

Abstract
Mitochondrial DNA (mtDNA) diseases are multi‐systemic disorders caused by mutations affecting a fraction or the entirety of mtDNA copies. Currently, there are no approved therapies for the majority of mtDNA diseases. Challenges associated with engineering mtDNA have in fact hindered the study of mtDNA defects. Despite these difficulties, it has been possible to develop valuable cellular and animal models of mtDNA diseases. Here, we describe recent advances in base editing of mtDNA and the generation of three‐dimensional organoids from patient‐derived human‐induced pluripotent stem cells (iPSCs). Together with already available modeling tools, the combination of these novel technologies could allow determining the impact of specific mtDNA mutations in distinct human cell types and might help uncover how mtDNA mutation load segregates during tissue organization. iPSC‐derived organoids could also represent a platform for the identification of treatment strategies and for probing the in vitro effectiveness of mtDNA gene therapies. These studies have the potential to increase our mechanistic understanding of mtDNA diseases and may open the way to highly needed and personalized therapeutic interventions.
Keywords: iPSCs, mitochondria, mitochondrial diseases, mitochondrial genome editing, organoids
Subject Categories: Genetics, Gene Therapy & Genetic Disease; Molecular Biology of Disease; Stem Cells & Regenerative Medicine
Mitochondrial DNA diseases are multi‐systemic disorders caused by mtDNA mutations. This review highlights recent advances in base editing of mtDNA and in the generation of organoids from patient‐derived iPSCs, opening the way to personalized therapies.
Introduction—mitochondria and mitochondrial DNA diseases
Mitochondria are double‐membrane organelles essential for the execution of key cellular functions including bioenergetics, redox balance, calcium homeostasis, and overall intercellular signal transduction (Picard & Shirihai, 2022). The vast majority of genes needed to produce more than 1,000 proteins important for mitochondrial activities are encoded in the nucleus (Pagliarini et al, 2008; Rath et al, 2021). In addition, mitochondria harbor their own genome. Mitochondrial DNA (mtDNA) is a small, circular molecule approximately 16,500 base pairs (bp) in size, encoding 13 proteins, 22 tRNAs, and two rRNAs (Anderson et al, 1981). These 37 mtDNA genes are vital to ensure proper oxidative phosphorylation (OXPHOS) functionality, which thus depends on the concerted action of both nuclear and mitochondrial genomes. Unlike nuclear DNA, mtDNA is polyploid, which means that within each cell there are multiple copies of mtDNA (Magnusson et al, 2003; Gustafsson et al, 2016).
The number of mtDNA copies can vary greatly in different cell types and organs (Filograna et al, 2021). For example, in mammals, sperm may contain around 100 copies of mtDNA while oocytes contain 150,000 copies (Chen et al, 1995; Wai et al, 2010). In humans, blood leukocytes contain around 150–600 mtDNA copies (Picard, 2021; Rausser et al, 2021) and the heart around 4–6,000 (D'Erchia et al, 2015). mtDNA content may even vary within the same tissue, as in the case of mammalian brain, where region‐specific variability can be observed (Brinckmann et al, 2010; Fuke et al, 2011). Although tissues with greater energy demands appear to contain more mtDNA copies per cell (Picard, 2021), the underlying mechanisms are not fully understood. It has been suggested that methylation of the mitochondrial DNA polymerase gamma (PolG) may contribute to the regulation of mtDNA copy number in tissues (Kelly et al, 2012). The relation with cell cycle progression remains not clear, as contrasting evidence exists with respect to the correlation between mtDNA initiation/replication and cell cycle phases (Pica‐Mattoccia & Attardi, 1972; Antes et al, 2010; Chatre & Ricchetti, 2013).
Mitochondrial diseases are genetic conditions caused by mutations in either nuclear or mitochondrial genes, ultimately impairing OXPHOS and mitochondrial function (Vafai & Mootha, 2012). Mitochondrial diseases are multi‐systemic, but often affect tissues with high energy demands, such as the nervous system, the heart, and skeletal muscles (Wallace, 1999; Russell et al, 2020). In this review, we focus on those mitochondrial diseases that are specifically caused by mtDNA alterations, including point mutations and large‐scale deletions. Given the multi‐copy nature of mtDNA, mtDNA point mutations or large‐scale deletions can affect a portion of mtDNA copies (i.e. heteroplasmy) or virtually all mtDNA copies (i.e., homoplasmy). mtDNA diseases comprise different clinical syndromes and are estimated to occur in 1:5,000 individuals (Cree et al, 2009; Table 1).
Table 1.
mtDNA diseases.
| Clinical syndromes caused by mtDNA point mutations | |||||
|---|---|---|---|---|---|
| Disease | Main clinical features | Frequent genes affected | Frequent mutations | Gene function | Mutation load |
| Leigh Syndrome (LS) | Psychomotor regression, seizures, difficulty in breathing, hypotonia, ataxia, lactic acidosis, and fatigue |
MT‐ND1 MT‐ND2 MT‐ND3 MT‐ND4 MT‐ND5 MT‐ND6 MT‐ATP6 MT‐ATP8 |
m.3890G > A m.13513G > A m.8993T > C m.8933T > G m.9185T > C m.9176T > G |
Protein subunits of Complex I or Complex V | Heteroplasmy >20% for Complex I mutations; Heteroplasmy >90% or homoplasmy for Complex V mutations |
| Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke‐like episodes (MELAS) | Seizures, stroke‐like episodes with encephalopathy, possible presence of myopathy, cardiomyopathy, and ataxia |
MT‐TL1 MT‐TQ MT‐TH MT‐TK MT‐TS1 MT‐TS2 MT‐ND1 MT‐ND5 MT‐ND6 |
m.3243A > G m.5541C > T |
tRNAs and protein subunits of Complex I | Heteroplasmy 50–90% |
| Myoclonic Epilepsy and Ragged‐Red Fibers (MERRF) | Myoclonus, epilepsy, progressive ataxia, muscle weakness seizures, ataxia, and cardiomyopathy |
MT‐TL1, MT‐TH MT‐TK MT‐TS1 MT‐TF MT‐TH MT‐TI MT‐TP MT‐TS2 |
m.8344A > G | tRNAs | Heteroplasmy 20–90% |
| Neuropathy, Ataxia, and Retinitis Pigmentosa (NARP) | Pigmentary retinopathy, ataxia, and neuropathy |
MT‐ATP6 MT‐ATP8 |
m.8993T > C m.8933T > G |
Protein subunits of Complex V | Heteroplasmy <90% |
| Leber's Hereditary Optic Neuropathy (LHON) | Subacute visual failure |
MT‐ND1 MT‐ND4 MT‐ND5 MT‐ND6 |
m.11778G > A m.14484T > C m.3460G > A m.13513G > A |
Protein subunits of Complex I | Homoplasmy |
| Clinical syndromes caused by mtDNA large‐scale deletions | |||
|---|---|---|---|
| Disease | Main clinical features | Type of mtDNA deletions | Mutation load |
| Chronic Progressive External Ophthalmoplegia (CPEO) | Ptosis, ophthalmoparesis, and myopathy | Single large‐scale mtDNA deletions (SLSMDs) | Variable heteroplasmy (also < 50%) |
| Kearns–Sayre Syndrome (KSS) | Progressive External Ophthalmoplegia, ptosis, pigmentary retinopathy, ataxia, sensorineural hearing loss, and myopathy | SLSMDs | Variable heteroplasmy (usually > 50%) |
| Pearson Marrow–Pancreas syndrome (PMPS) | Sideroblastic anemia, neutropenia and thrombocytopenia, diabetes, and failure to thrive | SLSMDs | Variable heteroplasmy (usually > 70%) |
Point mutations in mtDNA constitute a large part of mtDNA diseases and result in various clinical entities. Leigh syndrome (LS, OMIM # 256000) is a pediatric progressive retardation of psychomotor function associated with incoordination of eye movements, recurrent vomiting, epilepsy, pyramidal and extrapyramidal signs, central abnormalities of pulmonary ventilation, and lactic acidosis (Baertling et al, 2014; Lim et al, 2022a). Magnetic resonance imaging showing bilateral calcification in the basal ganglia region is particularly important and is pathognomonic of the disease. The most common mtDNA mutations affect genes encoding for components of Complex I (e.g., mutation m.3890G > A in gene MT‐ND1 or mutation m.13513G > A in gene MT‐ND5) or components of Complex V (e.g., mutation m.8993T > G or m.8993T > C in gene MT‐ATP6). Forms due to Complex V defects are also referred to as maternally inherited Leigh syndrome (MILS; Ciafaloni et al, 1993). Although the heteroplasmy can be highly variable in LS patients, Complex I mutations have been reported to be causative of LS even at a level of 20%, while MILS develops for heteroplasmy higher than 90% (Thorburn et al, 1993; Stenton et al, 2022; Lim et al, 2022a). Neuropathy, ataxia, retinitis pigmentosa, and ptosis (NARP, OMIM #551500) includes epilepsy and mental decline in addition to the clinical signs that make up the acronym, and usually occur in adulthood (Holt et al, 1990). NARP is caused by the same mutations that would cause MILS, but present at lower heteroplasmy (Thorburn et al, 1993). Mitochondrial encephalopathy, lactic acidosis, and stroke‐like episodes (MELAS, OMIM #540000) is defined by the presence of stroke‐like episodes, lactic acidosis, and ragged‐red fibers (Pavlakis et al, 1984). Other signs of central nervous system involvement include mental decline, recurrent headaches, focal epilepsy, and sensorineural deafness. The onset of the disease is variable, ranging from early childhood to young adulthood. MELAS is typically due to mtDNA mutations in tRNA genes; the most frequent one is m.3243A > G in gene MT‐TL1 (Goto et al, 1990; Nesbitt et al, 2013). The heteroplasmy in MELAS patients is variable but typically around 50–90%, with higher levels in the central nervous system compared to peripheral muscles (Scholle et al, 2020). Myoclonus, epilepsy, and ragged‐red fibers (MERRF, OMIM #545000) is characterized by myoclonus, muscle hyposthenia, hypotrophy, cerebellar ataxia, hearing loss, and mental deterioration (Shoffner & Wallace, 1992). The extent of clinical manifestations can vary widely even within the same family. Like MELAS, MERRF is caused by mutations in tRNA genes (e.g., m.8344A > G in gene MT‐TK) with variable heteroplasmy around 20–90% that is typically higher in the central nervous system (Lertrit et al, 1992; Hameed & Tadi, 2022). Leber's hereditary optic neuropathy (LHON, OMIM #535000) is hallmarked by acute or sub‐acute loss of central vision with outcome in optic atrophy, with onset in youth and higher prevalence in males (Yu‐Wai‐Man et al, 2002). Visual impairment is usually the only clinical manifestation of the disease. The three most frequent mutations affect genes encoding for components of Complex I (i.e., m.11778G > A in gene MT‐ND4; m.3460G > A in gene MT‐ND1; and m.14484T > C in gene MT‐ND6) and are mainly present in the patients at homoplasmic level (Wallace et al, 1988).
