

Abstract
Management of hepatoblastoma (HB), the most frequent pediatric liver cancer, is based on surgical resection and perioperative chemotherapy regimens. In this study, we aimed to identify actionable targets in HB and assess the efficacy of molecular therapies in preclinical models of HB.
Paired tumor and adjacent tissues from 31 HBs and a validation set of 50 HBs were analyzed using RNA-seq, SNP and methylation arrays. IGF2 overexpression was identified as the top targetable HB driver, present in 71% of HBs (22/31). IGF2high tumors displayed progenitor cell features and shorter recurrence-free survival. IGF2 overexpression was associated in 91% of cases with fetal promoter hypomethylation, ICR1 deregulation, 11p15.5 loss of heterozygosity or miR483-5p overexpression.
The antitumor effect of xentuzumab (a monoclonal antibody targeting IGF1/2) alone or in combination with the conventional therapeutic agent cisplatin was assessed in HB cell lines, in PDX-derived HB organoids and in a xenograft HB murine model. The combination of xentuzumab with cisplatin showed strong synergistic antitumor effects in organoids and in IGF2high cell lines. In mice (n=55), the combination induced a significant decrease in tumor volume and improved survival compared to cisplatin alone.
These results suggest that IGF2 is an HB actionable driver and that, in preclinical models of HB, the combination of IGF1/2 inhibition with cisplatin induces superior antitumor effects than cisplatin monotherapy. Overall, our study provides a rationale for testing IGF2 inhibitors in combination with cisplatin in HB patients with IGF2 overexpression.
INTRODUCTION
Hepatoblastoma (HB) is the most common pediatric liver cancer, generally occurring in children under 3 years of age[1]. Although its incidence has substantially increased over the last 30 years, HB is a rare disease, with 1.8 cases per million children/year[2, 3]. Unlike hepatocellular carcinoma (HCC), the most common primary liver tumor in adults, HB develops in the absence of an underlying liver disease or viral etiology. Most HB tumors occur sporadically, and only 5% of cases are associated with genetic diseases such as Beckwith-Wiedemann syndrome or Familial adenomatous polyposis[4].
Despite adjuvant and neoadjuvant cisplatin-based chemotherapy improves clinical outcome after resection[5], recurrence-free survival at 3 years for patients with advanced stages is only of 34%[6]. Furthermore, chemotherapy regimens are associated with severe and lifelong side effects such as ototoxicity and cardiomyopathy[7], which have especially impactful consequences for patients who are treated at early ages. Considering this, new treatment strategies are urgently awaited, and the identification of novel actionable drivers in HB is a current unmet medical need.
HB is a heterogeneous disease, so a better understanding of the molecular features of HB tumors is a prerequisite for developing targeted therapies. In this line, new HB classifications are emerging and include the so-called hepatocellular neoplasms not otherwise specified (HCN-NOS)[8, 9]. Globally, HBs present one of the lowest somatic mutation rates of all cancer types[10, 11], being mutations or deletions in the gene encoding β-catenin (CTNNB1) the most recurrent alteration (70–80% of HB patients)[12, 13], followed by mutations in NFE2L2 (10%) or in the TERT promoter (6%)[10], the latter mainly representing HCN-NOS cases[9]. On the other hand, Insulin Like Growth Factor 2 (IGF2) overexpression in HB has been proposed as a potential oncogenic driver[14, 15]. In HCC, IGF signaling has been identified as an epigenetic driver[16, 17] and has shown great promise as an actionable target in preclinical HCC experimental models[18]. Sensitivity of HB cells to IGF1R/IGF2 pathway inhibition has been demonstrated in vitro[15]. In the clinical context, IGF2 inhibition with xentuzumab (BI 836845; see ref. [19] for full sequence and structure), a monoclonal antibody against IGF1 and IGF2, is under investigation in different tumor types (NCT02191891, NCT03659136, NCT03099174, NCT02123823 and NCT02204072). In this regard, preliminary results of phase 1 trials have revealed that xentuzumab has promising antitumor activity and is well tolerated[20]. However, its clinical efficacy and safety in children with HB have not yet been explored.
Here, we analyzed 31 HB tumors, including HB (n=29) and HCN-NOS (n=2), and identified IGF2 as the main targetable deregulated pathway in HB. We then showed that IGF2high HBs are associated with poor outcome, which was the basis for testing xentuzumab alone or in combination with cisplatin –the backbone of all neoadjuvant chemotherapies in HB patients and the standard of care for standard-risk HB– in HB cell lines, in a PDX (patient derived xenograft)-derived tumor organoid model and in a xenograft murine model mimicking IGF2high HBs. In vitro, the combination exerted a synergistic reduction in viability and, in vivo, it improved survival and inhibited tumor angiogenesis compared to cisplatin. Overall, our findings suggest that HB patients with high IGF2 levels could benefit from xentuzumab combined with chemotherapy, and are a first step towards the implementation of precision medicine in HB.
MATERIALS AND METHODS
Human HB cohorts
The study cohort included primary HB tumor and adjacent non-tumor samples obtained from 31 patients[21] who received resection after standard neo-adjuvant therapy. Data from RNA sequencing, methylation array, SNP-array and human transcriptomic array data (GSE132219) have been previously reported elsewhere[21] (Supplementary Figure 1). The patient’s clinicopathological data are summarized in Table 1. As validation cohort, we used an in-house cohort of 50 HB tumors and 2 non-tumor formalin-fixed paraffin-embedded (FFPE) tissue samples from patients enrolled in the SIOPEL-3 clinical trial[22]. A second validation cohort consisting of published RNA sequencing data (GSE104766) from 25 HB tumors and their respective non-tumor adjacent tissue was used[23]. All samples were collected in accordance with European and Spanish law. Informed written consent was obtained from each patient in accordance with European guidelines for biomedical research. The study conformed to the ethical guidelines of the 1975 Declaration of Helsinki, and it was approved by the Human Ethics Committee of the Hospital Universitari Germans Trias i Pujol. All samples were histologically reviewed by two expert liver pathologists (R.A. and C.G.).
Table 1.
Clinico-pathological features of patients with IGF2-high and -low HB in the study cohort (n=31).
| IGF2high (n = 22) |
IGF2low (n = 9) |
p value | |
|---|---|---|---|
|
| |||
| Age (months), median (range) | 17.5 (1–180) | 13 (3–63) | 0.71 |
| >3 years, n (%) | 6 (27%) | 3 (33%) | 1 |
| Gender (male), n (%) | 13 (59%) | 6 (67%) | 1 |
| Serum AFP, ng/mL (range) | 620,000 (341–2,000,000) | 160,000 (663–2,186,461) | 0.27 |
| >1000 ng/ml, n (%) | 20 (95%) | 7 (78%) | 0.21 |
| >1,000,000 ng/ml, n (%) | 5 (23%) | 1 (11%) | 0.64 |
| CHIC* classification (High risk), n (%) | 9 (41%) | 2 (22%) | 0.43 |
| PRETEXT classification | |||
| PRETEXT I | 0 (0%) | 2 (22%) | 0.08 |
| PRETEXT II | 9 (41%) | 2 (22%) | 0.43 |
| PRETEXT III | 8 (36%) | 4 (44%) | 0.70 |
| PRETEXT IV | 5 (23%) | 1 (11%) | 0.64 |
| Neo-adjuvant chemotherapy, n (%) | 21 (95%) | 9 (100%) | 1 |
| Distant metastasis at diagnosis, n (%) | 7 (32%) | 2 (22%) | 0.69 |
| Vascular invasion, n (%) | 10 (45%) | 2 (22%) | 0.42 |
| Multifocality | 10 (45%) | 1 (11%) | 0.11 |
| HCN-NOS $ , n (%) | 2 (9%) | 0 | 1 |
| SCUD # , n (%) | 3 (13%) | 1 (11%) | 1 |
| Tumor histology | |||
| Epithelial, n (%) | 12 (55%) | 3 (33%) | 0.43 |
| Mixed epithelial and mesenchymal, n (%) | 10 (45%) | 6 (97%) | 0.43 |
| Main Epithelial Component | |||
| Fetal, n (%) | 8/22 (36%) | 5 (56%) | 0.43 |
| Non-Fetal, n (%) | 9/22 (41%) | 1 (11%) | 0.20 |
CHIC, Children’s Hepatic Tumors International Collaboration-Hepatoblastoma Stratification (CHIC-HS).
HCN-NOS: Hepatocellular Malignant Neoplasm, Not Otherwise Specified.
SCUD: Small cell undifferentiated hepatoblastoma.
Transcriptomic analysis
RNA-seq data from 31 HB tumor and adjacent non-tumor pairs from the study cohort[21] was used. Raw expression counts were normalized with the weighted “trimmed mean method” (TMM) implemented in the edgeR package[24] to correct library size differences between samples. The gene expression profile in HB tumors was analyzed using the NTP, Pre ranked GSEA and ssGSEA modules from GenePattern (www.genepattern.org) and gene signatures from MSigDB or previously reported (Supplementary Table 1).
Secondly, the in-house validation cohort of 50 HB tumors and 2 adjacent tissues was profiled using the NanoString nCounter Technology with a manually curated list of 65 genes, including HB markers and genes of key signaling pathways (Supplementary Table 2). RNA from tumor FFPE unstained sections was extracted using truXTRAC FFPE RNA microTUBE purification Kits (COVARIS, Woburn, MA). RNA quality control was assessed using Tape Station RNA chips (Agilent) and RNA quality was measured using DV200 metrics. RHOT2 and PNN genes were used as a housekeeping genes. NanoString output files were analyzed by nSolver Analysis Software 4.0.
For the validation cohort, RNA-sequencing data (GSE104766) from tumor and non-tumor HB samples from 25 HB patients were used[23]. Raw expression counts were normalized following the same procedure as previously described[21].
Weighted gene co-expression network analysis
To identify groups of correlated genes that are enriched in tumors compared to non-tumor samples, we used the Weighted gene co-expression network analysis (WGCNA) R package[25]. Networks enriched in tumors were selected based on FDR < 0.001 and module differential connectivity (MDC) ratio < 0.1. Subsequently, to identify the main signaling pathways in each network, we applied the Pathway enrichment module using the Gene Ontology database and selected the top significantly enriched term[26] (all FC > 2 and BH-adjusted p < 0.05). Hub genes (i.e. the top 5 most interconnected genes of the network) were identified based on intra-module connectivity[25]. The selected genes were filtered for cancer-related genes according to the OncoKB Cancer Gene List (www.oncokb.org) and for targetable alterations using the PANDRUGS database (www.pandrugs.org). Only drugs with high interaction evidence level (i.e. direct targets) were considered.
Classification of IGF2high tumor samples
IGF2 overexpression was defined as FC > 4 versus mean tumor adjacent tissue based on qRT-PCR data. For RNA-seq data, a cutoff of FC > 2 versus median tumor adjacent tissue was established based on ROC curve calculations (Supplementary Figure 2, Supplementary Table 3).
