

Abstract
Cytomegalovirus (CMV) infection alters natural killer (NK) cell phenotype and function toward a more memory-like immune state. These cells, termed adaptive NK cells, typically express CD57 and NKG2C but lack expression of the Fc receptor γchain (Gene: FCER1G, FcRγ), PLZF, and SYK. Functionally, adaptive NK cells display enhanced antibody-dependent cellular cytotoxicity (ADCC) and cytokine production. However, the mechanism behind this enhanced function is unknown. To understand what drives enhanced ADCC and cytokine production in adaptive NK cells, we optimized a CRISPR/Cas9 system to ablate genes from primary human NK cells. We ablated genes that encode molecules in the ADCC pathway, such as FcRγ, CD3ζ, SYK, SHP-1, ZAP-70, and the transcription factor PLZF, and tested subsequent ADCC and cytokine production. We found that ablating the FcRγ chain caused a modest increase in TNFα production. Ablation of PLZF did not enhance ADCC or cytokine production. Importantly, SYK kinase ablation significantly enhanced cytotoxicity, cytokine production, and target cell conjugation, while ZAP-70 kinase ablation diminished function. Ablating the phosphatase SHP-1 enhanced cytotoxicity but reduced cytokine production. These results indicate that the enhanced cytotoxicity and cytokine production of CMV-induced adaptive NK cells is more likely due to the loss of SYK than the lack of FcRγ or PLZF. The lack of SYK expression could improve target cell conjugation through enhanced CD2 expression or limit SHP-1-mediated inhibition of CD16A signaling, leading to enhanced cytotoxicity and cytokine production.
Keywords: Adaptive Natural Killer (NK) cells, Fc receptor, CD16a (FcγRIIIA), Antibody Dependent Cellular Cytotoxicity (ADCC), Cytokine production, SYK, Fcer1g, PLZF, cytomegalovirus (CMV)
Introduction
Natural killer (NK) cells are innate lymphocytes that can kill both cancerous and infected cells (1, 2). In peripheral blood, NK cells comprise 5 to 20% of lymphocytes, mainly surveying the bloodstream for potential target cells (3–5). NK cells kill target cells and signal neighboring cells through a variety of mechanisms, including natural cytotoxicity (e.g., missing self), FasL- or TRAIL-induced apoptosis, antibody-dependent cellular cytotoxicity (ADCC), and cytokine production. During ADCC and cytokine production, NK cells first recognize the Fc region of IgG bound to a target cell with CD16A (FcγRIIIA). CD16A utilizes signaling adapter molecules including the Fc receptor γ chain (Gene: FCER1G, FcRγ) and/or the CD3 ζ chain (CD3ζ) for signal transduction (6). Despite their seemingly redundant functions, FcRγ and CD3ζ have distinct features. For example, FcRγ contains one immunoreceptor tyrosine-based activation motif (ITAM), whereas CD3ζ contains three ITAMs (7). Crosslinking of CD16A by antibodies bound to a target cell recruits Src-family kinases, such as Lck, to phosphorylate ITAMs on FcRγ and/or CD3ζ. This phosphorylation recruits the tyrosine kinases SYK and/or ZAP-70 to ITAMs for subsequent signal transduction. SYK preferentially binds FcRγ, whereas ZAP-70 preferentially binds CD3ζ (8). SYK and ZAP-70 then phosphorylate their respective downstream targets (e.g., PI3K, VAV1, and phospholipase Cγ isoforms) to further propagate the CD16A signaling pathway, ultimately culminating in cytotoxicity and cytokine production (9, 10). While the molecules participating in CD16A-mediated signaling pathway are defined, the specific contributions of these signaling proteins to expanded primary human NK cell function were unclear.
Circulating NK cells are not a homogenous population. Various factors, such as infection, can drive NK cell phenotypic and functional diversity (11, 12). Cytomegalovirus (CMV) is the epitome of a pathogen that alters both NK cell phenotype and function (12–15). In mice, murine CMV (mCMV) infection results in expansion of a subset of NK cells expressing Ly49H, a receptor that binds mCMV peptide on MHC (12). This NK population persists, specifically expands upon mCMV reactivation, and has enhanced effector function, displaying adaptive-like qualities (12). In humans, CMV infection also results in expansion of NK cells expressing NKG2C and CD57 (13–15). These cells proliferate after recognizing CMV in vitro (16) and upon CMV reactivation in humans (17). After CMV reactivation, CD57+/NKG2C+/NKG2A- NK cells also exhibit enhanced cytokine production (17). These results led to the classification of CD57+/NKG2C+/NKG2A- NK cells as adaptive NK cells due to their adaptive qualities, such as longevity and antigen-specific expansion (albeit through a limited germline-encoded receptor). Adaptive NK cells are also hypothesized to play a protective role in cancer and transplant patients as a higher frequency of adaptive NK cells correlates with better clinical outcomes in individuals with reactivated CMV (18).
In addition to phenotypic differences, adaptive NK cells also show increases in ADCC and cytokine production compared to conventional NK cells (19, 20). Zhang et al. showed that CMV infection was associated with loss of the FcRγ chain. FcRγ-negative (FcRγ-) NK cells showed increased degranulation (CD107a), IFNγ, and TNFα production in response to opsonized infected cells over conventional NK cells (20). In addition to FcRγ deficiency, adaptive NK cells have been shown to lack both SYK kinase and PLZF (21, 22). PLZF is a transcription factor that binds the promoter of genes encoding FcRγ and SYK and likely plays a role in regulation of these genes (22, 23). In addition to CMV, other infectious diseases, such as HIV and malaria, can induce adaptive NK cell formation (24–26). HIV- and malaria-induced adaptive NK cells also show enhanced ADCC and cytokine production (24–26). However, these cells have a different phenotype than their CMV-induced counterparts, such as maintained SYK expression, highlighting adaptive NK cell heterogeneity (24–26).
The absence of FcRγ, PLZF, and/or SYK is hypothesized to be the mechanism responsible for enhancing the ADCC function of CMV-induced adaptive NK cells (17–22). Yet it was unclear whether these molecules are responsible for enhanced function or only correlate with it. Here, we investigated the role of molecules associated with CMV-induced adaptive NK cells—including FcRγ, PLZF, and SYK—in the ADCC signaling apparatus. We tested for redundancy in the ADCC signaling pathway by targeting molecules with similar functions as FcRγ and SYK, such as CD3ζ and ZAP-70, respectively. We also sought to disentangle which molecules are associated with cytotoxic activity versus cytokine production after CD16A activation. Though using a murine model is a potential approach to answer these questions, the murine CD3ζ chain does not associate well with CD16, limiting the utility of murine signaling protein knockouts for ADCC studies (27). For humans, until recently, it has not been possible to ablate genes in expanded primary human NK cells. Therefore, to accomplish these goals, we developed a CRISPR/Cas9 gene-editing protocol for thawed human expanded primary NK cells, ablated genes that encoded molecules in the CD16A pathway, and tested the effect of gene ablation on ADCC and cytokine production.
We show that SYK kinase ablation enhances ADCC, cytokine production, and target cell conjugation, while ZAP-70 ablation results in the opposite phenotype. SYK ablation enhanced killing more so than degranulation and the increased killing capacity was maintained for four weeks post-expansion. These results point toward a mechanism involving SYK for the enhanced function of CMV-induced adaptive NK cells. Overall, this study provides a mechanistic understanding of the enhanced ADCC function observed in CMV-induced adaptive NK cells and provides a new strategy for improving NK cell therapies by leveraging the ADCC pathway.
Methods
Resource Availability
Lead Contact
Requests for information or resources should be directed to the lead contact, Geoffrey Hart (hart0792@umn.edu).
Materials Availability
This study did not generate unique reagents. All reagents used are listed in Table 1.
Table 1.
List of reagents, resources, and tools used for the manuscript.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Biotinylated CD3 | STEMCELL Technologies | 18051 |
| Anti-RBC | Rockland | 1094139 |
| CD107a | Biolegend | 328625 |
| Viability | Tonbo | 13-0865-T500 |
| FcRγ | Millipore | FCABS400F |
| PLZF | Invitrogen | 17-9322-82 |
| CD247 (CD3ζ) | Biolegend | 644105 |
| IFNγ | BD Biosciences | 562392 |
| TNFα | Biolegend | 502938 |
| SYK | BD Horizon | 624144 |
| ZAP-70 | Biolegend | 691206 |
| CD16A | Biolegend | 360728 |
| SHP-1 | abcam | Ab209913 |
| CD56 | Beckman Coulter | A79388 |
| CD3 | Biolegend | 317322 |
| CD45 #1 BV510 | Biolegend | 304036 |
| CD45 #2 BV650 | Biolegend | 304043 |
| CD45 #3 BV785 | Biolegend | 304048 |
| CD45 AF647 | Biolegend | 304056 |
| CD11a (TS2/4) | Biolegend | 350606 |
| CD11a/CD18 (m24) | Biolegend | 363408 |
| CD18 (TS1/18) | Biolegend | 302108 |
| CD2 | Biolegend | 300224 |
| Trastuzumab (anti-HER2) | N/A | |
| Rituximab (anti-CD20) | N/A | |
| Biological samples | ||
| Healthy adult whole blood | Memorial Blood Bank | N/A |
| RPMI-1640 | Kd Medical | 50-101-8907 |
| Ficoll | MP Biomedicals | ICN50494 |
| ACK Lysis Buffer | Lonza | BP10-548E |
| Fetal bovine serum | PEAK Serum | PS-FB1 |
| EDTA | Fisher | BP2482-1 |
| RPMI-1640 | FIsher | SH3002701 |
| Gentamicin | Sigma-Aldrich | G1272 |
| Trypsin EDTA | Gibco | 25300062 |
| McCoy’s 5A Medium | Gibco | 166600082 |
| Pen/Strep | Gibco | 1514022 |
| MEM-alpha | Gibco | 12561 |
| HI-FBS | Gibco | 16140071 |
| HI-Horse Serum | Gibco | 26050088 |
| IL-2 | NCI | N/A |
| 2-Mercaptoethanol | Gibco | 21985023 |
| X-VIVO 15 | Lonza | BE02-060F |
| IL-15 | NCI | N/A |
| Cas9 | IDT | 1081059 |
| P3 Primary Cell Solution | Lonza | V4XP-3032 |
| BFA | Millipore Sigma | 20350-15-6 |
| Monensin | Millipore Sigma | M5273 |
| Formaldehyde | Thermo | 28908 |
| Triton | Fisher | BP151-500 |
| BSA | MP Biomedicals | 810681 |
| Critical commercial assays | ||
| NK Cell Isolation Kit | STEMCELL Technologies | 17955 |
| DNEasy Kit | Qiagen | 69506 |
| LEGENDScreen Human PE Kit | Biolegend | 700007 |
| Cell Trace Violet Labeling Kit | Thermo Fischer | C34571 |
| Cell Trace Far Red Labeling Kit | Thermo Fischer | C34564 |
| DELFIA | PerkinElmer | AD0116 |
| Experimental models: Cell lines | ||
| K562-aAPC | Branden Moriarity laboratory | K562-aAPC |
| SKOV-3 | Jeff Miller laboratory | SKOV-3 |
| SKOV-3 NLR | Jeff Miller laboratory | SKOV-3 NLR |
| Raji NLR | Jeff Miller laboratory | Raji NLR |
| NK-92-GFP | Bruce Walcheck laboratory | NK-92-GFP |
| NK-92-GFP-CD16A | Bruce Walcheck laboratory | NK-92-GFP-CD16A |
| NK-92-GFP-CD64/16A | Bruce Walcheck laboratory | NK-92-GFP-CD64/16A |
| Oligonucleotides | ||
| See supplementary table 1 | ||
| Software and algorithms | ||
| CRISPR Design Tool | Synthego | https://design.synthego.com/#/ |
| FlowJo (10.8.0) | FlowJo | N/A |
| ICE Analysis | Synthego | https://ice.synthego.com/#/ |
| Incucyte base analysis software | Sartorius | https://www.sartorius.com/en/products/live-cell-imaging-analysis/live-cell-analysis-software/incucyte-base-software#id-786768 |
| Excel (16.54) | Microsoft | N/A |
| RStudo (1.4.1717) | N/A | N/A |
| Prism (9.1.2) | GraphPad | N/A |
| Illustrator (25.4.1) | Adobe | N/A |
| Other | ||
| Red blood cell purification filter | Memorial Blood Center | 4C4300 |
| Mr. Frosty | Thermo-FIsher | 5100-0001 |
| G-Rex 6-well plates | Wilson Wolf | 80240M |
| G-Rex 24-well plates | Wilson Wolf | 80192M |
Data and code availability
R 4.1 (28) was used to generate Figure S1G. The code was adapted from https://biocorecrg.github.io/CRG_RIntroduction/volcano-plots.html (29) and is available at https://github.com/jamesdahlvang/volcano. All raw data is available upon request.
