

Abstract
Purpose:
A phase Ib/II clinical trial was conducted to evaluate the safety and efficacy of the combination of all-trans retinoic acid (ATRA) with pembrolizumab in stage IV melanoma patients.
Patients and methods:
Anti-PD-1 naïve stage IV melanoma patients were treated with pembrolizumab plus supplemental ATRA for three days surrounding each of the first four pembrolizumab infusions. The primary objective was to establish the maximum tolerated dose (MTD) and recommended phase II dose (RP2D) of the combination. The secondary objectives were to describe the safety and toxicity of the combined treatment and to assess antitumor activity in terms of a) the reduction in circulating myeloid derived suppressor cell (MDSC) frequency and b) progression free survival (PFS).
Results:
Twenty-four patients were enrolled, 46% diagnosed with M1a and 29% with M1c stage disease at enrollment. All patients had an ECOG status ≤1 and 75% had received no prior therapies. The combination was well tolerated, with the most common ATRA-related adverse events being headache, fatigue, and nausea. The RP2D was established at 150mg/m2 ATRA + 200 mg Q3W pembrolizumab. Median PFS was 20.3 months, and the overall response rate was 71%, with 50% of patients experiencing a complete response, and the 1-year overall survival was 80%. The combination effectively lowered the frequency of circulating MDSCs.
Conclusions:
With a favorable tolerability and high response rate, this combination is a promising frontline treatment strategy for advanced melanoma. Targeting MDSCs remains an attractive mechanism to enhance the efficacy of immunotherapies and this combination merits further investigation.
Keywords: MDSC, immunosuppression, anti-PD-1, ATRA, PFS, melanoma
Introduction
While immune checkpoint inhibitors (ICIs) have improved the outlook for many melanoma patients, a significant proportion of patients exhibit resistance to these therapies. A variety of mechanisms have been associated with resistance to ICIs (1). One resistance mechanism of particular interest is the accumulation of a heterogeneous population of immunosuppressive myeloid lineage cells called myeloid-derived suppressor cells (MDSCs) (2,3). MDSCs are potent suppressors of antitumor immunity and represent a significant obstacle to the success of ICI treatment in several cancer types (3,4). Therefore, overcoming the suppressive activity of MDSCs has the potential to enhance the efficacy of ICIs.
MDSCs diminish the efficacy of ICIs by inhibiting several different mechanisms, including direct cell:cell interactions as well as through the production of immunosuppressive factors (3–6). By secreting high levels of the immunosuppressive cytokines interleukin (IL)-10, transforming growth factor (TGF)-β, and generating reactive oxygen species (ROS) MDSCs inhibit antitumor immunity (3–6). In addition, MDSCs secrete arginase-1 (ARG1) and indoleamine 2,3-dioxygenase 1 (IDO1), depleting important nutrients required for effective T cell functions (3–6). By inhibiting the functions of CD8+ T cells, MDSCs eliminate a highly effective mechanism of antitumor immunity, as CD8+ T cells express the targets of ICIs and are important effectors of ICI activity.
MDSCs are distinguished from other myeloid cells based on the expression of HLA-DR and are characterized by their highly immunosuppressive functions (4,5). MDSCs can be divided into three subpopulations: monocytic MDSCs (MO-MDSCs, CD14+CD15−), polymorphonuclear-like MDSCs (PMN-MDSCs, CD14−CD15+Lox1+), and early MDSCs (eMDSCs, CD14−CD15−) (4,5). Due to their immature nature, MDSCs are susceptible to modification or reduction by differentiating agents such as all-trans retinoic acid (ATRA) (4,6,7). ATRA, a vitamin A derivative, is part of the standard of care therapy for acute promyelocytic leukemia. Although the mechanism resulting in ATRA-mediated MDSC differentiation is still under investigation, ATRA has been shown to decrease both the frequency and function of MDSCs through activation of ERK1/2, upregulation of glutathione synthase, and generation of glutathione (7). Increased glutathione production decreases production of ROS and results in subsequent terminal differentiation of MDSCs (7).
In previous clinical trials combining ATRA with a dendritic cell (DC) vaccine (8), IL-2 (9), or the anti-CTLA-4 ICI ipilimumab (6), ATRA decreased the frequency of circulating MDSCs and improved the antitumor functions of T cells. In addition, our prior clinical trial combining ipilimumab with ATRA found that the 150mg/m2 dose of ATRA appeared to be safe and well tolerated (6). Thus, in the current clinical trial, we hypothesized that combining ATRA with the anti-PD-1 monoclonal antibody pembrolizumab, would decrease the frequency of MDSCs and thereby improve the efficacy of pembrolizumab. The timing and initial dose of ATRA in relation to pembrolizumab was determined based on our prior experience with ATRA in similar patient populations (6). The primary objective of this study was to establish the maximum tolerated dose (MTD) and recommended phase II dose (RP2D) of ATRA in metastatic melanoma patients treated with pembrolizumab. The secondary objectives were to describe the safety and toxicity of combined treatment with pembrolizumab and ATRA in melanoma patients and to assess the antitumor activity in terms of a) the reduction in MDSC frequency and suppressive function in peripheral blood of metastatic melanoma patients and b) progression-free survival (PFS). Further experimental post-hoc analyses included overall response rate (ORR), disease control rate (DCR), clinical benefit rate, PFS at six months (six-month PFS rate), and overall survival (OS).
Materials and methods
Study design and patients
This open label, phase I/II clinical trial in 24 stage IV melanoma patients was conducted at the University of Colorado Cancer Center (ClinicalTrials.gov #NCT03200847). Eligible patients were over the age of 18 and had not been previously treated with anti-PD-1 therapy. Other inclusion/exclusion criteria are listed in Supplementary Table 1. The primary endpoints were to establish the MTD and RP2D and evaluate the effect on MDSCs at RP2D. Secondary endpoints were safety, overall response rate (ORR), disease control rate (DCR), and PFS according to RECIST v1.1.
A fixed dose regimen was selected for this study, 200 mg Q3W pembrolizumab plus the supplemental treatment of 150 mg/m2 ATRA (brand name VESANOID) orally for three days surrounding each of the first four infusions of pembrolizumab (day −1, day 0, day +1) for a total of 12 days of ATRA treatment. The dose selected is based on extensive simulations performed using the population pharmacokinetic (PK) model of pembrolizumab showing that the fixed dose of 200 mg every three weeks will provide exposures that 1) are optimally consistent with those obtained with the 2 mg/kg dose every three weeks, 2) will maintain individual patient exposures in the exposure range established in melanoma as associated with maximal efficacy response, and 3) will maintain individual patients exposure in the exposure range established in melanoma that is well tolerated and safe. ATRA is a standard treatment for patients with acute promyelocytic leukemia, it has also been used to treat patients with solid tumors. We have selected the dose of ATRA based on two prior clinical trials that combined ATRA with other treatments based on the pharmacokinetics and effects on MDSCs observed in these trials (7,8).
The study design includes a safety lead in with 12 patients. If there are four or fewer DLTs (defined in Supplementary table 3) in the first 12 patients, the dose will be considered as the RP2D, and remaining 12 patients will be accrued to the study to further evaluate the safety of the dose. However, if there are more than four DLTs among the first 12 patients, the dose of ATRA will be de-escalated. In our prior trial combining three days of ATRA (150mg/m2/day) around administration of ipilimumab, we noted headache as a significant common side effect that resolved with discontinuation of the ATRA (6). The de-escalation design was intended to aim for the effect on MDSC’s yet allow for a dose reduction for side effects with the hope of still observing at least some effect on the intended target of MDSC’s. The treatment effect of the RP2D on MDSCs will also be evaluated.
The study protocol and all amendments were reviewed and approved by the Colorado Multiple Institutional Review Board (COMIRB #16-1080). The trial was conducted in compliance with the International Conference on Harmonization Good Clinical Practice guidelines and the Declaration of Helsinki. All patients provided written informed consent.
Primary objective
To identify the MTD and RP2D of the combination of pembrolizumab and ATRA.
Secondary objectives
Describe the safety and toxicity of combined treatment with pembrolizumab and ATRA in melanoma patients. To assess the anti-tumor activity in terms of a) The reduction in MDSC frequency and suppressive function (measured as a continuous variable) in peripheral blood of advanced melanoma patients undergoing pembrolizumab and ATRA combination therapy, b) PFS.
Exploratory objective
To determine the clinical outcomes with tumor-specific T cell responses.