Methylation analysis
Methylation data from 27 HB tumors and 22 adjacent non-tumor pairs obtained from a Infinium MethylationEPIC 850K array (Illumina) were previously reported[21] and are accessible through GSE132219. Data were normalized and converted to B values using the ChAMP pipeline[27]. The differential methylation status of the IGF2 gene promoters in HB tumors and adjacent tissue was conducted using the ChAMP R package (v 2.22.0) by analyzing the array probes located within the maternal allele of adult promoter (P1), fetal promoters (P2, P3 and P4), and ICR1 regulatory region (CTCF-binding site) according to Ensembl Genome Browser (Ensembl.org) and previous studies[18] (Supplementary Table 4). Statistical comparisons for differential methylation analysis were performed using M values[28]. IGF2 hypomethylation was defined as the mean fold-change of the B-values in the fetal promoter region compared to adjacent non-tumor samples minus standard deviation as previously defined[18].
Loss of heterozygosity analysis
Loss of Heterozygosity (LOH) in HB samples was established from SNP array data of 30 HB tumor samples. SNP array and LOHs data was previously reported[21] and is accessible through GSE132037. Samples with LOH in the chromosomal region 11:2,129,112–2,149,603 from the GRCh38.p12 human genome version were considered positive for LOH in the IGF2 gene.
Exon-level gene expression and splicing analysis
The IGF2 exon-level expression analysis was performed using the data of 36 probes of the Human Transcriptome Array data (GSE132219)[21]. Specifically, expression of isoforms containing the adult-specific exons 1 (hg19, chr 11: 2,170,833–2,170,356) and 2 (11: 2,169,037–2,168,796) and fetal-specific exon 6 (11: 2,160,619–2,159,459) was assessed[18]. The supervised differential splicing between tumor and non-tumor samples as well as IGF2high vs. IGF2low tumors was conducted using Transcriptome Analysis Console software (ThermoFisher Scientific, Waltham, MA).
Mutational analysis
Mutations were called from paired tumor/normal RNA-Seq data. Mapped RNA-seq reads were subject to splitting, trimming, local indel realignment, and base-score recalibration pre-processing with the IndelRealigner and Table Recalibration tools from Genome Analysis Tool Kit (GATK)[29] under the GATK Best Practices for RNA-seq paradigm (https://gatkforums.broadinstitute.org/gatk/discussion/3892/the-gatk-best-practices-forvariant-calling-on-rnaseq-in-full-detail). MuTect[30] was then used with default settings to quantify somatic mutation burden. CTNNB1 and AXIN1 alterations were validated by PCR and Sanger sequencing, as previously described[21].
Identification of TERT promoter mutations:
DNA was extracted from macrodissected fresh frozen (FF) or formalin-fixed paraffin-embedded (FFPE) tumor tissues using the MagMAX™ DNA Multi-Sample kit (Thermo Fisher Scientific) or truXTRAC FFPE DNA microTUBE Kit-Column Purification (Covaris). Qubit was used to quantify DNA concentration and integrity. Then, we performed polymerase chain reaction (PCR) using specific primers (Forward: GGTGAAGGGGCAGGACGGGTGC; Reverse: GGCTTCCCACGTGCGCAGCAGGA) and 50 ng of purified genomic DNA. The PCR reaction was performed by PCR MasterMix (Thermo Fisher Scientific) including a negative and a positive control. The PCR reaction conditions were: initial denaturalization 95°C for 2 min, 40 cycles of denaturalization 95°C for 30 sec and annealing 60°C for 30 sec and extension 72°C for 2 min, and a 10 min final extension at 72°C. The PCR product was sequenced by Sanger Sequencing. Sequencing reactions of both DNA strands were performed with the BigDye® Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific), run on a Genetic Analyzer ABI 3130 and analyzed with SeqScape v5.3.1 (both from Applied Biosystems). Briefly, 5 μL of PCR product was incubated with 2 μL of illustra ExoProStar 1-Step (GE Healthcare Life Sciences) for 45 min at 37°C and 15 min at 80°C. After purification, 2 μL of purified PCR product was mixed with 2 μL of BigDye Buffer, 1 μL of BigDye, 1.6 μL of 1 μM of primer and 3.4 μL of DNase RNase-free water. The sequencing reaction conditions were: 95°C for 5 min, 96°C for 10 sec, 30 cycles of 50°C for 50 sec, and a 4-min extension at 60°C.
Cell lines and reagents
HepG2 (ATCC, Manassas, VA) and Huh6 (Japanese Collection of Research Bioresources, Osaka, Japan) HB cell lines were cultured in Dulbecco’s Modified Eagle Medium (ThermoFisher, Waltham, MA) supplemented with 10% heat-inactivated fetal bovine serum. Cells were maintained at 37°C in a 5% CO2 atmosphere. The anti-IGF1/2 monoclonal antibody xentuzumab (BI 836845; see ref. [19] for full sequence and structure) was produced and provided by Boehringer Ingelheim (Ingelheim, Germany) and cisplatin was purchased from Selleckchem (S1166) (Houston, TX). PBS was used as the control.
To generate the 2D PDX-derive cell line, PDX tissue wase minced and digested in PBS + 1 mg/mL collagenase IV for 90 minutes at 37°C. Tumor dissociate was strained though a 70 μm strainer and washed with complete RPMI (20% FBS, 1% glutamine, 1% penicillin/streptomycin). 100,000 cells were plated on collagen-coated 35 mm plates in human-2D media (complete RPMI, 40 ng/mL recombinant human EGF, 10 uM Y-27632, and 5 uM A83-01). Cells were cultured and maintained in human-2D media 37°C in a 5% CO2 atmosphere.
In vitro functional cell assays
Cell proliferation and colony formation capacity were evaluated in HepG2 (ATCC, Manassas, VA), Huh6 (Japanese Collection of Research Bioresources, Osaka, Japan) and 2D PDX-derived cells treated with xentuzumab (Boehringer Ingelheim, Ingelheim, Germany), cisplatin (Selleckchem, Houston, TX), its combination, or vehicle. For the matrix viability assay, 4000 cells/well were seeded in 96-well plates for 24h in a humidified atmosphere at 37°C and 5% CO2. Thereafter, cells were incubated for 72h with increasing concentrations of cisplatin and xentuzumab. Cell viability was measured with absorbance at 560 and 590 nm from resazurin. Combination index values were calculated for each column and averaged across each matrix. To assess colony formation capacity, 600 cells/well were seeded in 6-well plates. After 24h, Huh6 and HepG2 cells were treated with cisplatin (0.3 μM), xentuzumab (1 μM) or their combination and incubated for 12 days, with drug-containing media changed three times per week. Thereafter, cells were rinsed with cold PBS, fixed with methanol and stained with 0.5% Crystal Violet (Sigma-Aldrich, Saint Louis, MO). The number of colonies was manually counted. All experiments were performed in triplicate.
Establishment of the PDX-derived organoid model
A patient-derived HB tumor with high IGF2 expression provided by Xentech (Evry, France)[31] was implanted and grown subcutaneously in 6–8 week-old female NOD/SCID immunosuppressed mice (n = 3; Charles River Laboratories, Wilmington, MA). Animals were weighed and tumor volume was assessed three times per week using bilateral caliper measurements according to the formula volume = length × width2 × 0.5. Once tumors reached 1000 mm3, the animals were sacrificed and tumors were collected to generate three independent organoid lines (PDX-derived organoids). Histological and molecular characterization of the PDX have been reported elsewhere (PDX ID: HB-235)[31]. To generate the organoids[32], PDX tumors were minced and digested in sterile digestion media (PBS, 0.125 mg/mL collagenase from clostridium histolyticum, 0.125 mg/mL dispase II, and 0.1 mg/mL DNaseI) for two hours at 37°C. Tumor dissociates were strained through a 70 μm strainer and washed with basal media (Advanced DMEM/F-12, 1% glutamine, 1% penicillin/streptomycin, 10 mM HEPES). Cell dissociates were cultured at 50,000 cells per 50 μL Matrigel (Corning) in 24 well plates in human tumor organoid media (basal media, 1:50 B27 no vitamin A, 1:100 N2, 1 mM N-acetylcysteine, 10% Rspo1-conditioned media, 10 mM nicotinamide, 10 nM recombinant human [Leu15]-gastrin I, 50 ng/mL recombinant human EGF, 100 ng/mL recombinant human FGF10, 25 ng/mL recombinant human HGF, 10 μM forskolin, and 5 μM A83-01). Studies were performed in compliance with the “Guide for the Care and Use of Laboratory Animals” and they were approved by the Institutional Animal Care and Use Committee (IACUC-2016-0296).
Antitumor effect of IGF2 inhibition in an IGF2high organoid model
Organoids were seeded and treated with serial dilutions of cisplatin (Selleckchem, Houston, TX) or xentuzumab (Boehringer Ingelheim, Ingelheim, Germany) for cell viability analyses and IC50 calculations. After 3 days of incubation, cell viability was measured with absorbance at 560 and 590 nm from resazurin. Combination index values were calculated for each column and averaged across each matrix.
Caspase-dependent apoptosis was assessed by measuring caspase-3 and −7 activity (Caspase-Glo 3/7 Assay, Promega). Organoids were treated with or without 100 nM of IGF2 (Prepotech, Rocky Hill, NJ) and increasing concentrations of xentuzumab (0, 1, 10, 100 μM) and cisplatin (0, 1, 5 μM). All organoid experiments were performed using the 3 independent organoid lines and in technical triplicates.
IGF2high xenograft murine model
To generate the xenograft murine model of IGF2high HB, 5×106 HepG2 cells were subcutaneously injected into the right flank of immunocompromised 6–8 weeks-old athymic (nu/nu) (Crl:NU-Foxn1nu) female mice (n = 55 mice; Charles River Laboratories, Wilmington, MA). Animals were weighed and tumor volume was assessed 3 times per week using bilateral caliper measurements according to the following formula: volume = length × width2 × 0.5. When tumors reached 150 mm3, animals were randomly assigned to receive xentuzumab (n = 14), cisplatin (n = 13), combination therapy (xentuzumab plus cisplatin, n = 14) or drug vehicle (n = 14). Xentuzumab (100 mg/kg) was administered twice per week; and cisplatin (2 mg/kg) was administered daily. Both drugs were administered intraperitoneally. Body weight, tumor growth and survival (defined as time to reach 1500 mm3) were monitored in all the mice. Once animals reached the survival endpoint, they were sacrificed following Institutional Animal Care and Use Committee guidelines. Studies were performed in compliance with the “Guide for the Care and Use of Laboratory Animals” and they were approved by the Institutional Animal Care and Use Committee (IACUC-2016-0296).
