

Abstract
Numerous viruses infecting bacteria and archaea encode CRISPR-Cas system inhibitors, known as anti-CRISPR proteins (Acr). The Acrs typically are highly specific for particular CRISPR variants, resulting in remarkable sequence and structural diversity and complicating accurate prediction and identification of Acrs. In addition to their intrinsic interest for understanding the coevolution of defense and counter-defense systems in prokaryotes, Acrs could be natural, potent on-off switches for CRISPR-based biotechnological tools, so their discovery, characterization and application are of major importance. Here we discuss the computational approaches for Acr prediction. Due to the enormous diversity and likely multiple origins of the Acrs, sequence similarity searches are of limited use. However, multiple features of protein and gene organization have been successfully harnessed to this end including small protein size and distinct amino acid compositions of the Acrs, association of acr genes in virus genomes with genes encoding helix-turn-helix proteins that regulate Acr expression (Acr-associated proteins, Aca), and presence of self-targeting CRISPR spacers in bacterial and archaeal genomes containing Acr-encoding proviruses. Productive approaches for Acr prediction also involve genome comparison of closely related viruses, of which one is resistant and the other one is sensitive to a particular CRISPR variant, and “guilt by association” whereby genes adjacent to a homolog of a known Aca are identified as candidate Acrs. The distinctive features of Acrs are employed for Acr prediction both by developing dedicated search algorithms and through machine learning. New approaches will be needed to identify novel types of Acrs that are likely to exist.
Graphical Abstract
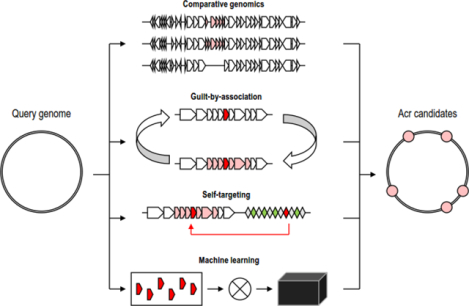
Introduction
CRISPR-Cas (Clustered Regularly Interspaced Short Palindromic Repeats and CRISPR-Associated proteins), the only known adaptive immunity systems in prokaryotes, are present in about 40% of bacteria and about 80% of archaea [1–3]. Upon encountering a foreign nucleic acid, in a process known as adaptation, dedicated Cas proteins incorporate a fragment of the foreign DNA fragment or of a cDNA copy of a foreign RNA (known as spacer) into an array of direct repeats, CRISPR [4]. The CRISPR locus is transcribed, and the long transcript is processed into mature CRISPR (cr) RNA that consists of the spacers flanked by portions of adjacent repeats [5, 6]. In the process known as interference, the crRNAs are then engaged by effector Cas proteins to detect and cleave the cognate nucleic acid [1, 2, 7]. The CRISPR-Cas systems are currently classified into two classes, six types, 33 subtypes and numerous variants within the subtypes that substantially differ from each other with respect to the organization of the effector complexes and the specific functional mechanisms [3].
Typically, CRISPR-Cas systems target different mobile genetic elements (MGE), primarily, viruses and plasmids [8]. The highly efficient CRISPR immunity puts MGE under a strong pressure to evade the CRISPR response which locks them into the arms race evolutionary dynamics with CRISPR-Cas systems. The evasion of CRISPR involves either escape through mutation of the sequence targeted by CRISPR (protospacer) or evolution of dedicated anti-CRISPR mechanisms. The best characterized of such mechanisms involves dedicated, MGE-encoded CRISPR inhibitors, anti-CRISPR proteins (Acr) [9]. The diversity of the CRISPR-Cas systems and the arms race dynamics dictate the diversity and fast evolution of the Acrs that are highly specific towards individual CRISPR-Cas subtypes and variants [3, 10–12]. Indeed, many bacterial and archaeal viruses encode multiple Acrs that target different varieties of CRISPR-Cas systems. Due to these factors, prediction of Acrs by virus genome analysis presents a major challenge that requires the development of dedicated computational approaches.
The acr genes were originally identified in CRISPR-Cas resistant Pseudomonas phages [13]. Genome comparison of resistant and sensitive phages revealed a variable locus that was located between conserved phage genes and encoded several small proteins. Subsequent experimental testing of each of these genes led to the identification of five Acrs inhibiting type I-F CRISPR-Cas systems, AcrIF1–5 [13]. Immediately downstream of the variable acr genes, there is a conserved gene encoding a putative helix-turn-helix protein, later shown to be a transcriptional regulator of the acr operon (denoted anti-CRISPR associated gene 1, aca1) [14, 15]. Unlike Acr proteins, Aca1 homologs were found both within and outside of Pseudomonas bacteria and their viruses. Thus, it has been reasoned that identification of this regulator in other viruses could point to additional putative Acrs that would be encoded next to Aca1, even if the respective genes were located outside of the initially defined putative Acr locus [14]. Indeed, more Acr candidates were predicted using this “guilt by association” approach and several of these were subsequently experimentally validated [16].
The next observation that resulted in a distinct computational approach for identification of Acr candidates came from the analysis of bacterial and archaeal genomes for self-targeting spacers in CRISPR arrays. If a CRISPR-cas locus contains a self-targeting spacer(s), the respective genome should be expected to encode an Acr, likely, within a prophage, that allows the organism to avoid autoimmunity. The first study implementing this approach was published in 2017 and reported a discovery of AcrIIA1–4 against type II-A CRISPR-Cas systems in Listeria [17]. It was found that the spacer targeted a prophage, which was hypothesized to encode an Acr against type II-A. A comparison of this prophage with closely related prophages from other Listeria genomes lacking self-targeting spacers revealed several regions of interest. Uncharacterized ORFs from those regions were tested experimentally. This procedure resulted in discovery of several Acrs acting against type II-A system.
By 2018, more than 100 Acrs were discovered [18–20], and the first dedicated database, Anti-CRISPRdb, was created [21]. Furthermore, general understanding of typical features of Acr sequences, their spread in bacterial and archaeal viruses and proviruses, organization of the respective loci, presence of Acas and self-targeting spacers, together with the fast accumulation of diverse experimentally characterized Acrs, provided information for mining genomes using expert-defined [22] or machine-learned features [23–25]. Most recently, a deep neural network algorithm DeepAcr [26], was developed to predict Acrs based on the protein sequences only. Applications of these methods revealed thousands of Acr candidates, of which only a few have been experimentally validated.
Along with computational methods, “agnostic” strategies of experimental Acr discovery have been developed as well. Generally, these approaches are applied when hundreds of proteins or genomic fragments have to be tested. One of such approaches involves cloning cas genes from the CRISPR-Cas system of interest along with a gene or genomic fragment that might encode Acr and a reporter plasmid coding for GFP and targeted by a crRNA. These constructs are tested in a cell-free transcription-translation system [27]. This approach was used for identification of Acr against type V-A systems [28] and for testing multiple Acr candidates fort VI-B system predicted by DeepAcr [26]. Genetic high-throughput systems also have been used for Acr discovery. Under this approach, DNA fragments from genomes or metagenomes are cloned in a plasmid along with antibiotic resistance genes, which are targeted by the CRISPR-Cas system of interest [29, 30]. The plasmid DNA from cells that survive in the presence of the respective antibiotic is sequenced and proteins encoded on this particular plasmid are tested for CRISPR-Cas inhibition.
The interest in Acr discovery remains high, both considering the intrinsic interest of the coevolution of defense and counter-defense systems and because Acrs are considered to be natural off-switches of CRISPR-Cas systems activity and therefore potentially can be used in multiple biotechnological applications ([31–34] and references therein). Yet, the challenges faced by Acr prediction are many including the fast evolution and generally low evolutionary conservation of these proteins, the wide spread of Aca homologs which complicates their use for Acr identification, and the poor annotation of many virus and provirus genomes. Here we describe computational approaches used for Acr discovery and discuss the common complications and potential improvements.
Comparative genomics
As mentioned above, discovery of Acr proteins began with the comparison of the genomes of phages that are sensitive and resistant to type I-F CRISPR-Cas systems [13]. The more similar the genomes are, the easier is the identification of the key differences that are likely to be relevant for the different phenotypes (Figure 1A). The same approach was used to discover the first Acr in archaea [35]. In this work a deletion mutant of Sulfolobus lytic rudivirus SIRV2 was isolated that, unlike the wild type, was sensitive to the host CRISPR-Cas systems. Comparison of this mutant genome sequence with those of the wild type SIRV2 and another closely related, CRISPR-resistant virus SIRV3 narrowed down the list of the Acr candidates to three genes, which were then tested separately for Acr activity, and a gene solely responsible for the CRISPR resistance, AcrID1, was discovered. One of the first Acrs for type II-A, AcrIIA6, was found by systematic screening for Streptococcus thermophilus phages resistant to type II-A CRISPR system followed by comparison of their genomes with the genomes of sensitive phages [36]. Similarly, screening of Listeria phages and prophages followed by comparison of the genomes of resistant phages to their close relative led to the identification of AcrVIA1 that inhibits a type VI-A CRISPR-Cas system [37]. The common limitation of the comparative genomics approach is the absence of closely related genomes with contrasting phenotypes for many viruses.