Procedures
A total of twelve subjects with a cohort size of six were tested during the safety lead in. If more than two subjects experience DLTs within the first six subjects, the next lower dose level of ATRA will be given to the subsequent cohort. If four or fewer subjects experience DLTs among the twelve subjects during the safety lead in, the remaining subjects will continue the same dose level. However, if more than four DLTs occur among the first 12 patients, the dose of ATRA will be de-escalated. A Pocock-type boundary was used to continuously monitor for toxicity (10), where the toxicity probability was set at 25%, and the desired probability of early stopping was set at 0.05. The RP2D was established at 200mg Q3W pembrolizumab plus the supplemental treatment of 150 mg/m2 ATRA orally for three days surrounding each of the first four infusions of pembrolizumab (day −1, 0, and +1), with patients continuing pembrolizumab for up to two years or until confirmed disease progression or unacceptable toxicity (Fig. 1A). All patients received the RP2D.
Figure 1. Clinical trial design and efficacy measures.
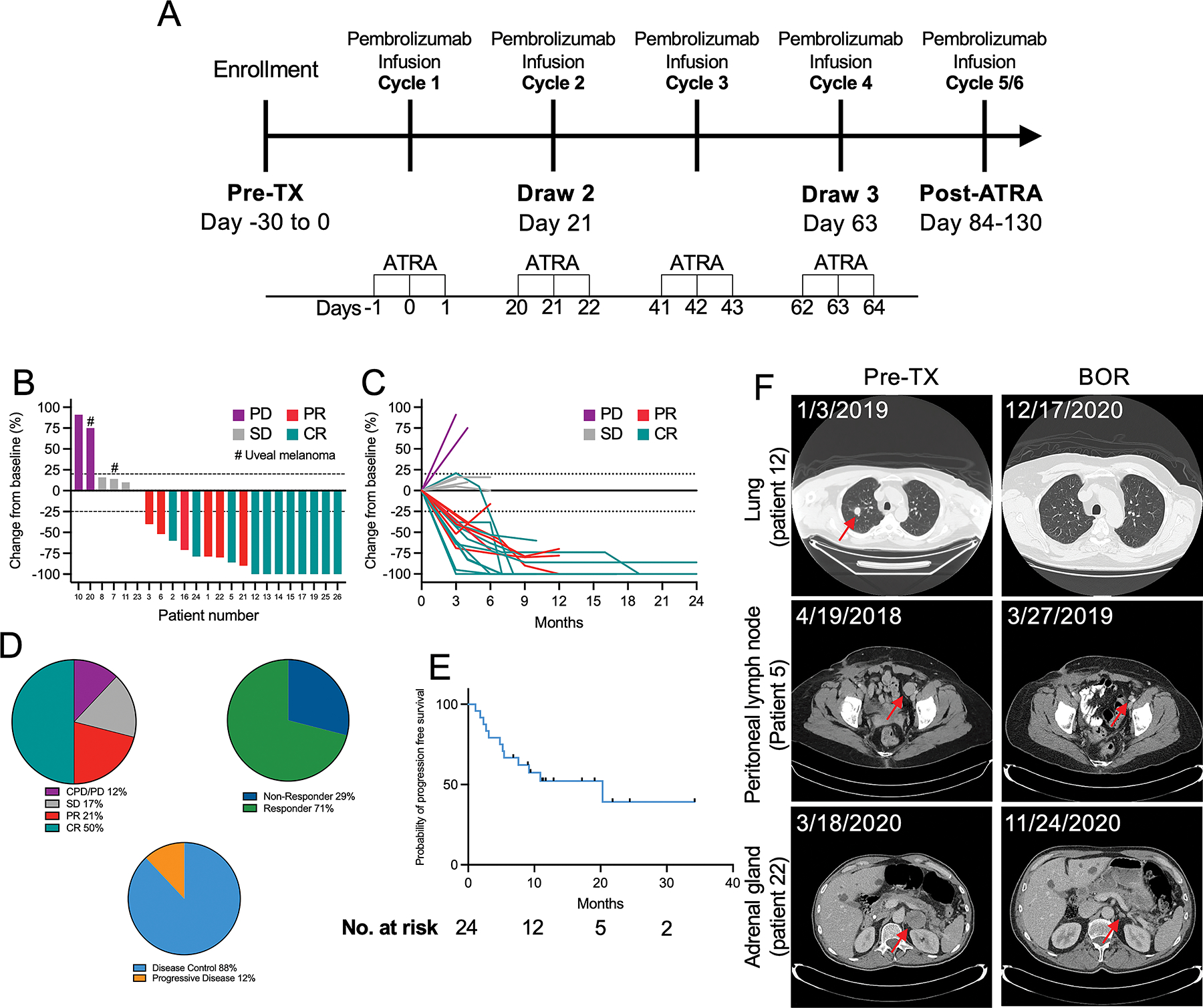
(A) Schematic depiction of the treatment and correlative sample collection schedule implemented in this clinical trial. (B) Waterfall plot depicting best overall responses (BOR) according to RECIST v1.1 criteria. (C) Spider plot depicting changes in target lesion size over time. (D) Comparisons of best of overall response, overall response rate, and disease control rate. (E) Survival curve showing the probability of progression free survival analyzed using the Kaplan-Meyer method, with data censored at first sign of progression, death, or last known follow-up. (F) Representative images from computerized tomography (CT) scans showing responses in the labeled organs.
Assessments
Primary outcome measures included safety assessments. Safety assessments included numbers of patients experiencing DLTs, (serious) adverse events, and changes from baseline clinical laboratory parameters. Adverse events were categorized and monitored according to CTCAE v4 guidelines. The secondary objectives were to describe the safety and toxicity of combined treatment with pembrolizumab and ATRA and to assess the antitumor activity in terms of a) the reduction in circulating MDSCs and b) PFS. Exploratory efficacy assessments included: ORR, DCR, clinical benefit rate (CBR), PFS at six months, and OS. Efficacy assessments were measured by standard of care clinical imaging, interpreted by a certified radiologist, and verified by the treating physician. Radiologic responses were categorized based on RECISTv1.1 criteria.
Sample collection
Peripheral blood from each patient was collected into tubes containing acid citrate dextrose anticoagulant (BD Biosciences). Research blood draws were performed at four time points: pre-treatment (0 to 30 days prior to the first ATRA administration), draw 2 (day 21), draw 3 (day 63), and post-ATRA treatment (84–130 days), as illustrated in Fig. 1A. Each research blood draw corresponded with standard of care clinical blood testing including complete blood counts (CBC) with automated differential count.
Flow cytometric analysis of circulating MDSCs
Peripheral blood was collected, and plasma was removed after centrifugation at 340 ×g for 10 min. Peripheral blood mononuclear cells (PBMCs) were isolated by ficoll (Cytiva) density centrifugation. Freshly isolated PBMCs were stained using the following fluorescently labeled monoclonal antibodies: APC-Lineage (CD3, CD19, CD20, CD56), BV570-CD45 (clone HI30, RRID:AB_2563426), BV421-CD11b (clone ICRF44, RRID: AB_11219589), APC-Cy7-CD14 (clone HCD14, RRID: AB_830693), PE-Cy7-CD15 (clone W6D3, RRID: AB_2561670), FITC-CD33 (clone HIM3-4, RRID: AB_314344), PerCP-Cy5.5-HLA-DR (clone L243, RRID: AB_893567), PE-Lox1 (clone 15C4, RRID: AB_2562181) (BioLegend). Live/dead discrimination was performed using Zombie Red dye (Biolegend). Stained cells were analyzed using the Beckman Coulter Gallios flow cytometer at the University of Colorado Cancer Center Flow Cytometry Shared Resource. Data were analyzed using FlowJo software Version 10.7 (Treestar, RRID:SCR_008520).
T cell response assay
PBMCs were isolated as described above and incubated for five hours in the presence of 1 μg/mL of NyESO1, tyrosinase, and gp100 peptide pools (PROIMMUNE) with FITC-anti-CD107a (clone H4A3, Biolegend, RRID: AB_1186036) and 1X Protein Transport Inhibitor (eBioscience) in RPMI1640 containing 10% FBS (Gemini Bio-Products), 2 mM l-glutamine (Mediatech), 100 μg/mL streptomycin (Mediatech), 100 IU/mL penicillin (Mediatech), 25 mM HEPES (Mediatech). After incubation, the cells were washed and stained with APC-Cy7-CD8 (clone HIT8a, RRID:AB_10613636), PerCP-Cy5.5-CD19 (clone HIB19, RRID:AB_2073119), PerCP-Cy5.5-HLA-DR (clone L243, RRID: AB_893567), and Zombie Red (Biolegend). The cells were then washed, fixed, and permeabilized using the BD Bioscience Cytoperm/Cytofix (AB_2869008) kit per the manufacturer’s protocol, and stained with BV421-IFNγ (clone 4S.B3, Biolegend, RRID: AB_2561398). Flow cytometric analysis was performed, and data analyzed as above.