Single large‐scale mtDNA deletions (SLSMDs) cause clinical syndromes comprising three overlapping phenotypes that are rarely observed in more than one member of the same family: chronic progressive external ophthalmoplegia (CPEO), Kearns–Sayre syndrome (KSS), and Pearson marrow–pancreas syndrome (PMPS; Harding & Hammans, 1992; Goldstein & Falk, 1993; Pitceathly et al, 2012; Broomfield et al, 2015). CPEO is characterized mainly by weakness or paralysis of the muscles that move the eye (ophthalmoplegia), and myopathy may also be present. Besides SLSMDs, CPEO can also be caused by single‐nucleotide substitutions in specific transfer (t)RNA genes (Moraes et al, 1993; Taylor et al, 2002) and pathogenic mutations in nuclear genes leading to multiple mtDNA deletions (Heighton et al, 2019). KSS (OMIM #530000) is characterized by a combination of ophthalmoplegia, retinal pigmentary degeneration, and cardiomyopathy (Tsang et al, 2018). PMPS (OMIM #557000) presents as refractory sideroblastic anemia with vacuolization of bone marrow precursors and exocrine pancreatic dysfunction (Rotig et al, 1989). Affected patients, who recovered spontaneously from infantile sideroblastic anemia, may later develop features of KSS (Larsson et al, 1990). SLSMDs are usually around 5,000 bp long. The most frequent deletion is the so‐called “common deletion” which is 4,977 bp long and starts from position 8,470 until position 13,446 (Cortopassi & Arnheim, 1990). The level of heteroplasmy for SLSMDs is variable and is typically higher in KSS and PMPS than in CPEO (López‐Gallardo et al, 2009; Broomfield et al, 2015).
Here, we provide an overview of the challenges associated with studying mtDNA diseases. In addition to clinical complexity, mtDNA diseases have been hard to model due to specific difficulties in mtDNA engineering, which have hampered the generation of a vast repertoire of in vitro and in vivo models. We describe how recent breakthrough findings could offer unprecedented opportunities for tackling mtDNA diseases. The possibility to perform base editing of mtDNA and significant advances in induced pluripotent stem cells (iPSCs) modeling using three‐dimensional (3D) organoids could lead to the development of innovative model platforms for assessing the impact of specific mtDNA mutations in different human tissues, and for evaluating the effectiveness of targeted treatments or mtDNA gene therapy in vitro. We anticipate that the combination of such technologies could pave the way to highly needed disease‐modifying and personalized interventional strategies for families affected by mtDNA diseases.
Challenges in handling and treating mtDNA diseases
A major challenge in developing treatments for mtDNA diseases is the extreme variability in the genotype–phenotype correlation (Wallace, 1999; Gorman et al, 2016). Besides LHON, which only affects the optic nerve, mtDNA diseases are clinically heterogeneous and typically involve multiple organs. Each defined clinical syndrome can be caused by different mtDNA mutations (Table 1). At the same time, certain individual mtDNA mutations can be associated with various clinical presentations. For example, mutations m.8993T > G or m.8993T > C in the gene MT‐ATP6 can cause LS or NARP depending on the heteroplasmy level, and mutation m.13513G > A in the gene MT‐ND5 can be associated with MELAS, LS, or LHON (Bannwarth et al, 2013). Furthermore, individuals affected by the same clinical syndrome might show distinct symptomatology and severity, irrespective of the underlying mtDNA mutation (Gorman et al, 2016).
Given this complexity, it has been challenging to identify treatments that are effective for a defined clinical phenotype or a defined causative genotype. Patients affected by mtDNA diseases may be pharmacologically treated with a combination of vitamins and antioxidants known as “mitochondrial cocktail,” although the efficacy of such intervention appears to be limited and unspecific (Bottani et al, 2020; Russell et al, 2020; Weissig, 2020). In addition, given the variable organ impairment, different patients may require organ‐specific therapies.
One notable exception is LHON, which is a unique mtDNA disease with restricted symptomatology (i.e., degeneration of retinal ganglion cells in the optic nerve). The antioxidant drug Idebenone has been officially approved for the treatment of LHON patients by the European Medicines Agency (EMA; Klopstock et al, 2011). LHON is the first mtDNA disease treated with mtDNA‐based gene therapy. The allotopic expression of wild‐type MT‐ND4 through intra‐ocular delivery using adeno‐associated viruses (AAV) has been proposed as a means to ameliorate vision in LHON (Guy et al, 2017). Remarkably, a phase III trial based on this gene therapy approach indeed reported improved eye function in treated LHON patients (Yu‐Wai‐Man et al, 2020; Falabella et al, 2022). For other mtDNA diseases, however, there are currently no approved therapies. Nonetheless, several treatment strategies are being pursued and different clinical trials are ongoing. These interventions include the modulation of mitochondrial biogenesis, NAD/NADH ratio, mitochondrial turnover, redox balance, or hypoxia (Bottani et al, 2020; Russell et al, 2020; Weissig, 2020).
One positive recent development stemming from advances in in vitro fertilization technologies is the possibility to prevent the transmission of pathogenic mtDNA to offspring. This procedure, known as mitochondrial replacement therapy (MRT), is based on transferring the nuclear DNA from an affected mother carrying mtDNA mutations into an enucleated oocyte or zygote of a donor woman with healthy mtDNA (Greenfield et al, 2017). In this way, the nuclear DNA of the offspring comes from the actual mother, while the mtDNA comes from the healthy female donor. There have been positive studies for MRT in rodents and primates (Tachibana et al, 2013; Kang et al, 2016b). MRT has apparently already been successful in a Mexican family (Zhang et al, 2017). However, this technology is currently only approved in a small number of countries, and some countries may never approve it because of ethical reasons. Moreover, it has been shown that a reversion to pathogenic mtDNA heteroplasmy can occur in some cases (Hudson et al, 2019). The reason for this reversion is not clear and may be potentially linked to unwanted carryover of the maternal mutated mtDNA taken up in the process of extracting the nucleus, or to possible selection of some mutations or mtDNA haplotypes (Chinnery et al, 2014). There are also concerns related to the long‐term consequences of MRT. Studies in mice and flies implied that subtle nuclear–mitochondrial DNA mismatch could cause late‐onset cardiac defects or infertility (Latorre‐Pellicer et al, 2016). Lastly, MRT cannot be used as a means to treat patients who are already affected by mtDNA diseases.
Challenges in understanding mtDNA diseases
The diagnosis and clinical handling of mtDNA diseases are hindered by the complexity of mtDNA genetics. mtDNA mutations (point mutations and single large‐scale deletions) are typically functionally recessive. This means that in order to cause a biochemical and functional impairment, they need to be present in a significant amount of mtDNA molecules (Gorman et al, 2016). Higher heteroplasmy is usually associated with more severe conditions, but the critical threshold for pathogenicity is different for distinct mtDNA mutations. For example, MT‐ATP6 mutations cause NARP at low level of heteroplasmy and MILS when the level is higher than 90% (Thorburn et al, 1993). The mutation load can also vary between individuals carrying the same mtDNA defect and even among different tissues in the same individual. For example, the mutation m.3243A > G causative of MELAS can be present at higher levels in the central nervous system than in the periphery, and with varying heteroplasmy in different brain regions of the same patients (Scholle et al, 2020). At the cellular level, increasing mutation load may lead to progressive changes in transcriptional response (Picard et al, 2014). At the same time, it has been proposed that mtDNA mutation load alone may not be sufficient to explain a defined clinical phenotype, as the individual nuclear background could modulate the presentation of mtDNA diseases (D'Aurelio et al, 2010; Pickett et al, 2018).
How mtDNA mutations are inherited is a matter adding further complexity to the study of mtDNA diseases. mtDNA mutations in humans are considered to be almost invariably only maternally inherited, as sperm mitochondria are degraded upon fertilization (Stewart & Chinnery, 2015). However, there have been reports suggesting bi‐parental inheritance of mtDNA mutations (Luo et al, 2018). The occurrence of such paternal inheritance of mtDNA is highly debated. One alternative explanation involves the transmission of nuclear‐encoded mitochondrial sequences (NUMTs) that may create the impression of mtDNA heteroplasmy (Wei & Chinnery, 2020). Indeed, recent findings showed that NUMTs are frequent in the general population (Wei et al, 2022a).