Experimental Model and Subject Details
Primary Cell Purification and Freezing
Primary red blood cells (RBCs) and peripheral blood mononuclear cells (PBMCs) were obtained and purified from deidentified adult blood donors (Memorial Blood Center). The work done in this study was approved by the Institutional Review Board (IRB) of the University of Minnesota. The cytomegalovirus (CMV) status of each donor was reported by Memorial Blood Center. RBCs were purified from whole blood by filtration (Memorial Blood Center, 4C4300) and resuspended at 50% hematocrit in RPMI-1640 + 25 mM HEPES, L-Glutamine, and 50 mg/L Hypo-Xanthine (Kd Medical, 50–101-8907). PBMCs were isolated using Ficoll (MP Biomedicals, ICN50494) centrifugation and the remaining RBCs were lysed using ACK (Lonza, BP10–548E). Once isolated, PBMCs were counted and resuspended at 2×108 cells/mL in 10mL of PBS + 2% FBS (PEAK Serum, PS-FB1) + 1 mM EDTA (SC Buffer). NK cells were then isolated from these PBMCs using a magnetic negative selection kit (STEMCELL Technologies, 17955). Briefly, PBMCs were stained with 500 μl of the manufacturer’s antibody cocktail, which is less antibody than the manufacturer recommends, and 30 μl of additional biotinylated anti-CD3 (STEMCELL Technologies, 18051) was added to the sample to improve purity. Samples were incubated at RT for 10 minutes. After incubation, 1 mL of magnetic beads was added to the sample, which was then vortexed 2–3 times and incubated for 10 minutes at RT. Post-incubation, 35 mL of SC Buffer was added to the cells, which were then placed on a magnet and incubated at RT for 10 minutes. After incubation, the liquid—which contained purified NK cells—was removed from the tube without touching the magnet. NK cells were then added to a new 50 mL conical and placed back on the magnet for 10 minutes at RT. After 10 minutes, purified NK cells were removed and counted. For cryopreservation, NK cells were resuspended in RMPI-1640 (Fisher, SH3002701) + 50% FBS (PEAK Serum, PS-FB1) at 107 cells/mL. 500 μL of the suspension was transferred to a cryovial. When all tubes were ready to freeze, 500 μl of FBS (PEAK Serum, PS-FB1) + 15% DMSO was added to cells in cryovials. The cells were immediately transferred to a pre-cooled Mr. Frosty (Thermo-Fisher, 5100–0001) and then placed in a −80° C freezer. After 24–48 hours, the cells were transferred to a vapor-phase liquid nitrogen freezer for later use.
K562-artificial Antigen Presenting Cell (aAPC) Cell Line Maintenance
Modified K562-aAPC cells were graciously provided by Dr. Branden Moriarity’s lab. These cells express 4–1BB ligand and IL-21 to aid NK cell proliferation (30). K562-aAPCs were maintained between 1×105 to 1.5×106 cells/mL in RPMI-1640 (Fisher, SH3002701) + 10% FBS (PEAK Serum, PS-FB1) (RP10) + 1 μg/mL gentamicin (Sigma-Aldrich, G1272) and incubated at 37 °C with 5% CO2. Unless listed otherwise, all 37 °C incubations are conducted at 5% CO2.
SKOV-3 Cell Line Maintenance
Both conventional SKOV-3 and fluorescent SKOV-3 NucLight Red cells (SKOV-3 NLR) were graciously provided by Dr. Jeffrey Miller’s lab. Once the cells became 80–95% confluent, the medium was aspirated from the flask and the cells were washed with PBS. After washing the cells, 5 mL of pre-warmed 0.05% Trypsin-EDTA (Gibco, 25300062) was added to the flask. The cells were then incubated at 37 °C for 5 minutes. Following incubation, the flask was tapped to dislodge cells from the plastic. The cells were collected and washed with 10 mL of pre-warmed McCoy’s 5A Medium (Gibco, 166600082) + 10% FBS (PEAK Serum, PS-FB1) + 1% Pen/Strep (Gibco, 1514022) + 1 μg /mL puromycin (Thermo Fisher, A1113830) to maintain NucLight Red expression (SKOV-3 Media). The resulting suspension was centrifuged, resuspended in pre-warmed SKOV-3 Media, and counted. Half of the cell suspension was used for counting. The SKOV-3 cells were then diluted to 6.5×104 cells/mL with SKOV-3 media, transferred to a new flask, and incubated at 37 °C.
Raji Cell Line Maintenance
Raji cells expressing NucLight Red (Raji NLR) were graciously provided by Dr. Jeffrey Miller’s lab. Every two days, Raji NLR cells were resuspended and centrifuged. After centrifugation, the cells were resuspended with the same volume in pre-warmed RP10 and counted. The Raji NLR cells were then diluted to 2×105 cells/mL with RP10 + 1 μg /mL of puromycin (Thermo Fisher, A1113830) to maintain NucLight Red expression, transferred to a new flask, and incubated at 37 °C.
NK-92 Cell Line Maintenance
NK-92 cells expressing GFP (parent line, NK-92-GFP), CD16A and GFP (NK-92-GFP-CD16A), and CD64/16A and GFP (NK-92-GFP-CD64/16A) were graciously provided by Dr. Bruce Walcheck’s lab. NK-92-GFP-CD16A cells stably express GFP and CD16A (31), whereas NK-92-GFP-CD64/16A cells stably express GFP and the cytoplasmic tail of CD16A with the extracellular domain of CD64 (32). Every two days, NK-92 cells were resuspended and centrifuged. After centrifugation, the supernatant from the cell culture was saved. The cells were then resuspended with the same volume of pre-warmed MEM-alpha (Gibco, 12561) + 10% HI-FBS (Gibco, 16140071) + 10% HI-Horse Serum (Gibco, 26050088) + 0.8% Pen/Strep (Gibco, 1514022) + 200 IU/mL IL-2 (NCI) + 0.08 mM 2-Mercaptoethanol (Gibco, 21985023) (NK-92 Media) and counted. After counting, cells were resuspended at 2×105 cells/mL in 50% NK-92 media and 50% of the previous culture’s supernatant, then incubated at 37 °C. Cells were maintained between 2×105 and 1.5×106 cells/mL.
NK Cell Expansion
Cryopreserved vials of primary NK cells were thawed by placing them in a water bath at 37 °C until the vial was 95% thawed. The cells were then transferred to a 15 mL conical. Pre-warmed RP10 was added dropwise at a rate of 1 mL/minute while the conical was swirled. After adding 3 mL of RP10, 6 more mL of pre-warmed RP10 was added and the cells were centrifuged at 650 × g. The cells were washed once with RP10, centrifuged, and resuspended in 5 mL of X-VIVO™ 15 (Lonza, BE02–060F) + 10% FBS (PEAK Serum, PS-FB1) (X-Vivo 10) + 2 ng/mL IL-15 (NCI). Cells were then transferred to a 6-well plate and incubated overnight. After incubation, 3×106 NK cells were mixed with 6×106 irradiated K562-aAPC cells in 30 mL X-Vivo 10 + 50 IU/mL IL-2 (NCI). K562-aAPCs were irradiated at 100 grays (RS-2000 X-Ray irradiator). The cells were added to a G-Rex© 6-well plate (Wilson Wolf, 80240M) and incubated at 37 °C for six days.
Methods Details
CRISPR/Cas9 Gene-editing
Single-guide RNAs (sgRNA) were designed using Synthego’s CRISPR Design tool (https://design.synthego.com/#/). sgRNA sequences are listed in Supplementary Table 1. Ribonucleoprotein complexes (RNPs) were made by combining 100 pmol of sgRNA (IDT) with 30 pmol of Cas9 (IDT, 1081059). If the volume of the RNP complex was less than 5 μL/sample, nuclease-free H2O was added after combining the RNP complex to bring the volume to 5 μL/sample. The RNP complexes were incubated at RT for 15 minutes. If multiple genes were ablated simultaneously, individual RNP complexes were made and combined after incubating for 15 minutes.
NK cells were centrifuged and resuspended in P3 Primary Cell Nucleofector© Solution (Lonza, V4XP-3032) at 3.33×108 cells/mL. 5×106 cells were mixed with RNPs and transferred without forming bubbles to a 16-well Nucleocuvette© Strip (Lonza, V4XP-3032). Unless otherwise stated, the cells were nucleofected with an Amaxa 4D-Nucleofector™ (Lonza) using code CA137. Post-nucleofection, the cells were rested for 15 minutes at RT. 80 μL of pre-warmed X-Vivo 10 was added to each nucleofection well. The cells were rested for 30 more minutes at RT, then the total volume was transferred to a G-Rex© 24-well plate (Wilson Wolf, 80192M) with 1.9 mL of pre-warmed X-Vivo 10. Each well was then washed with 100 μl of media, which was transferred to the cell suspension. The cells were rested once more for 30 minutes at RT before 4 mL of X-Vivo 10 + 4.5 ng/mL of IL-15 (NCI) was added to each well (3 ng/mL IL-15 final). The cells were then incubated at 37 °C for six days.
Degranulation and Cytokine Production Assay with Red Blood Cells as NK Targets
RBCs were opsonized with polyclonal anti-RBC rabbit antibody (Rockland, 1094139) and combined with NK cells at a ratio of 1 NK:5 RBC in RP10 + 1 μg/mL brefeldin A (BFA) + 2 μM monensin + 1:200 anti-CD107a, which measures degranulation (Biolegend, 328625). As a negative control, NK cells were also incubated with non-opsonized RBCs. The cells were then incubated at 37 °C for 5 hours. After five hours, the cells were centrifuged, washed, and stained for flow cytometry.
Flow Cytometry
Cells were resuspended in a master mix made of PBS, viability dye (Tonbo, 13–0865-T500), and surface stain antibodies, then incubated at RT in the dark for 20 minutes. The cells were washed and incubated in 2% formaldehyde (Thermo, 28908) at 37 °C for 10 minutes. After incubation, the cells were washed and resuspended in 0.04% Triton (Fisher, BP151–500) at RT in the dark for exactly 7 minutes. After 7 minutes, the cells were washed in PBS + 2% FBS (PEAK Serum, PS-FB1) + 2 mM EDTA (Fisher, BP2482–1) + 2% BSA (MP Biomedicals, 810681) (FACS buffer) and stained with an internal master mix in FACS buffer. Depending on the antibodies used, the cells were then incubated at RT in the dark for 30–120 minutes. After incubation, the cells were washed with FACS buffer, washed again with PBS, and resuspended in PBS for flow cytometry. Samples were run on a CytoFLEX flow cytometer (Beckman-Coulter) and analyzed via FlowJo 10.8.0 (FlowJo) to determine the extent of gene ablation and the effect of gene ablation on cell function.
ICE Analysis
Synthego’s Inference of CRISPR Edits (ICE) (https://ice.synthego.com/) was used to analyze gene ablation. For ICE analysis, DNA from gene-ablated and control samples was isolated using QIAGEN’s DNAeasy kit (Qiagen, 69506) according to the manufacturer’s instructions. PCR primers were designed to amplify a region of 500 base pairs (bp) around the cut site. The forward primer was designed 100–200 bp upstream from the cut site to capture it with high-quality sequencing. Additionally, we designed sequencing primers that were nested 5–50 base pairs inside the amplicon to improve sequencing efficiency. After amplifying the sgRNA cut site, approximately 100 ng of the PCR product was submitted to the UMN Genomics center along with 6.4 pmol of a sequencing primer for Sanger sequencing. The primers used are listed in Supplementary Table 1.
350 Surface Protein Analysis (LEGENDScreen™)
To assess the expression of approximately 350 surface proteins at once, cells were analyzed using Biolegend’s LEGENDScreen™ Human PE Kit (Biolegend, 700007). Subjects were barcoded with combinations of CD45 antibodies conjugated to three fluorophores, allowing us to test 7 subjects in one well (A, B, C, AB, AC, BC, ABC). After barcoding and pooling, samples were stained for CD56, CD3, live/dead, and CD14, then acquired on a BD Fortessa flow cytometer according to manufacturer instructions.
Cell-cell conjugation assay
Expanded primary NK cells were labeled with Cell Trace Violet (CTV, Thermo Fischer Scientific, C34571) and SKOV-3 cells (HER-2 +) were labeled with Cell Trace Far Red (Thermo Fischer Scientific, C34564) per the manufacturer’s instructions. After labeling, the cells were counted and resuspended in RP10. SKOV-3 and Raji NLR cells were resuspended at 2×106 cells/mL while NK cells were resuspended at 1×106/mL. Prior to combining target cells with NK cells, Raji NLR were incubated with 1 μg /mL rituximab and SKOV-3 cells were incubated with 10 μg /mL anti-HER2 (trastuzumab) antibody. For a 1:2 Effector: Target (E:T) ratio, 100 μL of each cell type was mixed together, centrifuged for 1 min at 20 × g and then incubated in a 37 °C water bath for the indicated time points (0, 2, 10, 20, and 30 min). After each time point, the cells were vortexed for 3 sec and immediately fixed with ice cold 2% paraformaldehyde in PBS. Fixed cells were then used to acquire 5×104 events on a flow cytometer. The percentage of conjugated NK cells was calculated by gating on the NLR and CTV double positive events (33).
DELFIA Killing Assay with SKOV-3 cells as NK Targets
DELFIA killing assays were performed according to manufacturer instructions (PerkinElmer, AD0116). Briefly, SKOV-3 cells were resuspended at 106 cells/mL in SKOV-3 Media. To label the cells, 1.5 μL/mL of Bis(acetoxymethyl)-2–2:6,2 terpyridine 6,6 dicarboxylate (BATDA) was added to the suspension. The cell suspension was then incubated for 30 minutes at 37 °C. After labeling the cells, they were washed and transferred to a round-bottom 96-well plate at a concentration of 8×103 cells/well. Anti-HER2 was then added to the well at a concentration of 3 or 10 μg/mL along with either expanded primary NK or NK-92 (effector) cells at various E:T ratios. The plate was spun at 400 × g for one minute and incubated at 37 °C for 2 hours with 5% CO2. After 2 hours, the plate was centrifuged at 500 × g for five minutes. 20 μL of supernatant was then transferred to a 96-well DELFIA Yellow Plate and combined with 200 μl of europium. Signal was measured by time-resolved fluorescence using a BioTek Synergy 2. To measure maximum release, BADTA-labeled target cells were incubated with 10μL of lysis buffer per the manufacturer’s instruction. To measure spontaneous lysis, BADTA-labeled SKOV-3 cells were cultured in parallel without effector cells or antibodies. When possible, samples were run in technical triplicates.