IHC analysis
Tumor samples from the mouse model were fixed in buffered 4% paraformaldehyde for 24 hours and embedded in paraffin to create formalin-fixed paraffin-embedded (FFPE) blocks. An expert pathologist blinded to the treatment arms (CM) assessed the percentage of viable and necroapoptic tissue in H&E-stained slides. This was used together with the ex-vivo tumor volume to calculate the viable and necroapoptotic tumor volume. CD31 staining (Abcam ab28364) was assessed by IHC on 3 μm-thick FFPE tissue sections after heat-induced antigen retrieval and quantified by the digital pathology imaging software QuPath (version 0.2.0). To ensure representative sampling of the entire tumor, 5 regions of interest were analyzed.
Western blotting
Huh6 and HepG2 cells and organoids were stimulated with 100 nM of IGF2 (PeproTech, East Windsor, NJ) to induce IGF1R pathway activation, along with cisplatin (0.3 μM for cell lines and 1 μM for organoids), xentuzumab (0.1 μM), its combination or vehicle. For Huh6 and HepG2 cell lines, after 24h of stimulation, cells were washed with PBS, collected by scraping and lysed in lysis buffer (50 mM Tris pH=7.4, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 0.25 mM EDTA, 1% Sodium deoxycholate) containing protease and phosphatase inhibitors. For organoids, after 24h of stimulation, organoids were collected in Cell Recovery Solution (Corning, Corning, NY), and matrigel was dissolved by rotation for 1h at 4°C. Pelleted organoids were then lysed in 2X Laemmli sample buffer containing 4% of 2-mercaptoethanol, heated at 95°C for 10min, sonicated, and heated again at 95°C for 5min. Protein concentration was determined using Protein Assay Dye Reagent Concentrate (Bio-Rad, Hercules, CA) and bovine serum albumin (BSA) was used as a standard. Protein extracts were resolved by SDS-PAGE and transferred onto polyvinylidene difluoride membranes by western blot. Membranes were blocked with 3% BSA and incubated overnight at 4°C with primary antibodies. Horseradish peroxidase-conjugated secondary antibodies were applied at room temperature for 1h. The specifications and dilutions of antibodies used for Western Blotting are listed in Supplementary Table 5. Blots were developed using ECL plus solution (GE Healthcare, Waukesha, IL) and imaged with the FUJI film Laser Image Analyzer.
Quantitative real-time PCR
The expression of IGF2 from promoter 1 (adult isoform), promoter 3 (fetal isoform), total IGF2, IGF1R, H19, and TGF-β mRNA levels were tested in the following sets of samples: HB tumor samples (n = 27), paired adjacent non-tumor liver samples (n = 25), HB cell lines (HepG2 and Huh6 cells), organoids and, as controls, healthy liver samples (n = 5, from patients with focal nodular hyperplasia or cystadenoma, without underlying liver disease). Total RNA from tissues and cell lines was isolated using the RNeasy Mini Kit (Qiagen, Hilden, Germany). The cDNA was reverse transcribed from 1 μg of total RNA using RNA to cDNA EcoDry Premix Double Primed (Clontech, Takara Biotechnology, Dalian, China) in a total volume of 20 μl. Quantitative real time PCR was performed using TaqMan probes for IGF2-P1 (Hs01005962-m1), IGF2-P3 (Hs00171254-m1), IGF2-P1/2/3 (Hs01005963-m1), IGF1R (Hs00609573-m1), H19 (Hs00262142_g1), and TGF-β (Hs00998133-m1). Each sample was analyzed in triplicate and normalized to the internal control 18S ribosomal RNA (Hs99999901-s1). The fold change of mRNA expression in tumor tissue was calculated referring to the average mRNA expression levels in adjacent non-tumor tissue.
To assess miR-483-5p expression levels, microRNA from tumor and paired adjacent tissue was obtained using the TaqMan MicroRna Reverse Transcription Kit (ThermoFisher Scientific), and qRT-PCR was performed following the TaqMan Small RNA assay and TaqMan probes for miR-483-5p (ThermoFisher Scientific, 002338). Each sample was analyzed in triplicate and normalized to the internal control miR-361-5p (ThermoFisher Scientific, 000554). Fold change was calculated referring to the averaged mRNA expression levels in adjacent non-tumor tissue.
Statistical analysis
Statistical analyses were conducted using R (version 3.6.2, R Foundation for Statistical Computing, Vienna, Austria). Comparisons of continuous variables were performed using Kruskal-Wallis and Dunn’s tests for non-parametric distributions, or ANOVA and Tukey tests for parametric distributions. Correlations for categorical variables were analyzed using Fisher’s exact test. Survival and time to response were assessed with Kaplan-Meier estimates and log-rank test.
Data availability
The data generated in this study including RNA-sequencing, methylation and SNP array are available within the article, its supplementary data files, and in Gene Expression Omnibus (GSE104766, GSE132219 and GSE132037). Data from validation cohorts were obtained from Gene Expression Omnibus (GSE132219 and GSE104766).
RESULTS
IGF signaling is highly deregulated in HB
To identify molecular alterations driving oncogenesis in HB, we used RNA-seq data from a cohort of 31 HB tumors[21] (29 HBs and 2 HCN-NOS) and matched non-tumor tissues (study cohort, Supplementary Figure 1, Table 1). Through gene co-expression network analysis, we identified seven network modules that were significantly differentially expressed between tumor and non-tumor samples from the study cohort. The top enriched gene ontology terms in these network modules included IGF2 signaling pathway (module 1), cell cycle and survival (modules 6–7), and immune response (modules 2–3) (FDR<0.001, Supplementary Table 6). Notably, the Wnt signaling pathway, which is a commonly deregulated in HB[21], was also enriched in the network module 1 (GO:0016055, p = 0.009). We also explored the top interconnected genes within each network (hub genes) and identified several targetable cancer-related candidates including IGF2, TGFB1, and BIRC3 (Supplementary Figure 3). Notably, IGF2 was the targetable gene with the highest expression levels in the study cohort (Supplementary Figure 3, Supplementary Tables 7–8), making it the most suitable candidate for our study.
We then assessed the rate of HB tumors overexpressing IGF2 in the study cohort. Of the 31 HBs, 22 (71%) were classified as IGF2high based on RNA-seq data (FC > 2 vs non-tumor adjacent tissue), and this was aligned with the IGF2 levels measured by qRT-PCR (Figure 1A and 1B) and with previous studies[14, 15]. Additionally, IGF2 pathway deregulation was also observed at the level of IGF1R and H19 (Supplementary Figure 4), in line with other reports[33].
Figure 1. Characterization of IGF2high HB tumors.
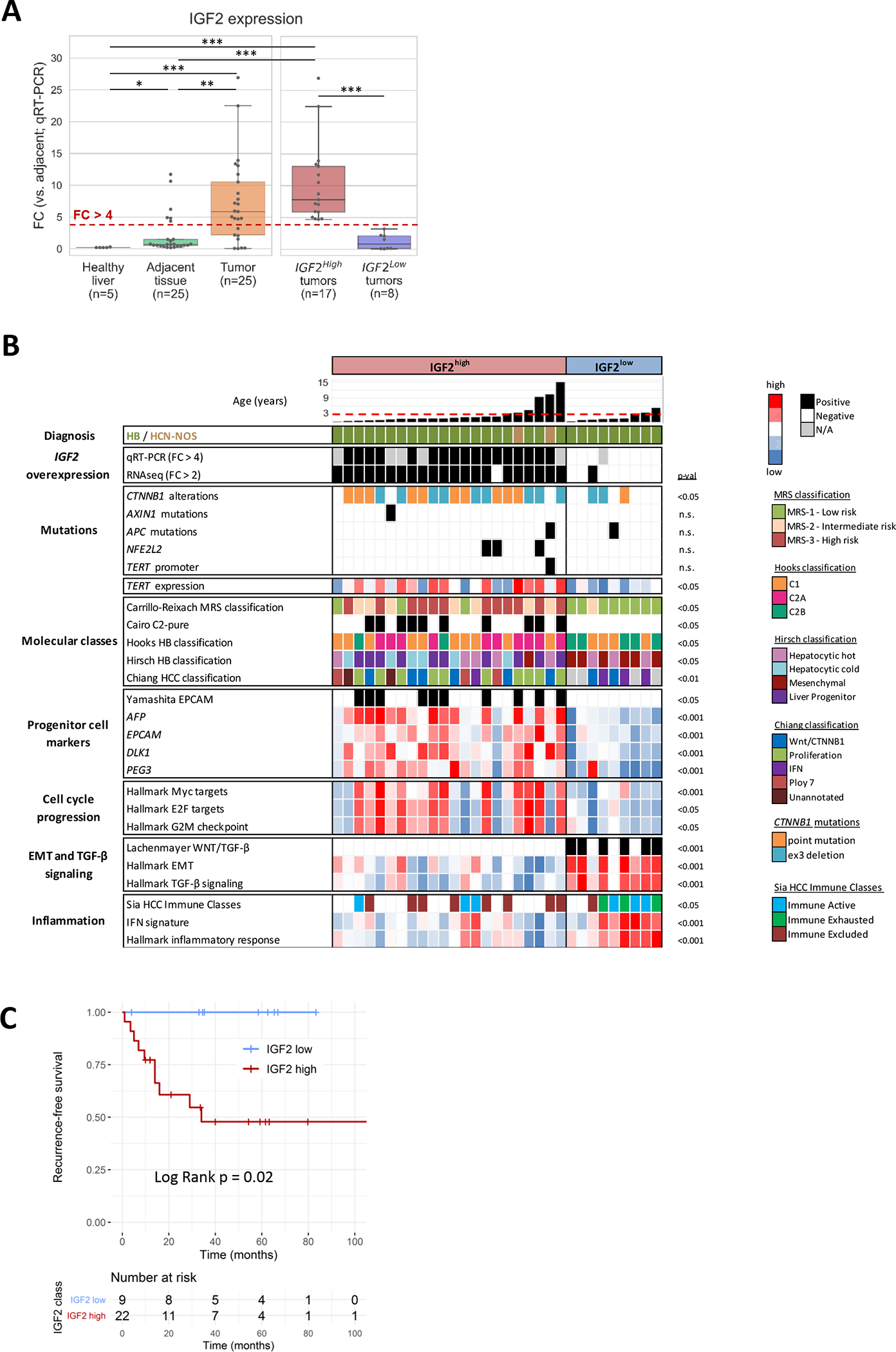
(A) IGF2 expression levels determined by quantitative RT-PCR in healthy liver, adjacent non-tumoral liver tissue and HB tumor samples (left). HB tumor samples classified as IGF2high and IGF2low (right). *p < 0.05, **p < 0.01, ***p < 0.001. (B) Molecular profile of IGF2high and IGF2low HB tumors. IGF2 expression levels were determined by RNA-seq and qRT-PCR. IGF2high was defined as fold-change (FC) > 4 versus mean adjacent non-tumoral tissue for qRT-PCR; and as FC > 2 versus median adjacent non-tumoral tissue for RNA-seq. The dashed red line marks the age of 3 years. HB: hepatoblastoma. HCN-NOS: Hepatocellular Malignant Neoplasm, Not Otherwise Specified. Statistical tests: t test and Fisher. Displayed p values were obtained comparing IGF2high and IGF2low HBs. (C) Kaplan-Meier’s plots for recurrence-free survival of IGF2high (n=9) and IGF2low (n=22) HB patients. Statistical test: log Rank.
