA1, an effect similar to that of inhibition of autophagy in the active-phase model. We also demonstrated that knock-out of the circadian rhythm gene Per1 abolished the circadian rhythmicity of the volume of infarction and also abolished GluA1 expression and autophagic activity in wild-type (WT) mice. Our results suggest an underlying mechanism by which the circadian rhythm participates in the autophagy-dependent regulation of GluA1 expression, which influences the volume of infarction in stroke.
SIGNIFICANCE STATEMENT Circadian rhythms affect the pathophysiological mechanisms of disease. Previous studies suggested that circadian rhythms affect the infarct volume in stroke, but the underlying mechanisms remain largely unknown. Here, we demonstrate that the smaller infarct volume after middle cerebral artery occlusion/reperfusion (MCAO/R) during the active phase is related to lower GluA1 expression and activation of autophagy. The decrease in GluA1 expression during the active phase is mediated by the p62–GluA1 interaction, followed by direct autophagic degradation. In short, GluA1 is the substrate of autophagic degradation, which mainly occurs after MCAO/R during the active phase but not the inactive phase.
Keywords: autophagy, circadian rhythms, GluA1, p62, stroke
Introduction
Circadian rhythms are ubiquitous in mammals (O'Neill et al., 2011) and affect the mechanisms of many diseases, including stroke. Previous studies of ischemic stroke in middle cerebral artery occlusion/reperfusion (MCAO/R) rodent models showed that the infarct volume in the inactive phase is larger than in the active phase (Esposito et al., 2020). In rodents, daytime is the inactive phase and nighttime is the active phase. In human patients, the infarct core is smaller in strokes that occur during the day than in those that occur at night (Reidler et al., 2021).
Glutamate is the main excitatory neurotransmitter in the brain and plays a crucial role in normal physiological processes. Ischemic stroke initiates a massive release of glutamate, resulting in extensive stimulation of glutamate receptors, which can ultimately be toxic to neurons (Dirnagl et al., 1999; Moskowitz et al., 2010; J. Yang et al., 2019; Y. Wang et al., 2021). The AMPA receptors (AMPARs), NMDA receptors (NMDARs), and kainate receptors are the main excitatory glutamate receptors. Accumulating evidence suggests that extrasynaptic NMDAR-induced cytotoxicity is dependent on Ca2+ and directly provokes further Ca2+ influx through receptor-gated ion channels (Tu et al., 2010). AMPARs are heteromeric or homomeric tetramers composed of subunits GluA1–4. Many studies have reported an association between the neurotoxic properties of glutamate and the activation of AMPARs. The expression of AMPAR subunits GluA1 and GluA2 is changed after ischemic injury. For instance, GluA1 but not GluA2 levels decrease following transient global brain ischemia/reperfusion (Dos-Anjos et al., 2009). Transient ischemia/reperfusion evokes an increase in the GluA1 subunit in the spine, and the increase in AMPARs can be blocked by NMDA receptor antagonists (Y. Li et al., 2020). In addition, ischemia/reperfusion induces the internalization and degradation of AMPAR subunits GluA1 and GluA2 by the oxidative stress signaling cascade (Achzet et al., 2021a). However, whether the toxic effects of AMPARs vary with circadian rhythm with ischemic insult is unclear.
Autophagy is an important process for the degradation of cytoplasmic proteins and organelles in the lysosomal pathway (Fleming et al., 2022; Foerster et al., 2022; Gao et al., 2022; Griffey and Yamamoto, 2022). An increasing number of studies have shown that autophagy plays a critical role in ischemic stroke (Cai et al., 2021; Wu et al., 2021; Chen et al., 2022; Huang et al., 2022; H. Li et al., 2022; M. Liu et al., 2022; Thiebaut et al., 2022; Xiao et al., 2022; Z. Yang et al., 2022; Zeng et al., 2022). However, whether autophagy is beneficial or harmful for the survival of neurons in ischemic stroke is controversial. The exact role and molecular mechanisms of autophagy in ischemic stroke have not been clarified. At present, autophagy is considered a double-edged sword (P. Wang et al., 2018; Nabavi et al., 2019; Shi et al., 2021).
Many proteins participate in circadian rhythms, including period circadian regulators 1–3 (Per1–3), B-aggressive lymphoma 1, clock circadian regulator, and cryptochrome circadian regulators 1 and 2 (Z. Liu and Chu, 2013). Clock genes, including Per1, are widely expressed in the brain and therefore may be crucial regulators of neuronal activity in many neuronal circuits. Indeed, Per1 knock-out mice show alterations in many forms of neural plasticity, including drug addiction, synaptic plasticity, and learning and memory processing (Abarca et al., 2002; Rawashdeh et al., 2014). It is also closely related to autophagic activity. After deletion of the Per1 gene, autophagy is inhibited and dysfunctional proteins cannot be degraded in a timely manner in vivo, thereby reducing the tolerance of neurons to ischemia (Rami et al., 2017). These results therefore demonstrate the crucial role of Per1 in the generation of circadian rhythms and suggest that Per1 is important for the phasing of gene expression and neural activity in neurons. Building on previous research, we investigated the relationship between the circadian rhythm, autophagy, and AMPAR expression in cerebral ischemic injury.
Materials and Methods
Animals
We maintained C57BL male or female mice (eight weeks old, weight 22–24 g) on a standard 12/12 h light/dark cycle. A 250-lux light was kept on between 7 A.M. and 7 P.M. The time when the light was turned on was defined as zeitgeber time (ZT)0; thus, daytime was from ZT0 to ZT12 and nighttime was from ZT12 to ZT24. Food and water were provided ad libitum. Per1−/− male mice were generated and bred by Kunshan Cyagen Animal Co. To avoid the possibility that operators' performance decreased during nighttime surgeries, eight-week-old mice were then housed in rooms with a normal or reversed light schedule for an extra five weeks, and all surgeries were performed during the daytime by operators blinded to light schedule. These experiments were approved by the Ethics Committee of the First Hospital of Soochow University (Suzhou, China). Sample sizes were determined by power analysis during the animal ethics application.
Middle cerebral artery occlusion/reperfusion injury (MCAO/R) model
We divided the mice into four groups: sham and MCAO/R groups for both the inactive phase (ZT5–ZT7) and the active phase (ZT17–ZT19). The MCAO/R groups underwent surgical ischemia/reperfusion injury. The neck fur of the mice was shaved and the skin beneath was disinfected with iodine. The animals were anesthetized with 4% chloral hydrate. An incision was made in the neck skin to identify and fix the right external carotid artery. A small incision was made in the proximal external carotid artery, and a thread plug was passed along the internal carotid artery. The thread plug was gently pushed along the internal artery for ≥10 mm until slight resistance was felt. After 75 min, we removed the thread plug, ligated the proximal internal carotid artery, and sutured the skin. Mice in the sham group underwent a control surgery. These animals were also anesthetized with 4% chloral hydrate, followed by ligation of the external carotid artery and insertion of the thread plug to a depth of <10 mm. After 75 min, the thread plug was removed and the skin was sutured (Xu et al., 2006; C. Wang et al., 2012; Miao et al., 2015). The mortality rate within 72 h is 15–20%, and the mortality rate within one week is >50%. Therefore, our experiments did not last longer than 72 h after ischemia.
During MCAO modeling, the regional cerebral blood flow was reduced by 70–90% after insertion of the filament and recovered to >70% of baseline after removal of the filament, suggesting adequate occlusion and reperfusion. If the blood flow changes were not in this range, the mice were excluded from additional experiments. Cerebral blood flow was monitored with a Doppler flowmeter (moorVMS-LDF, Moor Instruments).
Animals were anesthetized with 4% chloral hydrate and decapitated to remove the brain after 12, 24, or 72 h. The ischemic penumbra was sectioned and prepared for assay. The whole brain was prepared for staining with 2,3,5-triphenyltetrazolium chloride (TTC) to measure infarct volume.
Western blot analysis
Brain penumbral tissues were ground and homogenized in cell lysis buffer with complete protease inhibitor cocktail (Beyotime Biotechnology). After measuring the protein concentrations, loading buffer was added and mixtures were boiled to 100°C for 10 min. Proteins were separated by SDS-PAGE with equal amounts of protein per lane. Next, the proteins were transferred to a polyvinylidene fluoride or nitrocellulose membrane (both Merck Millipore, Ltd.) for immunoblotting. After blocking with 5% BSA in TBS containing 0.5% Tween 20 (TBS-T), the membranes were incubated with primary antibodies at 4°C overnight. The following primary antibodies were used: anti-LC3B [Cell Signaling Technology (CST), 1:1000 dilution, #83506S], anti-p62 (Sigma Germany, 1:1000 dilution, P0067), anti-Atg5 (CST, 1:1000 dilution, #12994S), anti-GluA1 (Abcam, 1:1000 dilution, #183797), and anti-GluA2 (Abcam, 1:1000 dilution, #133477). Anti-β-actin (CST, 1:2000 dilution, #4970S) was used as the internal reference. After incubation with the appropriate horseradish peroxidase (HRP)-conjugated secondary antibody (anti-rabbit, CST, 1:2000 dilution, #7074S; anti-mouse, CST, 1:2000 dilution, #7076S), protein bands were detected using a chemiluminescence imaging system [Clinx Science Instruments Co, Ltd. (CSI)] and Quantity One 1-D. Analysis software (Bio-Rad Laboratories). Signal intensities were analyzed using ImageJ (National Institutes of Health).
Immunofluorescence staining
Mice were anesthetized with 4% chloral hydrate and perfused intracardially with PBS followed by 4% paraformaldehyde in PBS. After perfusion, mice were decapitated and the brains were removed, fixed in 4% paraformaldehyde, and embedded in paraffin. Brain slides (paraffin slices, 6 µm) were placed in a paraffin drying machine for 1 h, sequentially dewaxed in xylene and alcohol, repaired with sodium citrate, permeated in 0.3% Triton X-100, washed with 1× PBS three times for 5 min each, and blocked with 5% BSA. We incubated the slides with primary antibodies against anti-LC3B (CST, 1:500 dilution, #83506S) and anti-NeuN (Abcam, 1:500 dilution, #104225) at 4°C overnight. Next, slides were incubated with a secondary antibody (AlexaFluor 488-conjugated anti-mouse or AlexaFluor 555-conjugated anti-rabbit) for 1 h at room temperature, followed by counterstaining with 4′,6-diamidino-2-phenylindole (DAPI; SouthernBiotech; #0100-20) for 10 min (Jiang et al., 2018). Images were obtained using a Nikon DSLR fluorescence scanning microscope (Nikon).
Surface receptor cross-linking with BS3
Protein cross-linking was conducted according to previous reports (Conrad et al., 2008; Mao et al., 2009; Van Dolah et al., 2011; Lu et al., 2014). Mice were decapitated and their brain penumbras were rapidly removed and collected in Eppendorf tubes (Eppendorf). Then, artificial cerebrospinal fluid spiked with 2 mm bis(sulfosuccinimidyl)suberate (BS3; AMOBE Biotechnology) was added to the tubes. The tissues were mashed and cross-linked for 180 min at 4°C with gentle agitation. This reaction was terminated by the addition of 100 mm glycine, followed by incubation at 4°C for 15 min. We subjected these samples to short-time centrifugation. The supernatant was resuspended in ice-cold cell lysis buffer. Samples were homogenized by sonication for 5 s and centrifuged at 12,000 × g for 15 min at 4°C. The supernatant was collected and stored at −80°C for Western blot (WB) analysis. The signal intensities for the surface, intracellular, and total (surface + intracellular) fractions were normalized to β-actin levels.
Co-immunoprecipitation
Supernatants (500 μg total protein per specimen) were incubated in immunoprecipitation buffer with anti-p62 (4 µg; Sigma) and anti-normal IgG (4 µg; CST, 1:1000 dilution, #2729S) for 3 h at 4°C, followed by incubation with 20 µl protein A/G agarose (Santa Cruz Biotechnology, #sc-2003) for 24 h at 4°C. The immunoprecipitation buffer (Beyotime Biotechnology) contained 20 mm Tris at pH 7.5, 150 mm of 1% Triton X-100, 1 mm ethylene glycol-bis(β-aminoethylether)-N,N,N',N'-tetraacetic acid, and 1 mm phenylmethylsulfonyl fluoride. Samples were boiled in 2× SDS loading buffer for 15 min and separated via SDS-PAGE. Anti-normal IgG was used as a negative control. It can bind to agarose but does not recognize specific antigens.