Incucyte Killing Assay with SKOV-3 cells as NK Targets
On the day prior to the killing assay, SKOV-3 NLR cells (HER-2 +) were resuspended at 4×104 cells/mL in RP10. 100 μl of the cell suspension was added to each well of an Incucyte-compatible flat-bottom 96-well plate. The SKOV-3 NLR cells were then incubated overnight at 37 °C to allow them to adhere to the bottom of the well.
The following day, NK cells were resuspended at 1.2×106 cells/mL in RP10. 60 μl of NK cells were added to a new 96-well plate. Anti-HER2 was then diluted to 20 μg/mL in RP10 and 60 μL of the solution was added to the NK cells. To prevent cells from localizing to the edge of the well, both the 96-well plate containing the NK cells/anti-HER2 and the plate containing the SKOV-3 NLR cells were incubated at 37 °C for 15 minutes. 100 μl of NK cells/anti-HER2 was then added to the plate containing SKOV-3 NLR cells for an effector: target (E: T) ratio around 15:1 and a final anti-HER2 concentration of 5 μg/mL. The cell mixture was then loaded into an Incucyte SX5 (Sartorius). Immediately after loading the plate, a t = 0-hour image was taken to ensure cells were not localized to the edge of the well. After confirming cells were dispersed throughout the well, four consecutive images were taken per well once per hour for 24 hours. As controls, SKOV-3 NLRs and NK cells without anti-HER2 were imaged as well as SKOV-3 NLRs without NK cells. Samples were run in technical triplicates.
Quantification and Statistical Analysis
Ablation Level Cutoffs
Data points from samples with less than 25% protein ablation were excluded from all analyses to ensure the gene-ablated population was substantial enough to analyze. For expansion experiments, the cutoff was only applied at day 6 (e.g., if a sample was above 25% protein ablation at day 6 but below 25% at day 13, the data from day 13 was included). Samples between 25–49% ablation were kept for ADCC assay analysis as the gene-ablated cells can be gated on via flow cytometry. Unlike flow cytometry-based ADCC assays, killing assays utilized a target cell count readout, precluding specific analysis of gene-ablated cells. Thus, killing assay data from samples with less than 49% protein ablation were excluded.
LEGENDScreen™ Cutoffs
Proteins that were expressed in fewer than 10% of cells from each subject at each timepoint were classified as not expressed in NK cells. These proteins were excluded from the final analysis.
Fold-change Data
Data were transformed using fold-change and log2 fold-change to ease the visualization of trends. Fold-change was calculated by dividing the gene-ablated value by the no RNP value for the measurement in question.
Gene Ablation Analysis
Flow cytometry was used to assess the extent of CRISPR/Cas9 gene-ablation at the protein level. To calculate the protein ablation percentage for single-gene ablations, the following formula was applied:
To calculate the protein ablation percentage for double-gene ablations, the following formula was used:
Incucyte Killing Assay Analysis
Incucyte Base Analysis Software (Satorius, v2019B) was used to analyze killing assays. Images with SKOV-3 NLRs + NK cells, SKOV-3 NLRs alone, and NK cells alone were chosen from the 0-hour, 12-hour, and 24-hour images. Image parameters were then adjusted to ensure the software detected SKOV-3 NLRs but not NK cells. Graphs were created by normalizing each well’s SKOV-3 NLR count at each timepoint to its time 0-hour count.
DELFIA Spontaneous Lysis Calculation
For each sample, specific lysis was used to report ADCC activity. The calculation for specific lysis is shown below:
Graphing and Statistical Analysis
Excel 16.54 (Microsoft) was used for data transformation and statistical analysis. Schematics were created with BioRender.com. R 4.1 (28) was used to generate Figure S1G. Prism 9.1.2 (GraphPad) was used to generate figures and run statistical analyses. Illustrator 25.4.1 (Adobe) was used to draft figures for publication. Sample sizes, independent experiments run, statistical tests, and significance thresholds are listed in each figure legend.
Results
Development of an effective CRISPR/Cas9 gene-editing protocol to ablate genes in expanded primary human NK cells
Previously published CRISPR/Cas9 protocols for expanded primary human NK cells have reported various platforms and settings (30, 34–37). We aimed to optimize a CRISPR/Cas9 protocol for the Amaxa 4D system as it can be scaled up for large numbers of cells, potentially allowing for future clinical applications. To optimize our CRISPR/Cas9 protocol, we first used 24 Amaxa 4D nucleofection codes (format: XX###) to ablate FcRγ (FCER1G) from expanded primary NK cells. Six days post-CRISPR, we measured gene ablation efficiency (Figure S1A) and cell recovery (Figure S1B) by flow cytometry. From these 24 electroporation codes, we picked six that balanced protein ablation and cell recovery. We also tested ‘CA137’, as it had been used for successful gene ablation in NK-92 cells (38). We observed >70% ablation of FcRγ (Figure S1C) for each code. However, viable cell recovery varied. Of the codes tested, CA137 showed the highest average recovery at 3.3×106 cells (Figure S1D). We concluded that using CA137 results in the best balance of effective gene ablation and cell recovery, leading us to use CA137 for the rest of the experiments. By evaluating a compilation of experiments that used CA137, we found that cell counts six days post-CRISPR often exceeded our starting cell count, indicating that the cells recovered well from nucleofection (Figure S1E). We also tested CRISPR/Cas9 deletion efficiency by molecular and protein-level methods to evaluate concordance. Indel analysis of sequencing data (Inference of CRISPR Edits (ICE)) and flow cytometry analysis of our target gene protein expression typically showed similar levels of gene ablation (Figure S1F). Representative gating schemes and protein ablation frequency for all data are shown in Figures S2A–M.
We then assessed the phenotype and function of expanded primary human NK cells post-CRISPR. To expand primary human NK cells, we incubated them for seven days by co-incubating them with IL-2, IL-15, and irradiated K562 artificial APC cells, which express 4–1BB ligand and IL-21. However, expanding human primary NK cells in vitro can affect NK cell phenotype (36). We were concerned that activation and expansion could trigger ADAM17 activity, which cleaves CD16A from the NK cell surface (39, 40). Thus, to evaluate the effect of our expansion protocol on cell phenotype, we barcoded expanded primary human NK cells from 7 subjects and examined the expression of 350 surface proteins pre- and post-expansion using Biolegend’s LEGENDScreen™. As expected, we observed that many proteins were significantly up- and down-regulated during expansion (Figure S1G). However, we did not see loss of CD16A expression, which indicated that our expanded cells would retain ADCC function. We confirmed this by measuring ADCC-induced degranulation and cytokine production with an anti-RBC ADCC assay, in which red blood cells (RBCs) coated with anti-human RBC polyclonal antibodies were used as target cells. On day 6 post-CRISPR, NK cells yielded high levels of degranulation (CD107a+) and IFNγ production (Figures S1H–I). Overall, these results show that our expansion and CRISPR/Cas9 protocol generates gene-ablated expanded primary NK cells that can be utilized in functional assays (Figure 1A).
Figure 1. FcRγ and PLZF ablation does not affect ADCC function.
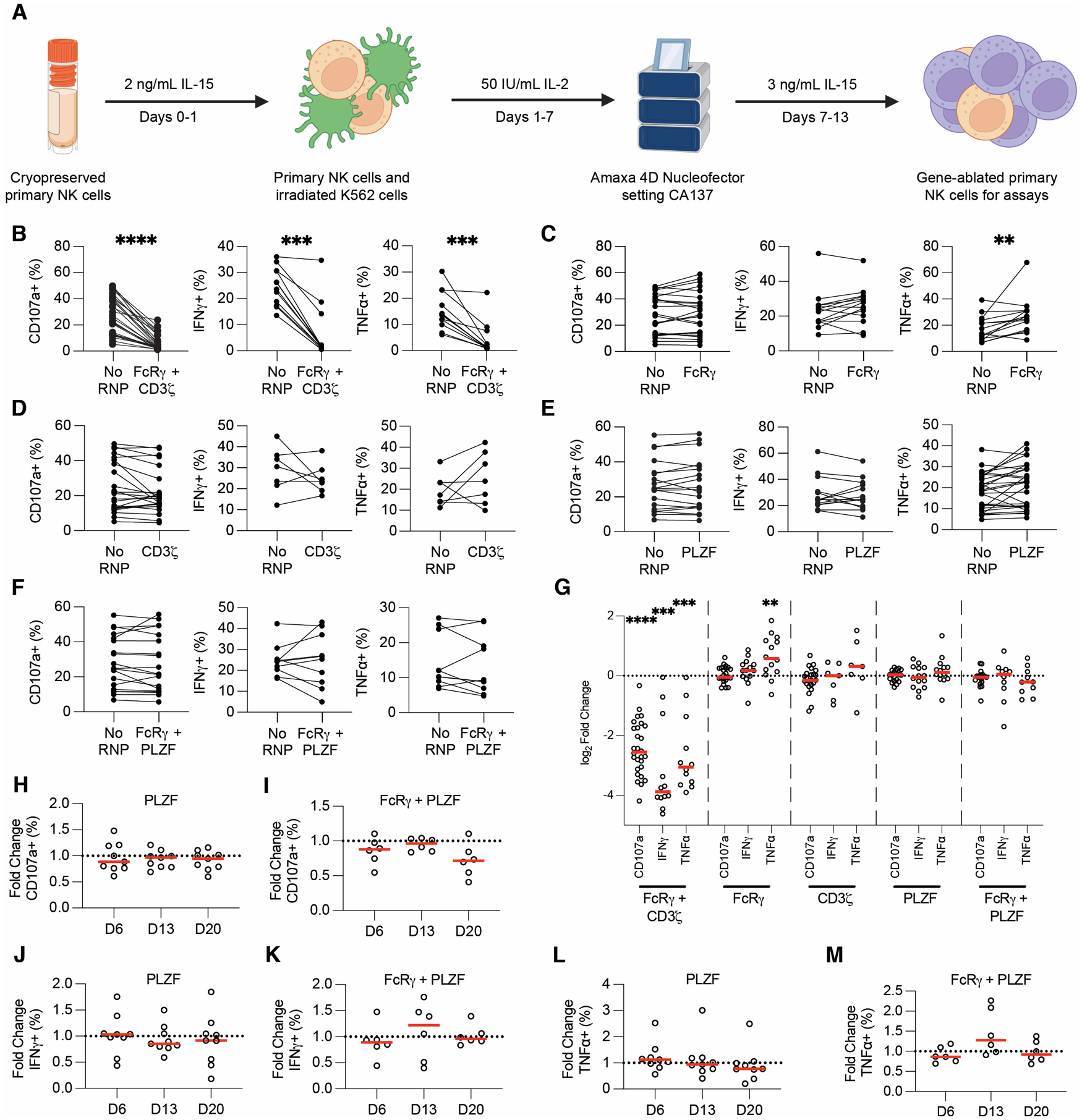
Schematic depiction of CRISPR/Cas9 protocol developed and used for this study (A). Anti-RBC degranulation assays were run six days post-CRISPR/Cas9 with FcRγ/CD3ζ - (B), FcRγ- (C), CD3ζ - (D), PLZF- (E), and FcRγ-/PLZF- (F) ablated expanded primary NK cells. CD107a, IFNγ, and TNFα production from gene-ablated and control expanded primary NK cells was measured by flow cytometry. Each dot is one subject donor. Lines connect samples from the same donor that were nucleofected without an RNP complex (control) or with an RNP complex. Values for No RNP groups are frequencies from all NK cells, whereas values for the gene-ablated sample are only from cells negative for the protein ablated. Data from 13 (CD107a) or six (IFNγ and TNFα) independent experiments. CD107a, IFNγ, and TNFα values for each subject shown in B-F were divided by the respective no RNP sample’s value and log2-transformed (G). CD107a (H-I), IFNγ (J-K), and TNFα (L-M) expression was measured in PLZF- (H, J, L) and FcRγ/PLZF- (I, K, M) ablated cells at days 6, 13, and 20 post-CRISPR/Cas9. The frequency of cells positive for CD107a, IFNγ, and TNFα in the gene-ablated group was divided by the frequency of cells positive for the functional marker in the subject-matched No RNP group. Dots represent subjects and red lines represents the median. Data from three independent experiments. Wilcoxon matched-pairs signed rank test. Significance shown in G carried over from tests run for B-F. **: p < 0.01; ***: p < 0.005; ****: p < 0.001; lack of stars indicates non-significance.
Ablating FcRγ increases TNFα, but PLZF ablation does not affect cytotoxicity or cytokine production in cellular ADCC assays.
We used our CRISPR/Cas9 gene-editing protocol to test whether the loss of proteins associated with adaptive NK cells affected ADCC and cytokine production. A representative gating scheme used for analyzing ADCC assays with gene-ablated NK cells is shown in Figure S2A. As a control, we first ablated both FcRγ and CD3ζ (CD247) from expanded primary NK cells and then tested the gene-ablated cells in anti-RBC ADCC assays. NK cells require expression of at least one of these adapter proteins for downstream CD16A signaling, so we predicted that ablation of both genes would preclude degranulation and cytokine production. As expected, we found that simultaneous ablation of FcRγ and CD3ζ decreased CD107a, IFNγ, and TNFα expression in every subject, with an average decrease of 80–90% for each marker (Figures 1B, G). We were also interested in ablating genes from the NK-92 cell line to validate our results (32, 38). NK-92 cells lack the FcRγ chain (38), meaning ablation of CD3ζ should preclude any functional activity. Thus, we ablated CD3ζ from two NK-92 cell lines: one expressing CD16A (NK-92-GFP-CD16A) and the other expressing a recombinant high-affinity Fc receptor consisting of the CD16A cytoplasmic tail and CD64 extracellular domain (NK-92-GFP-CD64/16A). We then performed DELFIA killing assays with CD3ζ-ablated NK cells and SKOV-3 cells coated with trastuzumab (anti-HER2) as targets. Ablation of CD3ζ abrogated target cell killing in both NK-92-GFP-CD16A (Figures S3A, E–H) and NK-92-GFP-CD64/16A (Figures S3C, I–L) cell lines. These controls show that our CRISPR/Cas9 sgRNAs were on-target and that differences in biological function could be revealed in both expanded primary NK and NK-92 cells.