These results were confirmed in two independent cohorts: in the first, our in-house validation cohort (n=50), 78% of patients (39/50) exhibited high levels of IGF2; and in the second[23], 76% (19/25) (Supplementary Figure 4).
Overall, our results indicate that IGF2 overexpression is the main targetable alteration among the top deregulated pathways in our HB cohort, and is present in 70–80% of cases.
IGF2high HB tumors are enriched in high risk HB molecular classes
We analyzed the transcriptomic and methylation profile of the study cohort to gain further insight into the molecular characteristics of IGF2high HB tumors (Figure 1B, Supplementary Figure 5). IGF2high tumors were enriched in high-risk molecular classes of HB including the MRS-3, the C2-pure, the C2A and the “Liver progenitor” classes of HB[15, 21, 23, 34]), as well as the EpiCB epigenetic cluster signature[21], and the HCC proliferation class[35]. IGF2high tumors also showed enrichment in progenitor cell markers (EPCAM, AFP, DLK1 and PEG3) and gene sets associated with cell cycle progression (Figure 1B, Supplementary Figure 5). Only 1 out of 31 cases were found to have TERT promoter mutations, and this single case corresponded to an HCN-NOS tumor. In terms of clinical features, IGF2high patients presented shorter recurrence-free survival after resection than IGF2low patients (median 34 months vs not reached for IGF2low; p = 0.02, Figure 1C), which align with their molecular features. No additional clinicopathological differences were observed between IGF2high and IGF2low tumors (Table 1).
In contrast, tumors with low IGF2 levels (IGF2low; 9/31, 29%) were enriched in low-risk and mesenchymal HB classes (MRS-1, C2B and “Mesenchymal” HB classes[15, 21, 23]), together with gene sets associated with inflammatory and interferon signaling, TGF-β pathway activation and epithelial-to-mesenchymal transition (Figure 1C). The high presence of inflammatory signaling was confirmed with gene signatures capturing specific immune cell populations including cytotoxic T cells (Supplementary Figure 5). The expression levels of TGFB1 and TGF-β pathway-related genes were higher in IGF2low tumors than in IGF2high and non-tumor samples (all p < 0.0001, Supplementary Figure 5). These results indicate that IGF2low tumors present a mesenchymal and highly infiltrated phenotype, which is in line with their association with a better outcome compared to IGF2high tumors.
These molecular features of IGF2high and IGF2low HBs were confirmed in an independent HB cohort of 25 patients[23]. In line with our data, gene expression analysis in the external cohort revealed enrichment of proliferation and cell cycle-related gene sets in IGF2high tumors, compared to inflammatory and interferon signaling in IGF2low cases (FDR < 0.05, Supplementary Figure 5).
Fetal promoter hypomethylation, ICR1 deregulation, LOH and miR-483-5p levels drive IGF2 overexpression in HB
Next, we investigated the mechanisms of IGF2 overexpression in HB. Previous reports have described aberrant IGF2 levels in HB associated with hypomethylation of the fetal P2, P3, and P4 promoters, as opposed to physiological P1-mediated expression[36]. Here, we used methylation 850K-array data to assess the methylation differences between IGF2high and IGF2low tumors. Tumors with high IGF2 expression levels presented significantly decreased methylation of the IGF2 fetal promoters compared to tumors with low IGF2 levels (Figure 2A, Supplementary Figure 6). In particular, IGF2 fetal promoter hypomethylation was present in 50% of IGF2high samples (9/18 vs 1/9 in IGF2low samples, p < 0.05; Figure 3). To assess whether the hypomethylation of the IGF2 fetal promoter correlated with higher expression of its fetal isoform, we expanded our analysis to the IGF2 splicing profile based on an HTA (Human Transcriptome Array) expression array at exon level. Our data demonstrated that the IGF2 fetal isoform was overexpressed in IGF2high tumors compared to IGF2low tumors (Figure 2B–C). Furthermore, overexpression of the fetal IGF2 isoform in IGF2high was confirmed by qRT-PCR in both the study cohort and in-house validation cohort (Supplementary Figure 6). Our results suggest that IGF2high HB tumors present a fetal IGF2 expression pattern through hypomethylation of the fetal promoters. In addition, we also identified gain of methylation in the IGF2/H19 imprinting region ICR1 of IGF2high tumors, in line with previous studies[15]. Particularly, ICR1 gain of methylation was present in 61% of IGF2high patients (11/18 vs 1/9 in IGF2low samples, p < 0.05; Figure 3, Supplementary Figure 6).
Figure 2. Epigenetic deregulations in IGF2high HB tumors.
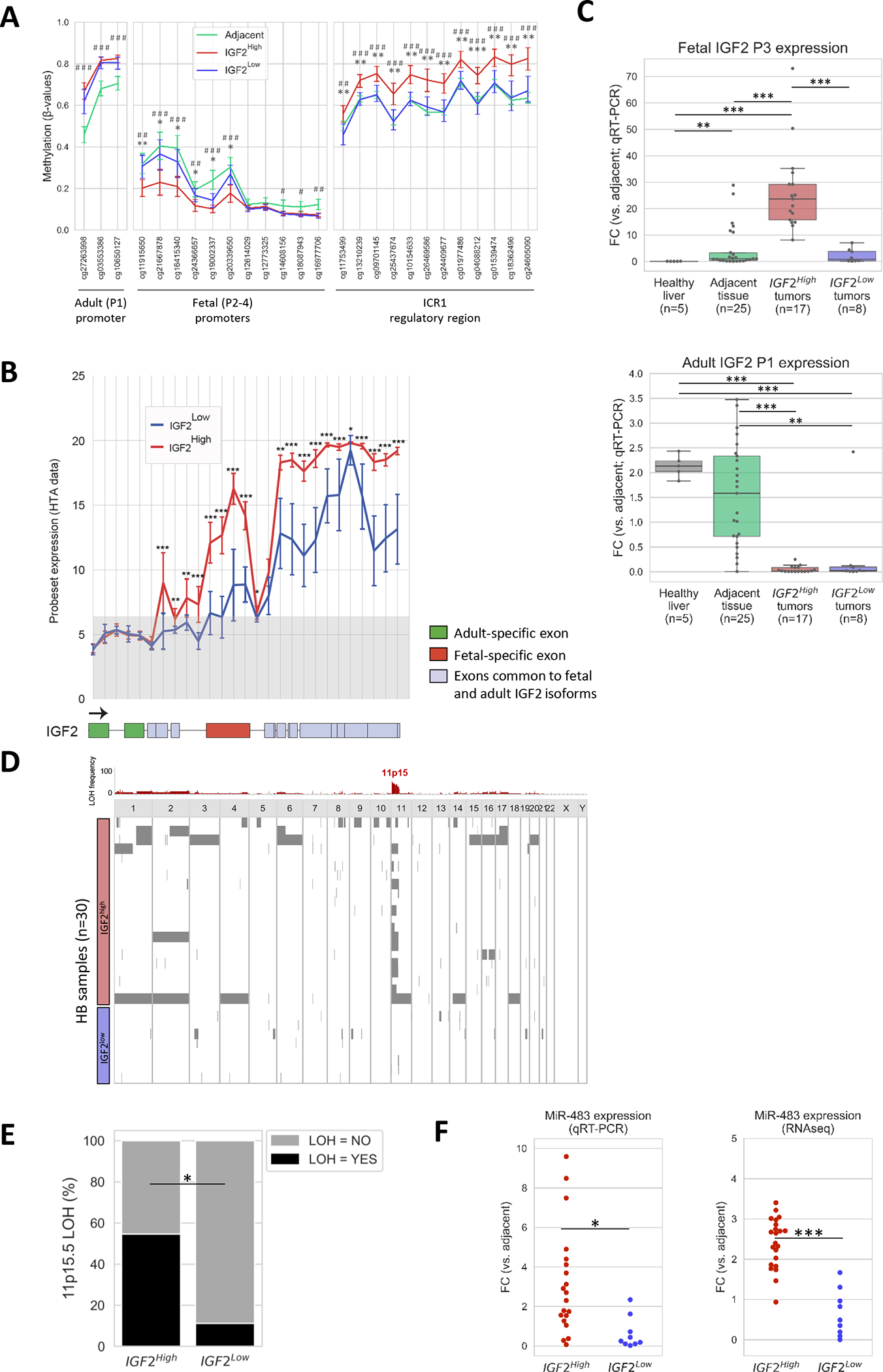
(A) Methylation levels in CpGs located within the IGF2 adult promoter (P1), fetal promoters (P2-P4), and ICR1 regulatory region of adjacent (n = 22), IGF2high tumors (n = 18) and IGF2low tumors (n = 9). Error bars indicate mean ± standard deviation (SD). (B) Human Transcriptome Array (HTA) data showing the expression of adult and fetal specific exons of IGF2 between IGF2high (red, n = 22) and IGF2low (blue, n = 9) tumors. The coding exons of the IGF2 gene are shown below the plot. The specific exons of the adult (hg19, chr 11: 2,170,833–2,170,356 and 2,169,037–2,168,796) and fetal (2,160,619–2,159,459) IGF2 isoforms are shown in green and red, respectively. The common coding exons in adult and fetal IGF2 isoforms are shown in gray. The gray shadow in the plot indicates no gene expression (values below the baseline level). (C) Expression levels of fetal P3-derived and adult P1-derived IGF2 isoforms determined by quantitative RT-PCR in healthy livers (n=5), adjacent liver tissue (n=25), and in IGF2high (n=17) and IGF2low (n=8) HB tumor samples. (D) Allelic imbalances in HB determined using CystoScan SNP array data. Chromosomal regions with loss of heterozygosity in HB tumors are shown in red, and the frequency of each alteration, in grey. (E) Frequency of LOH in 11p15.5 in IGF2high (n = 21) and IGF2low HB (n = 9) samples. (F) MiR-483-5p expression levels in IGF2high and IGF2low HB samples assessed by quantitative RT-PCR (left) and RNA sequencing (right). * P < 0.05, ** P < 0.01, *** P < 0.001 in IGF2high vs IGF2low unless otherwise indicated; # P < 0.05, ## P < 0.01, ### P < 0.001 in IGF2high vs Adjacent.
Figure 3. Integrated mechanisms of IGF2 overexpression in HB tumors.