Drug administration
We prepared a stock solution by dissolving 10 mg rapamycin [RAPA; Medchem Express (MCE), Monmouth Junction] in 1 ml dimethyl sulfoxide (DMSO; BioFroxx GmbH, #1084ML) and stored it at −20°C. Mice received intraperitoneal RAPA injections at a dose of 10 mg/kg, 1 h before MCAO/R, with equal volumes of the solvent in all experiments. We prepared another stock solution by dissolving 50 mg of 3-methyladenine (3-MA; MCE, Monmouth Junction) in 10 ml of 0.9% NaCl (Baxter Medical Supplies Co, Ltd.) and stored the solution at −20°C. Mice received intraperitoneal injections of 3-MA (15 mg/kg) 1 h before MCAO/R. The control group was treated with equal volumes of 0.9% NaCl in all experiments.
LV-Atg5-shRNA transfection, production, and stereotaxic injection
Mouse brains were injected with Atg5 short hairpin RNA lentivirus (LV-Atg5-shRNA; Shanghai Genechem Co, Ltd.), with green fluorescent protein (GFP) as a flag. Lentivirus was administered by injection in the right hemisphere, ipsilateral to the MCAO/R site, at three points. Point 1 was 0 mm anterior–posterior and 2 mm medial–lateral from the bregma; point 2 was −2 mm anterior–posterior and 3 mm medial–lateral from the bregma; and point 3 was −4 mm anterior–posterior and 2 mm medial–lateral from the bregma. Injection depth was 1.5 mm dorsal–ventral from the skull, as described previously (Hui et al., 2016). A total of 1.2 μl (0.4 μl per point) of GFP-labeled lentivirus suspension (1 × 109 TU/ml) was injected using a 10-μl Hamilton syringe (Hamilton Company) at a rate of 0.2 μl/min, and the needle was retained in place for an additional 10 min. One week after lentivirus pretreatment, we established an MCAO/R mouse model. Control mice were anesthetized with 4% chloral hydrate and their brains were perfused with 50 ml saline and then immediately harvested and frozen at −20°C until further analysis.
Polypeptide drug design and treatment
The designed polypeptide drugs were dissolved in 0.9% NaCl according to the instructions. The Tat-GluA1 (Tat-DISPRSLSGR, 10 mg/kg) and Tat-scr-GluA1 (Tat-IFNSDGAFMF, 10 mg/kg) were injected intraperitoneally once a day for one week before establishing the MCAO/R model. Tat-GluA1 was designed according to the GluA1 binding sequence, and the sequences close to the binding domains were synthesized as scrambled peptides. All peptides crossed the cell membrane by the domain of the HIV-1T protein (YGRKKRRQRRR; Ren et al., 2013). All peptides were manufactured by GL Biochem, Ltd.
Sample staining and calculation of infarct volume
At 12, 24, or 72 h after MCAO/R, the mouse brains were removed and placed in a freezer at −20°C for 15 min. Continuous 2-mm-thick coronal sections from the frontal to the occipital lobe were created using a sharp blade. Four brain slices were put in a Petri dish containing 0.5% TTC (Jiancheng Chemical Reagent) and incubated at 37°C. Brain slices were dyed for 20 min in the dark and intermittently inverted to ensure uniform staining. The volume of the infarct area, which appeared white in color, was measured in mm3 using ImageJ software with the morphometric method, as described previously.
Experimental design and statistical analysis
We performed all data analyses using SPSS 23 (Armonk) with a paired, two-tailed Student's t test, two-way ANOVA with Bonferroni's multiple comparison test, or one-way ANOVA with Tukey's post hoc adjustment test. Data are shown as the mean ± SEM. Normality tests were performed to confirm data were normally distributed. p < 0.05 was considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001). Quantification and all statistical analyses were performed by an observer who was blind to the experimental conditions.
Results
MCAO/R induced differences in infarct volume, GluA1/2 expression, and autophagic activity in active (ZT5–ZT7) and inactive (ZT17–ZT19) phases
To evaluate the influence of circadian rhythm on brain damage in stroke, we measured the infarct volume at 12, 24, and 72 h in male mice operated during the inactive phase (ZT5–ZT7) and the active phase (ZT17–ZT19; Fig. 1A). We did not find any significant difference in 12 h infarct volume between the ZT5–ZT7 and ZT17–ZT19 groups (F(5,30) = 25.855, p = 0.072, two-way ANOVA; Fig. 1B). However, 24 h (F(5,30) = 25.855, p = 0.008, two-way ANOVA; Fig. 1B) and 72 h (F(5,30) = 25.855, p = 0.002, two-way ANOVA; Fig. 1B) infarct volumes were larger in ZT5–ZT7 mice than in ZT17–ZT19 mice (Fig. 1B). These results suggest infarct volumes grew between 12 h and 72 h after occlusion, but the rate of infarct growth was lower in ZT17–ZT19 mice compared with ZT5–ZT7 mice. Since there was a difference in 24 h infarct volume between ZT5–ZT7 and ZT17–ZT19 male mice, we assessed whether such difference also existed in female mice. As expected, at 24 h postischemia, the infarct volumes of female mice were also smaller in the ZT17–ZT19 group than in the ZT5–ZT7 group (t(10) = 2.724, p = 0.0214, paired t test; Fig. 1C). These findings suggest circadian rhythm could affect stroke infarct volume without gender difference.
Figure 1.

Mouse brain infarct volume after cerebral ischemia. A, Overview of zeitgeber time (ZT). ZT0–ZT12 is the inactive phase and ZT12–24 is the active phase. MCAO/R was established at ZT5–ZT7 and ZT17–ZT19. B, TTC staining analysis of infarct volume at 12, 24, and 72 h post-MCAO at ZT5–ZT7 and ZT17–ZT19 in male mice (**p < 0.01, n = 6 mice per group). C, TTC staining analysis of infarct volume at 24 h post-MCAO at ZT5–ZT7 and ZT17–ZT19 in female mice (*p < 0.05, n = 6 mice per group). Data are shown as the mean ± SEM.
The role of glutamate signaling through AMPAR changes over time in ischemia stroke (Clarkson et al., 2011). To detect changes in AMPAR levels during cerebral ischemia, we collected penumbral protein from the mouse brains at ZT5–ZT7 and ZT17–ZT19. WB analysis of the penumbral protein supernatant showed GluA1 levels were lower in the ZT17–ZT19 (MCAO/R)24 h group than in the ZT5–ZT7 (MCAO/R)24 h (F(3,20) = 35.584, p < 0.0001, two-way ANOVA; Fig. 2A) and ZT17–ZT19 sham groups (F(3,20) = 35.584, p < 0.0001, two-way ANOVA; Fig. 2A). Figure 2B shows that GluA2 levels were higher in the ZT17–ZT19 group than in the ZT5–ZT7 group in both the sham group (F(3,20) = 21.443, p < 0.0001, two-way ANOVA; Fig. 2B) and the (MCAO/R)24 h group (F(3,20) = 21.443, p < 0.0001, two-way ANOVA; Fig. 2B).
Figure 2.
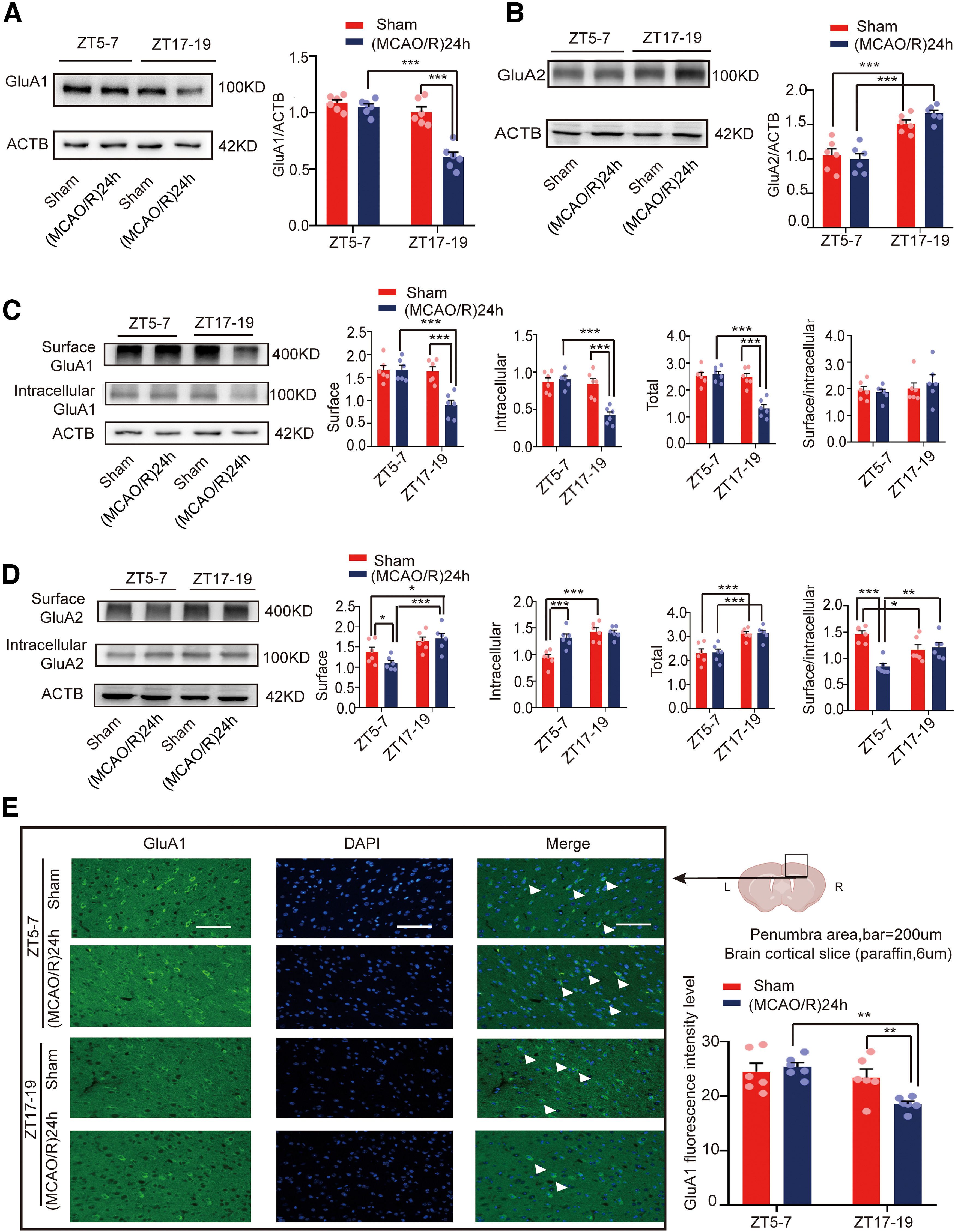
Mouse brain penumbra GluA1/2 expression in circadian rhythm in the sham and (MCAO/R)24 h groups. A, B, Western blot analysis and quantification of GluA1/2 at ZT5–ZT7 and ZT17–ZT19 in the sham and (MCAO/R)24 h groups (***p < 0.001, n = 6 mice per group). C, Cell surface, intracellular, and total (surface + intracellular) GluA1 levels and the surface/intracellular ratio at ZT5–ZT7 and ZT17–ZT19 (***p < 0.001, n = 6 mice per group). D, Cell surface, intracellular, and total (surface + intracellular) GluA2 levels and the surface/intracellular ratio at ZT5–ZT7 and ZT17–ZT19 (*p < 0.05, **p < 0.01, ***p < 0.001, n = 6 mice per group). E, Immunofluorescence analysis showing that GluA1 expression was significantly decreased in the (MCAO/R)24 h group at ZT17–ZT19 compared with the ZT5–ZT7 (MCAO/R)24 h and ZT17–ZT19 sham groups (**p < 0.01, n = 6 mice per group; scale bar = 200 µm). Data are shown as the mean ± SEM.