We next used CRISPR/Cas9 to test if adaptive NK cells have enhanced function because they lack the FcRγ chain. FcRγ only has one ITAM whereas CD3ζ contains three ITAMs. Thus, we hypothesized that if FcRγ is absent from NK cells, they will signal exclusively through CD3ζ, resulting in a stronger ADCC signal due to the increased ITAM number on CD3ζ. We also hypothesized that if the lack of the FcRγ chain enhances ADCC and cytokine production, lack of the CD3ζ chain may have the opposite effect. We ablated FcRγ and CD3ζ independently, then used the gene-ablated cells in anti-RBC ADCC assays. Surprisingly, we found that ablation of either the FcRγ or CD3ζ chain does not significantly affect degranulation or IFNγ production (Figures 1C–D, 1G). TNFα was expressed in a significantly higher frequency of FcRγ-negative cells (Figure 1C) but this was not true for CD3ζ-negative cells (Figure 1D). Overall, though ablating the FcRγ chain increases TNFα production during ADCC, neither ablation of the FcRγ nor the CD3ζ chain consistently alters ADCC function.
Adaptive NK cells also lack PLZF (ZBTB16) expression (22). PLZF controls expression of genes associated with adaptive NK cell activity, such as FcRγ and SYK (SYK), leading to some speculation that PLZF may be the regulator of adaptive NK cell development (22). Without PLZF, the FcRγ and other genes may not be transcribed, leading to stronger ADCC and cytokine production. We hypothesized that ablation of PLZF may trigger the adaptive NK cell transcriptional program, resulting in enhanced ADCC and cytokine production in PLZF-ablated expanded primary NK cells. To test this hypothesis, we ablated PLZF individually or PLZF together with FcRγ (FcRγ/PLZF). We found that there was no significant difference in cytotoxicity or cytokine production with PLZF or FcRγ/PLZF-ablated cells six days post-CRISPR/Cas9 in the anti-RBC ADCC assay (Figures 1E–G). However, we were concerned that testing PLZF-ablated cells six days post-CRISPR/Cas9 may be too soon to observe an effect. Since PLZF is a transcription factor, we speculated that changes in transcription could take longer than six days post-CRISPR/Cas9 to manifest. We therefore tested PLZF and FcRγ/PLZF-ablated cells for ADCC function 6, 13, and 20 days post-CRISPR/Cas9. Neither PLZF nor FcRγ/PLZF-ablated NK cells showed enhanced degranulation (Figures 1H–I), IFNγ (Figures 1J–K), or TNFα production (Figures 1L–M) at day 13 or 20 post-CRISPR. Overall, these results showed that ablation of PLZF did not significantly affect NK cell ADCC and cytokine production in this anti-RBC ADCC assay.
Ablations of SYK and ZAP-70 have opposite effects on degranulation and cytokine production
In addition to lacking FcRγ and PLZF, adaptive NK cells frequently lack the tyrosine kinase SYK (21, 22). Both SYK and the related ZAP-70 transduce signals from FcRγ and CD3ζ for ADCC. However, loss of SYK has been correlated with enhanced ADCC function in primary NK cells (21). We next tested if lack of SYK or ZAP-70 would affect ADCC and cytokine production. We ablated SYK or ZAP-70 (ZAP70) from expanded primary NK cells and then examined the function of the gene-ablated cells in anti-RBC ADCC assays. We found that SYK ablation significantly enhanced degranulation, with an average increase of 7% over no RNP controls (Figures 2A, D). Of the 35 samples tested, 26 showed enhanced degranulation with SYK ablation (Figures 2A, D). In contrast, ZAP-70 ablation significantly diminished NK cell degranulation, with an average decrease of 11.9% versus no RNP controls and decreases in 15 of 22 subjects tested (Figures 2A, D). For both SYK and ZAP-70-ablation, cytokine production followed the same pattern as degranulation. SYK-ablated cells produced significantly more IFNγ (Figures 2B, D) and TNFα (Figures 2C, D) than controls, whereas ZAP-70 ablated cells expressed significantly less IFNγ (Figures 2B, D) and TNFα (Figures 2C, D).
Figure 2. SYK ablation enhances ADCC function whereas ZAP-70 ablation hinders it.
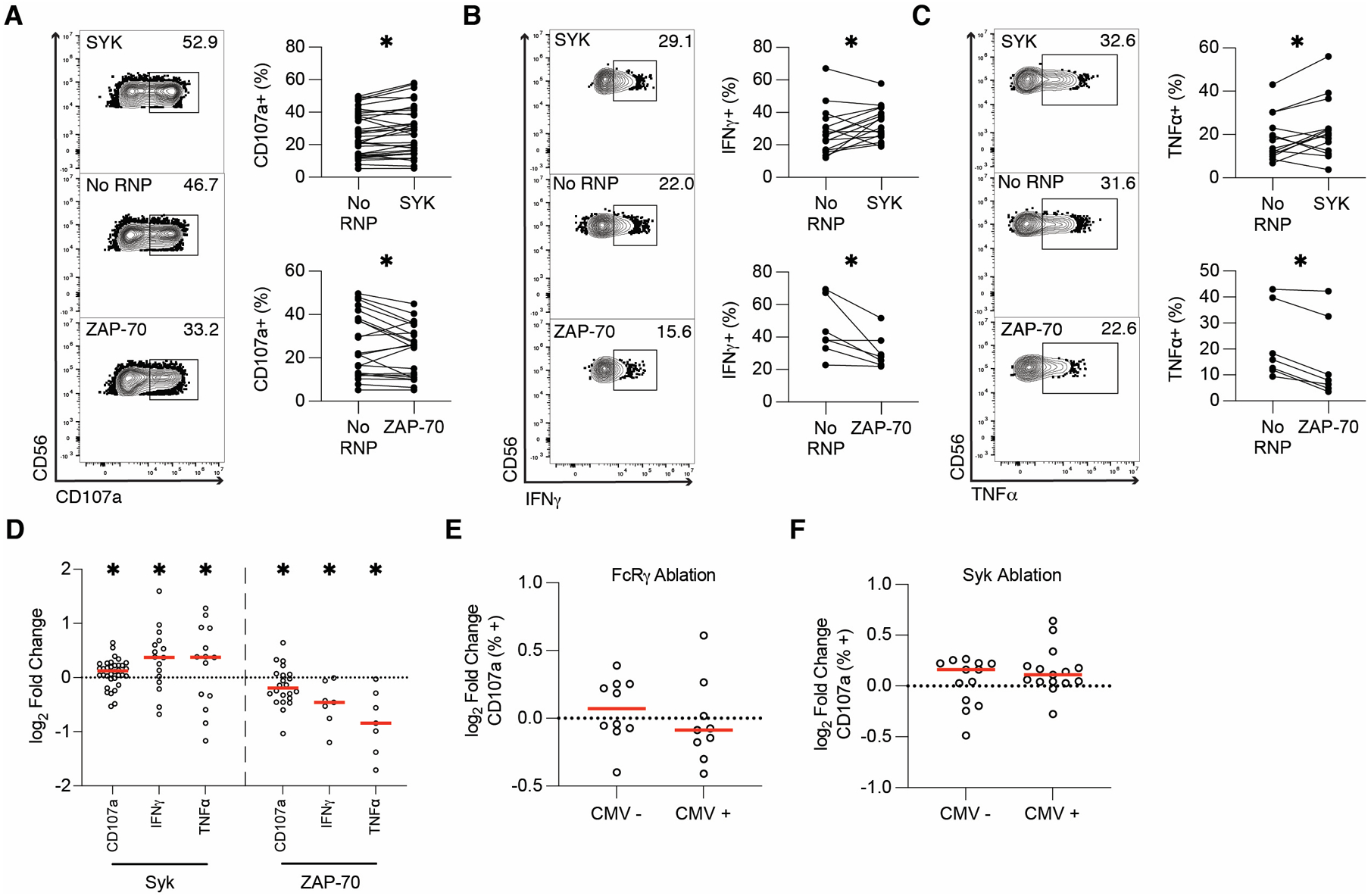
Six days post-CRISPR/Cas9, anti-RBC ADCC assays were run with SYK and ZAP-70-ablated expanded primary NK cells. CD107a (A), IFNγ (B), and TNFα (C) production from gene-ablated and control expanded primary NK cells was measured by flow cytometry. Flow plots show representative samples for SYK-ablated (top), No RNP (middle), and ZAP-70-ablated (bottom) expanded primary NK cells. Each dot is one subject/donor. Lines connect samples from one subject that were nucleofected without an RNP complex (control) or with an RNP complex. Values for no RNP groups are frequencies from all NK cells, whereas values for the gene-ablated sample are only from cells negative for the protein ablated. Data from 13 (CD107a) or 7 (IFNγ and TNFα) independent experiments. CD107a, IFNγ, and TNFα values for each subject shown in A-C were divided by the respective no RNP sample’s value and log-transformed (D). Data from SYK-ablated samples in Figure 2D and FcRγ-ablated samples in Figure 1G was stratified by CMV status (E-F). Wilcoxon matched-pairs signed rank test (A-C). Significance shown in D carried over from tests run for A-C. Unpaired t-test (E-F). *: p < 0.05; lack of stars indicates non-significance.
Though CRISPR/Cas9 gene-editing with expanded primary human NK cells is a valuable model for understanding NK cell function in vivo, high levels of inter-subject variability can cloud biological differences. We speculated that CMV status may account for some of this variability between donors, so we analyzed ADCC data by stratifying the tested donors based on CMV status. No significant differences were observed in ADCC between CMV-positive and CMV-negative subjects after FcRγ or SYK ablation (Figures 2E, F), indicating that CMV status did not affect the outcomes of gene ablation. Finally, to confirm the effects of SYK ablation on NK cell function, we ablated SYK in NK-92-GFP-CD16A and NK-92-GFP-CD64/16A cells and tested subsequent cytotoxicity using DELFIA killing assays with SKOV-3 as NK target cells. The results mirrored our expanded primary NK cell ADCC results, with SYK ablation enhancing killing activity in both cell lines (Figures S3B, D–L). Overall, these results indicate that SYK ablation enhances ADCC and cytokine production, whereas ZAP-70 ablation has the opposite effect.
SYK ablation maintains surface expression of CD16A in stimulated expanded primary NK cells
We followed these experiments by exploring the mechanism behind our results. First, we tested whether SYK or ZAP-70 ablation was altering baseline CD16A surface expression. Since CD16A is the receptor that binds the IgG Fc region, up or downregulation of CD16A could result in the enhanced or diminished function we observed in gene-ablated cells. We measured surface CD16A expression in FcRγ/CD3ζ-, SYK-, and ZAP-70-ablated expanded primary NK cells prior to ADCC. FcRγ/CD3ζ ablation diminished CD16A expression (Figure S3P). This was expected, as NK cells require either FcRγ or CD3ζ to express CD16A on the cell surface (41–43). However, the majority of SYK- and ZAP-70-ablated cells expressed CD16A at similar baseline frequencies (Figure S3Q–R).
Though SYK-ablated samples showed no difference from control cells in CD16A surface expression before stimulation, we were curious whether differences would be detectable after ADCC. Stimulation of NK cells can downregulate surface CD16A expression via A Disintegrin and Metalloprotease 17 (ADAM17) (39, 40, 44). To test this, we stained SYK-ablated samples for CD16A surface expression after ADCC assays were performed. Stimulated SYK-ablated samples showed a higher frequency of CD16A surface expression than No RNP controls (Figure S3S). This could indicate that SYK-ablated samples maintain CD16A surface expression better than canonical NK cells during stimulation, leading to enhanced ADCC function.
NK cell-target cell conjugation is enhanced by SYK ablation but diminished by ZAP-70 ablation
As another potential mechanism, we also tested whether SYK- or ZAP-70- ablated expanded primary NK cells differed in their ability to form stable cell-cell conjugates. We postulated that SYK-ablated cells may conjugate more effectively to target cells, leading to enhanced cytotoxicity and target cell killing (32, 45). Six days post-CRISPR/Cas9, SYK- and ZAP-70 ablated cells were labeled with CellTrace Violet (CTV), then incubated for various periods with fluorescent antibody-tagged SKOV-3 (CellTrace FarRed, CTFR, labeled) or Raji (Nuclight Red, NLR, expressing) cells. After incubation, samples were fixed and analyzed by flow cytometry as shown in Figure S3T (SKOV-3) and S3U (Raji). NK cell-target cell conjugates were detected as single events that were positive for both CTV and NLR/CTFR. At 30 minutes post-combination, SYK-ablated expanded primary NK cells showed enhanced conjugation with both anti-HER2-labeled SKOV-3 (Figure 3A) and Rituximab-labeled Raji (Figure 3B) cells as targets. In contrast, fewer ZAP-70-ablated expanded primary NK cells conjugated to target cells relative to the No RNP controls (Figures 3C–D). In NK-cell mediated ADCC, adhesion molecules, such as CD2 and LFA-1, mediate target cell conjugation to form the immunological synapse (46, 47). During ADCC, CD2 has also been shown synergize with CD16A, leading to enhanced cytotoxicity and cytokine production (46, 48). We hypothesized that since SYK-ablation enhanced cytotoxicity, cytokine production, and target cell conjugation during ADCC, SYK-ablated cells would show increased CD2 or LFA-1 expression at baseline. To test this, unstimulated SYK- and ZAP-70-ablated expanded primary NK cells were stained for CD2 or LFA-1 expression six days post-CRISPR/Cas9. SYK-ablated cells expressed CD2 at a significantly higher frequency than control samples (Figure 3E). Additionally, CD2 geometric median fluorescence intensity (gMFI) was trending higher in SYK-ablated samples, though this difference was insignificant (Figure 3F). ZAP-70-ablated samples showed the opposite phenotype of SYK-ablated cells with significantly fewer ZAP-70-ablated samples expressing CD2 (Figure 3G) and at a significantly lower gMFI (Figure 3H) than No RNP controls. In contrast, SYK- and ZAP-70-ablated samples showed no difference from control cells in expression of both the closed and open conformations of LFA-1 (data not shown). These results indicate that SYK ablation enhances target cell conjugation and that this could be due to increased CD2 expression.
