

Abstract
Our objective was to test whether p53 expression status is associated with survival for women diagnosed with the most common ovarian carcinoma histotypes (high‐grade serous carcinoma [HGSC], endometrioid carcinoma [EC], and clear cell carcinoma [CCC]) using a large multi‐institutional cohort from the Ovarian Tumor Tissue Analysis (OTTA) consortium. p53 expression was assessed on 6,678 cases represented on tissue microarrays from 25 participating OTTA study sites using a previously validated immunohistochemical (IHC) assay as a surrogate for the presence and functional effect of TP53 mutations. Three abnormal expression patterns (overexpression, complete absence, and cytoplasmic) and the normal (wild type) pattern were recorded. Survival analyses were performed by histotype. The frequency of abnormal p53 expression was 93.4% (4,630/4,957) in HGSC compared to 11.9% (116/973) in EC and 11.5% (86/748) in CCC. In HGSC, there were no differences in overall survival across the abnormal p53 expression patterns. However, in EC and CCC, abnormal p53 expression was associated with an increased risk of death for women diagnosed with EC in multivariate analysis compared to normal p53 as the reference (hazard ratio [HR] = 2.18, 95% confidence interval [CI] 1.36–3.47, p = 0.0011) and with CCC (HR = 1.57, 95% CI 1.11–2.22, p = 0.012). Abnormal p53 was also associated with shorter overall survival in The International Federation of Gynecology and Obstetrics stage I/II EC and CCC. Our study provides further evidence that functional groups of TP53 mutations assessed by abnormal surrogate p53 IHC patterns are not associated with survival in HGSC. In contrast, we validate that abnormal p53 IHC is a strong independent prognostic marker for EC and demonstrate for the first time an independent prognostic association of abnormal p53 IHC with overall survival in patients with CCC.
Keywords: ovarian cancer, high‐grade serous carcinoma, endometrioid, clear cell, TP53, p53, prognosis
Introduction
TP53 is universally mutated in tubo‐ovarian high‐grade serous carcinomas (HGSCs), and a lack of TP53 mutation is essentially inconsistent with a diagnosis of HGSC [1, 2]. TP53 mutations are broadly classified into gain‐of‐function (GOF) mutations (mutation type: nonsynonymous/missense, which are further subclassified into contact and conformational) versus loss‐of‐function (LOF) mutations (mutation type: frameshift [insertion/deletions], stop‐gain, and splicing). We have recently optimised p53 immunohistochemistry (IHC) to serve as an accurate surrogate for the TP53 mutation status with 96% sensitivity and perfect specificity [3]. The high, but not perfect, sensitivity was influenced by non‐functional p53 proteins caused by late truncating TP53 mutations that showed normal wild type pattern by IHC [3]. The abnormal patterns of p53 IHC also showed good agreement with the function of the mutation: overexpression (OE) for nonsynonymous GOF, and complete absence (CA) for LOF, the latter with some explainable exceptions [3]. The uncommon abnormal cytoplasmic (CY) pattern is associated with TP53 mutations in the nuclear localisation domain, adjacent to the tetramerisation domain or nuclear exclusion sequence [3, 4].
The prognostic associations of specific mutation types in HGSC have been controversial. Using p53 IHC, we previously reported that the LOF surrogate CA was associated with shorter survival in a combined cohort of 502 HGSC [5]. However, using TP53 mutation data from The Cancer Genome Atlas (TCGA) project, Kang et al showed no difference between GOF and other TP53 mutations with respect to overall survival [6]. Using a different classification in the TCGA data set, Brachova et al reported a higher risk of recurrence for GOF mutations [7]. More recently, Mandilaras et al showed no difference in overall survival using six different classification schemas for TP53 mutations; however, one cluster enriched in GOF mutations was associated with a worse prognosis [8]. Tuna et al reported that three hot‐spot GOF mutations (at G266, Y163, R282) were associated with shorter overall survival, but they did not find differences for different types of TP53 mutations [9].
In contrast to HGSC, only subsets of endometrioid carcinoma (EC) and clear cell carcinoma (CCC) harbour TP53 mutations [5]. Like its endometrial counterpart, in ovarian EC, abnormal p53 expression by IHC defines a prognostically adverse molecular subtype according to three studies totalling 749 cases [10, 11, 12]. However, the prognostic value in the low‐stage setting (The International Federation of Gynecology and Obstetrics [FIGO] stage I/II) has only been addressed in one study of 274 cases [11]. In CCC, two studies with 90 and 115 cases reported associations of abnormal p53 status assessed by IHC with shorter survival in univariate but not multivariate analysis [13, 14]. Larger genomic studies of 271 and 421 CCC showed associations of TP53 mutations with gene expression and methylation clusters that tend to show adverse clinical features but did not report an independent prognostic value for TP53 alone [15, 16]. Abnormal p53 function in endometrial carcinoma is associated with chromosomal instability (CIN) and adverse outcome and is now a strong indication for more aggressive management including adjuvant chemotherapy [17]. Accurate assessment of the impacts of abnormal p53 staining on outcome may be clinically important in low‐stage EC and CCC. The aims of the current study were to (1) clarify whether types of TP53 mutations inferred by three abnormal p53 IHC patterns are associated with prognosis in HGSC; (2) validate whether abnormal p53 IHC predicts higher risk of death in EC, particularly for FIGO stage I and II disease; and (3) elucidate the association of p53 status with overall survival in CCC using a large cohort from the Ovarian Tumor Tissue Analysis (OTTA) consortium [18, 19, 20].
Methods
Study cohort
Twenty‐five studies from the OTTA consortium contributed to this study. Each participating study received local ethics board approval (supplementary material, Table S1). Cases included in this study were recruited before the widespread use of diagnostic IHC (median year of diagnosis 2004, 25–75% quartiles 2000–2008, range 1978–2016). The 4‐μm‐thick sections of previously constructed tissue microarrays with each case represented by 1–3 cores were shipped to a central immunohistochemical laboratory at the University of Calgary, Alberta, Canada. Clinical covariates and follow‐up time and status were centrally collected at the University of New South Wales, Sydney, Australia. Histotype was assessed by various tiers of pathology review based on the 2014 World Health Organization (WHO) classification [21] (supplementary material, Table S1). Since the histotype review was heterogeneous, WT1 alone or in combination with p53 IHC was further used to identify potentially misclassified cases. Based on previous data that HGSC typically shows a combination of WT1 expression and abnormal p53 versus <1% of CCC and <2% of EC [22], we reassigned 23 WT1+/p53‐abnormal EC to HGSC, excluded 30 WT1+ CCC as they likely represent HGSC or low‐grade serous carcinoma, and excluded 121 WT1−/p53‐normal HGSC. Previously generated data and categorisation, where applicable, were used for CD8+ tumour infiltrating lymphocytes (TILs), CDKN2A, and TP53 mRNA expression [19, 20, 23].
Immunohistochemistry
Two previously validated p53 IHC assays were used [3, 24]. The first assay (pre‐treatment with BOND Epitope Retrieval Solution 2, Leica Biosystems, Wetzlar, Germany; p53 antibody clone DO‐7, dilution 1:2,500, Dako Omnis, Agilent, Santa Clara, CA, USA) was used to stain 55.4% of cases and 44.6% of cases were stained with a second assay (pre‐treatment for 30 min using heat‐induced antigen retrieval with Tris‐EDTA buffer, pH = 9.0; ready‐to‐use antibody DO‐7, Dako Omnis) due to a change in the automated IHC platform during the study period. The interpretation criteria for the three abnormal patterns were defined as follows: OE as strong nuclear staining of similar intensity in usually all but at least 80% of tumour cell nuclei; CA as complete absence of nuclear staining with retained staining in normal fibroblasts or lymphocytes serving as internal controls; and CY as unequivocal cytoplasmic staining of at least of moderate intensity and absent or variate nuclear staining [25]. In contrast, the normal (wild type) pattern was characterised by staining of variate intensity in tumour nuclei. After assessing the interobserver agreement with a kappa coefficient of 0.912 on a test set of n = 92 cases, one observer (MK) scored approximately 75% and a second observer (PR) scored 25% of the cases.