The difference in heteroplasmy observed among offspring of the same mother is usually explained by the presence of a genetic bottleneck taking place during oocyte development (Stewart & Chinnery, 2015). According to this hypothesis, there is a reduction in mtDNA copies in the early oocytes, which can inherit a different proportion of mutated mtDNA molecules from maternal mitochondria. During oocyte maturation and cell division, the mutation load can then increase, decrease, or remain the same, thereby leading to the development of offspring with variable heteroplasmy (Zaidi et al, 2019; van den Ameele et al, 2020). In addition, there is evidence suggesting the presence of selection for or against defined mtDNA variants (Wei & Chinnery, 2020). In particular, it has been reported that non‐synonymous changes in mtDNA protein‐coding genes are selected against in the germline, thereby preventing most mtDNA mutations from reaching homoplasmy (Stewart et al, 2008). A developmentally programmed mitochondrial degradation may be instrumental for such mtDNA selection (Palozzi et al, 2022). The mode of inheritance appears even more complex for large‐scale mtDNA deletions, as they seem to be rarely transmitted to offspring in humans (Chinnery et al, 2004).
To further complicate the study of pathological mtDNA variants, it has been reported that mtDNA defects also occur somatically in different tissues and organs later in life (Payne et al, 2011). The accumulation of mtDNA mutations may not be linked to oxidative‐mediated mtDNA damage (Kennedy et al, 2013). Conversely, as the mitochondrial genome undergoes continuous replication even in post‐mitotic cells (Magnusson et al, 2003; Gustafsson et al, 2016), mtDNA might accumulate mutations over time at higher rates compared to nuclear DNA, despite the high fidelity of PolG (Nissanka et al, 2018; Stewart & Chinnery, 2021). Among acquired mtDNA defects, multiple large‐scale mtDNA deletions have been found associated not only with genetic defects in nuclear genes encoding for proteins involved in mtDNA replication and mitochondrial dynamics but also with physiological aging (Payne et al, 2011; Vincent & Picard, 2018). Although the mechanisms underlying the tissue‐specific and temporal distribution of such somatic mtDNA mutations remain unclear, it has been suggested that the diversity in mtDNA heteroplasmy might contribute to the development of late‐onset disorders such as neurodegeneration, diabetes, and cancer (Hahn & Zuryn, 2019). Since mtDNA heteroplasmy can be a common phenomenon even in healthy conditions (Payne et al, 2013; Wei & Chinnery, 2020), it has been proposed that mtDNA mutations observed in older individuals might have been present since birth although at lower heteroplasmy and that they might have been clonally expanded throughout life (Keogh & Chinnery, 2013). Heteroplasmic mtDNA mutations seen in multiple tissues may thus represent a mixture of mutations that are germline inherited and mutations accumulated somatically upon aging due to continuous mtDNA replication (Stewart & Chinnery, 2021).
Challenges in engineering mtDNA
A major obstacle in the study of mtDNA mutations has been the difficulty of engineering mtDNA. While the quickly evolving clustered regularly interspaced short palindromic repeats, (CRISPR)/CRISPR‐associated proteins (Cas) field produces novel applications with astonishing speed, enabling nowadays the introduction of virtually any type of mutations in the nuclear DNA (Anzalone et al, 2020), manipulation of mtDNA remained difficult to achieve until very recently (Silva‐Pinheiro & Minczuk, 2022).
Several peculiar features of the mitochondrion and its genome complicate editing approaches. First, the organelle double membrane represents an important barrier that is not easily crossed by nucleic acids. This lack of efficient import into mitochondria represents a crucial challenge, as the delivery of a DNA template is essential for precise genome editing with CRISPR nucleases (Gammage et al, 2018a). Furthermore, all CRISPR/Cas systems require guide RNAs (gRNAs) in order to direct the genomic scissor to a specific target site. Thus, impaired nucleic acid import precludes the application to mtDNA editing even for CRISPR/Cas base editors that do not require template DNA (Jinek et al, 2012).
Even if DNA delivery might be achieved, it appears quite difficult for this DNA to integrate into the host mitochondrial genome. This is because mtDNA does not exhibit the same repairing mechanisms as nuclear DNA. Upon a DNA double‐strand break, mtDNA does not attempt to initiate a repair process, but rather undergoes degradation (Nissanka et al, 2018; Peeva et al, 2018). Although there is evidence of base excision repair (BER) and single‐strand break (SSB) repair in mtDNA, mitochondria do not seem to possess efficient double‐strand break (DSB) repair mechanisms, such as homologous recombination (HR) or non‐homologous end joining (NHEJ; Kazak et al, 2012). This lack of efficient recombination hampers the possibility for exogenously delivered DNA to be readily integrated into the host mtDNA (Hagström et al, 2014).
The multi‐copy nature of mtDNA adds an additional layer of complexity. In order to achieve effective correction, it is necessary to find a way to select the desired mtDNA variant among a high number of mtDNA molecules (Silva‐Pinheiro & Minczuk, 2022). The possibility of exogenously modifying the mutation load, known as “heteroplasmy shift,” has been exploited by several groups using different models (Jackson et al, 2020). The heteroplasmy shift is achieved by selectively eliminating mutant mtDNA molecules and allowing the remaining wild‐type mtDNA molecules to repopulate the mtDNA pool. Given the threshold effect of mtDNA diseases, shifting mtDNA heteroplasmy toward the wild‐type sequence could allow restoration of proper mitochondrial functionality.
Heteroplasmy shift has been accomplished following different strategies. First, scientists used mitochondrially targeted restriction endonucleases (mitoREs) that specifically cut mutant mtDNA copies and thereby induce their selective degradation (Srivastava & Moraes, 2001; Tanaka et al, 2002; Bacman et al, 2012; Reddy et al, 2015). Although effective, the use of mitoREs is, however, limited by the scarcity of unique restriction sites specific for pathogenic mtDNA mutations. A more versatile heteroplasmy shift can be obtained by employing mitochondrially targeted transcription activator‐like effector nucleases (mitoTALENs; Bacman et al, 2013; Hashimoto et al, 2015; Yang et al, 2018) and mitochondrially targeted zinc finger nucleases (mtZFNs; Minczuk et al, 2008; McCann et al, 2018). These approaches can target both point mutations and deletions by introducing a DSB. This targeted heteroplasmic shift was successfully demonstrated in various models (Bacman et al, 2013; Gammage et al, 2014, 2018b; Hashimoto et al, 2015; Reddy et al, 2015). Variations of these techniques include smaller‐sized constructs to facilitate packaging into adeno‐associated virus (AAV) vectors, mito I‐CreI homing endonuclease‐based designer meganuclease (mitoARCUS) that is smaller in size but with relatively long recognition site (Zekonyte et al, 2021), and mitoTALENickase that is capable of introducing single large‐scale mitochondrial deletions (Phillips et al, 2017).
Despite these progresses, heteroplasmy shift approaches cannot be applied to cells carrying mtDNA mutations at homoplasmic level or at very high levels of heteroplasmy, as they require the presence of a sufficient number of wild‐type mtDNA molecules to repopulate and restore function (Silva‐Pinheiro & Minczuk, 2022). Furthermore, none of the techniques described above are capable of introducing precise mitochondrial genome modifications.
Models of mtDNA diseases
The difficulty of engineering the mitochondrial genome significantly hindered the development of animal and cellular models of mtDNA diseases. Despite these challenges, relevant cellular and animal models of mtDNA diseases have been generated (Fig 1).
Figure 1. Modeling tools for mtDNA diseases.
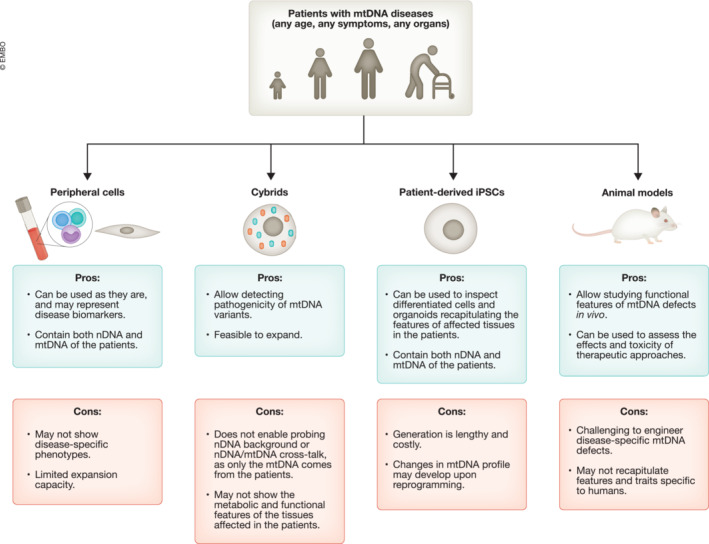
Despite the challenges associated with mtDNA engineering, it has been possible to develop approaches for modeling mtDNA diseases. Comparison (pros and cons) of these modeling strategies are presented here as a schematic drawing. See text for details. nDNA, nuclear DNA; mtDNA, mitochondrial DNA; iPSCs, induced pluripotent stem cells.
