

Abstract
Non-precious metal heterogeneous catalysts composed of first-row transition metals incorporated into nitrogen-doped carbon matrices (M-N-Cs) have been studied for decades as leading alternatives to Pt for the electrocatalytic O2 reduction reaction (ORR). More recently, similar M-N-C catalysts have been shown to catalyze the aerobic oxidation of organic molecules. This Focus Review highlights mechanistic similarities and distinctions between these two reaction classes and then surveys the aerobic oxidation reactions catalyzed by M-N-Cs. As the active-site structures and kinetic properties of M-N-C aerobic oxidation catalysts have not been extensively studied, the array of tools and methods used to characterize ORR catalysts are presented with the goal of supporting further advances in the field of aerobic oxidation.
Graphical Abstract
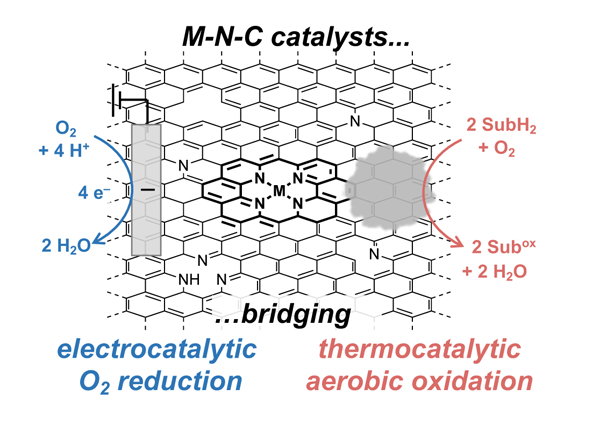
1. INTRODUCTION
Materials composed of metal ions integrated within nitrogen-doped carbon (M-N-Cs) are a class of "single-atom" catalysts1,2 that have received extensive attention for the electrochemical oxygen reduction reaction (ORR) in fuel cells.3–13 These catalysts often feature first-row transition-metal ions, such as iron and cobalt, and offer a compelling alternative to catalysts derived from platinum or other precious metals. Recent studies show that M-N-C catalysts are effective for many other applications, including CO2 electroreduction,14,15 N2 electroreduction,16 and a variety of reductions17–20 and oxidations21 of organic molecules. The electrochemical ORR is closely related to aerobic oxidation reactions, as both reaction classes lead to the net reduction of O2 to water using electrons and protons from a complementary electrochemical oxidation reaction (ORR) or from an organic molecule (aerobic oxidation) (Figure 1). The present Focus Review explores this relationship, with the premise that advances in M-N-C catalysts for electrochemical ORR provide a foundation for future advances in aerobic oxidation catalysis.
Figure 1.
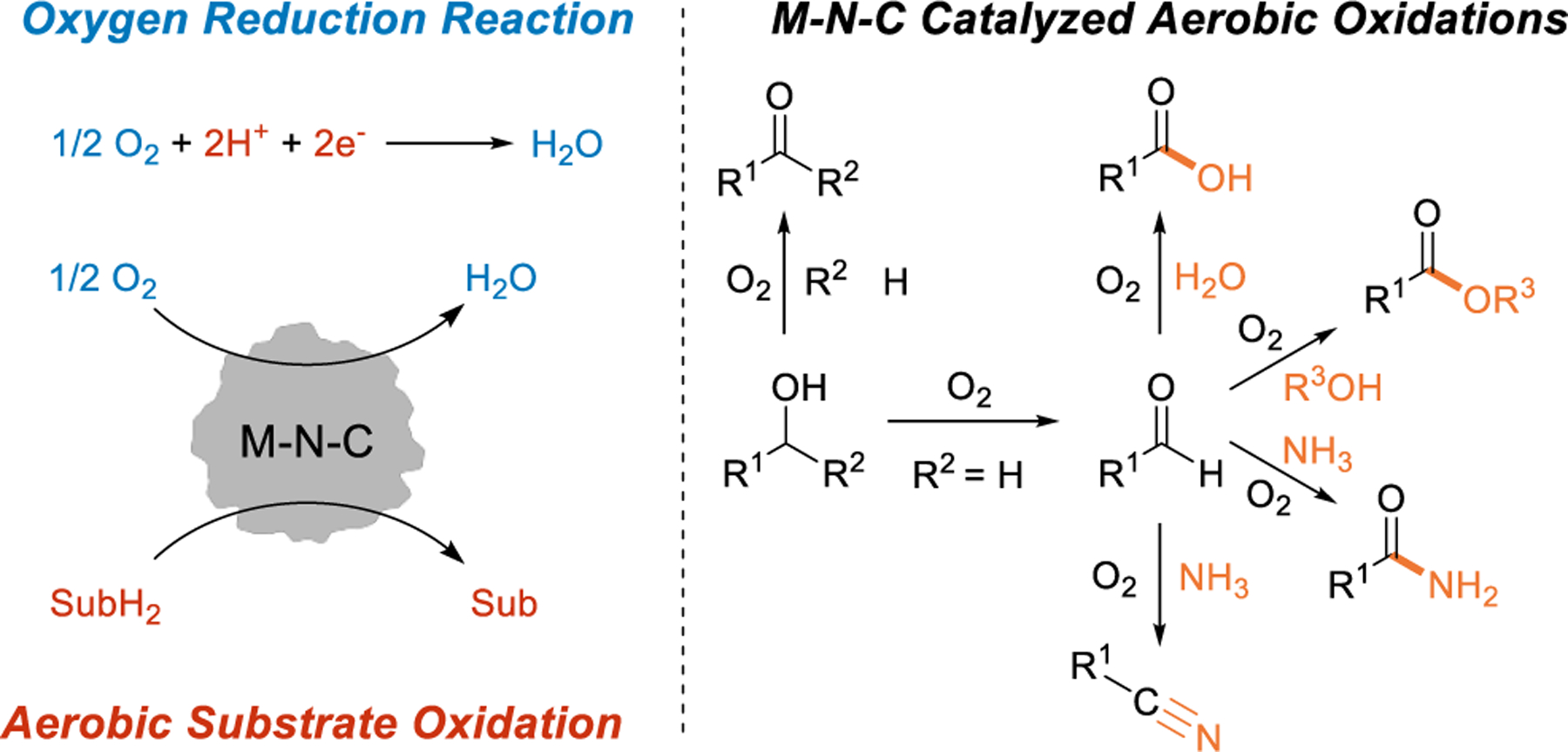
O2 accepts protons and electrons in the oxygen reduction reaction and aerobic oxidation catalysis (left). In the latter case, the protons and electrons derive from the bonds of molecules (SubH2), such as the O–H and C–H bonds of alcohols (right).
Aerobic oxidation reactions using M-N-C catalysts include the conversion of alcohols to aldehydes and ketones and oxidative coupling of primary alcohols with various partners to yield carboxylic acids, esters, and nitriles (Figure 1).21 Many of these transformations have been developed by using empirical approaches to identify the preferred catalyst and optimize the reactions. The applications are still somewhat limited relative to reactions that employ noble metal heterogeneous catalysts22–25 and homogeneous catalysts,26–30 however, at least partly reflecting the common requirement for high reaction temperatures, high O2 pressures, and/or long reaction times. A deeper understanding of the active sites in the M-N-C catalysts and the associated reaction mechanisms could play an important role in addressing these limitations. Uncovering the relationships between active-site structure and catalyst performance will require advances in structural characterization of the catalysts and quantitative studies of reaction kinetics. To date, few M-N-C-catalyzed aerobic oxidation reactions have been analyzed in this manner.
M-N-C catalysts are prepared by various pyrolysis-based synthetic routes (Figure 2a). These methods are reviewed in detail elsewhere,9,11 but the following overview provides important context for the content in this article. In most cases, a precursor mixture containing sources of the M, N, and C components is treated at temperatures of ~600–1000 °C under a flow of either an inert gas, such as Ar or N2, or a reactive gas, such as NH3 or H2. Some components of the precursor mixtures decompose and/or volatilize during this pyrolysis treatment, while others form carbon-based solids with varying degrees of graphitization and nitrogen incorporation. The metal ions either integrate within specific mononuclear binding sites in the nitrogen-doped carbon support31 or aggregate to form nanoparticles (e.g., metallic Fe, FeOx, FexCy, FexNy, etc.). The resulting pyrolyzed M-N-C solids feature mesoporous and/or microporous voids that regulate substrate access to active sites within them (Figure 2b). The structures of precursors and pyrolysis conditions, including the temperature, duration, spatial concentration gradients, and gas environment, influence the physicochemical properties of the catalyst, including the prevalence of N dopants and structural defects within the carbon, the speciation of metals as nanoparticles or single atoms, and the porosity of the material. Post-synthetic treatments are sometimes carried out with the intent to modify these properties. For example, metal aggregates may be removed by acid washing. The widely used pyrolysis-based synthesis routes may be grouped into four categories, defined according to the choice of precursors and/or post-synthetic treatments (Figure 2a): (i) carbon-supported molecular complexes (e.g., Co(porphyrin)/C3, Fe(Phen)3/C32), (ii) polymeric precursors (e.g., polyacrylonitrile,33 polyaniline34), (iii) unsupported organic mixtures (e.g., glucose, metal, and (NH3OH)Cl35), (iv) sacrificial supports36 that template mesopores after they are leached from the M-N-C (e.g., Fe-poly(ethyleneimine)-SiO2 followed by HF etch after pyrolysis37), and (v) decomposition of metal–organic frameworks (e.g., Fe(Phen)-impregnated ZIF-838, Fe-doped ZIF-839). The structural diversity of M-N-Cs that results from these synthetic routes requires further structural characterization to link the local structures of active centers to their kinetic function.
Figure 2.
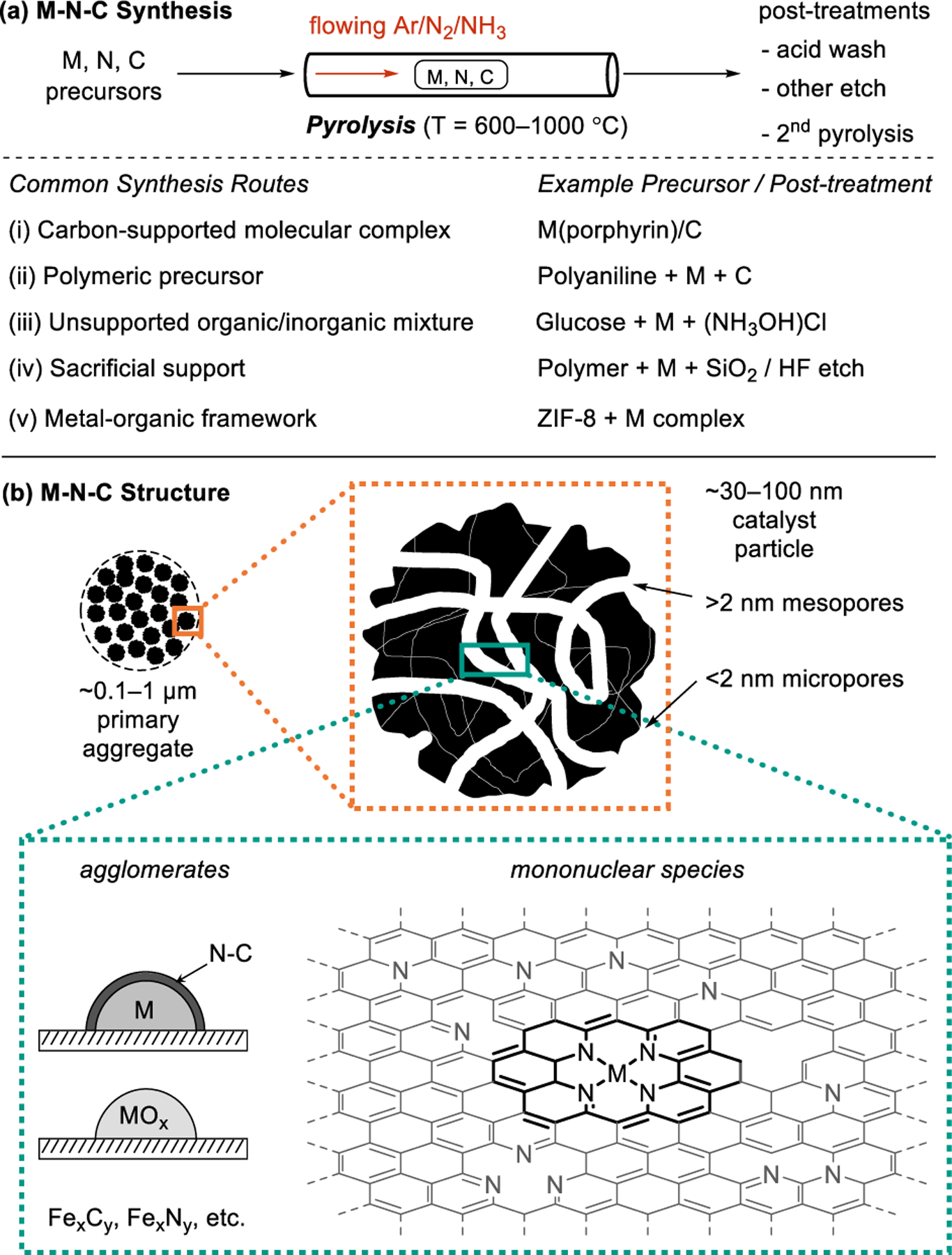
Overview of (a) synthetic methods and (b) structural features of M-N-C catalysts.
The thesis of this Focus Review is that methods and tools from the field of ORR electrocatalysis can transform the area of aerobic oxidation catalysis by M-N-Cs. The discussion begins by highlighting the mechanistic relationships between aerobic oxidation catalysis and ORR electrocatalysis in Section 2. Section 3 provides a comprehensive summary of the scope of aerobic oxidation reactions catalyzed by earth-abundant M-N-Cs. The intrinsic kinetic behavior, quantities of and structures of active centers responsible for aerobic oxidations are less well understood than they are in the ORR field. Therefore, Sections 4 and 5 discuss the approaches developed within the ORR field to assess reactivity and characterize the catalyst structures and the identity of M-N-C active sites. This content complements other reviews that focus on M-N-C electrocatalysts,6–13 the synthesis and characterization of M-N-Cs,9,11,40 and other applications of single-atom catalysts.1,41–43 Development of earth-abundant M-N-C catalysts for transformations of organic molecules is poised to benefit from cross-disciplinary efforts that combine organic chemistry, materials science, electrochemistry, and traditional heterogeneous catalysis approaches.
2. BRIDGING CATALYTIC AEROBIC OXIDATION AND ELECTROCHEMICAL ORR
Electrocatalytic O2 reduction and aerobic oxidation catalysis share several common features (Figure 3a). Both ORR and aerobic oxidations require O2 binding and reduction at the catalyst surface using protons and electrons either from an electrode and membrane (electrochemical ORR) or from the bonds of an organic molecule (aerobic oxidation). Two mechanistic pathways could be involved in thermochemical aerobic oxidation of organic molecules: (i) direct inner-sphere reaction (ISR) between the organic molecule and O2 (Figure 3b-i) or (ii) independent redox half-reactions (IHR) involving oxidation of the organic molecule and reduction of O2 (Figure 3b-ii). ISR mechanisms involve co-location of O2 and the substrate molecule, with direct transfer of protons and electrons (potentially via hydrogen-atom or hydride transfer) from the substrate to O2. ISR pathways of this type have precedent with molecular catalysts, including metal macrocycles that resemble the active sites of M-N-Cs.44–47 In contrast, IHR pathways feature separate elementary steps for O2 reduction and organic substrate oxidation. Thermochemical alcohol oxidations on noble metal catalysts have been proposed to feature independent half reactions involving surface hydrogen atoms,22 while more recent work has highlighted complementary electrochemical pathways involving independent transfer of electrons and protons in these steps.48–52
Figure 3.
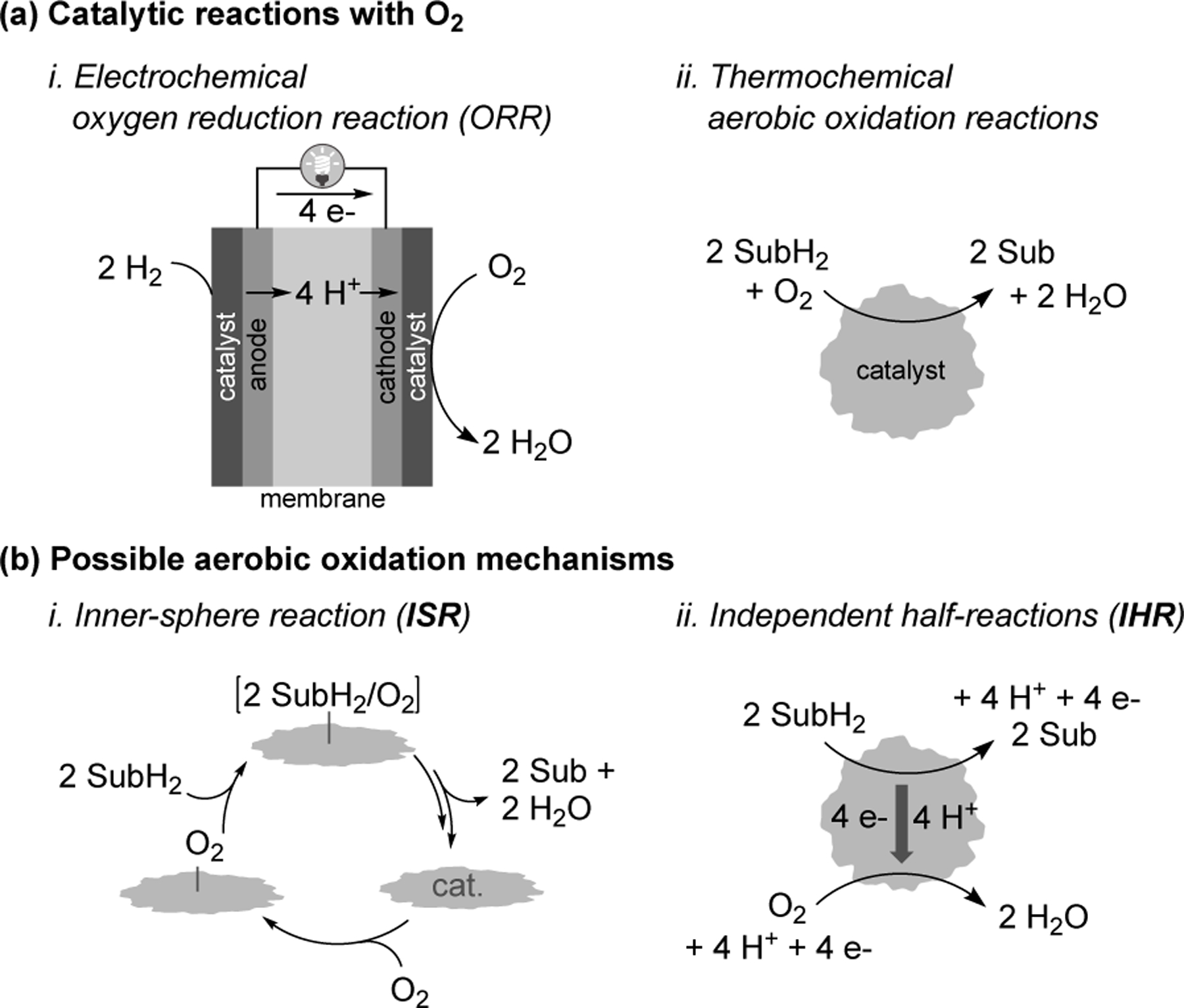
Representation of electrochemical and thermochemical reactions involving O2 reduction. (a) Relevance of O2 reactivity to fuel cells and aerobic oxidation which feature different sources of protons and electrons: H+/e− or chemical bonds in SubH2, and (b) two potential reaction pathways for thermochemical reactivity of O2 in aerobic oxidations. Adapted from Ref. 53. Copyright 2022 American Chemical Society.
Two recent studies investigating aerobic oxidation of a hydroquinone (HQ) on Fe- and Co-N-C under aqueous acidic conditions have found that M-N-Cs could support either ISR or IHR mechanisms (Figure 4a).53,54 One study measured reaction rates in semi-batch reactors containing M-N-C particles suspended as a slurry, whereas the second study measured reaction rates in a semi-batch reactor containing the M-N-C particles within a Nafion-bound film in contact with a glassy carbon electrode, with no applied potential (maintained at open circuit in order to measure the catalyst potential). Despite their similarities, the two systems exhibit different kinetic parameters, including rates, O2 and HQ reaction orders, HQ adsorption equilibrium constants, and linear free energy relationships with respect to HQ redox potential (Figure 4b). These differences correlate with different reaction mechanisms: the slurry-phase M-N-C mediates an ISR pathway, while the Nafion-bound M-N-C mediates an IHR pathway. The data indicate that slurry-phase conditions result in a high surface coverage of HQ, which supports direct hydrogen-atom transfer from HQ to O2 (i.e., ISR pathway, Figure 4c).53 In contrast, the data obtained with the Nafion-bound M-N-C indicate that O2 and HQ undergo electrochemical redox at distinct surface sites (i.e., IHR pathway, Figure 4d).54 It is possible that these differences may be reconciled by the different surface microenvironments of the two systems. For example, the ionic microenvironment of Nafion could account for a lower HQ adsorption equilibrium constant that disfavors the ISR mechanism without interfering with the IHR pathway. The generality of these observations for different organic substrates, such as those outlined in Section 3, remains to be tested.
Figure 4.
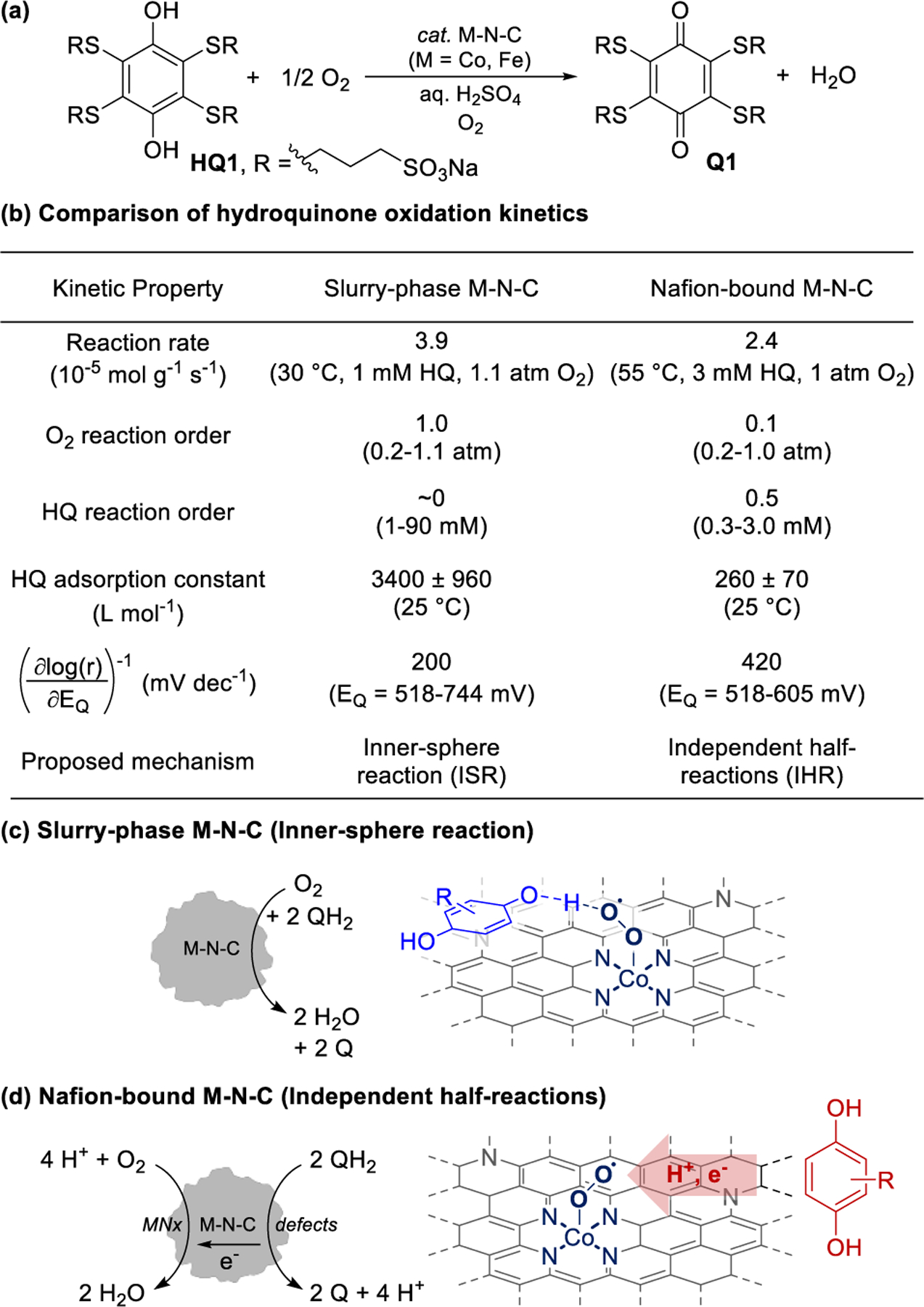
Inner-sphere and independent half-reaction modes of aerobic oxidation reactivity exhibited by hydroquinone substrates. Aqueous-phase hydroquinone oxidation (a) conditions and (b) kinetic parameters. (c) ISR reactivity of slurry-phase M-N-C and (d) IHR reactivity of Nafion-bound M-N-C. Adapted from Refs. 53 and 54. Copyright 2022 American Chemical Society.