Statistical analyses
The distributions of categorical or continuous variables were compared across histotypes using chi‐square or ANOVA tests, respectively. Time from diagnosis to death, due to any cause, was the primary endpoint. Left truncation was applied to mitigate potential survival bias introduced by the time between diagnosis and study enrolment and right censoring at 10 years from diagnosis was used to account for potential non‐cancer‐related deaths. Univariate Kaplan‐Meier survival analyses alongside log‐rank testing were used to assess overall survival. Cox proportional hazards regression models were applied to estimate hazard ratios (HRs) with 95% confidence intervals (CIs). Univariate Cox regression models evaluated associations with overall survival for covariates, including age (continuous), FIGO stage (III/IV versus reference I/II), the completeness of surgical cytoreduction (residual disease present versus absent/unknown), p53 status (normal versus abnormal) and, where applicable, grade (only appropriate for EC: 3 versus reference 1/2). Multivariate Cox regression models were adjusted for age (continuous), FIGO stage (III/IV versus reference I/II), grade (only appropriate for EC: 3 versus reference 1/2), the completeness of surgical cytoreduction (residual disease present versus absent/unknown), and stratified by the OTTA study contributing the sample to account for differing baseline hazards. Statistical analyses were carried out using SAS JMPv16.2.0 or RStudio v1.1.463. Statistical significance was defined by p < 0.05.
Results
Baseline clinicopathological features of study participants
The analysis included 6,678 patients with one of the three most common ovarian carcinoma histotypes (HGSC, EC, and CCC). By histotype, the 4,957 patients with HGSC were significantly older (mean age 60.2 years) compared to the 973 patients with EC (54.4 years) and 748 patients with CCC (55.3 years; p < 0.001; Table 1). Expected differences were observed for stage, residual disease, and 5‐year survival rate (5‐YSR) across histotypes. For example, 80.9% of HGSC were diagnosed at high stage (III or IV) versus only 14.1% of EC. The 5‐YSR was highest for patients diagnosed with EC (84.4%), intermediate for patients diagnosed with CCC (66.2%), and lowest for patients with HGSC (41.7%) (p < 0.001; Table 1).
Table 1.
Clinicopathological characteristics and p53 expression patterns across histotypes
| HGSC | % | EC | % | CCC | % | Total | % | P value | ||
|---|---|---|---|---|---|---|---|---|---|---|
| n | Total | 4,957 | 973 | 748 | 6,678 | |||||
| Age | Mean | 60.2 | 54.4 | 55.3 | 58.8 | <0.001 | ||||
| Min–Max | 22–93 | 21–91 | 27–84 | 21–93 | ||||||
| FIGO stage | I/II | 894 | 19.1 | 771 | 85.9 | 550 | 76.6 | 2,215 | 35.2 | <0.001 |
| III/IV | 3,775 | 80.9 | 127 | 14.1 | 168 | 23.4 | 4,070 | 64.8 | ||
| Unknown | 288 | 75 | 30 | 393 | ||||||
| Residual disease | Absent | 893 | 37.8 | 422 | 89.0 | 324 | 79.0 | 1,639 | 50.4 | <0.001 |
| Present | 1,472 | 62.2 | 52 | 11.0 | 86 | 21.0 | 1,610 | 49.6 | ||
| Unknown | 2,592 | 499 | 338 | 3,429 | ||||||
| 5‐YSR | %, SE | 41.7, 0.8 | 84.4, 1.2 | 66.2, 1.8 | 50.8, 0.6 | <0.001 | ||||
| Unknown | 200 | 41 | 23 | 264 | ||||||
| p53 | Normal | 327 | 6.6 | 857 | 88.1 | 662 | 88.5 | 1,846 | 27.6 | <0.001 |
| Abnormal | 4,630 | 93.4 | 116 | 11.9 | 86 | 11.5 | 4,832 | 72.4 | ||
| Abnormal OE | 3,221 | 69.6 | 85 | 73.3 | 68 | 79.1 | 3,374 | 69.8 | 0.23 | |
| Abnormal CA | 1,176 | 25.4 | 27 | 23.3 | 17 | 19.8 | 1,220 | 25.2 | ||
| Abnormal CY | 233 | 5.0 | 4 | 3.4 | 1 | 1.2 | 238 | 4.9 | ||
P values calculated excluding cases with ‘unknown’ information.
Prevalence of abnormal p53 IHC
HGSC showed the highest prevalence (4,630/4,957, 93.4%) of abnormal p53 staining compared to EC (116/973, 11.9%) and CCC (86/748, 11.5%). The distribution of different abnormal staining patterns was not significantly different across histotypes (Table 1). In HGSC, abnormal OE indicative of nonsynonymous/missense GOF mutations was the most common pattern (69.6%), followed by CA representing LOF mutations (25.4%) and the uncommon CY pattern reflecting mutations affecting the nuclear localisation of p53 (5.0%) (Table 1, Figure 1A).
Figure 1.
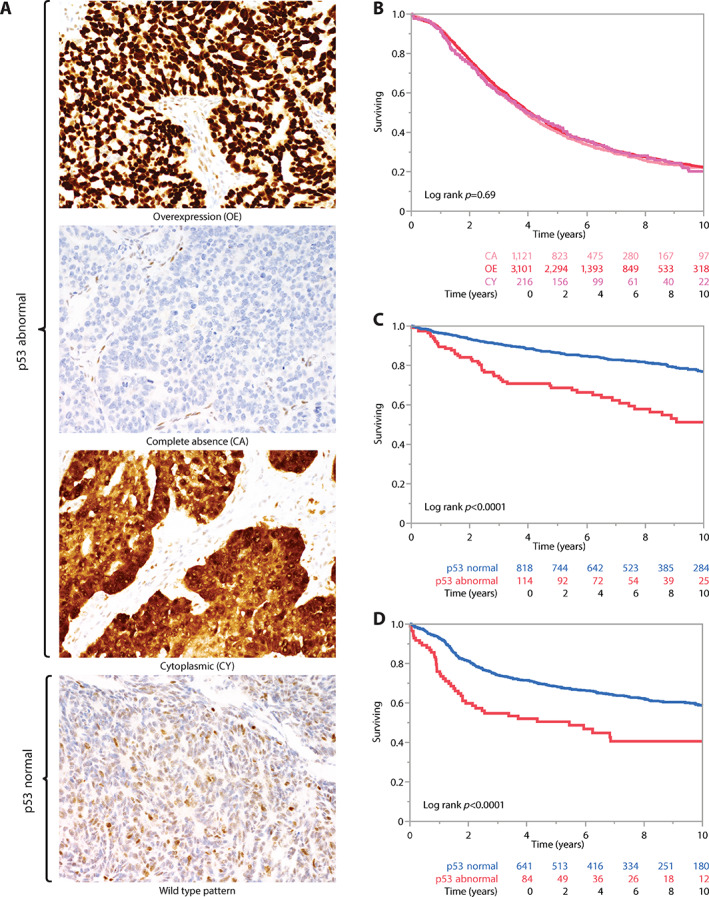
(A) p53 IHC patterns. (B) Kaplan‐Meier survival analyses of the three abnormal p53 IHC patterns in HGSC. (C) Kaplan‐Meier survival analyses of abnormal p53 versus normal p53 in EC. (D) Kaplan‐Meier survival analyses of abnormal p53 versus normal p53 in CCC.