Figure 3. SYK ablation improves target cell conjugation and ZAP-70 ablation diminishes it.
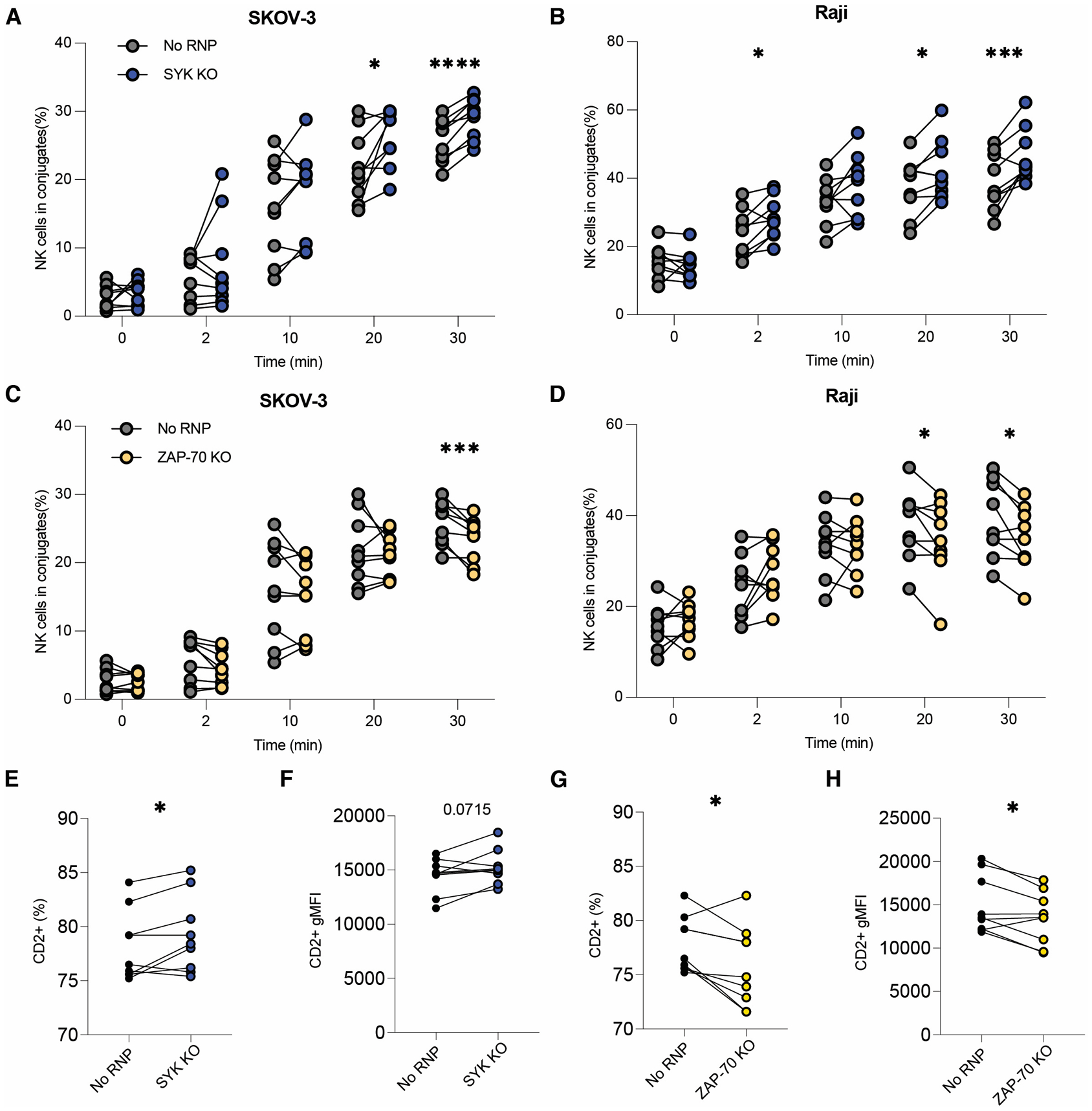
SYK- and ZAP-70-ablated expanded primary NK cells’ target cell conjugation capacity was tested six days post-CRISPR/Cas9 by flow cytometry. Fluorescently labeled and opsonized SKOV-3 (A, C) or Raji (B, D) cells were incubated with control or gene-edited CTV-labeled NK cells for the listed period, vortexed, then fixed and analyzed by flow cytometry. Shown is the percentage of NK cells that were also positive for the target cell fluorophore, indicating target cell conjugation (A-D). Each dot represents one subject. Lines connect samples nucleofected without an RNP complex (control) or with an RNP complex. Paired t test. *: p < 0.05; ***: p < 0.005; ****: p < 0.001; lack of stars indicates non-significance. Frequency (E-F) and geometric MFI (G-H) of CD2 expression on SYK- and ZAP-70-ablated samples. Each dot represents one subject. Lines connect samples nucleofected without an RNP complex (control) or with an RNP complex. Paired t test. *: p < 0.05; other p value listed. All data from three independent experiments.
SHP-1 ablation increases degranulation but decreases cytokine production
We next explored other molecules in the CD16A signaling pathway, including SHP-1. SHP-1 is a phosphatase that can dephosphorylate ZAP-70 (49, 50) and induce ITAM-mediated inhibition (ITAMi) (51–53). ITAMi is a process in which weakly ligated receptors recruit SHP-1 instead of SYK or ZAP-70 to partially phosphorylated ITAMs, allowing SHP-1 to dephosphorylate the ITAM and terminate signaling (54, 55). Interestingly, ITAMi can also be facilitated by SYK dissociating from the partially phosphorylated ITAM of a weakly occupied receptor, allowing SHP-1 to dephosphorylate the ITAM (56, 57). We thus hypothesized that SHP-1 ablation would enhance NK cell-mediated ADCC and cytokine production. To test this, we ablated SHP-1 (PTPN6) from expanded primary NK cells and tested the SHP-1-ablated cells’ ADCC function and cytokine production. In line with our hypothesis, SHP-1 ablation robustly enhanced degranulation in the anti-RBC ADCC assay. On average, 43.55% of SHP-1-ablated samples were CD107a+, compared to 34.43% of subject-matched, no RNP samples in the assay (Figures 4A, 4D). However, both IFNγ (Figures 4B, 4D) and TNFα (Figures 4C–D) were expressed at significantly lower frequencies in SHP-1 ablated samples relative to no RNP controls. Since cytokine production reflected degranulation in both SYK and ZAP-70-ablated cells, we were surprised that SHP-1 ablation resulted in enhanced ADCC but reduced cytokine production. Overall, while our CD107a data suggest that SHP-1 ablation increases cytotoxicity, the effect of SHP-1 ablation on cytokine production is both confounding and interesting.
Figure 4. SHP-1 ablation enhances degranulation but diminishes cytokine production.
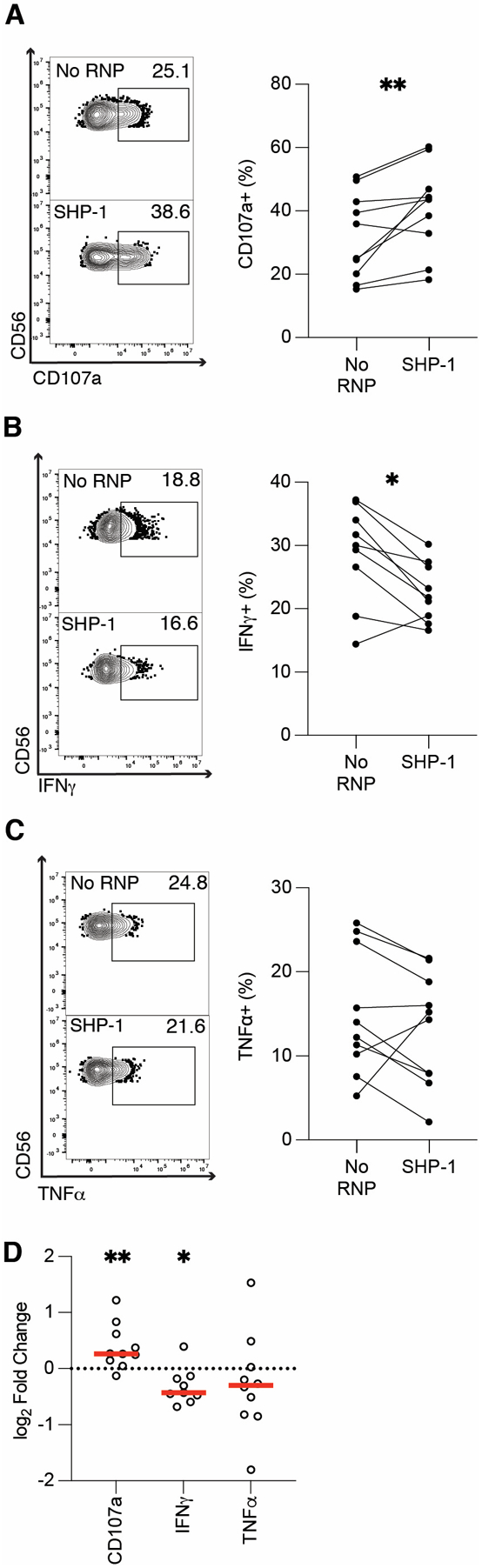
Six days post-CRISPR/Cas9, anti-RBC ADCC assays were run with SHP-1 ablated expanded primary NK cells. CD107a (A), IFNγ (B), and TNFα (C) production from gene-ablated and control expanded primary NK cells was measured by flow cytometry. Flow plots show representative samples for no RNP (top), and SHP-1-ablated (bottom) expanded primary NK cells. Each dot is one subject/donor. Lines connect subjects that were nucleofected without an RNP complex (control) or with an RNP complex. Values for no RNP groups are frequencies from all NK cells, whereas values for the gene-ablated sample are only from cells negative for the protein ablated. CD107a, IFNγ, and TNFα values for each subject shown in A-C were divided by the respective no RNP sample’s value and log-transformed (D). Data from five independent experiments. Wilcoxon matched-pairs signed rank test. *: p < 0.05; **: p < 0.01; lack of stars indicates non-significance.
SYK and SHP-1 ablation enhance expanded primary NK cell killing
Next, we tested whether our ADCC degranulation results in the anti-RBC assay would correlate with ADCC towards a SKOV-3 target cell line. We hypothesized that gene-ablated cell killing would reflect CD107a expression in the anti-RBC ADCC assays. We used two complementary assays to measure target cell killing— a molecule release ‘DELFIA’ assay and cell tracking with an ‘Incucyte’ machine. In DELFIA assays, SKOV-3 target cells are loaded with a chemical called BADTA, then incubated with effector cells (e.g., NK cells), for 2–6 hours. After incubation, BADTA levels in the supernatant are measured, which reflects pores generated in the target cell or necrotic death releasing the BADTA into the supernatant. For DELFIA assays, we ablated FcRγ/CD3ζ, SYK, ZAP-70, and SHP-1 from expanded primary NK cells. Six days post-CRISPR/Cas9, the gene-ablated cells were incubated with anti-HER2 and BADTA-loaded SKOV-3 cells. In parallel with the anti-RBC results, ablating FcRγ/CD3ζ significantly diminished ADCC function in DELFIA assays (Figures 5A–D). We found that SHP-1 ablation resulted in significantly more specific lysis of target cells with 10 μg/mL (Figures 5A–B) and 3 μg/mL anti-HER2 (Figures 5C–D). SYK-ablated cells killed significantly more target cells with 3 μg/mL anti-HER2 only (Figures 5A–D). In the 10 μg/mL DELFIA assays, SYK ablation increased target cell killing in 8/10 subjects with a median specific lysis of 86.6%, an 8.1% increase over the No RNP group. However, these increases were insignificant (Figures 5A–B). We also observed significantly less killing in the ZAP-70-ablated samples (Figures 5A–D). Although the 3 μg/mL and 10 μg/mL DELFIA assays showed slightly different results, the trends are similar for each ablated gene.
Figure 5. SYK and SHP-1 ablation increases NK cell killing.