Aerobic oxidations that operate by IHR mechanisms leverage the conductive nature of the catalytic solid to couple redox half-reactions in a manner akin to electrocatalysis. In such cases, it is reasonable to expect that these catalysts could serve as electrocatalysts for each of the two independent half-reactions. The degree of electronic coupling of MN4 active centers to the surrounding graphitic carbon structure has been proposed to play a key role in ORR electrocatalysis. For example, Mukerjee and coworkers55 found that a suite of Fe-N-Cs synthesized with different underlying carbon support structures feature ORR rates (0.8 VRHE) that correlate with the conductivity of the carbon estimated by the full-width at half maximum of the C 1s peak in the X-ray photoelectron spectra. In a complementary study, Surendranath and coworkers demonstrated the influence of electronic coupling between a molecular CoN4 catalyst and the support.56,57 In principle, electrochemical elementary steps could be paired with inner-sphere steps, thus impacting ISR mechanisms. To our knowledge, such pathways have not been demonstrated.
Collectively, the data outlined in this section highlight the conceptual relationship between M-N-C catalyzed aerobic oxidation and electrochemical ORR. The following sections expand on this relationship, showing how structural and mechanistic approaches now common in the field of ORR could benefit the field of aerobic oxidation catalysis.
3. M-N-C CATALYZED AEROBIC OXIDATION REACTIONS
This section outlines the scope of aerobic oxidations catalyzed by M-N-C materials, starting with alcohol oxidation and oxidative coupling reactions and then addressing other oxidative dehydrogenation and coupling reactions. For each reaction class, we document the scope of reactivity, summarize any catalyst structural characterization provided, and, to the extent possible, assess relevant reactivity metrics and kinetic and mechanistic studies. Catalyst source materials and salient details of the synthetic methods will be presented in the text; however, catalysts will then be abbreviated with the notation “M-N-C(i/ii/iii/iv/v)” in figures, with the value i–v referring to the relevant synthetic approach noted in Figure 2a. References to catalyst loadings in the different reactions reflect common usage by organic chemists, where "mol%" refers of total metal loading with respect to limiting reagent or substrate. It is acknowledged that this value represents an upper bound, as the fraction of catalytically relevant active centers per total metal is almost always less than unity (see Section 4 and Table 1 in Section 4.2.2). More rigorous active site densities have not been quantified for most of the systems outlined in this section.
Table 1.
Representative active site densities of Fe-N-C catalysts quantified by different methods.
| Catalyst | Bulk Fe content / wt% | Titrant | Site Densitya / μmol g−1 | FeNx/Fetotala |
|---|---|---|---|---|
| Fe0.5NC144,149 (v) | 1.5 | CO | 63 | 0.23 |
| NO | (31) 51 | (0.11) 0.19 | ||
| CN- | 56 | 0.21 | ||
| CNRS Fe-N-C138 (v) | 2.5 | CO | 96 | 0.22 |
| NO | 24 (40) | 0.05 (0.09) | ||
| PAJ Fe-N-C138 (iv) | 0.6 | CO | 33 | 0.31 |
| NO | 4 (7) | 0.04 (0.06) | ||
| UNM Fe-N-C138 (iv) | 0.8 | CO | 52 | 0.36 |
| NO | 10 (17) | 0.07 (0.12) | ||
| ICL Fe-N-C138 (ii) | 1.0 | CO | 37 | 0.20 |
| NO | 14 (24) | 0.08 (0.13) | ||
| FeNC-CVD-750154 (v) | 2.0 | NO | (102) 170 | (0.28) 0.46 |
| Fe-N-CΔ-DCDA145 (v) | 7.1 | CO | 130 | 0.10 |
| NO | 78 (130) | 0.06 (0.10) |
NO2− stripping quantities are listed for both 5 e− and 3 e− assumptions, in that order, with the value not reported by the authors enclosed in parentheses.
3.1. Alcohol Oxidation to Aldehydes and Ketones
Oxidation of alcohols to aldehydes and ketones over M-N-C catalysts has been demonstrated for benzylic and simple aliphatic substrates. Li and coworkers58 reported the oxidation of alcohols in 2016 over a Co-N-C(v) catalyst derived from the pyrolysis of a Co-carboxylate/triazine metal–organic framework. Substrates were oxidized under air in aqueous solvent without added base and required high catalyst loading (10 mol%), long reaction time (48 h), and high temperature (110 °C). Primary, secondary, and benzylic aldehydes and ketones were synthesized with GC-MS yields of 85–98%, whereas allylic and aliphatic ketones were afforded in 59–72% yields in 60 h reaction times (Figure 5a). A similar scope of reactivity has been achieved without added base using Co-N-C(i) catalysts synthesized with either 1-Butyl-3-methylimidazolium bromide59 or 1-methyl-3-cyanomethyl-1H-imidazolium chloride60 as additives. Xu and coworkers61 used a benzimidazole-derived Co-N-C(v) to expand the scope to include methoxy-functionalized benzylic substrates but required exogenous base. The broadest scope of aldehyde and ketone synthesis using exogenous base was reported in 2022 by Beller and coworkers62 over a Co-N-C(i) catalyst synthesized with piperazine and tartaric acid. This method required lower temperature (80 °C), catalyst loading (6.5 mol% Co), and reaction time (24 h) than base-free methodologies. Substituted aromatic and heterocyclic aldehydes and ketones containing a variety of pharmaceutically relevant functional groups could be synthesized with yields ≥75% (Figure 5b). The same catalyst was competent for a variety of other aerobic oxidations that further functionalize the aldehyde (vide infra). Base-free methodologies for aldehyde and ketone synthesis have yet to reach comparable scope or reactivity compared with those that use exogenous base.
Figure 5.
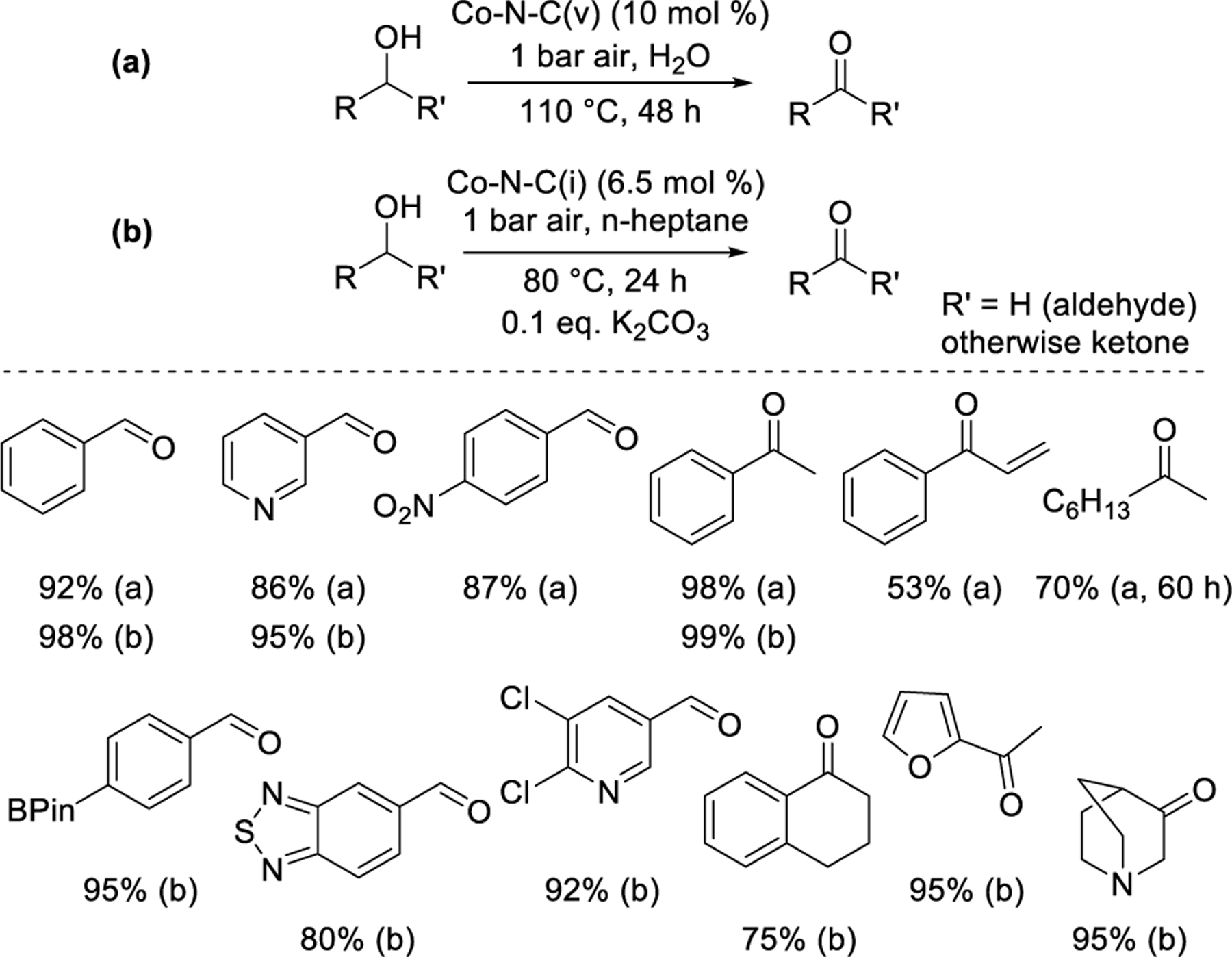
Aerobic oxidation of alcohols to aldehydes and ketones (a) without exogenous base,58 and (b) with exogenous base.62 Percentages reflect yield of the specified product.
Some structural information has been provided for M-N-C alcohol oxidation catalysts. Li and coworkers58 detected ~10 nm metallic Co nanoparticles using transmission electron microscopy (TEM) and X-ray diffraction (XRD), some of which were encapsulated within graphitic shells, whereas an N-free analog had particles ~100 nm in size and gave lower yields. Beller and coworkers62 similarly identified metallic Co nanoparticles encapsulated by N-doped graphitic shells and observed lower yields using a catalyst containing CoOx. These data point toward the importance of N-doping and Co but were not able to discriminate whether CoNx or Co aggregates were the active centers. The Co-N-C(v) catalyst studied by Li and coworkers58 showed qualitatively more types of basic sites than the N-free analog, inferred by the presence of additional peaks in its CO2 temperature-programmed desorption (TPD) profile. The authors speculated that these basic sites facilitated reactivity without added base. Quantitative characterizations of the active sites were not attempted in these studies. Davis and coworkers63 studied M-N-C(i) (M = Co, Cu) synthesized using 1,10-phenanthroline (Phen) as a nitrogen source that was proposed to have atomically dispersed MNx species on the basis of X-ray absorption spectroscopy (XAS) and scanning transmission electron microscopy (STEM) data. The rate of aqueous benzyl alcohol oxidation to benzaldehyde (per g, 80 °C, 0.05 M alcohol, 10 bar O2, 0.1 M NaOH) was unaffected by an acid washing treatment that removed nanoparticles from Co-N-C, suggesting that CoNx are the active sites for alcohol oxidation.
The kinetics of alcohol oxidation have been studied more broadly than most aerobic oxidations catalyzed by M-N-Cs. Because active-site densities have not been directly quantified, rates are typically reported and/or available only as quantities normalized with respect to the total M content. These values represent a lower bound for the turnover frequency (TOF) because they assume that all metal atoms are catalytically active. Davis and coworkers64 compared the rates of aqueous benzyl alcohol oxidation to benzaldehyde over M-N-C(iv) (M = Co, Cr, Fe, Ni, Cu). Normalized rate values (per total M, 80 °C, 0.05 M alcohol, 10 bar O2) were between 0.08–3.4×10−2 mol (mol M)−1 s−1 and were higher on Cu-N-C and Co-N-C than on the other catalysts by factors of ≥5 (Cu) and ≥2 (Co). Catalysts reused after 8 h of reaction had 40–90% lower rates, which could be recovered to different extents by treatment in flowing H2 at 300 °C for 3 h (Co = 74% recovery, Cu = 15% recovery, Fe ~ 100% recovery). The addition of 2,2,6,6-tetramethylpiperidine N-oxyl (TEMPO, 2 mol% relative to substrate) increased the rate over Fe-N-C by a factor of five. Zhang, Hu, and coworkers65 measured a similar normalized rate (3.5×10−2 mol (mol Co)−1 s−1) of benzyl alcohol oxidation to benzaldehyde under different conditions (90 °C, n-heptane solvent, 0.2 M alcohol, 1 bar O2) on a Co-N-C(iii) catalyst. Li et al.66 measured the rate of oxidation of cinnamyl alcohol to cinnamaldehyde in n-octane solvent (100 °C, 1 atm of O2) using a Cu-N-C(v) catalyst synthesized from a Cu-doped ZIF-8 MOF (ZIF = zeolitic imidazolate framework; MOF = metal organic framework). This corresponded to a normalized rate of 2 mol (mol Cu)−1 s−1. The different reaction conditions used in each of these studies together with the lack of active site quantitation prevent direct comparison of catalyst performance.
Mechanistic hypotheses for alcohol oxidation have been developed using kinetic isotope effect and reaction order measurements. Davis and coworkers64 reported H/D kinetic isotope effect values for aerobic oxidation of C6H5CD2OH substrates (80 °C, 0.05 M alcohol, 10 bar O2) of 4.8 and 2.2 for Fe-N-C(iv) and Cu-N-C(iv), respectively, to conclude that β-H elimination is a kinetically relevant step. The rate was zero-order in alcohol (0.025–0.1 M) on both Cu-N-C(iv) and Fe-N-C(iv). The rate was zero-order in O2 on Fe-N-C(iv) (3–20 bar), whereas the order in O2 transitioned from ~0.7 (3–10 bar) to zero-order at higher O2 pressures (10–20 bar) on Cu-N-C(iv). The authors concluded that the catalyst surface was saturated with both O2 and alcohol but did not propose a sequence of elementary steps to describe the mechanism. Zhang, Hu, and coworkers65 reported that the rate of aerobic benzyl alcohol oxidation (90 °C) on Co-N-C(iii) in heptane solvent was ~0.5-order in O2 (1–12 bar) and zero-order at higher O2 pressures (12–36 bar), consistent with the rate data of Davis and coworkers.64 In contrast, benzyl alcohol inhibited reaction rates (−0.3 order, 0.05–0.75 M, 2 bar O2; −0.5 order, 0.2–3.9 M, 12 bar O2), as did benzaldehyde (−0.4 order, 0.05–0.4 M, 0.01 M alcohol, 2 bar O2) and H2O (−0.5 order, 0.02–0.06 M, 0.01 M alcohol, 2 bar O2).65 The authors proposed a two-site mechanism, reflecting the ISR pathway in Figure 3b-i, wherein displacement of O2 at the metal center by competitive adsorption of benzyl alcohol leads to the observed inhibition.
3.2. Alcohol Oxidation to Carboxylic Acids
Beller and coworkers62 used H2O as a reactant and solvent to support oxidation of primary alcohols to the corresponding carboxylic acids using the same Co-N-C(i) catalyst noted above (cf. Figure 5b). More demanding reaction conditions were used, including higher temperature (110 °C), higher pressure (10 bar air), and 0.5 equivalents of KOH. A variety of substituted benzoic acids, including heterocycles, could be synthesized using this method (Figure 6). Rate data under well-defined conditions were not reported.
Figure 6.
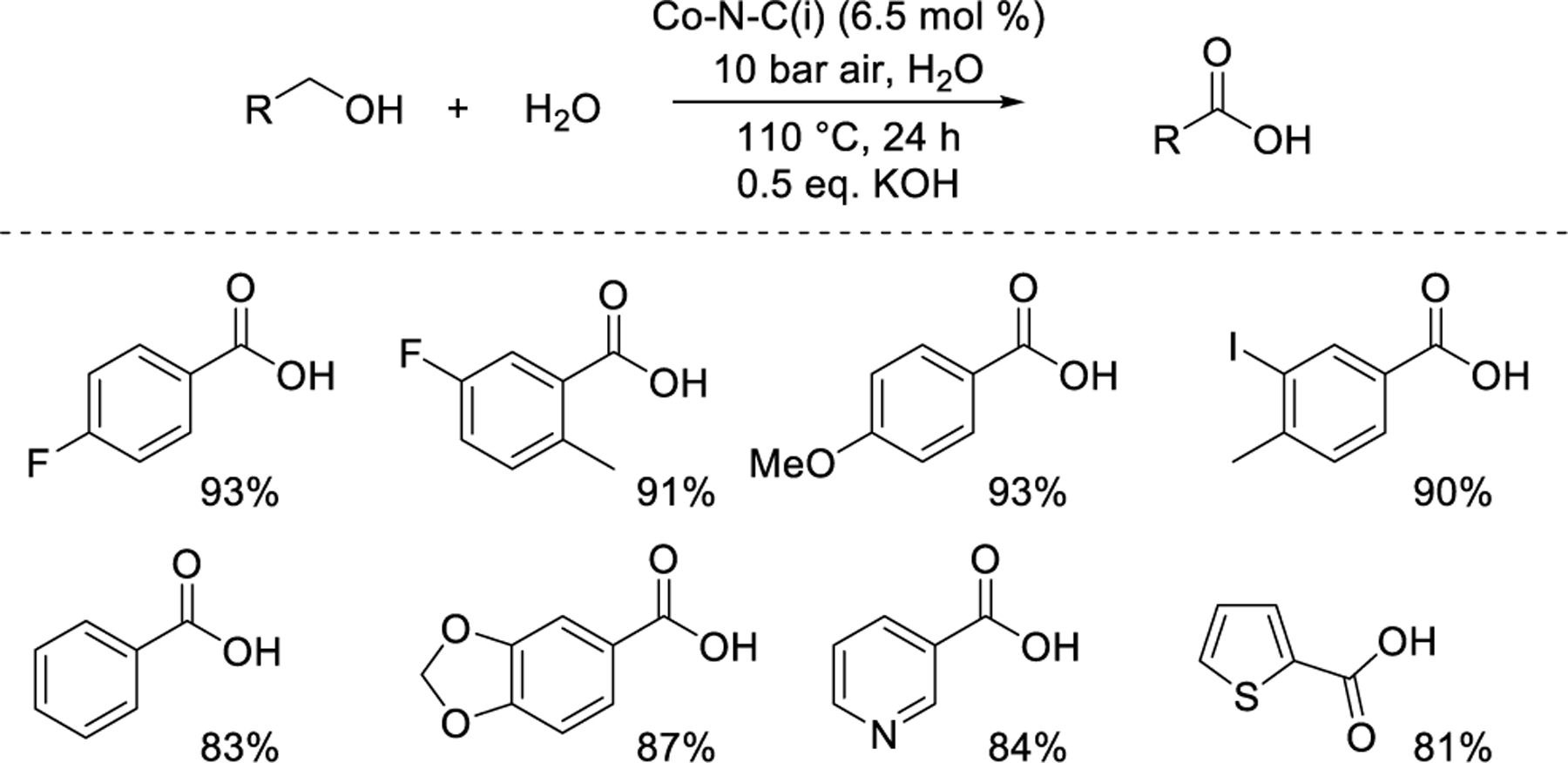
Oxidation of alcohols to carboxylic acids over M-N-C.62 Percentages reflect yield of the specified product.
3.3. Alcohol Oxidation to Esters
Aldehydes generated by alcohol oxidation can be oxidatively coupled with alcohols to generate ester products. Beller and coworkers67 reported alcohol esterification in 2013 using a Co-N-C(i) material synthesized using a carbon-supported Co(Phen)2 precursor. Benzylic, heterobenzylic, and allylic alcohols were aerobically cross esterified with methanol, which also served as the solvent. Substrates included those with functional groups such as halides, alkyl groups, ethers, and thioethers, and afforded methyl esters in yields of ≥80%. Longer-chain primary alcohol partners (C2–C6) were cross coupled with benzylic alcohols and homocoupled in n-heptane. In 2017, Huang and coworkers60 reported the methyl esterification of substituted benzyl alcohols using a Co-N-C(i) synthesized with a 1-methyl-3-cyanomethyl-1H-imidazolium chloride additive at lower temperatures (60 °C) but with a scope that lacked difficult substrates like S-containing molecules and coupling partners other than methanol. In 2022, Beller and coworkers62 reported ester synthesis over a Co-N-C(i) catalyst synthesized with piperazine and tartaric acid ligands with a scope similar to their original report67 but at lower temperature (55 °C), in air, and with lower base loading (0.1 eq., Figure 7). Other studies extended this method using similar M-N-C catalysts to synthesize lactones from diols68 and to oxidize biomass-derived furfural and 5-hydroxymethylfurfural.69,70,71 Reaction conditions were also developed to eliminate the use of exogenous base, albeit with more limited synthetic scope, by using catalysts such as Co-N-C(v) derived from the Co-imidazolate MOF ZIF-6768 and Co-N-C(i) using graphitic carbon nitride (g-C3N4) instead of carbon as a support for Co coordinated with 1,4-benzenedicarboxylic acid and triethylenediamine.72
Figure 7.
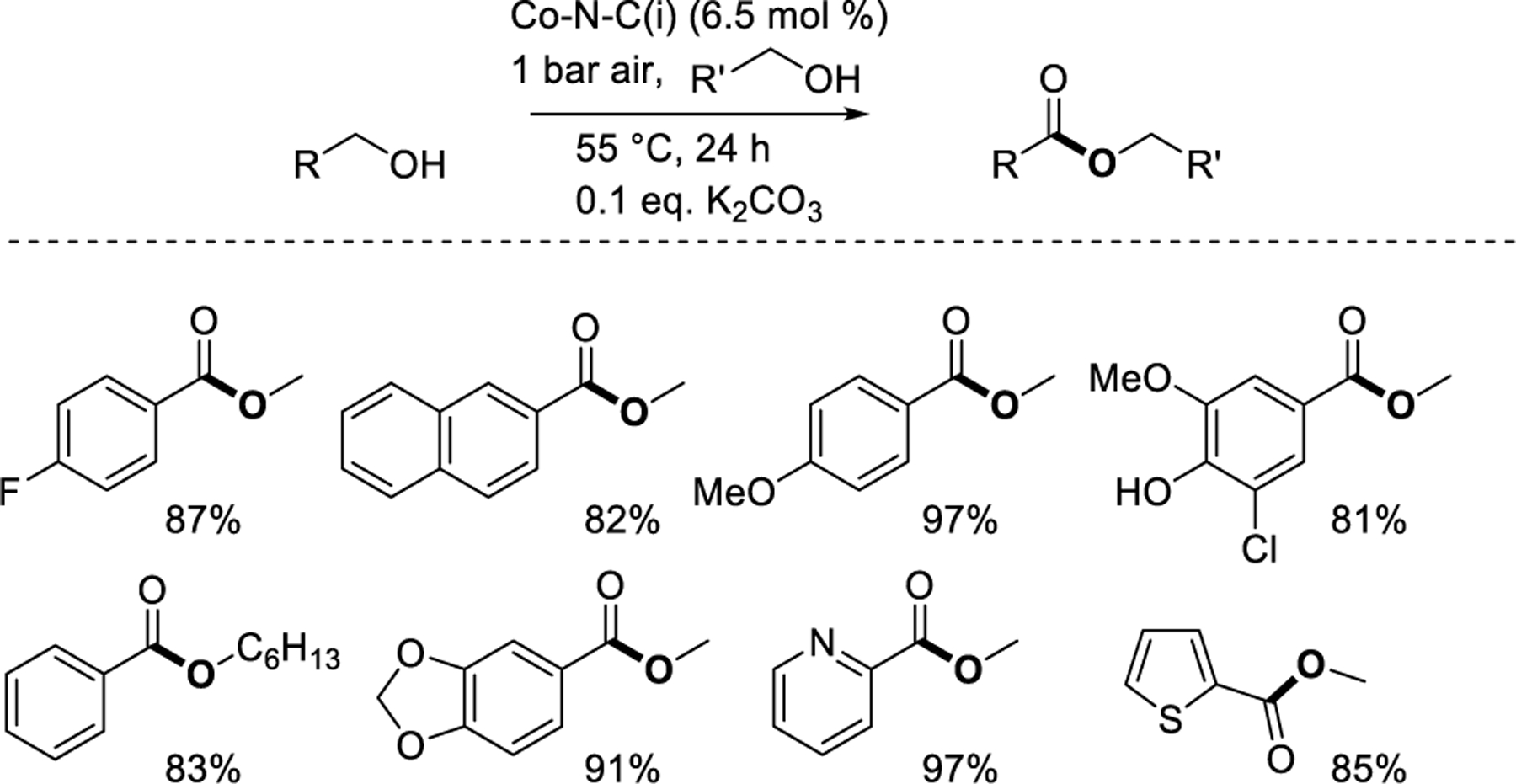
Oxidative alcohol coupling to form esters.62 Percentages reflect yield of the specified product.