Peripheral cells such as fibroblasts have been extensively used for investigating the pathogenesis of mtDNA diseases. Peripheral cells can be obtained with minimally invasive procedures, and can maintain patient‐specific mtDNA mutations (Bourgeron et al, 1993). Patient‐derived fibroblasts have been used to evaluate the functionality of the respiratory chain activity (Invernizzi et al, 2012; Saada, 2014), the integrity of the mitochondrial network (Caporali et al, 2020; Del Dotto et al, 2020), and to measure calcium metabolism or oxidative stress (Venco et al, 2015; Granatiero et al, 2016). Nonetheless, their restricted proliferative capacity and inter‐individual variability hamper the application of peripheral cells in mechanistic disease‐modeling studies.
A remarkable advance came with the development of transmitochondrial cytoplasmic hybrids (cybrids; King & Attardi, 1989). Cybrids are produced by fusing enucleated donor cells (cytoplasts) obtained from patients (e.g., fibroblasts or platelets) with recipient cells devoid of mtDNA (ρ0). Cybrids have been instrumental in dissecting the pathogenicity of individual mtDNA defects in vitro (King & Attardi, 1989). Using cybrids, it is possible to verify the consequences of mtDNA point mutations and large‐scale deletions and to correlate the heteroplasmy level of a specific mutation with the biochemical impairment (Picard et al, 2014; Cavaliere et al, 2022). Cybrids allow to determine the potential pathogenicity of any newly identified mtDNA mutations in patients. This information could play a role in genetic diagnosis: if the investigated mtDNA mutation does not show defects in cybrids, it is then possible that the patient phenotype may be caused by an unknown nuclear DNA mutation. Cybrids have been used in countless of in vitro works (Bugiardini et al, 2020; Hernández‐Ainsa et al, 2022; Schaefer et al, 2022). In addition, they have also been crucial for the derivation of animal models (see below). Nonetheless, cybrids may hold some limitations as in vitro models of mtDNA diseases. Recipient cells are often represented by immortalized cancer‐like cells to enable effective growth and expansion. Such cancer‐like immortalized cells mainly rely on glycolysis and not OXPHOS for energy production (Kondoh et al, 2005; Giang et al, 2013; Wilkins et al, 2014). Hence, cybrids, as well as peripheral fibroblasts, cannot faithfully reproduce the functional features of OXPHOS‐dependent neurons or cardiac cells, which are the cell types typically impaired in patients affected by mtDNA diseases (Pfeffer et al, 2013; Carelli & Chan, 2014).
Different mammalian animal models of mtDNA diseases have been successfully developed (Stewart, 2021). One strategy to generate mtDNA mutant mice has been to manipulate the mitochondrial replication machinery. By disrupting the catalytic subunit of the nuclear‐encoded PolG, it has been possible to obtain mice known as “mtDNA mutator mice,” which accumulate high levels of multiple mtDNA mutations that are transmissible in the germline (Trifunovic et al, 2004; Kujoth et al, 2005). These mice did not show typical signs of mtDNA diseases, but rather severe premature aging and early death. In a similar manner, the disruption of the nuclear‐encoded mtDNA replication‐associated helicase TWINKLE led to mice with multiple mtDNA deletions, known as “mtDNA deletor mice” (Tyynismaa et al, 2005). These mice developed features reminiscent of CPEO.
The first heteroplasmic mice harboring a pathogenic mtDNA mutation were the “mito‐mice” (Inoue et al, 2000; Nakada et al, 2001). They were generated by fusing cytoplasts carrying a pathogenic 4,696 bp mtDNA deletion with mouse embryos. The mice had a severe phenotype and died early because of renal failure. By microinjecting cybrid embryonic stem cells (ESCs) carrying a single mtDNA mutation in the gene MT‐ND1 into mouse embryos, it was then possible to generate mice carrying a homoplasmic pathogenic mtDNA mutation (Kasahara et al, 2006). These mice displayed a milder phenotype with no change in life span. A similar approach was used to generate mice carrying a mutation in gene MT‐ND6 associated with LS and LHON phenotypes (Yokota et al, 2010). These latter mice showed Complex I deficiency and lactate overproduction, but no other phenotype reminiscent of LS patients. Another mouse model of this mutation developed visual problems and degeneration of optic nerve cells (Lin et al, 2012). Heteroplasmic mice were derived using cybrid ESCs carrying a pathogenic mutation of a mtDNA‐encoded tRNA known to cause MELAS‐like clinical phenotype (Shimizu et al, 2014). These animals showed skeletal muscle defects and renal failure, thereby recapitulating some features of the human disease (Shimizu et al, 2015). Recently, it was possible to obtain mice showing metabolic defects carrying high heteroplasmy level of the pathogenic tRNA mutation m.2748A > G, which is orthologous to the human mutation m.3302A > G associated with MELAS (Tani et al, 2022).
As an alternative strategy, allotopic expression of mtDNA mutations has been attempted. This led to the development of mice with nuclear expression of the mitochondrial gene MT‐ATP6 harboring a mutation associated with NARP/LS syndromes (Dunn & Pinkert, 2012). These mice showed some behavioral defects. However, this approach remains controversial, as other works in vitro demonstrated that proteins expressed allotopically from the nucleus are not properly imported into the mitochondria (Perales‐Clemente et al, 2011).
Induced pluripotent stem cells (iPSCs) and mtDNA modifications
Modeling mtDNA diseases can benefit from the development of cellular models that not only carry specific pathogenic mtDNA mutations but also exhibit the functional and metabolic properties of the cells and tissues affected in the patients. A technology that allowed establishing models with such features came in 2006–2007 with the discovery of induced pluripotent stem cells (iPSCs; Takahashi & Yamanaka, 2006; Takahashi et al, 2007).
iPSCs are obtained from somatic cells through forced expression of transcription factors that are specific for ESCs. Like ESCs, iPSCs acquire the ability to proliferate indefinitely (i.e., self‐renewal) and to generate cell progenies belonging to all three germ layers (i.e., pluripotency). Unlike ESCs, however, iPSCs do not require embryos, and can be derived from the somatic cells of virtually every individual using a robust and relatively straightforward process known as cellular reprogramming (Takahashi et al, 2007; Shi et al, 2017). Effective reprogramming has been demonstrated using a number of different techniques and diverse parental somatic cells (Shi et al, 2017). Nowadays, iPSCs are considered a remarkable platform not only for investigating disease‐related mechanisms but also for the establishment of drug discovery pipelines (Shi et al, 2017; Rowe & Daley, 2019).
One relevant issue to take into consideration before applying the iPSC system for modeling mtDNA diseases is to determine whether the mtDNA profile can be effectively retained during the reprogramming process. In 2011, we investigated this aspect and found that human iPSCs carried mtDNA variants that were not observed in the parental fibroblasts (Prigione et al, 2011). Numerous works later confirmed these initial observations (Perales‐Clemente et al, 2016; Kang et al, 2016a; Zambelli et al, 2018; Deuse et al, 2019; Palombo et al, 2021; Wei et al, 2021). Detected mtDNA mutations affected various mtDNA genes (encoding for mRNAs, tRNAs, or rRNAs) as well as mtDNA haplogroup‐specific regions, and encompassed both synonymous and non‐synonymous modifications. Overall, it was estimated that in human iPSCs, the mutation rate for mtDNA is about 8.62 × 10−5 per base pair (Wei et al, 2021), while the mutation rate for the nuclear genome is about 1 × 10−9 per base pair (Kuijk et al, 2020). Importantly, the heteroplasmy of these mtDNA mutations could differ greatly among iPSC lines generated from the same donor cells. mtDNA mutations in iPSCs may have consequences on bioenergetics, redox balance, and overall cellular functions, and may also encode neoantigens potentially responsible for eliciting immune responses (Chen et al, 2018; Deuse et al, 2019). Hence, differences in mtDNA profiles might contribute to behavioral heterogeneity among iPSC clones (Kelly et al, 2013; Carelli et al, 2022). Several groups have, therefore, argued that screening for mtDNA modifications should become part of the routine quality control of all newly generated iPSC lines, in a similar manner as nuclear chromosomal rearrangements are currently monitored (Hämäläinen, 2016; Lorenz & Prigione, 2016; Carelli et al, 2022; Rossi et al, 2022).
The mechanisms underlying mtDNA changes upon reprogramming of somatic cells into iPSCs are not known (Sercel et al, 2021). During the generation of iPSCs, there is a reduction in the mtDNA copy number that can in turn increase upon differentiation (Facucho‐Oliveira & St John, 2009; Prigione et al, 2010; St John, 2016). For this reason, we initially interpreted changes in mtDNA mutation profile in iPSCs as a “genetic bottleneck” (Prigione et al, 2011), a phenomenon reminiscent of the events occurring during germ line development where a restriction in the mtDNA amount can cause a different distribution of mutated mtDNA to the daughter cells (Stewart & Chinnery, 2015). This interpretation still holds true, but it remains to be determined whether it is a pure random phenomenon or whether there are mechanistic drivers (Sercel et al, 2021; Carelli et al, 2022).
To simplify this phenomenon, we could imagine two potential scenarios underlying the mtDNA heteroplasmy shift in iPSCs reprogrammed from fibroblasts (Fig 2). The first possibility is that the mtDNA changes seen in iPSCs are a consequence of the mtDNA heterogeneity of the starting somatic cell population (Young et al, 2012). Since iPSCs are clonally derived from reprogramming one fibroblast cell, it is possible that this particular cell may be different from the rest of the fibroblast population and could carry rare mtDNA variants (Fig 2 left, yellow mitochondria). The derived iPSC lines will thus carry these rare mtDNA variants and not variants expressed by other fibroblast cells that were not reprogrammed (Fig 2 left, blue and red mitochondria). The rare mtDNA variants present in iPSC lines could also vary in heteroplasmy due to changes in mtDNA content that may lead to different ratios of mtDNA mutation load. The result will be that some of the mtDNA variants contained in the original fibroblast population are lost, while some are retained with similar or increased levels of heteroplasmy. The second possibility is that mtDNA mutations may occur de novo during iPSC reprogramming (Fig 2). Hence, in addition to rare mtDNA variants present in the original parental cell, iPSCs could harbor mtDNA mutations that were not present in the parental samples (Fig 2 right, purple mitochondria). These de novo mtDNA modifications could also show various heteroplasmy levels in different iPSC lines due the mentioned bottleneck effect.