Figure 1. Comparative genomic approaches for Acr prediction.
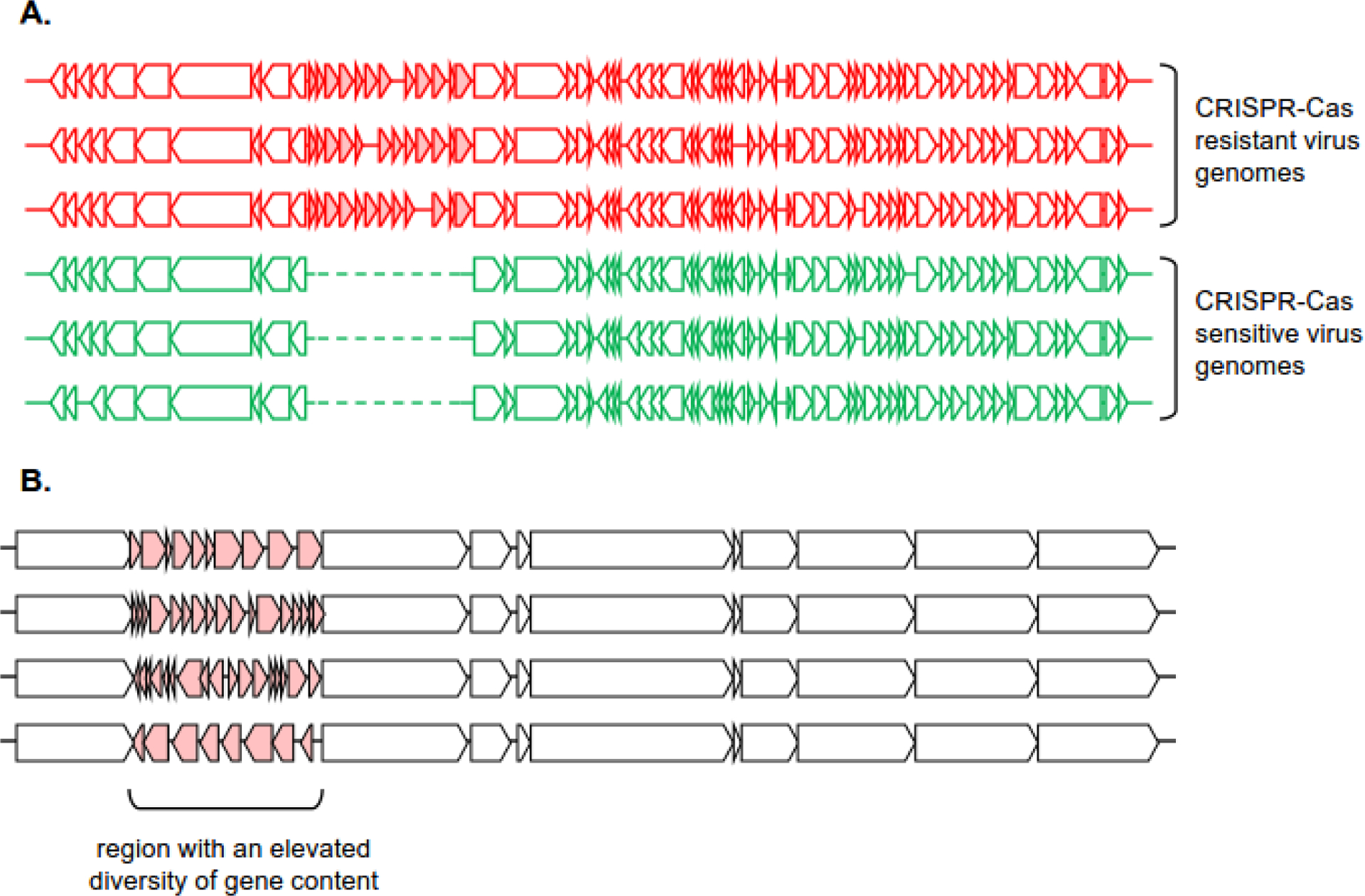
A. A comparison between a set of related CRISPR-Cas-resistant virus genomes (outlined in red) and CRISPR-Cas-sensitive virus genomes (outlined in green) A region that is consistently present in resistant genomes and absent in sensitive genomes, is likely to contain Acr genes (candidate genes highlighted in red).
B. A comparison between a set of related virus or provirus genomes. A region that displays unusually high diversity of gene content, flanked by highly syntenic regions, is likely to contain Acr genes (candidate genes highlighted in red)
In a broader view, comparative genomics may be useful to identify regions in viral genomes that are prone to gene gain, loss and shuffling, and thus can be inferred not to contain genes for essential virion components, proteins involved in genome replication, host cell attachment, injection, or lysis (Figure 1B). Typically, such regions include multiple anti-defense genes that are expressed early in infection and inhibit various host defense systems, including CRISPR-Cas. For instance, in Pseudomonas Mu-like phages where Acrs were originally discovered, acr genes are located between two conserved viral genes coding for phage Mu G gene and protease/scaffold protein [13]. The sets of genes located in this region differ in otherwise closely related viral genomes. Thus, this region shows short-term evolutionary dynamics and therefore is detectable by comparative genomics, even in the absence of any functional evidence. Similarly, terminal regions of archaeal SIRV viruses differ widely and have been shown to encode the first characterized Acr for type III-B system, AcrIIIB1 [38], which has been shown to be expressed early, along with another Acr protein, AcrID1 [39, 40]. Thus, genes located in variable virus regions, especially when the virus host typically encodes CRISPR-Cas systems, are candidates for the Acr function that merit experimental validation solely on the basis of comparative genomic observations.
Guilt by association approaches
The approach for identification of functional connections between proteins by analysis of their gene context has grown from the seminal discovery of bacterial operons by Jakob and Monod [41], and started to be actively applied when the first several complete genomes of bacteria and archaea became available. This approach to gene function prediction is often called “guilt by association” (GBA) [42–44].
The first acr genes, which inhibit type I-F CRISPR-Cas systems, were discovered in clusters together with other small hypothetical protein-coding genes and followed by an HTH transcriptional regulator, aca1 [13]. Further experimental validation showed that the remaining hypothetical proteins in these clusters had anti-CRISPR activity against type I-E CRISPR-Cas systems, establishing that Acrs targeting different CRISPR-Cas system subtypes are often encoded in operons and often with an aca gene [14]. Thus, the computational procedure is simple: homologs of known Acrs or Acas (seeds) are identified in other viral (or plasmid, or chromosome) genomic sequences, and proteins encoded next to the respective genes are tested for Acr activity (Figure 2A).
Figure 2. Guilt-by-association and self-targeting spacers approaches for Acr prediction.