The structures and reactivity of M-N-C catalysts for esterification have been characterized to varying extents. In their initial report, Beller and coworkers67 detected Co, CoO, and Co3O4 phases by XRD, and concluded that the surfaces of the particles were enriched in Co3O4 by STEM and EDS analysis, whereas only metallic Co particles with N-doped graphitic shells were detected in their later report.62 In these studies cases, atomically dispersed CoNx species were not considered as an alternative. Co appears to be privileged for this reactivity on the basis of screening studies, which evaluated benzyl alcohol methyl esterification over a range of M-N-C(iv) catalysts73 (M = Co, Fe, Cu, V, Cr, Ni, Mn) and M-N-C(i) catalysts62 (M = Fe, Mn, Co Cu). Co-N-C catalysts competent for esterification have been synthesized by a variety of methods noted in Figure 2a, including (i),60,62,67,69,72 (ii),74 (iii),75,76 (iv),73 and (v).68,71,77 These studies have observed CoOx nanoparticles,60,67,69,74,77 metallic Co nanoparticles60,74,77 (often with N-C shells62,67,68,71,72,76), and an absence of large metal aggregates that suggests the presence of mononuclear CoNx species.73,75 Quantitative relationships between active site structures and reactivity have not been established.
Normalized rates (per total Co, 60 °C, 1 bar O2) for the methyl esterification of benzyl alcohol are higher under conditions with added base. Among the three catalysts that have been tested in the presence and absence of base,67,72,73 the Co-N-C(iv) of Gao and coworkers has the highest normalized rate with added base (9×10−2 mol (mol Co)−1 s−1),732 whereas the Co-N-C(i) of Li et al. has the highest normalized rate in the absence of exogenous base (2×10−3 mol (mol Co)−1 s−1).72 That the optimal catalyst is different in the presence and absence of base implicates mechanistic distinctions that have not yet been elucidated. Mechanistic pathways involving the oxidation of alcohol to aldehyde followed by hemiacetal formation and oxidation to the ester were proposed based on analogy with known homogeneous chemistry67 and the observation of aldehydes as byproducts.68 Some studies62,71,72,75 have suggested that a surface-bound superoxide radical anion is involved based on lower yields in the presence of the free-radical scavenger butylated hydroxytoluene and EPR signals for 5,5-dimethyl-1-pyrroline N-oxide after radical capture; however, this evidence shows only the formation of radical species but not their abundance or catalytic relevance. The dearth of well-defined rate data and limited mechanistic understanding of the elementary steps involved in catalysis hamper the development of structure-reactivity relationships for esterification with M-N-C catalysts.
3.4. Alcohol Oxidation to Nitriles
Aldehydes generated by alcohol oxidation can be oxidatively coupled with ammonia to generate nitrile products. Beller and coworkers78 reported nitrile synthesis using M-N-C(i) catalysts (M = Co, Fe) synthesized using Co(Phen)2 and Fe(Phen)3 precursors (Figure 8a). The two catalysts afforded similar yields for most nitriles, but Fe required a slightly higher catalyst loading (4.5 mol%) than Co (4 mol%). M-N-C(i) catalysts derived from other M(Phen)2 complexes (M = Cu, Mn, Cr, V, Ni) gave lower yields in screening reactions with benzyl alcohol. Substituted and halogenated benzylic, heterocyclic, allylic, and aliphatic alcohols were oxidized to nitriles with yields ≥60% under 5 bar O2 in t-amyl alcohol solvent at 130 °C in 20–30 h. A similar scope can be accessed using a Co-N-C(iv)79 synthesized with a 11,11′-Bis(dipyrido[3,2-a:2′,3′-c]phenazinyl) polymer, a Co-N-C(i) synthesized with a 1-methyl-3-cyanomethyl-1H-imidazolium chloride additive,60 a Cu-Fe-N-C(ii)80 synthesized with carbon-supported polyvinylpyridine, or a Co-N-C(i)62 synthesized with piperazine and tartaric acid ligands. Primary amine substrates can also be oxidized instead of alcohols to afford nitriles using Fe-N-C(i)81 and Co-N-C(i)82 catalysts. Aqueous conditions83,84 that avoid the use of organic solvents have also been reported using a Co-N-C(v)83 derived from a Co-imidazolate MOF core with a Zn-imidazolate shell or an Fe-N-C(v)84 derived from an Fe-doped Zn-imidazolate MOF impregnated with benzylamine. In an exemplary case84 (Figure 8b), substituted and halogenated benzylic, heterocyclic, allylic, and aliphatic nitriles were synthesized in water at lower temperature (35 °C), although higher temperatures were required for less reactive substrates.
Figure 8.
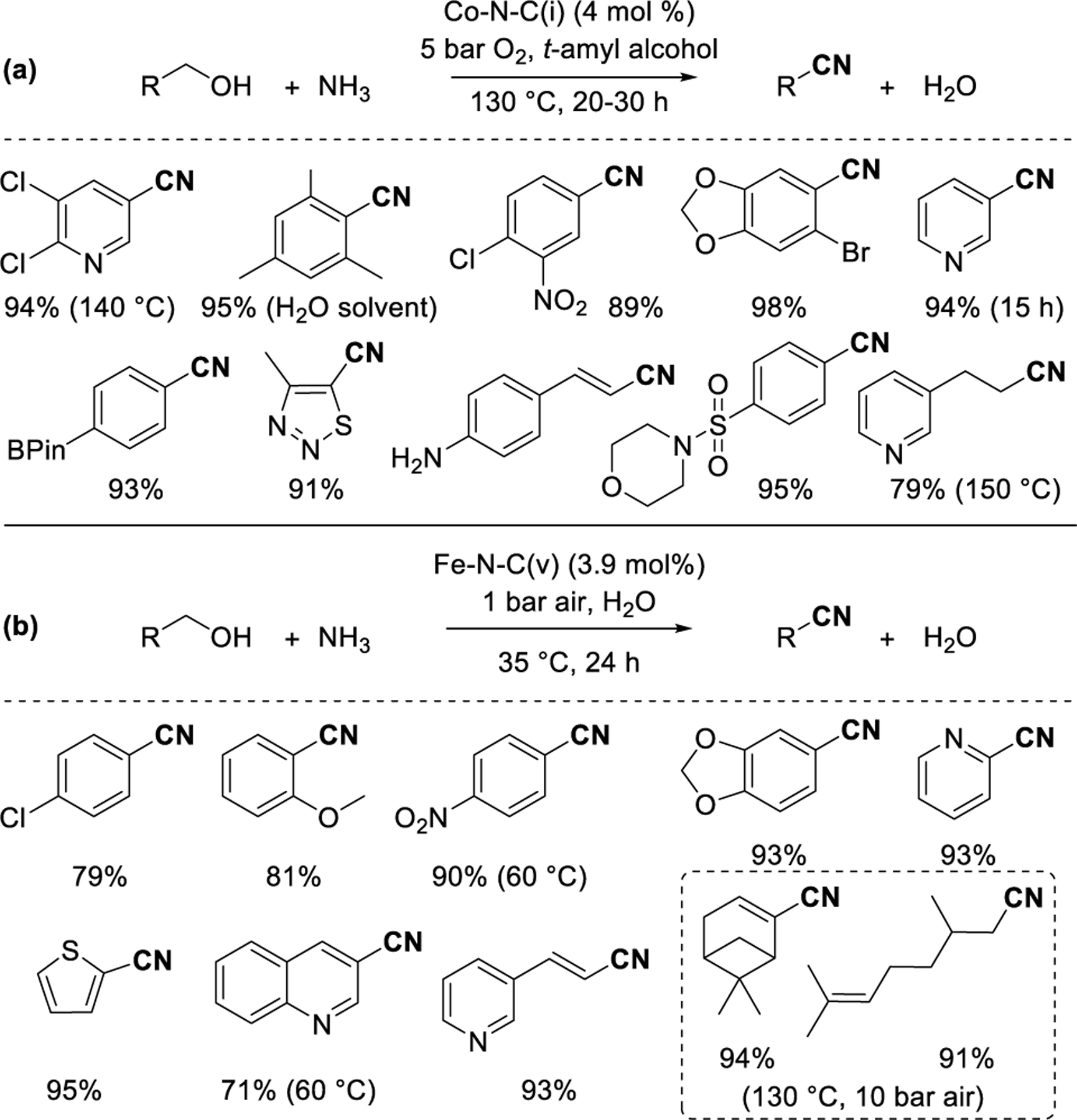
Synthesis of nitriles from alcohols and ammonia, (a) in organic solvent78 and (b) in aqueous solvent.84 Percentages reflect yield of the specified product.
Different metal species have been characterized in catalysts used for nitrile synthesis. In their initial report,78 Beller and coworkers attributed reactivity to CoOx and FeOx nanoparticles encapsulated within N-doped graphitic shells, while Huang and coworkers60 observed metallic Co aggregates by XRD and ionic Co by XPS consistent with CoOx surfaces. In a more recent study, Beller and coworkers62 attributed reactivity to metallic Co particles within N-doped graphitic shells. Cai, Lu, and coworkers83 similarly observed metallic Co nanoparticles (6 nm average diameter) in TEM images. On the other hand, metal nanoparticles were not detected in a Co-N-C(iv)79 by XRD or TEM, but XPS detected Co–N bonds, which is consistent with CoNx species. FeNx were also detected in STEM images of a Fe-N-C(v) catalyst84 that lacked Fe nanoparticles in XRD patterns and STEM images. The bimetallic Cu-Fe-N-C(ii) studied by Kobayashi and coworkers80 had both mono- and bimetallic nanoparticles according to XRD and STEM, and analysis after acid washing treatments suggested that active Fe species resisted acid washing whereas active Cu species were diminished. Metal-free reactivity for this transformation has also been claimed,85 although it is difficult to exclude the role of adventitious metal species.86 Overall, studies that have attributed reactivity to aggregates did not conclusively rule out the presence of MNx species, while studies detecting MNx species could not rigorously exclude the presence of small aggregates. A quantitative basis for the reactivity of either species is currently lacking.
The proposed reaction pathway78 for nitrile synthesis involves alcohol oxidation to an aldehyde, which undergoes condensation with ammonia to generate an imine via a hemiaminal intermediate.79,84 The imine can then undergo oxidation to the nitrile. Consistent with this proposal, time course studies79 detected benzaldehyde as an intermediate that was later consumed, and quantified rates of benzaldehyde conversion to the nitrile revealed an order of magnitude higher rate than benzyl alcohol conversion. Other studies have independently reported nitrile syntheses from aldehydes over M-N-C catalysts.87–89
3.5. Alcohol Oxidation to Primary Amides
Beller and coworkers62 showed that conditions for nitrile synthesis could be adapted to form amides by including H2O as a reactant (and solvent). These reactions used the previously noted Co-N-C(i) catalyst synthesized with piperazine and tartaric acid ligands. The reaction required high temperatures (120 °C) and high pressures (10 bar air). A variety of substituted benzoic amides including heterocycles could be synthesized using this method (Figure 9). Quantitative conversion of benzonitrile to benzamide was observed when benzonitrile was subjected to the reaction conditions. A similar scope of amides can also be synthesized starting from aldehyde substrates using an Fe-N-C(i) catalyst and similar conditions (H2O solvent, 120 °C, 10 bar air, 24 h).87 Well-defined rate data were not reported in these studies.
Figure 9.
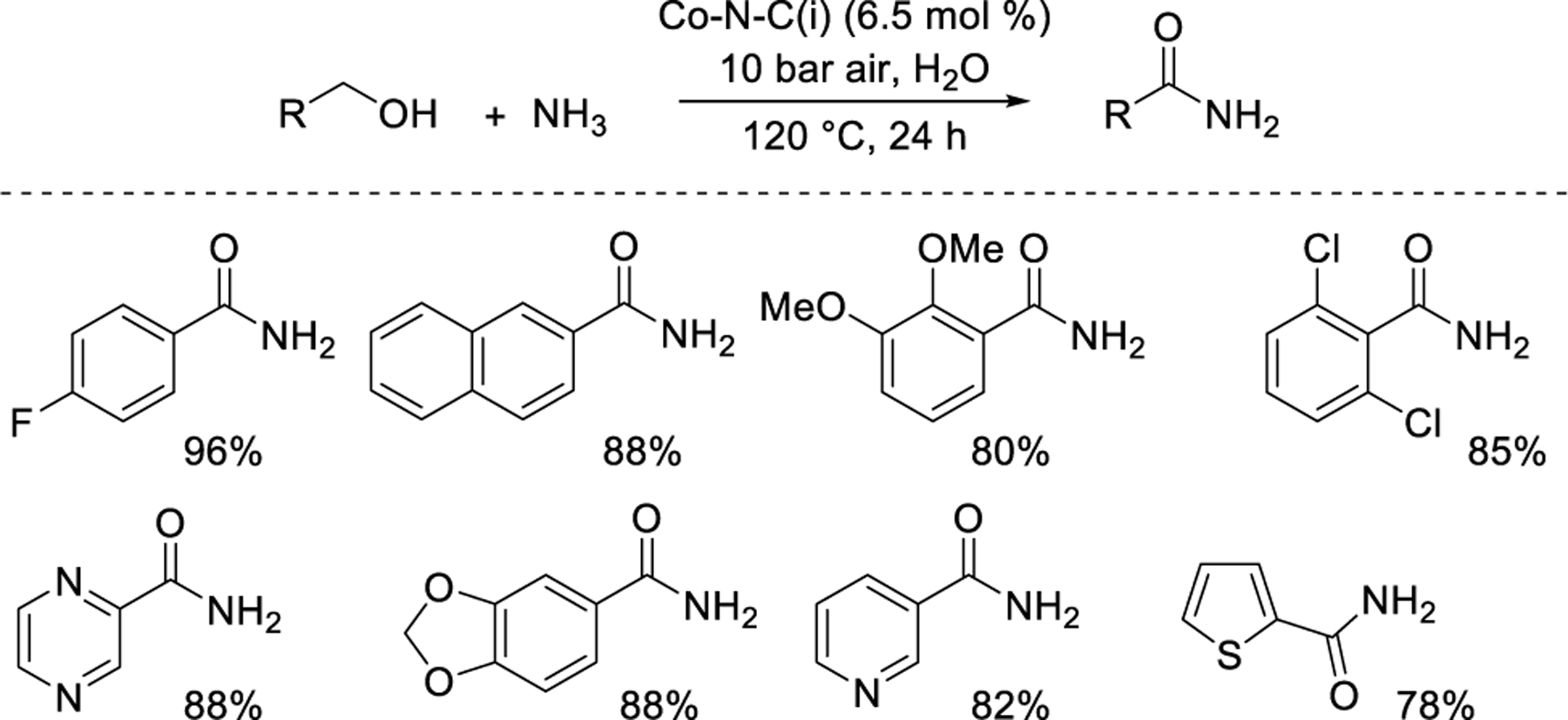
Aerobic oxidation of alcohols to amides over M-N-C.62 Percentages reflect yield of the specified product.
3.6. Oxidative Dehydrogenation of N-heterocycles
Iosub and Stahl90 reported the dehydrogenation of partially saturated N-heterocycles using a Co-N-C(i) catalyst synthesized via the pyrolysis of carbon-supported Co(Phen)2 complex (Figure 10a). A similar scope was reported concurrently by Beller and coworkers91 using an Fe-N-C(i) catalyst synthesized with a carbon-supported Fe(Phen)3 precursor (Figure 10b). Substituted tetrahydroquinolines and other N-heterocycles were dehydrogenated under different conditions in each report: the Fe-N-C(i) catalyst was used at lower loading than the Co-N-C(i) catalyst and without exogenous base, but with higher temperature, O2 pressure, and duration (100 °C, 15 bar O2, 12 h). Metallic Co and Co3O4 nanoparticles were detected in the Co-N-C(i) material by XRD, whereas the Fe-N-C(i) material did not have nanoparticles large enough to give XRD features, but FeOx nanoparticles encapsulated within graphitic sheets were observed in TEM images. Selectivity improved when some of the nanoparticles were leached in an acid treatment step. The identity of the active centers was not probed further in these reports, and MNx centers were not considered. Co-N-C(ii),92 Co-N-C(v),93 and Fe-N-C(iv)94 catalysts with similar structural features have been reported to promote this reaction with similar scope. Zhang and coworkers95 reported that an Fe-N-C(iii) catalyst synthesized with FeCl3 and a 1-butyl-3-methylimidazolium bromide ionic liquid facilitated the oxidation of three biologically relevant N-heterocycles and 1,2-diphenylhydrazine in phosphate-buffered (pH = 7.4) aqueous solution under O2 at ambient temperature. Including NaSCN in the reaction mixture eliminated reactivity, implicating FeNx sites as the active centers. In a complementary study, Li, Jaouen, and coworkers96 showed that an Fe-N-C(v) catalyst derived from an Fe(Phen)-impregnated ZIF-8 MOF catalyzed the oxidative dehydrogenation of NADPH to NADP+. In 2022, Beller and coworkers97 reported N-heterocycle dehydrogenation using a Zn-N-P-C(v) catalyst synthesized with a ZIF-8 MOF impregnated with triphenylphosphine as the precursor. Quinoline yields after 6 h (110 °C, 1 bar air, H2O solvent) were weakly affected by the inclusion of KSCN (2–5 eq. with respect to Zn), suggesting that the active sites were not metallic.
Figure 10.
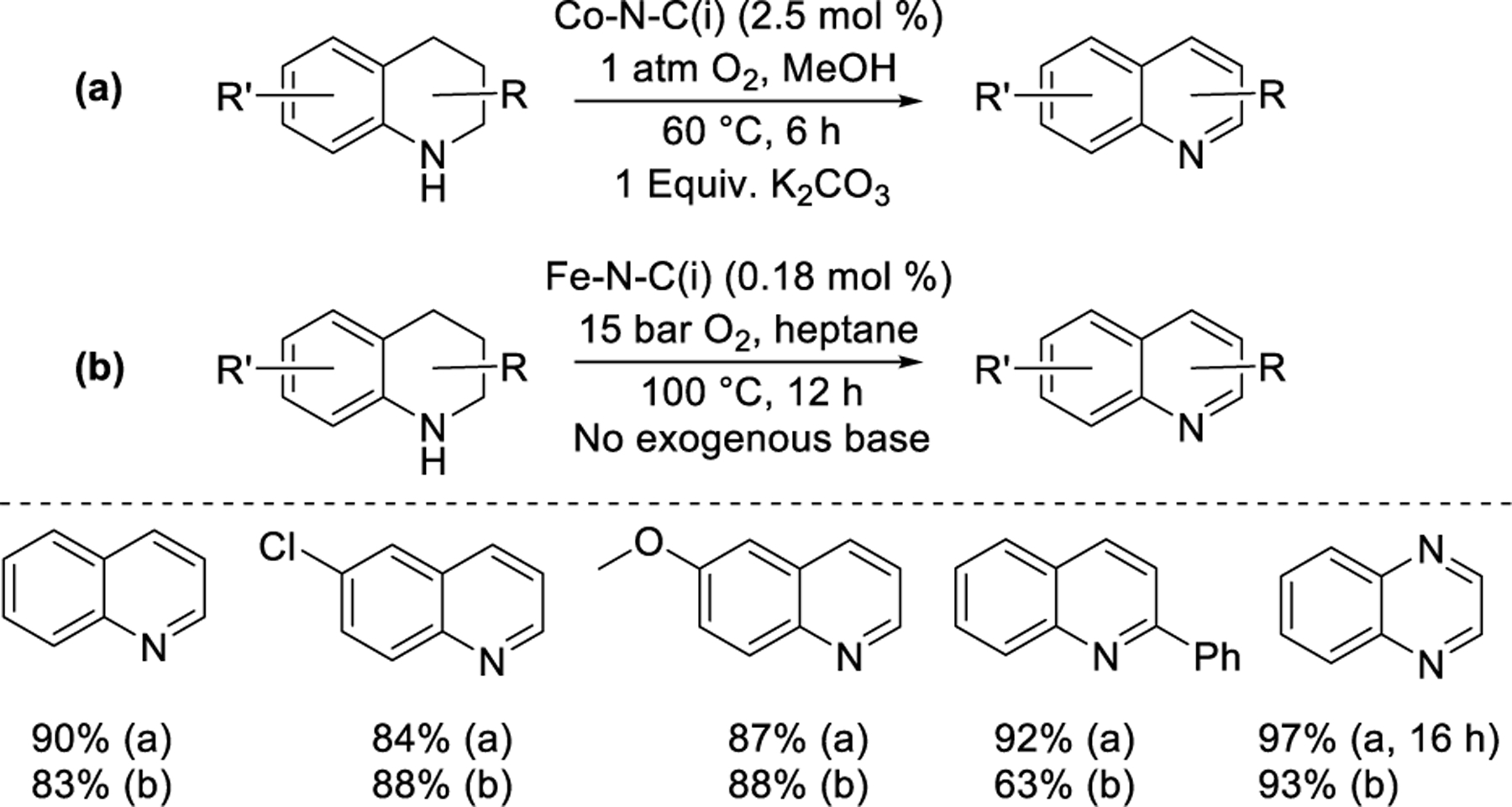
Dehydrogenation of N-heterocycles developed by Iosub and Stahl90 (a) and Beller and coworkers91 (b). Percentages reflect yield of the specified product.
3.7. Other Relevant Aerobic Oxidations
Aerobic oxidations related to those described above have also been developed. Intramolecular oxidative cyclization of an o-aminophenol-derived imine was reported to form a benzoxazole ring, using a Co-N-C catalyst synthesized by the pyrolysis of SiO2-supported Co(Phen)2 complex.98Xu et al.99 reported benzylamine oxidation and subsequent reaction with primary amines to form imines over a V-N-C(v) catalyst synthesized by the pyrolysis of a V-containing MOF (NH2-MIL-101(V)). The scope of its oxidative homocoupling reactivity included heteroatom-containing benzylic amines and aliphatic amines, in addition to cross-coupling of benzylamine with aniline or aliphatic amines. The V-N-C(v) catalyst did not have nanoparticles detectible by XRD or TEM. The authors concluded that VN4 sites catalyzed the reaction, but suitable controls were not included to rule out the potential contributions of small VOx moieties. The normalized rate (per total V) of the V-N-C(v) catalyst to form N-benzylidene benzylamine was 54 mol (mol V)−1 s–1.
Gu and coworkers100 reported the preparation of conjugated diynes by a Glaser-Hay type reaction catalyzed by a Cu-N-C(iv) catalyst synthesized with dopamine hydrochloride and Cu precursors. At 40 °C in ambient air, 1,3-diynes were synthesized in 85–97% yields by oxidative homocoupling of terminal aryl and alkyl alkynes. The same catalyst under the same conditions achieved selective heterocoupling of aryl and alkyl alkynes with yields of 65–82% by using a slight excess of one substrate (1.3:1). Trifluoromethyl, halogen, methoxy, and phenoxy substituents, and S- and N-containing heterocycles were tolerated. The active centers were hypothesized to be mononuclear CuNx sites based on a low Cu-Cu scattering intensity in the EXAFS and density functional theory (DFT) calculations showing a low energy barrier for coupling of aromatic and aliphatic alkynes on the CuNx sites. Rate data under well-defined conditions were not quantified.
Ethylbenzene oxidation101–105 and other arylalkane oxidations106–108 have been reported in the presence of M-N-C catalysts, but the role of the M-N-C is unclear, as many studies report non-zero yields in blank reactions and with metal-free solids, suggesting a potential role of radical-chain pathways that could be modulated by the M-N-C. Oxidation of internal diols or esterification of benzylic ketones, both requiring C–C oxidative cleavage, also could feature radical pathways.109,110
4. METHODS FOR ASSESSING INTRINSIC KINETIC BEHAVIOR
Designing active site structures for an intended function requires quantitative and informative measurements of catalytic phenomena. The primary experimental observables of catalysis (substrate consumption, product formation, current) are only indirectly related to the active centers required for their appearance, but they can be interpreted as reaction rates that reflect the structures and numbers of active centers when they are measured under conditions wherein convoluting factors are properly controlled. A generalized rate expression that includes such factors can be written as follows (Figure 11a):
| (1) |
where r is the rate of reaction per mass of catalyst (mol g−1 s−1), EF is the overall effectiveness factor111 (EF = measured rate/ diffusion-free rate), and for each site type i in the bulk material, Ni is the bulk density of site i (mol g−1), Di is the fraction of sites i accessible to substrate (i.e., the dispersion), and TOFi is the turnover frequency at site i (s−1). In eq 1, the TOF (or turnover rate) is the quantity of interest for fundamental understanding of active centers because it enables the fair comparison of catalysts made by different synthetic approaches and in different laboratories, and provides a framework to evaluate whether higher mass-based rates are the result of higher site densities or higher intrinsic reactivity of different site types.112–114 Attempts to assess the reactivity of M-N-Cs in aerobic oxidation at the level of TOFs are rare,63,64 and we are not aware of studies that fully account for convoluting factors such as product inhibition, catalyst deactivation, transport, active-site density and accessibility, and active-site diversity. In general, TOF values are most reliable when obtained by using initial-rate methods. Previous reports of M-N-C catalyzed aerobic oxidation have typically prioritized reaction yields, limiting the ability to determine TOF values owing to the potential kinetic influence of changes in concentrations and/or catalyst deactivation during the reaction. Kinetic characterization of M-N-C catalysts represents a significant opportunity for future development of the field of aerobic oxidations. In this section, methods and concepts relevant to the measurement of site-specific TOFs in M-N-Cs are summarized, largely drawing from studies of M-N-C ORR electrocatalysts.