T cell suppression assay
The suppressive function of MDSCs was determined as previously described (11). Total MDSCs were isolated using stepwise magnetic bead isolation. T cells for stimulation were negatively selected using the Miltenyi Biotec Pan T Cell Isolation Kit according to the manufacturer’s protocol. To purify MDSCs, in a separate aliquot, T cells were first positively selected using CD3 microbeads (Miltenyi Biotec), followed by removal of B cells (CD19 microbeads, Miltenyi Biotec), and NK cells (CD56 microbeads, Miltenyi Biotec). Remaining unlabeled cells were then separated based on HLA-DR (HLA-DR microbeads, Milteny Biotec), with HLA-DR− MDSCs and HLA-DR+ mature myeloid cells. All separations were performed on a Miltenyi Biotec AutoMACS Pro separator.
T cells from the pan T cell isolation kit were first labeled with 5 μM carboxyfluorescein succinimidyl ester (CFSE, Sigma Aldrich). Labeled T cells were stimulated using plate bound anti-CD3 (clone OKT3, Biolegend, RRID: AB_571927, 5μg/mL) and soluble anti-CD28 (clone 28.2, Biolegend, AB_314304, 5μg/mL) in complete RPMI as described above in round bottom 96 well plates. T cells were mixed with HLA-DR+ cells or HLA-DR− MDSCs at a 1:1 ratio with 105 T cells and 105 of either population of myeloid cells. Unstimulated T cells and stimulated T cells were included as controls.
After 96 hours of incubation, cells were stained with PerCP-Cy5.5 anti-CD3 (Clone SK7, RRID: AB_10640736), Pacific Blue anti-CD4 (clone OKT4, RRID:AB_1595438), APC-Cy7 anti-CD8 (clone HIT8a, RRID: AB_2044006), and Zombie Red from Biolgend. Flow cytometric analysis was analyzed as above. Only samples with sufficient cell numbers to perform this assay with all controls were included in this analysis.
Circulating cytokine analysis
The concentrations of IL-1β, IL-2, IL-4, IL-6, IL-8, IL-10, IL-12p70, IL-13, tumor necrosis factor-α (TNFα), and IFNγ in the plasma were measured using the V-PLEX Proinflammatory Panel 1 Human Kit (Meso Scale Discovery) according to the manufacturer’s protocol. The concentrations of each cytokine were quantified on a QuickPlex SQ 120 instrument (Meso Scale Discovery) located in the Human Immune Monitory Shared Resource at the University of Colorado School of Medicine.
Statistical analysis
The primary goal of this phase I/II study is to characterize the safety and establish the RP2D. The dose selected was based on the pharmacokinetics and effects on MDSCs observed in previous trials (4,6,7). A total of 24 evaluable subjects provide a two-sided 95%-exact confidence interval with a width no wider than 0.37 if the true toxicity rate is at most 25%.
The study is powered on the endpoint of MDSCs, as this is the primary rationale for combining ATRA with pembrolizumab. Our previous study showed that the mean reduction of MDSCs was 16.5 with a variance of 121 among four subjects. Due to the small sample size, we assume conservatively that the mean reduction is 70% and the variance is 30% more than the observed values, resulting in a mean reduction of 11.58 and a variance of 157.3. We assume the response rate of this study is no lower than 50%, thus we will have twelve or more responding subjects among the 24 enrolled. These twelve subjects provide 83% power to detect a mean reduction of 11.58 with a variance of the reduction of 157.3 using a paired t-test with a two-sided alpha of 0.05.
Comparison of pre- and post-ATRA treatment data was performed using the Wilcoxon signed-rank test. Comparisons across more than two timepoints were performed using a mixed effects model with Greenhouse–Geisser correction and Dunnet’s multiple comparisons test. All statistical tests were performed using GraphPad Prism version 9.2.0 or SAS version 9.4 (RRID:SCR_008567).
Data availability
The datasets used in the current analysis are available from the corresponding authors upon reasonable request.
Results
Patients
A total of 24 patients were enrolled in this prospective, single arm, single institution clinical trial between 10/31/2017 and 07/30/2020 (Table 1). At enrollment, the median age was 66 years (42 – 94), median BMI was 28.5 (19 – 36), and 87% (n = 21) of the patients were male. All patients had an ECOG performance status ≤1, and 17% (n = 4) had elevated lactate dehydrogenase (LDH) levels on the date of informed consent. Among enrolled patients, 75% (n = 18) had received no previous treatment for melanoma, with 13% (n = 3) receiving prior ipilimumab, 4% (n = 1) receiving prior radiation (brain metastases ~3 years prior to enrollment), 8% (n = 2) receiving prior plaque brachytherapy, and none of the patients receiving prior targeted therapy (Table 1). Sixty seven percent (n = 16) of patients had a cutaneous primary melanoma, 8% (n = 2) had a uveal primary melanoma, 4% (n = 1) had a mucosal primary melanoma, and 21% (n = 5) had an unknown primary melanoma. BRAF V600 mutations were observed in 50% (n = 12) of patients, and NRAS mutations were observed in 20% (n = 5) of patients. The median time from initial melanoma diagnosis to trial screen was 11.5 months (range, 0 – 62 months). At trial screen, the patients had an average of two metastatic sites with the most common sites of metastases being lung (50%, n = 12), lymph node (38%, n = 9), soft tissue (33%, n = 8), and liver (17%, n = 4). At the time of enrollment, the average sum of the longest axis of target lesions for RECIST v1.1 assessment was 51 mm (8 – 163 mm). Follow-up began at the time of enrollment, lasting until death, withdrawal due to enrollment in a different clinical trial, change in treatment, or data cut off, with a median duration of follow-up at the time of data cut-off of 15 months (3 – 35 months), whichever occurred first.
Table 1.
Patient demographics and disease characteristics at baseline
| Characteristic | All patients (n = 24) |
|---|---|
|
| |
| Median age, years (range) | 66 (42 – 94) |
| Male sex, n (%) | 21 (87) |
| Median BMI (range) | 28.5 (19 – 36) |
| ECOG performance status, n (%) | |
| 0 | 14 (58) |
| 1 | 10 (42) |
| Median time from diagnosis to trial screen, months (range) | 11.5 (0 – 62) |
| Median follow-up, months (range)a | 15 (3 – 34) |
| Elevated LDH level, n (%) | 4 (17) |
| Prior therapies, n (%) | |
| Ipilimumab | 3 (13) |
| Radiation | 1 (4) |
| Plaque brachytherapy | 2 (8) |
| No prior therapies | 18 (75) |
| Primary melanoma subtype, n (%) | |
| Cutaneous | 16 (67) |
| Uveal | 2 (8) |
| Mucosal | 1 (4) |
| Unknown | 5 (21) |
| BRAFV600 mutational status, n (%) | |
| WT | 9 (38%) |
| Mutant | 12 (50%) |
| Undetermined | 3 (12%) |
| Sites of metastasis at trial screen, n (%) | |
| Lung | 12 (50%) |
| Lymph nodes | 9 (38%) |
| Soft tissue | 8 (33%) |
| Liver | 4 (17%) |
| Adrenal gland | 2 (8%) |
| Subcutaneous | 2 (8%) |
| Skin | 2 (8%) |
| Brain | 2 (8%) |
| Breast | 1 (4%) |
| Bone | 1 (4%) |
| M Stage, n (%) | |
| M1a | 11 (46%) |
| M1b | 4 (17%) |
| M1c | 7 (29% |
| M1d | 2 (8%) |
Follow-up was defined as the time from trial enrollment until one of the following: death, withdrawal due to enrollment in a different clinical trial or change in treatment, two years after completing treatment (two-year regiment), or data cut-off, whichever occurred first.
Safety
The mean duration of exposure to treatment was 8 months (2 – 24 months) from the date of the first dose of ATRA to the off-treatment date. Grade 3 or 4 adverse events that were attributed to pembrolizumab by the investigators occurred in 4 (17%) patients, and grade 3 or 4 events that were attributed to ATRA occurred in 1 (4%) patient (Table 2). The rate of permanent discontinuation of a study drug because of treatment-related adverse events was 25% (n = 6). Four patients (17%) discontinued the study due to pembrolizumab-related side effects; two patients (8%) experienced immune-related (IR) colitis, one patient (4%) experienced IR hepatitis, and one patient (4%) experienced polymyalgia rheumatica-like myalgias, and two patients discontinued the study due to ATRA-related side effects including intolerable headaches (n = 1, 4%) and stroke (n = 1, 4%).