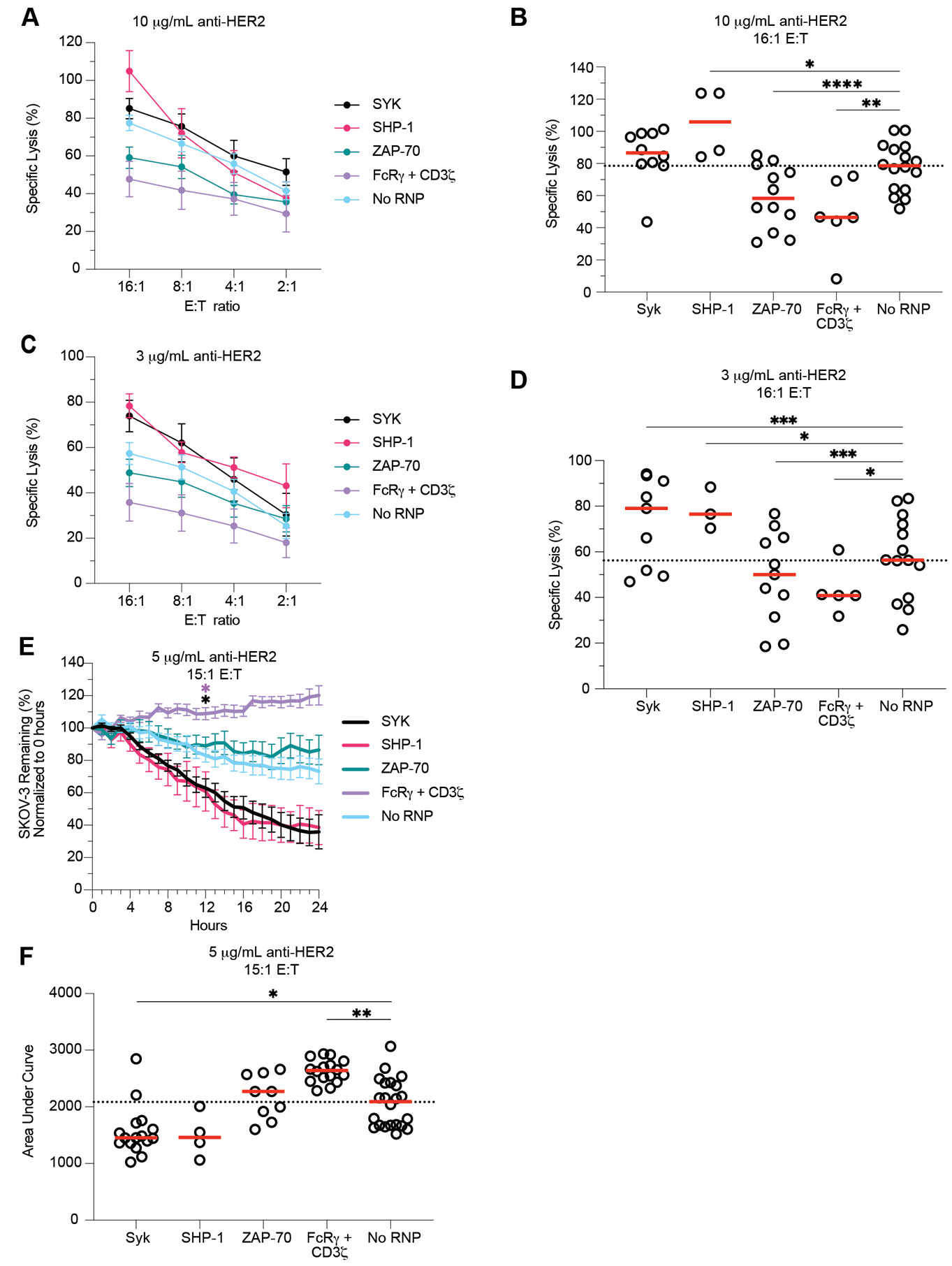
SYK-, SHP-1-, ZAP-70-, and FcRγ/CD3ζ-ablated expanded primary NK cells were used in DELFIA and Incucyte killing assays six days post-CRISPR/Cas9. DELFIA assays were run by combining NK cells with opsonized SKOV-3 cells. SKOV-3 cells were opsonized with 10 μg/mL (A) and 3 μg/mL (C) anti-HER2, then incubated with NK cells at various E:T ratios. Specific lysis values from both 10 μg/mL (B) and 3 μg/mL (D) samples tested at the 16:1 E:T ratios were plotted. For Incucyte killing assays, NLR-SKOV-3 cells were opsonized with 5 μg/mL anti-HER2 and incubated with expanded primary NK cells at an E:T ratio of about 15:1. Wells were imaged every hour to quantify the percentage of SKOV-3 cells remaining from the original count (E). For each subject, the area under the SKOV-3 % remaining curve was calculated (F). Lines indicate the mean, whereas error bars represent SE (A, C, E). Dots represent individual subjects, whereas red lines represent the median (B, D, F). Data from 7 independent experiments. Dunnet’s multiple comparison’s test for mixed effects model (B, D-F). For E, the cumulative area under the curve was calculated for each subject and group at each timepoint, then compared to the No RNP group. Position of the star indicates the first timepoint that differences became statistically significant, and the color of star indicates the group that was significantly different (E). Numerical values indicate p values for samples with 0.1 > p > 0.05. *: p < 0.05; **: p < 0.01; ***: p < 0.005; ****: p < 0.001. Lack of stars indicates non-significance.
To measure target cell killing over time, we used a microscopy-based live cell tracking Incucyte machine. Fluorescent SKOV-3 cells were opsonized with anti-HER2, incubated with gene-ablated and no RNP NK cells, then imaged every hour for 24 hours. We hypothesized that SKOV-3 death would correlate with degranulation and DELFIA readouts. Once again, we used FcRγ/CD3ζ−, SYK-, ZAP-70-, and SHP-1-ablated cells in Incucyte assays six days post-CRISPR/Cas9. FcRγ/CD3ζ-ablated samples killed significantly fewer target cells (Figures 5E–F). In line with our other results, we saw that SYK-ablation significantly enhanced killing function (Figure 5E–F). While SHP-1-ablated samples followed the same trends as in the DELFIA assays, these differences were not significant (Figures 5E–F). ZAP-70-ablated samples showed no difference from control cells in the Incucyte assay (Figures 5E–F). Overall, degranulation, DELFIA specific release, and Incucyte AUC results correlated with each other (Figures S3M–O). Of note, the magnitude of difference between SYK- and SHP-1 ablated cell killing of SKOV-3 cells versus no RNP cells was greater than the difference between the anti-RBC degranulation assay results (Figures 2D, 4D, 5B, D, F). Together, these data support our hypothesis that SYK ablation enhances cytotoxic function in CMV-induced adaptive NK cells.
SYK-ablated cells retain enhanced function post-expansion
Ex vivo expansion of NK cells and subsequent transplantation is an exciting avenue for cancer treatment (58). However, the success of these therapies often depends on the ability of NK cells to survive and retain function post-expansion and post-transplant (59). Though our main goal was to understand the mechanism by which adaptive NK cells gain enhanced ADCC function, we also wanted to test if SYK-ablated NK cells could survive and retain enhanced function post-expansion. Therefore, we ablated SYK from expanded primary human NK cells, then used them in anti-RBC degranulation and cytokine production assays at days 6, 13, 20, and 27 post-CRISPR. Across nearly all time points, SYK ablation resulted in enhanced CD107a (Figure 6A), IFNγ (Figure 6B), and TNFα (Figure 6C) expression. SYK-ablated samples also retained enhanced killing in DELFIA (Figures 6D–G) and Incucyte (Figures 6H–I) assays with SKOV-3 cells as NK targets. Overall, these results show that SYK ablation’s effect on ADCC function is sustained after expanding the cells for four weeks post-CRISPR, indicating that SYK-negative cells could be considered for NK cell-based ADCC therapies.
Figure 6. SYK-ablated expanded primary NK cells retain enhanced function.
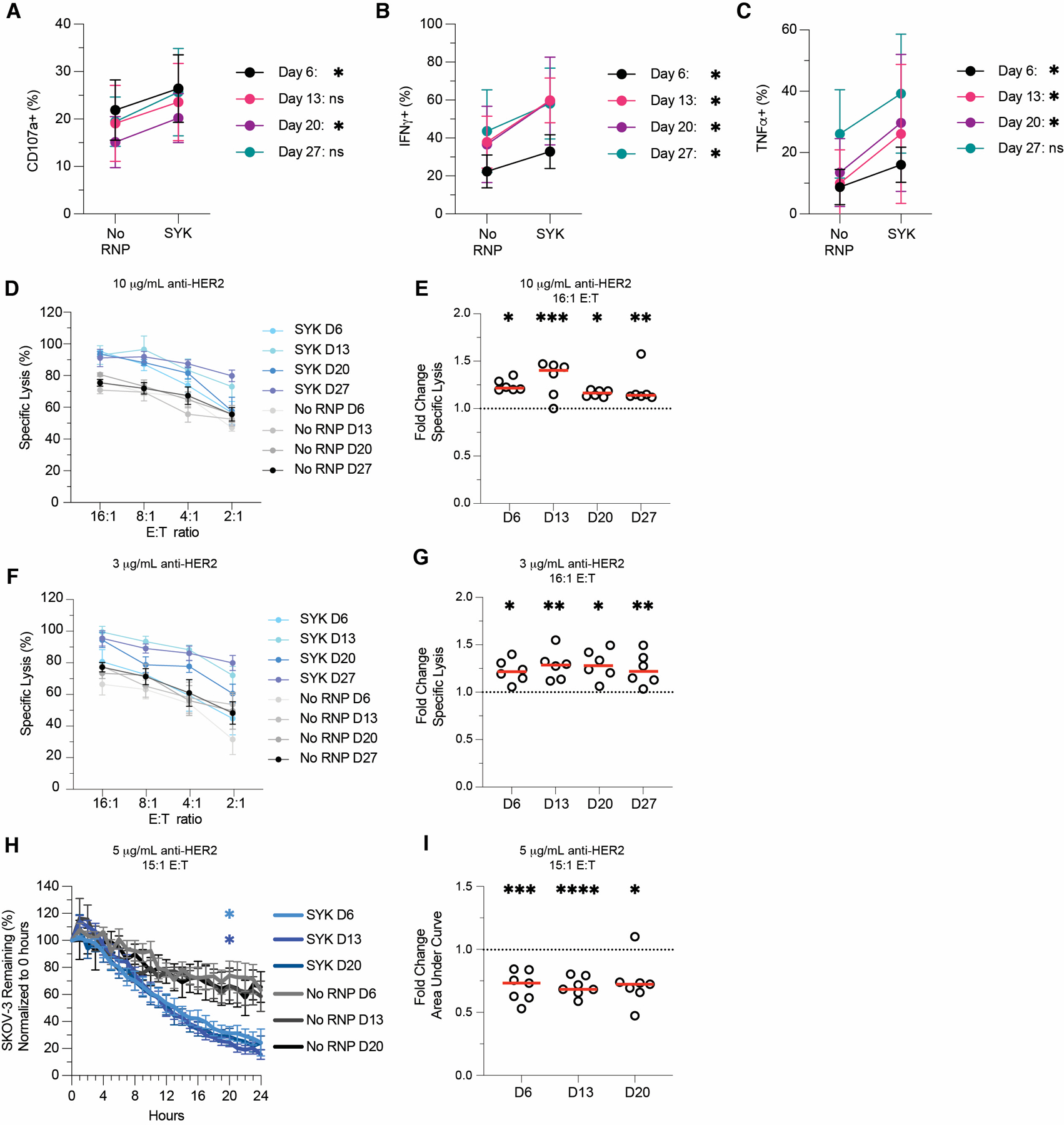
SYK-ablated NK cells were tested in ADCC (A-C), DELFIA (D-G), and Incucyte (H-I) assays at days 6, 13, 20, and 27 post-CRISPR/Cas9. For visual comparison, Day 6 data is from Figures 3 and 5. For anti-RBC ADCC assays, CD107a (A), IFNγ (B), and TNFα (C) production in gene-ablated and control expanded primary NK cells was measured by flow cytometry. Plots indicate mean and SD. DELFIA assays were run by incubating NK cells with opsonized SKOV-3 cells. ADCC-specific lysis of SKOV-3 cells opsonized with 10 μg/mL anti-HER2 (D) or 3 μg/mL anti-HER2 (F) at various E:T ratios. SYK-ablated fold change in specific lysis over No RNP group for 16:1 E:T at 10 μg/mL anti-HER2 (E) and 3 μg/mL anti-HER2 (G). Percentage of SKOV-3 cells from t = 0 remaining in Incucyte killing assay (H) and SYK-ablated fold change in SKOV-3 AUC relative to the No RNP group (I). Lines indicate the mean and error bars represent SE (D, F, H). Dots indicate individual subjects and red lines indicate the median (E, G, I). Data from three independent experiments. Wilcoxon matched-pairs signed rank test (A-C). Paired t-tests of raw values (E, G, I). For H, the cumulative area under the curve was calculated for each subject and group at each timepoint, then compared to the No RNP group with Dunnet’s multiple comparison’s test for mixed effects models. Position of the star indicates the first timepoint that differences became statistically significant, and the color of star indicates the group that was significantly different (H). *: p < 0.05; **: p < 0.01; ***: p < 0.005; ****: p < 0.001; lack of stars indicates non-significance.
Discussion
Adaptive NK cells show enhanced function relative to canonical NK cells (12, 19, 21, 22), and higher frequencies of adaptive NK cells correlate with protection against diseases such as CMV, HIV, and malaria (18, 24, 26). However, it was unclear which intracellular molecules are required to mediate enhanced adaptive NK cell function and whether the same molecules control both cytotoxicity and cytokine production. Here, we show that ablating SYK kinase in expanded primary NK cells enhances ADCC, cytokine production, and target cell conjugation. SYK-ablated expanded primary NK cells retained their enhanced function up to 27 days post-CRISPR. Overall, these results indicate that a mechanism involving SYK kinase, including but not limited to increased conjugation through CD2 and perhaps involving decreased ITAM inhibition through SHP-1, could explain why CMV-induced adaptive NK cells show enhanced ADCC and cytokine production relative to conventional NK cells (17–22). These results also suggest that SYK-ablated NK cells may have increased ADCC function in vivo.