Figure 11.
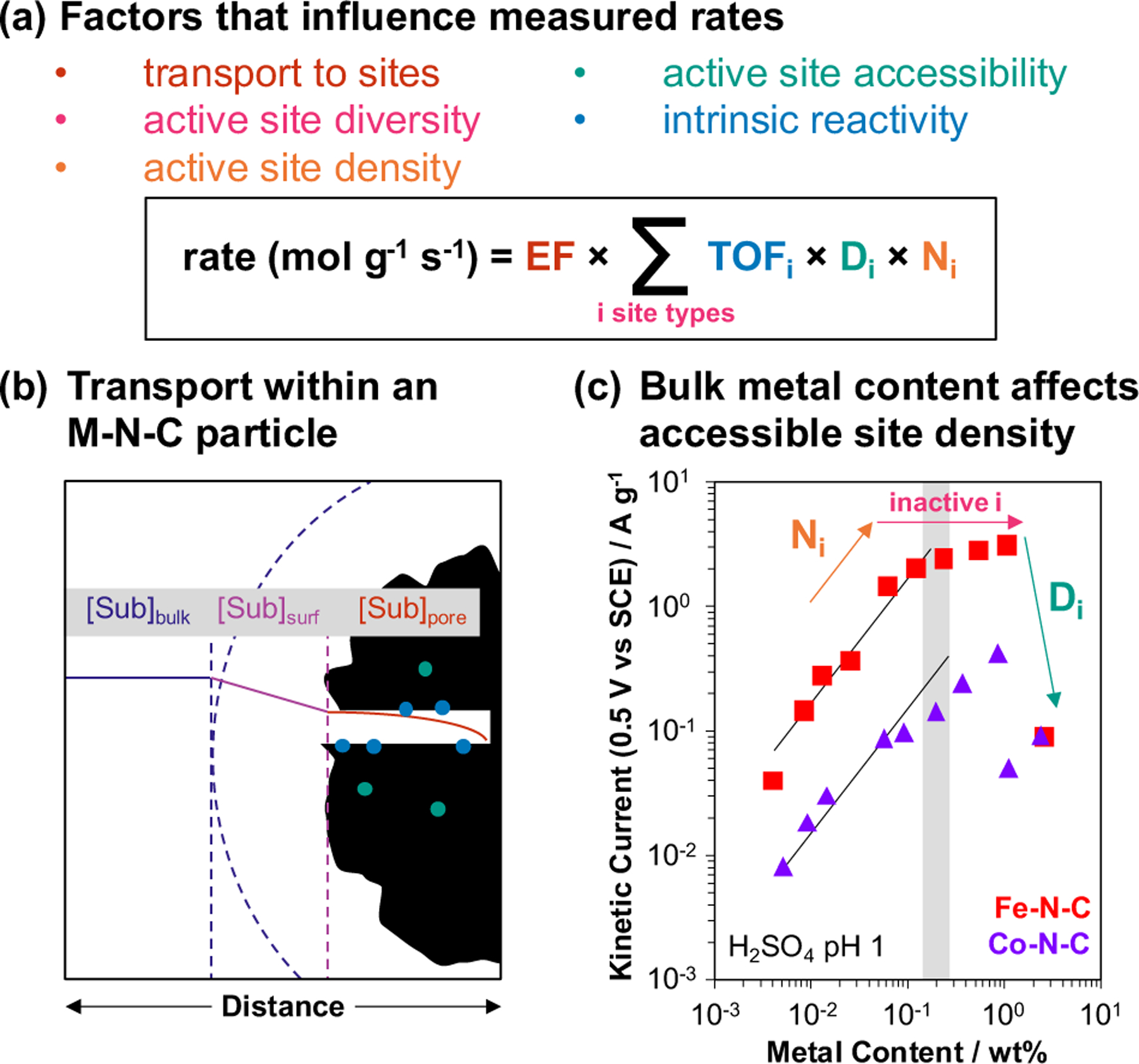
Comprehensive description of factors that influence kinetic behavior of M-N-C catalysts. (a) Contributions of transport, site quantity, accessibility, and intrinsic reactivity to overall rates, (b) substrate concentration profile in bulk solution, at the particle surface, and within a catalyst particle under transport-controlled conditions, and (c) systematic studies across bulk metal content in ORR.123 Panel (c) is adapted with permission from Ref. 123. Copyright 2007 Elsevier.
4.1. Assessing Transport Contributions
Prior to surface reaction, gas-phase substrates (e.g., O2) must diffuse across the gas-liquid interface to dissolve into the liquid phase (gas–liquid), and together with other liquid-phase substrates (e.g., organic molecules), they must diffuse from bulk fluid to the surface of catalyst particles (external), then diffuse within particles to active sites (internal). When these processes are kinetically relevant (Figure 11b), they give rise to spatial gradients in substrate concentration in the bulk fluid phase ([Sub]bulk), at the catalyst surface ([Sub]surf), and within the catalyst particle ([Sub]pore). A reaction rate measurement designed to illuminate surface-mediated elementary steps that depend on [Sub]pore must not be corrupted by kinetic contributions from these diffusion processes, such that the measured quantity of [Sub]bulk is equivalent to [Sub]pore.
In electrocatalytic ORR studies, the M-N-C catalysts are typically deposited onto an electrode surface within an ionomer film, and this feature both simplifies and complicates the analysis of transport. The studies often use a rotating disc electrode, which enables understanding the convective transport rate to the catalyst surface and the kinetic relevance of external diffusion limitations, as described by the Koutecký–Levich equation.115 Nevertheless, internal diffusion within the catalyst film may limit rates at lower overpotentials before the onset of external diffusion limitations.116 The kinetic relevance of internal diffusion limitations in heterogeneous catalysts is predicted by a dimensionless parameter known as the Thiele modulus (ϕTh; EF = tanh(ϕTh)/ϕTh), which depends on the characteristic length of catalyst particles or the deposited catalyst layer, the diameter of pores, the concentration and diffusion coefficients of substrates, the reaction rate (per particle). In electrocatalytic systems, additional factors include the ohmic resistance and ionic conductivity of the catalyst layer.116,117 The onset of internal diffusion limitations is reflected as an increase in the apparent Tafel slope at increased current densities and thinner catalyst films are less likely to suffer from internal transport limitations.116 Internal diffusion resistances can also influence selectivity when product molecules are given time to react further as they exit the catalyst layer, as can arise in H2O2 reduction to H2O in the so-called “2×2” ORR pathway.13,118,119 Materials design can also be leveraged to facilitate transport within catalysts. For example, Müllen, Feng, and coworkers120 used SiO2 colloid templating to synthesize a polyaniline-derived Fe-N-C(iv) catalyst with ~14 nm mesopores. This catalyst showed an ORR polarization curve consistent with kinetically limited rates until the onset of external diffusion limitations, whereas its non-mesoporous analog showed ORR reactivity consistent with internal transport limitations.
Transport limitations are seldom considered in aerobic oxidation studies using M-N-C catalysts. O2–liquid transport rates were considered by Davis and coworkers63,64 to select M-N-C loadings sufficiently low that this transport process was not limiting, but external and internal transport were not discussed. External diffusion limitations can be probed by varying the solution agitation rate and aggregate size of the catalyst, both of which influence the width of stagnant boundary layers at the external surface of particles. In a kinetic study of aerobic hydroquinone oxidation using slurry-phase M-N-Cs, Stahl, Root and coworkers showed that suitable reactor agitation was required to achieve agitation-independent rates unaffected by external transport limitations.53 The rate of internal diffusion was estimated and compared with measured reaction rates using the Mears criterion121 to conclude that internal diffusion limitations were not corrupting measured rates. The Madon–Boudart criterion,122 wherein TOF values do not vary with site density (Ni), is particularly effective, but this strategy is inaccessible without accurate site quantification methodologies or synthetic strategies to generate a single type of site. The kinetic relevance of both O2 and organic substrate diffusion should be considered on a case-by-case basis. Low O2 solubility in aqueous solvent often leads to a lower overall mass transport coefficient for O2 compared to higher-concentration organic substrates. Organic solvents have higher O2 solubility, which can lead to mass transport limitationas associated with large organic substrate molecules, which may have slower diffusion and/or be present in lower concentrations.
The discussion above shows that the consideration of mass transport, established within the field of heterogeneous catalysis and adopted in electrocatalytic studies of M-N-Cs, provides a framework for assessing the role of transport in kinetic studies of aerobic oxidations.
4.2. Assessing Active Site Density
The remaining terms in eq 1 highlight the importance of assessing the diversity (Σsites i), number (Ni), and accessibility (Di) of active sites in M-N-C catalysts. Although direct methods to quantify accessible active sites are desirable, early studies can begin to assess the importance of these parameters through systematic variation of the bulk metal content. Jaouen and Dodelet123 studied the ORR on a suite of Co- and Fe-N-C(i) catalysts with 0.005–2.7 wt% metal contents. The ORR kinetic currents measured on these catalysts (per g, ambient T, 1 bar O2, H2SO4 pH 1, 0.5 VSCE) varied linearly with the metal loadings at the lowest values (Figure 11c) but reached a maximum at higher metal loadings (~0.1–1 wt%), and sharply decreased at the highest loadings (>1 wt%). The linear rate data measured on Fe-N-C at low Fe loadings were assumed to reflect catalysts that contained predominantly mononuclear FeNx species and used to estimate that the turnover rate for ORR at 0.5 VSHE is 0.14 ± 0.03 e− site−1 s−1. This value represents a lower bound for the TOF, which would increase if this suite of Fe-N-C contained similar fractions of FeNx per total Fe less than unity. The ORR rate (per g catalyst) departed from the linear dependence at Fe loadings above ~0.2 wt% (gray bar, Figure 11c), indicating that additional Fe species different from FeNx with lower TOF were incorporated into the material, likely as agglomerates. Finally, a sharp decrease in the rate corresponded to a decrease in the material’s micropore specific area, meaning that a greater fraction of sites was likely inaccessible. The general trends observed on Fe-N-C(i) hold for Co-N-C(i), but at low loadings the linear dependence is not as strong (Figure 11c), indicating that for the synthesis method used, Co is prone to forming multiple species even at low loadings. This study illustrates several key concepts: (i) M-N-C catalysts frequently contain multiple types of metal species with different intrinsic reactivity, (ii) higher metal loadings tend to lead to the formation of less active species, and (iii) the synthesis of materials with widely varying metal loadings is an important approach to probe metal speciation. This approach has not yet been employed in studies of M-N-C catalyzed aerobic oxidation.
The systematic variation of site densities relies on approaching low enough quantities that speciation becomes similar; however, this approach is not suitable to understand the behavior of materials made by widely different synthetic routes or with intrinsically diverse site speciation. Instead, independent methods for the quantification of accessible active sites are needed. Quantitative knowledge of active site densities can further combine with spectroscopic knowledge of their structure to form a bridge between structure and function, in turn guiding the design of precise synthetic methods. Here, we summarize site quantification methods developed primarily in ORR studies that could be used to study aerobic oxidation catalysts. Techniques that appear to be limited to electrochemical ORR applications124 are not reviewed. The site quantification techniques discussed below focus on MNx centers because these are broadly accepted as highly active species for ORR but this discussion does not imply that MNx should be assumed to be the active centers for aerobic oxidations. Instead, the techniques below establish rigorous protocols for testing the validity of this hypothesis.
4.2.1. Site Counting Using Irreversible Poisons During Catalysis.
Simultaneously measuring an active site quantity and a reaction rate directly links the number of sites to catalytic function and may be achieved by irreversible poisoning of active sites. In the ideal case, a titrant molecule is selectively and irreversibly adsorbed to the material at a putative active site structure. Then, the simultaneous quantification of the site abundance on the solid and the associated decrease in reaction rate can be expressed as a TOF that is necessarily ascribed to the poisoned sites. The challenge, in practice, is to achieve these ideal conditions by titrant selection for specific materials and reaction conditions. Lack of selectivity leads to underestimation of the TOF when titrants bind to inactive sites, and lack of irreversibility requires excess titrant to suppress rates as dictated by chemical equilibrium and can lead to underestimation of the TOF.
A variety of molecules reversibly inhibit the reactivity of M-N-C catalysts during ORR but do not poison them irreversibly. Although such reversibility precludes quantifying active sites, it has enabled their qualitative identification. Early studies demonstrated that CO binds to supported Fe(porphyrin)125 complexes and inhibits ORR reversibly. This molecular reactivity motivated studies of CO poisoning of Fe-N-C. Dodelet and coworkers126 observed a lack of irreversible poisoning of Fe-N-C(i) by CO at 0.1 VSCE and pH 1 in constant potential polarization curves. Separately, Ozkan and coworkers127 observed partially reversible inhibition by CO on Fe-N-C(i) in 0.5 M H2SO4 and ν(C≡O) peaks redshifted from gas-phase CO by 130 cm−1 in DRIFTS that reflect Fe-bound CO. No CO inhibition is observed with N-C ORR catalysts that lack metal centers.128,129 Anions expected to coordinate axially to MNx centers (e.g., SCN−,130 halides,131) inhibit reactivity but their binding irreversibility has not been demonstrated, and they are known to inhibit catalysis partially or weakly under certain conditions.63 For instance, SCN− may be washed away by H2O,130 so most studies demonstrate only reversible inhibition of MNx sites by equilibrating the surface with SCN− in bulk aqueous solution.104,132,133 In other cases, poisons are unselective and cause restructuring of the active sites (H2S)134 or are unable to access active sites sequestered within pores (H3PO4).135 Collectively, these observations implicate some form of M as the active site for ORR and highlight the challenge of identifying irreversible poisons.
Poisoning studies of M-N-C catalysts have received only limited consideration in aerobic oxidation catalysis. Davis and coworkers63 reported poisoning of Co-N-C(i) and Cu-N-C(i) catalysts by benzoic acid during benzyl alcohol oxidation (80 °C, 10 bar O2, 0.1 M benzyl alcohol in H2O). M-N-Cs were exposed to benzoic acid prior to reaction and separated from the solution, and the filtrate was analyzed to quantify the amount of adsorbed benzoic acid. The decrease in rate did not depend linearly on benzoic acid uptake, and the authors proposed the existence of distinct sites, some resistant to poisoning by benzoic acid. Irreversible selective titration of MNx sites was not verified, however, suggesting that reversible inhibition by benzoic acid under reaction conditions could also rationalize the observations.
Irreversible poisoning studies have been applied more broadly in ORR studies. Kucernak and coworkers136,137 studied the effects of nitrite exposure on Fe-N-C(ii) and discovered that certain pretreatments could lead to poisoning of ORR reactivity. Systematic variations in pH and potential demonstrated that nitrite adsorbed at Fe centers could be reduced to nitrosyl in acid medium; Fe–NO could also be formed by disproportionation of NH2OH.137 Fe–NO species are stable under ORR conditions (Figure 12a), supporting irreversible titration. Quantifying the charge passed during a reductive stripping of the Fe–NO species (Qstrip, Figure 12b), and assuming the product of the reduction (NH4+ = 5 e− per Fe–NO, according to Kucernak and coworkers), enables quantification of active site density.136 The NO2− stripping technique has been adopted more broadly in studies of ORR on Fe-N-C.138,139 Yet, Maillard, Jaouen, and coworkers140 reported that the NO2− stripping method quantifies the exposed surface of Fe2O3 nanoparticles supported on N-doped carbon. Moreover, Choi and coworkers141 provided evidence that NO reduction generates NH2OH (3 e− per Fe–NO), not NH4+, indicating that all values based on NH4+ underestimate the FeN density by a factor of 3/5. Modifications to the method of NO adsorption should also be considered carefully: Kucernak and coworkers142 have reported that gas-phase NO adsorbs on the carbon support143 at locations unassociated with FeNx sites, leading to overestimates of active site densities. Given these uncertainties, applying this technique in future studies requires careful comparison with control samples and complementary site quantification techniques.138
Figure 12.
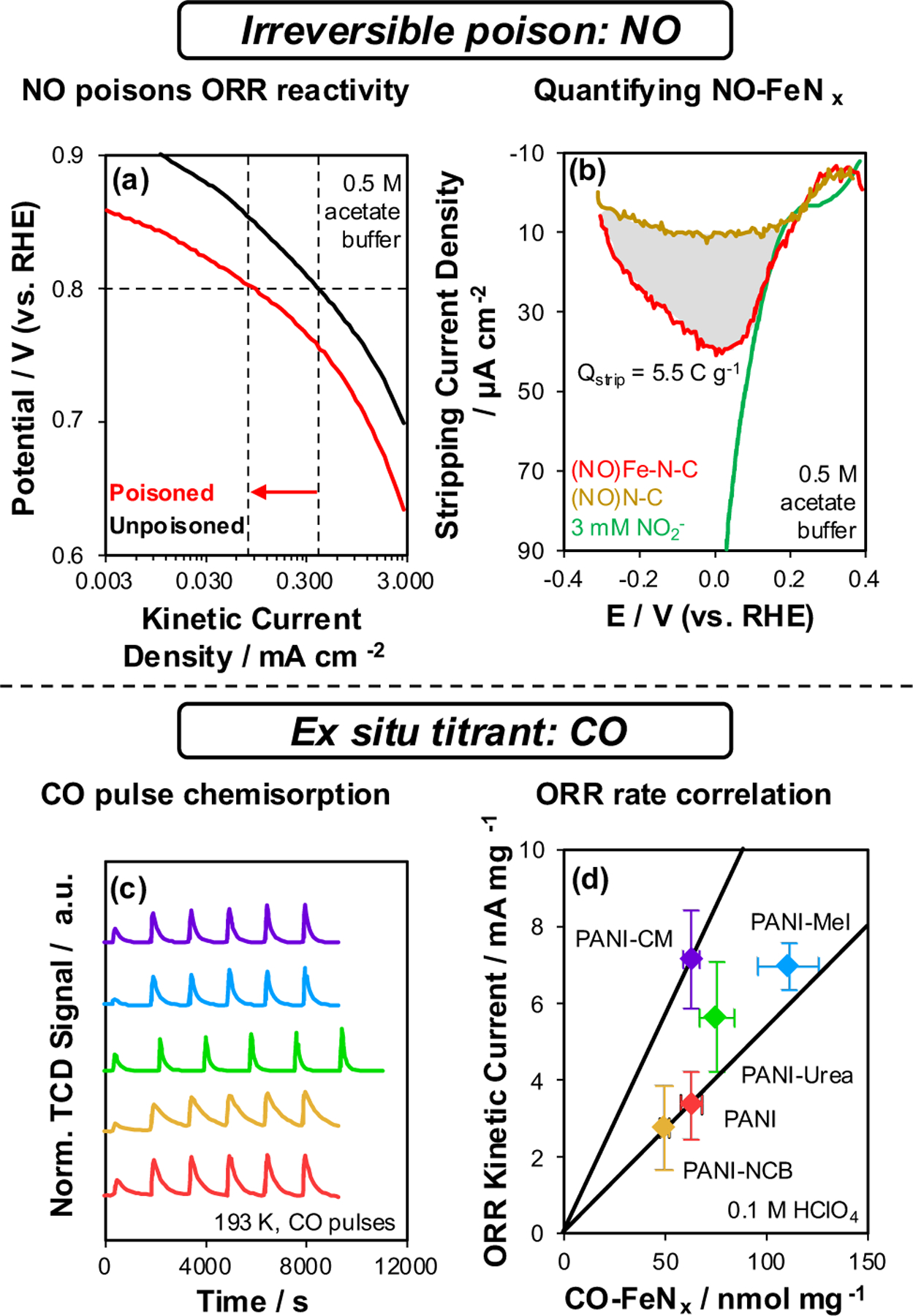
Methods for active site quantification in Fe-N-C catalysts reported in the ORR literature: (a, b) irreversible poisons136 and (c, d) ex situ chemical titrations.150 Panels (a) and (b) are adapted from Ref. 136. Copyright 2016 Nature Publishing Group. Panels (c) and (d) are adapted from Ref. 150. Copyright 2018 American Chemical Society.
In 2021, Choi, Jaouen, and coworkers144 reported a complementary site quantification method based on poisoning by cyanide. The key considerations that enabled this technique to succeed were the exclusion of O2, oxidation of the catalyst at an electrode prior to the adsorption of cyanide, and careful quantification of cyanide uptake by its disappearance from solution rather than assuming complete uptake. Subsequent ORR polarization curve measurements within a narrow potential range (0.70–1.05 VRHE) and at a slow scan rate (1 mV s−1) quantified the decrease in rate, which correlated linearly with a range of cyanide uptakes and quantified the total number of active sites upon extrapolation to zero rate. For the Fe–N–C(v) investigated, CN−, NO2− (assuming 3 e− reduction to NH2OH), and CO chemisorption (vide infra) gave approximately the same site density value (Table 1, Fe0.5NC). The CN− poisoning method could also be extended to Co, Mn, and Ni-containing catalysts, suggesting generality, and quantifying a ranking of intrinsic ORR TOFs (Fe > Co > Mn ~ Ni). Although safety considerations and sensitivity to O2 limit the ease of adopting this method, it involves fewer assumptions, such as the product formed by NO2− stripping. Validations among a range of more diverse M-N-C catalysts and by different research groups would further establish the importance of this techniqu. In summary, both CN− and NO2− poisoning quantify MNx sites in situ under aqueous conditions. It is untested whether these methodologies can be extended from ORR in aqueous media to aerobic oxidation catalysis in organic solvents.
4.2.2. Site Counting Using Ex Situ Chemical Titration Methods.
Correlative relationships between measured rates and independently measured site quantities can provide alternative evidence for the identity of active sites when they span a range of catalyst materials with different properties. These relationships are well established for nanoparticle-based precious metal catalysts, where either gas-phase chemisorption of a probe molecule (e.g., H2, CO)146,147 or electrochemical underpotential deposition (of hydrogen) or stripping (of CO)116 enable quantifying the number of surface metal sites and calculation of turnover rates. Such relationships and titration methods have recently been established for M-N-Cs in ORR.
Strasser and coworkers148 applied the technique of CO pulse chemisorption at cryogenic temperature (193 K) to quantify the active sites of Fe-N-C(ii) and Mn-N-C(ii) ORR catalysts. Low temperature (193 K) was required to ensure saturation of MNx sites that bind CO more weakly than Pt or other noble metals. The method quantifies the disappearance of CO from calibrated pulses in a stream of flowing inert gas until saturation of the surface can be verified by identical pulse areas (Figure 12c). CO was hypothesized to selectively adsorb at accessible MNx sites in these materials. A later report by Strasser and coworkers149 further highlighted that thermal pretreatments in flowing inert gas to 600 °C are required to ensure accuracy and reproducibility of the method because they desorb O2 or other adventitious adsorbates from FeNx.
The utility of the CO pulse chemisorption technique was demonstrated using a series of Fe-N-C(ii) materials prepared using different additives with a polyaniline N source (cyanamide, melamine, urea, nicarbazin).150 CO-quantified FeNx site densities accounted for ORR kinetic current densities (per mg, 0.8 VRHE) that varied by a factor of ~4 (with one outlier, Figure 12d). In a later study,151 Strasser and coworkers concluded that CO does not bind to NiN4 sites, suggesting that the utility of CO may not be general for any given M-N-C. To date, CO pulse chemisorption data have been reported for Fe, Co, and Sn moieties.152 Despite its potential limitations, adoption of the CO pulse chemisorption technique has provided methods to benchmark catalysts prepared by diverse methods among different research groups and to compare their turnover frequencies (per CO-quantified FeNx) and site densities.153 Such comparisons highlight opportunities to improve catalyst rates (per mass) by either increasing their site densities or intrinsic turnover frequencies.