Table 2.
Most commonly occurring treatment-related adverse events
| Event | Pembrolizumab | ATRA | ||
|---|---|---|---|---|
| Any | Grade 3 or 4 | Any | Grade 3 or 4 | |
| Number of patients with event (percent) | ||||
| Any adverse event | 24 (100) | 11 (46) | 24 (100) | 11 (46) |
| Treatment-related adverse event | 24 (100) | 4 (17) | 24 (100) | 1 (4) |
| General disorders | ||||
| Fatigue | 16 (67) | 1 (4) | 11 (46) | 0 |
| Decreased appetite | 2 (8) | 0 | 0 | 0 |
| Eye and nervous system disorders | ||||
| Headache | 2 (8) | 0 | 22 (92) | 0 |
| Meningitis/stroke | 0 | 1 (4) | 0 | 1 (4) |
| Gastrointestinal disorders | ||||
| Nausea | 4 (17) | 0 | 7 (29) | 0 |
| Diarrhea | 5 (21) | 1 (4) | 2 (8) | 0 |
| Dry mouth | 4 (17) | 0 | 4 (17) | 0 |
| Gastroesophageal reflux disease | 2 (8) | 0 | 0 | 0 |
| Skin and subcutaneous tissue disorders | ||||
| Rash maculo-papular | 5 (21) | 0 | 4 (17) | 0 |
| Pruritus | 2 (8) | 0 | 0 | 0 |
| Hepatic disorders | ||||
| Aspartate aminotransferase increased | 4 (17) | 0 | 2 (8) | 0 |
| Alanine aminotransferase increased | 3 (13) | 0 | 1 (4) | 0 |
| Hematologic and lymphatic disorders | ||||
| Anemia | 4 (17) | 0 | 2 (8) | 0 |
| Endocrine disorders | ||||
| Hypothyroidism | 2 (8) | 0 | 0 | 0 |
| Adrenal insufficiency | 0 | 1 (4) | 0 | 0 |
All adverse events assessed using CTCAE v.4.
The most common adverse events of any grade likely related to ATRA were headache (n = 22, 92%), fatigue (n = 11, 46%), nausea (n = 7, 29%), vomiting (n = 5, 21%), dry mouth (n = 4, 17%), and dry skin (n = 4, 17%) (Table 2). The most common adverse events of any grade likely related to pembrolizumab were fatigue (n = 16, 67%), diarrhea (n = 5, 21%), rash maculo-papular (n = 5, 21%), and dry mouth (n = 4, 17%). Of the events related to ATRA, one patient experienced a grade 4 event, and of the events related to pembrolizumab, all were grade 3 or lower. A full description of all adverse events observed during the study can be found in Supplementary Table 2.
Of the 24 patients in the study, only one patient (4%) experienced a dose limiting toxicity (DLT, as defined in Supplementary Table 3) at the dose of 150mg/m2 ATRA + 200 mg Q3W pembrolizumab. Therefore, this dose was determined to be the RP2D and MTD, and all patients received this dose.
Efficacy
With a median follow-up time of 15 months (3 – 34 months), the ORR was 71% (n = 17) and the DCR was 88% (n = 21) (Fig. 1B–2D, Table 3). At the time of data cut-off, the six-month PFS rate for all patients was 63% and the median PFS (calculated from treatment start date to data cut-off) was 20.3 months (95% CI, 5.2 – 12.9) (Fig. 2E). Using RECIST v1.1 criteria, complete responses (CR) were observed in 50% (n = 12) of patients, partial responses (PR) in 21% (n = 5), stable disease (SD) in 17% (n = 4), and progressive disease (PD) or clinical progressive disease (CPD) observed in 12% (n = 3) (Fig. 1B and 1D).
Figure 2. The combination of ATRA and pembrolizumab decreases the number of circulating PMN-MDSCs.
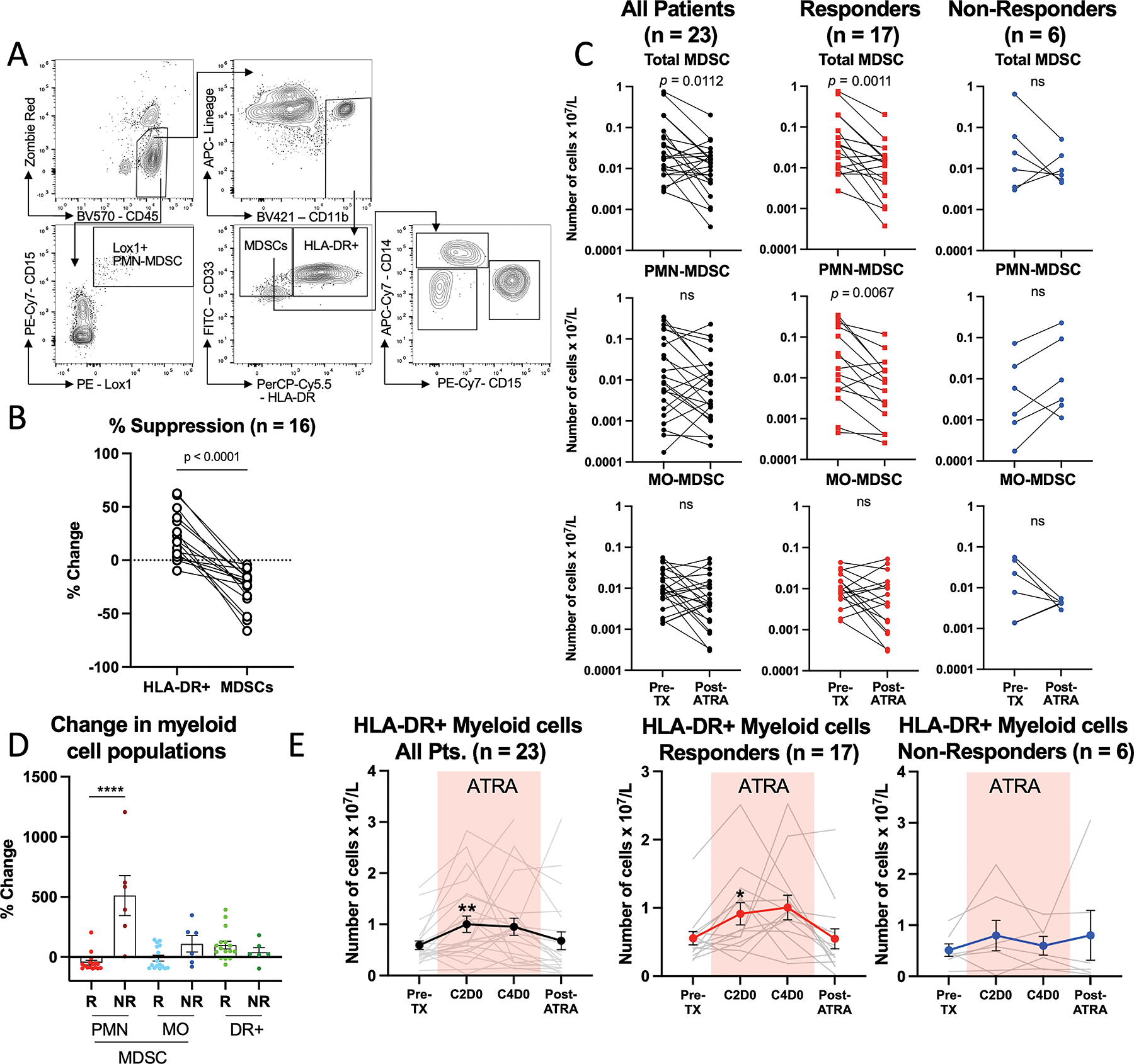
(A) Representative flow cytometric gating strategy to identify and quantify circulating MDSC subsets. (B) The suppressive function of MDSCs depicted as a percent change in T cell proliferation from T cells stimulated with anti-CD3/CD28 comparing HLA-DR+ myeloid cells (DR+) to MDSCs. Connecting lines indicate paired analysis from each patient. (C) Comparisons of the number of circulating MDSC subsets in pre-treatment (pre-TX) and post-ATRA blood samples, from all patients (pts), responding (R) patients, and non-responding (NR) patients. Connecting lines indicate paired analysis from each patient. (D) Summary of the percent change from pre- to post-ATRA timepoints in myeloid cell populations. (E) Comparisons of number of circulating HLA-DR+ mature myeloid cells in all patients (pts), responding (R) patients, and non-responding (NR) patients. Colored box denotes the time when the patients were being treated with ATRA. * Denotes p < 0.05, ** p < 0.01, *** p < 0.001. Cycle 2 Day 0 (C2D0), Cycle 4 Day 0 (C4D0).