AMPAR incorporation into and removal from the cell surface is one of the main molecular mechanisms underlying AMPAR signaling. AMPAR has been suggested to be trafficked to the cell surface in the form of GluA1/2 (Adesnik and Nicoll, 2007; Gray et al., 2007). Thus, it makes more sense to detect the AMPAR levels on the cell membrane. Therefore, brain penumbral supernatant was extracted using the BS3 cross-linking method, which selectively cross-links surface membrane-bound receptors to form high-molecular-weight aggregates that could be readily separated from unlinked intracellular receptors by gel electrophoresis. Figure 2C shows that cell surface (F(3,20) = 13.42, p < 0.0001, two-way ANOVA; Fig. 2C), intracellular (F(3,20) = 16.835, p < 0.0001, two-way ANOVA; Fig. 2C), and total (surface + intracellular; F(3,20) = 21.219, p < 0.0001, two-way ANOVA; Fig. 2C) GluA1 levels in the (MCAO/R)24 h group at ZT17–ZT19 were significantly lower compared with the ZT5–ZT7 (MCAO/R)24 h and ZT17–ZT19 sham groups. No changes were found in the surface/intracellular ratio between the (MCAO/R)24 h group at ZT17–ZT19 and the ZT5–ZT7 (MCAO/R)24 h group or the ZT17–ZT19 sham group. However, Figure 2D shows that the cell surface GluA2 levels were increased in the ZT17–ZT19 (MCAO/R)24 h group compared with the ZT5–ZT7 (MCAO/R)24 h group (F(3,20) = 7.234, p < 0.0001, two-way ANOVA; Fig. 2D). The intracellular GluA2 levels were increased in the ZT5–ZT7 (MCAO/R)24 h group compared with the ZT5–ZT7 sham group (F(3,20) = 13.18, p < 0.0001, two-way ANOVA; Fig. 2D). In addition, intracellular GluA2 levels were increased in the ZT17–ZT19 sham group compared with the ZT5–ZT7 sham groups (F(3,20) = 13.18, p < 0.0001, two-way ANOVA; Fig. 2D). Total GluA2 levels in the sham and (MCAO/R)24 h groups at ZT17–ZT19 were significantly increased compared with the sham (F(3,20) = 11.925, p < 0.0001, two-way ANOVA; Fig. 2D) and (MCAO/R)24 h (F(3,20) = 11.925, p < 0.0001, two-way ANOVA; Fig. 2D) groups at ZT5–ZT7. Finally, the surface/intracellular ratio of GluA2 was decreased in the ZT5–ZT7 (MCAO/R)24 h group compared with the ZT5–ZT7 sham group (F(3,20) = 10.779, p < 0.0001, two-way ANOVA; Fig. 2D) and the ZT17–ZT19 (MCAO/R)24 h group (F(3,20) = 10.779, p = 0.003, two-way ANOVA; Fig. 2D). Moreover, immunofluorescence analysis showed that GluA1 levels in the (MCAO/R)24 h group at ZT17–ZT19 were significantly lower than in the ZT5–ZT7 (MCAO/R)24 h (F(3,20) = 6.444, p = 0.001, two-way ANOVA; Fig. 2E) and ZT17–ZT19 sham groups (F(3,20) = 6.444, p = 0.009, two-way ANOVA; Fig. 2E).
To detect autophagic activity during cerebral ischemia, we determined the expression of the autophagy marker proteins light chain 3 (LC3) and p62. Figure 3A shows that the LC3BII/LC3BI ratio was higher in the (MCAO/R)24 h group at ZT17–ZT19 than in the ZT5–ZT7 (MCAO/R)24 h (F(3,20) = 37.322, p < 0.0001, two-way ANOVA; Fig. 3A) and ZT17–ZT19 sham (F(3,20) = 37.322, p < 0.0001, two-way ANOVA; Fig. 3A) groups. Conversely, Figure 3B shows that the relative expression of p62 in the (MCAO/R)24 h group at ZT17–ZT19 was significantly lower than in the ZT5–ZT7 (MCAO/R)24 h (F(3,20) = 44.113, p < 0.0001, two-way ANOVA; Fig. 3B) and ZT17–ZT19 sham (F(3,20) = 44.113, p < 0.0001, two-way ANOVA; Fig. 3B) groups. Moreover, immunofluorescence analysis showed an increase in the proportion of LC3B colocalized with NeuN in cortical neurons in the ZT17–ZT19 (MCAO/R)24 h group compared with the ZT5–ZT7 (MCAO/R)24 h (F(3,20) = 9.889, p < 0.0001, two-way ANOVA; Fig. 3C) and ZT17–ZT19 sham (F(3,20) = 9.889, p < 0.0001, two-way ANOVA; Fig. 3C) groups. These results suggest that GluA1 expression was decreased and autophagic activity was increased 24 h after stroke onset during the active phase.
Figure 3.
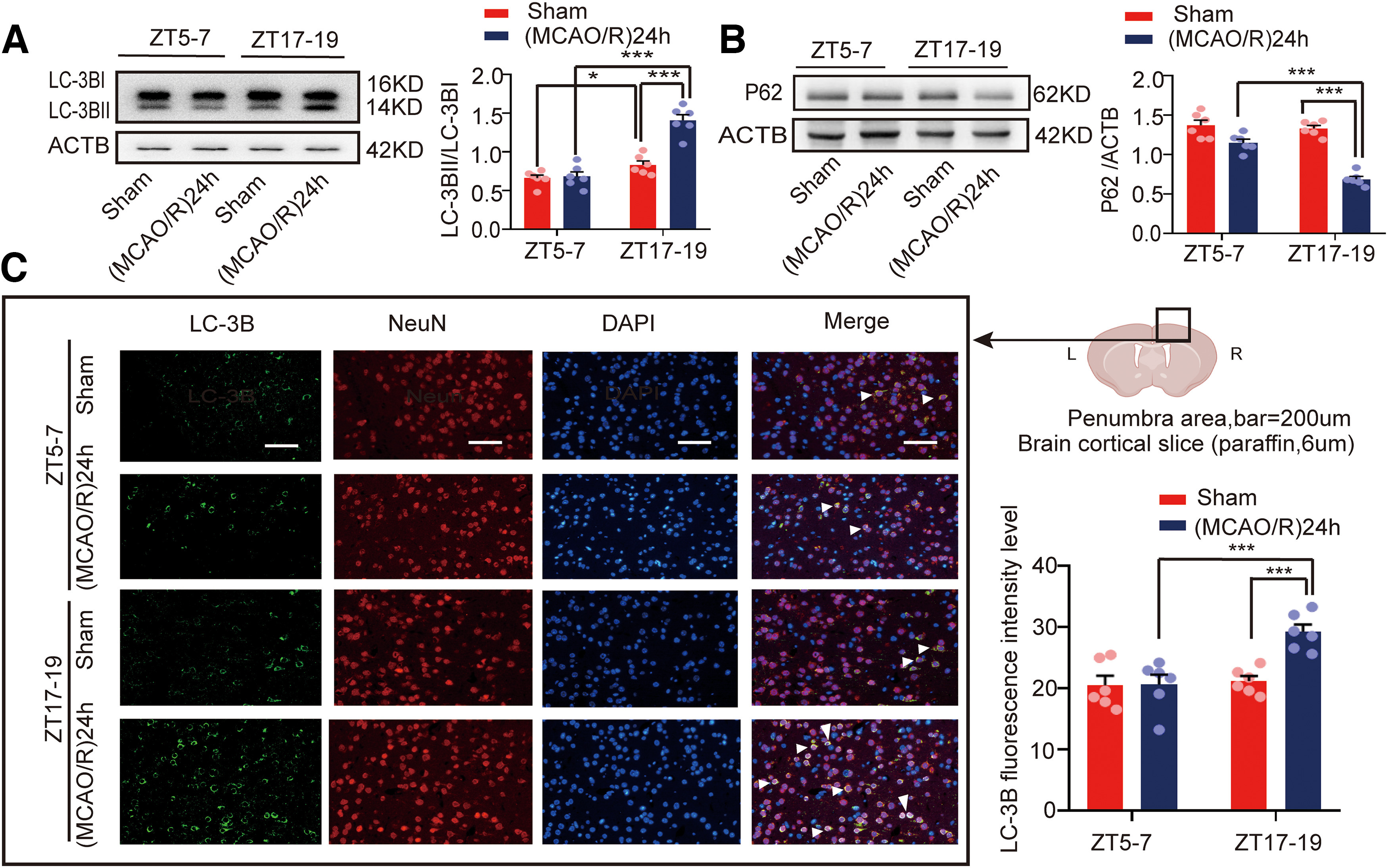
Mouse brain penumbra autophagic activity in circadian rhythm in the sham and (MCAO/R)24 h groups. A, B, Western blot analysis and quantification of LC3BII/LC3BI (***p < 0.001, *p < 0.05, n = 6 mice per group) and p62 (***p < 0.001, n = 6 mice per group) levels in penumbra tissue in the ZT17–ZT19 and ZT5–ZT7 groups. C, Immunofluorescence analysis showed that LC3B expression was significantly increased in the (MCAO/R)24 h group at ZT17–ZT19 compared with the ZT5–ZT7 (MCAO/R)24 h and ZT17–ZT19 sham groups (***p < 0.001, n = 6 mice per group; scale bar = 200 µm). Data are shown as the mean ± SEM.
Because tissue was collected at 24 h after MCAO/R, i.e., in the same circadian phase as the stroke, we also analyzed GluA1 expression and autophagic activity in the opposing circadian phase. Penumbral protein was collected from the mouse brains at 12 h after stroke onset in the ZT5–ZT7 and ZT17–ZT19 groups. GluA1 levels were not different between the ZT5–ZT7 (MCAO/R)12 h and ZT17–ZT19 (MCAO/R)12 h groups (Fig. 4A). However, the LC3BII/LC3BI ratio was higher in the (MCAO/R)12 h group at ZT17–ZT19 than in the ZT5–ZT7 (MCAO/R)12 h group (F(3,20) = 4.372, p = 0.005, two-way ANOVA; Fig. 4B). On the contrary, Figure 4C shows that the relative expression of p62 in the ZT17–ZT19 (MCAO/R)12 h group was significantly lower than in the ZT5–ZT7 (MCAO/R)12 h (F(3,20) = 4.373, p = 0.005, two-way ANOVA; Fig. 4C) and ZT17–ZT19 sham (F(3,20) = 4.373, p = 0.015, two-way ANOVA; Fig. 4C) groups. These findings indirectly suggest there may be an interval between the activation of autophagy and the degradation of GluA1 within 12 h after ischemia.
Figure 4.

Mouse brain penumbra GluA1 expression and autophagic activity in circadian rhythm in the sham and (MCAO/R)12 h groups. A, Western blot analysis and quantification of GluA1 at ZT5–ZT7 and ZT17–ZT19 in the sham and (MCAO/R)12 h groups (n = 6 mice per group). B, C, Western blot analysis and quantification of LC3BII/LC3BI (**p < 0.01, n = 6 mice per group) and p62 (**p < 0.01, *p < 0.05, n = 6 mice per group) levels in penumbra tissue in the ZT17–ZT19 and ZT5–ZT7 groups. Data are shown as the mean ± SEM.
Regulation of autophagy influenced GluA1 expression
To determine whether autophagy regulated the expression of GluA1 24 h after stroke onset, we employed RAPA to induce autophagy and 3-MA to inhibit it. The results showed that the LC3BII/LC3BI ratio was increased in the RAPA group (F(5,30) = 15.51, p = 0.0237, one-way ANOVA; Fig. 5A) and decreased in the 3-MA group (F(5,30) = 15.51, p = 0.0026, one-way ANOVA; Fig. 5A) compared with the control group at ZT17–ZT19. The relative expression level of p62 was decreased in the RAPA group (F(5,36) = 16.69, p = 0.0069, one-way ANOVA; Fig. 5B) and increased in the 3-MA group (F(5,36) = 16.69, p = 0.0015, one-way ANOVA; Fig. 5B) compared with the control group at ZT17–ZT19. These results suggest that intraperitoneal injection of RAPA increased autophagic activity, whereas 3-MA inhibited it. In addition, we tested the effects of the drugs during active-phase stroke to determine differences in infarct volume. Staining with TTC revealed that infarct volume was smaller in the RAPA group than in the DMSO control group (F(3,20) = 18.25, p = 0.0258, one-way ANOVA; Fig. 5C), and infarct volume was larger in the 3-MA group than in the 0.9% NaCl group (F(3,20) = 18.25, p = 0.0017, one-way ANOVA; Fig. 5C). Moreover, statistical analysis revealed that infarct volume was negatively correlated with the LC3BII/LC3BI ratio (R2 = 0.673, p = 0.001; Fig. 5D).