Figure 2. mtDNA bottleneck and heteroplasmy shift in human iPSCs.
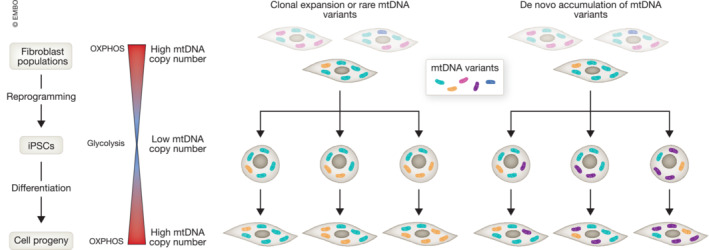
During reprogramming of somatic fibroblasts to induced pluripotent stem cells (iPSCs), the cells acquire a glycolytic metabolism and reduce their number of mtDNA copies. Upon differentiation of iPSCs into energy‐demanding progenies, the metabolism shifts toward oxidative phosphorylation (OXPHOPS) and the mtDNA copy number increases. These processes are accompanied by changes in the mtDNA profile that are not fully understood. mtDNA profile changes could be due to clonal expansion of rare mtDNA variants (left, yellow mitochondria) or to de novo introduction of mtDNA alterations (right, purple mitochondria). In the first case, the heterogeneity of the somatic cell population may play a role. Since iPSCs are clonally derived, it could be that the fibroblast cell from which iPSCs are generated would carry mtDNA variants that are different from the rest of the fibroblast cell population. In the second case, there may be mtDNA alterations arising as a consequence of reprogramming (e.g., because of errors in mtDNA polymerase gamma). In all cases, the rare variants derived from the parental cells (left, yellow mitochondria) or introduced during reprogramming (right, purple mitochondria) could acquire different heteroplasmy in various iPSC clones. The mtDNA profile of each iPSC line appears to be overall maintained during differentiation, despite changes in metabolism and mtDNA copy number (Perales‐Clemente et al, 2016; Kang et al, 2016a; Palombo et al, 2021; Wei et al, 2021).
Overall, it remains to be determined whether the mtDNA bottleneck effect and related heteroplasmy shift in iPSCs are simply stochastic or rather driven by biological mechanisms, for example, due to altered PolG activity or redox imbalance. Some mtDNA variants might have a selective advantage (Yoneda et al, 1992; Russell et al, 2018) or may promote the metabolic–epigenetic reconfiguration associated with cellular reprogramming (Shakiba et al, 2019; Sercel et al, 2021). During the generation of iPSCs, in parallel to epigenetic changes leading to the establishment of an embryonic‐like gene expression program, there are modifications occurring at the mitochondrial and metabolic levels (Prigione et al, 2010; Folmes et al, 2011; Agathocleous & Harris, 2013; Lisowski et al, 2018). iPSCs acquire a glycolytic‐driven metabolic state that is then converted to OXPHOS upon differentiation. Hence, specific mtDNA modifications might potentially be needed to support this metabolic reconfiguration. However, multiple groups demonstrated that mtDNA mutations were retained without significant heteroplasmic changes during subsequent cultivation and differentiation of the iPSC lines (Perales‐Clemente et al, 2016; Kang et al, 2016a; Palombo et al, 2021; Wei et al, 2021). Furthermore, mtDNA modifications including point mutations and large‐scale deletions have been observed also in human ESCs (Maitra et al, 2005; Van Haute et al, 2013). Thus, it remains unclear how reprogramming‐associated and differentiation‐associated metabolic reconfigurations could influence the heteroplasmy shift in iPSCs. Understanding these processes will likely have important implications for the application of iPSCs in biomedical research (Box 1).
Box 1. In need of answers.
What are the mechanisms underlying the heteroplasmy shift and bottleneck effect of mtDNA occurring upon reprogramming somatic cells to pluripotency? Can we modulate such phenomena?
If we cannot prevent the occurrence of mtDNA modifications in human iPSCs, can we design pipelines to effectively monitor the mtDNA profile and remove unwanted mutations for all iPSC lines used in medical applications and translational studies?
If we apply mtDNA base editing to introduce mtDNA mutations in healthy iPSCs, can we reach a level of mutation load high enough to cause functional defects and recapitulate disease‐related phenotypes? Similarly, if we apply mtDNA base editing to correct mtDNA mutations in patient‐derived iPSCs, can we decrease the mutation load significantly to the point that the functional defects are corrected, and the disease phenotypes ameliorated?
Is it possible to reliably predict which mtDNA base editing constructs and configurations are best suited to target each specific mtDNA mutation?
How can we further reduce unwanted off‐target effects in nuclear or mitochondrial genomes that might be associated with mtDNA base editing?
Will it be possible to develop novel mtDNA editing approaches to target pathogenic mtDNA mutations in sites that are currently not editable? And can there be editing approaches effective also for large‐scale deletions?
Can we obtain complex organoids and assembloid systems including vasculature and immune cells? And could such models be able to show the impact of mtDNA mutations and their segregation in different human tissues?
iPSCs and 3D organoids as next‐generation models for mtDNA diseases
iPSCs hold great potential for modeling mtDNA diseases given their ability to generate virtually any tissue of the human body. mtDNA diseases particularly affect tissues and cells that are highly dependent on mitochondrial energy production, such as neuronal cells, muscle, and cardiac cells (Pfeffer et al, 2013; Carelli & Chan, 2014). Differentiated cells from iPSCs do in fact show reliance on OXPHOS metabolism (Chung et al, 2007; Zheng et al, 2016b; Cliff & Dalton, 2017; Tanosaki et al, 2021). Hence, by using differentiated cells from patient‐derived iPSCs, it may be possible to obtain model systems that carry patient‐specific mtDNA mutations and exhibit appropriate metabolic and functional properties. Such models could shed light on the mechanisms underlying mtDNA diseases and could help identify targets of intervention (Inak et al, 2017; McKnight et al, 2021).
In addition to two‐dimensional (2D) differentiation of iPSCs, it is now possible to obtain three‐dimensional (3D) organized tissues known as organoids. Several organoid models have been obtained from human iPSCs, including brain, gut, liver, blood vessels, kidney, and retina (Nakano et al, 2012; Lancaster et al, 2013; Takebe et al, 2013; Taguchi et al, 2014; Workman et al, 2017; Wimmer et al, 2019; Cowan et al, 2020; Lewis‐Israeli et al, 2021). Organoids recapitulate features of human organs with respect to cellular organization and architectures. Their formation in vitro is accomplished by modulating signaling pathways that are known to guide and govern patterning and tissue morphogenesis in vivo (McCauley & Wells, 2017). In the context of mtDNA diseases, organoids might thus help address the impact of mtDNA mutations in different human tissues and organs.
Given the strong impact that mtDNA defects typically have on cells of the nervous system, for investigating mtDNA diseases it may be particularly interesting to employ 3D structures recapitulating features of the human brain (Chiaradia & Lancaster, 2020; Le et al, 2021). These organoids, defined as brain organoids or neural organoids, can be obtained to reproduce features of different brain regions, including spinal cord, midbrain, cortex, striatum, or cerebellum (Pașca et al, 2022). Such brain region‐specific organoids can then be mixed into so‐called “assembloids” that can be used to address the communication between different brain regions (Miura et al, 2022). Brain organoids typically consist of cells belonging to the neuro‐ectodermal lineage. For this reason, several groups are working on strategies to incorporate into brain organoids also endodermal‐derived blood vessel cells and mesodermal‐derived microglia. The presence of vasculature structure is necessary in vivo to enable effective neuronal commitment and maturation (Lange et al, 2016) and would be crucial for distributing oxygen and nutrients throughout the organoids. The incorporation of microglia into cerebral organoids would not only provide support for neuronal development but may also help unveil the impact of immune‐mediated mechanisms in the modeled diseases.
In the past few years, several groups employed iPSCs derived from patients affected by mtDNA diseases to generate 2D and 3D models (McKnight et al, 2021; Table 2). The iPSC‐based modeling works have so far mainly focused on diseases caused by single mtDNA mutations.