Previous work indicated that adaptive NK cells often lack expression of the transcription factor PLZF and FcRγ. In primary NK cells, loss of FcRγ has been associated with enhanced function, leading to hypotheses that the lack of FcRγ results in a stronger ADCC signaling (20, 22). Additionally, PLZF binds the FcRγ promoter region (22). Thus, it seemed possible that loss of PLZF may drive loss of FcRγ, which could lead to enhanced ADCC function. We found that FcRγ ablation led to a modest increase in TNFα production but no significant increase in degranulation or IFNγ expression. In PLZF-ablated samples, neither ADCC nor cytokine production was enhanced for up to 20 days post-CRISPR. Despite these results, PLZF may still be an important transcription factor for adaptive NK cell biology as other transcription factors could have compensated for its loss in our study. In other work, Liu et al. ablated FcRγ and PLZF individually in primary human NK cells and cross-linked CD16A to test ADCC function (35). Like Liu et al., we observed that ablating PLZF and FcRγ/PLZF did not enhance degranulation or cytokine production during ADCC. However, Liu et al. also showed that FcRγ-ablation directly enhanced both IFNγ and TNFα production, but not degranulation, during ADCC. By cross-linking CD16A, Liu et al. directly stimulated the CD16A pathway. In contrast to Liu et al., we used two cellular targets to assess ADCC and cytokine production: red blood cells and SKOV-3 cells. Interactions between surface proteins on target and NK cells can modulate NK cell ADCC activity (60, 61), which may explain the differences between our data and prior work. Given our findings, if gene-ablated NK cells are to be used in clinical settings, this emphasizes the need to test the target cell of interest in pre-clinical assays. Overall, our results show the potential for enhanced cytokine production in FcRγ-negative NK cells but also indicated that lack of the FcRγ chain does not universally enhance ADCC function.
In addition to lacking FcRγ, adaptive NK cells can lack SYK kinase (21, 22, 62). Loss of SYK was not observed in adaptive NK cells from malaria subjects in Mali (26), but has been observed regularly during CMV infection. Like FcRγ-negative cells, SYK-negative primary NK cells exhibit enhanced ADCC function (21, 22). In ADCC, SYK preferentially binds the FcRγ chain whereas ZAP-70 preferentially binds the CD3ζ chain (8). Thus, it seemed possible that lack of the FcRγ chain in adaptive NK cells results in less SYK-mediated ADCC signaling, indicating a potential role for SYK in controlling ADCC signal strength. SYK ablation enhanced ADCC and cytokine production, whereas ZAP-70 ablation decreased both, indicating that SYK and/or ZAP-70 mediates ADCC signal strength.
By evaluating potential mechanisms behind this phenomenon, we observed that SYK ablation also increases stable NK cell conjugate formation. In addition, SYK-ablated NK cells expressed the adhesion molecule CD2 at a higher frequency than controls. Previous reports have shown that CD2 expression predicts NK cell-mediated ADCC levels (46, 48). Thus, our findings could indicate that increased CD2 expression in SYK-ablated cells enhances target cell binding, contributing to increased cytotoxicity, cytokine production, and killing.
Beyond conjugation, we examined SHP-1 regulation of ADCC signal strength as a potential contributor to the functional behavior we observed when altering CD16A signaling partners. SHP-1 is a phosphatase that can modulate signaling in multiple pathways. Relevant to this study, SHP-1 can dephosphorylate ZAP-70 in T cells (49, 50). In addition, SHP-1 has been shown to mediate ITAM inhibition (ITAMi), in which SHP-1 is recruited to weakly-ligated Fc receptor ITAMs and dephosphorylates them, dampening signaling (51, 53, 57). We show that SHP-1 ablation enhances degranulation and killing, in line with the phenotype observed for SYK-ablated NK cells. Since both these components can inhibit ADCC signaling, it is probable that SHP-1 is partially responsible for the SYK-ablation functional phenotype, which warrants further investigation. Interestingly, IFNγ production from SHP-1 ablated NK cells was consistently lower than control NK cells—the opposite result of SYK ablation. One explanation could be different CD16A binding thresholds to initiate degranulation versus cytokine production. Fauriat et al. proposed a model for NK cell function in which CD107a signaling requires a low threshold and triggers granule release immediately after stimulation whereas TNFα secretion requires a moderate threshold and longer stimulation (~3 hours), and IFNγ secretion requires the highest threshold and longest stimulation (~5.5 hours) (63). Concurrently, strong activation through CD16A yields a self-regulation loop in which CD16A is enzymatically cleaved by ADAM17 (39, 40, 64, 65). Thus, it is possible that without SHP-1 negatively regulating CD16A signaling, SHP-1-ablated cells are rapidly activated and degranulate. This could lead to ADAM17 cleaving CD16A from the cell surface, resulting in a lack of signal for eventual IFNγ production. Lack of SHP-1 has been associated with overactive immune cells (66). Thus, we hypothesize that SHP-1 ablation eliminates inhibition of both SYK (through ITAMi) and ZAP-70 (by dephosphorylation) activity, resulting in robust CD16A signaling, rapid ADAM17-induced cleavage of CD16A, and loss of the hours-long CD16A-stimulation required for IFNγ production. Adhesion molecules could also contribute to this phenotype since SHP-1 regulates adhesion in T cells (67) and SHP-1 phosphorylation is associated with more sequential contacts in NK cells (68). Additional studies beyond the scope of this study are needed to test these hypotheses. Lastly, one limitation of the CRIPSR/Cas9 method is that the cells need to be proliferating for the CRISPR/Cas9 to effectively ablate genes. No gene-ablation was detected when the CRISPR was done on freshly isolated resting NK cells. This limits our study of testing the effect of gene-ablation to only proliferating NK cells. In the future, to test the function of SYK and ZAP-70 in resting NK cells, one could use drugs specific for SYK or ZAP-70 or use a novel CRISPR method that is able to ablate genes in resting NK cells. We hypothesize the results would be similar to the proliferating cells. However, this would need to be directly tested.
Our results reveal that mechanisms involving a lack of SYK kinase, such as enhanced target cell conjugation or reduced SHP-1-mediated inhibition, could explain the enhanced ADCC function in CMV-induced adaptive NK cells. Yet they do not explain adaptive NK cell behavior in all settings. Adaptive NK cells from malaria patients in Mali lacked FcRγ chain and PLZF expression and showed enhanced ADCC and cytokine production in vitro (26). However, in contrast to CMV-induced adaptive NK cells, over 90% of adaptive NK cells in the Malian cohort were SYK-positive and only 50% expressed NKG2C (26). The differences indicate that malaria-induced adaptive NK cells may contain novel origins and phenotypes and highlight the potential heterogeneity of adaptive NK cells induced by different infections (26). While our study shows how SYK ablation can enhance degranulation and cytokine production with a CMV-induced adaptive NK cell phenotype, there may be multiple mechanisms driving this enhanced function. Understanding other mechanisms that increase cytotoxicity and cytokine production will be important toward understanding NK cell function and improving their use as a therapeutic intervention.
The CRISPR/Cas9 method we developed allowed us to ablate molecules in the CD16A pathway and define the consequences of the ablations on CD16A signaling in expanded primary human NK cells. These findings build on previous work in the NK and Fc receptor signaling fields as well as adaptive NK cell biology. With gene-editing in expanded primary human NK cells, the field can address new questions on how adaptive NK cells function. Future studies will focus on understanding why SYK ablation enhances NK cell function, ZAP-70 ablation decreases function, and SHP-1 ablation does both. In summary, these findings help define what drives enhanced cytotoxicity and cytokine production in CMV-induced adaptive NK cells and show that SYK ablation may be a viable therapy for CAR NK cells that utilize the CD16A signaling.
Supplementary Material
Key Points.
Ablating FcRγ or PLZF from expanded primary human NK cells does not affect ADCC
Ablating SYK increases ADCC in NK cells whereas ablating ZAP-70 decreases ADCC
The enhanced function of SYK-ablated cells is retained up to 27 days post-CRISPR
Acknowledgements
Special thanks to Patrick Willey and the rest of the staff at the University of Minnesota University Imaging Centers (UIC, SCR_020997), the University of Minnesota flow cytometry core, and the University of Minnesota Genomics Center. Special thanks to Peter Hinderlie (Dr. Jeffrey Miller’s laboratory) for technical support in setting up Incucyte assays and analyzing results. Thanks to Dr. Peter Morawski (Benaroya Research Institute) for helping with a confirmatory analysis (Infinity Flow R program) for our Biolegend LegendScreen results. Special thanks to the NCI for supplying IL-2 and IL-15 and to Synthego for their guide RNA design tool for CRISPR/Cas9. Thanks to Dr. Jai Rutella and Dr. Miller’s NK cell Immunology Research Forum (NIRF) group for their expertise and advice. Thanks to Dr. Joshua Baller for computational and statistical assistance with the data and manuscript. Thanks to Sloane Fussell for providing model figure graphics.
Grant support:
This work was funded by the National Institutes of Health (R01 AI146031; R01 AI143828; R21 AI149659), the department of defense (DOD) (CA200922), and by the University of Minnesota Clinical and Translational Science Institute (CTSI) and Medical School.
Footnotes
Declaration of interests
The authors declare no competing interests related to the content of this work.
References
- 1.Abel AM, Yang C, Thakar MS, and Malarkannan S. 2018. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front Immunol 9: 1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lanier LL 2008. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol 9: 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mujal AM, Delconte RB, and Sun JC. 2021. Natural Killer Cells: From Innate to Adaptive Features. Annu Rev Immunol 39: 417–447. [DOI] [PubMed] [Google Scholar]
- 4.Perera Molligoda Arachchige AS 2021. Human NK cells: From development to effector functions. Innate Immun 27: 212–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burrack KS, Hart GT, and Hamilton SE. 2019. Contributions of natural killer cells to the immune response against Plasmodium. Malar J 18: 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lanier LL, Yu G, and Phillips JH. 1991. ANALYSIS OF FcγRIII (CD16) MEMBRANE EXPRESSION AND ASSOCIATION WITH CD3ζ AND FcεRI-γ BY SITE-DIRECTED MUTATION Journal of Immunology 146: 1571–1576. [PubMed] [Google Scholar]
- 7.Love PE, and Hayes SM. 2010. ITAM-mediated signaling by the T-cell antigen receptor. Cold Spring Harb Perspect Biol 2: a002485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shiue L, Green J, Green OM, Karas JL, Morgenstern JP, Ram MK, Taylor MK, Zoller MJ, Zydowsky LD, Bolen JB, and Brugge JS. 1995. Interaction of p72syk with the gamma and beta Subunits of the High-Affinity Receptor for Immunoglobulin E, FcεRI. Molecular and Cellular Biology 15: 272–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watzl C, and Long EO. 2010. Signal transduction during activation and inhibition of natural killer cells. Curr Protoc Immunol Chapter 11: Unit 11 19B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freedman TS, Tan YX, Skrzypczynska KM, Manz BN, Sjaastad FV, Goodridge HS, Lowell CA, and Weiss A. 2015. LynA regulates an inflammation-sensitive signaling checkpoint in macrophages. Elife 4:e09183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang C, Siebert JR, Burns R, Gerbec ZJ, Bonacci B, Rymaszewski A, Rau M, Riese MJ, Rao S, Carlson KS, Routes JM, Verbsky JW, Thakar MS, and Malarkannan S. 2019. Heterogeneity of human bone marrow and blood natural killer cells defined by single-cell transcriptome. Nat Commun 10: 3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun JC, Beilke JN, and Lanier LL. 2009. Adaptive immune features of natural killer cells. Nature 457: 557–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lopez-Verges S, Milush JM, Schwartz BS, Pando MJ, Jarjoura J, York VA, Houchins JP, Miller S, Kang SM, Norris PJ, Nixon DF, and Lanier LL. 2011. Expansion of a unique CD57(+)NKG2Chi natural killer cell subset during acute human cytomegalovirus infection. Proc Natl Acad Sci U S A 108: 14725–14732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guma M, Angulo A, Vilches C, Gomez-Lozano N, Malats N, and Lopez-Botet M. 2004. Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood 104: 3664–3671. [DOI] [PubMed] [Google Scholar]
- 15.Monsivais-Urenda A, Noyola-Cherpitel D, Hernandez-Salinas A, Garcia-Sepulveda C, Romo N, Baranda L, Lopez-Botet M, and Gonzalez-Amaro R. 2010. Influence of human cytomegalovirus infection on the NK cell receptor repertoire in children. Eur J Immunol 40: 1418–1427. [DOI] [PubMed] [Google Scholar]
- 16.Guma M, Budt M, Saez A, Brckalo T, Hengel H, Angulo A, and Lopez-Botet M. 2006. Expansion of CD94/NKG2C+ NK cells in response to human cytomegalovirus-infected fibroblasts. Blood 107: 3624–3631. [DOI] [PubMed] [Google Scholar]
- 17.Foley B, Cooley S, Verneris MR, Pitt M, Curtsinger J, Luo X, Lopez-Verges S, Lanier LL, Weisdorf D, and Miller JS. 2012. Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C+ natural killer cells with potent function. Blood 119: 2665–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cichocki F, Taras E, Chiuppesi F, Wagner JE, Blazar BR, Brunstein C, Luo X, Diamond DJ, Cooley S, Weisdorf DJ, and Miller JS. 2019. Adaptive NK cell reconstitution is associated with better clinical outcomes. JCI Insight 4(2):e125553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hwang I, Zhang T, Scott JM, Kim AR, Lee T, Kakarla T, Kim A, Sunwoo JB, and Kim S. 2012. Identification of human NK cells that are deficient for signaling adaptor FcRgamma and specialized for antibody-dependent immune functions. Int Immunol 24: 793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang T, Scott JM, Hwang I, and Kim S. 2013. Cutting edge: antibody-dependent memory-like NK cells distinguished by FcRgamma deficiency. J Immunol 190: 1402–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee J, Zhang T, Hwang I, Kim A, Nitschke L, Kim M, Scott JM, Kamimura Y, Lanier LL, and Kim S. 2015. Epigenetic modification and antibody-dependent expansion of memory-like NK cells in human cytomegalovirus-infected individuals. Immunity 42: 431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schlums H, Cichocki F, Tesi B, Theorell J, Beziat V, Holmes TD, Han H, Chiang SC, Foley B, Mattsson K, Larsson S, Schaffer M, Malmberg KJ, Ljunggren HG, Miller JS, and Bryceson YT. 2015. Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity 42: 443–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Correia MP, Stojanovic A, Bauer K, Juraeva D, Tykocinski LO, Lorenz HM, Brors B, and Cerwenka A. 2018. Distinct human circulating NKp30(+)FcepsilonRIgamma(+)CD8(+) T cell population exhibiting high natural killer-like antitumor potential. Proc Natl Acad Sci U S A 115: E5980–E5989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou J, Amran FS, Kramski M, Angelovich TA, Elliott J, Hearps AC, Price P, and Jaworowski A. 2015. An NK Cell Population Lacking FcRgamma Is Expanded in Chronically Infected HIV Patients. J Immunol 194: 4688–4697. [DOI] [PubMed] [Google Scholar]
- 25.Arora G, Hart GT, Manzella-Lapeira J, Doritchamou JY, Narum DL, Thomas LM, Brzostowski J, Rajagopalan S, Doumbo OK, Traore B, Miller LH, Pierce SK, Duffy PE, Crompton PD, Desai SA, and Long EO. 2018. NK cells inhibit Plasmodium falciparum growth in red blood cells via antibody-dependent cellular cytotoxicity. Elife 7:e36806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hart GT, Tran TM, Theorell J, Schlums H, Arora G, Rajagopalan S, Sangala ADJ, Welsh KJ, Traore B, Pierce SK, Crompton PD, Bryceson YT, and Long EO. 2019. Adaptive NK cells in people exposed to Plasmodium falciparum correlate with protection from malaria. J Exp Med 216: 1280–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aguilar OA, Fong LK, Ishiyama K, DeGrado WF, and Lanier LL. 2022. The CD3zeta adaptor structure determines functional differences between human and mouse CD16 Fc receptor signaling. J Exp Med 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.R. 2021. R: A language environment for statistical computing, 4.1 ed. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 29.Bonnin S 2020. Introduction to R 2021: 19.11 Volcano Plots.