Figure 5.

Mouse brain infarct volume and LC3B, p62, and GluA1/2 expression in penumbra tissue in the (MCAO/R)24 h group at ZT17–ZT19 following regulation with RAPA and 3-MA. A, B, Western blot analysis and quantification of levels of LC3BII/LC3BI (*p < 0.05, **p < 0.01, n = 6 mice per group) and p62 (*p < 0.05, **p < 0.01, n = 7 mice per group). C, RAPA (10 mg/kg) or 3-MA (15 mg/kg) was administered 1 h before MCAO/R model establishment at ZT17–ZT19. Mice in the sham group received an equal amount of solvent. TTC staining analysis of infarct volume (*p < 0.05, **p < 0.01, n = 6 mice per group). D, Linear regression analysis was conducted to determine infarct volume and LC3BII/LC3BI expression (**p < 0.01). E, Cell surface, intracellular, and total (surface + intracellular) GluA1/2 levels and the surface/intracellular ratio after RAPA treatment (**p < 0.01, n = 6 mice per group). F, Cell surface, intracellular, and total (surface + intracellular) GluA1/2 levels and the surface/intracellular ratio after 3-MA treatment (*p < 0.05, **p < 0.01, n = 6 mice per group). G, Western blot analysis and quantification of GluA1 levels (*p < 0.05, **p < 0.01, n = 6 mice per group). Data are shown as the mean ± SEM.
We further evaluated the effects of activation or inhibition of autophagy on AMPAR expression in the penumbra during active-phase stroke. We determined the AMPAR distribution with the BS3 protein cross-linking assay. Surface (t(10) = 3.834, p = 0.0033, paired t test; Fig. 5E), intracellular (t(10) = 3.698, p = 0.0041, paired t test; Fig. 5E), and total (t(10) = 4.302, p = 0.0016, paired t test; Fig. 5E) GluA1 levels, but not the GluA1 surface/intracellular ratio, were significantly decreased in the RAPA group compared with the control group. By contrast, surface, intracellular, and total GluA2 levels and the GluA2 surface/intracellular ratio showed no significant difference between these two groups (Fig. 5E). Moreover, surface (t(12) = 3.384, p = 0.0054, paired t test; Fig. 5F) and total (t(12) = 2.278, p = 0.0418, paired t test; Fig. 5F) GluA1 levels but not intracellular GluA1 levels or the GluA1 surface/intracellular ratio at ZT17–ZT19 were significantly increased in the 3-MA group compared with the 0.9% NaCl group. However, surface, intracellular, and total GluA2 levels and the GluA2 surface/intracellular ratio did not differ between the 3-MA and 0.9% NaCl groups (Fig. 5F). Furthermore, WB results showed that GluA1 levels in the RAPA (F(5,30) = 14.01, p = 0.0135, one-way ANOVA; Fig. 5G) and 3-MA (F(5,30) = 14.01, p = 0.0019, one-way ANOVA; Fig. 5G) groups were consistent with the total GluA1 expression from BS3 cross-linking experiments (Fig. 5E,F).
To assess the generalizability of these results, we conducted a series of stroke experiments during the inactive phase (ZT5–ZT7). Similarly, the results showed that RAPA induced autophagy [F(5,30) = 11.4, p = 0.007, one-way ANOVA, for LC3BII/LC3BI (Fig. 6A); F(5,30) = 19.19, p = 0.026, one-way ANOVA, for p62 (Fig. 6B)], whereas 3-MA inhibited it [F(5,30) = 11.4, p = 0.0027, one-way ANOVA, for LC3BII/LC3BI (Fig. 6A); F(5,30) = 19.19, p = 0.0186, one-way ANOVA, for p62 (Fig. 6B)]. In addition, RAPA decreased the expression of GluA1 (F(5,30) = 14.24, p = 0.0334, one-way ANOVA; Fig. 6C), whereas 3-MA increased it (F(5,30) = 14.24, p = 0.0018, one-way ANOVA; Fig. 6C). To rule out the influence of ischemia itself, we further examined GluA1 expression regulated by RAPA or 3-MA in nonischemic conditions at ZT5–ZT7. Likewise, under these different conditions, RAPA decreased the expression of GluA1 (F(4,25) = 14.87, p = 0.0335, one-way ANOVA; Fig. 6D), whereas 3-MA increased it compared with corresponding control groups (F(4,25) = 14.87, p = 0.0054, one-way ANOVA; Fig. 6D). As expected, in nonischemic conditions, RAPA also induced autophagy [F(4,25) = 13.23, p = 0.0109, one-way ANOVA, for LC3BII/LC3BI (Fig. 6E); F(4,25) = 13.09, p = 0.0127, one-way ANOVA, for p62 (Fig. 6F)], whereas 3-MA inhibited it [F(4,25) = 13.23, p = 0.0466, one-way ANOVA, for LC3BII/LC3BI (Fig. 6E); F(4,25) = 13.09, p = 0.0036, one-way ANOVA, for p62 (Fig. 6F)].
Figure 6.
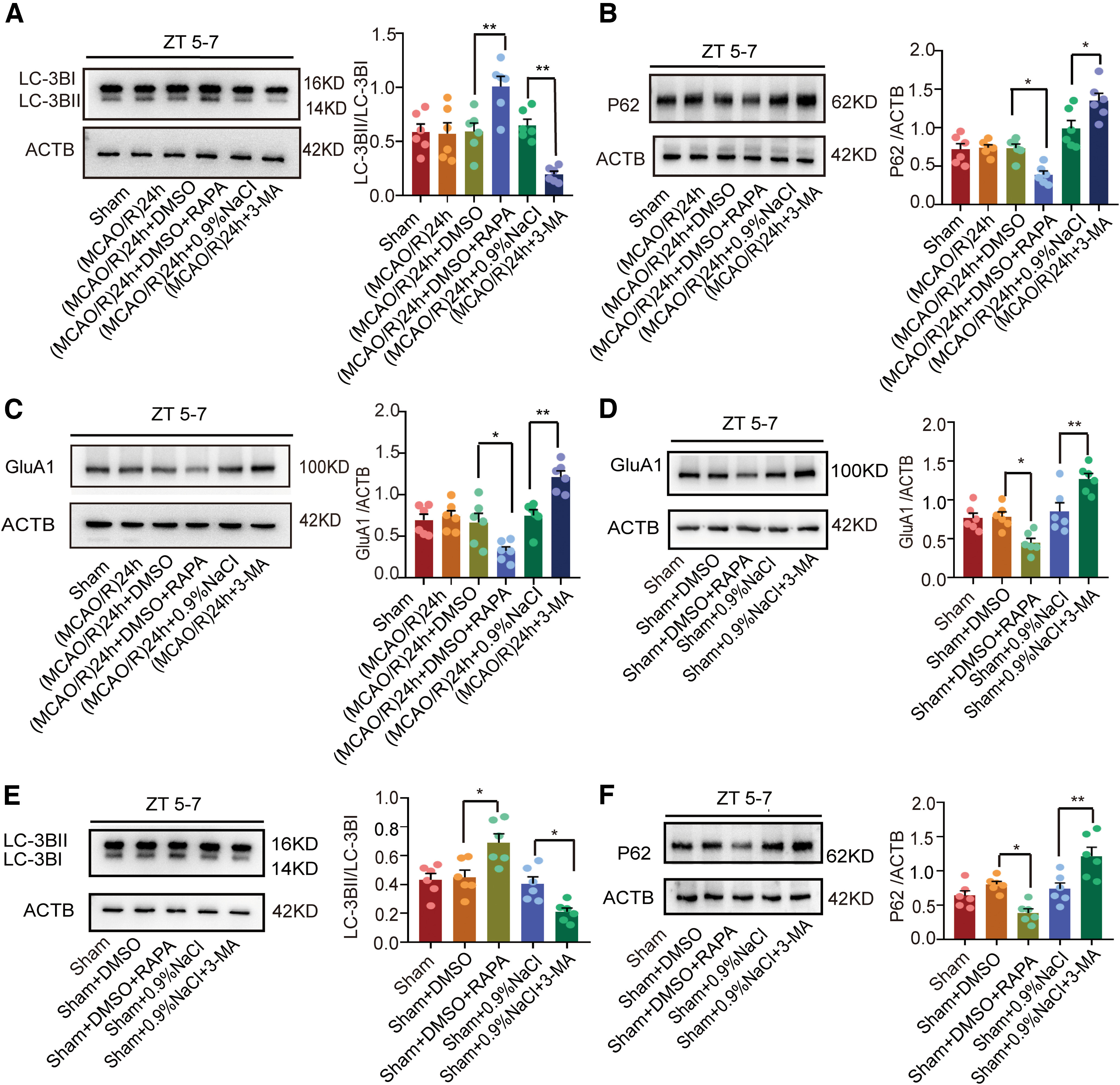
LC3B, p62, and GluA1 expression after administration of RAPA and 3-MA in (MCAO/R)24 h ZT5–ZT7 and nonischemic conditions. A, B, Western blot analysis and quantification of LC3BII/LC3BI (**p < 0.01, n = 6 mice per group) and p62 (*p < 0.05, n = 6 mice per group) expression in penumbra tissue at ZT5–ZT7. C, Western blot analysis and quantification of GluA1 in penumbra tissue at ZT5–ZT7 (*p < 0.05, **p < 0.01, n = 6 mice per group). D, Western blot analysis and quantification of GluA1 expression (*p < 0.05, **p < 0.01, n = 6 mice per group). E, F, Western blot analysis and quantification of LC3BII/LC3BI (*p < 0.05, n = 6 mice per group) and p62 (*p < 0.05, **p < 0.01, n = 6 mice per group) expression. Data are shown as the mean ± SEM.
Taken together, these data suggest that regulation of GluA1 by autophagy is a common phenomenon, in both ischemic and nonischemic states and in both the active and inactive phases.
Inhibition of autophagy increased GluA1 expression
To exclude the possibility that pharmacological methods were nonspecific, we used LV-Atg5-shRNA to knock down Atg5 and thereby inhibit autophagy. The lentivirus was injected intracranially, and the viral flag GFP was successfully detected in the cortex (Fig. 7A). First, we examined the effect during active-phase stroke. WB results showed that the levels of the autophagy-related protein Atg5 were significantly decreased in the MCAO/R + LV-Atg5-shRNA group compared with the MCAO/R + vector group (F(3,24) = 10.05, p = 0.0002, one-way ANOVA; Fig. 7B). Meanwhile, compared with the vector group, the expression of the autophagic protein LC3 was decreased (F(3,20) = 9.921, p = 0.0045, one-way ANOVA; Fig. 7C) and p62 expression was increased (F(3,20) = 10.420, p = 0.0021, one-way ANOVA; Fig. 7D).
Figure 7.

Mouse brain infarct volume and LC3B, p62, and GluA1/2 expression in penumbra tissue in the (MCAO/R)24 h group at ZT17–ZT19 after Atg5 knock-down. A, One week after LV-Atg5-shRNA or vector was injected into the cortex. Viral flag GFP showed successful injection of the virus (scale bar = 50 and 400 µm). B, Atg5 protein levels were decreased in the LV-Atg5-shRNA group compared with the vector group one week after LV-Atg5-shRNA administration (**p < 0.01, n = 7 mice per group). C, D, Western blot analysis and quantification of LC3BII/LC3BI (**p < 0.01, n = 6 mice per group) and p62 (**p < 0.01, n = 6 mice per group). E, Cell surface, intracellular, and total (surface + intracellular) GluA1/2 levels and surface/intracellular ratios (**p < 0.01, n = 6 mice per group). F, Western blot analysis and quantification of GluA1 expression (**p < 0.01, n = 8 mice per group). G, TTC staining analysis of infarct volume (*p < 0.05, n = 6 mice per group). Data are shown as the mean ± SEM.