Table 2.
iPSC models of mtDNA diseases.
| Disease | Genes and mutations studied | Mutation levels in iPSC lines | Differentiated cell models investigated | Publications |
|---|---|---|---|---|
| Leigh Syndrome (LS) |
MT‐ND5 (m.13513G > A) MT‐ATP6 (m.9185T > C; m.8993T > G) |
Homoplasmy or high heteroplasmy for MT‐ATP6. Low heteroplasmy for MT‐ND5 | Neural progenitors, neurons, and brain organoids | Ma et al (2015), Lorenz et al (2017), Zheng et al (2016a), Galera‐Monge et al (2020), Romero‐Morales et al (2022) |
| Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke‐like episodes (MELAS) |
MT‐TL1 (m.3243A > G) MT‐ND1 (m.5541C > T) |
Variable heteroplasmy, including mutation‐free iPSCs | Neurons, skeletal muscles, endothelial cells, retinal pigment epithelium, and spinal cord organoids | Hämäläinen et al (2013), Hatakeyama et al (2015), Winanto et al (2020), Pek et al (2019), Chichagova et al (2017), Gunnewiek et al (2020) |
| Myoclonic Epilepsy and Ragged‐Red Fibers (MERRF) | MT‐TK (m.8344A > G) | Variable heteroplasmy, including mutation‐free iPSCs | Cardiomyocytes and neural progenitors | Chou et al (2016) |
| Leber's Hereditary Optic Neuropathy (LHON) | MT‐ND4 (m.11778G > A) | Homoplasmy | Retinal ganglion cells | Wu et al (2018), Yang et al (2019), Yang et al (2020) |
| Kearns–Sayre Syndrome (KSS) | Common deletion | Variable heteroplasmy | Neural progenitors, fibroblast‐like cells, and cardiomyocytes | Lester Sequiera et al (2021) |
| Pearson Marrow–Pancreas syndrome (PMPS) | Common deletion | Variable heteroplasmy | Hematopoietic progenitors | Cherry et al (2013) |
Several studies focused on MELAS, particularly, on the common mtDNA mutation m.3243A > G in the tRNA‐encoding MT‐TL1 gene. In agreement with the heteroplasmy shift and bottleneck effect of iPSC generation, MELAS iPSC lines were found to carry the mutation at variable heteroplasmy (Folmes et al, 2013; Hämäläinen et al, 2013; Kodaira et al, 2015; Gunnewiek et al, 2020). Mutation‐low or mutation‐free iPSC lines could be obtained and used as isogenic controls (i.e., carrying the same nuclear DNA background). Differentiated neurons with high m.3243A > G mutation load showed defective mitochondrial complex activity and mitophagy (Hämäläinen et al, 2013), and aberrant neuronal network functionality and synchronicity (Gunnewiek et al, 2020). The MELAS mutation m.5541C > T in MT‐TW gene caused defective neuronal differentiation without apparent impairment of neural progenitors or skeletal muscle cells (Hatakeyama et al, 2015). Other cell types were investigated as MELAS models, including endothelial cells that exhibited pro‐atherogenic properties (Pek et al, 2019) and retinal pigment epithelium cells that showed dysfunctional phagocytosis (Chichagova et al, 2017). Lastly, by using 3D spinal cord organoids carrying the mutation m.3243A > G at high heteroplasmy, it was possible to uncover neurogenesis delays and impaired neurite outgrowth as mechanistic disease features of MELAS (Winanto et al, 2020).
LS iPSCs have been obtained from patients harboring homoplasmic mutations in gene MT‐ATP6. Neural cells differentiated from these LS iPSCs exhibited defects in bioenergetics (Ma et al, 2015) and calcium homeostasis (Lorenz et al, 2017), and increased susceptibility to glutamate toxicity (Zheng et al, 2016a). Neurons derived from an LS individual carrying relatively low heteroplasmy level of a mutation in gene MT‐ND5 also showed defects in calcium signaling (Galera‐Monge et al, 2020). In addition, 3D models of LS have been generated. Brain organoids carrying the mutation m.8993T > G in gene MT‐ATP6 at high heteroplasmy developed abnormally, showing reduced size and altered cortical architecture (Romero‐Morales et al, 2022). Interestingly, similar defects in size and cellular organization were also observed in brain organoids derived from LS patients carrying mutations in the nuclear gene SURF1 (Inak et al, 2021). These works collectively suggest that mitochondrial diseases such as LS could affect the physiological process of human neurogenesis (Brunetti et al, 2021).
To model LHON, iPSCs carrying mutations in the gene MT‐ND4 were differentiated into neurons (Danese et al, 2022) and retinal ganglion cells (Yang et al, 2020). These latter cells showed defective neurite outgrowth and increased mitochondrial biogenesis (Wu et al, 2018), disrupted action potentials (Yang et al, 2019), and increased oxidative stress and apoptosis with aberrant mitochondrial transport (Yang et al, 2019).
MERFF has also been modeled with iPSCs derived from patients carrying the mutation m.8344A > G in the gene MT‐TK (Chou et al, 2016). There, differentiated cardiomyocytes and neural progenitors showed oxidative stress and defective mitochondrial dynamics. Neurons directly derived from fibroblasts of MERFF patients exhibited oxidative stress with reduced bioenergetics and increased mitophagy (Villanueva‐Paz et al, 2019).
Human iPSCs carrying large‐scale mtDNA deletions have been generated from patients affected by KSS and PMPS (Cherry et al, 2013; Russell et al, 2018; Lester Sequiera et al, 2021; Peron et al, 2021; Hernández‐Ainsa et al, 2022). Initial studies showed defects in hematopoietic progenitors (Cherry et al, 2013), but the contribution of mtDNA deletions to the disease pathogenesis in differentiated progenies has not been addressed in detail.
Besides mechanistic studies, some iPSC studies focused on using the derived models to test therapeutic strategies. The drug Sonlicromanol, currently employed in a stage IIB clinical trial for MELAS (Janssen et al, 2019), was found beneficial on the activity and transcriptional signature of neuronal cells derived from MELAS patients (Gunnewiek et al, 2021). Rapamycin improved the bioenergetics of neurons carrying homoplasmic MT‐ATP6 mutations and enhanced their resistance to glutamate toxicity (Zheng et al, 2016a). Somatic cell nuclear transfer (SCNT) was employed in iPSCs as a means to replace mutant mitochondria carrying a homoplasmic mutation in the gene MT‐ATP6 with healthy donor oocyte mitochondria (Ma et al, 2015). iPSC‐derived neural cells from patients with mtDNA diseases can also be used to perform drug screenings to identify new treatments. In a proof‐of‐concept work, iPSC‐derived neural cells harboring MT‐ATP6 mutations associated with LS were used to screen selected FDA‐approved drugs, leading to the identification of potential compounds to be repositioned for LS (Lorenz et al, 2017).
The base editing revolution for mtDNA
A groundbreaking finding for the mtDNA disease field came in 2020 with the development by Mok et al (2020) of a genome engineering method that allows targeted C‐to‐T base conversion in the mtDNA. This approach is reminiscent of the CRISPR/Cas base editor system since it does not generate DSB and does not require a DNA template. In contrast to CRISPR/Cas, however, this new method does not depend on gRNAs for guidance to the target site. Instead, it modifies the mitochondrial genome through a deaminase enzyme guided by proteins termed transcription activator‐like effectors (TALEs).
Mok et al (2020) identified a cytidine deaminase from Burkholderia cenocepacia that can act on double‐stranded DNA and dubbed this enzyme “double‐stranded DNA deaminase toxin A” (DddA). The discovery was remarkable as other deaminases known to date could only manipulate single‐stranded DNA and were therefore dependent on enzymes capable of unwinding DNA double strands, such as Cas proteins. This limited the application of such base editing systems to nuclear DNA. Conversely, DddA can act directly on double‐stranded DNA and enables transitions of C·G to T·A bp through deamination of cytidines at TC sites in the mtDNA. In the first step, cytidine is deaminated to uridine. In a second step during DNA replication, uridine is read as thymidine by the DNA polymerase and adenine is incorporated on the opposite strand. Uridine is then replaced by thymidine, thus completing the base editing process (Fig 3A). The DddA enzyme is split into two halves to avoid widespread deamination in unwanted locations, and each half is fused to TALEs designed to bind left and right of the desired target site. In order to reconstitute the deaminase at the target locus, two constructs are required for each manipulation, bringing both halves in close proximity. The constructs are imported into mitochondria via mitochondrial targeting sequences (MTS) fused to each N‐terminus. Editing efficiency is improved through the fusion of one copy of uracil glycosylase inhibitor (UGI) to each C‐terminus, which should prevent uracil excision during BER (Nilsen et al, 1997). This novel genome editing tool was coined “DddA‐derived cytosine base editor” (DdCBE; Mok et al, 2020; Fig 3A).
Figure 3. Base editing of mtDNA.