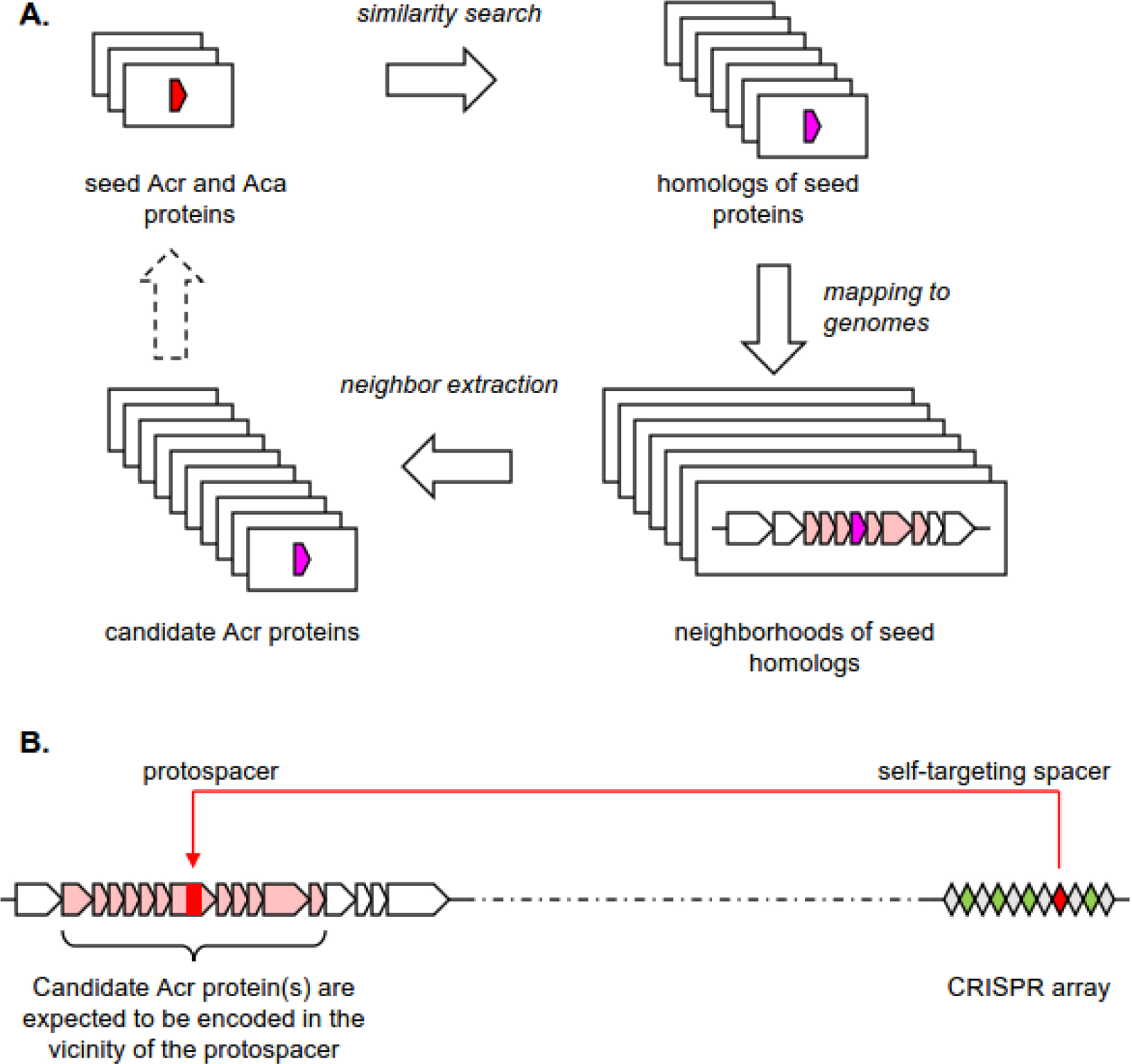
A. The guilt-by-association approach starts with a group of seed Acr/Aca genes. Genes in the neighborhoods of reliably identified homologs are also presumed to be involved in CRISPR-Cas-resistance, producing a set of candidate Acr/Aca genes. After filtering using additional criteria (such as length and amino acid composition), the procedure can be iterated to further expand the candidate gene set.
B. A self-targeting spacer in a CRISPR-Cas bearing genome is presumed to target a provirus that was integrated in the host genome despite the presence of targeting spacer. Thus, the neighborhood of such protospacer is likely to contain (remnants of) a provirus, bearing Acr genes.
The guilt by association procedure can be run for several iterations with as many seeds as needed, either until convergence or until the desirable number of candidates for experimental testing is found (Figure 2A). This approach became an integral part of many Acr discovery pipelines. Along with the self-targeting approach, guilt by association was used for the discovery of AcrIIA1–4 [17]; for identification of 12 acr genes inhibiting four CRISPR subtypes: three for I-E, four for I-F, one for I-C and three for V-A [45]; for identification of two additional anti-type V Acrs [28] and two additional Acrs for type II-A [46]. More recently, analysis of the neighbors of genes encoding the putative transcriptional regulator Aca5 in selected genomes led to the identification of additional 13 Acrs against I-E and I-F systems, namely AcrIE8, AcrIF11–22 [16], and subsequently, AcrIIA24–32 from Streptococcus phages and prophages [47].
The GBA strategy faces several challenges. The first one is the “banana conundrum” (“I know how to spell banana, I don’t know where to stop”), which is related to the fact that iterative searches of genomic neighborhoods rarely converge. Typically, some of the acr neighboring genes are common across a broad diversity of prokaryotic and virus genomes, which results in identification of hundreds or thousands of neighbors after several iterations, and this number of candidates is unmanageable for most of the experimental procedures for Acr testing. This problem is especially relevant when Aca (transcriptional regulator) is selected as a seed for GBA as recently demonstrated for Aca5 protein, which has hundreds of homologs in viral and plasmid genomes and on bacterial chromosomes [16], so that the authors had to manually select a small subset of the aca5 loci for their further experimental analysis of aca5 neighbors. Hundreds of Aca homologs were identified by systematic search for such proteins in the vicinity of known Acrs in genomic databases, and then, the GBA approach yielded thousands of candidate Acrs encoded in the vicinity of all those aca genes [48]. The second problem is that Acrs are often encoded next to genes involved in inhibition of other defense systems, such as restriction-modification and abortive infection [16], and therefore, using acr gene neighbors as seeds can result in fast departure from the acr context. Finally, because Acrs for different CRISPR-Cas subtypes encoded next to each other [14, 16, 37, 45], GBA is generally unable to accurately predict which CRISPR-Cas system will be inhibited by an Acr candidate, complicating experimental validation. This latter difficulty, however, can be at least partially overcome by analysis of co-occurrence of Acr candidates with particular variants of CRISPR-Cas systems in the genomes of hosts of the respective viruses.
Self-targeting spacers
Self-targeting spacers (STS) occur in roughly 20% of microbial genomes and the majority of them target a provirus [8, 49], providing ample opportunity to search these proviruses for Acrs targeting the CRISPR-Cas system(s) present in the same genome [28] (Figure 2B). In many cases, the searches for Acrs in such targeted proviruses proved successful. The first type II-A Acrs from Listeria monocytogenes [17] were identified using the STS approach. In this case, the search was initiated by finding a spacer derived from CRISPR array of type II-A CRISPR-Cas system which targeted a prophage present in the same genome. It was hypothesized that this prophage encoded an Acr for this system. Next, it was shown that the prophage indeed conferred resistance to this particular CRISPR-Cas variant. Then, this prophage was compared to a closely related prophage from a non-self-targeting L. monocytogenes strain, to identify specific loci that were unique to the prophage from the self-targeting strain. These loci were tested one-by-one and a locus conferring CRISPR-Cas resistance was identified, followed by the demonstration that expression of any of the two genes in this locus, acrIIA1 and acrIIA2, was sufficient to inhibit the type II-A CRISPR-Cas. GBA analysis of the neighbors of homologous genes in other related prophages, followed by experimental validation, resulted in characterization of two more Acrs for type II-A systems, AcrIIA3 and AcrIIA4.
A high-throughput modification of the STS approach was employed for the discovery of AcrVA proteins in three self-targeting Moraxella bovoculi strains [28]. For all predicted prophages from these strains, 2 to 10 kb PCR fragments (67 in total) were obtained, cloned and tested for inhibitory activity against type V-A system. Next, all individual ORFs from the four fragments with inhibitory activity were tested, resulting in the identification of three Acrs, AcrVA1, AcrVA4 and AcrVA5 [28]. Independently, application of the STS approach combined with GBA approach was also used for identification of type V-A and I-C inhibitors by other researchers [45]. In this work, the acrVA1 gene was identified by testing gene neighbors of verified acrIF11 homolog in the genome of the same M.bovoculi strains containing a self-targeting spacer encoded in the CRISPR array of type V-A system. Two more type V-A inhibitors, AcrVA2 and AcrVA3, were found in the same work using GBA approach.
In the above study, a computational pipeline for the discovery of self-targeting in prophage regions was developed which was subsequently applied for interrogation of Staphylococcus genomes for acrIIA genes [46]. In this work, 48 genomes were found to encode a type II-A CRISPR-Cas system with at least one STS. However, 36 of these genomes carried mutations in the corresponding PAM and therefore were excluded from the further analysis. The authors then focused on two genomes that encoded Cas9 proteins most closely similar to Streptococcus pyogenes Cas9 (SpyCas9) which is actively employed in genome editing. The prophage genomic regions were identified, and their fragments were cloned and tested using via in vitro cell-free transcription-translation assays [46]. Only one fragment of the 27 tested was found to inhibit Staphylococcus aureus Cas9 (SauCas9), and for only two ORFs in this fragment, there was no functional prediction. One of the two, AcrIIA13, when tested separately, showed strong inhibitory activity against SauCas9.