To further verify whether autophagy inhibition by Atg5 knock-down increased GluA1 expression, we performed a BS3 cross-linking experiment. The results showed that surface (t(10) = 4.382, p = 0.0014, paired t test; Fig. 7E) and total (t(10) = 3.909, p = 0.0029, paired t test; Fig. 7E) GluA1 levels but not intracellular GluA1 levels or the surface/intracellular ratio were higher in the MCAO/R + LV-Atg5-shRNA group than in the vector group. However, as illustrated in the same figure, there was no difference between the two groups in surface, intracellular, and total GluA2 levels and surface/intracellular ratio. WB results also showed that GluA1 expression was higher in the MCAO/R + LV-Atg5-shRNA group than in the vector group (F(3,28) = 9.089, p = 0.0048, one-way ANOVA; Fig. 7F). Moreover, infarct volume was larger in the LV-Atg5-shRNA group compared with the vector group during active-phase stroke (t(10) = 3.146, p = 0.0104, paired t test; Fig. 7G).
Subsequently, we conducted similar tests to determine whether autophagy regulates GluA1 expression during the inactive phase (ZT5–ZT7). As expected, GluA1 expression was increased in the MCAO/R + LV-Atg5-shRNA group compared with the vector group (F(3,20) = 8.797, p = 0.0025, one-way ANOVA; Fig. 8A). In agreement with the 3-MA experiments, expression of the autophagic protein p62 was increased (F(3,20) = 7.063, p = 0.0146, one-way ANOVA; Fig. 8B) and LC3 expression was decreased (F(3,20) = 4.688, p = 0.0389, one-way ANOVA; Fig. 8C) after Atg5 knock-down. To rule out the influence of ischemia itself, similar to the above pharmacological intervention experiments, we detected GluA1 expression after Atg5 knock-down, which decreased LC3 expression (F(2,15) = 6.82, p = 0.023, one-way ANOVA; Fig. 8D) and increased p62 expression (F(2,15) = 11.64, p = 0.0053, one-way ANOVA; Fig. 8E), in nonischemic ZT5–ZT7 mice. The results showed that Atg5 knock-down still upregulated the expression of GluA1 in nonischemic conditions (F(2,15) = 10.87, p = 0.009, one-way ANOVA; Fig. 8F). These data further proved that autophagy regulates GluA1 levels in both active and inactive circadian phases, in both ischemic and nonischemic conditions.
Figure 8.
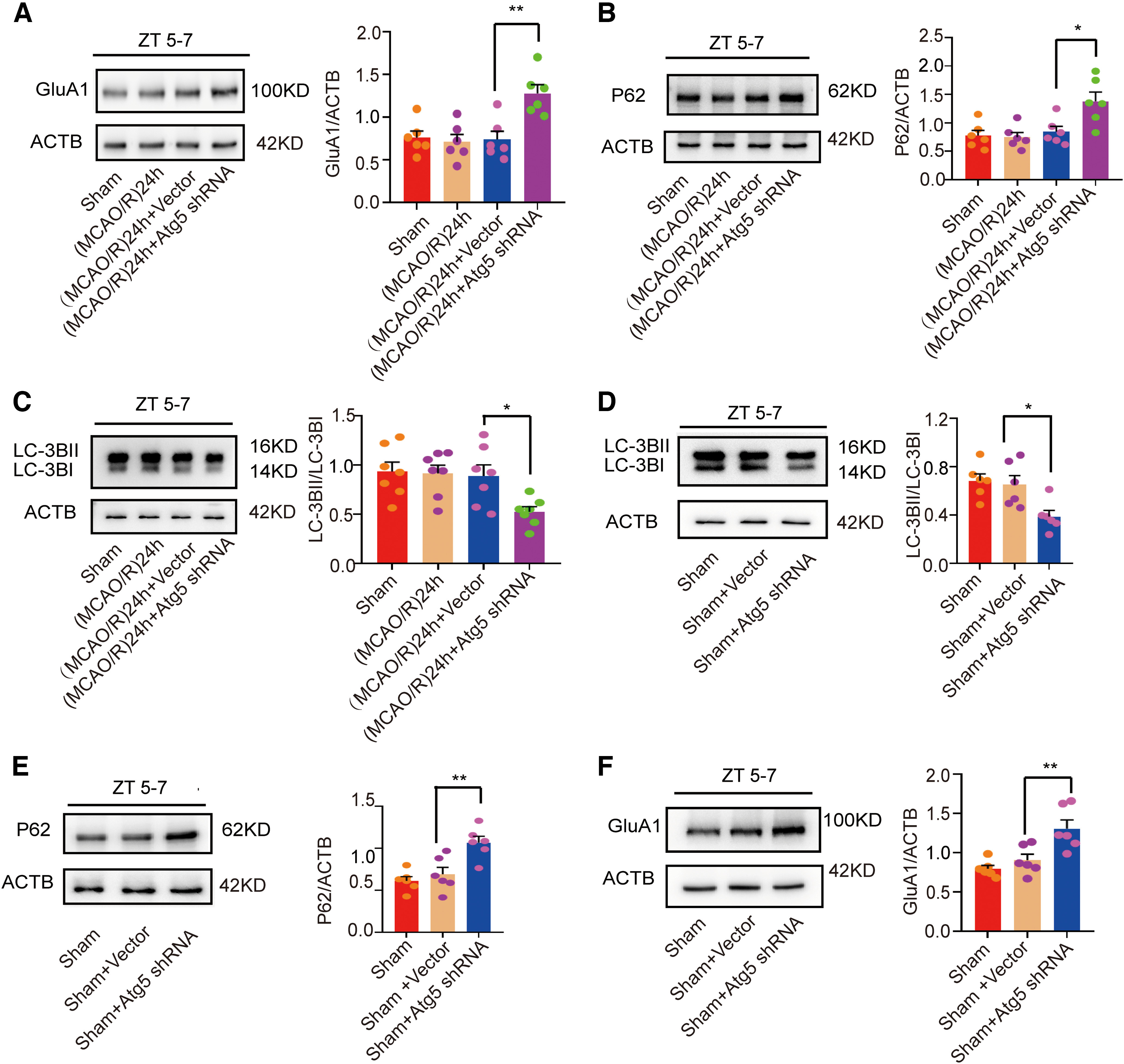
LC3B, p62, and GluA1 expression after administration of LV-Atg5-shRNA in the (MCAO/R)24 h ZT5–ZT7 group and under nonischemic conditions. A, Western blot analysis and quantification of GluA1 expression in penumbra tissue at ZT5–ZT7 (**p < 0.01, n = 6 mice per group). B, C, Western blot analysis and quantification of p62 (*p < 0.05, n = 6 mice per group) and LC3BII/LC3BI (*p < 0.05, n = 7 mice per group) expression in penumbra tissue at ZT5–ZT7. D, E, Western blot analysis and quantification of LC3BII/LC3BI (*p < 0.05, n = 6 mice per group) and p62 (**p < 0.01, n = 6 mice per group) expression in the sham group. F, Western blot analysis and quantification of GluA1 expression in the sham groups (**p < 0.01, n = 6 mice per group). Data are shown as the mean ± SEM.
Disruption of GluA1–p62 interaction blocked GluA1 degradation and aggravated brain damage
p62 functions as an autophagic adapter in mammals and has been shown to be active in autophagic degradation of misfolded proteins (Komatsu et al., 2007). In addition, it interacts with AMPAR subunits and influences their trafficking and phosphorylation (Jiang et al., 2009). To verify whether interactions between p62 and GluA1 were changed in the ZT17–ZT19 (MCAO/R)24 h (active-phase stroke) group, we performed a co-immunoprecipitation experiment. Figure 9A illustrates that the interaction between p62 and GluA1 was increased in the (MCAO/R)24 h group compared with the sham group (t(10) = 3.213, p = 0.0093, paired t test; Fig. 9A). We further evaluated whether direct interactions between p62 and GluA1 were required for the autophagy-induced decrease in GluA1 expression. To determine the effect of increased binding of p62 to GluA1, we used the specific blocking peptide Tat-GluA1 (Tat-DISPRSLSGR) to block the p62–GluA1 interaction. Tat-scrambled GluA1 (Tat-Scr-GluA1) was used as a control (Tat-IFNSDGAFMF; Ren et al., 2013). The polypeptides were administered intraperitoneally once a day for one week. GFP was used as a flag. The drug was successfully absorbed in the brain (Fig. 9B). As expected, the results showed Tat-GluA1 significantly inhibited the binding between GluA1 and p62 (F(3,20) = 10.78, p = 0.0051, one-way ANOVA; Fig. 9C). Moreover, we evaluated the effects of Tat-GluA1 on brain damage. The results showed that infarct volume was larger in the Tat-GluA1 group than in the Tat-Scr-GluA1 group at ZT17–ZT19 (t(10) = 4.088, p = 0.0022, paired t test; Fig. 9D).
Figure 9.

Mouse brain infarct volume and LC3B, p62, and GluA1/2 expression in penumbra tissue of the (MCAO/R)24 h ZT17–ZT19 group after injection of Tat-GluA1 or Tat-Scr-GluA1. A, Co-immunoprecipitation of p62 and GluA1 at ZT17–ZT19 (**p < 0.01, n = 6 mice per group). B, Polypeptide drugs were administered intraperitoneally once a day for one week. GFP showed that the drug was successfully absorbed in the mouse brain (scale bar = 200 and 50 µm). C, Tat-GluA1 significantly abolished the binding between p62 and GluA1 (**p < 0.01, n = 6 mice per group). D, TTC staining analysis of infarct volume (**p < 0.01, n = 6 mice per group). E, F, Western blot analysis and quantification of LC3BII/LC3BI (*p < 0.05, n = 6 mice per group) and p62 (**p < 0.01, n = 6 mice per group). G, Cell surface, intracellular, and total (surface + intracellular) GluA1/2 levels and surface/intracellular ratios (**p < 0.01, *p < 0.05, n = 6 mice per group). Data are shown as the mean ± SEM.
Next, we studied whether disruption of the GluA1–p62 interaction affected autophagic activity. At ZT17–ZT19, autophagic proteins LC3 (Fig. 9E) and p62 (Fig. 9F) were detected. The results showed no difference between the Tat-GluA1 and Tat-Scr-GluA1 groups. These findings indicate that inhibition of the GluA1–p62 interaction in the penumbra abolished the protective effects of autophagy in active-phase stroke.
Then, we conducted a BS3 cross-linking experiment to detect GluA1 expression in the stroke groups. We found that the surface (t(10) = 4.334, p = 0.0015, paired t test; Fig. 9G) and total (t(10) = 4.030, p = 0.0024, paired t test; Fig. 9G) GluA1 levels and the surface/intracellular ratio (t(10) = 2.868, p = 0.0167, paired t test; Fig. 9G) were increased in the Tat-GluA1 group compared with the Tat-Scr-GluA1 control group at ZT17–ZT19. However, intracellular GluA1 levels did not significantly differ between the two groups. We further examined whether Tat-GluA1 affected the expression of GluA2. We found that the surface, intracellular, and total GluA2 levels and the surface/intracellular ratio did not significantly differ between the two groups (Fig. 9G).
These results suggest that interactions between p62 and GluA1 are required for the autophagy-induced decrease in GluA1 expression.