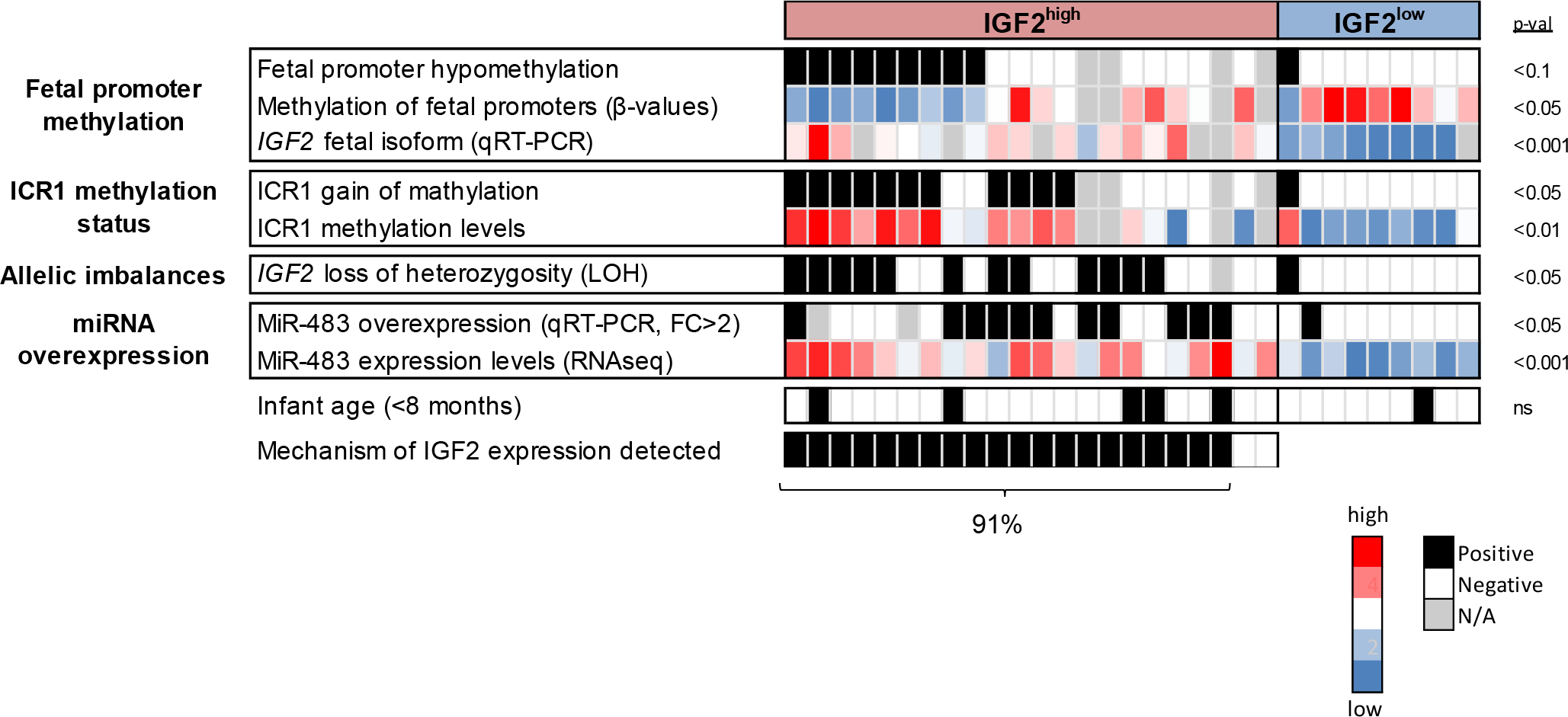
Heatmap presenting the epigenetic and genomic deregulations affecting each HB tumor. Fetal promoter hypomethylation occurred in 50% of IGF2high cases; ICR1 gain of methylation in 61% IGF2-related allelic imbalances, in 57% of IGF2high cases; and miR483 overexpression, in 55% of IGF2high cases. Statistical significance between IGF2high and IGF2low was calculated by Fisher and t tests.
Additionally, the 11p15.5 imprinted locus, which contains IGF2, has also recently been reported as the second most frequently altered locus in hepatoblastoma[15], mostly through copy-neutral loss of heterozygosity (LOH). Since IGF2 presents monoallelic expression in normal conditions, LOH in this genomic region increases its expression[14, 37]. We used our SNP array data to determine the rates of this allelic imbalance in IGF2high vs IGF2low HB tumors. IGF2high tumors were significantly enriched in 11p15.5 LOH compared to IGF2low tumors [12/21 (57%) vs 1/9 (11%), p<0.05, Figure 2D–E, Figure 3]. Interestingly, the four IGF2high HB samples corresponding to infant patients (< 8 months of age) showed 11p15.5 LOH (Figure 3), which suggests that their IGF2 overexpression was not due to physiological regulation in fetal and early postnatal stages[38], but due to genetic aberrations.
Finally, the expression of the micro-RNA miR-483-5p has been reported to promote fetal IGF2 transcription by binding to its 5′ untranslated region (UTR)[39] and as a mechanism of IGF2 overexpression in HCC[18]. In our study cohort, miR-483-5p levels were significantly higher in IGF2high HB tumors than in IGF2low HB tumors, both according to the FC vs adjacent non-tumor tissue in RNA-seq (2.4 vs 0.7, p < 0.05) and qRT-PCR data (3.2 vs 0.6, p < 0.001) (Figure 2F). Overall, miR-483-5p overexpression, defined as FC > 2 vs adjacent non-tumor samples, was detected in 55% (11/20) of IGF2high HB tumors and only in 11% of IGF2low tumors (p < 0.05; Figure 3).
Altogether, our results indicate that IGF2 overexpression in HB is driven by hypomethylation of its fetal promoters, ICR1 deregulation, LOH in 11p15.5, and miR-483-5p expression. Most IGF2high HB patients [91% (20/22)] presented at least one of these mechanisms of IGF2 overexpression (Figure 3).
IGF2high HB cells present high sensitivity to IGF2-inhibition
The presence of IGF2 overexpression in 71% of HB patients and their poor outcome prompted us to test IGF2 blockage in HB. In addition, the monoclonal antibody against IGF1 and IGF2, named xentuzumab, has shown promising antitumor effects in other cancer types[20]. To this end, the antitumor effects of xentuzumab were evaluated in vitro (HB cell lines and HB PDX-organoids) and in an in vivo murine model of HB. The effects were compared to those of a conventional therapeutic agent for HB patients (cisplatin)[5]. IGF2 expression levels in HB cell lines (HepG2 and Huh6) were assessed by qRT-PCR (Supplementary Figure 7). HepG2 cells presented high IGF2 expression, defined as FC > 4 compared to non-tumor adjacent tissue (median FC = 19.60), and were used as a model to recapitulate IGF2high HB tumors. Conversely, Huh6 cells showed low IGF2 expression (median FC = 0.04; Supplementary Figure 7) and thus were considered to phenocopy the IGF2low HBs. As observed in IGF2high human HB tumors, HepG2 cells also showed high expression of the receptor IGF1R, low H19 expression, and high expression of the fetal isoform of IGF2 (P2–4 derived) (Supplementary Figure 7). Previous studies[14] have reported that HepG2 cells present fetal promoter hypomethylation and LOH genomic imbalances as opposed to Huh6. For validation purposes, we generated a cell line derived from a human PDX model with high expression of IGF2 (hereinafter referred to as PDX-derived cell line).
We tested the effect of IGF2 blockade and chemotherapy in IGF2high cells (HepG2 and PDX-derived cell line) and IGF2low cells (Huh6). In IGF2high cells, xentuzumab strongly decreased cell viability in combination with low doses of cisplatin (0.1 – 1 μM), where cisplatin was not effective as monotherapy, eliciting a strong synergistic effect (Combination Indexes: HepG2, CI = 0.47; PDX-derived cells, CI = 0.44; Figure 4A, Supplementary Figure 7). In contrast, the synergism of IGF2 blockade and chemotherapy was not observed in Huh6 cells (IGF2low), where the anti-proliferative effects were mainly driven by cisplatin, with a mild added effect of xentuzumab (CI = 0.94, Figure 4A).
Figure 4. Anti-proliferative and pro-apoptotic effect of IGF2 inhibition in combination with cisplatin in HB cell lines and organoids.
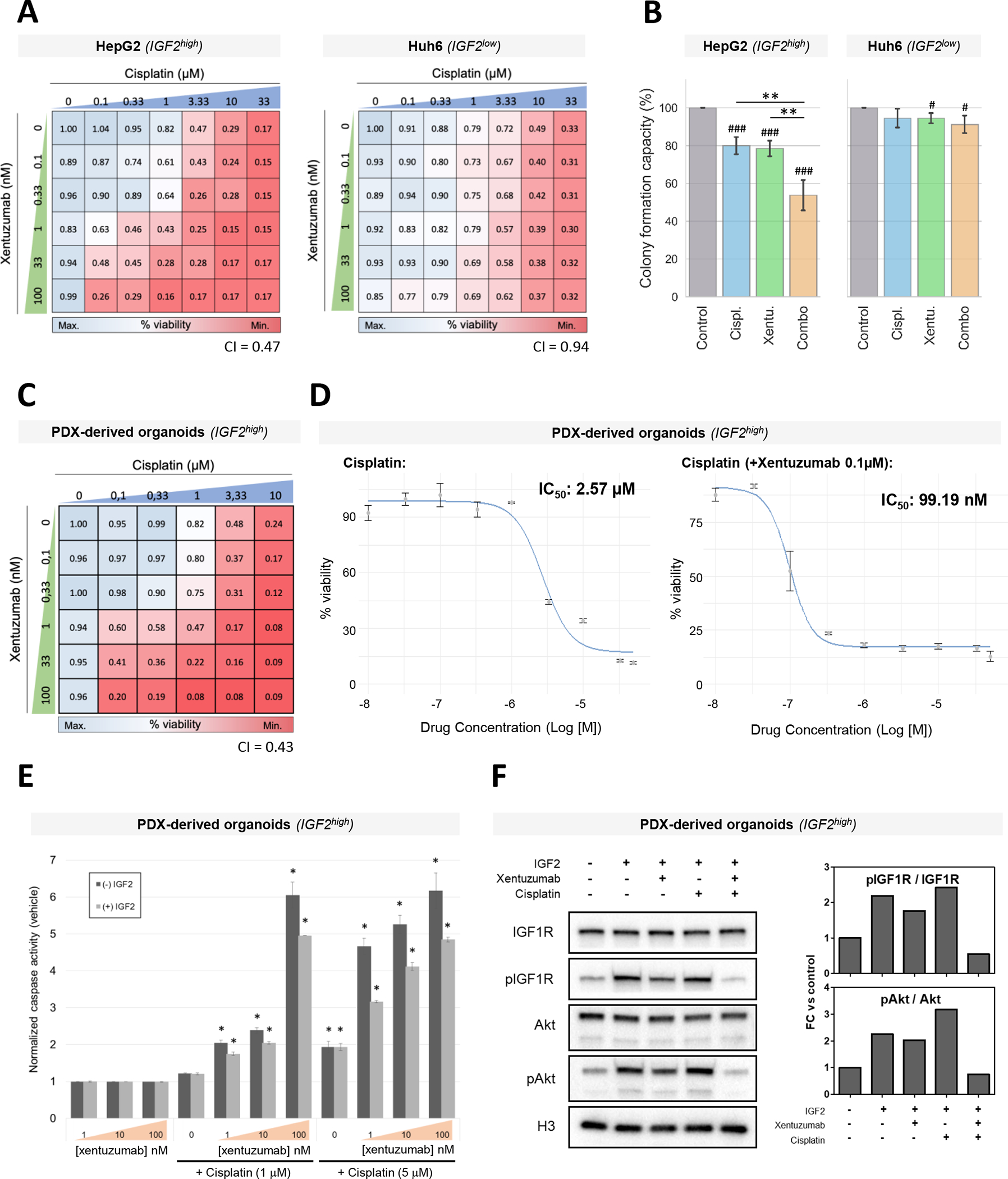
(A-B) Cell viability (A) and colony formation capacity (B) in HepG2 and Huh6 cell lines treated with increasing concentrations of cisplatin and xentuzumab (A) or with cisplatin (0.3 μM), xentuzumab (1 μM) or its combination (combo) (B). (C) Cell viability in IGF2high tumor organoids treated with increasing concentrations of cisplatin (0–10 μM) and xentuzumab (0–100 nM). (D) IC50 curves for cisplatin alone (left) and cisplatin combined with xentuzumab (right). IC50 values for cisplatin are indicated. (E) Caspase C3-dependent apoptosis analysis in tumor organoids treated with growing concentrations of cisplatin (0–5 μM) and xentuzumab (0–100 nM). Error bars indicate mean ± standard deviation (SD). (F) Representative Western blot analysis and quantification of the IGF2/IGF1R pathway in tumor organoids stimulated by IGF2 and treated with xentuzumab and/or cisplatin. Histone H3 was used as a loading control. * P < 0.05, ** P < 0.01.