Thus, although STS appears to be the most straightforward approach for Acr prediction, allowing identification of Acrs specific for a given CRISPR-Cas system subtype, in practice, this approach still requires heavy additional filtering to reduce the experimental testing burden. This filtering can be facilitated by comparative genomic analysis and discarding functionally annotated genes involved in essential aspects of viral reproduction. An additional caveat of the STS approach for Acr prediction is that many bacterial and archaeal genomes encompass multiple proviruses so that the detection of a STS match does not necessarily point to the presence of an acr gene in the same genomic region. All the caveats notwithstanding, STS is a highly relevant feature that is often used in computational pipelines for large scale Acr prediction described below.
Feature analysis and machine learning
Acr genes are typically clustered, codirected, and frequently found next to a transcriptional regulator (Aca). Additionally, most of the Acrs share other notable features: typically, these are small proteins (roughly between 50 and 150 amino acids) that lack transmembrane segments or signal peptides. All these features can be used for Acr prediction. The first bioinformatic pipeline for Acr prediction was designed by Yin and colleagues [48]. This pipeline runs two iterations of GBA search. During the first iteration, homologs of known Acr are found in available prokaryotic and viral genomes and their gene neighbors collected according to several empirical rules. Specifically, to be considered an Acr candidate, a gene adjacent to a known Acr was required to be codirected with the latter and encode a predicted protein shorter than 200 aa; additionally, an HTH domain-containing gene (putative Aca) was required to be encoded within four genes of the known Acr. Neighbor genes for homologs of all known and putative new Aca genes were collected using the same set of rules except that the distance between co-directed genes was required to be less than 150 nt (considered to be the maximum distance between genes in an operon). The results of both iterations were combined and supplemented by information on STS, location of identified candidates within or in the vicinity of proviruses and CRISPR-Cas system subtypes present in the respective genomes. Altogether, more than 50,000 loci encoding candidate Acrs were identified. Analysis of this data led to several salient observations: 1) Homologs of Acrs inhibiting certain CRISPR-Cas subtypes are often encoded in genomes lacking this particular CRISPR-Cas subtype; 2) The majority of the Aca-Acr loci were identified in genomes with no STS, and some were found in genomes lacking CRISPR-Cas systems; 3) Many predicted Aca proteins are actually known antitoxins and some predicted Acrs are toxins. The latter observation illuminated a major challenge of GBA approach as DNA-binding proteins are ubiquitous in other phage regulatory systems, in particular, toxin-antitoxin modules, making them a non-exclusive feature of acr operons [50]. A very similar strategy with the same features is implemented in the AcrFinder webserver, with additional filtering of predicted Acr candidates to exclude functionally characterized or highly conserved domains [22]. However, AcrFinder runs only one iteration of GBA, with known Acr and Aca proteins employed as seeds.
Classical feature-based prediction methods require an expert decision on what features are biologically relevant and algorithmically informative for recognition of an Acr protein (gene). A broad class of machine learning (ML) techniques circumvents this step by automatically assessing a wide (sometimes, astronomically vast) range of possible features (gene context, gene and protein sequences, sequence alignments and more) and trains a model to distinguish between the positive and the negative subsets of the training set by adjusting the model parameters (see Figure 3 for the general organization of ML approaches). Some ML methods explicitly indicate the most informative features, whereas in others, these features remain hidden in the model’s “black box” and cannot be easily extracted. Like feature extraction methods, all ML methods ultimately rely on the expert input; however, in the latter case, it is supplied in the form of the curated positive and negative training sets rather than as an explicit list of features.
Figure 3. Machine learning-based approaches for Acr prediction.
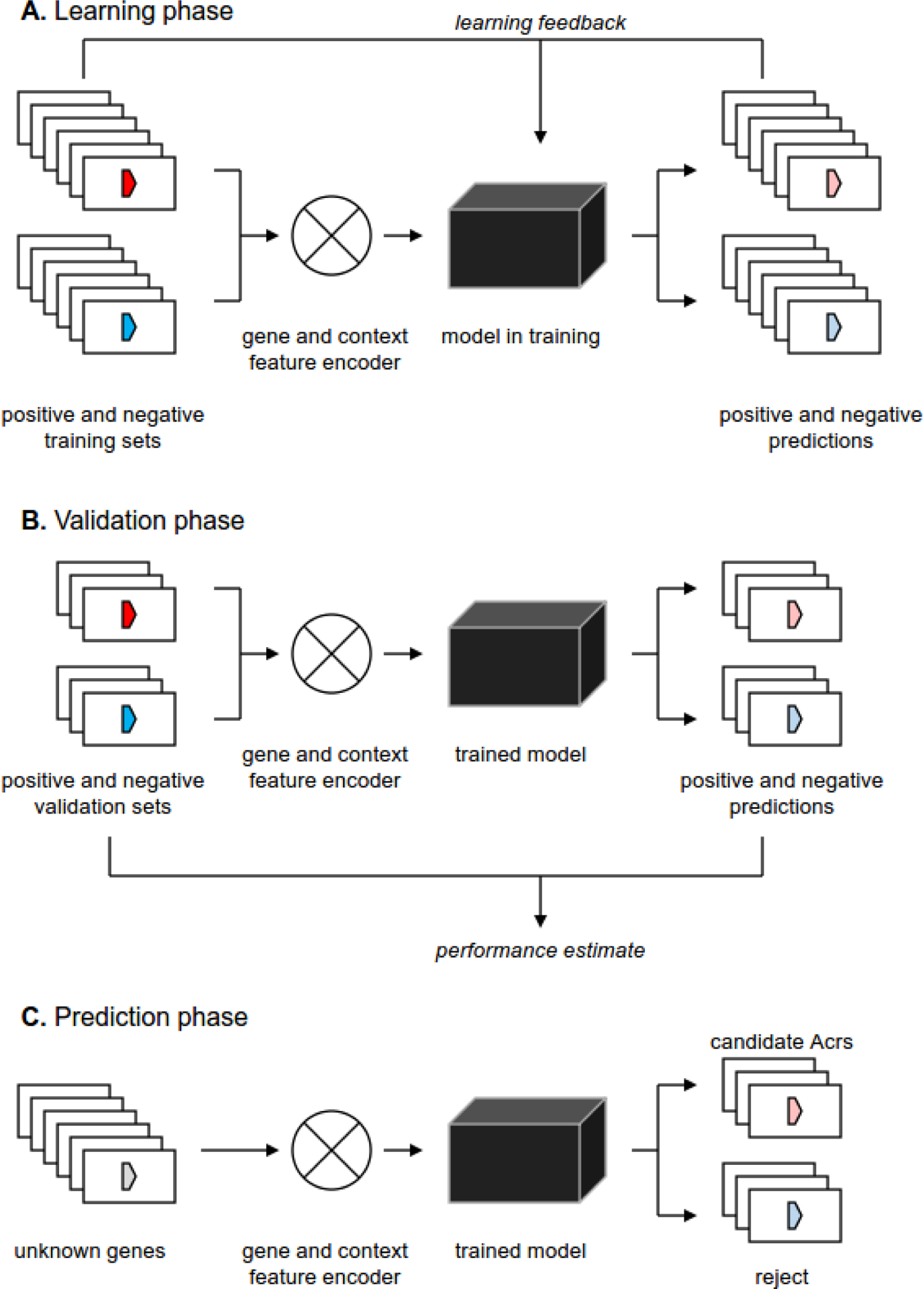
A. At the learning phase the gene and/or the gene context features of the training set of genes (Acrs and non-Acrs) are encoded and fed into the machine learning model (neural network, random forest, etc). Comparison between the predictions, generated by the model, and the known status of training set genes are fed back into the model to maximize the discrimination between the positive and the negative training sets.
B. At the validation phase a subset of known Acrs and non-Acrs, withdrawn from the training set, is fed into the trained model to assess its performance. Care should be taken to avoid cross-contamination between the training and the validation sets (e.g. by clustering the sequences and ensuring that all members of cluster are used either for training or for validation, but not for both).
C. At the prediction phase a set of unknown genes is encoded and fed into the trained model that classifies these genes as either positive or negative (often with a quantitative measure of confidence, associated with each prediction).
So far three distinct ML based models were built for Acr identification. One method [23] used a set of attributes assigned to each known (or candidate) Acr such as size, whether or not a protein is annotated in profile databases, whether or not an HTH protein is encoded in the vicinity, presence of self-targeting spacer, mean size of the array of co-directed genes (directon) containing the respective gene, and more. For training, a positive set of non-redundant Acrs and their verified homologs, and a negative set of randomly selected non-Acrs from viral and prokaryotic genomes were used. A random forest of extremely randomized trees was adopted as the ML technique. This method achieved precision of 78% and recall of 57% on the test set for the binary model (AUC of 0.83). The set of more than 230,000 detected Acr candidates was further trimmed using several heuristics such as considering only those that encoded an HTH protein in the neighborhood of an acr gene, self-targeting, maximum directon size of 5 genes, and a number of identified homologs below a defined threshold. This filtering allowed to narrow down the set to 2,526 best candidate families, from which candidates for actual testing were still selected using the empirically defined key features such as self-targeting and presence of HTH in the respective direction. Among 21 top candidates tested, four displayed inhibitory activity against I-C system, whereas 17 showed no activity against Pseudomonas systems I-C, I-E or I-F. Obviously, the failure to detect activity for the latter group does not imply that these candidates do not function as Acrs against other variants of same CRISPR-Cas subtypes, considering that they originated from different genomes. However, these results demonstrate limitations of the ML approaches, primarily, the lack of specific prediction of Acrs targets, and hence, the requirement for a comprehensive experimental validation setup.