Per1 knock-out abolished the effect of circadian rhythmicity on infarct volume, GluA1/2 expression, and autophagic activity in ischemic stroke
Per1 is an important clock gene (Reppert and Weaver, 2002), which plays a critical role in maintaining and regulating the stability of circadian rhythms (Bae et al., 2001; Zheng et al., 2001). To explore the relationship between PER1 and circadian rhythm effects on brain damage, we knocked out Per1 (Fig. 10A) and performed a series of experiments. First, we found that the infarct volume in Per1−/− mice was not significantly different between the active and inactive phases (F(3,20) = 23.897, p = 0.107, two-way ANOVA; Fig. 10B). In wild-type (WT) mice, the BS3 cross-linking experiment showed that cell surface, intracellular, and total (surface + intracellular) GluA1 levels in the (MCAO/R)24 h group at ZT17–ZT19 were significantly decreased compared with the ZT17–ZT19 sham group. No changes were found in the surface/intracellular ratio in the (MCAO/R)24 h group at ZT17–ZT19 compared with the ZT17–ZT19 sham group (Fig. 11A). In Per1−/− knock-out mice, different from WT mice, the surface (F(3,20) = 9.52, p < 0.0001, two-way ANOVA; Fig. 11B), intracellular (F(3,20) = 3.519, p = 0.031, two-way ANOVA; Fig. 11B), and total (F(3,20) = 10.607, p < 0.0001, two-way ANOVA; Fig. 11B) GluA1 levels and the surface/intracellular ratio (F(3,20) = 4.148, p = 0.002, two-way ANOVA; Fig. 11B) in the stroke group at ZT17–ZT19 were significantly increased compared with the sham group at ZT17–ZT19. Moreover, there was no difference in the surface, intracellular, and total GluA2 levels and the surface/intracellular ratio among the sham and stroke groups at ZT17–ZT19 (Fig. 11D). The WB results for GluA1 (F(3,28) = 24.651, p < 0.0001, two-way ANOVA; Fig. 11C) and GluA2 (Fig. 11E) in Per1−/− knock-out mice were similar to those of total GluA1/2 expression in the BS3 cross-linking experiment (Fig. 11B,D). Meanwhile, the LC3BII/LC3BI ratio was decreased (F(3,24) = 12.02, p < 0.0001, two-way ANOVA; Fig. 12A) and p62 expression was increased (F(3,24) = 7.309, p < 0.0001, two-way ANOVA; Fig. 12B) 24 h after MCAO/R in Per1−/− mice compared with WT mice (sham and MCAO/R) at ZT17–ZT19. No differences were found in autophagic activity among these groups at ZT5–ZT7 (Fig. 12A,B). Immunofluorescence analysis also showed that the proportion of LC3B colocalized with NeuN in cortical neurons did not significantly differ between groups in Per1−/− mice, regardless of the phase of the day or the presence of stroke injury (Fig. 12C). These results suggest that Per1 knock-out aggravated brain injury during active-phase stroke by blocking autophagy-dependent degradation of GluA1.
Figure 10.

Mouse brain infarct volume 24 h post-MCAO in circadian rhythm after Per1 knock-out. A, Nucleic acid electrophoresis tests showed successful knock-out of the Per1 gene (Per1−/− mouse homozygotes, one band at 459 bp; WT allele, one band at 637 bp). B, TTC staining analysis of infarct volume in Per1−/− and WT mice at ZT5–ZT7 and ZT17–ZT19 (***p < 0.001, n = 6 mice per group). Data are shown as the mean ± SEM.
Figure 11.
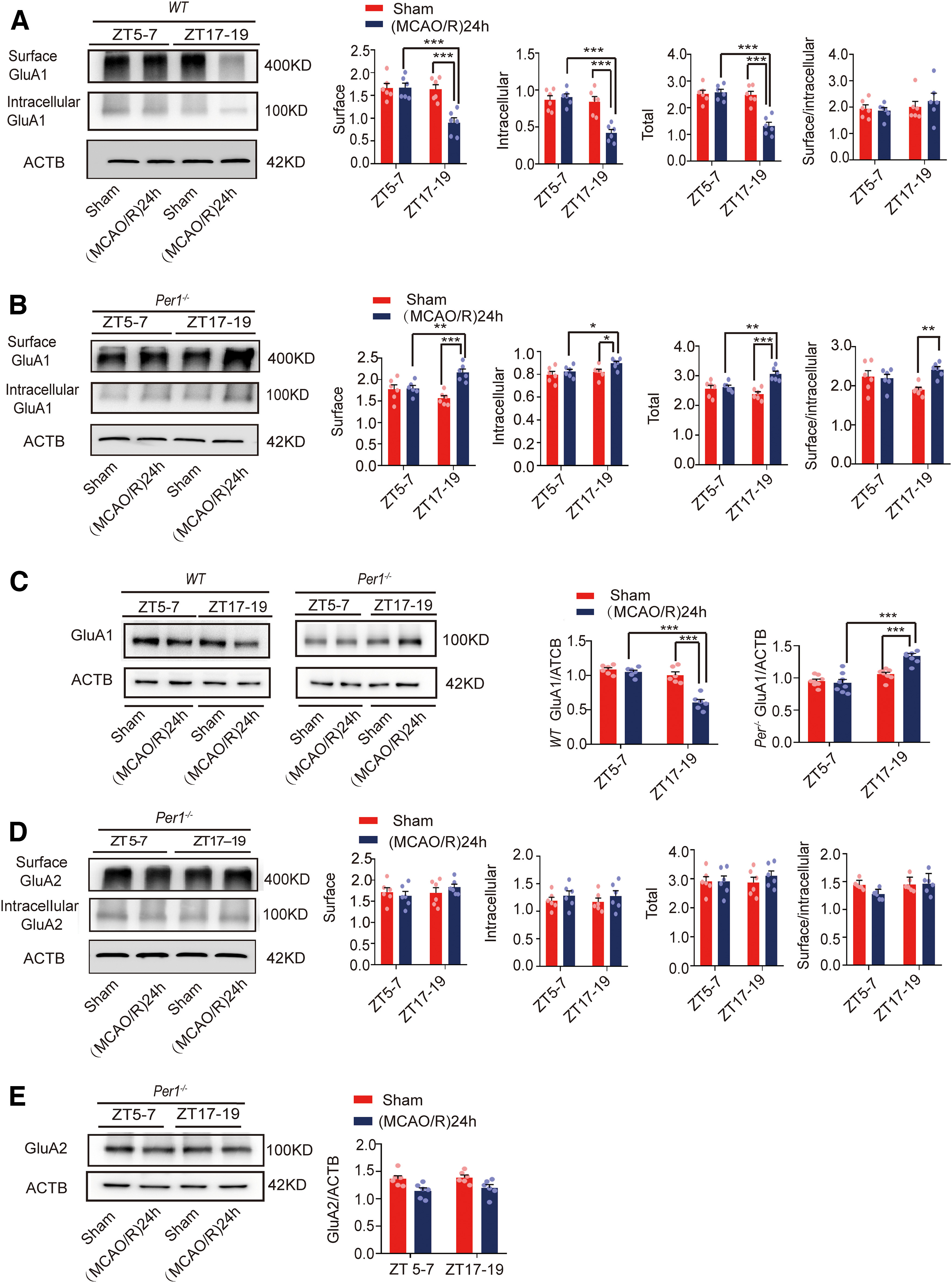
GluA1/2 expression in penumbra tissue in the (MCAO/R)24 h group in circadian rhythm after Per1 knock-out. A, B, Cell surface, intracellular, and total (surface + intracellular) GluA1 levels and the surface/intracellular ratio in WT and Per1−/−mice at ZT5–ZT7 and ZT17–ZT19 (*p < 0.05, **p < 0.01, ***p < 0.001, n = 6 mice per group). C, Western blot analysis and quantification of GluA1 in Per1−/− and WT mice at ZT5–ZT7 and ZT17–ZT19 (***p < 0.001, n = 6–8 mice per group). D, Cell surface, intracellular, and total (surface + intracellular) GluA2 levels and the surface/intracellular ratio in Per1−/−mice at ZT5–ZT7 and ZT17–ZT19. E, Western blot analysis and quantification of GluA2 in Per1−/− mice at ZT5–ZT7 and ZT17–ZT19. Data are shown as the mean ± SEM.
Figure 12.
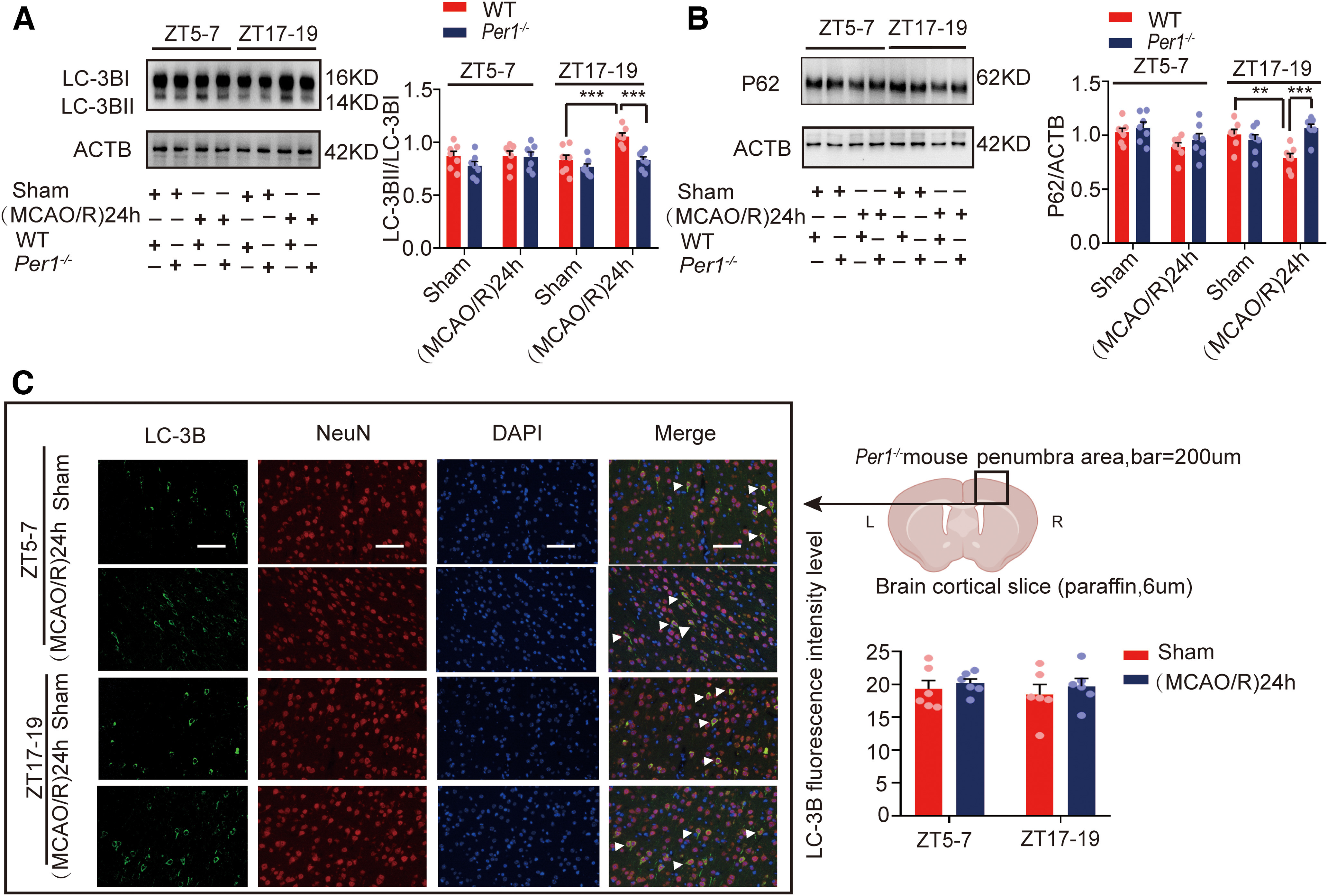
Autophagic activity in penumbra tissue in the (MCAO/R)24 h group in circadian rhythm after Per1 knock-out. A, B, Western blot analysis and quantification of LC3BII/LC3BI (***p < 0.001, n = 7 mice per group) and p62 (**p < 0.01, ***p < 0.001, n = 7 mice per group). C, Immunofluorescence analysis showed that LC3B levels did not significantly differ between groups in Per1−/− mice (scale bar = 200 µm). Data are shown as the mean ± SEM.
Discussion
We explored the circadian cycle of autophagic flux in an MCAO/R mouse model and the mechanism underlying regulation of brain injury by autophagy. We found that infarct volume was smaller, GluA1 expression was decreased, and autophagy was activated in active-phase (ZT17–ZT19) stroke compared with inactive-phase (ZT5–ZT7) stroke. We also showed that GluA1 expression under different autophagic activities (regulated by RAPA, 3-MA, or LV-Atg5-shRNA) was neither ischemia-dependent nor circadian rhythm-dependent. However, infarct volume was positively correlated with GluA1 expression and negatively correlated with autophagic activity. In addition, we found that autophagy-dependent degradation of GluA1 was mediated by interaction between GluA1 and p62. Disruption of GluA1–p62 binding appeared to eliminate the protective effects of autophagy, resulting in greater infarct volume and increased GluA1 expression in active-phase (ZT17–ZT19) stroke. Furthermore, knock-out of the Per1 gene abolished the neuroprotective effects of autophagy in active-phase stroke.