The in situ NO2− stripping and ex situ CO titration approaches have been compared in a cross-laboratory study138 that features four Fe-N-C(ii/iv/v) catalysts with ORR rates (per g, 0.8 VRHE, 0.8 mg cm−2 loading) that varied by a factor of ~4. NO2− quantified 12–39% of the number of sites that CO quantified. This discrepancy was ascribed to the inability of NO2− to access surface sites that are not exposed to electrolyte, whereas CO can access all surface sites in the gas phase. The NO2− stripping value was proposed to reflect a lower bound for the active site density, while CO titration reflects an upper bound. TOFs calculated using these values varied by up to a factor of 11 (NO2−, a factor of 3.6 in the case of CO). The differences could result either from titration of less-reactive sites included in rate normalizations, the influence of transport limitations, or differences in the intrinsic reactivity of sites. CO and NO titrants have begun to be applied more broadly in the ORR literature (Table 1) but have yet to be reported in any aerobic oxidation catalysis study. To our knowledge, the highest reported fraction of FeNx per total Fe quantified by a widely accepted chemical titration is 46%154 (Table 1), which corresponds to ~1 wt% FeNx, highlighting that substantial improvements in reactivity could be realized if samples were synthesized to contain greater fractions of accessible atomically dispersed FeNx. The application of these site titration methodologies in aerobic oxidation catalysis studies could facilitate benchmarking of turnover rates between samples prepared by different methodologies and by different research groups.
5. METHODS FOR STRUCTURAL CHARACTERIZATION OF CATALYTICALLY RELEVANT ACTIVE SITES
A wide array of characterization techniques is available to probe the bulk structural features of carbonaceous materials and detailed monographs155,156 exist on this topic. Material properties such as pore geometry157–159 and hydrophobicity160–163 define the solvating environments of active centers but their effects cannot be quantitatively probed until the types and numbers of active centers that reside within them are known. Thus, this section surveys the techniques used to characterize the metal species of M-N-C catalysts that are well-established in the ORR field and have relevance for aerobic oxidation catalysis. Techniques are emphasized that enable discriminating active centers from spectator species within a pool of different site configurations that are commonly encountered in M-N-Cs. Nascent or infrequently used techniques in ORR studies (e.g., ssNMR,164 NRVS,165,166 EPR,166,167 WAXS,168 XES167) are not reviewed in detail as their relevance to aerobic oxidation catalysis is not yet clear.
5.1. X-Ray Absorption Spectroscopy
X-ray absorption spectroscopy (XAS) consists of the excitation of core-level electrons that results in the emission of photoelectrons by the photoelectric effect. The different absorption edges associated with this process (e.g., K-edge = 1s, L-edge = 2p) are well-separated for most elements, which makes XAS an element-specific technique. The transmission configuration commonly employed using synchrotron-generated X-ray radiation further makes XAS a bulk technique that probes all atoms in a material at the chosen absorption edge. The spectrum in the vicinity of the absorption edge is referred to as the X-ray absorption near edge structure (XANES). XANES is sensitive to the oxidation state of the metal atoms being probed and can also show features characteristic of their local environment. The scattering of emitted photoelectrons caused by the atoms surrounding the excited atom leads to oscillations in the absorption coefficient as the incident energy increases beyond the absorption edge, which is a phenomenon known as the extended X-ray absorption fine structure (EXAFS). Careful analysis of the EXAFS can give useful information about the local environment of the absorbing atom. The use of these techniques to characterize ORR-relevant active sites in M-N-Cs is discussed below.
5.1.1. XANES.
The position of the main absorption edge of the XANES is sensitive to the oxidation state of the metal being probed. Comparison with well-defined molecular references typically suggests that Fe-N-C materials include a mixture of FeII and FeIII species under ambient conditions. The XANES includes two general features in addition to the main absorption edge: the pre-edge, which appears at energies below the main absorption edge, and the white line intensity, which is the absorption maximum after the absorption edge. Pre-edge features reflect transitions to unoccupied valence orbitals (e.g., at the K-edge, 1s→3d), and these features can be rigorously interpreted within the framework of ligand field theory in the case of molecular complexes,169 which demonstrate that pre-edge peak positions and intensities are sensitive to a variety of factors including spin state, oxidation state, geometry, and ligation environment. In the case of M-N-Cs, pre-edge features tend to be less intense compared with molecular complexes like porphyrins and phthalocyanines,170 likely reflecting the heterogeneity of ligating environments in M-N-Cs that broaden pre-edge features and preclude their interpretation at the same level of detail as molecular complexes. The intensity of the white line can be used to diagnose the presence of MOx clusters, which show a more intense white line feature than MNx centers.171 Because the interpretation of the XANES is complicated by the potential heterogeneity of M-N-C materials, in situ methods have been developed to selectively probe sites that change during catalysis.172
Mukerjee and coworkers55 applied a differential XANES method, referred to as “Δμ”, to analyze the difference between XANES spectra of an Fe-N-C(i) catalyst measured in situ at 0.9 VRHE and 0.1 VRHE (Ar-saturated 0.1 M NaOH, Figure 13a,b). The difference spectrum reflects only the surface FeNx moieties that undergo a redox transformation in response to the change in potential from FeIINx (0.1 V) to (H)O-FeIIINx (0.9 V), while any bulk signals for catalytically irrelevant sites are eliminated by subtraction. Two samples were compared: Fe(TPP)Cl/C pyrolyzed at either 300 °C (Figure 13a) or 800 °C (Figure 13b). Pyrolysis at 300 °C leaves the porphyrin moiety intact and the Δμ signal was accurately modeled173 using the local structure of Fe(TPP) with a bound O atom (O-FeN4C12, Figure 13a). The Δμ signal (Figure 13b) for the Fe-N-C pyrolyzed at 800 °C differed from the Fe(TPP) precursor because it decomposed to form the Fe-N-C structure. The structures that fit this Δμ signal best corresponded to either O-FeN4C10 or O-FeN4C8, which correspond to pyridinic N4 ligation environments with or without missing C atoms (such as at an edge plane), respectively.
Figure 13.
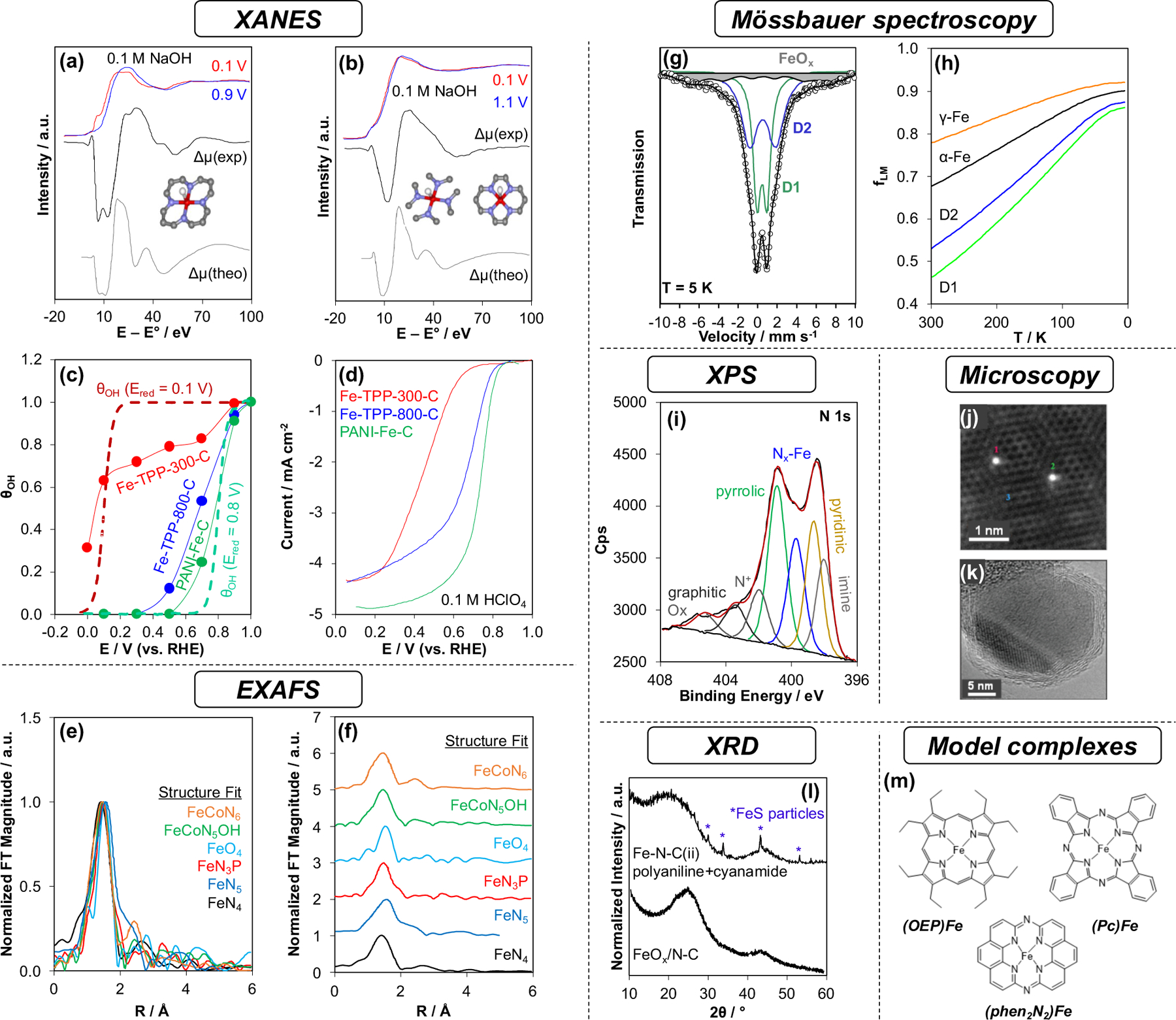
Commonly used approaches to characterize metal sites in M-N-C materials: (a-d) in situ XANES Δμ technique,55,174 (e, f) EXAFS,178–183 (g, h) Mössbauer spectroscopy, (i) XPS,208 (j, k) electron microscopy,211,215 (l) XRD, 211,171 and (m) model complexes.170 The EXAFS spectra in (e) and (f) are normalized to their maximum intensity for comparative purposes. The XRD patterns in (l) are normalized to their maximum intensity for comparative purposes. Panels (a) and (b) are adapted from Ref. 55. Copyright 2013 American Chemical Society. Panels (c) and (d) are adapted from Ref. 174. Copyright 2015 American Chemical Society. Panels (e) and (f) contain spectra adapted with permission from Refs. 15,179–183. Copyright 2019 American Chemical Society, 2019 Wiley-VCH, 2019 Royal Society of Chemistry, 2019 American Chemical Society, 2020 Wiley-VCH, 2020 American Chemical Society. Panels (g) and (h) are adapted with permission from Ref. 198. Copyright 2016 Elsevier. Panel (i) is adapted with permission from Ref. 208. Copyright 2016 Elsevier. Panel (j) is adapted with permission from Ref. 211. Copyright 2017 American Association for the Advancement of Science. Panel (k) is adapted with permission from Ref. 215. Copyright 2011 American Association for the Advancement of Science. The top trace in panel (l) is adapted with permission from Ref. 211. Copyright 2017 American Association for the Advancement of Science. The bottom trace in panel (l) is adapted from Ref. 171. Copyright 2021 American Chemical Society.
Mukerjee and coworkers174 extended the applications of the in situ Δμ technique to these Fe-N-C(i) samples and a benchmark polyaniline-derived Fe-N-C(ii) catalyst34 as a function of potential in acid electrolyte. The signal was interpreted to reflect the reaction: Nx–FeII+H2O ⇌ Nx–FeIII–OHads + H+ + e−. Fitting was therefore used to quantify the fractional coverage of active sites by hydroxide (θOH) as a function of potential (solid lines, Figure 13c). This measured dependence was reasonably modeled as a surface-mediated electrocatalytic redox process (dashed lines, Figure 13c) and correlated with the onset of ORR catalysis measured in separate rotating disc electrode experiments (Figure 13d). This technique was also successfully applied later175 to demonstrate that CoN4 sites do not undergo this distinct redox transformation as a function of potential that FeN4 sites experience. Recently, advances in the time resolution of XAS have enabled the use of transient XAS measurements in combination with modulation excitation spectroscopy176 to improve the resolution of Δμ spectra and to probe the kinetics of FeII/III redox processes.177 In summary, in situ XANES studies provide active-site-specific structural information in cases where conditions can be designed to effect a selective change in their structure or adsorbate binding. These methods have not yet been applied to M-N-C aerobic oxidation catalysts, but they have the potential to provide valuable insights into the active centers.
5.1.2. EXAFS.
The EXAFS of atomically dispersed M-N-C catalysts is typified by a strong first-shell scattering from nearest-neighbor atoms and weaker second-shell scattering from the surrounding carbon support. These features can be seen in the radial-space EXAFS representation obtained by Fourier transform analysis according to the EXAFS equation (Figure 13e,f). Fitting of the EXAFS is an art that requires judicious control of the many fitting parameters involved and consideration of a variety of candidate structures. Often, many potential structures will fit the same EXAFS signal and complementary characterization is necessary to distinguish among them. The EXAFS signal of Fe-N-C(v) ORR catalysts15,178 (black trace, Figure 13e,f) assigned to the canonical FeN4 structure is difficult to distinguish from the EXAFS signals of Fe-N-C materials assigned to a variety of other structures.179–183 The challenge in fitting the EXAFS is that it averages contributions from all structures in the sample, but frequently the EXAFS is fit to a single structure without quantitative corroborating evidence that other species do not coexist with a more reasonable species, such as MN4. Chief among such convoluting species are metal aggregates that frequently coexist with atomically dispersed moieties. Feng et al.184 showed that EXAFS may not detect nanoparticles when they are present as a mixture with single-atom sites. As a result, caution should be taken in the interpretation of EXAFS without additional evidence for a lack of nanoparticle species.185 In general, the second shell scattering of the EXAFS is difficult to interpret and could result from a variety of factors, including different carbon support environments; nanoparticles of different size, abundance, and chemical composition (oxide, nitride, carbide, etc); and/or binuclear site configurations. The interpretation of the first-shell ligands of metals in M-N-Cs by EXAFS is also prone to pitfalls186 because light elements tend to scatter similarly, making it difficult to differentiate neighbors on the periodic table from one another (e.g., C vs. N vs. O). The coordination number is also subject to error186 when mononuclear species coexist with others that aren’t included in the fit. Nevertheless, EXAFS provides valuable structural information when samples are well controlled154,178 or when it is used in situ to track major structural changes across a range of conditions.31,176,187 The EXAFS of M-N-Cs have often been interpreted in aerobic oxidation studies without consideration of the aforementioned pitfalls. In situ XAS has not been used to follow the evolution of MNx structures during aerobic oxidation catalysis.
5.2. Mössbauer Spectroscopy
Mössbauer spectroscopy is especially useful for the characterization of Fe-N-C catalysts. The content in this section complements other reports that survey the broader application of this technique in heterogeneous catalysis,188,189 fundamental principles of Mössbauer spectroscopy,190 and practical recommendations.191 Briefly, the Mössbauer effect describes the recoilless emission of γ-rays and their resonant absorption by a nucleus of the same element in the sample. This absorption occurs with recoil, as described by momentum conservation, meaning that variations in the relative doppler velocity of the emitter and absorber tunes the energy of absorption, which is further influenced by nuclear hyperfine interactions specific to the sample. Due to these features, Mössbauer spectroscopy is element-specific and highly sensitive to metal oxidation state and its ligand field; however, it is limited to only a few Mössbauer-active elements. Most Mössbauer spectra in ORR studies are reported on Fe-N-C materials, although it has also been used to characterize Sn-N-C.152 Emission Mössbauer is also possible with 57Co-enriched Co-N-C.5
Early 57Fe Mössbauer studies4,192 of Fe-N-C detected a variety of Fe species, including FeII, FeIII, Fe0, and carbide- and oxide-based Fe aggregates. Structures of FeNx doublet species were proposed by comparison with macrocyclic complexes.193–195 Dodelet and coworkers196 studied five Fe-N-C(i,ii) catalysts, which featured many different 57Fe species present in the Mössbauer spectrum. The peak area of a doublet termed “D1” (ambient T, δ = 0.3 mm s−1, ΔEQ = 0.86 mm s−1) in these materials correlated with the ORR fuel cell current (0.8 VRHE). The advent of MOF-precursor synthesis methods has enabled the characterization of Fe-N-C(v) materials with less heterogeneity when Fe loadings are kept relatively low. Even with these samples, however, a mixture of two species is typically observed in the 57Fe Mössbauer spectra178 (Figure 13g). Jaouen and coworkers197 studied an Fe-N-C(v) with only “D1” and “D2” doublet Fe species at low temperature (4.5 K, 80 K) under an external magnetic field and compared the spectra with those calculated for a variety of periodic and cluster-based FeN4C10–12 models. DFT calculations indicated that the D1 doublet (80 K, δ = 0.46 mm s−1, ΔEQ = 0.94 mm s−1) is consistent with a porphyrinic high-spin FeIIIN4C12 moiety and the D2 doublet (80 K, δ = 0.49 mm s−1, ΔEQ = 2.25 mm s−1) is consistent with a pyridinic low- or intermediate-spin FeIIN4C10. The D1 site is proposed to be accessible to O2 and remains oxidized under the conditions of the measurement, whereas D2 is either occluded within inaccessible regions or binds O2 weakly such that it is reduced to FeII during the measurement. Jaouen and coworkers198 measured the temperature dependence of the Lamb-Mössbauer factors (fLM, Figure 13h) of the D1 and D2 species, enabling their accurate quantification. The value of fLM becomes similar for all species at low temperatures, indicating that where possible, spectra should be collected at <10 K for the measurement to be rigorously quantitative. In addition, Kramm and coworkers166 showed that nanoparticulate FeOx species are frequently present in Fe-N-C materials and can be deconvoluted as a magnetic sextet in low-temperature 57Fe Mössbauer spectra (4.2 K). Such FeOx moieties lose their magnetic ordering at higher temperatures and appear as a doublet similar to D1. Thus, low-temperature 57Fe Mössbauer measurements are crucial for accurate assignment and quantification of FeN4 species. Yet, very few examples have been published of Fe-N-C with low-temperature 57Fe Mössbauer data that unambiguously show a lack of FeOx aggregates.154 This type of evidence is essential to justify claims that Fe species exist solely as mononuclear sites.
In situ 57Fe Mössbauer spectroscopy has also contributed to understanding which FeNx sites are catalytically relevant. In an early study, van der Kraan and coworkers199 observed by in situ 57Fe Mössbauer spectroscopy that a fraction of Fe sites are reduced when an Fe(porphyrin)-derived Fe-N-C(i) is polarized at −0.05 VRHE. Neidig, Holby, Zelenay, and coworkers165 compared the 57Fe Mössbauer spectra (80 K) of a polyaniline-derived Fe-N-C(ii) catalyst exposed to ambient conditions, after reduction at −0.1 VRHE, and after the reduced catalyst was exposed to 1 atm NO(g). A doublet with similar characteristics (δ = 0.53 mm s−1, ΔEQ = 0.83 mm s−1) to the FeIIIN4 D1 species identified by others (referred to as Doublet 4 in this study) was reduced to a new species (δ = 1.54 mm s−1, ΔEQ = 2.72 mm s−1), and then shifted back after exposure to NO(g) (δ = 0.60 mm s− 1, ΔEQ = 0.56 mm s−1). The authors concluded that the shift after NO exposure indicated that the FeIINx site was reoxidized to FeIIINx. Although all the doublets that the authors deconvoluted were reduced electrochemically, only this D1 doublet shifted after NO exposure, indicating that this is likely the only gas-accessible and catalytically relevant Fe site, consistent with later assignments197 made based on ex situ spectra and DFT calculations described above. Jaouen and coworkers200 studied the in situ 57Fe Mössbauer spectra of an Fe-N-C(v) during fuel cell operation and found that this D1 FeNx center catalyzed ORR with the highest TOF, but that it was less stable than the D2 site. The authors speculated that either a more graphitic local structure changes the FeII/III redox potential of the D2 site which makes it more difficult to demetalate, or that they may be protected beneath a layer of N-doped carbon that protects them from demetallation but enable ORR catalysis at N-based active sites in that layer. The importance of either type of FeNx site for aerobic oxidation catalysis is unknown because in situ 57Fe Mössbauer spectroscopy has not yet been used in these studies.
5.3. X-Ray Photoelectron Spectroscopy
XPS is a surface-sensitive technique conducted under high-vacuum conditions (with the exception of specially designed equipment for “near-ambient pressure” XPS). The XPS spectra of the metal species incorporated in M-N-C materials are uninformative, beyond providing a verification that MII and possibly MIII species exist in the material.201 The N 1s binding energy is characteristic of different surface functional groups in carbon, including: oxygenated N (e.g., –NO2), graphitic N, N+ (e.g., in pyridine-N-oxide, N+–O−), pyrrolic N, pyridinic N, and imine N (Figure 13i). Early assignments for these N environments202 have been refined using more accurate references. Specifically, the N1s spectra of reference materials such as polypyrrole and polypyridine have demonstrated that hydrogenated pyridinic species appear in the same region assigned to pyrrolic species, which are sometimes collectively referred to as “hydrogenated species.”203 XPS practitioners have warned that care should be taken with binding energy referencing, especially when the C1s peak is used.204,205
A feature assigned to N bound to M (399.5 eV for Nx-Fe in Figure 13i) is relevant to the characterization of active sites in M-N-C materials. This feature is referred to as FeNx because the spectra do not have sufficient resolution to differentiate between FeN4 and other defective species such as FeN3 or FeN3C, which have similar binding energies according to DFT calculations.206,207 The DFT-calculated spectra of these different pyridinic FeNxCy site configurations and the XPS spectrum of a model pyridinic molecular complex170 both agree that the ligating N atoms in the FeNx site are pyridinic. Because of these ambiguities, expert practitioners often refrain from quantitative interpretation of MNx speciation based on XPS alone, but qualitatively infer trends with respect to ORR reactivity and the relative abundance of pyridinic N in families of samples.205,208,209,210 Although XPS has been widely used in aerobic oxidation studies, its lack of precision for quantitatively characterizing MNx speciation requires complementary techniques.
5.4. Electron Microscopy
Transmission electron microscopy (TEM) and scanning transmission electron microscopy (STEM) have become increasingly common in the characterization of M-N-C materials, with improvements in resolution and the growing accessibility of advanced instrumentation. Specifically, high resolution high-angle annular dark-field imaging (HAADF-STEM) with aberration correction can provide atomic-resolution images of heavy atoms in light-atom matrices because contrast is a function of atomic number. HAADF-STEM images reported by Zelenay and coworkers211 first identified FeNx sites incorporated within graphitic sheets (Figure 13j). Since this report, many others have observed atomically dispersed metal sites in N-doped carbons by this technique.212–214 Zelenay and coworkers211 combined their HAADF-STEM imaging with electron energy loss spectroscopy (EELS) to conclude that the elemental composition of the atomically dispersed sites was, on average, FeN4. Care must be taken in acquiring EELS or energy-dispersive x-ray spectroscopy (EDX) signals in order to avoid beam damage during longer collection time. HRTEM images showing carbon-encapsulated metal nanoparticles have also been reported215 (Figure 13k). TEM imaging has shown that carbon-encapsulated particles are resistant to washing by concentrated HF.216
Electron microscopy can be subject to a few artifacts. A bright spot can indicate that multiple lighter atoms are aligned on top of one another, rather than the presence of a heavier metal atom. Imaging thin regions of the sample and corroborating elemental composition with EELS or EDX can help avoid this pitfall. Further, STEM images may represent a statistically insignificant portion of the bulk material. Efforts can be made to minimize this uncertainty by imaging more overall area and estimating the surface density of single atoms, which should match that of a bulk elemental analysis technique together with the material’s surface area.217 Electron microscopy has been applied in aerobic oxidation studies to suggest that either aggregates or MNx sites are responsible for catalysis, but complementary techniques are required for quantitative assessment of their relative abundance and catalytic relevance.
5.5. X-Ray Diffraction
X-Ray diffraction (XRD) is broadly used in studies of aerobic oxidations catalyzed by M-N-Cs but is rarely combined with other techniques that quantitatively characterize active sites. There are several pitfalls in the interpretation of XRD patterns218 and the most prominent in the aerobic oxidation literature is to infer that presence or absence of an XRD feature is representative of all the metal sites in the material. On one hand, the detection of metal aggregates (e.g., in polyaniline-derived Fe-N-C(ii),211 Figure 13l) does not preclude the existence of atomically dispersed sites (imaged in Fe-N-C(ii) in Figure 13j). Or, aggregates may be inaccessible to substrates within the bulk of the material and, therefore, catalytically irrelevant (e.g., Figure 13k). On the other hand, features for aggregates may be completely absent in XRD patterns collected on materials known to contain aggregates, such as intentionally synthesized FeOx/N-C materials171 (Figure 13l). This ambiguity is an inherent limitation of the XRD technique because line broadening is inversely proportional to aggregate size, and nanoparticles smaller than ~3 nm cannot be detected.218 As a result, XRD can be used in aerobic oxidation studies as a diagnostic for the presence of aggregates but must be combined with other techniques to provide evidence for the identity of active sites.