Two other ML methods relied only on amino acid composition of known Acrs to build predictive models. One of these, AcRanker, employed EXtreme Gradient Boosting (XGBoost) ranking as the learning technique [25]. For running the learning procedure, the amino acids sequences were recoded into 7 classes based on their physicochemical properties and data on amino acid composition, as well as dipeptide and tripeptide frequency counts was transformed into a multidimensional feature vector for a given protein sequence. All non-Acr proteins in a given proteome were used as the negative set. A variant of this training method was designed to include only prophage regions identified in available genomes. For method validation, Acrs that were not present in the learning set and encoded in the prophage regions were employed. BLASTP ranking (sequence similarity searches were performed for known Acrs against the same protein set as in AcRanker) was used for comparison. If an Acr ranked within top 10 candidates, it was considered as a success. The method performed relatively poorly on entire proteomes but significantly better performance was achieved when only prophage proteins were considered. Overall, AcRanker performed better than sequence similarity search, which is not surprising because in many test cases there were no detectable homologs of known Acrs in the analyzed protein sets. As in the work by Gussow et al described above, only self-targeting genomes with protospacers associated with a correct PAM were considered as candidates for experimental testing [25]. In total, ~200 Acr candidates were predicted in the prophage regions of 21 interrogated genomes. Among those, 10 candidates were selected manually based on additional considerations such as being part of a cluster of genes encoding small proteins and prediction of a strong promotor and terminator. These candidates were tested for inhibitory activity against SpyCas9 and SauCas9. Only three were found to inhibit either of the two Cas9s, with a relatively low efficiency [25].
Another machine learning Acr classifier, PaCRISPR, extracts various features from multiple alignments of Acr homologs (sequence composition, local order, autocorrelation, and more) and uses a machine learning technique, support vector machine (SVM), to train an ensemble of models. An elaborate approach to design negative test sets consisting of small proteins encoded in MGEs, but not similar to Acrs, was used to avoid overtraining; the authors report a substantial performance advantage over AcRanker [24]. This tool is integrated into a user-friendly web server which, in addition to Acr prediction, provides detailed information and graphic visualization for each query genome and Acr candidate [24]. PaCRISPR is currently used by dedicated Acr databases, such as AcrDB [51] and AcrHub [52] to predict potential Acrs.
The most recent machine learning approach, DeepAcr, utilizes a set of distinct features derived from amino acid sequence [26]. First, amino acids in a given protein are converted into numerical values and fed into a bidirectional recurrent cell incorporating three neural networks as the learning structures: long short-term memory (LSTM), linear, and gated recurrent unit (GRU). The output of this network is combined with additional 12 features, capturing properties (e.g., protein length and instability index) of the entire protein. Next, a multilayer perceptron (MLP) converts this input into a confidence score reflecting the certainty that the input protein is an Acr. The resulting model was trained on 420 known Acrs; 420 diverse small accessory non-Acr proteins, associated with type III CRISPR-Cas systems, were chosen as the negative set. DeepAcr achieved a performance accuracy of 96% using the LSTM network, 94% using the linear network, and 95% using the GRU network when testing on 15% of the positive and negative sets not included in learning. Application of the method for available prokaryotic genomes resulted in identification of more than one million Acr candidates. Notably, DeepAcr yielded a 96% overlap with the list of Acr candidates obtained by Gussow et al, although contextual information was not employed for learning in the case of DeepAcr. For experimental validation of DeepAcr, 77 Acr candidates for type VI-B CRISPR-Cas system were selected and tested using the cell-free transcription-translation assay with three phylogenetically distinct Cas13b effectors. Among these candidates, one protein, AcrVIB1, showed strong inhibitory activity against one of the Cas13b effectors. AcrVIB1 is encoded within a predicted prophage region and next to HTH-containing regulator although no self-targeting in this genome was reported. Thus, at this point, although DeepAcr showed excellent performance on test sets of known Acrs and negative controls, experimental validation so far has been quite limited, and therefore, the true potential of this method remains to be explored.
On the whole, as in many areas of computational biology, it appears likely that the future of Acr prediction belongs to machine learning approaches. However, as discussed above, the current approaches yield very long lists of candidates that remain to be pared down using various heuristics and/or direct experimentation. Arguably, a new generation of machine learning methods is likely to be required to make prediction highly efficient.
Potential new directions of anti-CRISPR gene prediction
The evolutionary origins of most Acrs remains a puzzle. A plausible possibility seems to be that viruses “borrow” components of CRISPR-Cas systems and repurpose them to function as Acrs, possibly via the dominant negative mechanism, which is a common route of evolution of viral antidefense systems [53, 54]. Two genes found both in viral genomes and in CRISPR-cas loci were recently characterized. One includes members of several subfamilies of Cas4 nucleases, which are encoded in many viral genomes [55]. It has been recently shown that viral Cas4 functions as an Acr inhibiting spacer acquisition [56]. Considering that several Acrs are often encoded in the same viral genome, the presence of a cas4 gene in a viral genome improve the chances of finding other Acrs, even if they are not encoded close to the cas4 gene, as is the case in some viruses of Sulfolobales (Figure 4A). This approach can be considered as relaxed “guilt-by-association”. Thus, clusters of genes for small proteins from viral genomes encoding Cas4 can be promising candidates to test for anti-CRISPR activity against CRISPR-Cas systems frequently found in the genomes of the respective host group (Figure 4B).
Figure 4. Viral Cas4 as a seed for a relaxed guilt-by-association approach.

A.Cas4 for in context of other characterized Acrs and their homologs in three different viruses of Sulfolobales. Note high density of Acr genes and their homologs in SIRV2. Only three Acrs from this genome where so far experimentally characterized.
B. Cas4 and clusters of small unknown proteins in other viral genomes. In can be expected that in addition to cas4 these viruses encode other Acrs within the clusters of small genes, like in SIRV2 genome shown in the panel A.
Genes are shown by block arrows, roughly to scale. Color code is explained at the bottom of the Figure. Gene ID is indicated below each arrow. Gray dashes lines show omitted part of viral genome. Complete SIRV2 genome is shown on panel A; two halves of the genome are separated by arrows. CRISPR-Cas system types found in respective taxa (blue) are indicated in the boxed on the right. Brief description of the genes is shown for annotated proteins or Acrs above the respective arrows. Abbreviations: SIRV2 and SIRV8 - Sulfolobus islandicus rod-shaped virus 2 and 8 respectively; AFV1 - Acidianus filamentous virus 1; SSV3 - Sulfolobus spindle-shaped virus 3; ACSV - Aquificae Conch Spring virus; HTH – helix turn helix; RHH – ribbon helix helix.
The second example is the ring nuclease of the DUF1874 family which cleaves cyclic tetra-adenylate, the second messenger synthesized by the Cas10 polymerase-cyclase, preventing activation of CARF-associated effectors in type III CRISPR-Cas systems [57]. This ring nuclease is encoded by many bacteriophages and functions as a broad spectrum AcrIII-1 that inhibits diverse type III systems by targeting a widespread second messenger rather than a specific Cas protein. Homologs of DUF1874 genes are often found in type III systems where they apparently control this additional defense mechanism, preventing cell death [58].
The only known case of structural mimicry of Cas proteins by an Acr is AcrF3, which mimics the Cas8f structural elements responsible for the recruitment of Cas3 nuclease [59]. This is also in line with idea of repurposing components of CRISPR-Cas system for inhibitory functions. However, it remains unclear whether AcrF3 protein originated from Cas8f directly or convergently evolved to fold into a helix resembling the recruitment helix of Cas8f.
Another growing theme among Acrs is DNA mimicking whereby several Acrs, namely AcrIF2, 6, 7, 10, 13, AcrIIA2 and AcrIIA4, have been shown subvert CRISPR effector complexes by blocking their binding to the target DNA [60–65]. Systematic prediction of viral DNA-mimicry proteins could become an approach for Acr identification to complement the existing ones.