Abnormal p53 IHC patterns and univariate associations with overall survival, clinicopathological parameters, and selected biomarkers in HGSC
For HGSC, the frequency of normal p53 expression was higher than expected compared to previous smaller studies (current 6.6% versus expected 2–4%), suggesting possible misclassification of p53‐normal non‐HGSC as ‘HGSC’ in the current study [3, 26]. Therefore, analyses for HGSC were restricted to cases with abnormal p53 expression. Kaplan‐Meier survival analyses of 4,435 HGSC with follow‐up data showed overlapping survival curves for the three abnormal p53 patterns (log‐rank p = 0.69; Figure 1B). Univariate associations with clinical parameters such as age, stage, and residual disease did not show any significant differences across the abnormal p53 patterns (Table 2). No significant differences across selected biomarkers such as CD8+ TILs, CDKN2A, and germline BRCA1/2 mutation status were observed.
Table 2.
Clinicopathological characteristics and selected biomarkers across abnormal p53 expression patterns in HGSC
| Abnormal OE | % | Abnormal CA | % | Abnormal CY | % | Total | % | P value | ||
|---|---|---|---|---|---|---|---|---|---|---|
| HGSC | Total | 3,221 | 1,176 | 233 | 4,630 | |||||
| Age | Mean | 60.5 | 60 | 61.2 | 0.121 | |||||
| Range | 26–93 | 28–91 | 35–87 | |||||||
| Stage | I/II | 583 | 19.2 | 207 | 19.0 | 42 | 19.7 | 832 | 19.1 | 0.968 |
| III/IV | 2,461 | 80.8 | 884 | 81.0 | 171 | 80.3 | 3,516 | 80.9 | ||
| Unknown | 177 | 85 | 20 | 282 | ||||||
| Residual disease | Absent | 580 | 37.5 | 214 | 38.0 | 36 | 38.3 | 830 | 37.7 | 0.255 |
| Present | 965 | 62.5 | 349 | 62.0 | 58 | 61.7 | 1,372 | 62.3 | ||
| Unknown | 1,676 | 613 | 139 | 2,428 | ||||||
| CD8+ TILs | Negative | 386 | 15.7 | 148 | 16.5 | 26 | 14.3 | 560 | 15.8 | 0.818 |
| Low | 385 | 15.7 | 154 | 17.2 | 28 | 15.4 | 567 | 16.0 | ||
| Moderate | 1,068 | 43.5 | 385 | 43.0 | 84 | 46.2 | 1,537 | 43.5 | ||
| High | 618 | 25.2 | 208 | 23.2 | 44 | 24.2 | 870 | 24.6 | ||
| CDKN2A | Normal | 874 | 32.6 | 329 | 33.7 | 52 | 26.3 | 1,255 | 32.5 | 0.102 |
| Abnormal block | 1,637 | 61.0 | 601 | 61.6 | 135 | 68.2 | 2,373 | 61.5 | ||
| Abnormal absent | 171 | 6.4 | 46 | 4.7 | 11 | 5.6 | 228 | 5.9 | ||
| BRCA1/2 | Mutated | 197 | 24.9 | 70 | 24.1 | 11 | 22.0 | 278 | 24.6 | 0.880 |
| Wild type | 594 | 75.1 | 220 | 75.9 | 39 | 78.0 | 853 | 75.4 |
P values calculated excluding cases with ‘unknown’ information.
Binary p53 IHC status and uni‐ and multivariate associations with overall survival, clinicopathological parameters, and selected biomarkers in EC and CCC
Analyses were grouped for a binary comparison of cases with abnormal versus normal p53 IHC separately for EC and CCC [27]. Kaplan‐Meier survival analyses showed a significantly different overall survival between the groups for both EC and CCC (log‐rank p < 0.0001 for both; Figure 1C,D). The 5‐YSR for p53‐abnormal EC was 69% compared to 87% for p53‐normal EC, while the 5‐YSR for p53‐abnormal CCC was 50% compared to 68% for p53‐normal CCC.
For EC, univariate survival analyses showed significant associations for stage, residual disease, binary p53 IHC status, grade, and age (supplementary material, Table S2). Except residual disease, these parameters remained significant in multivariate analysis (supplementary material, Table S2). Abnormal p53 IHC showed an HR of 2.18 (95% CI 1.36–3.47, p = 0.0011) after adjustment for age, stage, residual disease, grade, and stratified by OTTA study (supplementary material, Table S2). A Kaplan‐Meier survival analysis for stage I/II EC cases without residual disease showed p53‐abnormal cases having a 5‐YSR of 80% versus 91% for p53‐normal cases (log‐rank p = 0.0016; Figure 2A). In this low‐stage setting, the univariate HR for p53‐abnormal EC was 2.14 (95% CI 1.32–3.47, p = 0.0021, n = 737) compared to the reference group of p53‐normal EC, and the univariate HR was 2.59 for grade 3 (95% CI 1.65–4.07, p < 0.0001, n = 571) compared to the reference group grade 1 and 2 combined (Figure 2B). Stage III/IV cases with p53 abnormalities had a lower 5‐YSR (35%) than those who were p53 normal (58%, log‐rank p = 0.008, Figure 2C). With respect to clinical parameters, significantly more p53‐abnormal cases were diagnosed at a higher stage (27.6% stage III/IV for p53‐abnormal cases compared to 12.4% for p53‐normal cases, p < 0.001; Table 3). However, there were no associations with age or residual disease. With respect to biomarkers, p53‐normal EC cases demonstrated higher proportions of CD8+ TILs. Conversely, p53‐abnormal EC cases more commonly showed abnormal CDKN2A expression patterns and were commonly grade 3. However, slightly more than half of p53‐abnormal EC were diagnosed as low grade (grade 1 or 2). Approximately a third of grade 3 EC were p53 abnormal (32.5%, n = 41/126). Hence, there was roughly the same number of p53‐normal grade 3 EC cases (n = 85) compared to p53‐abnormal EC of all grades (n = 88). To assess the prognostic relation between p53 status and grade in EC, we created a combined variable for cases with available grade information (75.3%, 733/973): 76.4% (560/733) of cases were p53 normal and low grade (grade 1/2), 11.6% (85/733) were p53 normal and grade 3, 6.4% (47/733) were p53 abnormal and low grade, and 5.6% (41/733) were p53 abnormal and grade 3. Next, we performed a Kaplan‐Meier survival analysis for these four groups restricted to stage I/II EC cases without residual disease. Patients with p53‐normal, low‐grade EC had a 5‐YSR of 91.9%, compared to 83.5% for p53‐abnormal, low‐grade EC, 79.3% for p53‐normal, grade 3 EC, and 76.8% for p53‐abnormal, grade 3 EC (Figure 2D).
Figure 2.