Table 3.
Summary of investigator-assessed clinical efficacy parameters
| Parameter | All Patients (n = 24) |
|---|---|
|
| |
| Best overall response, n (%) | |
| Complete response (CR) | 12 (50%) |
| Partial response (PR) | 5 (21%) |
| Stable disease (SD) | 4 (17%) |
| Progressive disease (PD) | 2 (8%) |
| Clinical progressive disease (CPD)a | 1 (4%) |
| Median treatment duration, months (range)b | 8 (2 – 24) |
| Median duration of response, months (range)c | 11 (0.4 – 34) |
| Clinical benefit rate (CBR), n (%)d | 14 (58%) |
| Objective response rate (ORR), n (%)e | 17 (71%) |
| Disease control rate (DCR), n (%)f | 21 (88%) |
| Median follow-up time, months (range) | 15 (3 – 34) |
| Median PFS, months | 20.3 |
| 6 month PFS rate, n (%)g | 15 (63%) |
| Progressive disease (PD)h | 12 (50%) |
| On treatmenti | 10 (42%) |
| Median time to progression, months (range) | 4 (1 – 10) |
| Off treatmentj | 2 (8%) |
| Median time to progression, months (range) | 15 (9 – 20) |
| Survival rate | 75% |
Abbreviations: CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease; CPD, clinical progressive disease; CBR, clinical benefit rate; ORR, objective response rate; DCR, disease control rate; PFS, progression free survival.
Progression without a scan (physician diagnosed progression based on symptoms prior to scan).
Defined as time between first ATRA dose and last treatment dose (pembrolizumab or ATRA).
Defined as measured from the time at which CR or PR criteria are first met until the first date of PD.
Defined as the proportion of patients demonstrating CR, PR, or SD for a minimum of four months from date of first CR, PR, or SD.
Patients who achieved a best overall response of CR or PR.
Patients who achieved a best overall response of CR, PR, or SD.
Patients without progression at six months from the date of the first treatment.
Patients diagnosed with PD or CPD.
Patients who progressed while on treatment.
Patients who progressed after coming off treatment.
Response to therapy was assessed based on RECIST v1.1 criteria.
In patients who responded to therapy (CR or PR) (n = 17), the median duration of response was 11 months (0.4 – 34 months) (Fig. 1C, and Table 3). At the time of data cut off, 12 patients (50%) were still responding to therapy. Overall, 58% (n = 14) had clinical benefit (CR, PR, or SD for a minimum of four months from their initial radiographically demonstrated response) from the combination. Responses to therapy were observed across metastatic sites, including lung, brain (previously untreated), skin, adrenal gland, and lymph nodes (Fig. 1F). Of the 58% (n = 14) of patients that showed clinical benefit, 11 patients had a known driver mutation (BRAF, n = 6; NRAS, n = 4; BRAF+NRAS, n = 1; mutational analysis was unavailable from two patients) (Supplementary Table 4). In addition, of patients who showed clinical benefit, ten were diagnosed with cutaneous primary melanomas, and four were melanomas of unknown primary. Finally, of the patients who showed clinical benefit, 14% (n = 2) received prior ipilimumab monotherapy and 86% (n = 12) received no prior therapies. Alternatively, of the patients who progressed while off treatment (n = 2, 8%), both patients experienced a BOR of SD. Median OS was not reached; however, the OS rate (calculated from treatment start date to data cut-off) was 71%, with a one-year OS of 80%.
MDSC analysis
MDSCs were identified in PBMCs as CD45+CD3−CD19−CD56−CD11b+CD33+HLA-DR−/low cells (Fig. 2A). There is strong evidence in the literature and from our group showing that this population and its subsets are highly suppressive of T cell proliferation (3–6,12–14), and pre-treatment blood samples showed that this was also the case in our patient cohort (Fig. 2B) Paired analysis comparing pretreatment (Pre-TX) to post-ATRA timepoints showed sustained decreases in absolute numbers of circulating total MDSCs (CD45+CD3−CD19−CD56−CD11b+CD33+HLA-DR−/low) four to six weeks after stopping ATRA (Fig. 2C). This was more striking in responding patients (CR or PR) (n = 17), where the average percent change was −49%, compared to non-responders (SD or PD) (n = 6), where the average percent change was +110% (p = 0.017) (Fig. 2C and 2D). While the absolute numbers of PMN-MDSCs were not significantly different when comparing all patients, circulating PMN-MDSCs (CD45+Lox1+CD15+) (Fig. 2A) decreased in responding patients (percent change −46%), but not in non-responders (percent change +427% [p = 0.003]) (Fig. 2C and 2D) compared at the Pre-TX and Post-ATRA timepoints. No statistically significant changes were observed in the frequency of MO-MDSCs in either responders or non-responders (Fig. 2C and 2D).
To assess whether the decrease in MDSC populations was due to ATRA-mediated differentiation into mature HLA-DR+ myeloid cells (6), we measured the frequency of HLA-DR+ myeloid cells (CD45+CD3−CD19−CD56−CD11b+CD33+HLA-DR+) (Fig. 2A). We observed a statistically significant increase in HLA-DR+ myeloid cells at the cycle 2 day 0 (C2D0) timepoint (p = 0.008, study day 21) and a trend toward increased levels at the C4D0 timepoint (p = 0.087, study day 63), while no differences were observed at the post-ATRA timepoint, when the patients were no longer on ATRA (Fig. 2D). This was driven by increased numbers of HLA-DR+ myeloid cells in the responding but not in the non-responding patients (Fig. 2E).
Multiplex cytokine analysis
To determine if the decreases in MDSCs were associated with alterations in the concentrations of circulating cytokines, we performed multiplex cytokine analysis. We found that concentrations of IL-6 and IL-10 were elevated while patients were on ATRA but returned to normal levels after the completion of ATRA treatment (Fig. 3A). IL-12p70, and TNFα were also significantly increased but rose over time and were not associated with ATRA treatment (Fig. 3A). There were no statistically significant differences or trends in the circulating concentrations of the other cytokines measured (IL-1B, IL-2, IL-4, IL-8, IL-13, and IFN-γ) (Supplementary Fig S1A).
Figure 3. The combination of ATRA and pembrolizumab promotes cytokine production and tumor-specific T cell activation.