Circadian rhythms are ubiquitous in mammals (O'Neill et al., 2011), and disruption of these rhythms affects mammals' physiological and pathologic processes (Zueva et al., 2016). In a human stroke study, patients with symptom onset in the daytime showed smaller ischemic core volumes compared with those with onset at night (Reidler et al., 2021). A recent study reported a smaller penumbra and reduced infarct growth in mouse stroke models in which the occlusion occurred during the active phase (ZT15–ZT21; Esposito et al., 2020). Consistent with previous studies, we also demonstrated that infarct volume was larger during the inactive phase (ZT5–ZT7) than during the active phase (ZT17–ZT19) in MCAO/R mice.
AMPAR is an ionic glutamate receptor consisting of four subunits (GluA1–4) that plays an important role in the ischemia-related excitotoxic pathway. Ischemic insult leads to altered GluA1/2 AMPAR subunit expression, which allows massive Ca2+ entry through AMPARs. The GluA2 subunit inhibits calcium influx, exerting neuroprotective effects against cerebral ischemia (Achzet et al., 2021b). Previous studies reported that oxygen glucose deprivation/reperfusion induced internalization and degradation of the GluA1 and GluA2 AMPAR subunits in vitro (Mazzocchetti et al., 2020). Our study revealed that GluA1 expression was dramatically decreased in the MCAO/R group compared with the sham group at ZT17–ZT19, while there was no significant difference at ZT5–ZT7.
However, the explanation for changes in GluA2 levels at different time points is complex. GluA2 surface expression was decreased in mice with stroke in the inactive phase (ZT5–ZT7). Surprisingly, total GluA2 expression was increased in both sham and MCAO/R mice in the active phase (ZT17–ZT19), which indicated that the decreased expression of GluA2 in the inactive phase was reversed in the active phase in both sham and MCAO/R mice. It could be inferred that the increase in GluA2 expression was likely regulated by other circadian rhythm mechanisms. One possible explanation is that the expression of adenosine deaminase acting on RNA 2b (ADAR2b), which catalyzes the editing of the GluA2 Q/R site (Wright and Vissel, 2012), is influenced by the circadian rhythm (Terajima et al., 2017).
Many studies have demonstrated the crucial role of autophagy in the regulation of MCAO/R-induced neuron injury. However, whether autophagy is helpful or harmful is still debated, and the role of autophagy in the pathophysiological changes in ischemic stroke is particularly complex (P. Wang et al., 2018). Previous research suggested that the role of autophagy in NMDAR-dependent long-term depression and chemical long-term depression is to degrade AMPARs or key synaptic protein substrates (Shehata et al., 2012). Another study found that autophagy degrades the T19-phosphorylated form of postsynaptic density-95 from synapses and causes an increase in AMPAR surface mobility (Compans et al., 2021). In the present study, we used RAPA and 3-MA to induce and inhibit autophagy, respectively. We observed that RAPA and 3-MA affected GluA1 expression in both ischemic and nonischemic states, and in stroke during both active and inactive phases. We also observed that infarct volume was smaller in the RAPA group and larger in the 3-MA group at ZT17–ZT19.
In addition to interfering with autophagy by pharmacological methods, we blocked it at the genetic level. We used LV-Atg5-shRNA to knock down Atg5 and found that GluA1 expression was higher and infarct volume was larger compared with the vector group. These results indicate that the role of autophagy in ischemic insult might be in large part related to regulating the degradation of GluA1. This result was in agreement with those of other studies in which autophagy protected against ischemia/reperfusion injury (Rami et al., 2017; Rao et al., 2017; Xin et al., 2017; X. Liu et al., 2018). However, there are limitations in the present study. LV-Atg5-shRNA could infect all types of cells in the brain, including neurons, microglial cells, and astrocytes. Therefore, the possibility that autophagic activity was regulated in non-neuronal cells cannot be ruled out. In the future, there is a need to use viruses that specifically interfere with Atg5 expression in neurons. We also found that the LC3BII/LC3BI ratio was increased at ZT17–ZT19 compared with ZT5–ZT7 in the sham group (Fig. 3A). This result is consistent with a previous study showing that the autophagy level is related to circadian rhythm, as it is higher at night than during the day in mouse models (Rami et al., 2017). Yet, possibly because of crosstalk between active-phase stroke and the stimulation of ischemia, we observed that expression of LC3BII/LC3BI was upregulated while that of p62 was downregulated in mouse brains after MCAO/R during the active phase (ZT17–ZT19), but not the inactive phase (ZT5–ZT7). We conjecture that autophagy promoted the degradation of GluA1 at ZT17–ZT19 in the MCAO/R group, protecting against increased cerebral infarct volume.
We also analyzed the mechanism of selective degradation of GluA1 by autophagy. A study revealed that p62 acts as an autophagy receptor to mediate selective autophagy (Geetha and Wooten, 2002). The ZZ-type zinc finger domain of p62 directly binds to the AMPAR GluA1 subunit intracellular loop L2–3 (Jiang et al., 2009; Ren et al., 2013). p62 regulates AMPAR trafficking and synaptic plasticity and also serves as a scaffolding protein to facilitate GluA1 Ser818 phosphorylation by protein kinase C λ (PKCλ; Ren et al., 2013). Our findings revealed that after MCAO/R during the active phase (ZT17–ZT19), the ability of p62 to bind to GluA1 was enhanced. Using polypeptides to selectively disrupt the interaction between p62 and GluA1, we found increased GluA1 expression and worsened ischemic brain injury, but unchanged autophagic activity. The decrease in GluA1 expression after MCAO/R during the active phase was likely mediated by the p62–GluA1 interaction, followed by direct autophagic degradation.
Previous research has reported decreased autophagic activity in the hippocampus and increased neuronal injury after cerebral ischemia after Per1 knock-out (Wiebking et al., 2013). Consistent with previous studies, we also found Per1−/− mice were susceptible to ischemia. However, we did not find reduced autophagy after Per1 knock-out in the cortex. Per1 mRNA levels were upregulated at ZT17–ZT19 and downregulated at ZT5–ZT7. We found that autophagy was induced by cerebral ischemia at ZT17–ZT19 but not at ZT5–ZT7. Yet, RAPA could induce autophagy in nonischemic states at any time period. It is possible that ischemia-induced activation of autophagy requires the presence of PER1, while the activation of autophagy by drugs may not require PER1.
In summary, we may need to consider the influence of the circadian rhythm on the pathophysiological mechanism of ischemic stroke in clinical treatment of ischemic stroke.
Footnotes
The work was supported by the Soochow Clinical Experts Introduction Team Project Grant No. 5201013206, the Basic Science (Natural Science) of Colleges and Universities in Jiangsu Province Grant No. 21KJB320012, the Key R&D Program of Jiangsu Province (Social Development) (key project-clinical frontier technology) Grant No. BE2019666, and the Natural Scientific Foundation of Jiangsu Province Grant No. BK20211075.
The authors declare no competing financial interests.
References
- Abarca C, Albrecht U, Spanagel R (2002) Cocaine sensitization and reward are under the influence of circadian genes and rhythm. Proc Natl Acad Sci U S A 99:9026–9030. 10.1073/pnas.142039099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achzet LM, Davison CJ, Shea M, Sturgeon I, Jackson DA (2021a) Oxidative stress underlies the ischemia/reperfusion-induced internalization and degradation of AMPA receptors. Int J Mol Sci 22:717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achzet LM, Astruc-Diaz F, Beske PH, Natale NR, Denton TT, DA J (2021b) Liposomal encapsulated FSC231, a PICK1 inhibitor, prevents the ischemia/reperfusion-induced degradation of GluA2-containing AMPA receptors. Pharmaceutics 13:636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adesnik H, Nicoll RA (2007) Conservation of glutamate receptor 2-containing AMPA receptors during long-term potentiation. J Neurosci 27:4598–4602. 10.1523/JNEUROSCI.0325-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae K, Jin X, Maywood ES, Hastings MH, Reppert SM, Weaver DR (2001) Differential functions of mPer1, mPer2, and mPer3 in the SCN circadian clock. Neuron 30:525–536. [DOI] [PubMed] [Google Scholar]
- Cai Y, Yang E, Yao X, Zhang X, Wang Q, Wang Y, Liu J, Fan W, Yi K, Kang C, Wu J (2021) FUNDC1-dependent mitophagy induced by tPA protects neurons against cerebral ischemia-reperfusion injury. Redox Biol 38:101792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Zhan J, Yan X, Mamun AA, Zhang Y, Xu Y, Zhang H, Li X, Zhou K, Xiao J (2022) FGF21 alleviates microvascular damage following limb ischemia/reperfusion injury by TFEB-mediated autophagy enhancement and anti-oxidative response. Signal Transduct Target Ther 7:349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson AN, Overman JJ, Zhong S, Mueller R, Lynch G, Carmichael ST (2011) AMPA receptor-induced local brain-derived neurotrophic factor signaling mediates motor recovery after stroke. J Neurosci 31:3766–3775. 10.1523/JNEUROSCI.5780-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compans B, Camus C, Kallergi E, Sposini S, Martineau M, Butler C, Kechkar A, Klaassen RV, Retailleau N, Sejnowski TJ, Smit AB, Sibarita JB, Bartol TM Jr, Perrais D, Nikoletopoulou V, Choquet D, Hosy E (2021) NMDAR-dependent long-term depression is associated with increased short term plasticity through autophagy mediated loss of PSD-95. Nat Commun 12:2849. 10.1038/s41467-021-23133-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, Marinelli M, Wolf ME (2008) Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature 454:118–121. 10.1038/nature06995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA (1999) Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci 22:391–397. [DOI] [PubMed] [Google Scholar]
- Dos-Anjos S, Martinez-Villayandre B, Montori S, Regueiro-Purriños MM, Gonzalo-Orden JM, Fernández-López A (2009) Global ischemia-induced modifications in the expression of AMPA receptors and inflammation in rat brain. Brain Res 1287:20–27. 10.1016/j.brainres.2009.06.065 [DOI] [PubMed] [Google Scholar]
- Esposito E, Li W, TM E, Park JH, Sencan I, Guo S, Shi J, Lan J, Lee J, Hayakawa K, Sakadzic S, Ji X, Lo EH (2020) Potential circadian effects on translational failure for neuroprotection. Nature 582:395–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming A, Bourdenx M, Fujimaki M, Karabiyik C, Krause GJ, Lopez A, Martin-Segura A, Puri C, Scrivo A, Skidmore J, Son SM, Stamatakou E, Wrobel L, Zhu Y, Cuervo AM, Rubinsztein DC (2022) The different autophagy degradation pathways and neurodegeneration. Neuron 110:935–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foerster EG, Mukherjee T, Cabral-Fernandes L, Rocha JDB, Girardin SE, Philpott DJ (2022) How autophagy controls the intestinal epithelial barrier. Autophagy 18:86–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W, Wang X, Zhou Y, Wang X, Yu Y (2022) Autophagy, ferroptosis, pyroptosis, and necroptosis in tumor immunotherapy. Signal Transduct Target Ther 7:196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geetha T, Wooten MW (2002) Structure and functional properties of the ubiquitin binding protein p62. FEBS Lett 512:19–24. [DOI] [PubMed] [Google Scholar]