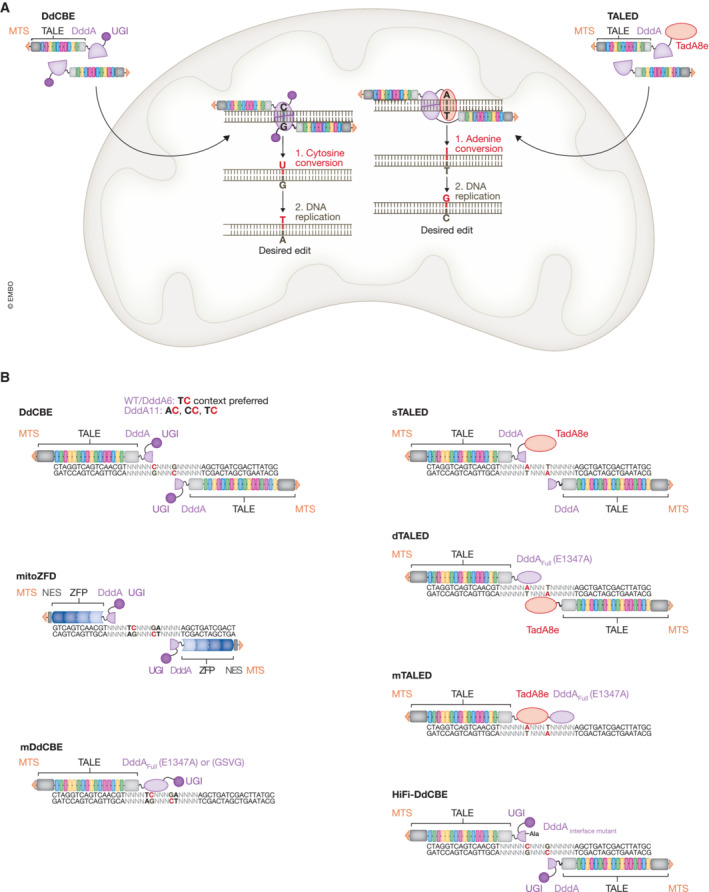
(A) Using double‐stranded DNA deaminase (DddA)‐derived cytosine base editors (DdCBE) or transcription activator‐like effector (TALE)‐linked deaminases (TALEDs), it is possible to introduce precise single gene modifications of the mtDNA. Nucleotides that are directly targeted are shown in red. The constructs are imported into the mitochondria via their mitochondrial targeting sequence (MTS) and are guided to the target site by their TALEs. In mitochondria, DddA halves are brought into close proximity and the enzyme is reconstituted. For DdCBE, in the first editing step, the nucleobase is deaminated, resulting in cytidine conversion to uridine (C > U). During DNA replication (second step), uridine is read as thymidine by the DNA polymerase and adenosine is incorporated on the opposite strand. In the next replication cycle, uridine is replaced by thymidine, ultimately resulting in the conversion of the mismatched U·G to a T·A bp. Analogously, in case of TALEDs, adenosine is first deaminated to inosine (A > I) and then the I·T bp is converted to a G·C bp during replication. Here, inosine is read as guanosine, and cytidine is incorporated on the opposite strand. In the next replication cycle, inosine is replaced by guanosine, thus completing the editing round. (B) Schematic representation of different mtDNA editing platforms. Both DNA strands can be edited. Targeted nucleotides are shown in red. DdCBE: DddA is split into two halves and thus two constructs per target site are required. Wild‐type (WT) DddA and DddA6 prefer TC contexts, while DddA11 can additionally deaminate cytidines in AC and CC contexts. Zinc finger deaminases (ZFD): DddA is also split into two halves and editing occurs preferentially at TC sites. The constructs are guided to the target site via zinc finger proteins (ZFP), while nuclear export signals (NES) reduce nuclear localization. Monomeric DdCBE (mDdCBE): a full‐length DddA mutant, either catalytically attenuated (E1347A) or with reduced cytotoxicity (GSVG), is fused to one single TALE protein with MTS and uracil glycosylase inhibitor (UGI). Only one construct per target site is required. TALEDs exist in three different configurations where adenine in any sequence context is converted to guanine. Split TALED (sTALED): DddA is split into two halves and a full‐length adenine deaminase (TadA8e) is fused to the C‐terminus of one construct. Dimeric TALED (dTALED): catalytically attenuated full‐length DddA (E1347A) is fused to one construct and the full‐length adenine deaminase is fused to another construct. Monomeric TALED (mTALED): only one construct per target site is required, and catalytically attenuated full‐length DddA (E1347A) and full‐length adenine deaminase TadA8e are fused to one single TALE protein with MTS. High‐fidelity DdCBE (HiFi‐DdCBE): DddA is split into two halves. An interface mutant with reduced spontaneous self‐assembly due to an alanine mutation (−Ala) is fused to one construct; cytidines at TC, AC, and CC sites can be targeted with reduced off‐target editing. Additional abbreviations: TadA8e: adenine deaminase; DddAFull(E1347A): catalytically impaired full‐length DddA; DddAFull(GSVG): full‐length DddA mutant with reduced cytotoxicity. C, cytidine; G, guanosine; U, uridine; A, adenosine; T, thymidine; I, inosine.
Since the original publication in 2020, multiple groups have demonstrated the feasibility of this new DdCBE technique. Employed models included mice (Lee et al, 2021, 2022b; Guo et al, 2022; Silva‐Pinheiro et al, 2022b; Wei et al, 2022b), rats (Qi et al, 2021), zebrafish (Guo et al, 2021; Sabharwal et al, 2021), and human zygotes (Chen et al, 2022; Wei et al, 2022c). Most of the works used mRNA microinjection as delivery system (Lee et al, 2021, 2022b; Qi et al, 2021; Sabharwal et al, 2021; Chen et al, 2022; Guo et al, 2022; Wei et al, 2022b, 2022c). Additionally, it was also possible to deliver DdCBE constructs into mouse heart via AAV, providing a proof of concept of in vivo mtDNA editing of post‐mitotic tissues (Silva‐Pinheiro et al, 2022b).
Besides being applied in a variety of organisms, the DdCBE technique is already evolving (Fig 3B). The tool has been adapted for use with zinc fingers as targeting proteins, a platform dubbed “zinc finger deaminases” (ZFD; Lim et al, 2022b). Besides achieving base editing with up to 30% efficiency in mtDNA, ZFD/DdCBE hybrids were shown to modify mtDNA in unique mutation patterns, sometimes even avoiding unwanted bystander editing (Lim et al, 2022b). Another improvement was the development of two mutant enzymes engineered from wild‐type DddA: DddA6 for enhanced editing at TC sites, and DddA11 for editing in AC and CC sequence contexts in addition to TC sites (Mok et al, 2022a). Both DddA6 and DddA11 exhibited increased off‐target editing of mtDNA, but enabled greater editing efficiency for difficult‐to‐edit loci and broaden the substrate scope (Mok et al, 2022a). Additionally, it has been possible to engineer a monomeric DdCBE (mDdCBE), a non‐toxic full‐length variant of DddA that does not need to be split into two halves (Mok et al, 2022b). The small size of mDdCBE facilitates the packaging into AAV, and the presence of a single TALE‐binding site increases the number of targetable sites, adding more versatility to the mtDNA base editing method (Mok et al, 2022b).
Another important milestone for mtDNA base editing has been the development of TALE‐linked deaminases (TALEDs; Cho et al, 2022). This technology allows A‐to‐G base conversion in the mtDNA through adenine deamination. This is achieved by fusing a known adenine deaminase (TadA8e) to a DdCBE construct. The possibility of modifying A·T to G·C bp significantly expands the number of potentially targeted disease‐associated mtDNA mutations. Different TALED configurations have been reported, thereby increasing the options available for mtDNA base editing design with respect to the choice of delivery vehicle (e.g., AAV) and accessibility of target sites.
DdCBE has been already used to generate a mouse model carrying the pathogenic mutation m.13513G > A in MT‐ND5 gene (m.12918G > A in mice) that is associated in humans with LS, MELAS, and LHON syndromes (Lee et al, 2022b). The authors demonstrated improved editing efficiency at the target site by co‐injecting DdCBE with mito‐TALENs into mouse embryos. While DdCBE introduced the desired mutation, mito‐TALENs were designed to specifically cut unedited copies. The resulting mice exhibited damaged mitochondria in tissues, as well as abnormal brain structures resulting in premature death (Lee et al, 2022b). Moreover, the development of a library of DdCBEs to knock out mouse mtDNA protein‐coding genes led the Minczuk group to obtain a mouse model carrying the mutation m.8069G > A in gene mt‐Apt6 at high heteroplasmy (Silva‐Pinheiro et al, 2022a).
One major concern for mtDNA base editing remains safety. Whereas off‐target editing in mtDNA was shown to be minimal in the original work (Mok et al, 2020), increasing evidence suggests that it can be more substantial (Guo et al, 2021, 2022; Qi et al, 2021; Chen et al, 2022; Lee et al, 2022a; Silva‐Pinheiro et al, 2022b; Wei et al, 2022b, 2022c). No off‐target editing was observed in nuclear pseudogenes (Mok et al, 2020; Chen et al, 2022; Guo et al, 2022; Lee et al, 2022a; Silva‐Pinheiro et al, 2022b; Wei et al, 2022c). However, the studies that analyzed genome‐wide specificity on the nuclear genome detected significant off‐target editing (Lei et al, 2022; Lee et al, 2022b; Wei et al, 2022b). It has been suggested that the split halves may spontaneously reassemble rather than depending on both TALE arrays for guidance (Lei et al, 2022; Lee et al, 2022b). To avoid this spontaneous reassembly, high‐fidelity DdCBE (HiFi‐DdCBE) was developed (Lee et al, 2022a). HiFi‐DdCBE was obtained by mutating key residues at the split interface of DddA, thereby leading to decreased off‐target assembly of both halves, while still enabling guided reconstitution at target site. Off‐target editing could also be markedly reduced by including nuclear export signals (NES) on the editing constructs (Lei et al, 2022; Lee et al, 2022b), or by co‐transfection of the DddA inhibitor DddIA (Lei et al, 2022; Lee et al, 2022b).
In less than 3 years since the original discovery of DdCBE, much has happened in the field of precise mtDNA manipulation (Table 3). Nevertheless, more work is needed to reliably predict which constructs and configurations are best suited for any given target site. More safety studies are also required before translating mtDNA base editing technology into human applications. Furthermore, not all types of mutations are targetable. With base editors, mtDNA manipulations are restricted to transitions (i.e., C<‐>T and G<‐>A), whereas transversions (i.e., interchanges of purines with pyrimidines), insertions, deletions, or mutations encompassing more than one nucleotide are not achievable (Box 1).