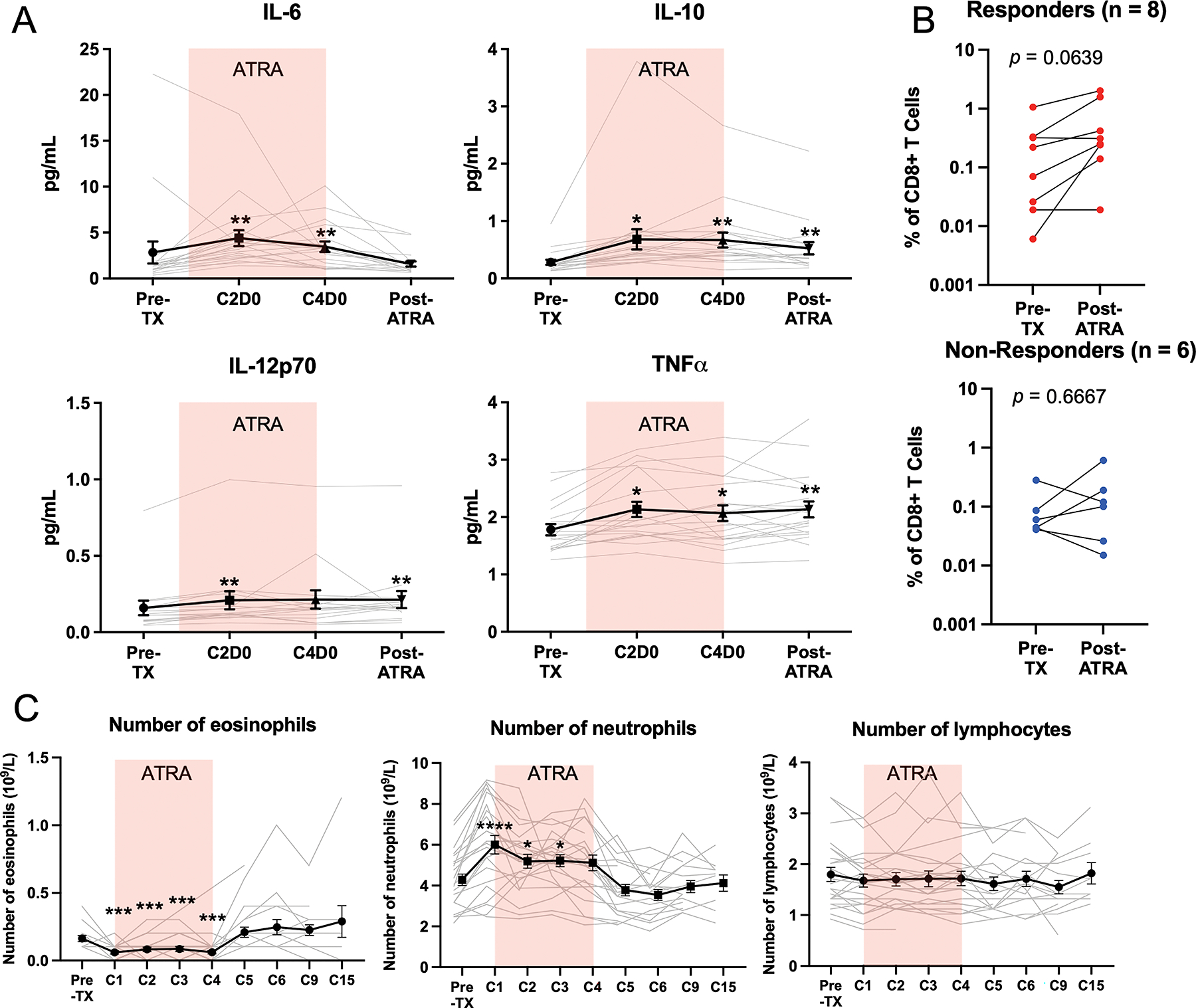
(A) Analysis of circulating cytokine concentrations. (B) Comparisons of the frequency of tumor antigen-specific (NyESO, tyrosinase, and gp100) T cells, identified as CD8+CD107+IFNγ+ T cells. (C) Analysis of the number of circulating white blood cell populations from clinical complete blood count (CBC) testing. Colored box denotes the time when the patients were being treated with ATRA. * Denotes p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
Tumor-specific T cell analysis
Because we have previously observed that targeting MDSCs with ATRA improves the activation of circulating tumor-specific T cells (6), we compared the frequency of tumor antigen (NY-ESO-1, tyrosinase, gp100)-specific T cells between pre-TX and post-ATRA timepoints in responding vs. non-responding patients. We found a trend towards modest increases in antigen specific CD8+ T cells in responding patients but not in non-responding patients (Fig. 3B and Supplementary Fig. S1B).
Other hematologic alterations
As in our previous study (6), ATRA significantly reduced the number of circulating eosinophils (Fig. 3C). This observation was expected as ATRA prevents the differentiation of eosinophil hemopoietic precursors into mature eosinophils by downregulating the expression of the IL-5 receptor (15). In addition, we noted a significant increase in the number of circulating neutrophils associated with the timing of ATRA treatment (Fig. 3C). No changes in the number of circulating lymphocytes were noted (Fig. 3C). No other clinically significant changes were noted from the clinical laboratory testing (Supplementary Fig. S2).
Discussion
Improving ICI-response rates remains an important goal in the immunoncology field. Because MDSCs are an important mediator of resistance to ICIs (3–6), targeting these highly immunosuppressive cells in combination with ICIs is a rational therapeutic combination. In this single arm, single institution clinical trial we found that the combination of ATRA and pembrolizumab was safe and well tolerated. Furthermore, the combination effectively lowers the frequency of circulating MDSCs. This is the first time this combination has been tested in metastatic melanoma patients, and the encouraging clinical outcomes found, point to the importance of MDSCs in ICI responses and the potential of this combination as an effective first line therapy for metastatic melanoma patients.
The primary objective of this study was to determine the MTD and RP2D of this combination. These were established to be 150mg/m2 ATRA + 200mg Q3W pembrolizumab. Regarding the safety and tolerability of the combination (secondary study objective), we observed that 21% (n = 5) of patients experienced grade 3 or 4 treatment-related adverse events. The most common treatment-related adverse event of any grade was headache (92%, n = 22), which was limited to and corresponded with the three-day course of ATRA treatment. Overall, this combination was well tolerated, with 25% (n = 6) of patients discontinuing treatment due to treatment-related adverse events, a rate similar to that reported for pembrolizumab monotherapy (16). Of note, one patient presented with a complex multifactorial picture of encephalitis complicated by a history of cerebral microangiopathy. The ATRA was discontinued as it was felt that it may have contributed to the onset of their symptoms, which eventually resolved. In addition, one patient elected to discontinue ATRA due to intolerable headache.
The median PFS in this study was 20.3 months (secondary study objective). Further exploratory post-hoc analysis showed that the ORR was 71%, with 50% of patients achieving a CR. The responses were durable, with the median duration of response lasting 11 months. These results are notably higher than previously published clinical trials using single agent pembrolizumab (16). When observed in the context of other currently available immunotherapies in similar patient populations, such as combination ipilimumab (anti-CTLA-4) and nivolumab (anti-PD-1) (17) or relatlimab (anti-Lag3) and nivolumab (18), the favorable safety profile, high response rate, and extended median PFS make this a promising combination therapy in the frontline setting for stage IV melanoma patients. While the continued addition of new immune checkpoint inhibitors to current combinations promotes enhanced immune cell activation, this can have the unintended effect of increasing the risk of treatment-limiting immune-related adverse events. Furthermore, the use of ATRA may particularly benefit patients with high frequencies of MDSCs who may be more prone to ICI-resistance. Therefore, targeting the multiple immunosuppressive functions of MDSCs using this approach offers the potential for improved responses while decreasing the risk of treatment limiting and often life altering autoimmune toxicities.
While we noted high response rates in both cutaneous and melanomas of unknown primary, the combination was less effective in patients diagnosed with rare melanoma subtypes (uveal or mucosal). Uveal melanomas are noted for their low ICI response rates, with low mutational burden and predisposition for liver metastasis making them particularly difficult to treat (19–21). However, it is important to clearly state that the current study was not powered to compare response rates across melanoma subtype or metastatic sites, and larger clinical trials specifically designed to test these hypotheses will be required to make final conclusions.
Targeting MDSCs is an attractive way to enhance the efficacy of ICIs, with few significant side effects (22–25). In the present study, we found that the combination of ATRA and pembrolizumab effectively reduced the frequency of total MDSCs, mainly driven by a significant reduction in circulating PMN-MDSCs (secondary study objective). This was more apparent in patients who responded to the therapy than those who did not. The PMN-MDSC population is highly immunosuppressive and plays an important role in ICI resistance (4,26). We did not observe statistically significant changes in the frequency of circulating MO-MDSCs. This may be due to the changes in MO-MDSCs being more observable in the tumor microenvironment (13). However, as the present study did not include tumor biopsies, this hypothesis will be tested in future studies. The decrease in circulating MDSCs corresponded with an increase in HLA-DR+ myeloid cells during the ATRA treatment portion of the study. Pre-treatment levels of these cells have previously been observed to strongly correlate with responses to ICIs (27). ATRA may increase their frequency, promoting antitumor immunity as proposed in this prior study (27).
The levels of several circulating cytokines have been associated with differential clinical outcomes in patients with metastatic melanoma (12,28,29). While the changes we observed were modest, the levels of circulating IL-6 and IL-10 increased during the time when patients were on ATRA and returned to near baseline levels during the pembrolizumab monotherapy portion of the study. We also observed consistent, but small, increases in the concentrations of IL-12p70 and TNFα that remained elevated after ATRA completion. These cytokines have mixed associations with responses to ICIs reported in the literature. IL-6 and IL-10 in particular can be associated with both positive and negative outcomes (12,28–30). Our previous work suggests that high levels of IL-6 are associated with MDSC accumulation (12). However, other results show that changes in IL-6 levels can be associated with positive clinical outcomes (31). Likewise, while IL-10 is an immunosuppressive cytokine produced by MDSCs (4), high levels of IL-10 have been correlated with positive ICI responses and clinical outcomes (30,32). Separate previous reports have found that ATRA can induce IL-6 (33) and IL-10 (34) expression in multiple cell types involved in antitumor immunity, limiting our ability to determine the source of these cytokines in the circulation of these patients. The mechanisms by which this combination may alter the expression and circulating concentrations of these cytokines and the cell types that are producing them merits further investigation.