5.6. Model Complexes
The data provided by the preceding tools can be augmented via comparisons with well-defined molecular complexes hypothesized to represent the local coordination environment of M-N-Cs. Surendranath and coworkers170 synthesized an N4 macrocycle (phen2N2, Figure 13m) containing ligating pyridinic N atoms that contrast the pyrrolic N atoms of commonly used molecular analogs such as phthalocyanine (Pc) and octaethylporphyrin (OEP). The (phen2N2)Fe complex showed spectroscopic features (N 1s XPS, Fe k-edge XANES and EXAFS) and ORR reactivity (onset potential, H2O2 selectivity) that were a closer match with an Fe-N-C(v) than the other molecular analogs. Nbae and coworkers219 corroborated these findings and showed in a complementary study220 that a similar pyridinic FeN4 complex was more stable than FePc toward demetallation when both complexes adsorbed onto carbon black underwent ORR cycling in 0.5 M H2SO4. A similar CoN4 complex221 without bridgehead N atoms has also been reported to catalyze CO2 electroreduction with a higher TOF than pyrrolic CoN4 complexes. These studies demonstrate that the structure of the macrocyclic unit that surrounds the atomically dispersed metal components in M-N-C catalysts can strongly influence their reactivity, even when they are nominally ligated within an N4 environment. These effects have not been explored in the context of aerobic oxidation catalysis on M-N-Cs.
6. OUTSTANDING CHALLENGES AND FUTURE DIRECTIONS
The foregoing sections juxtapose the development of M-N-Cs as aerobic oxidation catalysts with the state-of-the-art in characterizing the active centers of M-N-C catalysts in the ORR field. Below, we highlight four key areas where insights from the ORR field is positioned to drive new developments in aerobic oxidation.
6.1. Quantitative Structural Characterizations of Active Centers
Studies reporting aerobic oxidation chemistries on M-N-C catalysts have typically not reported quantitative assessment of active site densities beyond determining the bulk metal content of the catalysts. As a result, characterization methods that reveal metal structures remain correlational at best with respect to catalyst reactivity, and most studies fail to conclusively demonstrate whether MNx species or metal aggregates are the dominant active centers. Adoption of active site quantification techniques like the ones developed by ORR researchers would facilitate comparison of catalyst materials within studies and between laboratories. Active site densities, which may be a minority of the bulk metal, can then be related more directly to characterized structures and reaction rates. Chemisorption studies and in situ poisoning of active centers are promising for this purpose and should be applied in aerobic oxidation studies. The combination of these techniques will help clarify whether MNx centers or metal aggregates are the dominant active centers for aerobic oxidations, which is highlighted as an unresolved gap in knowledge by our survey in Section 3. Finally, because bulk materials properties are often only indirectly related to catalytic performance, such analyses will improve reproducibility in materials synthesis and enable meaningful comparisons between different materials classes in terms of their catalytically relevant active sites.
6.2. Kinetic Benchmarking with Turnover Rates
Comparing catalytic materials made by different synthetic routes or from different laboratories requires benchmarking with turnover rates.112–114 Most aerobic oxidation studies do not compare M-N-C catalysts on the basis of turnover rates as adopted by the ORR field.138,153 The diverse range of chemistries performed using M-N-Cs (cf. Section 3) has thus far limited the adoption of a standard reaction for turnover rate benchmarking. This weakness, combined with a dearth of active site quantification, makes the quantification of turnover rates rare. Meaningful kinetic data must reflect measured rates that are free from transport artifacts. Further work is required to assess the viability of a proposed benchmark reaction by correlating its rates on a wide variety of M-N-Cs with quantitative measurements of their site densities.
6.3. Mechanistic Insights into Aerobic Oxidations
The range of aerobic oxidation chemistries catalyzed by M-N-C materials belies a lack of knowledge about the elementary steps that comprise the catalytic cycle of each transformation. The ways in which the active centers of M-N-C catalysts facilitate the activation of O2 and organic molecules within one catalytic cycle may be diverse. Mechanistic analyses can suggest ways to improve rates or to develop new chemistries by modifying active centers, targeting a kinetic branch point, or hypothesizing a particular step with a new coupling partner. Beyond the elucidation of elementary reaction steps, there is also limited understanding of whether M-N-Cs catalyze aerobic oxidations by the ISR or IHR pathways described in studies of hydroquinone oxidation (cf. Figure 3b).53,54
6.4. Harnessing Materials Synthesis for Well-Defined Catalyst Structures
Most M-N-C catalysts employed for aerobic oxidation chemistries are ill-defined materials made by the pyrolysis of supported molecular precursors. The palette of M-N-C materials,9,11 developed for ORR has yet to be fully leveraged in studies of aerobic oxidation reactions. Precise materials synthesis has the potential to access a broader range of active site structures and/or more uniform active site structures. Uniform active centers can simplify the interpretation of rate data by making the identity of the active site less ambiguous. Collaborations between ORR researchers and aerobic oxidation researchers could facilitate knowledge sharing in materials synthesis and characterization and catalytic applications.
ACKNOWLEDGMENTS
The authors acknowledge Abdulhadi Al-Zahrani for assistance compiling site titration data. Funding to support research from our lab included in this article and manuscript preparation was provided by the Center for Molecular Electrocatalysis, an Energy Frontier Research Center, funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences (J.S.B., M.R.J., T.W.R., S.S.S. – research focused on M-N-C catalyzed reactions for the ORR and mediated electrochemistry), U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, DE-FG02–05ER15690 (J.S.B., F.K., S.S.S. – research focused on preparation and characterization of single-site M-N-C catalysts), and Ruth L. Kirschstein NRSA fellowship from the NIH, F32GM137472 (J.S.B.).
Biographies
Jason S. Bates received his B.S. in Chemical Engineering at the University of Kansas in 2014 and a Ph.D. in Chemical Engineering at Purdue University in 2019, under the supervision of Rajamani Gounder. He is currently an NIH postdoctoral fellow at the University of Wisconsin–Madison in the department of Chemistry, under the supervision of Shannon S. Stahl. His research explores the fundamentals of heterogeneous (electro)catalysis in areas relevant to decarbonization of the energy and chemical industries.
Mathew R. Johnson received his B.S. in Chemistry from Whittier College in 2016. He is currently a graduate student at the University of Wisconsin–Madison in the department of Chemistry, under the supervision of Shannon S. Stahl. His research focuses on the integration of heterogeneous and homogeneous catalysts into electrochemical flow systems.
Fatemeh Khamespanah received her B.S in Chemistry at Sharif University of Technology in 2014 and a Ph.D. in Chemistry at Louisiana State University in 2020, under the supervision of Prof. Andrew W. Maverick. She is currently a postdoctoral researcher at the University of Wisconsin–Madison under the supervision of Prof. Shannon S. Stahl. Her research has included catalysis with homogeneous complexes toward activation of CO2 and the synthesis and characterization of atomically dispersed Fe-N-C heterogeneous catalysts using strategies from synthesis of molecular complexes.
Thatcher Root obtained degrees in Chemistry and Chemical Engineering at Massachusetts Institute of Technology and the University of Minnesota. After a postdoctoral appointment at AT&T Bell Laboratories, he joined the Chemical Engineering faculty at the University of Wisconsin–Madison where he is now Professor of Chemical and Biological Engineering. He has investigated catalysis in systems ranging from automobile exhaust emissions to fine chemicals and pharmaceuticals.
Shannon S. Stahl obtained his B.S. in Chemistry at the University of Illinois Urbana–Champaign and a Ph.D from Caltech in 1997, under the supervision of Prof. John Bercaw. He was an NSF postdoctoral fellow with Prof. Stephen Lippard at Massachusetts Institute of Technology from 1997–1999. He is currently a Steenbock Professor of Chemical Sciences at the University of Wisconsin–Madison, where he began his independent career in 1999. His research group specializes in catalysis, with an emphasis on the catalytic chemistry of molecular oxygen and aerobic oxidation reactions, and electrocatalytic reactions related to chemical synthesis and energy conversion.
REFERENCES
- (1).Cui X; Li W; Ryabchuk P; Junge K; Beller M Bridging Homogeneous and Heterogeneous Catalysis by Heterogeneous Single-Metal-Site Catalysts. Nat. Catal 2018, 1, 385. [Google Scholar]
- (2).Kaiser SK; Chen Z; Faust Akl D; Mitchell S; Pérez-Ramírez J Single-Atom Catalysts across the Periodic Table. Chem. Rev 2020, 120, 11703–11809. [DOI] [PubMed] [Google Scholar]
- (3).Bagotzky VS; Tarasevich MR; Radyushkina KA; Levina OA; Andrusyova SI Electrocatalysis of the Oxygen Reduction Process on Metal Chelates in Acid Electrolyte. J. Power Sources 1978, 2, 233–240. [Google Scholar]
- (4).van Veen JAR; van Baar JF; Kroese KJ Effect of Heat Treatment on the Performance of Carbon-Supported Transition-Metal Chelates in the Electrochemical Reduction of Oxygen. J. Chem. Soc., Faraday Trans January 1981, 77, 2827–2843. [Google Scholar]
- (5).Scherson DA; Gupta SL; Fierro C; Yeager EB; Kordesch ME; Eldridge J; Hoffman RW; Blue J Cobalt Tetramethoxyphenyl Porphyrin—Emission Mossbauer Spectroscopy and O2 Reduction Electrochemical Studies. Electrochim. Acta 1983, 28, 1205–1209. [Google Scholar]
- (6).Gewirth AA; Varnell JA; DiAscro AM Nonprecious Metal Catalysts for Oxygen Reduction in Heterogeneous Aqueous Systems. Chem. Rev 2018, 118, 2313–2339. [DOI] [PubMed] [Google Scholar]
- (7).Wang W; Jia Q; Mukerjee S; Chen S Recent Insights into the Oxygen-Reduction Electrocatalysis of Fe/N/C Materials. ACS Catal 2019, 9, 10126–10141. [Google Scholar]
- (8).Zhao C-X; Li B-Q; Liu J-N; Zhang Q Intrinsic Electrocatalytic Activity Regulation of M–N–C Single-Atom Catalysts for the Oxygen Reduction Reaction. Angew. Chem. Int. Ed 2021, 60, 4448–4463. [DOI] [PubMed] [Google Scholar]
- (9).He Y; Liu S; Priest C; Shi Q; Wu G Atomically Dispersed Metal–Nitrogen–Carbon Catalysts for Fuel Cells: Advances in Catalyst Design, Electrode Performance, and Durability Improvement. Chem. Soc. Rev 2020, 49, 3484–3524. [DOI] [PubMed] [Google Scholar]
- (10).Sun T; Tian B; Lu J; Su C Recent Advances in Fe (or Co)/N/C Electrocatalysts for the Oxygen Reduction Reaction in Polymer Electrolyte Membrane Fuel Cells. J. Mater. Chem. A 2017, 5, 18933–18950. [Google Scholar]
- (11).Martinez U; Komini Babu S; Holby EF; Chung HT; Yin X; Zelenay P Progress in the Development of Fe-Based PGM-Free Electrocatalysts for the Oxygen Reduction Reaction. Adv. Mater 2019, 31, 1806545. [DOI] [PubMed] [Google Scholar]
- (12).Shi Q; Hwang S; Yang H; Ismail F; Su D; Higgins D; Wu G Supported and Coordinated Single Metal Site Electrocatalysts. Mater. Today 2020, 37, 93–111. [Google Scholar]
- (13).Asset T; Atanassov P Iron-Nitrogen-Carbon Catalysts for Proton Exchange Membrane Fuel Cells. Joule 2020, 4, 33–44. [Google Scholar]
- (14).Varela AS; Ranjbar Sahraie N; Steinberg J; Ju W; Oh H-S; Strasser P Metal-Doped Nitrogenated Carbon as an Efficient Catalyst for Direct CO2 Electroreduction to CO and Hydrocarbons. Angew. Chem. Int. Ed 2015, 54, 10758–10762. [DOI] [PubMed] [Google Scholar]
- (15).Li J; Pršlja P; Shinagawa T; Martín Fernández AJ; Krumeich F; Artyushkova K; Atanassov P; Zitolo A; Zhou Y; García-Muelas R; et al. Volcano Trend in Electrocatalytic CO2 Reduction Activity over Atomically Dispersed Metal Sites on Nitrogen-Doped Carbon. ACS Catal 2019, 9, 10426–10439. [Google Scholar]
- (16).Geng Z; Liu Y; Kong X; Li P; Li K; Liu Z; Du J; Shu M; Si R; Zeng J Achieving a Record-High Yield Rate of 120.9 for N2 Electrochemical Reduction over Ru Single-Atom Catalysts. Adv. Mater 2018, 30, 1803498. [DOI] [PubMed] [Google Scholar]
- (17).Jagadeesh RV; Wienhöfer G; Westerhaus FA; Surkus A-E; Pohl M-M; Junge H; Junge K; Beller M Efficient and Highly Selective Iron-Catalyzed Reduction of Nitroarenes. Chem. Commun 2011, 47, 10972. [DOI] [PubMed] [Google Scholar]
- (18).Westerhaus FA; Jagadeesh RV; Wienhöfer G; Pohl M-M; Radnik J; Surkus A-E; Rabeah J; Junge K; Junge H; Nielsen M; et al. Heterogenized Cobalt Oxide Catalysts for Nitroarene Reduction by Pyrolysis of Molecularly Defined Complexes. Nat. Chem 2013, 5, 537–543. [DOI] [PubMed] [Google Scholar]
- (19).Jagadeesh RV; Surkus A-E; Junge H; Pohl M-M; Radnik J; Rabeah J; Huan H; Schünemann V; Brückner A; Beller M Nanoscale Fe2O3-Based Catalysts for Selective Hydrogenation of Nitroarenes to Anilines. Science 2013, 342, 1073–1076. [DOI] [PubMed] [Google Scholar]
- (20).Jagadeesh RV; Murugesan K; Alshammari AS; Neumann H; Pohl M-M; Radnik J; Beller M MOF-Derived Cobalt Nanoparticles Catalyze a General Synthesis of Amines. Science 2017, 358, 326–332. [DOI] [PubMed] [Google Scholar]
- (21).He L; Weniger F; Neumann H; Beller M Synthesis, Characterization, and Application of Metal Nanoparticles Supported on Nitrogen-Doped Carbon: Catalysis beyond Electrochemistry. Angew. Chem. Int. Ed 2016, 55, 12582–12594. [DOI] [PubMed] [Google Scholar]
- (22).Mallat T; Baiker A Oxidation of Alcohols with Molecular Oxygen on Solid Catalysts. Chem. Rev 2004, 104, 3037–3058. [DOI] [PubMed] [Google Scholar]
- (23).Liu X; Madix RJ; Friend CM Unraveling Molecular Transformations on Surfaces: A Critical Comparison of Oxidation Reactions on Coinage Metals. Chem. Soc. Rev 2008, 37, 2243–2261. [DOI] [PubMed] [Google Scholar]
- (24).Davis SE; Ide MS; Davis RJ Selective Oxidation of Alcohols and Aldehydes over Supported Metal Nanoparticles. Green Chem. 2012, 15, 17–45. [Google Scholar]
- (25).Miyamura H; Kobayashi S Tandem Oxidative Processes Catalyzed by Polymer-Incarcerated Multimetallic Nanoclusters with Molecular Oxygen. Acc. Chem. Res 2014, 47, 1054–1066. [DOI] [PubMed] [Google Scholar]
- (26).Stahl SS Palladium Oxidase Catalysis: Selective Oxidation of Organic Chemicals by Direct Dioxygen-Coupled Turnover. Angew. Chem. Int. Ed 2004, 43, 3400–3420. [DOI] [PubMed] [Google Scholar]
- (27).Piera J; Bäckvall J-E Catalytic Oxidation of Organic Substrates by Molecular Oxygen and Hydrogen Peroxide by Multistep Electron Transfer—A Biomimetic Approach. Angew. Chem. Int. Ed 2008, 47, 3506–3523. [DOI] [PubMed] [Google Scholar]
- (28).Wendlandt AE; Suess AM; Stahl SS Copper-Catalyzed Aerobic Oxidative C–H Functionalizations: Trends and Mechanistic Insights. Angew. Chem. Int. Ed 2011, 50, 11062–11087. [DOI] [PubMed] [Google Scholar]
- (29).Allen SE; Walvoord RR; Padilla-Salinas R; Kozlowski MC Aerobic Copper-Catalyzed Organic Reactions. Chem. Rev 2013, 113, 6234–6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ryland BL; Stahl SS Practical Aerobic Oxidations of Alcohols and Amines with Homogeneous Copper/TEMPO and Related Catalyst Systems. Angew. Chem. Int. Ed 2014, 53, 8824–8838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Li J; Jiao L; Wegener E; Richard LL; Liu E; Zitolo A; Sougrati MT; Mukerjee S; Zhao Z; Huang Y; et al. Evolution Pathway from Iron Compounds to Fe1(II)–N4 Sites through Gas-Phase Iron during Pyrolysis. J. Am. Chem. Soc 2020, 142, 1417–1423. [DOI] [PubMed] [Google Scholar]
- (32).Lefèvre M; Proietti E; Jaouen F; Dodelet J-P Iron-Based Catalysts with Improved Oxygen Reduction Activity in Polymer Electrolyte Fuel Cells. Science 2009, 324, 71–74. [DOI] [PubMed] [Google Scholar]
- (33).Gupta S; Tryk D; Bae I; Aldred W; Yeager E Heat-Treated Polyacrylonitrile-Based Catalysts for Oxygen Electroreduction. J. Appl. Electrochem 1989, 19, 19–27. [Google Scholar]
- (34).Wu G; More KL; Johnston CM; Zelenay P High-Performance Electrocatalysts for Oxygen Reduction Derived from Polyaniline, Iron, and Cobalt. Science 2011, 332, 443–447. [DOI] [PubMed] [Google Scholar]
- (35).Han L; Cheng H; Liu W; Li H; Ou P; Lin R; Wang H-T; Pao C-W; Head AR; Wang C-H; et al. A Single-Atom Library for Guided Monometallic and Concentration-Complex Multimetallic Designs. Nat. Mater 2022, 21, 681–688. [DOI] [PubMed] [Google Scholar]
- (36).Serov A; Artyushkova K; Niangar E; Wang C; Dale N; Jaouen F; Sougrati M-T; Jia Q; Mukerjee S; Atanassov P Nano-Structured Non-Platinum Catalysts for Automotive Fuel Cell Application. Nano Energy 2015, 16, 293–300. [Google Scholar]
- (37).Serov A; Robson MH; Artyushkova K; Atanassov P Templated Non-PGM Cathode Catalysts Derived from Iron and Poly(Ethyleneimine) Precursors. Appl. Catal., B 2012, 127, 300–306. [Google Scholar]
- (38).Proietti E; Jaouen F; Lefèvre M; Larouche N; Tian J; Herranz J; Dodelet J-P Iron-Based Cathode Catalyst with Enhanced Power Density in Polymer Electrolyte Membrane Fuel Cells. Nat. Commun 2011, 2, 416. [DOI] [PubMed] [Google Scholar]
- (39).Zhang H; Osgood H; Xie X; Shao Y; Wu G Engineering Nanostructures of PGM-Free Oxygen-Reduction Catalysts Using Metal-Organic Frameworks. Nano Energy 2017, 31, 331–350. [Google Scholar]
- (40).Wang H-F; Chen L; Pang H; Kaskel S; Xu Q MOF-Derived Electrocatalysts for Oxygen Reduction, Oxygen Evolution and Hydrogen Evolution Reactions. Chem. Soc. Rev 2020, 49, 1414–1448. [DOI] [PubMed] [Google Scholar]
- (41).Wang A; Li J; Zhang T Heterogeneous Single-Atom Catalysis. Nat. Rev. Chem 2018, 2, 65–81. [Google Scholar]
- (42).Gawande MB; Fornasiero P; Zbořil R Carbon-Based Single-Atom Catalysts for Advanced Applications. ACS Catal 2020, 10, 2231–2259. [Google Scholar]
- (43).Singh B; Gawande MB; Kute AD; Varma RS; Fornasiero P; McNeice P; Jagadeesh RV; Beller M; Zbořil R Single-Atom (Iron-Based) Catalysts: Synthesis and Applications. Chem. Rev 2021, 121, 13620–13697. [DOI] [PubMed] [Google Scholar]
- (44).Anson CW; Ghosh S; Hammes-Schiffer S; Stahl SS Co(Salophen)-Catalyzed Aerobic Oxidation of p-Hydroquinone: Mechanism and Implications for Aerobic Oxidation Catalysis. J. Am. Chem. Soc 2016, 138, 4186–4193. [DOI] [PubMed] [Google Scholar]
- (45).Anson CW; Stahl SS Cooperative Electrocatalytic O2 Reduction Involving Co(Salophen) with p-Hydroquinone as an Electron–Proton Transfer Mediator. J. Am. Chem. Soc 2017, 139, 18472–18475. [DOI] [PubMed] [Google Scholar]
- (46).Singha A; Mondal A; Nayek A; Dey SG; Dey A Oxygen Reduction by Iron Porphyrins with Covalently Attached Pendent Phenol and Quinol. J. Am. Chem. Soc 2020, 142, 21810–21828. [DOI] [PubMed] [Google Scholar]
- (47).Hooe SL; Cook EN; Reid AG; Machan CW Non-Covalent Assembly of Proton Donors and p-Benzoquinone Anions for Co-Electrocatalytic Reduction of Dioxygen. Chem. Sci 2021, 12, 9733–9741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Zope BN; Hibbitts DD; Neurock M; Davis RJ Reactivity of the Gold/Water Interface During Selective Oxidation Catalysis. Science 2010, 330, 74–78. [DOI] [PubMed] [Google Scholar]
- (49).Wieckowski A; Neurock M Contrast and Synergy between Electrocatalysis and Heterogeneous Catalysis. Adv. in Phys. Chem 2011, 2011, 1–18. [Google Scholar]
- (50).Ide MS; Davis RJ The Important Role of Hydroxyl on Oxidation Catalysis by Gold Nanoparticles. Acc. Chem. Res 2014, 47, 825–833. [DOI] [PubMed] [Google Scholar]
- (51).Adams JS; Kromer ML; Rodríguez-López J; Flaherty DW Unifying Concepts in Electro- and Thermocatalysis toward Hydrogen Peroxide Production. J. Am. Chem. Soc 2021, 143, 7940–7957. [DOI] [PubMed] [Google Scholar]
- (52).Ryu J; Bregante DT; Howland WC; Bisbey RP; Kaminsky CJ; Surendranath Y Thermochemical Aerobic Oxidation Catalysis in Water Can Be Analysed as Two Coupled Electrochemical Half-Reactions. Nat. Catal 2021, 4, 742–752. [Google Scholar]
- (53).Bates JS; Biswas S; Suh S-E; Johnson MR; Mondal B; Root TW; Stahl SS Chemical and Electrochemical O2 Reduction on Earth-Abundant M-N-C Catalysts and Implications for Mediated Electrolysis. J. Am. Chem. Soc 2022, 144, 922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Howland WC; Gerken JB; Stahl SS; Surendranath Y Thermal Hydroquinone Oxidation on Co/N-doped Carbon Proceeds by a Band-Mediated Electrochemical Mechanism. J. Am. Chem. Soc 2022, 144, 11253–11262. [DOI] [PubMed] [Google Scholar]
- (55).Ramaswamy N; Tylus U; Jia Q; Mukerjee S Activity Descriptor Identification for Oxygen Reduction on Nonprecious Electrocatalysts: Linking Surface Science to Coordination Chemistry. J. Am. Chem. Soc 2013, 135, 15443–15449. [DOI] [PubMed] [Google Scholar]
- (56).Kaminsky CJ; Wright J; Surendranath Y Graphite-Conjugation Enhances Porphyrin Electrocatalysis. ACS Catal 2019, 9, 3667–3671. [Google Scholar]