In the section on comparative genomics above, we describe a version of the comparative genomic approach that can be employed to identify variable loci in viral genomes encoding small uncharacterized proteins flanked by conserved viral genes (Figure 1B). Combined with the predictions produced by ML classifiers, systematic application of such comparisons appears to be a promising future direction for large-scale Acr identification.
Numerous Acrs resist identification of any homologs even with the most sensitive sequence analysis methods. Recently, protein structure prediction has been revolutionized by a new generation of computational methods, in particular, AlphaFold, DeepFold and RoseTTAFold, that take advantage of protein sequence conservation within a deep learning framework [66–68]. These approaches are expected to be particularly productive when applied to fast evolving proteins, such as Acrs, providing for the systematic identification of homologs of known Acrs that are undetectable by sequence similarity searches, elucidation of the origins of Acrs via structural similarity and prediction of interactions of Acrs with CRISPR effectors.
Furthermore, entire CRISPR systems, or stand-alone CRISPR arrays, in particular virus-encoded mini arrays for which no target was identified and single repeat units [69], also can be involved in anti-CRISPR functions by substituting crRNA in effector complexes and yielding unproductive effectors. Comprehensive computational identification of such cases in viruses and other mobile genetic elements, followed by experimental investigation, appears promising and can lead to discovery of novel anti-CRISPR mechanisms.
Concluding remarks
The computational approaches discussed in this review have helped to predict many Acrs that were subsequently validated experimentally. However, identification of Acrs remains a complicated, challenging task. At the time of this writing, 103 distinct families of Acrs and 13 Acas are recorded in anti-CRISPR database [18]. For only a handful of these, a detailed mechanism of inhibition has been characterized [9, 31, 61, 70–76]. Furthermore, for some of these proteins, the originally demonstrated inhibitory activity was not reproducible when a different experimental testing method was applied [77]. Thus, upon deeper experimental study, some of these proteins might be excluded from the Acr list. Some of the Acrs have thousands of homologs in protein databases including those encoded in non-MGE chromosomal regions and in genomes lacking CRISPR-Cas systems [48]. Most of the Aca proteins also have thousands of homologs. Thus, a pressing question is: are all those homologs true Acr and Aca or could they have other functions including, perhaps, inhibition of non-CRISPR defense systems? The lack of clarity on this issue hampers, to different extents, all the methods for Acr prediction described here because they all rely on sequence similarity, explicitly or implicitly. Furthermore, using the same search parameters for different protein families leads to considerable over- or under-detection of homologs for many of those families. The potential solutions to these problems involve phylogenetic analysis of Acr and Aca homolog families for identification of branches that are strongly associated with virus and MGE genomes and, in the case of Acas, to known Acrs. Such subsets of the respective families are likely to include bona fide Acrs and Acas. For these groups of proteins, specific profiles should be constructed from multiple sequence alignments and similarity search parameters should be tuned to minimize the false positive and maximize the true positive rate.
Another outstanding question is how to choose a negative set for machine learning methods. Considering that Acrs are anti-defense proteins, it is important to include inhibitors of other defense systems to discriminate against. Indeed, restriction-modification (RM) or, more generally, self vs nonself recognition innate immunity systems are the most abundant defense systems in bacteria and archaea [78], and therefore, it appears plausible that the majority of inhibitors encoded in viral genomes target RM systems and/or various abortive infection mechanisms. Few such inhibitors have been identified so far [79], so concerted efforts to identify such inhibitors experimentally and/or computationally (primarily, by comparative genome analysis of viruses resistant and sensitive to a particular type of defense systems) can be expected to substantially boost the predictive power of machine learning methods for all these inhibitors, and Acrs in particular.
The next non-trivial issue is prediction of the specificity of Acrs towards types and subtypes of CRISPR-Cas systems. One complication is that Acrs targeting different CRISPR subtypes and even types are often encoded in the same viral directon [16, 45]. Another difficulty stems from the frequent turnover of CRISPR-Cas systems, including in situ effector genes shuffling [55], leading to decoupling between Acrs and CRISPR-Cas systems present in the same genome such that some known Acrs are encoded in CRISPR-less genomes [48]. Therefore, in order to increase the success rate in Acr identification, expansion of the experimental testing setup to include all varieties of CRISPR-Cas encoded in genomes of related organisms is highly desirable.
Overall, progress in prediction of Acr genes suffers from problems at both ends of the pipeline. At present, it seems that only a small fraction of the actual diversity of Acrs is known which severely hampers the learning stage, both for machine learning methods and for human experts. Current machine learning approaches show relatively low performance in real life predictions [23, 25, 26] because there is little understanding of the most relevant diagnostic features of Acrs (or even if such features exist). Conversely, experimental testing of Acr prediction is currently time and labor consuming and is not always reliable, with uncertainty plaguing both negative and positive results. Some of the sources of this uncertainty are the possibility that a candidate Acr acts against a different CRISPR variant under different conditions [77] and pleiotropy, which could be the cause of the identification of a conserved phage head-tail adaptor protein as AcrIIA8 [30].
All these caveats notwithstanding, major progress should be expected in the prediction and experimental investigation of Acrs as databases grow and both computational and experimental methods improve. Already, multiple software packages are now available to give any method of choice a try to predict and characterize novel Acrs for any CRISPR-Cas system of interest and user-friendly Acr database provide detailed information on known and predicted anti-CRISPR proteins and their gene neighborhoods (Table 1 and 2).
Table 1.
Computational pipelines for Acr prediction
| Name | Availability | Main approaches and features | Reference |
|---|---|---|---|
| Yin et al. | Suppl | GBA, STS | [48] |
| STSS | SP, Suppl | STS | [28] |
| AcRanker | Web, SP | ML:AA | [25] |
| AcrFinder | Web, SP | GBA, STS, FUNC* | [22] |
| Gussow et al. | SP, Suppl | ML: AA, context, STS, Number of homologs | [23] |
| DeepAcr | SP | ML: AA | [26] |
| PreAcrs | SP | ML: AA | [80] |
Abbreviations: Suppl – supplementary data; Web – web version of the program, SP – software package; ML – machine learning; STS – self-targeting spacers; GBA – guilt-by-association; AA – amino acid sequence features; FUNC* – discarding functionally characterized proteins
Table 2.
Acr databases and their features
| Name | URL | Main features | Reference |
|---|---|---|---|
| Anti-CRISPR | https://tinyurl.com/anti-CRISPR | Expert curated; provides information and nomenclature for experimentally characterized Acrs and Acas only | [18] |
| AcrDB | https://bcb.unl.edu/AcrDB/ | Provides detailed information for computationally predicted anti-CRISPR (Acr) and Acr-associated (Aca), their homologs and analyzes their gene neighborhoods | [51] |
| AcrHub | https://pacrispr.erc.monash.edu/AcrHub/ | Provides comprehensive list of experimentally validated and predicted Acrs and their gene neighborhoods | [52] |
| Anti-CRISPRdb | http://guolab.whu.edu.cn/anti-CRISPRdb | Provides detailed information for characterized and predicted anti-CRISPR (Acr) and their neighborhoods | [73] |
Highlights.