Consistently, xentuzumab in combination with cisplatin reduced the colony formation capacity of IGF2high HepG2 cells compared to monotherapies, including cisplatin (all p < 0.05, Figure 4B). Conversely, the combination treatment did not show significant differences in the Huh6 cell line compared to monotherapies. When comparing the effect of the combination, this was 5.1-fold stronger in HepG2 cells (IGF2high) than in Huh6 cells (IGF2low), corresponding to a 46% vs 9% reduction, respectively (p < 0.001, Supplementary Figure 7). On the other hand, xentuzumab alone induced significant differences in colony formation compared to the control (all p < 0.01), but it did not induce superior antitumor effects compared to cisplatin monotherapy (Figure 4B). The impact of xentuzumab alone in HepG2 cells was between 4.5-fold stronger than in Huh6 cells (22% vs 5%, p < 0.01; Figure 4B and Supplementary Figure 7).
Overall, the IGF1/2 inhibitor xentuzumab in combination with cisplatin strongly reduced the proliferation and colony formation capacity of HB cells, and this effect was more pronounced in IGF2high cells than in IGF2low cells.
The combination of IGF2 inhibition and cisplatin elicits synergistic effects in IGF2high HB organoids
Considering the antitumor capacity of IGF2 inhibition with xentuzumab combined with cisplatin observed in IGF2high HB cell lines, we proceeded to further evaluate the antitumor and molecular effects of this combination in more complex experimental models such as organoids derived from a PDX murine model generated from an IGF2high HB tumor[31]. First, we confirmed by RT-PCR that the high IGF2 levels in the original HB tumor were maintained in the organoids. Indeed, IGF2 levels in the organoid model were 19.5-fold higher when compared to mean IGF2 levels of the adjacent human liver tissue (Supplementary Table 9).
Next, organoids were treated with xentuzumab, cisplatin, xentuzumab + cisplatin, or control IgG, and organoid viability was assessed. A strong synergistic reduction in cell viability was observed with the combination of low doses of xentuzumab (1 nM) and cisplatin (< 1 μM), where monotherapies were not effective (combination index: 0.43; Figure 4C). A synergistic peak was observed for 1 μM cisplatin and 100 nM xentuzumab. This was further confirmed by calculating the IC50 for each condition. Indeed, the IC50 obtained for cisplatin combined with 0.1 μM xentuzumab was 25-fold lower than the one obtained for cisplatin as monotherapy (99.19 nM vs 2.57 μM), while the IC50 for xentuzumab alone was not reached with the assessed concentrations (>100 nM) (Figure 4D, Supplementary Figure 8).
Next, we investigated the effect of the combination of xentuzumab and cisplatin at low concentrations. The combination of xentuzumab and cisplatin activated apoptosis in a dose-dependent manner in organoids (Figure 4E), while monotherapies did not. The combination also reduced the activation of the IGF1R pathway, as measured by western blotting of IGF1R and Akt phosphorylation, in both organoids and cell lines stimulated with IGF2 (Figure 4F, Supplementary Figure 9). Intriguingly, xentuzumab reduced IGF2 pathway activation, but this effect was markedly enhanced by the combination, partially explaining the synergism of the combination and stressing the importance of combining both treatments to obtain antitumoral responses.
Therefore, these data validate the results observed in cell lines, suggesting that the efficacy of cisplatin could be greatly improved by the combination with IGF2 inhibition in IGF2high HB patients.
The combination of IGF2 inhibition and cisplatin is more effective than cisplatin alone in a murine model of IGF2high HB
The antitumor potential of IGF2 inhibition in combination with chemotherapy was tested in a xenograft murine model of IGF2high HB, generated through subcutaneous implantation of HepG2 cells. Mice bearing HB tumors were randomized to receive cisplatin, xentuzumab, cisplatin + xentuzumab, or control IgG. Mice treated with the combination of xentuzumab and cisplatin showed significantly increased survival (median survival: 16 days) compared to those treated with cisplatin alone (10 days, p = 0.038 vs combination) or IgG control (13 days, p = 0.027 vs combination) (Figure 5A). Conversely, the monotherapy treatment arms were unable to significantly reduce the viable tumor volume compared to the control. No significant differences in body weight or other toxicity signs were observed, indicating that all treatments were well-tolerated (Supplementary Figure 10).
Figure 5. Antitumor effect of xentuzumab plus cisplatin in an IGF2high xenograft model.
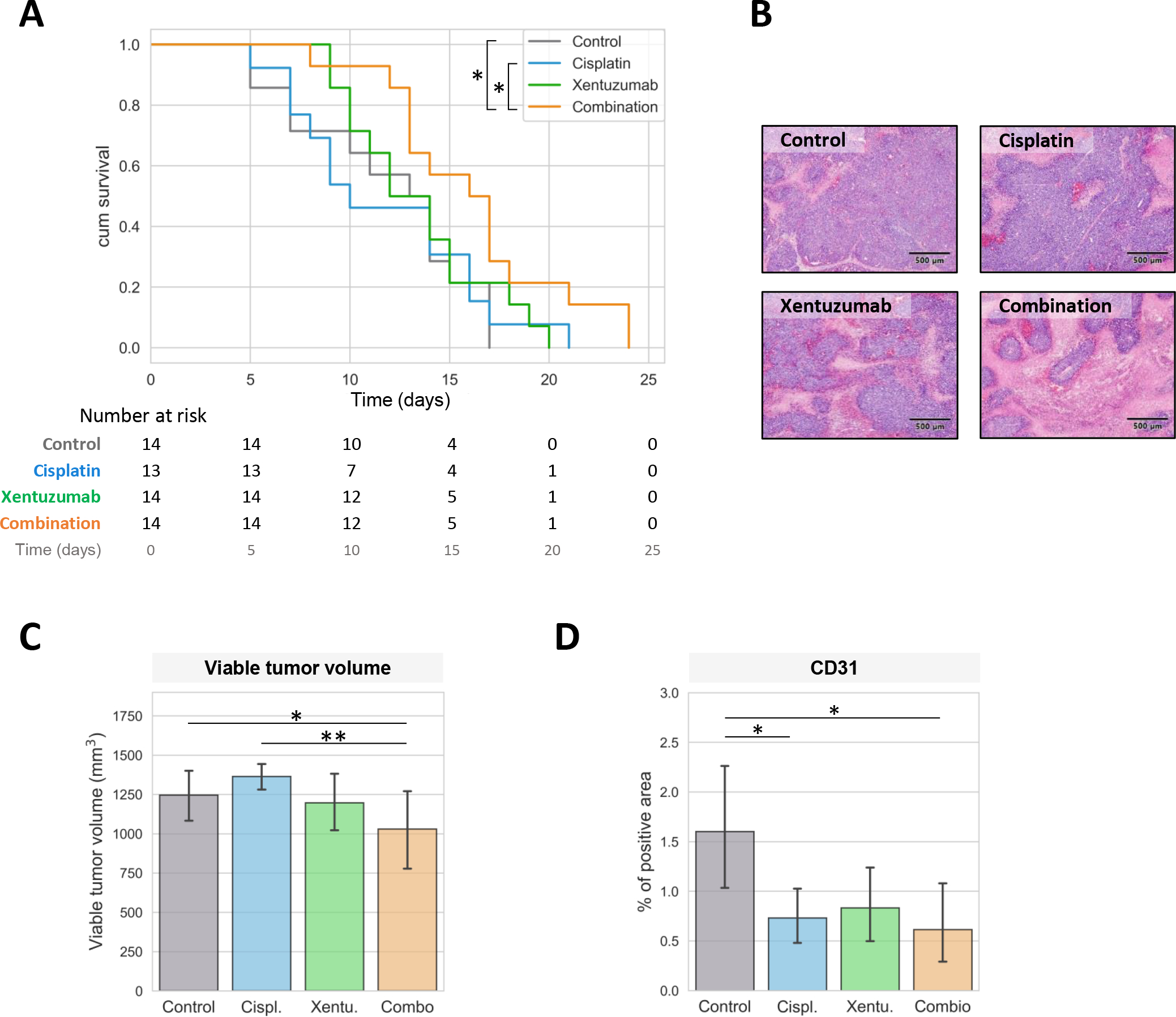
(A) Survival, measured as time to reach 1500 mm3, in animals treated with control IgG (n = 14), cisplatin (n = 13), xentuzumab (n = 14) or its combination (n = 14). (B) Representative images of H&E slides in mice treated with control IgG, cisplatin, xentuzumab or its combination. Images were captured at 40x. (C) Representative H&E stainings for each treatment regimen. (D) CD31 staining in tumors from treated mice. Error bars indicate mean ± standard deviation (SD). * P < 0.05; ** P < 0.01, *** P < 0.001.
Next, we analyzed the tumors and the antitumor effects of the treatments at the histological level. The combination of cisplatin and xentuzumab significantly decreased the fraction of viable tumor volume compared to cisplatin alone (p < 0.01) and the control (p < 0.05; Figure 5B–C). Furthermore, CD31 expression, a well-defined marker of angiogenesis, was significantly decreased in tumors from the combination treatment and cisplatin alone, likely because of the previously reported anti-angiogenic effect of cisplatin treatment and IGF1/2 blockade[18, 40] (Figure 5D).