- (57).Kaminsky CJ; Weng S; Wright J; Surendranath Y Adsorbed Cobalt Porphyrins Act like Metal Surfaces in Electrocatalysis. Nat. Catal 2022, 5, 430–442. [Google Scholar]
- (58).Bai C; Li A; Yao X; Liu H; Li Y Efficient and Selective Aerobic Oxidation of Alcohols Catalysed by MOF-Derived Co Catalysts. Green Chem 2016, 18, 1061–1069. [Google Scholar]
- (59).Zhao X; Zhou Y; Jin A-L; Huang K; Liu F; Tao D-J Co-N-C Catalysts Synthesized by Pyrolysis of Co-Based Deep Eutectic Solvents for Aerobic Oxidation of Alcohols. New J. Chem 2018, 42, 15871–15878. [Google Scholar]
- (60).Mao F; Qi Z; Fan H; Sui D; Chen R; Huang J Heterogeneous Cobalt Catalysts for Selective Oxygenation of Alcohols to Aldehydes, Esters and Nitriles. RSC Adv 2017, 7, 1498–1503. [Google Scholar]
- (61).Sun Y; Ma H; Luo Y; Zhang S; Gao J; Xu J Activation of Molecular Oxygen Using Durable Cobalt Encapsulated with Nitrogen-Doped Graphitic Carbon Shells for Aerobic Oxidation of Lignin-Derived Alcohols. Chem. Eur. J 2018, 24, 4653–4661. [DOI] [PubMed] [Google Scholar]
- (62).Senthamarai T; Chandrashekhar VG; Rockstroh N; Rabeah J; Bartling S; Jagadeesh RV; Beller MA “Universal” Catalyst for Aerobic Oxidations to Synthesize (Hetero)Aromatic Aldehydes, Ketones, Esters, Acids, Nitriles, and Amides. Chem 2022, 8, 508–531. [Google Scholar]
- (63).Xie J; Kammert JD; Kaylor N; Zheng JW; Choi E; Pham HN; Sang X; Stavitski E; Attenkofer K; Unocic RR; et al. Atomically Dispersed Co and Cu on N-Doped Carbon for Reactions Involving C–H Activation. ACS Catal 2018, 8, 3875–3884. [Google Scholar]
- (64).Xie J; Yin K; Serov A; Artyushkova K; Pham HN; Sang X; Unocic RR; Atanassov P; Datye AK; Davis RJ Selective Aerobic Oxidation of Alcohols over Atomically-Dispersed Non-Precious Metal Catalysts. ChemSusChem 2017, 10, 359–362. [DOI] [PubMed] [Google Scholar]
- (65).Huang K; Fu H; Shi W; Wang H; Cao Y; Yang G; Peng F; Wang Q; Liu Z; Zhang B; et al. Competitive Adsorption on Single-Atom Catalysts: Mechanistic Insights into the Aerobic Oxidation of Alcohols over Co–N–C. J. Catal 2019, 377, 283–292. [Google Scholar]
- (66).Li Z; Fan T; Li H; Lu X; Ji S; Zhang J; Horton JH; Xu Q; Zhu J Atomically Defined Undercoordinated Copper Active Sites over Nitrogen‐Doped Carbon for Aerobic Oxidation of Alcohols. Small 2022, 18, 2106614. [DOI] [PubMed] [Google Scholar]
- (67).Jagadeesh RV; Junge H; Pohl M-M; Radnik J; Brückner A; Beller M Selective Oxidation of Alcohols to Esters Using Heterogeneous Co3O4–N@C Catalysts under Mild Conditions. J. Am. Chem. Soc 2013, 135, 10776–10782. [DOI] [PubMed] [Google Scholar]
- (68).Zhong W; Liu H; Bai C; Liao S; Li Y Base-Free Oxidation of Alcohols to Esters at Room Temperature and Atmospheric Conditions Using Nanoscale Co-Based Catalysts. ACS Catal 2015, 5, 1850–1856. [Google Scholar]
- (69).Deng J; Song H-J; Cui M-S; Du Y-P; Fu Y Aerobic Oxidation of Hydroxymethylfurfural and Furfural by Using Heterogeneous CoxOy–N@C Catalysts. ChemSusChem 2014, 7, 3334–3340. [DOI] [PubMed] [Google Scholar]
- (70).Lv G; Chen S; Zhu H; Li M; Yang Y Pyridinic-Nitrogen-Dominated Nitrogen-Doped Graphene Stabilized Cu for Efficient Selective Oxidation of 5-Hydroxymethfurfural. Appl. Surf. Sci 2018, 458, 24–31. [Google Scholar]
- (71).Feng Y; Jia W; Yan G; Zeng X; Sperry J; Xu B; Sun Y; Tang X; Lei T; Lin L Insights into the Active Sites and Catalytic Mechanism of Oxidative Esterification of 5-Hydroxymethylfurfural by Metal-Organic Frameworks-Derived N-Doped Carbon. J. Catal 2020, 381, 570–578. [Google Scholar]
- (72).Su H; Zhang K-X; Zhang B; Wang H-H; Yu Q-Y; Li X-H; Antonietti M; Chen J-S Activating Cobalt Nanoparticles via the Mott-Schottky Effect in Nitrogen-Rich Carbon Shells for Base-Free Aerobic Oxidation of Alcohols to Esters. J. Am. Chem. Soc 2017, 139, 811–818. [DOI] [PubMed] [Google Scholar]
- (73).Li N; Shang S; Wang L; Niu J; Lv Y; Gao S Superior Performance of Co-N/m-C for Direct Oxidation of Alcohols to Esters under Air. Chin. J. Catal 2018, 39, 1249–1257. [Google Scholar]
- (74).Panwar V; Ray SS; Jain SL Highly Efficient (CoOx-N@C, PANI) Nanopowder Derived from Pyrolysis of Polyaniline Grafted Cobalt Acetate for Oxidative Methyl Esterification of Benzyl Alcohols. Mol. Catal 2017, 427, 31–38. [Google Scholar]
- (75).Long Z; Chen X; Lu P; Liu S; Zhang Y; Chen G; Tong M; Sun L; Zhan W; Huang F A Bi-Functional Cobalt and Nitrogen Co-Doped Carbon Catalyst for Aerobic Oxidative Esterification of Benzyl Alcohol with Methanol and Oxygen Reduction Reaction. Catal. Lett 2019, 149, 3160–3168. [Google Scholar]
- (76).Zhu Q; Wang F; Zhang F; Dong Z Renewable Chitosan-Derived Cobalt@N-Doped Porous Carbon for Efficient Aerobic Esterification of Alcohols under Air. Nanoscale 2019, 11, 17736–17745. [DOI] [PubMed] [Google Scholar]
- (77).Zhou Y-X; Chen Y-Z; Cao L; Lu J; Jiang H-L Conversion of a Metal–Organic Framework to N-Doped Porous Carbon Incorporating Co and CoO Nanoparticles: Direct Oxidation of Alcohols to Esters. Chem. Commun 2015, 51, 8292–8295. [DOI] [PubMed] [Google Scholar]
- (78).Jagadeesh RV; Junge H; Beller M Green Synthesis of Nitriles Using Non-Noble Metal Oxides-Based Nanocatalysts. Nat. Commun 2014, 5, 4123. [DOI] [PubMed] [Google Scholar]
- (79).Shang S; Wang L; Dai W; Chen B; Lv Y; Gao S High Catalytic Activity of Mesoporous Co–N/C Catalysts for Aerobic Oxidative Synthesis of Nitriles. Catal. Sci. Technol 2016, 6, 5746–5753. [Google Scholar]
- (80).Yasukawa T; Yang X; Kobayashi S Earth-Abundant Bimetallic Nanoparticle Catalysts for Aerobic Ammoxidation of Alcohols to Nitriles. J. Org. Chem 2020, 85, 7543–7548. [DOI] [PubMed] [Google Scholar]
- (81).Jagadeesh RV; Junge H; Beller M “Nanorust”-Catalyzed Benign Oxidation of Amines for Selective Synthesis of Nitriles. ChemSusChem 2015, 8, 92–96. [DOI] [PubMed] [Google Scholar]
- (82).Natte K; Jagadeesh RV; Sharif M; Neumann H; Beller M Synthesis of Nitriles from Amines Using Nanoscale Co3O4-Based Catalysts via Sustainable Aerobic Oxidation. Org. Biomol. Chem 2016, 14, 3356–3359. [DOI] [PubMed] [Google Scholar]
- (83).Sun K; Sun J; Lu G-P; Cai C Enhanced Catalytic Activity of Cobalt Nanoparticles Encapsulated with an N-Doped Porous Carbon Shell Derived from Hollow ZIF-8 for Efficient Synthesis of Nitriles from Primary Alcohols in Water. Green Chem 2019, 21, 4334–4340. [Google Scholar]
- (84).Sun K; Shan H; Neumann H; Lu G-P; Beller M Efficient Iron Single-Atom Catalysts for Selective Ammoxidation of Alcohols to Nitriles. Nat. Commun 2022, 13, 1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Shang S; Dai W; Wang L; Lv Y; Gao S Metal-Free Catalysis of Nitrogen-Doped Nanocarbons for the Ammoxidation of Alcohols to Nitriles. Chem. Commun 2017, 53, 1048–1051. [DOI] [PubMed] [Google Scholar]
- (86).Wang L; Ambrosi A; Pumera M “Metal-Free” Catalytic Oxygen Reduction Reaction on Heteroatom-Doped Graphene Is Caused by Trace Metal Impurities. Angew. Chem. Int. Ed 2013, 52, 13818–13821. [DOI] [PubMed] [Google Scholar]
- (87).Murugesan K; Senthamarai T; Sohail M; Sharif M; Kalevaru NV; Jagadeesh RV Stable and Reusable Nanoscale Fe2O3-Catalyzed Aerobic Oxidation Process for the Selective Synthesis of Nitriles and Primary Amides. Green Chem 2018, 20, 266–273. [Google Scholar]
- (88).Pan L; Fu W; Zhang L; Wang S; Tang T Highly Dispersed Co Species in N-Doped Carbon Enhanced the Aldehydes Ammoxidation Reaction Activity. Mol. Catal 2022, 518, 112087. [Google Scholar]
- (89).Zhao H; Sun X; Xu D; Zhu Q; Zhu Y; Dong Z Fe-Based N-Doped Dendritic Catalysts for Catalytic Ammoxidation of Aromatic Aldehydes to Aromatic Nitriles. J. Colloid Interface Sci 2020, 565, 177–185. [DOI] [PubMed] [Google Scholar]
- (90).Iosub AV; Stahl SS Catalytic Aerobic Dehydrogenation of Nitrogen Heterocycles Using Heterogeneous Cobalt Oxide Supported on Nitrogen-Doped Carbon. Org. Lett 2015, 17, 4404–4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Cui X; Li Y; Bachmann S; Scalone M; Surkus A-E; Junge K; Topf C; Beller M Synthesis and Characterization of Iron–Nitrogen-Doped Graphene/Core–Shell Catalysts: Efficient Oxidative Dehydrogenation of N-Heterocycles. J. Am. Chem. Soc 2015, 137, 10652–10658. [DOI] [PubMed] [Google Scholar]
- (92).Liao C; Li X; Yao K; Yuan Z; Chi Q; Zhang Z Efficient Oxidative Dehydrogenation of N-Heterocycles over Nitrogen-Doped Carbon-Supported Cobalt Nanoparticles. ACS Sustain. Chem. Eng 2019, 7, 3646–13654. [Google Scholar]
- (93).Wu Y; Chen Z; Cheong W-C; Zhang C; Zheng L; Yan W; Yu R; Chen C; Li Y Nitrogen-Coordinated Cobalt Nanocrystals for Oxidative Dehydrogenation and Hydrogenation of N-Heterocycles. Chem. Sci 2019, 10, 5345–5352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Sun S; Liu Z; Yang F; Qiu T; Wang M; Feng A; Wang Y; Li Y FexC Enhancing the Catalytic Activity of FeNx in Oxidative Dehydration of N-Heterocycles. Green Chemical Engineering 2021. DOI: 10.1016/j.gce.2021.12.007 [DOI]
- (95).He F; Mi L; Shen Y; Mori T; Liu S; Zhang Y Fe–N–C Artificial Enzyme: Activation of Oxygen for Dehydrogenation and Monoxygenation of Organic Substrates under Mild Condition and Cancer Therapeutic Application. ACS Appl. Mater. Interfaces 2018, 10, 35327–35333. [DOI] [PubMed] [Google Scholar]
- (96).Wu D; Li J; Xu S; Xie Q; Pan Y; Liu X; Ma R; Zheng H; Gao M; Wang W; et al. Engineering Fe–N Doped Graphene to Mimic Biological Functions of NADPH Oxidase in Cells. J. Am. Chem. Soc 2020, 142, 19602–19610. [DOI] [PubMed] [Google Scholar]
- (97).Sun K; Shan H; Ma R; Wang P; Neumann H; Lu G-P; Beller M Catalytic Oxidative Dehydrogenation of N-Heterocycles with Nitrogen/Phosphorus Co-Doped Porous Carbon Materials. Chem. Sci 2022, 13, 6865–6872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).He J; Lin F; Yang X; Wang D; Tan X; Zhang S Sustainable Synthesis of 2-Arylbenzoxazoles over a Cobalt-Based Nanocomposite Catalyst. Org. Process Res. Dev 2016, 20, 1093–1096. [Google Scholar]
- (99).Xu Q; Feng B; Ye C; Fu Y; Chen D-L; Zhang F; Zhang J; Zhu W Atomically Dispersed Vanadium Sites Anchored on N-Doped Porous Carbon for the Efficient Oxidative Coupling of Amines to Imines. ACS Appl. Mater. Interfaces 2021, 13, 15168–15177. [DOI] [PubMed] [Google Scholar]
- (100).Zhang Y; Wu Y; Su Y; Cao Y; Liang Z; Yang D; Yu R; Zhang D; Wu J; Xiao W; et al. In Situ Synthesis of CuN4/Mesoporous N‐Doped Carbon for Selective Oxidative Crosscoupling of Terminal Alkynes under Mild Conditions. Small 2022, 18, 2105178. [DOI] [PubMed] [Google Scholar]
- (101).Fu L; Chen Y; Zhao S; Liu Z; Zhu R Sulfur-Mediated Synthesis of N-Doped Carbon Supported Cobalt Catalysts Derived from Cobalt Porphyrin for Ethylbenzene Oxidation. RSC Adv 2016, 6, 19482–19491. [Google Scholar]
- (102).Qiu Y; Yang C; Huo J; Liu Z Synthesis of Co-N-C Immobilized on Carbon Nanotubes for Ethylbenzene Oxidation. J. Mol. Catal. A Chem 2016, 424, 276–282. [Google Scholar]
- (103).Yang C; Fu L; Zhu R; Liu Z Influence of Cobalt Species on the Catalytic Performance of Co-N-C/SiO2 for Ethylbenzene Oxidation. Phys. Chem. Chem. Phys 2016, 18, 4635–4642. [DOI] [PubMed] [Google Scholar]
- (104).Liu W; Zhang L; Liu X; Liu X; Yang X; Miao S; Wang W; Wang A; Zhang T Discriminating Catalytically Active FeNx Species of Atomically Dispersed Fe–N–C Catalyst for Selective Oxidation of the C–H Bond. J. Am. Chem. Soc 2017, 139, 10790–10798. [DOI] [PubMed] [Google Scholar]
- (105).Jie S; Yang C; Chen Y; Liu Z Facile Synthesis of Ultra-Stable Co-N-C Catalysts Using Cobalt Porphyrin and Peptides as Precursors for Selective Oxidation of Ethylbenzene. Mol. Catal 2018, 458, 1–8. [Google Scholar]
- (106).Tu D-H; Li Y; Li J; Gu Y-J; Wang B; Liu Z-T; Liu Z-W; Lu J Selective Oxidation of Arylalkanes with N-Graphitic-Modified Cobalt Nanoparticles in Water. Catal. Commun 2017, 97, 130–133. [Google Scholar]
- (107).Lin X; Jie S; Liu Z Sulfur and Nitrogen-Doped Porous Cobalt Carbon Catalyst for High Efficient Aerobic Oxidation of Hydrocarbons. Mol. Catal 2018, 455, 143–149. [Google Scholar]
- (108).Wang X; Jie S; Liu Z The Influence of Encapsulated Cobalt Content within N-Doped Bamboo-like Carbon Nanotubes Catalysts for Arylalkanes Oxidation. Mater. Chem. Phys 2019, 232, 393–399. [Google Scholar]
- (109).Luo H; Wang L; Shang S; Niu J; Gao S Aerobic Oxidative Cleavage of 1,2-Diols Catalyzed by Atomic-Scale Cobalt-Based Heterogeneous Catalyst. Commun. Chem 2019, 2, 17. [Google Scholar]
- (110).Xie C; Lin L; Huang L; Wang Z; Jiang Z; Zhang Z; Han B Zn-Nx Sites on N-Doped Carbon for Aerobic Oxidative Cleavage and Esterification of C(CO)-C Bonds. Nat. Commun 2021, 12, 4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Fogler HS Elements of Chemical Reaction Engineering; Prentice Hall, 2005. [Google Scholar]
- (112).Boudart M Turnover Rates in Heterogeneous Catalysis. Chem. Rev 1995, 95, 661–666. [Google Scholar]
- (113).Ribeiro FH; Wittenau AESV; Bartholomew CH; Somorjai GA Reproducibility of Turnover Rates in Heterogeneous Metal Catalysis: Compilation of Data and Guidelines for Data Analysis. Catal. Rev 1997, 39, 49–76. [Google Scholar]
- (114).Davis R Turnover Rates on Complex Heterogeneous Catalysts. AIChE J 2018, 64, 3778–3785. [Google Scholar]
- (115).Bard AJ; Faulkner LR Electrochemical Methods: Fundamentals and Applications, 2nd ed.; Wiley: New York, 2001. [Google Scholar]
- (116).Dix ST; Lu S; Linic S Critical Practices in Rigorously Assessing the Inherent Activity of Nanoparticle Electrocatalysts. ACS Catal 2020, 10, 10735–10741. [Google Scholar]
- (117).Gloaguen F; Convert P; Gamburzev S; Velev OA; Srinivasan S An Evaluation of the Macro-Homogeneous and Agglomerate Model for Oxygen Reduction in PEMFCs. Electrochim. Acta 1998, 43, 3767–3772. [Google Scholar]
- (118).Bonakdarpour A; Lefevre M; Yang R; Jaouen F; Dahn T; Dodelet J-P; Dahn JR Impact of Loading in RRDE Experiments on Fe–N–C Catalysts: Two- or Four-Electron Oxygen Reduction? Electrochem. Solid-State Lett 2008, 11, B105. [Google Scholar]
- (119).Robson MH; Serov A; Artyushkova K; Atanassov P A Mechanistic Study of 4-Aminoantipyrine and Iron Derived Non-Platinum Group Metal Catalyst on the Oxygen Reduction Reaction. Electrochim. Acta 2013, 90, 656–665. [Google Scholar]
- (120).Liang H-W; Wei W; Wu Z-S; Feng X; Müllen K Mesoporous Metal–Nitrogen-Doped Carbon Electrocatalysts for Highly Efficient Oxygen Reduction Reaction. J. Am. Chem. Soc 2013, 135, 16002–16005. [DOI] [PubMed] [Google Scholar]
- (121).Mears D Tests for Transport Limitations in Experimental Catalytic Reactors. Ind. Eng. Chem. Process Des. Dev 1971, 10, 541–547. [Google Scholar]
- (122).Madon RJ; Boudart M Experimental Criterion for the Absence of Artifacts in the Measurement of Rates of Heterogeneous Catalytic Reactions. Ind. Eng. Chem. Fund 1982, 21, 438–447. [Google Scholar]
- (123).Jaouen F; Dodelet J-P Average Turn-over Frequency of O2 Electro-Reduction for Fe/N/C and Co/N/C Catalysts in PEFCs. Electrochim. Acta 2007, 52, 5975–5984. [Google Scholar]
- (124).Snitkoff-Sol RZ; Friedman A; Honig HC; Yurko Y; Kozhushner A; Zachman MJ; Zelenay P; Bond AM; Elbaz L Quantifying the Electrochemical Active Site Density of Precious Metal-Free Catalysts in Situ in Fuel Cells. Nat. Catal 2022, 5, 163–170. [Google Scholar]
- (125).Bae IT; Scherson DA In Situ X-Ray Absorption of a Carbon Monoxide−Iron Porphyrin Adduct Adsorbed on High-Area Carbon in an Aqueous Electrolyte. J. Phys. Chem. B 1998, 102, 2519–2522. [Google Scholar]
- (126).Birry L; Zagal JH; Dodelet J-P Does CO Poison Fe-Based Catalysts for ORR? Electrochem. Commun 2010, 12, 628–631. [Google Scholar]
- (127).Zhang Q; Mamtani K; Jain D; Ozkan U; Asthagiri A CO Poisoning Effects on FeNC and CNx ORR Catalysts: A Combined Experimental–Computational Study. J. Phys. Chem. C 2016, 120, 15173–15184. [Google Scholar]
- (128).Maldonado S; Stevenson KJ Influence of Nitrogen Doping on Oxygen Reduction Electrocatalysis at Carbon Nanofiber Electrodes. J. Phys. Chem. B 2005, 109, 4707–4716. [DOI] [PubMed] [Google Scholar]
- (129).von Deak D; Singh D; King JC; Ozkan US Use of Carbon Monoxide and Cyanide to Probe the Active Sites on Nitrogen-Doped Carbon Catalysts for Oxygen Reduction. Appl. Catal., B 2012, 113–114, 126–133.
- (130).Thorum MS; Hankett JM; Gewirth AA Poisoning the Oxygen Reduction Reaction on Carbon-Supported Fe and Cu Electrocatalysts: Evidence for Metal-Centered Activity. J. Phys. Chem. Lett 2011, 2, 295–298. [Google Scholar]
- (131).Wang Q; Zhou Z-Y; Lai Y-J; You Y; Liu J-G; Wu X-L; Terefe E; Chen C; Song L; Rauf M; et al. Phenylenediamine-Based FeNx/C Catalyst with High Activity for Oxygen Reduction in Acid Medium and Its Active-Site Probing. J. Am. Chem. Soc 2014, 136, 10882–10885. [DOI] [PubMed] [Google Scholar]
- (132).Cheng T; Yu H; Peng F; Wang H; Zhang B; Su D Identifying Active Sites of CoNC/CNT from Pyrolysis of Molecularly Defined Complexes for Oxidative Esterification and Hydrogenation Reactions. Catal. Sci. Technol 2016, 6, 1007–1015. [Google Scholar]
- (133).Tang C; Surkus A-E; Chen F; Pohl M-M; Agostini G; Schneider M; Junge H; Beller M A Stable Nanocobalt Catalyst with Highly Dispersed CoNx Active Sites for the Selective Dehydrogenation of Formic Acid. Angew. Chem. Int. Ed 2017, 56, 16616–16620. [DOI] [PubMed] [Google Scholar]
- (134).Singh D; Mamtani K; Bruening CR; Miller JT; Ozkan US Use of H2S to Probe the Active Sites in FeNC Catalysts for the Oxygen Reduction Reaction (ORR) in Acidic Media. ACS Catal 2014, 4, 3454–3462. [Google Scholar]
- (135).Jain D; Gustin V; Basu D; Gunduz S; Deka DJ; Co AC; Ozkan US Phosphate Tolerance of Nitrogen-Coordinated-Iron-Carbon (FeNC) Catalysts for Oxygen Reduction Reaction: A Size-Related Hindrance Effect. J. Catal 2020, 390, 150–160. [Google Scholar]
- (136).Malko D; Kucernak A; Lopes T In Situ Electrochemical Quantification of Active Sites in Fe–N/C Non-Precious Metal Catalysts. Nat. Commun 2016, 7, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Malko D; Kucernak A; Lopes T Performance of Fe–N/C Oxygen Reduction Electrocatalysts toward NO2–, NO, and NH2OH Electroreduction: From Fundamental Insights into the Active Center to a New Method for Environmental Nitrite Destruction. J. Am. Chem. Soc 2016, 138, 16056–16068. [DOI] [PubMed] [Google Scholar]
- (138).Primbs M; Sun Y; Roy A; Malko D; Mehmood A; Sougrati M-T; Blanchard P-Y; Granozzi G; Kosmala T; Daniel G; et al. Establishing Reactivity Descriptors for Platinum Group Metal (PGM)-Free Fe–N–C Catalysts for PEM Fuel Cells. Energy Environ. Sci 2020, 13, 2480–2500. [Google Scholar]
- (139).Liu S; Wang M; Yang X; Shi Q; Qiao Z; Lucero M; Ma Q; More KL; Cullen DA; Feng Z; et al. Chemical Vapor Deposition for Atomically Dispersed and Nitrogen Coordinated Single Metal Site Catalysts. Angew. Chem. Int. Ed 2020, 59, 21698–21705. [DOI] [PubMed] [Google Scholar]
- (140).Kumar K; Dubau L; Mermoux M; Li J; Zitolo A; Nelayah J; Jaouen F; Maillard F On the Influence of Oxygen on the Degradation of Fe‐N‐C Catalysts. Angew. Chem. Int. Ed 2020, 59, 3235–3243. [DOI] [PubMed] [Google Scholar]
- (141).Kim DH; Ringe S; Kim H; Kim S; Kim B; Bae G; Oh H-S; Jaouen F; Kim W; Kim H; et al. Selective Electrochemical Reduction of Nitric Oxide to Hydroxylamine by Atomically Dispersed Iron Catalyst. Nat. Commun 2021, 12, 1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Boldrin P; Malko D; Mehmood A; Kramm UI; Wagner S; Paul S; Weidler N; Kucernak A Deactivation, Reactivation and Super-Activation of Fe-N/C Oxygen Reduction Electrocatalysts: Gas Sorption, Physical and Electrochemical Investigation Using NO and O2. Appl. Catal., B 2021, 292, 120169. [Google Scholar]
- (143).Teng H; Suuberg EM Chemisorption of Nitric Oxide on Char. 1. Reversible Nitric Oxide Sorption. J. Phys. Chem 1993, 97, 478–483. [Google Scholar]
- (144).Bae G; Kim H; Choi H; Jeong P; Kim DH; Kwon HC; Lee K-S; Choi M; Oh H-S; Jaouen F; et al. Quantification of Active Site Density and Turnover Frequency: From Single-Atom Metal to Nanoparticle Electrocatalysts. JACS Au 2021, 1, 586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Mehmood A; Gong M; Jaouen F; Roy A; Zitolo A; Khan A; Sougrati M-T; Primbs M; Bonastre AM; Fongalland D; et al. High Loading of Single Atomic Iron Sites in Fe–NC Oxygen Reduction Catalysts for Proton Exchange Membrane Fuel Cells. Nat. Catal 2022, 5, 311–323. [Google Scholar]
- (146).Boudart M; Aldag A; Benson JE; Dougharty NA; Girvin Harkins C On the Specific Activity of Platinum Catalysts. J. Catal 1966, 6, 92–99. [Google Scholar]
- (147).Hughes TR; Houston RJ; Sieg RP Flow Adsorption Method for Catalyst Metal Surface Measurements. Ind. Eng. Chem. Proc. Des. Dev 1962, 1, 96–102. [Google Scholar]
- (148).Sahraie NR; Kramm UI; Steinberg J; Zhang Y; Thomas A; Reier T; Paraknowitsch J-P; Strasser P Quantifying the Density and Utilization of Active Sites in Non-Precious Metal Oxygen Electroreduction Catalysts. Nat. Commun 2015, 6, 8618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Luo F; Choi CH; Primbs MJM; Ju W; Li S; Leonard ND; Thomas A; Jaouen F; Strasser P Accurate Evaluation of Active-Site Density (SD) and Turnover Frequency (TOF) of PGM-Free Metal–Nitrogen-Doped Carbon (MNC) Electrocatalysts Using CO Cryo Adsorption. ACS Catal 2019, 4841–4852.