In addition to its effects on MDSCs, ATRA has been shown to activate the innate immune sensing pathway in several types of tumor cells, including melanoma (35). Additionally, activation of the innate immune sensing pathway can induce the expression of many cytokines, including IL-6, IL-10, IL-12, and TNFα (36,37) that we found to be increased in the circulation of the patients in this study. While the current study was not designed to analyze this pathway, it is possible that the dual action of targeting MDSCs and inducing a more immunogenic tumor microenvironment may account for the high response rates in this patient cohort. Future studies that include on treatment tumor biopsies will be required to test this hypothesis.
Overall, this pilot study demonstrates that the combination of ATRA and pembrolizumab appears to be a promising approach to enhance the efficacy of anti-PD-1 and further supports the role of MDSCs in preventing effective antitumor immunity. Several new therapies designed to target tumor-induced MDSCs are currently entering clinical trials (38–40). These include therapies that induce MDSC differentiation (39), target MDSC chemotaxis (38), and those that target the suppressive functions of MDSCs (IL-10, TGFβ, Arg1, IDO1)(40–42). In addition, a recent study found that adding radiation therapy to ATRA and anti-PD-1 therapy may be an effective mechanism to further induce the differentiation of MDSCs towards a pro-inflammatory, antitumor myeloid cell phenotype (43).
Several study limitations were noted. First, this is a single arm study lacking pembrolizumab monotherapy as a control, limiting the investigators’ ability to analyze the contributions of each compound to clinical responses or the correlative analyses (Supplementary table 5). Of note, previous studies have observed reductions in circulating MDSCs in patients treated with single agent anti-PD-1 (44–46). Second, the low numbers of patients from a single institution and relatively short follow-up time limit the generalizability to the larger melanoma population. Furthermore, most patients in the current study had a low disease burden, received few prior treatments, and none had previously received anti-PD-1 therapies, possibly accounting for the high ORR and CR rates. Future studies will include more heavily pretreated patients and patients from multiple institutions to test the generalizability of these results. In addition, there are many previously reported limitations in the RECIST v1.1 criteria used to measure clinical responses (47,48). Some of these limitations may account for some of high response rates observed in this study. While the current proof of principal study only had sufficient funding to treat patients with ATRA during the first four cycles of pembrolizumab, future studies will extend the treatment duration. Finally, while the study was originally intended to investigate changes in MDSC suppressive functions at each investigational timepoint, the decrease in overall MDSC numbers limited our ability to perform the T cell suppression assays at the timepoints indicated in our clinical trial protocol. Future studies will include later blood draw timepoints to assess the repopulation and functional status of circulating MDSCs following discontinuation of ATRA.
Supplementary Material
Translational relevance.
Myeloid-derived suppressor cells (MDSCs) limit the efficacy of immune checkpoint inhibitors (ICIs) including anti-PD-1. We sought to target MDSCs using the differentiating agent all-trans retinoic acid (ATRA) to enhance the efficacy of the anti-PD-1 antibody pembrolizumab in anti-PD-1 naïve stage IV melanoma patients. In this phase I/II clinical trial, we found that the combination was well tolerated, and the recommended phase II dose was established. The combination effectively lowered the number of circulating MDSCs. The median progression-free survival was 20.3 months. When compared to prior clinical trials with single agent pembrolizumab, or clinical trials testing combinations of ICIs, the combination of ATRA and pembrolizumab has a favorable overall response rate (ORR) of 71%, with 50% of patients experiencing a complete response (CR). These results provide initial evidence for the safety and efficacy of this approach in the frontline setting for stage IV melanoma patients.
Acknowledgements
This study (ClinicalTrials.gov # NCT03200847) was funded by Merck & Co Inc. (MISP 54054) to MDM. Further funding support was provided to the University of Cancer Center under NCI Cancer Center Support Grant P30CA046934. We are grateful to the patients and their families for their participation in the study; the funders; and all investigators and their teams who participated in the trial.
Footnotes
Conflict of interest statement: M.M. has received research grants from Merck Sharpe & Dohme. K.L. has received grants and personal fees from Amgen, Bristol-Myers Squibb, Genentech, GlaxoSmithKline, Merck Sharp & Dohme, Novartis, and Roche. T.M. has participated in advisory boards for Array BioPharma, Bristol Myers Squibb, Checkmate Pharmaceuticals, and Iovance Biotherapeutics. R.G. has received consulting or advisory fees from Array BioPharma, Bristol Myers Squibb, Incyte, Lumos Pharma (formerly NewLink Genetics), Pharmatech, Novartis, Roche/Genentech, and Vavotar Life Sciences. All other authors report no potential conflicts of interest.
References
- 1.Morrison C, Pabla S, Conroy JM, Nesline MK, Glenn ST, Dressman D, et al. Predicting response to checkpoint inhibitors in melanoma beyond PD-L1 and mutational burden. J Immunother Cancer 2018;6(1):32 doi 10.1186/s40425-018-0344-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tobin RP, Davis D, Jordan KR, McCarter MD. The clinical evidence for targeting human myeloid-derived suppressor cells in cancer patients. J Leukoc Biol 2017;102(2):381–91 doi 10.1189/jlb.5VMR1016-449R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weber R, Fleming V, Hu X, Nagibin V, Groth C, Altevogt P, et al. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front Immunol 2018;9:1310 doi 10.3389/fimmu.2018.01310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol 2021;21(8):485–98 doi 10.1038/s41577-020-00490-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Groth C, Hu X, Weber R, Fleming V, Altevogt P, Utikal J, et al. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br J Cancer 2019;120(1):16–25 doi 10.1038/s41416-018-0333-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tobin RP, Jordan KR, Robinson WA, Davis D, Borges VF, Gonzalez R, et al. Targeting myeloid-derived suppressor cells using all-trans retinoic acid in melanoma patients treated with Ipilimumab. Int Immunopharmacol 2018;63:282–91 doi 10.1016/j.intimp.2018.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res 2007;67(22):11021–8 doi 10.1158/0008-5472.CAN-07-2593. [DOI] [PubMed] [Google Scholar]
- 8.Iclozan C, Antonia S, Chiappori A, Chen DT, Gabrilovich D. Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer Immunol Immunother 2013;62(5):909–18 doi 10.1007/s00262-013-1396-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mirza N, Fishman M, Fricke I, Dunn M, Neuger AM, Frost TJ, et al. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res 2006;66(18):9299–307 doi 10.1158/0008-5472.CAN-06-1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ivanova A, Qaqish BF, Schell MJ. Continuous toxicity monitoring in phase II trials in oncology. Biometrics 2005;61(2):540–5 doi 10.1111/j.1541-0420.2005.00311.x. [DOI] [PubMed] [Google Scholar]
- 11.Davis RJ, Silvin C, Allen CT. Avoiding phagocytosis-related artifact in myeloid derived suppressor cell T-lymphocyte suppression assays. J Immunol Methods 2017;440:12–8 doi 10.1016/j.jim.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tobin RP, Jordan KR, Kapoor P, Spongberg E, Davis D, Vorwald VM, et al. IL-6 and IL-8 Are Linked With Myeloid-Derived Suppressor Cell Accumulation and Correlate With Poor Clinical Outcomes in Melanoma Patients. Front Oncol 2019;9:1223 doi 10.3389/fonc.2019.01223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tcyganov E, Mastio J, Chen E, Gabrilovich DI. Plasticity of myeloid-derived suppressor cells in cancer. Curr Opin Immunol 2018;51:76–82 doi 10.1016/j.coi.2018.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun 2016;7:12150 doi 10.1038/ncomms12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Upham JW, Sehmi R, Hayes LM, Howie K, Lundahl J, Denburg JA. Retinoic acid modulates IL-5 receptor expression and selectively inhibits eosinophil-basophil differentiation of hemopoietic progenitor cells. J Allergy Clin Immunol 2002;109(2):307–13 doi 10.1067/mai.2002.121527. [DOI] [PubMed] [Google Scholar]