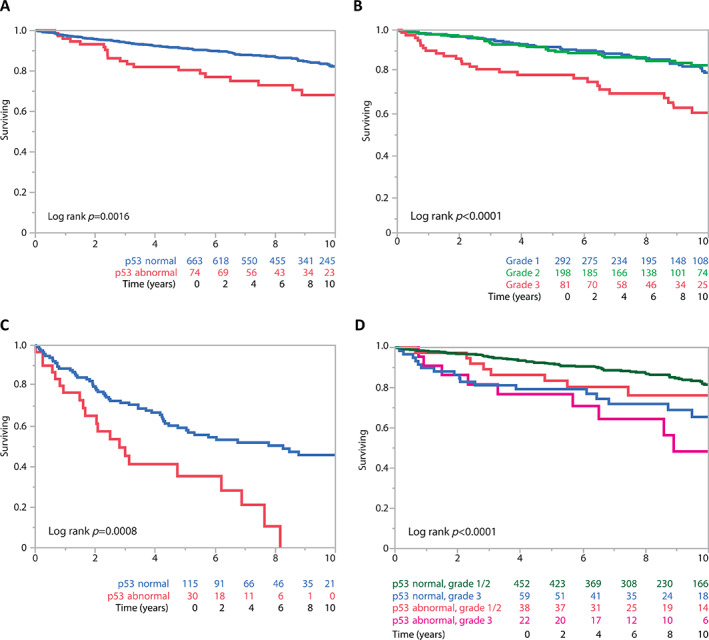
EC: stage‐stratified Kaplan‐Meier survival analyses. (A) By abnormal versus normal p53 at low FIGO stage (I/II) without residual disease. (B) By grade at low FIGO stage (I/II) without residual disease. (C) Abnormal versus normal p53 at high FIGO stage (III/IV). (D) By combined grade and abnormal versus normal p53 at low FIGO stage (I/II) without residual disease.
Table 3.
Clinicopathological characteristics and selected biomarkers across binary p53 expression status in EC and CCC
| p53 abnormal | % | p53 normal | % | Total | % | P value | ||
|---|---|---|---|---|---|---|---|---|
| EC | Total | 116 | 857 | 973 | ||||
| Age | Mean | 55 | 54.3 | 54.4 | 0.597 | |||
| Range | 22–86 | 21–91 | 21–91 | |||||
| FIGO Stage | I/II | 76 | 72.4 | 695 | 87.6 | 771 | 85.9 | <0.001 |
| III/IV | 29 | 27.6 | 98 | 12.4 | 127 | 14.1 | ||
| Residual disease | Absent | 51 | 44.0 | 371 | 43.3 | 422 | 43.4 | 0.697 |
| Unknown | 57 | 49.1 | 442 | 51.6 | 499 | 51.3 | ||
| Present | 8 | 6.9 | 44 | 5.1 | 52 | 5.3 | ||
| CD8+ TILs | Negative | 26 | 30.6 | 173 | 26.2 | 199 | 26.7 | 0.026 |
| Low | 25 | 29.4 | 118 | 17.9 | 143 | 19.2 | ||
| Moderate | 24 | 28.2 | 259 | 39.2 | 283 | 38.0 | ||
| High | 10 | 11.8 | 110 | 16.7 | 120 | 16.1 | ||
| CDKN2A | Normal | 40 | 41.2 | 619 | 82.1 | 659 | 77.4 | <0.001 |
| Abnormal block | 40 | 41.2 | 36 | 4.8 | 76 | 8.9 | ||
| Abnormal absent | 17 | 17.5 | 99 | 13.1 | 116 | 13.6 | ||
| Grade | I/II | 47 | 53.4 | 560 | 86.8 | 607 | 82.8 | <0.001 |
| III | 41 | 46.6 | 85 | 13.2 | 126 | 17.2 | ||
| CCC | Total | 86 | 662 | 748 | ||||
| Age | Mean | 56.1 | 55.2 | 0.452 | ||||
| Range | 28–83 | 27–84 | ||||||
| Stage | I/II | 50 | 61.0 | 500 | 78.6 | 550 | 76.6 | <0.001 |
| III/IV | 32 | 39.0 | 136 | 21.4 | 168 | 23.4 | ||
| Residual disease | Absent | 32 | 37.2 | 292 | 44.1 | 324 | 43.3 | <0.001 |
| Unknown | 33 | 38.4 | 305 | 46.1 | 338 | 45.2 | ||
| Present | 21 | 24.4 | 65 | 9.8 | 86 | 11.5 | ||
| CD8+ TILs | Negative | 20 | 29.4 | 266 | 50.3 | 286 | 47.9 | <0.001 |
| Low | 12 | 17.6 | 122 | 23.1 | 134 | 22.4 | ||
| Moderate | 21 | 30.9 | 81 | 15.3 | 102 | 17.1 | ||
| High | 15 | 22.1 | 60 | 11.3 | 75 | 12.6 | ||
| CDKN2A | Normal | 32 | 41.0 | 381 | 67.1 | 413 | 63.9 | <0.001 |
| Abnormal block | 34 | 43.6 | 70 | 12.3 | 104 | 16.1 | ||
| Abnormal absent | 12 | 15.4 | 117 | 20.6 | 129 | 20.0 |
For CCC, univariate analyses showed significant associations of overall survival with stage, age, residual disease, and binary p53 IHC status (supplementary material, Table S2). Stage, residual disease, and binary p53 IHC status remained significant in multivariate analysis (supplementary material, Table S2). Abnormal p53 IHC showed an HR of 1.57 (95% CI 1.11–2.22, p = 0.012) adjusted for age, stage, and residual disease and stratified by OTTA study (supplementary material, Table S2). A Kaplan‐Meier survival analysis for stage I/II CCC cases without residual disease revealed significant survival differences with p53‐abnormal cases having a 5‐YSR of 71% versus 82% for p53‐normal cases (log‐rank p = 0.0066; Figure 3A). No significant differences were observed for stage III/IV disease (Figure 3B). Univariate associations with clinical parameters showed that significantly more p53‐abnormal cases were diagnosed at a higher stage (39.0% stage III/IV for p53‐abnormal CCC compared to 21.4% for p53‐normal CCC, p < 0.001) and more likely to have residual disease after debulking surgery (Table 3). However, there were no associations with age. With respect to biomarkers, p53‐abnormal CCC cases showed higher proportions of CD8+ TILs and more commonly abnormal CDKN2A expression patterns.
Figure 3.
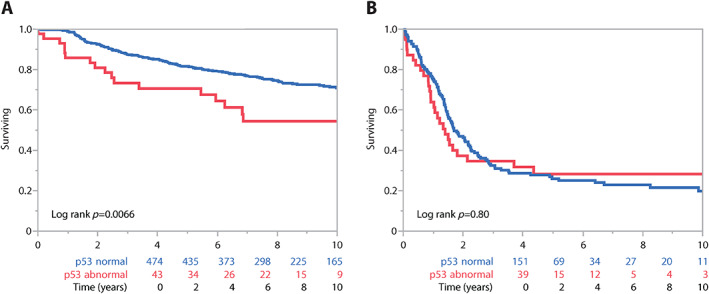
CCC: stage‐stratified Kaplan‐Meier survival analyses. (A) Abnormal p53 versus normal p53 at low FIGO stage (I/II) without residual disease. (B) Abnormal p53 versus normal p53 at high FIGO stage (III/IV).
Correlation of p53 IHC patterns with TP53 mRNA expression
Combining all three histotypes, there was a significant association of TP53 mRNA expression with the four p53 IHC patterns for 2,111 analysed cases (p < 0.0001; Figure 4). The relative mean TP53 mRNA expression normalised to housekeeping genes was (−2.54). Cases with abnormal p53 OE showed the highest mRNA expression (−2.06), followed by cases with normal p53 pattern (−2.56), while cases with abnormal CY (−3.09) and abnormal CA (−3.74) showed the lowest TP53 mRNA expression.
Figure 4.