- 30.Pomeroy EJ, Hunzeker JT, Kluesner MG, Lahr WS, Smeester BA, Crosby MR, Lonetree CL, Yamamoto K, Bendzick L, Miller JS, Geller MA, Walcheck B, Felices M, Webber BR, Starr TK, and Moriarity BS. 2020. A Genetically Engineered Primary Human Natural Killer Cell Platform for Cancer Immunotherapy. Mol Ther 28: 52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Binyamin L, Alpaugh RK, Hughes TL, Lutz CT, Campbell KS, and Weiner LM. 2008. Blocking NK cell inhibitory self-recognition promotes antibody-dependent cellular cytotoxicity in a model of anti-lymphoma therapy. J Immunol 180: 6392–6401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Snyder KM, Hullsiek R, Mishra HK, Mendez DC, Li Y, Rogich A, Kaufman DS, Wu J, and Walcheck B. 2018. Expression of a Recombinant High Affinity IgG Fc Receptor by Engineered NK Cells as a Docking Platform for Therapeutic mAbs to Target Cancer Cells. Front Immunol 9: 2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burshtyn DB D. C 2009. Natural Killer Cell Conjugate Assay Using Two-Color Flow Cytometry. In Natural Killer Cell Protocols. 89–96. [DOI] [PubMed] [Google Scholar]
- 34.Rautela J, Surgenor E, and Huntington ND. 2018. Efficient genome editing of human natural killer cells by CRISPR RNP. bioRxiv. [DOI] [PubMed] [Google Scholar]
- 35.Liu W, Scott JM, Langguth E, Chang H, Park PH, and Kim S. 2020. FcRgamma Gene Editing Reprograms Conventional NK Cells to Display Key Features of Adaptive Human NK Cells. iScience 23: 101709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang RS, Lai MC, Shih HA, and Lin S. 2021. A robust platform for expansion and genome editing of primary human natural killer cells. J Exp Med 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Naeimi Kararoudi M, Dolatshad H, Trikha P, Hussain SA, Elmas E, Foltz JA, Moseman JE, Thakkar A, Nakkula RJ, Lamb M, Chakravarti N, McLaughlin KJ, and Lee DA. 2018. Generation of Knock-out Primary and Expanded Human NK Cells Using Cas9 Ribonucleoproteins. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hintz HM, Snyder KM, Wu J, Hullsiek R, Dahlvang JD, Hart GT, Walcheck B, and LeBeau AM. 2021. Simultaneous Engagement of Tumor and Stroma Targeting Antibodies by Engineered NK-92 Cells Expressing CD64 Controls Prostate Cancer Growth. Cancer Immunol Res 9: 1270–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Romee R, Foley B, Lenvik T, Wang Y, Zhang B, Ankarlo D, Luo X, Cooley S, Verneris M, Walcheck B, and Miller J. 2013. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood 121: 3599–3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y, Wu J, Newton R, Bahaie NS, Long C, and Walcheck B. 2013. ADAM17 cleaves CD16b (FcgammaRIIIb) in human neutrophils. Biochim Biophys Acta 1833: 680–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lanier LL, Yu G, and Phillips JH. 1989. Co-association of CD3ζ with a receptor (CD16) for IgG Fc on human natural killer cells Nature 342: 803–805. [DOI] [PubMed] [Google Scholar]
- 42.Hibbs ML, Selvaraj P, Carpen O, Springer TA, Kuster H, Jouvin M-HE, and Kinet J-P. 1989. Mechanisms for Regulating Expression of Membrane Isoforms of FcyRIII (CD16). Science 246: 1608–1611. [DOI] [PubMed] [Google Scholar]
- 43.Kurosaki T, and Ravetch JV. 1989. A single amino acid in the glycosyl phosphatidylinositol attachment domain determines the membrane topology of Fc gamma RIII. Nature 342: 805–807. [DOI] [PubMed] [Google Scholar]
- 44.Goodier MR, Lusa C, Sherratt S, Rodriguez-Galan A, Behrens R, and Riley EM. 2016. Sustained Immune Complex-Mediated Reduction in CD16 Expression after Vaccination Regulates NK Cell Function. Front Immunol 7: 384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang W, Erbe AK, Hank JA, Morris ZS, and Sondel PM. 2015. NK Cell-Mediated Antibody-Dependent Cellular Cytotoxicity in Cancer Immunotherapy. Front Immunol 6: 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu LL, Landskron J, Ask EH, Enqvist M, Sohlberg E, Traherne JA, Hammer Q, Goodridge JP, Larsson S, Jayaraman J, Oei VYS, Schaffer M, Tasken K, Ljunggren HG, Romagnani C, Trowsdale J, Malmberg KJ, and Beziat V. 2016. Critical Role of CD2 Co-stimulation in Adaptive Natural Killer Cell Responses Revealed in NKG2C-Deficient Humans. Cell Rep 15: 1088–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Orange JS, Harris KE, Andzelm MM, Valter MM, Geha RS, and Strominger JL. 2003. The mature activating natural killer cell immunologic synapse is formed in distinct stages. Proc Natl Acad Sci U S A 100: 14151–14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tang JJ, Sung AP, Guglielmo MJ, Navarrete-Galvan L, Redelman D, Smith-Gagen J, and Hudig D. 2020. Natural Killer (NK) Cell Expression of CD2 as a Predictor of Serial Antibody-Dependent Cell-Mediated Cytotoxicity (ADCC). Antibodies (Basel) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brockdorff J, Williams S, Couture C, and Mustelin T. 1999. Dephosphorylation of ZAP‐70 and inhibition of T cell activation by activated SHP1. Eur J Immunol 29: 2539–2550. [DOI] [PubMed] [Google Scholar]
- 50.Plas DR, Johnson R, Pingel JT, Matthews RJ, Dalton M, Roy G, Chan AC, and Thomas ML. 1996. Direct regulation of ZAP-70 by SHP-1 in T cell antigen receptor signaling. Science 272: 1173–1176. [DOI] [PubMed] [Google Scholar]
- 51.Pinheiro da Silva F, Aloulou M, Skurnik D, Benhamou M, Andremont A, Velasco IT, Chiamolera M, Verbeek JS, Launay P, and Monteiro RC. 2007. CD16 promotes Escherichia coli sepsis through an FcR gamma inhibitory pathway that prevents phagocytosis and facilitates inflammation. Nat Med 13: 1368–1374. [DOI] [PubMed] [Google Scholar]
- 52.Kanamaru Y, Pfirsch S, Aloulou M, Vrtovsnik F, Essig M, Loirat C, Deschenes G, Guerin-Marchand C, Blank U, and Monteiro RC. 2008. Inhibitory ITAM Signaling by FcαRI-FcRγ Chain Controls Multiple Activating Responses and Prevents Renal Inflammation. The Journal of Immunology 180: 2669–2678. [DOI] [PubMed] [Google Scholar]
- 53.Aloulou M, Ben Mkaddem S, Biarnes-Pelicot M, Boussetta T, Souchet H, Rossato E, Benhamou M, Crestani B, Zhu Z, Blank U, Launay P, and Monteiro RC. 2012. IgG1 and IVIg induce inhibitory ITAM signaling through FcgammaRIII controlling inflammatory responses. Blood 119: 3084–3096. [DOI] [PubMed] [Google Scholar]
- 54.Abram CL, and Lowell CA. 2007. The expanding role for ITAM-based signaling pathways in immune cells. Sci STKE 2007: 1–6. [DOI] [PubMed] [Google Scholar]
- 55.Ivashkiv LB 2011. How ITAMs inhibit signaling. Sci Signal 4: 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ben Mkaddem S, Hayem G, Jonsson F, Rossato E, Boedec E, Boussetta T, El Benna J, Launay P, Goujon JM, Benhamou M, Bruhns P, and Monteiro RC. 2014. Shifting FcgammaRIIA-ITAM from activation to inhibitory configuration ameliorates arthritis. J Clin Invest 124: 3945–3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mkaddem SB, Murua A, Flament H, Titeca-Beauport D, Bounaix C, Danelli L, Launay P, Benhamou M, Blank U, Daugas E, Charles N, and Monteiro RC. 2017. Lyn and Fyn function as molecular switches that control immunoreceptors to direct homeostasis or inflammation. Nat Commun 8: 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu S, Galat V, Galat Y, Lee YKA, Wainwright D, and Wu J. 2021. NK cell-based cancer immunotherapy: from basic biology to clinical development. J Hematol Oncol 14: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, McKenna D, Le C, Defor TE, Burns LJ, Orchard PJ, Blazar BR, Wagner JE, Slungaard A, Weisdorf DJ, Okazaki IJ, and McGlave PB. 2005. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 105: 3051–3057. [DOI] [PubMed] [Google Scholar]
- 60.Lewis GK, Ackerman ME, Scarlatti G, Moog C, Robert-Guroff M, Kent SJ, Overbaugh J, Reeves RK, Ferrari G, and Thyagarajan B. 2019. Knowns and Unknowns of Assaying Antibody-Dependent Cell-Mediated Cytotoxicity Against HIV-1. Front Immunol 10: 1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Temming AR, de Taeye SW, de Graaf EL, de Neef LA, Dekkers G, Bruggeman CW, Koers J, Ligthart P, Nagelkerke SQ, Zimring JC, Kuijpers TW, Wuhrer M, Rispens T, and Vidarsson G. 2019. Functional Attributes of Antibodies, Effector Cells, and Target Cells Affecting NK Cell-Mediated Antibody-Dependent Cellular Cytotoxicity. J Immunol 203: 3126–3135. [DOI] [PubMed] [Google Scholar]
- 62.Zaghi E, Calvi M, Puccio S, Spata G, Terzoli S, Peano C, Roberto A, De Paoli F, van Beek JJ, Mariotti J, De Philippis C, Sarina B, Mineri R, Bramanti S, Santoro A, Le-Trilling VTK, Trilling M, Marcenaro E, Castagna L, Di Vito C, Lugli E, and Mavilio D. 2021. Single-cell profiling identifies impaired adaptive NK cells expanded after HCMV reactivation in haploidentical HSCT. JCI Insight 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fauriat C, Long EO, Ljunggren HG, and Bryceson YT. 2010. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood 115: 2167–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jing Y, Ni Z, Wu J, Higgins L, Markowski TW, Kaufman DS, and Walcheck B. 2015. Identification of an ADAM17 cleavage region in human CD16 (FcgammaRIII) and the engineering of a non-cleavable version of the receptor in NK cells. PLoS One 10: e0121788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu J, Mishra HK, and Walcheck B. 2019. Role of ADAM17 as a regulatory checkpoint of CD16A in NK cells and as a potential target for cancer immunotherapy. J Leukoc Biol 105: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mahmood S, Kanwar N, Tran J, Zhang ML, and Kung SK. 2012. SHP-1 phosphatase is a critical regulator in preventing natural killer cell self-killing. PLoS One 7: e44244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Azoulay-Alfaguter I, Strazza M, Peled M, Novak HK, Muller J, Dustin ML, and Mor A. 2017. The tyrosine phosphatase SHP-1 promotes T cell adhesion by activating the adaptor protein CrkII in the immunological synapse. Sci Signal 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kennedy PR, Barthen C, Williamson DJ, Pitkeathly WTE, Hazime KS, Cumming J, Stacey KB, Hilton HG, Carrington M, Parham P, and Davis DM. 2019. Genetic diversity affects the nanoscale membrane organization and signaling of natural killer cell receptors. Sci Signal 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
R 4.1 (28) was used to generate Figure S1G. The code was adapted from https://biocorecrg.github.io/CRG_RIntroduction/volcano-plots.html (29) and is available at https://github.com/jamesdahlvang/volcano. All raw data is available upon request.