Overall, these results indicate that the combination of xentuzumab and cisplatin improved survival and showed an increased antitumor effect compared to cisplatin alone. In addition, our data provide the rationale for testing IGF2 inhibitors in combination with cisplatin in trials with HB patients that express high levels of IGF2.
DISCUSSION
HB is a rare pediatric liver cancer that has largely been understudied at the molecular level. Recently, the molecular profile of these tumors has been deciphered using multi-omics approaches[9, 15, 21, 23, 41]. Despite these efforts, there are still no available targeted therapies in HB. Hence, HB patients receive standard preoperative platin-based chemotherapy regimens[3], associated with serious adverse effects, which could be partially avoided with other therapeutic approaches[7]. Herein, we provide evidence suggesting that IGF2 overexpression is a targetable alteration in HB (considering HBs and HCN-NOS), and describe the epigenetic mechanisms of IGF2 overexpression. Additionally, we showed remarkable antitumor effects of the combination of IGF2 inhibition and cisplatin in preclinical models.
Gene co-expression network analysis revealed that IGF2/IGF1R signaling is one of the most deregulated pathways in HB. In line with previous studies showing high IGF2 levels in HB[14, 42], IGF2 overexpression was detected in 71% of HB cases, being the most overexpressed targetable gene in tumors compared to non-tumor tissue. IGF2 overexpression is associated with progenitor cell markers (EPCAM, AFP, DLK1 and PEG3) and HB molecular classes of high-risk tumors (C2[21, 34], MRS-3[21]). In line with this, it has been reported that genes with epigenetic regulation such as IGF2 are abundantly expressed in the fetal liver[43] and that HB tumors with liver progenitor features are highly proliferative and have poor survival[15, 34]. On the other hand, IGF2low tumors were associated with a mesenchymal HB phenotype, with higher presence of EMT and TGF-β signaling markers, together with higher immune infiltrate. Importantly, patients with IGF2high tumors presented poorer recurrence-free survival compared to IGF2low tumors, highlighting the need for new therapies for this subgroup of patients[44].
IGF2 is a well-known paternally imprinted gene that encodes a fetal peptide hormone that regulates cellular proliferation and differentiation during the early stages of development[45]. It is highly expressed in the liver during fetal stages from the promoters P2, P3, and P4, and in adulthood, it is expressed from both alleles of promoter P1, albeit at much lower levels[36]. Reactivation of IGF2 expression through fetal promoter demethylation has been described as a crucial mechanism for IGF2 overexpression in liver cancer[18, 41, 46]. Other mechanisms of IGF2 overexpression in cancer involve ICR1 deregulation[15, 47], LOH of the 11p15.5 region[14, 15] and miR-483-5p expression[18, 48]. Our integrative analysis revealed that these four mechanisms are pivotal in driving IGF2 overexpression in HB, explaining 91% of the cases of IGF2high HB. Others have previously suggested a complex interaction between IGF and Wnt/β-catenin signaling and potential activation of IGF1R signaling due to CTNNB1 mutations in colon cancer[49, 50].
The high prevalence of IGF2 overexpression in HB and its association with poor outcomes make it an excellent candidate as a targetable alteration to develop personalized therapeutic approaches, since targeted therapeutic options for these patients are urgently needed. Targeted inhibition of IGF1/2 ligands with xentuzumab abrogates proliferative signaling through IGF1R, without affecting metabolic cell functions[18, 51]. Earlier studies in HCC pointed to the receptor IGF1R as a potential target[17]; however, subsequent clinical trials blocking this receptor using IGF1R mAb or IGF1R/insulin receptor (INSR) tyrosine kinase inhibitors failed to demonstrate beneficial outcomes[44] and resulted in metabolic-related toxic effects due to the inhibition of insulin metabolic signaling. Therefore, xentuzumab has important advantages over previous attempts to inhibit the IGF2/IGF1R pathway.
In HCC and colon cancer in vivo models, xentuzumab has shown encouraging antitumor activity and a favorable safety profile, specific for IGF2high tumors[18, 51]. A potent antiproliferative effect of xentuzumab has also been reported in cell lines from several cancer types, including HCC and HB[15, 18]. This promising background prompted us to further evaluate the potential of xentuzumab as a new targeted therapy for IGF2high HB tumors by assessing its effects on tumor organoids and a murine xenograft model. In this regard, xentuzumab combined with cisplatin significantly improved the effects of the compounds administered as monotherapy. Specifically, xentuzumab in combination with cisplatin was able to improve by 25-fold the efficacy of cisplatin alone in HB organoids and demonstrated an enhanced antitumor effect in IGF2high cells compared to IGF2low, indicating a new treatment strategy that would increase survival in IGF2high HB patients. In addition, the combination was effective at low cisplatin doses, suggesting that - in this setting - chemotherapy doses could be reduced and the chemotherapy-related adverse events diminished. This is especially important considering that chemotherapy causes serious adverse effects in HB patients such as ototoxicity[7, 52], which is detrimental to language and social development, particularly in young children[53]. Of note, patient stratification based on IGF2 tumor levels at time of diagnosis would be required in clinical studies testing xentuzumab in HB.
On the other hand, in other tumor types, treatment with IGF2/IGF1R inhibitors has been demonstrated to reactivate the antitumor effect of DNA-damaging agents such as cisplatin[54]. Our results support the idea that IGF1R pathway inhibition enhances sensitivity to DNA-damaging agents, as we see reactivation of the efficacy of cisplatin in the combination setting both in our HepG2 xenograft model and in our in vitro models.
The combination reduced aberrant angiogenesis, as observed by CD31 staining, similarly than monotherapies, and in line with the previously described anti-angiogenic effects of xentuzumab and cisplatin[18, 40] in other tumor types. Specifically, both IGF2 blockage and cisplatin have been reported to inhibit aberrant vasculature formation by downregulating VEGF signaling[55] and promoting the release of matrix metalloproteinases[40], respectively.
One limitation of the current study lies in the lack of specific cellular models for each of the newly described HB subtypes. Thus, despite HepG2 cells lines have been recently suggested to be derived from an HCN-NOS hepatoblastoma tumors[10], in the present study were used as model overall representing HB cases with high levels of IGF2. In this sense, clinical studies are awaited to clarify whether patients with HCN-NOS tumors need to be managed different from patients with HB tumors.
A second limitation of the study was the use of post-chemotherapy specimens. Here, we acknowledge that the implementation of our findings into the clinical setting will require further validation in diagnostic biopsies and the evaluation of the safety and antitumoral effects of the combination in ad-hoc pre-clinical models.
Overall, this study defines IGF2 as a promising molecular target in HB and provides evidence that IGF2 inhibition with xentuzumab enhances the efficacy of cisplatin. Therefore, combining xentuzumab with cisplatin could exert a greater antitumor effect than cisplatin alone in a subset of HB patients (~70%) presenting IGF2 overexpression. This provides a preclinical rationale for exploring this targeted therapy in combination with cisplatin in patients with IGF2high HB.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Catherine Guettier, from the Université Paris-Saclay, for the pathological review of the samples included in the study.
This study was funded by a grant from Boehringer Ingelheim. J. Abril-Fornaguera was supported by the PREDOCS-UB grant from the University of Barcelona (UB) and by a mobility grant from Fundació Universitària Agustí Pedro i Pons. J. Carrillo-Reixach and A. Del Río-Álvarez were supported by the Catalan Agency for the Management of University and Research Grants (AGAUR, 2019 FI_B01024 and 2022_FI_B00528). U. Balaseviciute was supported by the EILF-EASL Juan Rodés PhD Studentship from the European Association for the Study of the Liver (EASL) and the EASL International Liver Foundation (EILF). R. Pinyol was supported by the University of Barcelona (UB) and IDIBAPS/Fundació Clínic Barcelona. C. Montironi was supported by a Rio Hortega grant from Instituto de Salud Carlos III (ISCIII), Fondo Social Europeo (CM19/00039). P. K. Haber was supported by the fellowship grant of the German Research Foundation (DFG/HA 8754/1–1). M. Puigvehi received a Juan Rodés scholarship grant from Asociación Española para el Estudio del Hígado (AEEH). C. E. Willoughby was supported by a Sara Borrell fellowship (CD19/00109) from the ISCIII and Fondo Social Europeo. J. Peix received support from the Generalitat de Catalunya (PERIS, SLT017/20/000206). E. Guccione was supported by Alex’s Lemonade Stand Foundation. D. Sia was supported by the Gilead Sciences Research Scholar Program in Liver Disease. C. Armengol received funding from Instituto de Salud Carlos III (PI10/02082, PI13/02340), the European Union’s Horizon 2020 research and innovation program under grant agreement N° 668596 (ChiLTERN) and N° 826121 (iPC), CIBERehd (CB06/04/0033), AGAUR (2017-SGR-490) and the Ramón y Cajal (RYC-2010-07249) program of the Spanish Ministry of Science and Innovation. J. M. Llovet was supported by grants from the Samuel Waxman Cancer Research Foundation; the Spanish National Health Institute (MICINN, PID2019-105378RB-I00); NIH (R01 DK128289-01); through a partnership between Cancer Research UK (CRUK), Fondazione AIRC per la Ricerca sul Cancro and Fundación Científica de la Asociación Española Contra el Cáncer (FAECC) (Accelerator Award, HUNTER, Ref. C9380/A26813); and by grants from the Acadèmia de Ciències Mèdiques i de la Salut de Catalunya i Balears; and the Fundación Científica de la Asociación Española Contra el Cáncer (FAECC; Ref. PRYGN223117LLOV).
Footnotes
Authors’ Disclosures
This study was supported by a research grant from Boehringer Ingelheim. JML is receiving research support from Bayer HealthCare Pharmaceuticals, Eisai Inc, Ipsen, Boehringer-Ingelheim and consulting fees from Eli Lilly, Bayer HealthCare Pharmaceuticals, Eisai Inc, Merck, Bristol-Myers Squibb, Ipsen, Glycotest, Nucleix, Genentech, Roche, and AstraZeneca. The remaining authors declare no competing interests.