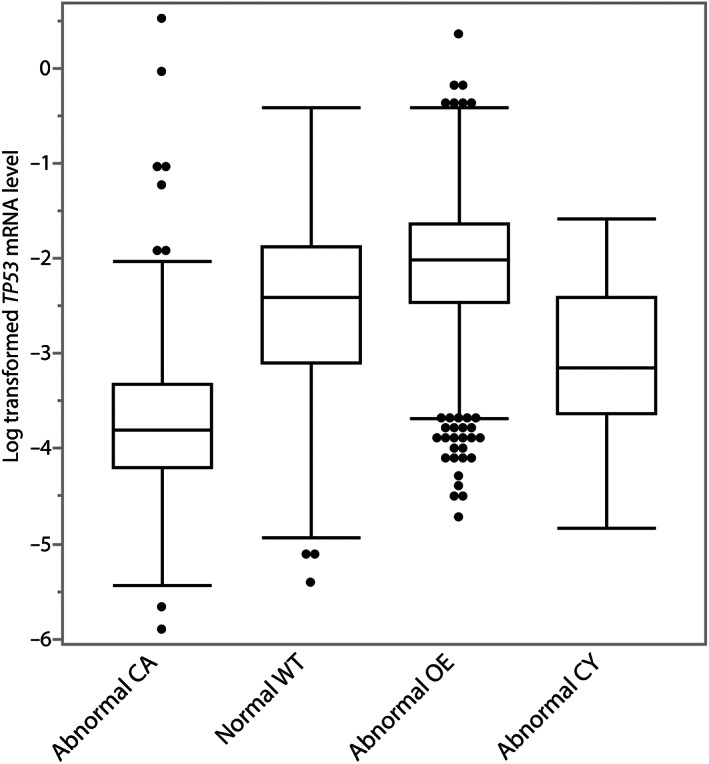
Association of p53 IHC patterns with TP53 mRNA expression, including all histotypes.
Discussion
Our study provides further evidence that the functional groups of TP53 mutations assessed by abnormal surrogate p53 IHC patterns are not associated with survival in HGSC. For endometriosis‐associated ovarian carcinomas, we validate that abnormal p53 IHC is a strong independent prognostic marker for EC and demonstrate for the first time an independent prognostic association of abnormal p53 IHC with overall survival in CCC, especially for stage I/II CCC.
There is controversy on the prognostic role of the types or functional groups of TP53 mutations in HGSC. Our data are in line with most of the studies that did not find differences between GOF and LOF TP53 mutations in HGSC [6, 8, 9]. However, the results contradict our own earlier report that suggested an adverse prognostic association for the LOF surrogate pattern CA, although the association was only significant in one of three cohorts and the combined cohort [5]. The differences may be due to increased power in the current study (4,630 versus 502) and improved interpretation of p53 IHC with better delineation of abnormal patterns. The earlier study did not report the CY pattern and had a higher rate of the normal staining (12.0% versus 6.6% in the current study). The frequency of normal staining observed in the current study (6.6%) was still higher than expected: 2–4% of HGSC can show the normal/wild type p53 pattern by IHC due to late truncating TP53 mutations that do not result in nonsense‐mediated mRNA decay of TP53 [3, 26]. Poor antigenicity due to less standardised processing and longer ischemia times historically could have led to false negative interpretations of abnormal OE as a normal pattern in older specimens. However, since we could not exclude the possibility of some misclassified p53‐normal non‐HGSC, we restricted all HGSC analyses to abnormal patterns. In contrast to a previous study [28], we saw no difference across the abnormal p53 IHC patterns with respect to clinical parameters or selected biomarkers including BRCA1/2 germline mutations. TP53 mutations are a ubiquitous driver in HGSC and may be permissive for the development of a diverse range of mutational processes driving CIN in HGSC [29]. The prognosis is primarily determined by the extreme CIN with some prognostic effects from homologous recombination DNA repair deficiency, focal amplifications, or the tumour microenvironment [19, 30, 31, 32]. Given the very high prevalence of CIN in HGSC irrespective of type of TP53 mutation, it is not surprising that p53 IHC patterns per se have no prognostic effects [29]. Since the development of p53 autoantibodies is associated with GOF mutations and decreased p53 protein degradation, we expected a higher CD8‐mediated immune response in HGSC with the abnormal OE pattern [33]. However, we did not observe an association between abnormal OE and CD8+ TILs. The expected association of p53 IHC patterns with TP53 mRNA expression serves as cross‐validation of the assays, particularly, the low mRNA expression of the CA group with LOF mutation due to nonsense‐mediated decay of TP53 mRNA. In a previous study, TP53 mRNA expression was not associated with overall survival in HGSC (HR = 0.98, 95% CI 0.95–1.02, p = 0.57) [23].
For EC, our results are comparable to three earlier studies validating the unfavourable association of abnormal p53 IHC with overall survival [10, 11, 12]. Particularly clinically relevant is our finding in support of Krämer et al that abnormal p53 IHC can identify patients with low‐stage (I/II) disease at higher risk of death. These women require adjuvant therapy, likely in the form of platinum‐based chemotherapy, which is effective in women with p53‐abnormal endometrial ECs [17]. This would potentially render p53 not only a prognostic but also a predictive marker in EC. However, p53 IHC does not identify all women with low‐stage (I/II) disease at an increased risk of mortality. In contrast to the study by Krämer et al, we show that grade is also prognostic in the low‐stage setting. This difference between Krämer et al and our current study may be explained by interobserver variability in the assignment of grade [34] or by differences in the grading system used. Although we have no information on the specific grading system for individual cases, 95% of the EC cases in the current study were diagnosed between 1996 and 2011, during a time when the Silverberg grading system was the universal standard [35]. The Silverberg grading consists of a sum score of architectural complexity, degree of nuclear atypia, and mitotic count. In 2008, Malpica proposed a histotype‐specific grading system, and the FIGO grading system for endometrial ECs was adopted for use in ovarian ECs in the 2014 WHO classification [21, 36]. The FIGO/WHO grading ranks based on the percentage of solid architecture with the option of increase by one based on high nuclear atypia. Hence, relatively newer cases in the study by Krämer et al might have been graded by the WHO/FIGO system, which was not prognostic in the stage I/II setting. In line with this observation, Parra‐Herran et al recently showed a superior prognostic stratification by the Silverberg system, particularly for grade 3, compared to the WHO/FIGO system in EC [37]. Although p53‐abnormal EC is more likely grade 3, the majority of grade 3 EC are p53 normal, and both (p53‐abnormal, grade 3 EC and p53‐normal, grade 3 EC) have a similar but shorter survival compared to p53‐normal, low‐grade EC. The p53‐normal, grade 3 EC may include the recently described uncommon histotypes of dedifferentiated ovarian carcinomas or mesonephric‐like adenocarcinomas, which were historically diagnosed as EC and are associated with shorter survival [38, 39]. Excluding them would likely have resulted in even larger survival differences between p53‐abnormal and p53‐normal EC. It may be premature to dismiss grade, yet the limitations of interobserver reproducibility with grade are likely unresolvable. But future studies with large case numbers comparing Silverberg and FIGO/WHO grade could address this issue. Notably, in the setting of stage I/II without residual disease, the combination of p53 normal and low grade did not reach a 5‐YSR of 95% generally considered as threshold to withhold adjuvant chemotherapy [40]. In order to expand the spectrum of p53‐normal, low‐risk EC to substages beyond IA/IB, other biomarker combinations such as hormone receptor PR and CTNNB1 should be further validated [18, 20, 41, 42, 43, 44, 45].
Another interesting observation is the intermediate survival of p53‐abnormal, low‐grade EC. Because low‐grade tumours by Silverberg grading usually have low nuclear atypia, these tumours might not have developed CIN in the context of a TP53 mutation, which was the rationale to use p53 as a surrogate for the copy number high genotype originally described by TCGA in endometrial carcinomas [46]. Future studies may refine the interplay between TP53 mutations and copy number status in ovarian EC. A related question is then how to identify p53‐abnormal EC because the prevalence is relatively low and universal testing might not be justified. Perhaps, a similar approach of pathology‐driven selective testing based on nuclear features, which has shown high sensitivity in endometrial carcinoma, could be used [47].