- 16.Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann Oncol 2019;30(4):582–8 doi 10.1093/annonc/mdz011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hodi FS, Chesney J, Pavlick AC, Robert C, Grossmann KF, McDermott DF, et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol 2016;17(11):1558–68 doi 10.1016/S1470-2045(16)30366-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tawbi HA, Schadendorf D, Lipson EJ, Ascierto PA, Matamala L, Castillo Gutierrez E, et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N Engl J Med 2022;386(1):24–34 doi 10.1056/NEJMoa2109970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Algazi AP, Tsai KK, Shoushtari AN, Munhoz RR, Eroglu Z, Piulats JM, et al. Clinical outcomes in metastatic uveal melanoma treated with PD-1 and PD-L1 antibodies. Cancer 2016;122(21):3344–53 doi 10.1002/cncr.30258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bol KF, Ellebaek E, Hoejberg L, Bagger MM, Larsen MS, Klausen TW, et al. Real-World Impact of Immune Checkpoint Inhibitors in Metastatic Uveal Melanoma. Cancers (Basel) 2019;11(10) doi 10.3390/cancers11101489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chua V, Mattei J, Han A, Johnston L, LiPira K, Selig SM, et al. The Latest on Uveal Melanoma Research and Clinical Trials: Updates from the Cure Ocular Melanoma (CURE OM) Science Meeting (2019). Clin Cancer Res 2021;27(1):28–33 doi 10.1158/1078-0432.CCR-20-2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sarhan D, Cichocki F, Zhang B, Yingst A, Spellman SR, Cooley S, et al. Adaptive NK Cells with Low TIGIT Expression Are Inherently Resistant to Myeloid-Derived Suppressor Cells. Cancer Res 2016;76(19):5696–706 doi 10.1158/0008-5472.CAN-16-0839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Limagne E, Richard C, Thibaudin M, Fumet JD, Truntzer C, Lagrange A, et al. Tim-3/galectin-9 pathway and mMDSC control primary and secondary resistances to PD-1 blockade in lung cancer patients. Oncoimmunology 2019;8(4):e1564505 doi 10.1080/2162402X.2018.1564505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bae J, Accardi F, Hideshima T, Tai YT, Prabhala R, Shambley A, et al. Targeting LAG3/GAL-3 to overcome immunosuppression and enhance anti-tumor immune responses in multiple myeloma. Leukemia 2021. doi 10.1038/s41375-021-01301-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murciano-Goroff YR, Warner AB, Wolchok JD. The future of cancer immunotherapy: microenvironment-targeting combinations. Cell Res 2020;30(6):507–19 doi 10.1038/s41422-020-0337-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Condamine T, Dominguez GA, Youn JI, Kossenkov AV, Mony S, Alicea-Torres K, et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol 2016;1(2) doi 10.1126/sciimmunol.aaf8943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krieg C, Nowicka M, Guglietta S, Schindler S, Hartmann FJ, Weber LM, et al. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat Med 2018;24(2):144–53 doi 10.1038/nm.4466. [DOI] [PubMed] [Google Scholar]
- 28.Laino AS, Woods D, Vassallo M, Qian X, Tang H, Wind-Rotolo M, et al. Serum interleukin-6 and C-reactive protein are associated with survival in melanoma patients receiving immune checkpoint inhibition. J Immunother Cancer 2020;8(1) doi 10.1136/jitc-2020-000842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berraondo P, Sanmamed MF, Ochoa MC, Etxeberria I, Aznar MA, Perez-Gracia JL, et al. Cytokines in clinical cancer immunotherapy. Br J Cancer 2019;120(1):6–15 doi 10.1038/s41416-018-0328-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oft M IL-10: master switch from tumor-promoting inflammation to antitumor immunity. Cancer Immunol Res 2014;2(3):194–9 doi 10.1158/2326-6066.CIR-13-0214. [DOI] [PubMed] [Google Scholar]
- 31.Reitter EM, Ay C, Kaider A, Pirker R, Zielinski C, Zlabinger G, et al. Interleukin levels and their potential association with venous thromboembolism and survival in cancer patients. Clin Exp Immunol 2014;177(1):253–60 doi 10.1111/cei.12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamazaki N, Kiyohara Y, Uhara H, Iizuka H, Uehara J, Otsuka F, et al. Cytokine biomarkers to predict antitumor responses to nivolumab suggested in a phase 2 study for advanced melanoma. Cancer Sci 2017;108(5):1022–31 doi 10.1111/cas.13226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alatshan A, Kovacs GE, Aladdin A, Czimmerer Z, Tar K, Benko S. All-Trans Retinoic Acid Enhances both the Signaling for Priming and the Glycolysis for Activation of NLRP3 Inflammasome in Human Macrophage. Cells 2020;9(7) doi 10.3390/cells9071591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X, Allen C, Ballow M. Retinoic acid enhances the production of IL-10 while reducing the synthesis of IL-12 and TNF-alpha from LPS-stimulated monocytes/macrophages. J Clin Immunol 2007;27(2):193–200 doi 10.1007/s10875-006-9068-5. [DOI] [PubMed] [Google Scholar]
- 35.Bolis M, Paroni G, Fratelli M, Vallerga A, Guarrera L, Zanetti A, et al. All-Trans Retinoic Acid Stimulates Viral Mimicry, Interferon Responses and Antigen Presentation in Breast-Cancer Cells. Cancers (Basel) 2020;12(5) doi 10.3390/cancers12051169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duewell P, Steger A, Lohr H, Bourhis H, Hoelz H, Kirchleitner SV, et al. RIG-I-like helicases induce immunogenic cell death of pancreatic cancer cells and sensitize tumors toward killing by CD8(+) T cells. Cell Death Differ 2014;21(12):1825–37 doi 10.1038/cdd.2014.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heidegger S, Wintges A, Stritzke F, Bek S, Steiger K, Koenig PA, et al. RIG-I activation is critical for responsiveness to checkpoint blockade. Sci Immunol 2019;4(39) doi 10.1126/sciimmunol.aau8943. [DOI] [PubMed] [Google Scholar]
- 38.Bilusic M, Heery CR, Collins JM, Donahue RN, Palena C, Madan RA, et al. Phase I trial of HuMax-IL8 (BMS-986253), an anti-IL-8 monoclonal antibody, in patients with metastatic or unresectable solid tumors. J Immunother Cancer 2019;7(1):240 doi 10.1186/s40425-019-0706-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sharma MD, Rodriguez PC, Koehn BH, Baban B, Cui Y, Guo G, et al. Activation of p53 in Immature Myeloid Precursor Cells Controls Differentiation into Ly6c(+)CD103(+) Monocytic Antigen-Presenting Cells in Tumors. Immunity 2018;48(1):91–106 e6 doi 10.1016/j.immuni.2017.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ciardiello D, Elez E, Tabernero J, Seoane J. Clinical development of therapies targeting TGFbeta: current knowledge and future perspectives. Ann Oncol 2020;31(10):1336–49 doi 10.1016/j.annonc.2020.07.009. [DOI] [PubMed] [Google Scholar]
- 41.Tang K, Wu YH, Song Y, Yu B. Indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors in clinical trials for cancer immunotherapy. J Hematol Oncol 2021;14(1):68 doi 10.1186/s13045-021-01080-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR, et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J Immunother Cancer 2017;5(1):101 doi 10.1186/s40425-017-0308-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rao E, Hou Y, Huang X, Wang L, Wang J, Zheng W, et al. All-trans retinoic acid overcomes solid tumor radioresistance by inducing inflammatory macrophages. Science Immunology 2021;6(60):eaba8426 doi doi: 10.1126/sciimmunol.aba8426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pico de Coana Y, Wolodarski M, van der Haar Avila I, Nakajima T, Rentouli S, Lundqvist A, et al. PD-1 checkpoint blockade in advanced melanoma patients: NK cells, monocytic subsets and host PD-L1 expression as predictive biomarker candidates. Oncoimmunology 2020;9(1):1786888 doi 10.1080/2162402X.2020.1786888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun SH, Benner B, Savardekar H, Lapurga G, Good L, Abood D, et al. Effect of Immune Checkpoint Blockade on Myeloid-Derived Suppressor Cell Populations in Patients With Melanoma. Front Immunol 2021;12:740890 doi 10.3389/fimmu.2021.740890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weber J, Gibney G, Kudchadkar R, Yu B, Cheng P, Martinez AJ, et al. Phase I/II Study of Metastatic Melanoma Patients Treated with Nivolumab Who Had Progressed after Ipilimumab. Cancer Immunol Res 2016;4(4):345–53 doi 10.1158/2326-6066.CIR-15-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grimaldi S, Terroir M, Caramella C. Advances in oncological treatment: limitations of RECIST 1.1 criteria. Q J Nucl Med Mol Imaging 2018;62(2):129–39 doi 10.23736/S1824-4785.17.03038-2. [DOI] [PubMed] [Google Scholar]
- 48.Shafiei A, Bagheri M, Farhadi F, Apolo AB, Biassou NM, Folio LR, et al. CT Evaluation of Lymph Nodes That Merge or Split during the Course of a Clinical Trial: Limitations of RECIST 1.1. Radiol Imaging Cancer 2021;3(3):e200090 doi 10.1148/rycan.2021200090. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used in the current analysis are available from the corresponding authors upon reasonable request.