REFERENCES
- [1].Schnater JM, Köhler SE, Lamers WH, et al. Where do we stand with hepatoblastoma? Cancer 2003; 98: 668–678. [DOI] [PubMed] [Google Scholar]
- [2].Linabery AM, Ross JA. Trends in childhood cancer incidence in the U.S. (1992–2004). Cancer 2008; 112: 416–432. [DOI] [PubMed] [Google Scholar]
- [3].Feng J, Polychronidis G, Heger U, et al. Incidence trends and survival prediction of hepatoblastoma in children: a population-based study. Cancer Commun 2019; 39: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Nussbaumer G, Benesch M. Hepatoblastoma in molecularly defined, congenital diseases. Am J Med Genet A 2022; 188: 2527–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kremer N, Walther AE, Tiao GM. Management of hepatoblastoma. Curr Opin Pediatr 2014; 26: 362–369. [DOI] [PubMed] [Google Scholar]
- [6].Semeraro M, Branchereau S, Maibach R, et al. Relapses in hepatoblastoma patients: Clinical characteristics and outcome – Experience of the International Childhood Liver Tumour Strategy Group (SIOPEL). Eur J Cancer 2013; 49: 915–922. [DOI] [PubMed] [Google Scholar]
- [7].Knight KR, Chen L, Freyer D, et al. Group-Wide, Prospective Study of Ototoxicity Assessment in Children Receiving Cisplatin Chemotherapy (ACCL05C1): A Report From the Children’s Oncology Group. J Clin Oncol 2017; 35: 440–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].López-Terrada D, Alaggio R, De Dávila MT, et al. Towards an international pediatric liver tumor consensus classification: proceedings of the Los Angeles COG liver tumors symposium. Mod Pathol 2014; 27: 472–491. [DOI] [PubMed] [Google Scholar]
- [9].Sumazin P, Peters TL, Sarabia SF, et al. Hepatoblastomas with carcinoma features represent a biological spectrum of aggressive neoplasms in children and young adults. J Hepatol 2022; 77: 1026–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Eichenmüller M, Trippel F, Kreuder M, et al. The genomic landscape of hepatoblastoma and their progenies with HCC-like features. J Hepatol 2014; 61: 1312–1320. [DOI] [PubMed] [Google Scholar]
- [11].Gröbner SN, Worst BC, Weischenfeldt J, et al. The landscape of genomic alterations across childhood cancers. Nature 2018; 555: 321–327. [DOI] [PubMed] [Google Scholar]
- [12].Koch A, Denkhaus D, Albrecht S, et al. Childhood hepatoblastomas frequently carry a mutated degradation targeting box of the beta-catenin gene. Cancer Res 1999; 59: 269–73. [PubMed] [Google Scholar]
- [13].Cairo S Activation of Wnt and Myc signaling in hepatoblastoma. Frontiers in Bioscience 2012; E4: 480. [DOI] [PubMed] [Google Scholar]
- [14].Honda S, Arai Y, Haruta M, et al. Loss of imprinting of IGF2 correlates with hypermethylation of the H19 differentially methylated region in hepatoblastoma. Br J Cancer 2008; 99: 1891–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hirsch TZ, Pilet J, Morcrette G, et al. Integrated Genomic Analysis Identifies Driver Genes and Cisplatin-Resistant Progenitor Phenotype in Pediatric Liver Cancer. Cancer Discov 2021; 11: 2524–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Breuhahn K, Longerich T, Schirmacher P. Dysregulation of growth factor signaling in human hepatocellular carcinoma. Oncogene 2006; 25: 3787–800. [DOI] [PubMed] [Google Scholar]
- [17].Tovar V, Alsinet C, Villanueva A, et al. IGF activation in a molecular subclass of hepatocellular carcinoma and pre-clinical efficacy of IGF-1R blockage. J Hepatol 2010; 52: 550–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Martinez-Quetglas I, Pinyol R, Dauch D, et al. IGF2 Is Up-regulated by Epigenetic Mechanisms in Hepatocellular Carcinomas and Is an Actionable Oncogene Product in Experimental Models. Gastroenterology 2016; 151: 1192–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].World Health Organization. International Nonproprietary Names for Pharmaceutical Substances (INN) RECOMMENDED International Nonproprietary Names: List 76. WHO Drug Information 2016; 30: 477–544. [Google Scholar]
- [20].de Bono J, Lin C-C, Chen L-T, et al. Two first-in-human studies of xentuzumab, a humanised insulin-like growth factor (IGF)-neutralising antibody, in patients with advanced solid tumours. Br J Cancer 2020; 122: 1324–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Carrillo-Reixach J, Torrens L, Simon-Coma M, et al. Epigenetic footprint enables molecular risk stratification of hepatoblastoma with clinical implications. J Hepatol 2020; 73: 328–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Perilongo G, Maibach R, Shafford E, et al. Cisplatin versus cisplatin plus doxorubicin for standard-risk hepatoblastoma. N Engl J Med 2009; 361: 1662–70. [DOI] [PubMed] [Google Scholar]
- [23].Hooks KB, Audoux J, Fazli H, et al. New insights into diagnosis and therapeutic options for proliferative hepatoblastoma. Hepatology 2018; 68: 89–102. [DOI] [PubMed] [Google Scholar]
- [24].Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010; 26: 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008; 9: 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ashburner M, Ball CA, Blake JA, et al. Gene Ontology: tool for the unification of biology. Nat Genet 2000; 25: 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Borad MJ, Champion MD, Egan JB, et al. Integrated genomic characterization reveals novel, therapeutically relevant drug targets in FGFR and EGFR pathways in sporadic intrahepatic cholangiocarcinoma. PLoS Genet 2014; 10: e1004135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Xie C, Leung Y-K, Chen A, et al. Differential methylation values in differential methylation analysis. Bioinformatics 2019; 35: 1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Piskol R, Ramaswami G, Li JB. Reliable Identification of Genomic Variants from RNA-Seq Data. The American Journal of Human Genetics 2013; 93: 641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature Biotechnology 2013 31:3 2013; 31: 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Nicolle D, Fabre M, Simon-Coma M, et al. Patient-derived mouse xenografts from pediatric liver cancer predict tumor recurrence and advise clinical management. Hepatology 2016; 64: 1121–1135. [DOI] [PubMed] [Google Scholar]
- [32].Broutier L, Mastrogiovanni G, Verstegen MM, et al. Human primary liver cancer–derived organoid cultures for disease modeling and drug screening. Nat Med 2017; 23: 1424–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hartmann W, Waha A, Koch A, et al. p57(KIP2) is not mutated in hepatoblastoma but shows increased transcriptional activity in a comparative analysis of the three imprinted genes p57(KIP2), IGF2, and H19. Am J Pathol 2000; 157: 1393–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Cairo S, Armengol C, de Reyniès A, et al. Hepatic Stem-like Phenotype and Interplay of Wnt/β-Catenin and Myc Signaling in Aggressive Childhood Liver Cancer. Cancer Cell 2008; 14: 471–484. [DOI] [PubMed] [Google Scholar]
- [35].Chiang DY, Villanueva A, Hoshida Y, et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res 2008; 68: 6779–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Vu TH, Hoffman AR. Promoter-specific imprinting of the human insulin-like growth factor-II gene. Nature 1994; 371: 714–7. [DOI] [PubMed] [Google Scholar]
- [37].Bell AC, Felsenfeld G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 2000; 405: 482–485. [DOI] [PubMed] [Google Scholar]
- [38].Li X, Cui H, Sandstedt B, et al. Expression levels of the insulin-like growth factor-II gene (IGF2) in the human liver: developmental relationships of the four promoters. Journal of Endocrinology 1996; 149: 117–124. [DOI] [PubMed] [Google Scholar]
- [39].Liu M, Roth A, Yu M, et al. The IGF2 intronic miR-483 selectively enhances transcription from IGF2 fetal promoters and enhances tumorigenesis. Genes Dev 2013; 27: 2543–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ramer R, Schmied T, Wagner C, et al. The antiangiogenic action of cisplatin on endothelial cells is mediated through the release of tissue inhibitor of matrix metalloproteinases-1 from lung cancer cells. Oncotarget 2018; 9: 34038–34055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Nagae G, Yamamoto S, Fujita M, et al. Genetic and epigenetic basis of hepatoblastoma diversity. 2021; 12: 5423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Gray SG, Eriksson T, Ekström C, et al. Altered expression of members of the IGF-axis in hepatoblastomas. Br J Cancer 2000; 82: 1561–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Venkatraman A, He XC, Thorvaldsen JL, et al. Maternal imprinting at the H19-Igf2 locus maintains adult haematopoietic stem cell quiescence. Nature 2013; 500: 345–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer 2012; 12: 159–169. [DOI] [PubMed] [Google Scholar]
- [45].Bergman D, Halje M, Nordin M, et al. Insulin-like growth factor 2 in development and disease: a mini-review. Gerontology 2013; 59: 240–9. [DOI] [PubMed] [Google Scholar]
- [46].Li X, Nong Z, Ekström C, et al. Disrupted IGF2 promoter control by silencing of promoter P1 in human hepatocellular carcinoma. Cancer Res 1997; 57: 2048–54. [PubMed] [Google Scholar]
- [47].Rovina D, La Vecchia M, Cortesi A, et al. Profound alterations of the chromatin architecture at chromosome 11p15.5 in cells from Beckwith-Wiedemann and Silver-Russell syndromes patients. Sci Rep 2020; 10: 8275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Li X, Nadauld L, Ootani A, et al. Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat Med 2014; 20: 769–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Belharazem D, Magdeburg J, Berton AK, et al. Carcinoma of the colon and rectum with deregulation of insulin-like growth factor 2 signaling: clinical and molecular implications. J Gastroenterol 2016; 51: 971–984. [DOI] [PubMed] [Google Scholar]
- [50].Lee H, Kim N, Yoo YJ, et al. β-catenin/TCF activity regulates IGF-1R tyrosine kinase inhibitor sensitivity in colon cancer. Oncogene 2018; 37: 5466–5475. [DOI] [PubMed] [Google Scholar]
- [51].Friedbichler K, Hofmann MH, Kroez M, et al. Pharmacodynamic and Antineoplastic Activity of BI 836845, a Fully Human IGF Ligand-Neutralizing Antibody, and Mechanistic Rationale for Combination with Rapamycin. Mol Cancer Ther 2014; 13: 399–409. [DOI] [PubMed] [Google Scholar]
- [52].Sivaprakasam P, Gupta AA, Greenberg ML, et al. Survival and long-term outcomes in children with hepatoblastoma treated with continuous infusion of cisplatin and doxorubicin. J Pediatr Hematol Oncol 2011; 33: e226–e230. [DOI] [PubMed] [Google Scholar]
- [53].Wei M, Yuan X. Cisplatin-induced Ototoxicity in Children With Solid Tumor. J Pediatr Hematol Oncol 2019; 41: E97–E100. [DOI] [PubMed] [Google Scholar]
- [54].Ferté C, Loriot Y, Clémenson C, et al. IGF-1R targeting increases the antitumor effects of DNA-damaging agents in SCLC model: An opportunity to increase the efficacy of standard therapy. Mol Cancer Ther 2013; 12: 1213–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Yao N, Yao D, Wang L, et al. Inhibition of autocrine IGF-II on effect of human HepG2 cell proliferation and angiogenesis factor expression. Tumor Biology 2012; 33: 1767–76. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study including RNA-sequencing, methylation and SNP array are available within the article, its supplementary data files, and in Gene Expression Omnibus (GSE104766, GSE132219 and GSE132037). Data from validation cohorts were obtained from Gene Expression Omnibus (GSE132219 and GSE104766).