For CCC, our study is the first to demonstrate that abnormal p53 status is an independent prognostic factor. Due to the relatively low prevalence of p53‐abnormal cases (12%) and the smaller effect compared to EC, large numbers were needed to show an independent prognostic association. Cunningham et al described two methylation clusters, whereby one was associated with a higher rate of TP53 mutations and adverse clinical factors such as higher stage and residual disease, while the other cluster was characterised by ARID1A/PIK3CA co‐mutations, low‐stage disease, and aneuploidy [15]. Using targeted DNA and whole transcriptome RNA sequencing, Bolton et al also showed two distinct clusters with TP53 and ARID1A as their respective lead alterations [16]. ARID1A mutations may appear relatively favourable, but this may be due to the inverse relationship with poor prognostic p53 because ARID1A by itself was not independently prognostic in a large series of CCC [48].
Both endometriosis‐associated ovarian carcinomas, EC and CCC, showed the same prevalence (12%) of abnormal p53 cases and similar prognostic associations. In both histotypes, abnormal p53 and abnormal CDKN2A often coexist. This finding should be taken into consideration when interpreting the association of block‐like p16 expression and shorter survival in EC and CCC [20]. However, differences between EC and CCC were also observed. In EC, the survival differences persisted in stage III/IV, while there were none in stage III/IV CCC, in line with the larger effect of p53 abnormal in EC compared to CCC in the multivariate model. Given the apparent efficacy of platinum‐based chemotherapy in p53‐abnormal endometrial ECs [17], abnormal p53 may also serve as a predictive marker for platinum‐based chemotherapy in ovarian EC while high‐stage p53‐normal EC may undergo alternative treatments (e.g. immune check point blockade for mismatch repair deficient [MMRd] cases). In contrast, p53 does not predict chemotherapy response in the generally chemoresistant CCC [49]. Another difference between EC and CCC is the change in directionality for the association of abnormal p53 status and CD8+ TILs. p53‐normal ECs have higher levels of CD8+ TIL infiltration. p53‐normal EC includes ultra‐(POLE) mutated [POLEmut] and hypermutated (MMRd) molecular subtypes, which due to their increased expression of neoantigens attract more CD8+ TILs [48]. CCC has generally lower levels of CD8+ TILs compared to EC [19], which is related to the absence of immunogenic POLEmut and MMRd molecular subtypes in CCC [19, 50]. TP53 mutated CCC shows enriched expression of genes involved in immune‐related pathways [16].
Limitations of our study include incomplete data annotations for some covariates such as grade. For EC, the molecular subtype (i.e. POLEmut or MMRd) status was not available. However, the frequency of the prognostically favourable POLEmut is three times lower in ovarian EC (3.5%) compared to endometrial carcinomas (~10%) [11]. According to León‐Castillo et al, p53 abnormalities might be secondary to POLEmut; however, most are subclonal [51]. Truncal p53 abnormalities in the context of POLEmut were only rarely seen in 1 of 177 (0.6%) endometrial carcinomas [24]. Considering that POLEmut is three times less common in ovarian compared to endometrial EC, the possibility of co‐occurrence of truncal TP53 and POLE mutations in ovarian EC is remote. Our data suggest that there is misclassification of p53‐normal non‐HGSC into the HGSC category; however, we restricted analyses to p53‐abnormal HGSC. On the other hand, we used WT1 alone or in combination with p53 to ensure that there was no misclassification of HGSC as ‘EC or CCC’. Additional overlay with TP53 mutations would have strengthened the findings and potentially validated whether certain GOF mutations as identified by Tuna et al are associated with an unfavourable outcome in HGSC [9].
In conclusion, we provide strong evidence that abnormal p53 by IHC is an independent unfavourable prognostic marker in ovarian EC and CCC, which in conjunction with other biomarkers could inform clinical management in the low‐stage setting. Abnormal patterns of p53 IHC show no association with survival in HGSC. Hence, in HGSC, its role remains in the diagnostic and not the prognostic realm.
Author contributions statement
MK and JDB conceived, designed, and supervisedthe study. PFR, EYK and MK collected immunohistochemical protein scores. MK and AW performed analyses. MK and EYK drafted the manuscript and JDB revised the manuscript. All authors contributed through collection, curation, and maintenance of respective consortia based, or local institution, collections of patient samples including recruitment and consenting of patients, clinical care, abstraction of clinical data, and updating of outcome and follow‐up data. All authors revised the manuscript and approved submission of the final version.
Disclosures
JDB is a founder of Tailor Bio.
Supporting information
Table S1. Study information
Table S2. Uni‐ and multivariate Cox regression analyses for EC and CCC
Acknowledgements
For all participating studies, the authors thank all the women who participated in research, study staff, study participants, doctors, nurses, health care providers, and health information sources who have contributed to the study. The authors thank Shuhong Liu and Young Ou from the Anatomical Pathology Research Laboratory at the University of Calgary for performing mmunohistochemistry, and Thomas Kryton, image specialist, for compiling the composite figures. The authors thank the Australian Ovarian Cancer Study (AOCS) Group that can be found at http://www.aocstudy.org.
The study was funded by Alberta Precision Laboratory research support fund (RS17‐601, RS19‐609, RS10‐526). AOCS was supported by the U.S. Army Medical Research and Materiel Command under DAMD17‐01‐1‐0729, the Cancer Council Victoria, Queensland Cancer Fund, the Cancer Council New South Wales, the Cancer Council South Australia, the Cancer Foundation of Western Australia, the Cancer Council Tasmania, and the National Health and Medical Research Council of Australia (NHMRC; ID199600, ID400413, and ID400281). AOCS gratefully acknowledges additional support from the Peter MacCallum Foundation and Ovarian Cancer Australia (OCA); AOV: Canadian Institutes of Health Research (MOP‐86727), we thank Mie Konno, Michelle Darago, Faye Chambers; BAV: ELAN Funds of the University of Erlangen‐Nuremberg; BGS: Breast Cancer Now and the Institute of Cancer Research (ICR). ICR acknowledges NHS funding to the NIHR Biomedical Research Centre; BRZ: Brazilian National Council for Scientific and Technological Development, grant No. 478416/2009‐1; CAL: Cancer Research Society (19319) and CLS Internal Research support RS14‐508; CNI: CNI funded by Instituto de Salud Carlos Tercero (AES grant PI19/01730) and Fondo Europeo de Desarrollo Regional, FEDER. Instituto de Salud Carlos III (PI 12/01319); Ministerio de Economía y Competitividad (SAF2012). AO is partially funded by FIS PI19/00640 supported by FEDER funds and the Spanish Network on Rare Diseases (CIBERER); DOV: NCI/NIH R01 CA168758, NCI/NIH R01 CA112523, and NCI/NIH R01 CA087538. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH; GER: German Federal Ministry of Education and Research, Programme of Clinical Biomedical Research (01GB9401) and the German Cancer Research Center (DKFZ); HAW: US National Institutes of Health (R01‐CA58598, N01‐CN‐55424, and N01‐PC‐67001); HOP: Department of Defense (DAMD17‐02‐1‐0669) and NCI (K07‐CA080668, R01‐CA95023, MO1‐RR000056). This project used the UPMC Hillman Cancer Center and Tissue and Research Pathology/Pitt Biospecimen Core shared resource, which is supported in part by award P30CA047904; LAX: American Cancer Society Early Detection Professorship (SIOP‐06‐258‐01‐COUN) and the National Center for Advancing Translational Sciences (NCATS), Grant UL1TR000124; MAY: National Institutes of Health (R01‐CA122443, R01‐CA243483, P30‐CA15083, P50‐CA136393); Mayo Foundation; Minnesota Ovarian Cancer Alliance; Fred C. and Katherine B. Andersen Foundation; POC: Pomeranian Medical University; SEA: Cancer Research UK C490/A16561, the UK National Institute for Health Research Biomedical Research Centres at the University of Cambridge, Cambridge Cancer Centre. The University of Cambridge has received salary support for PDPP from the NHS in the East of England through the Clinical Academic Reserve; STA: National Institutes of Health (U01‐CA71966, U01‐CA69417); SWE: Swedish Cancer Foundation (CAN 2018/384); TVA: This work was supported by Canadian Institutes of Health Research grant (MOP‐86727) and by NIH/NCI 1 R01CA160669‐01A1; UKO: The UKOPS study was funded by The Eve Appeal (the Oak Foundation) with contribution to U. Menon and A. Gentry‐Maharaj's salary through MRC core funding (MC_UU_00004/01) and the National Institute for Health Research University College London Hospitals Biomedical Research Centre; VAN: BC's Gynecological Cancer Research Team (OVCARE) receives core funding from The BC Cancer Foundation and the VGH and UBC Hospital Foundation; WMH: National Health and Medical Research Council of Australia, Enabling Grants ID 310670 and ID 628903. Cancer Institute NSW Grants 12/RIG/1‐17 and 15/RIG/1‐16. The Westmead GynBiobank acknowledges financial support from the Sydney West Translational Cancer Research Centre, funded by the Cancer Institute NSW. SJR is supported by National Health and Medical Research Council of Australia (NHMRC) grant APP2009840. The contents of the published material are solely the responsibility of the authors and do not reflect the views of NHMRC.