- (150).Leonard ND; Wagner S; Luo F; Steinberg J; Ju W; Weidler N; Wang H; Kramm UI; Strasser P Deconvolution of Utilization, Site Density, and Turnover Frequency of Fe–Nitrogen–Carbon Oxygen Reduction Reaction Catalysts Prepared with Secondary N-Precursors. ACS Catal 2018, 8, 1640–1647. [Google Scholar]
- (151).Luo F; Wagner S; Onishi I; Selve S; Li S; Ju W; Wang H; Steinberg J; Thomas A; Kramm UI; et al. Surface Site Density and Utilization of Platinum Group Metal (PGM)-Free Fe–NC and FeNi–NC Electrocatalysts for the Oxygen Reduction Reaction. Chem. Sci 2021, 12, 384–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (152).Luo F; Roy A; Silvioli L; Cullen DA; Zitolo A; Sougrati MT; Oguz IC; Mineva T; Teschner D; Wagner S; et al. P-Block Single-Metal-Site Tin/Nitrogen-Doped Carbon Fuel Cell Cathode Catalyst for Oxygen Reduction Reaction. Nat. Mater 2020, 19, 1215–1223. [DOI] [PubMed] [Google Scholar]
- (153).Specchia S; Atanassov P; Zagal JH Mapping Transition Metal–Nitrogen–Carbon Catalyst Performance on the Critical Descriptor Diagram. Curr. Opin. Electrochem 2021, 27, 100687. [Google Scholar]
- (154).Jiao L; Li J; Richard LL; Sun Q; Stracensky T; Liu E; Sougrati MT; Zhao Z; Yang F; Zhong S; et al. Chemical Vapour Deposition of Fe–N–C Oxygen Reduction Catalysts with Full Utilization of Dense Fe–N4 Sites. Nat. Mater 2021, 20, 1385–1391. [DOI] [PubMed] [Google Scholar]
- (155).Kinoshita K Carbon: Electrochemical and Physicochemical Properties; Wiley: New York, 1988. [Google Scholar]
- (156).Schlögl R Carbons. In Handbook of Heterogeneous Catalysis; Ertl G, Knözinger H, Schüth F, Weitkamp J, Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; pp 357–427. [Google Scholar]
- (157).Derouane EG Shape Selectivity in Catalysis by Zeolites: The Nest Effect. J. Catal 1986, 100, 541–544. [Google Scholar]
- (158).Bhan A; Iglesia E A Link between Reactivity and Local Structure in Acid Catalysis on Zeolites. Acc. Chem. Res 2008, 41, 559–567. [DOI] [PubMed] [Google Scholar]
- (159).Gounder R; Iglesia E The Catalytic Diversity of Zeolites: Confinement and Solvation Effects within Voids of Molecular Dimensions. Chem. Commun 2013, 49, 3491–3509. [DOI] [PubMed] [Google Scholar]
- (160).Sievers C; Noda Y; Qi L; Albuquerque EM; Rioux RM; Scott SL Phenomena Affecting Catalytic Reactions at Solid–Liquid Interfaces. ACS Catal 2016, 6, 8286–8307. [Google Scholar]
- (161).Harris JW; Bates JS; Bukowski BC; Greeley J; Gounder R Opportunities in Catalysis over Metal-Zeotypes Enabled by Descriptions of Active Centers Beyond Their Binding Site. ACS Catal 2020, 10, 9476–9495. [Google Scholar]
- (162).Bates JS; Gounder R Kinetic Effects of Molecular Clustering and Solvation by Extended Networks in Zeolite Acid Catalysis. Chem. Sci 2021, 12, 4699–4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Potts DS; Bregante DT; Adams JS; Torres C; Flaherty DW Influence of Solvent Structure and Hydrogen Bonding on Catalysis at Solid–Liquid Interfaces. Chem. Soc. Rev 2021, 50, 12308–12337. [DOI] [PubMed] [Google Scholar]
- (164).Wei Z; Becwar SM; Chmelka BF; Sautet P Atomic Environments in N-Containing Graphitic Carbon Probed by First-Principles Calculations and Solid-State Nuclear Magnetic Resonance. J. Phys. Chem. C 2021, 125, 8779–8787. [Google Scholar]
- (165).Kneebone JL; Daifuku SL; Kehl JA; Wu G; Chung HT; Hu MY; Alp EE; More KL; Zelenay P; Holby EF; et al. A Combined Probe-Molecule, Mössbauer, Nuclear Resonance Vibrational Spectroscopy, and Density Functional Theory Approach for Evaluation of Potential Iron Active Sites in an Oxygen Reduction Reaction Catalyst. J. Phys. Chem. C 2017, 121, 16283–16290. [Google Scholar]
- (166).Wagner S; Auerbach H; Tait CE; Martinaiou I; Kumar SCN; Kübel C; Sergeev I; Wille H-C; Behrends J; Wolny JA; et al. Elucidating the Structural Composition of an Fe–N–C Catalyst by Nuclear- and Electron-Resonance Techniques. Angew. Chem. Int. Ed 2019, 58, 10486–10492. [DOI] [PubMed] [Google Scholar]
- (167).Saveleva VA; Ebner K; Ni L; Smolentsev G; Klose D; Zitolo A; Marelli E; Li J; Medarde M; Safonova OV; et al. Potential-Induced Spin Changes in Fe/N/C Electrocatalysts Assessed by In Situ X-Ray Emission Spectroscopy. Angew. Chem. Int. Ed 2021, 60, 11707–11712. [DOI] [PubMed] [Google Scholar]
- (168).Sgarbi R; Kumar K; Saveleva VA; Dubau L; Chattot R; Martin V; Mermoux M; Bordet P; Glatzel P; Ticianelli EA; et al. Electrochemical Transformation of Fe-N-C Catalysts into Iron Oxides in Alkaline Medium and Its Impact on the Oxygen Reduction Reaction Activity. Appl. Catal., B 2022, 311, 121366. [Google Scholar]
- (169).Westre TE; Kennepohl P; DeWitt JG; Hedman B; Hodgson KO; Solomon EI A Multiplet Analysis of Fe K-Edge 1s → 3d Pre-Edge Features of Iron Complexes. J. Am. Chem. Soc 1997, 119, 6297–6314. [Google Scholar]
- (170).Marshall-Roth T; Libretto NJ; Wrobel AT; Anderton KJ; Pegis ML; Ricke ND; Voorhis TV; Miller JT; Surendranath Y A Pyridinic Fe-N4 Macrocycle Models the Active Sites in Fe/N-Doped Carbon Electrocatalysts. Nat. Commun 2020, 11, 5283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (171).Li J; Zitolo A; Garcés-Pineda FA; Asset T; Kodali M; Tang P; Arbiol J; Galán-Mascarós JR; Atanassov P; Zenyuk IV; et al. Metal Oxide Clusters on Nitrogen-Doped Carbon Are Highly Selective for CO2 Electroreduction to CO. ACS Catal 2021, 11, 10028–10042. [Google Scholar]
- (172).Mukerjee S; Arruda T In-Situ Synchrotron Spectroscopic Studies of Electrocatalysis on Highly Dispersed Nano-Materials. Theory and Experiment in Electrocatalysis 2010, 503–572.
- (173).Ankudinov AL; Ravel B; Rehr JJ; Conradson SD Real-Space Multiple-Scattering Calculation and Interpretation of x-Ray-Absorption near-Edge Structure. Phys. Rev. B 1998, 58, 7565–7576. [Google Scholar]
- (174).Jia Q; Ramaswamy N; Hafiz H; Tylus U; Strickland K; Wu G; Barbiellini B; Bansil A; Holby EF; Zelenay P; et al. Experimental Observation of Redox-Induced Fe–N Switching Behavior as a Determinant Role for Oxygen Reduction Activity. ACS Nano 2015, 9, 12496–12505. [DOI] [PubMed] [Google Scholar]
- (175).Zitolo A; Ranjbar-Sahraie N; Mineva T; Li J; Jia Q; Stamatin S; Harrington GF; Lyth SM; Krtil P; Mukerjee S; et al. Identification of Catalytic Sites in Cobalt-Nitrogen-Carbon Materials for the Oxygen Reduction Reaction. Nat. Commun 2017, 8, 957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (176).Urakawa A; Bürgi T; Baiker A Sensitivity Enhancement and Dynamic Behavior Analysis by Modulation Excitation Spectroscopy: Principle and Application in Heterogeneous Catalysis. Chem. Eng. Sci 2008, 60, 4902–4909. [Google Scholar]
- (177).Ebner K; Clark AH; Saveleva VA; Smolentsev G; Chen J; Ni L; Li J; Zitolo A; Jaouen F; Kramm UI; et al. Time‐ Resolved Potential‐Induced Changes in Fe/N/C‐Catalysts Studied by In Situ Modulation Excitation X‐Ray Absorption Spectroscopy. Adv. Energy Mater 2022, 12, 2103699. [Google Scholar]
- (178).Zitolo A; Goellner V; Armel V; Sougrati M-T; Mineva T; Stievano L; Fonda E; Jaouen F Identification of Catalytic Sites for Oxygen Reduction in Iron- and Nitrogen-Doped Graphene Materials. Nat. Mater 2015, 14, 937–942. [DOI] [PubMed] [Google Scholar]
- (179).Zhang H; Li J; Xi S; Du Y; Hai X; Wang J; Xu H; Wu G; Zhang J; Lu J; et al. Graphene-Supported Single-Atom FeN5 Catalytic Site for Efficient Electrochemical CO2 Reduction. Angew. Chem. Int. Ed 2019, 58, 14871–14876. [DOI] [PubMed] [Google Scholar]
- (180).Zhang G; Jia Y; Zhang C; Xiong X; Sun K; Chen R; Chen W; Kuang Y; Zheng L; Tang H; et al. A General Route via Formamide Condensation to Prepare Atomically Dispersed Metal–Nitrogen–Carbon Electrocatalysts for Energy Technologies. Energy Environ. Sci 2019, 12, 1317–1325. [Google Scholar]
- (181).Xiao M; Chen Y; Zhu J; Zhang H; Zhao X; Gao L; Wang X; Zhao J; Ge J; Jiang Z; et al. Climbing the Apex of the ORR Volcano Plot via Binuclear Site Construction: Electronic and Geometric Engineering. J. Am. Chem. Soc 2019, 141, 17763–17770. [DOI] [PubMed] [Google Scholar]
- (182).Zhang S; Jin M; Shi T; Han M; Sun Q; Lin Y; Ding Z; Zheng LR; Wang G; Zhang Y; et al. Electrocatalytically Active Fe-(O-C2)4 Single-Atom Sites for Efficient Reduction of Nitrogen to Ammonia. Angew. Chem. Int. Ed 2020, 59, 13423–13429. [DOI] [PubMed] [Google Scholar]
- (183).Yuan K; Lützenkirchen-Hecht D; Li L; Shuai L; Li Y; Cao R; Qiu M; Zhuang X; Leung MKH; Chen Y; et al. Boosting Oxygen Reduction of Single Iron Active Sites via Geometric and Electronic Engineering: Nitrogen and Phosphorus Dual Coordination. J. Am. Chem. Soc 2020, 142, 2404–2412. [DOI] [PubMed] [Google Scholar]
- (184).Feng K; Zhang H; Gao J; Xu J; Dong Y; Kang Z; Zhong J Single Atoms or Not? The Limitation of EXAFS. Appl. Phys. Lett 2020, 116, 191903. [Google Scholar]
- (185).Resasco J; Christopher P Atomically Dispersed Pt-Group Catalysts: Reactivity, Uniformity, Structural Evolution, and Paths to Increased Functionality. J. Phys. Chem. Lett 2020, 11, 10114–10123. [DOI] [PubMed] [Google Scholar]
- (186).Wang M; Feng Z Pitfalls in X-Ray Absorption Spectroscopy Analysis and Interpretation: A Practical Guide for General Users. Curr. Opin. Electrochem 2021, 30, 100803. [Google Scholar]
- (187).He Y; Shi Q; Shan W; Li X; Kropf AJ; Wegener EC; Wright J; Karakalos S; Su D; Cullen DA; et al. Dynamically Unveiling Metal–Nitrogen Coordination during Thermal Activation to Design High-Efficient Atomically Dispersed CoN4 Active Sites. Angew. Chem. Int. Ed 2021, 60, 9516–9526. [DOI] [PubMed] [Google Scholar]
- (188).Delgass WN; Boudart M Application of Mössbauer Spectroscopy to the Study of Adsorption and Catalysis. Catalysis Reviews 1969, 2, 129–160. [Google Scholar]
- (189).Dumesic JA; Topsøe H Mössbauer Spectroscopy Applications to Heterogeneous Catalysis. In Advances in Catalysis; Eley DD, Pines H, Weisz PB, Eds.; Academic Press, 1977; Vol. 26, pp 121–246. [Google Scholar]
- (190).Gütlich P; Bill E; Trautwein A Mössbauer Spectroscopy and Transition Metal Chemistry: Fundamentals and Application; Springer: Berlin; ; Heidelberg, 2011. [Google Scholar]
- (191).Grandjean F; Long GJ Best Practices and Protocols in Mössbauer Spectroscopy. Chem. Mater 2021, 33, 3878–3904. [Google Scholar]
- (192).Schulenburg H; Stankov S; Schünemann V; Radnik J; Dorbandt I; Fiechter S; Bogdanoff P; Tributsch H Catalysts for the Oxygen Reduction from Heat-Treated Iron(III) Tetramethoxyphenylporphyrin Chloride: Structure and Stability of Active Sites. J. Phys. Chem. B 2003, 107, 9034–9041. [Google Scholar]
- (193).Koslowski UI; Abs-Wurmbach I; Fiechter S; Bogdanoff P Nature of the Catalytic Centers of Porphyrin-Based Electrocatalysts for the ORR: A Correlation of Kinetic Current Density with the Site Density of Fe−N4 Centers. J. Phys. Chem. C 2008, 112, 15356–15366. [Google Scholar]
- (194).Kramm UI; Abs-Wurmbach I; Herrmann-Geppert I; Radnik J; Fiechter S; Bogdanoff P Influence of the Electron-Density of FeN4-Centers Towards the Catalytic Activity of Pyrolyzed FeTMPPCl-Based ORR-Electrocatalysts. J. Electrochem. Soc 2010, 158, B69. [Google Scholar]
- (195).Kramm UI; Herranz J; Larouche N; Arruda TM; Lefèvre M; Jaouen F; Bogdanoff P; Fiechter S; Abs-Wurmbach I; Mukerjee S; et al. Structure of the Catalytic Sites in Fe/N/C-Catalysts for O2-Reduction in PEM Fuel Cells. Phys. Chem. Chem. Phys 2012, 14, 11673–11688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (196).Kramm UI; Lefèvre M; Larouche N; Schmeisser D; Dodelet J-P Correlations between Mass Activity and Physicochemical Properties of Fe/N/C Catalysts for the ORR in PEM Fuel Cell via 57Fe Mössbauer Spectroscopy and Other Techniques. J. Am. Chem. Soc 2014, 136, 978–985. [DOI] [PubMed] [Google Scholar]
- (197).Mineva T; Matanovic I; Atanassov P; Sougrati M-T; Stievano L; Clémancey M; Kochem A; Latour J-M; Jaouen F Understanding Active Sites in Pyrolyzed Fe–N–C Catalysts for Fuel Cell Cathodes by Bridging Density Functional Theory Calculations and 57Fe Mössbauer Spectroscopy. ACS Catal 2019, 9, 9359–9371. [Google Scholar]
- (198).Sougrati MT; Goellner V; Schuppert AK; Stievano L; Jaouen F Probing Active Sites in Iron-Based Catalysts for Oxygen Electro-Reduction: A Temperature-Dependent 57Fe Mössbauer Spectroscopy Study. Catal. Today 2016, 262, 110–120. [Google Scholar]
- (199).Bouwkamp-Wijnoltz AL; Visscher W; van Veen JAR; Boellaard E; van der Kraan AM; Tang SC On Active-Site Heterogeneity in Pyrolyzed Carbon-Supported Iron Porphyrin Catalysts for the Electrochemical Reduction of Oxygen: An In Situ Mössbauer Study. J. Phys. Chem. B 2002, 106, 12993–13001. [Google Scholar]
- (200).Li J; Sougrati MT; Zitolo A; Ablett JM; Oğuz IC; Mineva T; Matanovic I; Atanassov P; Huang Y; Zenyuk I; et al. Identification of Durable and Non-Durable FeNx Sites in Fe–N–C Materials for Proton Exchange Membrane Fuel Cells. Nat. Catal 2021, 4, 10–19. [Google Scholar]
- (201).Artyushkova K; Levendosky S; Atanassov P; Fulghum J XPS Structural Studies of Nano-Composite Non-Platinum Electrocatalysts for Polymer Electrolyte Fuel Cells. Top. Catal 2007, 46, 263–275. [Google Scholar]
- (202).Pels JR; Kapteijn F; Moulijn JA; Zhu Q; Thomas KM Evolution of Nitrogen Functionalities in Carbonaceous Materials during Pyrolysis. Carbon 1995, 33, 1641–1653. [Google Scholar]
- (203).Matanovic I; Artyushkova K; Strand MB; Dzara MJ; Pylypenko S; Atanassov P Core Level Shifts of Hydrogenated Pyridinic and Pyrrolic Nitrogen in the Nitrogen-Containing Graphene-Based Electrocatalysts: In-Plane vs Edge Defects. J. Phys. Chem. C 2016, 120, 29225–29232. [Google Scholar]
- (204).Greczynski G; Hultman L Compromising Science by Ignorant Instrument Calibration—Need to Revisit Half a Century of Published XPS Data. Angew. Chem. Int. Ed 2020, 59, 5002–5006. [DOI] [PubMed] [Google Scholar]
- (205).Artyushkova K Misconceptions in Interpretation of Nitrogen Chemistry from X-Ray Photoelectron Spectra. J. Vac. Sci. Technol., A 2020, 38, 031002. [Google Scholar]
- (206).Artyushkova K; Kiefer B; Halevi B; Knop-Gericke A; Schlogl R; Atanassov P Density Functional Theory Calculations of XPS Binding Energy Shift for Nitrogen-Containing Graphene-like Structures. Chem. Commun 2013, 49, 2539–2541. [DOI] [PubMed] [Google Scholar]
- (207).Kabir S; Artyushkova K; Kiefer B; Atanassov P Computational and Experimental Evidence for a New TM–N3/C Moiety Family in Non-PGM Electrocatalysts. Phys. Chem. Chem. Phys 2015, 17, 17785–17789. [DOI] [PubMed] [Google Scholar]
- (208).Jia Q; Ramaswamy N; Tylus U; Strickland K; Li J; Serov A; Artyushkova K; Atanassov P; Anibal J; Gumeci C; et al. Spectroscopic Insights into the Nature of Active Sites in Iron–Nitrogen–Carbon Electrocatalysts for Oxygen Reduction in Acid. Nano Energy 2016, 29, 65–82. [Google Scholar]
- (209).Dzara MJ; Artyushkova K; Sougrati MT; Ngo C; Fitzgerald MA; Serov A; Zulevi B; Atanassov P; Jaouen F; Pylypenko S Characterizing Complex Gas–Solid Interfaces with in Situ Spectroscopy: Oxygen Adsorption Behavior on Fe–N–C Catalysts. J. Phys. Chem. C 2020, 124, 16529–16543. [Google Scholar]
- (210).Artyushkova K; Serov A; Rojas-Carbonell S; Atanassov P Chemistry of Multitudinous Active Sites for Oxygen Reduction Reaction in Transition Metal–Nitrogen–Carbon Electrocatalysts. J. Phys. Chem. C 2015, 119, 25917–25928. [Google Scholar]
- (211).Chung HT; Cullen DA; Higgins D; Sneed BT; Holby EF; More KL; Zelenay P Direct Atomic-Level Insight into the Active Sites of a High-Performance PGM-Free ORR Catalyst. Science 2017, 357, 479–484. [DOI] [PubMed] [Google Scholar]
- (212).Zhang H; Hwang S; Wang M; Feng Z; Karakalos S; Luo L; Qiao Z; Xie X; Wang C; Su D; et al. Single Atomic Iron Catalysts for Oxygen Reduction in Acidic Media: Particle Size Control and Thermal Activation. J. Am. Chem. Soc 2017, 139, 14143–14149. [DOI] [PubMed] [Google Scholar]
- (213).Li J; Chen M; Cullen DA; Hwang S; Wang M; Li B; Liu K; Karakalos S; Lucero M; Zhang H; et al. Atomically Dispersed Manganese Catalysts for Oxygen Reduction in Proton-Exchange Membrane Fuel Cells. Nat. Catal 2018, 1, 935–945. [Google Scholar]
- (214).Wang XX; Cullen DA; Pan Y-T; Hwang S; Wang M; Feng Z; Wang J; Engelhard MH; Zhang H; He Y; et al. Nitrogen-Coordinated Single Cobalt Atom Catalysts for Oxygen Reduction in Proton Exchange Membrane Fuel Cells. Adv. Mater 2018, 30, 1706758. [DOI] [PubMed] [Google Scholar]
- (215).Wu G; More KL; Johnston CM; Zelenay P High-Performance Electrocatalysts for Oxygen Reduction Derived from Polyaniline, Iron, and Cobalt. Science 2011, 332, 443–447. [DOI] [PubMed] [Google Scholar]
- (216).Matter PH; Wang E; Millet J-MM; Ozkan US Characterization of the Iron Phase in CNx-Based Oxygen Reduction Reaction Catalysts. J. Phys. Chem. C 2007, 111, 1444–1450. [Google Scholar]
- (217).Kunwar D; Zhou S; DeLaRiva A; Peterson EJ; Xiong H; Pereira-Hernández XI; Purdy SC; ter Veen R; Brongersma HH; Miller JT; et al. Stabilizing High Metal Loadings of Thermally Stable Platinum Single Atoms on an Industrial Catalyst Support. ACS Catal 2019, 9, 3978–3990. [Google Scholar]
- (218).Holder CF; Schaak RE Tutorial on Powder X-Ray Diffraction for Characterizing Nanoscale Materials. ACS Nano 2019, 13, 7359–7365. [DOI] [PubMed] [Google Scholar]
- (219).Moriya M; Takahama R; Kamoi K; Ohyama J; Kawashima S; Kojima R; Okada M; Hayakawa T; Nabae Y Fourteen-Membered Macrocyclic Fe Complexes Inspired by FeN4-Center-Embedded Graphene for Oxygen Reduction Catalysis. J. Phys. Chem. C 2020, 124, 20730–20735. [Google Scholar]
- (220).Ohyama J; Moriya M; Takahama R; Kamoi K; Kawashima S; Kojima R; Hayakawa T; Nabae Y High Durability of a 14-Membered Hexaaza Macrocyclic Fe Complex for an Acidic Oxygen Reduction Reaction Revealed by In Situ XAS Analysis. JACS Au 2021, 1, 1798–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (221).Sun L; Huang Z; Reddu V; Su T; Fisher AC; Wang X A Planar, Conjugated N4-Macrocyclic Cobalt Complex for Heterogeneous Electrocatalytic CO2 Reduction with High Activity. Angew. Chem. Int. Ed 2020, 59, 17104–17109. [DOI] [PubMed] [Google Scholar]