- Gray EE, Fink AE, Sarinana J, Vissel B, O'Dell TJ (2007) Long-term potentiation in the hippocampal CA1 region does not require insertion and activation of GluR2-lacking AMPA receptors. J Neurophysiol 98:2488–2492. [DOI] [PubMed] [Google Scholar]
- Griffey CJ, Yamamoto A (2022) Macroautophagy in CNS health and disease. Nat Rev Neurosci 23:411–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Chen Y, Liu R, Li B, Fei X, Li X, Liu G, Li Y, Xu B, Fang W (2022) P-Glycoprotein aggravates blood brain barrier dysfunction in experimental ischemic stroke by inhibiting endothelial autophagy. Aging Dis 13:1546–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui H, Rao W, Zhang L, Xie Z, Peng C, Su N, Wang K, Wang L, Luo P, Hao YL, Zhang S, Fei Z (2016) Inhibition of Na(+)-K(+)-2Cl(-) Cotransporter-1 attenuates traumatic brain injury-induced neuronal apoptosis via regulation of Erk signaling. Neurochem Int 94:23–31. [DOI] [PubMed] [Google Scholar]
- Jiang J, Parameshwaran K, Seibenhener ML, Kang MG, Suppiramaniam V, Huganir RL, Diaz-Meco MT, Wooten MW (2009) AMPA receptor trafficking and synaptic plasticity require SQSTM1/p62. Hippocampus 19:392–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Dai J, Cui H (2018) Vitexin reverses the autophagy dysfunction to attenuate MCAO-induced cerebral ischemic stroke via mTOR/Ulk1 pathway. Biomed Pharmacother 99:583–590. [DOI] [PubMed] [Google Scholar]
- Komatsu M, et al. (2007) Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131:1149–1163. [DOI] [PubMed] [Google Scholar]
- Li H, Li X, Xu Z, Lu J, Cao C, You W, Yu Z, Shen H, Chen G (2022) Unbalanced regulation of Sec22b and Ykt6 blocks autophagosome axonal retrograde flux in neuronal ischemia-reperfusion injury. J Neurosci 42:5641–5654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Ding R, Wang F, Guo C, Liu A, Wei L, Yuan S, Chen F, Hou S, Ma Z, Zhang Y, Cudmore RH, Wang X, Shen H (2020) Transient ischemia-reperfusion induces cortical hyperactivity and AMPAR trafficking in the somatosensory cortex. Aging (Albany NY) 12:4299–4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Zhou X, Li Y, Ma S, Pan L, Zhang X, Zheng W, Wu Z, Wang K, Ahsan A, Wu J, Jiang L, Lu Y, Hu W, Qin Z, Chen Z, Zhang X (2022) TIGAR alleviates oxidative stress in brain with extended ischemia via a pentose phosphate pathway-independent manner. Redox Biol 53:102323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Tian F, Wang S, Wang F, Xiong L (2018) Astrocyte autophagy flux protects neurons against oxygen-glucose deprivation and ischemic/reperfusion injury. Rejuvenation Res 21:405–415. [DOI] [PubMed] [Google Scholar]
- Liu Z, Chu G (2013) Chronobiology in mammalian health. Mol Biol Rep 40:2491–2501. 10.1007/s11033-012-2330-4 [DOI] [PubMed] [Google Scholar]
- Lu HF, Wu PF, Yang YJ, Xiao W, Fan J, Liu J, Li YL, Luo Y, Hu ZL, Jin Y, Wang F, Chen JG (2014) Interactions between N-ethylmaleimide-sensitive factor and GluR2 in the nucleus accumbens contribute to the expression of locomotor sensitization to cocaine. J Neurosci 34:3493–3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao LM, Wang W, Chu XP, Zhang GC, Liu XY, Yang YJ, Haines M, Papasian CJ, Fibuch EE, Buch S, Chen JG, Wang JQ (2009) Stability of surface NMDA receptors controls synaptic and behavioral adaptations to amphetamine. Nat Neurosci 12:602–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzocchetti P, Mancini A, Sciaccaluga M, Megaro A, Bellingacci L, Di Filippo M, Cesarini EN, Romoli M, Carrano N, Gardoni F, Tozzi A, Calabresi P, Costa C (2020) Low doses of Perampanel protect striatal and hippocampal neurons against in vitro ischemia by reversing the ischemia-induced alteration of AMPA receptor subunit composition. Neurobiol Dis 140:104848. 10.1016/j.nbd.2020.104848 [DOI] [PubMed] [Google Scholar]
- Miao Z, He Y, Xin N, Sun M, Chen L, Lin L, Li J, Kong J, Jin P, Xu X (2015) Altering 5-hydroxymethylcytosine modification impacts ischemic brain injury. Hum Mol Genet 24:5855–5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskowitz MA, Lo EH, Iadecola C (2010) The science of stroke: mechanisms in search of treatments. Neuron 67:181–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabavi SF, Sureda A, Sanches-Silva A, Pandima DK, Ahmed T, Shahid M, Sobarzo-Sanchez E, Dacrema M, Daglia M, Braidy N, Vacca RA, Berindan-Neagoe I, Gulei D, Barreca D, Banach M, Nabavi SM, Dehpour AR, Shirooie S (2019) Novel therapeutic strategies for stroke: the role of autophagy. Crit Rev Clin Lab Sci 56:182–199. [DOI] [PubMed] [Google Scholar]
- O'Neill JS, van Ooijen G, Dixon LE, Troein C, Corellou F, Bouget FY, Reddy AB, Millar AJ (2011) Circadian rhythms persist without transcription in a eukaryote. Nature 469:554–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rami A, Fekadu J, Rawashdeh O (2017) The hippocampal autophagic machinery is depressed in the absence of the circadian clock protein PER1 that may lead to vulnerability during cerebral ischemia. Curr Neurovasc Res 14:207–214. [DOI] [PubMed] [Google Scholar]
- Rao Z, Pan X, Zhang H, Sun J, Li J, Lu T, Gao M, Liu S, Yu D, Ding Z (2017) Isoflurane preconditioning alleviated murine liver ischemia and reperfusion injury by restoring AMPK/mTOR-mediated autophagy. Anesth Analg 125:1355–1363. [DOI] [PubMed] [Google Scholar]
- Rawashdeh O, Jilg A, Jedlicka P, Slawska J, Thomas L, Saade A, Schwarzacher SW, Stehle JH (2014) PERIOD1 coordinates hippocampal rhythms and memory processing with daytime. Hippocampus 24:712–723. [DOI] [PubMed] [Google Scholar]
- Reidler P, Brehm A, Sporns PB, Burbano VG, Stueckelschweiger L, Broocks G, Liebig T, Psychogios MN, Ricke J, Dimitriadis K, Dichgans M, Kunz WG, Tiedt S (2021) Circadian rhythm of ischaemic core progression in human stroke. J Neurol Neurosurg Psychiatry 94:70–73. [DOI] [PubMed] [Google Scholar]
- Ren SQ, Yan JZ, Zhang XY, Bu YF, Pan WW, Yao W, Tian T, Lu W (2013) PKCλ is critical in AMPA receptor phosphorylation and synaptic incorporation during LTP. EMBO J 32:1365–1380. 10.1038/emboj.2013.60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shehata M, Matsumura H, Okubo-Suzuki R, Ohkawa N, Inokuchi K (2012) Neuronal stimulation induces autophagy in hippocampal neurons that is involved in AMPA receptor degradation after chemical long-term depression. J Neurosci 32:10413–10422. 10.1523/JNEUROSCI.4533-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Cheng Q, Chen C (2021) The role of autophagy in the pathogenesis of ischemic stroke. Curr Neuropharmacol 19:629–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terajima H, Yoshitane H, Ozaki H, Suzuki Y, Shimba S, Kuroda S, Iwasaki W, Fukada Y (2017) ADARB1 catalyzes circadian A-to-I editing and regulates RNA rhythm. Nat Genet 49:146–151. [DOI] [PubMed] [Google Scholar]
- Thiebaut AM, Buendia I, Ginet V, Lemarchand E, Boudjadja MB, Hommet Y, Lebouvier L, Lechevallier C, Maillasson M, Hedou E, Déglon N, Oury F, Rubio M, Montaner J, Puyal J, Vivien D, Roussel BD (2022) Thrombolysis by PLAT/tPA increases serum free IGF1 leading to a decrease of deleterious autophagy following brain ischemia. Autophagy 18:1297–1317. 10.1080/15548627.2021.1973339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu W, Xu X, Peng L, Zhong X, Zhang W, Soundarapandian MM, Balel C, Wang M, Jia N, Zhang W, Lew F, Chan SL, Chen Y, Lu Y (2010) DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell 140:222–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dolah DK, Mao LM, Shaffer C, Guo ML, Fibuch EE, Chu XP, Buch S, Wang JQ (2011) Reversible palmitoylation regulates surface stability of AMPA receptors in the nucleus accumbens in response to cocaine in vivo. Biol Psychiatry 69:1035–1042. 10.1016/j.biopsych.2010.11.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Pei A, Chen J, Yu H, Sun ML, Liu CF, Xu X (2012) A natural coumarin derivative esculetin offers neuroprotection on cerebral ischemia/reperfusion injury in mice. J Neurochem 121:1007–1013. [DOI] [PubMed] [Google Scholar]
- Wang P, Shao BZ, Deng Z, Chen S, Yue Z, Miao CY (2018) Autophagy in ischemic stroke. Prog Neurobiol 163–164:98–117. [DOI] [PubMed] [Google Scholar]
- Wang Y, Lu S, Chen Y, Li L, Li X, Qu Z, Huang J, Fan L, Yuan C, Song N, Zhang J, Xu W, Yang S, Wang Y (2021) Smoothened is a therapeutic target for reducing glutamate toxicity in ischemic stroke. Sci Transl Med 13:eaba3444. [DOI] [PubMed] [Google Scholar]
- Reppert SM, Weaver DR (2002) Coordination of circadian timing in mammals. Nature 418:935–941. [DOI] [PubMed] [Google Scholar]
- Wiebking N, Maronde E, Rami A (2013) Increased neuronal injury in clock gene Per-1 deficient-mice after cerebral ischemia. Curr Neurovasc Res 10:112–125. [DOI] [PubMed] [Google Scholar]
- Wright A, Vissel B (2012) The essential role of AMPA receptor GluR2 subunit RNA editing in the normal and diseased brain. Front Mol Neurosci 5:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Zheng Y, Liu M, Li Y, Ma S, Tang W, Yan W, Cao M, Zheng W, Jiang L, Wu J, Han F, Qin Z, Fang L, Hu W, Chen Z, Zhang X (2021) BNIP3L/NIX degradation leads to mitophagy deficiency in ischemic brains. Autophagy 17:1934–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao P, Gu J, Xu W, Niu X, Zhang J, Li J, Chen Y, Pei Z, Zeng J, Xing S (2022) RTN4/Nogo-A-S1PR2 negatively regulates angiogenesis and secondary neural repair through enhancing vascular autophagy in the thalamus after cerebral cortical infarction. Autophagy 18:2711–2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin H, Katakowski M, Wang F, Qian JY, Liu XS, Ali MM, Buller B, Zhang ZG, Chopp M (2017) MicroRNA cluster miR-17-92 cluster in exosomes enhance neuroplasticity and functional recovery after stroke in rats. Stroke 48:747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Chua CC, Gao J, Hamdy RC, Chua BH (2006) Humanin is a novel neuroprotective agent against stroke. Stroke 37:2613–2619. 10.1161/01.STR.0000242772.94277.1f [DOI] [PubMed] [Google Scholar]
- Yang J, Vitery MDC, Chen J, Osei-Owusu J, Chu J, Qiu Z (2019) Glutamate-releasing SWELL1 channel in astrocytes modulates synaptic transmission and promotes brain damage in stroke. Neuron 102:813–827.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Huang C, Wen X, Liu W, Huang X, Li Y, Zang J, Weng Z, Lu D, Tsang CK, Li K, Xu A (2022) Circular RNA circ-FoxO3 attenuates blood-brain barrier damage by inducing autophagy during ischemia/reperfusion. Mol Ther 30:1275–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X, Zhang YD, Ma RY, Chen YJ, Xiang XM, Hou DY, Li XH, Huang H, Li T, Duan CY (2022) Activated Drp1 regulates p62-mediated autophagic flux and aggravates inflammation in cerebral ischemia-reperfusion via the ROS-RIP1/RIP3-exosome axis. Mil Med Res 9:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Albrecht U, Kaasik K, Sage M, Lu W, Vaishnav S, Li Q, Sun ZS, Eichele G, Bradley A, Lee CC (2001) Nonredundant roles of the mPer1 and mPer2 genes in the mammalian circadian clock. Cell 105:683–694. 10.1016/s0092-8674(01)00380-4 [DOI] [PubMed] [Google Scholar]
- Zueva MT, Manko OM, Smoleevsky AE (2016) Alterations of physiological rhythms in neurodegenerative disorders: problems and prospects of light therapy. Klin Med (Mosk) 94:427–432. [PubMed] [Google Scholar]