Conflict of interest statement: The authors have no relevant conflicts of interest regarding this publication. AD has received grant funding from AstraZeneca, unrelated to work described in this publication. AH has received honoraria from BMS, MSD, Roche, AstraZeneca, Boehringer Ingelheim, Abbvie, Jansen‐Cilag, and Ipsen. DDB has received honoraria from AstraZeneca, has a consulting role with Exo Therapeutics Research, and receives funding from Roche/Genentech, AstraZeneca, and BeiGene. UM has received an Honorarium from NY Obstetrical Society and held personal shares between 1 April 2011 and 30 October 2021. PG has received honoraria from AstraZeneca, GSK, and Eisai.
Contributor Information
Martin Köbel, Email: mkoebel@ucalgary.ca.
James D Brenton, Email: James.Brenton@cruk.cam.ac.uk.
Data availability statement
Individual patient data and related tumor information underlying this article cannot be shared publicly due to data privacy protection laws. However, grouped data will be shared on reasonable request to the corresponding author.
References
- 1. Ahmed AA, Etemadmoghadam D, Temple J, et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J Pathol 2010; 221: 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vang R, Levine DA, Soslow RA, et al. Molecular alterations of TP53 are a defining feature of ovarian high‐grade serous carcinoma: a rereview of cases lacking TP53 mutations in The Cancer Genome Atlas Ovarian study. Int J Gynecol Pathol 2016; 35: 48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Köbel M, Piskorz AM, Lee S, et al. Optimized p53 immunohistochemistry is an accurate predictor of TP53 mutation in ovarian carcinoma. J Pathol Clin Res 2016; 2: 247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rabban JT, Garg K, Ladwig NR, et al. Cytoplasmic pattern p53 immunoexpression in pelvic and endometrial carcinomas with TP53 mutation involving nuclear localization domains: an uncommon but potential diagnostic pitfall with clinical implications. Am J Surg Pathol 2021; 45: 1441–1451. [DOI] [PubMed] [Google Scholar]
- 5. Köbel M, Reuss A, du Bois A, et al. The biological and clinical value of p53 expression in pelvic high‐grade serous carcinomas. J Pathol 2010; 222: 191–198. [DOI] [PubMed] [Google Scholar]
- 6. Kang HJ, Chun SM, Kim KR, et al. Clinical relevance of gain‐of‐function mutations of p53 in high‐grade serous ovarian carcinoma. PLoS One 2013; 8: e72609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brachova P, Mueting SR, Carlson MJ, et al. TP53 oncomorphic mutations predict resistance to platinum and taxane‐based standard chemotherapy in patients diagnosed with advanced serous ovarian carcinoma. Int J Oncol 2015; 46: 607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mandilaras V, Garg S, Cabanero M, et al. TP53 mutations in high grade serous ovarian cancer and impact on clinical outcomes: a comparison of next generation sequencing and bioinformatics analyses. Int J Gynecol Cancer 2019; 29: 346–352. [DOI] [PubMed] [Google Scholar]
- 9. Tuna M, Ju Z, Yoshihara K, et al. Clinical relevance of TP53 hotspot mutations in high‐grade serous ovarian cancers. Br J Cancer 2020; 122: 405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Parra‐Herran C, Lerner‐Ellis J, Xu B, et al. Molecular‐based classification algorithm for endometrial carcinoma categorizes ovarian endometrioid carcinoma into prognostically significant groups. Mod Pathol 2017; 30: 1748–1759. [DOI] [PubMed] [Google Scholar]
- 11. Krämer P, Talhouk A, Brett MA, et al. Endometrial cancer molecular risk stratification is equally prognostic for endometrioid ovarian carcinoma. Clin Cancer Res 2020; 26: 5400–5410. [DOI] [PubMed] [Google Scholar]
- 12. Leskela S, Romero I, Rosa‐Rosa JM, et al. Molecular heterogeneity of endometrioid ovarian carcinoma: an analysis of 166 cases using the endometrial cancer subrogate molecular classification. Am J Surg Pathol 2020; 44: 982–990. [DOI] [PubMed] [Google Scholar]
- 13. Parra‐Herran C, Bassiouny D, Lerner‐Ellis J, et al. p53, mismatch repair protein, and POLE abnormalities in ovarian clear cell carcinoma: an outcome‐based clinicopathologic analysis. Am J Surg Pathol 2019; 43: 1591–1599. [DOI] [PubMed] [Google Scholar]
- 14. Similä‐Maarala J, Soovares P, Pasanen A, et al. TCGA molecular classification in endometriosis‐associated ovarian carcinomas: novel data on clear cell carcinoma. Gynecol Oncol 2022; 165: 577–584. [DOI] [PubMed] [Google Scholar]
- 15. Cunningham JM, Winham SJ, Wang C, et al. DNA methylation profiles of ovarian clear cell carcinoma. Cancer Epidemiol Biomarkers Prev 2022; 31: 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bolton KL, Chen D, Corona de la Fuente R, et al. Molecular subclasses of clear cell ovarian carcinoma and their impact on disease behavior and outcomes. Clin Cancer Res 2022; 28: 4947–4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. León‐Castillo A, de Boer SM, Powell ME, et al. Molecular classification of the PORTEC‐3 trial for high‐risk endometrial cancer: impact on prognosis and benefit from adjuvant therapy. J Clin Oncol 2020; 38: 3388–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sieh W, Köbel M, Longacre TA, et al. Hormone‐receptor expression and ovarian cancer survival: an Ovarian Tumor Tissue Analysis consortium study. Lancet Oncol 2013; 14: 853–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ovarian Tumor Tissue Analysis (OTTA) Consortium , Goode EL, Block MS, et al. Dose‐response association of CD8+ tumor‐infiltrating lymphocytes and survival time in high‐grade serous ovarian cancer. JAMA Oncol 2017; 3: e173290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rambau PF, Vierkant RA, Intermaggio MP, et al. Association of p16 expression with prognosis varies across ovarian carcinoma histotypes: an Ovarian Tumor Tissue Analysis consortium study. J Pathol Clin Res 2018; 4: 250–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kurman RJ, Carcangiu ML, Herrington CS, et al. WHO Classification of Tumours of Female Reproductive Organs (4th edn). Lyon, France: IARC, 2014. [Google Scholar]
- 22. Köbel M, Rahimi K, Rambau PF, et al. An immunohistochemical algorithm for ovarian carcinoma typing. Int J Gynecol Pathol 2016; 35: 430–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Millstein J, Budden T, Goode EL, et al. Prognostic gene expression signature for high‐grade serous ovarian cancer. Ann Oncol 2020; 31: 1240–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Singh N, Piskorz AM, Bosse T, et al. p53 immunohistochemistry is an accurate surrogate for TP53 mutational analysis in endometrial carcinoma biopsies. J Pathol 2020; 250: 336–345. [DOI] [PubMed] [Google Scholar]