Numerous viruses infecting bacteria and archaea encode one or more proteins that protect them against CRISPR-mediated immunity (anti-CRISPR proteins, Acrs)
Most Acrs are small proteins with low sequence conservation which hampers their identification by computational methods
Several computational pipelines for Acr prediction have been developed based on their diagnostic features
Acrs have been predicted using comparative genomics, guilt-by-association approach, identification of self-targeting spacers and machine learning techniques
Progress in Acr prediction hinges on the availability of efficient wet lab pipelines to expand the repertoire of known acr genes and validate the computationally discovered candidates
Funding information
The authors’ research is supported by the Intramural Research Program at the National Library of Medicine.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CRediT author statement
Kira Makarova: Conceptualization, original draft preparation; Yuri Wolf: Visualization, manuscript writing and editing; Eugene Koonin: Manuscript writing, reviewing and editing,
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Mohanraju P, Makarova KS, Zetsche B, Zhang F, Koonin EV, van der Oost J. Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science. 2016;353:aad5147. [DOI] [PubMed] [Google Scholar]
- [2].Hille F, Richter H, Wong SP, Bratovic M, Ressel S, Charpentier E. The Biology of CRISPR-Cas: Backward and Forward. Cell. 2018;172:1239–59. [DOI] [PubMed] [Google Scholar]
- [3].Makarova KS, Wolf YI, Iranzo J, Shmakov SA, Alkhnbashi OS, Brouns SJJ, et al. Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol. 2020;18:67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].McGinn J, Marraffini LA. Molecular mechanisms of CRISPR-Cas spacer acquisition. Nat Rev Microbiol. 2019;17:7–12. [DOI] [PubMed] [Google Scholar]
- [5].Hochstrasser ML, Doudna JA. Cutting it close: CRISPR-associated endoribonuclease structure and function. Trends Biochem Sci. 2015;40:58–66. [DOI] [PubMed] [Google Scholar]
- [6].Liao C, Beisel CL. The tracrRNA in CRISPR Biology and Technologies. Annu Rev Genet. 2021;55:161–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Liu TY, Doudna JA. Chemistry of Class 1 CRISPR-Cas effectors: Binding, editing, and regulation. J Biol Chem. 2020;295:14473–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Shmakov SA, Sitnik V, Makarova KS, Wolf YI, Severinov KV, Koonin EV. The CRISPR Spacer Space Is Dominated by Sequences from Species-Specific Mobilomes. mBio. 2017;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Davidson AR, Lu WT, Stanley SY, Wang J, Mejdani M, Trost CN, et al. Anti-CRISPRs: Protein Inhibitors of CRISPR-Cas Systems. Annu Rev Biochem. 2020;89:309–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Stanley SY, Maxwell KL. Phage-Encoded Anti-CRISPR Defenses. Annu Rev Genet. 2018;52:445–64. [DOI] [PubMed] [Google Scholar]
- [11].Peng X, Mayo-Munoz D, Bhoobalan-Chitty Y, Martinez-Alvarez L. Anti-CRISPR Proteins in Archaea. Trends Microbiol. 2020;28:913–21. [DOI] [PubMed] [Google Scholar]
- [12].Malone LM, Birkholz N, Fineran PC. Conquering CRISPR: how phages overcome bacterial adaptive immunity. Curr Opin Biotechnol. 2021;68:30–6. [DOI] [PubMed] [Google Scholar]
- [13].Bondy-Denomy J, Pawluk A, Maxwell KL, Davidson AR. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature. 2013;493:429–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Pawluk A, Staals RH, Taylor C, Watson BN, Saha S, Fineran PC, et al. Inactivation of CRISPR-Cas systems by anti-CRISPR proteins in diverse bacterial species. Nat Microbiol. 2016;1:16085. [DOI] [PubMed] [Google Scholar]
- [15].Stanley SY, Borges AL, Chen KH, Swaney DL, Krogan NJ, Bondy-Denomy J, et al. Anti-CRISPR-Associated Proteins Are Crucial Repressors of Anti-CRISPR Transcription. Cell. 2019;178:1452–64 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pinilla-Redondo R, Shehreen S, Marino ND, Fagerlund RD, Brown CM, Sorensen SJ, et al. Discovery of multiple anti-CRISPRs highlights anti-defense gene clustering in mobile genetic elements. Nat Commun. 2020;11:5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rauch BJ, Silvis MR, Hultquist JF, Waters CS, McGregor MJ, Krogan NJ, et al. Inhibition of CRISPR-Cas9 with Bacteriophage Proteins. Cell. 2017;168:150–8 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bondy-Denomy J, Davidson AR, Doudna JA, Fineran PC, Maxwell KL, Moineau S, et al. A Unified Resource for Tracking Anti-CRISPR Names. CRISPR J. 2018;1:304–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Pawluk A, Davidson AR, Maxwell KL. Anti-CRISPR: discovery, mechanism and function. Nat Rev Microbiol. 2018;16:12–7. [DOI] [PubMed] [Google Scholar]
- [20].Maxwell KL. The Anti-CRISPR Story: A Battle for Survival. Mol Cell. 2017;68:8–14. [DOI] [PubMed] [Google Scholar]
- [21].Dong C, Hao GF, Hua HL, Liu S, Labena AA, Chai G, et al. Anti-CRISPRdb: a comprehensive online resource for anti-CRISPR proteins. Nucleic Acids Res. 2018;46:D393–D8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yi H, Huang L, Yang B, Gomez J, Zhang H, Yin Y. AcrFinder: genome mining anti-CRISPR operons in prokaryotes and their viruses. Nucleic Acids Res. 2020;48:W358–W65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gussow AB, Park AE, Borges AL, Shmakov SA, Makarova KS, Wolf YI, et al. Machine-learning approach expands the repertoire of anti-CRISPR protein families. Nat Commun. 2020;11:3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wang J, Dai W, Li J, Xie R, Dunstan RA, Stubenrauch C, et al. PaCRISPR: a server for predicting and visualizing anti-CRISPR proteins. Nucleic Acids Res. 2020;48:W348–W57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Eitzinger S, Asif A, Watters KE, Iavarone AT, Knott GJ, Doudna JA, et al. Machine learning predicts new anti-CRISPR proteins. Nucleic Acids Res. 2020;48:4698–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wandera KG, Alkhnbashi OS, Bassett HVI, Mitrofanov A, Hauns S, Migur A, et al. Anti-CRISPR prediction using deep learning reveals an inhibitor of Cas13b nucleases. Mol Cell. 2022;82:2714–26 e4. [DOI] [PubMed] [Google Scholar]
- [27].Marshall R, Maxwell CS, Collins SP, Jacobsen T, Luo ML, Begemann MB, et al. Rapid and Scalable Characterization of CRISPR Technologies Using an E. coli Cell-Free Transcription-Translation System. Mol Cell. 2018;69:146–57 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Watters KE, Fellmann C, Bai HB, Ren SM, Doudna JA. Systematic discovery of natural CRISPR-Cas12a inhibitors. Science. 2018;362:236–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Forsberg KJ, Bhatt IV, Schmidtke DT, Javanmardi K, Dillard KE, Stoddard BL, et al. Functional metagenomics-guided discovery of potent Cas9 inhibitors in the human microbiome. Elife. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Uribe RV, van der Helm E, Misiakou MA, Lee SW, Kol S, Sommer MOA. Discovery and Characterization of Cas9 Inhibitors Disseminated across Seven Bacterial Phyla. Cell Host Microbe. 2019;25:233–41 e5. [DOI] [PubMed] [Google Scholar]
- [31].Jia N, Patel DJ. Structure-based functional mechanisms and biotechnology applications of anti-CRISPR proteins. Nat Rev Mol Cell Biol. 2021;22:563–79. [DOI] [PubMed] [Google Scholar]
- [32].Zhang Y, Marchisio MA. Type II anti-CRISPR proteins as a new tool for synthetic biology. RNA Biol. 2021;18:1085–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Calvache C, Vazquez-Vilar M, Selma S, Uranga M, Fernandez-Del-Carmen A, Daros JA, et al. Strong and tunable anti-CRISPR/Cas activities in plants. Plant Biotechnol J. 2022;20:399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Marino ND, Pinilla-Redondo R, Csorgo B, Bondy-Denomy J. Anti-CRISPR protein applications: natural brakes for CRISPR-Cas technologies. Nat Methods. 2020;17:471–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].He F, Bhoobalan-Chitty Y, Van LB, Kjeldsen AL, Dedola M, Makarova KS, et al. Anti-CRISPR proteins encoded by archaeal lytic viruses inhibit subtype I-D immunity. Nat Microbiol. 2018;3:461–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hynes AP, Rousseau GM, Agudelo D, Goulet A, Amigues B, Loehr J, et al. Widespread anti-CRISPR proteins in virulent bacteriophages inhibit a range of Cas9 proteins. Nat Commun. 2018;9:2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Meeske AJ, Jia N, Cassel AK, Kozlova A, Liao J, Wiedmann M, et al. A phage-encoded anti-CRISPR enables complete evasion of type VI-A CRISPR-Cas immunity. Science. 2020;369:54–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bautista MA, Black JA, Youngblut ND, Whitaker RJ. Differentiation and Structure in Sulfolobus islandicus Rod-Shaped Virus Populations. Viruses. 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bhoobalan-Chitty Y, Johansen TB, Di Cianni N, Peng X. Inhibition of Type III CRISPR-Cas Immunity by an Archaeal Virus-Encoded Anti-CRISPR Protein. Cell. 2019;179:448–58 e11. [DOI] [PubMed] [Google Scholar]