- 25. Köbel M, Kang EY. The many uses of p53 immunohistochemistry in gynecological pathology: proceedings of the ISGyP companion society session at the 2020 USCAP annual meeting. Int J Gynecol Pathol 2021; 40: 32–40. [DOI] [PubMed] [Google Scholar]
- 26. Köbel M, Luo L, Grevers X, et al. Ovarian carcinoma histotype: strengths and limitations of integrating morphology with immunohistochemical predictions. Int J Gynecol Pathol 2019; 38: 353–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Köbel M, Kalloger SE, Boyd N, et al. Ovarian carcinoma subtypes are different diseases: implications for biomarker studies. PLoS Med 2008; 5: e232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Boyarskikh UA, Gulyaeva LF, Avdalyan AM, et al. Spectrum of TP53 mutations in BRCA1/2 associated high‐grade serous ovarian cancer. Front Oncol 2020; 10: 1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Macintyre G, Goranova TE, De Silva D, et al. Copy number signatures and mutational processes in ovarian carcinoma. Nat Genet 2018; 50: 1262–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Köbel M, Huntsman D, Gilks CB. Critical molecular abnormalities in high‐grade serous carcinoma of the ovary. Expert Rev Mol Med 2008; 10: e22. [DOI] [PubMed] [Google Scholar]
- 31. Bolton KL, Chenevix‐Trench G, Goh C, et al. Association between BRCA1 and BRCA2 mutations and survival in women with invasive epithelial ovarian cancer. JAMA 2012; 307: 382–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chan AM, Enwere E, McIntyre JB, et al. Combined CCNE1 high‐level amplification and overexpression is associated with unfavourable outcome in tubo‐ovarian high‐grade serous carcinoma. J Pathol Clin Res 2020; 6: 252–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Anderson KS, Wong J, Vitonis A, et al. p53 autoantibodies as potential detection and prognostic biomarkers in serous ovarian cancer. Cancer Epidemiol Biomarkers Prev 2010; 19: 859–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gilks CB, Ionescu DN, Kalloger SE, et al. Tumor cell type can be reproducibly diagnosed and is of independent prognostic significance in patients with maximally debulked ovarian carcinoma. Hum Pathol 2008; 39: 1239–1251. [DOI] [PubMed] [Google Scholar]
- 35. Shimizu Y, Kamoi S, Amada S, et al. Toward the development of a universal grading system for ovarian epithelial carcinoma: testing of a proposed system in a series of 461 patients with uniform treatment and follow‐up. Cancer 1998; 82: 893–901. [DOI] [PubMed] [Google Scholar]
- 36. Malpica A. Grading of ovarian cancer: a histotype‐specific approach. Int J Gynecol Pathol 2008; 27: 175–181. [DOI] [PubMed] [Google Scholar]
- 37. Parra‐Herran C, Bassiouny D, Vicus D, et al. FIGO versus Silverberg grading systems in ovarian endometrioid carcinoma: a comparative prognostic analysis. Am J Surg Pathol 2019; 43: 161–167. [DOI] [PubMed] [Google Scholar]
- 38. Tessier‐Cloutier B, Coatham M, Carey M, et al. SWI/SNF‐deficiency defines highly aggressive undifferentiated endometrial carcinoma. J Pathol Clin Res 2021; 7: 144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. da Silva EM, Fix DJ, Sebastiao APM, et al. Mesonephric and mesonephric‐like carcinomas of the female genital tract: molecular characterization including cases with mixed histology and matched metastases. Mod Pathol 2021; 34: 1570–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Köbel M, Kalloger SE, Santos JL, et al. Tumor type and substage predict survival in stage I and II ovarian carcinoma: insights and implications. Gynecol Oncol 2010; 116: 50–56. [DOI] [PubMed] [Google Scholar]
- 41. Rambau P, Kelemen LE, Steed H, et al. Association of hormone receptor expression with survival in ovarian endometrioid carcinoma: biological validation and clinical implications. Int J Mol Sci 2017; 18: 515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hollis RL, Stanley B, Iida Y, et al. Hormone receptor expression patterns define clinically meaningful subgroups of endometrioid ovarian carcinoma. Gynecol Oncol 2019; 155: 318–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hollis RL, Thomson JP, Stanley B, et al. Molecular stratification of endometrioid ovarian carcinoma predicts clinical outcome. Nat Commun 2020; 11: 4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang L, Rambau PF, Kelemen LE, et al. Nuclear β‐catenin and CDX2 expression in ovarian endometrioid carcinoma identify patients with favourable outcome. Histopathology 2019; 74: 452–462. [DOI] [PubMed] [Google Scholar]
- 45. Hollis RL, Stanley B, Thomson JP, et al. Integrated molecular characterisation of endometrioid ovarian carcinoma identifies opportunities for stratification. NPJ Precis Oncol 2021; 5: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cancer Genome Atlas Research Network , Kandoth C, Schultz N, et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013; 497: 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kang EY, Wiebe NJ, Aubrey C, et al. Selection of endometrial carcinomas for p53 immunohistochemistry based on nuclear features. J Pathol Clin Res 2022; 8: 19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Heinze K, Nazeran TM, Lee S, et al. Validated biomarker assays confirm that ARID1A loss is confounded with MMR deficiency, CD8+ TIL infiltration, and provides no independent prognostic value in endometriosis‐associated ovarian carcinomas. J Pathol 2022; 256: 388–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Takenaka M, Köbel M, Garsed DW, et al. Survival following chemotherapy in ovarian clear cell carcinoma is not associated with pathological misclassification of tumor histotype. Clin Cancer Res 2019; 25: 3962–3973. [DOI] [PubMed] [Google Scholar]
- 50. Rodriguez M, Kang EY, Farrington K, et al. Accurate distinction of ovarian clear cell from endometrioid carcinoma requires integration of phenotype, immunohistochemical predictions, and genotype: implications for Lynch syndrome screening. Am J Surg Pathol 2021; 45: 1452–1463. [DOI] [PubMed] [Google Scholar]
- 51. León‐Castillo A, Gilvazquez E, Nout R, et al. Clinicopathological and molecular characterisation of 'multiple‐classifier' endometrial carcinomas. J Pathol 2020; 250: 312–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Study information
Table S2. Uni‐ and multivariate Cox regression analyses for EC and CCC
Data Availability Statement
Individual patient data and related tumor information underlying this article cannot be shared publicly due to data privacy protection laws. However, grouped data will be shared on reasonable request to the corresponding author.