- [40].Okutan E, Deng L, Mirlashari S, Uldahl K, Halim M, Liu C, et al. Novel insights into gene regulation of the rudivirus SIRV2 infecting Sulfolobus cells. RNA Biol. 2013;10:875–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Jacob F, Monod J. Genetic regulatory mechanisms in the synthesis of proteins. J Mol Biol. 1961;3:318–56. [DOI] [PubMed] [Google Scholar]
- [42].Galperin MY, Koonin EV. Who’s your neighbor? New computational approaches for functional genomics. Nat Biotechnol. 2000;18:609–13. [DOI] [PubMed] [Google Scholar]
- [43].Aravind L Guilt by association: contextual information in genome analysis. Genome Res. 2000;10:1074–7. [DOI] [PubMed] [Google Scholar]
- [44].Hanson AD, Pribat A, Waller JC, de Crecy-Lagard V. ‘Unknown’ proteins and ‘orphan’ enzymes: the missing half of the engineering parts list--and how to find it. Biochem J. 2009;425:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Marino ND, Zhang JY, Borges AL, Sousa AA, Leon LM, Rauch BJ, et al. Discovery of widespread type I and type V CRISPR-Cas inhibitors. Science. 2018;362:240–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Watters KE, Shivram H, Fellmann C, Lew RJ, McMahon B, Doudna JA. Potent CRISPR-Cas9 inhibitors from Staphylococcus genomes. Proc Natl Acad Sci U S A. 2020;117:6531–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Song G, Zhang F, Tian C, Gao X, Zhu X, Fan D, et al. Discovery of potent and versatile CRISPR-Cas9 inhibitors engineered for chemically controllable genome editing. Nucleic Acids Res. 2022;50:2836–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yin Y, Yang B, Entwistle S. Bioinformatics Identification of Anti-CRISPR Loci by Using Homology, Guilt-by-Association, and CRISPR Self-Targeting Spacer Approaches. mSystems. 2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Nobrega FL, Walinga H, Dutilh BE, Brouns SJJ. Prophages are associated with extensive CRISPR-Cas auto-immunity. Nucleic Acids Res. 2020;48:12074–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Jensen RB, Gerdes K. Programmed cell death in bacteria: proteic plasmid stabilization systems. Mol Microbiol. 1995;17:205–10. [DOI] [PubMed] [Google Scholar]
- [51].Huang L, Yang B, Yi H, Asif A, Wang J, Lithgow T, et al. AcrDB: a database of anti-CRISPR operons in prokaryotes and viruses. Nucleic Acids Res. 2021;49:D622–D9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wang J, Dai W, Li J, Li Q, Xie R, Zhang Y, et al. AcrHub: an integrative hub for investigating, predicting and mapping anti-CRISPR proteins. Nucleic Acids Res. 2021;49:D630–D8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Koonin EV, Makarova KS, Wolf YI, Krupovic M. Evolutionary entanglement of mobile genetic elements and host defence systems: guns for hire. Nat Rev Genet. 2020;21:119–31. [DOI] [PubMed] [Google Scholar]
- [54].Koonin EV, Dolja VV, Krupovic M. The logic of virus evolution. Cell Host Microbe. 2022;30:917–29. [DOI] [PubMed] [Google Scholar]
- [55].Hudaiberdiev S, Shmakov S, Wolf YI, Terns MP, Makarova KS, Koonin EV. Phylogenomics of Cas4 family nucleases. BMC Evol Biol. 2017;17:232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zhang Z, Pan S, Liu T, Li Y, Peng N. Cas4 Nucleases Can Effect Specific Integration of CRISPR Spacers. J Bacteriol. 2019;201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Athukoralage JS, McMahon SA, Zhang C, Gruschow S, Graham S, Krupovic M, et al. An anti-CRISPR viral ring nuclease subverts type III CRISPR immunity. Nature. 2020;577:572–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Makarova KS, Timinskas A, Wolf YI, Gussow AB, Siksnys V, Venclovas C, et al. Evolutionary and functional classification of the CARF domain superfamily, key sensors in prokaryotic antivirus defense. Nucleic Acids Res. 2020;48:8828–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Rollins MF, Chowdhury S, Carter J, Golden SM, Miettinen HM, Santiago-Frangos A, et al. Structure Reveals a Mechanism of CRISPR-RNA-Guided Nuclease Recruitment and Anti-CRISPR Viral Mimicry. Mol Cell. 2019;74:132–42 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Liu L, Yin M, Wang M, Wang Y. Phage AcrIIA2 DNA Mimicry: Structural Basis of the CRISPR and Anti-CRISPR Arms Race. Mol Cell. 2019;73:611–20 e3. [DOI] [PubMed] [Google Scholar]
- [61].Wang H, Gao T, Zhou Y, Ren J, Guo J, Zeng J, et al. Mechanistic insights into the inhibition of the CRISPR-Cas surveillance complex by anti-CRISPR protein AcrIF13. J Biol Chem. 2022;298:101636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Guo TW, Bartesaghi A, Yang H, Falconieri V, Rao P, Merk A, et al. Cryo-EM Structures Reveal Mechanism and Inhibition of DNA Targeting by a CRISPR-Cas Surveillance Complex. Cell. 2017;171:414–26 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Chowdhury S, Carter J, Rollins MF, Golden SM, Jackson RN, Hoffmann C, et al. Structure Reveals Mechanisms of Viral Suppressors that Intercept a CRISPR RNA-Guided Surveillance Complex. Cell. 2017;169:47–57 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Kim I, Koo J, An SY, Hong S, Ka D, Kim EH, et al. Structural and mechanistic insights into the CRISPR inhibition of AcrIF7. Nucleic Acids Res. 2020;48:9959–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Shin J, Jiang F, Liu JJ, Bray NL, Rauch BJ, Baik SH, et al. Disabling Cas9 by an anti-CRISPR DNA mimic. Sci Adv. 2017;3:e1701620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Pearce R, Li Y, Omenn GS, Zhang Y. Fast and accurate Ab Initio Protein structure prediction using deep learning potentials. PLoS Comput Biol. 2022;18:e1010539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Wang J, Lisanza S, Juergens D, Tischer D, Watson JL, Castro KM, et al. Scaffolding protein functional sites using deep learning. Science. 2022;377:387–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Faure G, Shmakov SA, Yan WX, Cheng DR, Scott DA, Peters JE, et al. CRISPR-Cas in mobile genetic elements: counter-defence and beyond. Nat Rev Microbiol. 2019;17:513–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Yang L, Zhang L, Yin P, Ding H, Xiao Y, Zeng J, et al. Insights into the inhibition of type I-F CRISPR-Cas system by a multifunctional anti-CRISPR protein AcrIF24. Nat Commun. 2022;13:1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Hwang S, Pan C, Garcia B, Davidson AR, Moraes TF, Maxwell KL. Structural and Mechanistic Insight into CRISPR-Cas9 Inhibition by Anti-CRISPR Protein AcrIIC4Hpa. J Mol Biol. 2022;434:167420. [DOI] [PubMed] [Google Scholar]
- [72].Zhu Y, Gao A, Zhan Q, Wang Y, Feng H, Liu S, et al. Diverse Mechanisms of CRISPR-Cas9 Inhibition by Type IIC Anti-CRISPR Proteins. Mol Cell. 2019;74:296–309 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Dong C, Wang X, Ma C, Zeng Z, Pu DK, Liu S, et al. Anti-CRISPRdb v2.2: an online repository of anti-CRISPR proteins including information on inhibitory mechanisms, activities and neighbors of curated anti-CRISPR proteins. Database (Oxford). 2022;2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Dong L, Guan X, Li N, Zhang F, Zhu Y, Ren K, et al. An anti-CRISPR protein disables type V Cas12a by acetylation. Nat Struct Mol Biol. 2019;26:308–14. [DOI] [PubMed] [Google Scholar]
- [75].Forsberg KJ, Schmidtke DT, Werther R, Uribe RV, Hausman D, Sommer MOA, et al. The novel anti-CRISPR AcrIIA22 relieves DNA torsion in target plasmids and impairs SpyCas9 activity. PLoS Biol. 2021;19:e3001428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Mejdani M, Pawluk A, Maxwell KL, Davidson AR. Anti-CRISPR AcrIE2 Binds the Type I-E CRISPR-Cas Complex But Does Not Block DNA Binding. J Mol Biol. 2021;433:166759. [DOI] [PubMed] [Google Scholar]
- [77].Johnson MC, Hille LT, Kleinstiver BP, Meeske AJ, Bondy-Denomy J. Lack of Cas13a inhibition by anti-CRISPR proteins from Leptotrichia prophages. Mol Cell. 2022;82:2161–6 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Tesson F, Herve A, Mordret E, Touchon M, d’Humieres C, Cury J, et al. Systematic and quantitative view of the antiviral arsenal of prokaryotes. Nat Commun. 2022;13:2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Hampton HG, Watson BNJ, Fineran PC. The arms race between bacteria and their phage foes. Nature. 2020;577:327–36. [DOI] [PubMed] [Google Scholar]
- [80].Zhu L, Wang X, Li F, Song J. PreAcrs: a machine learning framework for identifying anti-CRISPR proteins. BMC Bioinformatics. 2022;23:444. [DOI] [PMC free article] [PubMed] [Google Scholar]