Table 3.
mtDNA base editing platforms.
| Platform | Type of mutation targetable | Pros | Cons | Publications |
|---|---|---|---|---|
| DdCBE | C > T and G > A in TC motifs |
|
|
Mok et al (2020), Chen et al (2022), Wei et al (2022c), Lei et al (2022), Qi et al (2021), Lee et al (2021), Silva‐Pinheiro et al (2022a), Silva‐Pinheiro et al (2022b), Guo et al (2022), Wei et al (2022b), Lee et al (2022b), Guo et al (2021), Sabharwal et al (2021) |
| DddA6 | C > T and G > A in TC motifs |
|
|
Mok et al (2022a) |
| DddA11 | C > T and G > A in TC, AC, and CC motifs |
|
|
Mok et al (2022a) |
| ZFD | C > T and G > A in TC motifs |
|
|
Lim et al (2022b) |
| TALED | A > G and T > C |
|
|
Cho et al (2022) |
| mDdCBE | C > T and G > A in TC motifs |
|
|
Mok et al (2022b) |
| HiFi‐DdCBE | C > T and G > A in TC, AC, and CC motifs |
|
|
Lee et al (2022a) |
Combining mtDNA editing with iPSCs and organoid models
To properly dissect the contribution of specific gene defects, it is important to use cell models that harbor the same genetic background in order to reduce variations that are not due to gene‐specific changes. In the iPSC field, the gold standard for modeling diseases caused by nuclear mutations is to employ CRISPR/Cas systems to produce isogenic iPSC lines. Isogenic iPSCs carry the same genetic background and differ only at the site of a specific gene mutation. Isogenic iPSCs can be obtained either by introducing the disease mutation into control healthy iPSCs or by correcting the mutation in patient‐derived iPSCs. By comparing 2D and 3D models derived from the unedited iPSCs and their engineered isogenic counterparts, it is then possible to identify defects that are caused specifically by the studied nuclear gene mutation (Ben Jehuda et al, 2018).
As mentioned above, such approaches are unfortunately more challenging for mtDNA defects. However, some strategies can be envisioned. In the investigation of pathogenic heteroplasmic mtDNA mutations, it is possible to take advantage of the spontaneous heteroplasmy shift occurring upon reprogramming to iPSCs. This can be achieved by screening for iPSC clones carrying very low levels of heteroplasmy without the need for external mtDNA manipulation (Fujikura et al, 2012; Hämäläinen et al, 2013; Ma et al, 2015; Lin et al, 2019; Gunnewiek et al, 2020; Fig 4A). However, given that the iPSC‐associated heteroplasmy shift cannot be effectively controlled or predicted, the identification of isogenic iPSC lines may require extensive screening of numerous iPSC clones. In order to more precisely manipulate the heteroplasmy level of patient‐derived iPSCs, it is possible to employ mitoREs, mito‐TALENS, or mtZFNs (Yahata et al, 2017, 2021; Yang et al, 2018, 2020). These approaches cause heteroplasmy shifts that have the potential to lead to the generation of isogenic iPSC lines containing low levels of mtDNA mutations. Lastly, mtDNA base editing strategies such as DdCBE might also be applied to directly insert precise site‐specific modifications into the mitochondrial genome.
Figure 4. Modeling mtDNA diseases combining iPSCs and mtDNA editing.
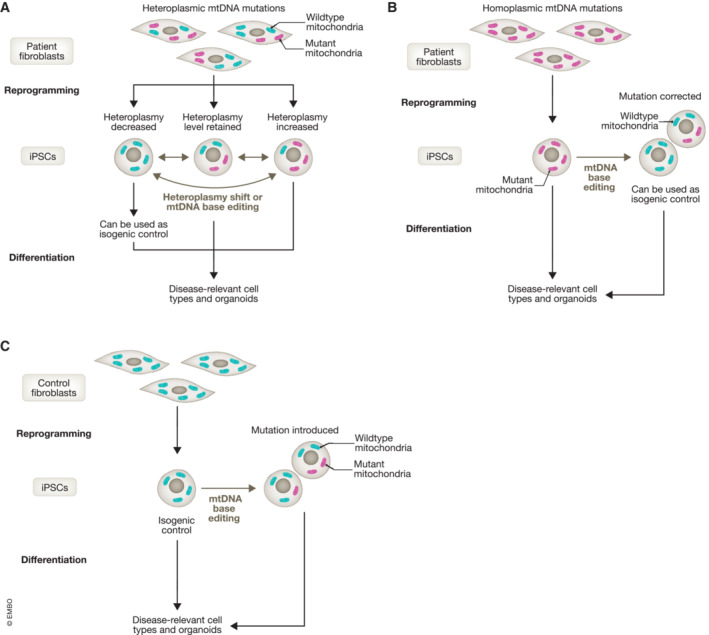
(A) Reprogramming to pluripotency of somatic cells carrying heteroplasmic mtDNA mutations may result in three different outcomes: (i) maintenance of the same heteroplasmy level, (ii) increase in heteroplasmy, and (iii) reduction in heteroplasmy. In case of decreased heteroplasmy, the obtained iPSC lines may be used as isogenic control without the need for further genetic manipulation. Mutation load might also be altered using heteroplasmy shift approaches, such as mitoREs, mito‐TALENS, or mtZFNs. Lastly, mtDNA base editing like DdCBE could be employed to target the mutation and thereby modify its heteroplasmy. The resulting iPSCs carrying a pathogenic mtDNA mutation at various heteroplasmy levels in the same patient‐specific nuclear and mitochondrial genetic background can then be differentiated into disease‐relevant cell types or organoids for disease modeling. (B) During reprogramming to pluripotency of somatic cells carrying homoplasmic mtDNA mutations, homoplasmy is retained at the same level as the parental cells. Hence, heteroplasmy shift approaches are not effective in modulating the mutation load. In order to edit homoplasmic mtDNA mutations in iPSCs, mtDNA base editing would be the only option available. The edited iPSCs can be further differentiated into disease‐relevant cell types or organoids for disease modeling. (C) mtDNA base editing might also be applied to modify the mtDNA profile of iPSCs derived from healthy control individuals. By introducing pathogenic mtDNA mutations, it may be possible to obtain models of mtDNA diseases that do not require the use of patient‐derived cells. However, this approach will not allow for addressing the potential interplay between the nuclear and mitochondrial genomes in a patient‐specific manner.
The ability to modify mtDNA via base editing opens the way to the generation of effective disease models also for homoplasmic mtDNA mutations (Fig 4B). Homoplasmic mtDNA mutations have so far not been possible to introduce or correct, neither through cellular reprogramming‐associated modifications nor with conventional heteroplasmy shift approaches. DdCBE and other mtDNA base editing platforms may thus enable the derivation of isogenic iPSC lines, for example, by targeting a homoplasmic mtDNA mutation to significantly reduce its mutation load in order to revert the disease defects.
It is important to notice that the possibility to engineer mtDNA mutations in iPSCs could also enable the generation of mtDNA disease models without the need for obtaining somatic cells from patients (Fig 4C). This approach, already commonly accepted for nuclear gene defects, would allow investigating of cells and organoids for mutants and controls with identical nuclear genetic backgrounds. This strategy is particularly convenient for rare and pediatric conditions, as in the case of mtDNA diseases. Nevertheless, this approach would not allow for investigating the nuclear–mitochondrial genomes interplay. mtDNA alterations will only be related to a control nuclear background, thereby limiting the possibility to unveil the interconnection between patient‐specific nuclear DNA and the presence or absence of defined mtDNA alterations.
Lastly, mtDNA base editing in iPSCs may have potential utility also for modeling diseases caused by nuclear mutations. In fact, given that during cellular reprogramming changes in mtDNA profile can occur, one could envision the use of mtDNA editing to remove these unwanted mtDNA mutations from newly generated iPSC lines, before proceeding to use the iPSC lines in disease modeling and therapeutic applications (Box 1).
Concluding remarks and future outlook
The recent establishment of key remarkable technologies has brought important advances for mtDNA diseases. The multi‐copy nature of mtDNA, its lack of efficient double‐strand break repair mechanisms, and its cellular localization within a double‐membrane organelle have made editing mtDNA a major challenge. As a result, only a limited number of cellular and animal models for mtDNA diseases could be created. The discovery of mtDNA base editing combined with the possibility to build 3D organoid models from human iPSCs have the potential to lead to the development of new effective model platforms for mtDNA diseases. These technologies may also help establish therapies in a personalized medicine manner, which could be potentially highly beneficial for highly heterogeneous conditions such as mtDNA diseases.
In order to move forward using these technologies, crucial questions and technical aspects remain to be addressed (Box 1). For example, we still do not know the mechanisms underlying the heteroplasmy shift occurring in human iPSCs. Moreover, current mtDNA base editing approaches only work for point mutations and not for large‐scale deletions, and even for point mutations, they can only target few specific sites and cannot edit or correct all known pathogenic mtDNA mutations. Nevertheless, we expect that mtDNA‐engineered iPSCs could represent a decisive advance in the study of mtDNA diseases. The heterogeneous and complex nature of these diseases makes it inherently difficult to decipher the contribution of a mutation to the pathophysiology of the disease. Comparing both mutant and control cells under the same nuclear background conditions could allow elucidating the contribution of specific mtDNA defects to disease phenotypes in different human tissues. Moreover, the use of complex organoid models could shed light on how mtDNA heteroplasmy segregates during human tissue organization. This knowledge could in turn lead to a better understanding of genotype–phenotype correlation and heterogeneous organ specificity of mtDNA mutations in patients with mtDNA diseases.
Overall, by using novel mtDNA base editors and applying them to iPSC‐derived organoids, it may be possible to develop complex human models of mtDNA diseases. These new models could lead to the discovery of relevant pathological mechanisms and might pave the way to interventional strategies for these challenging diseases with high unmet medical needs.
Author contributions
Isabella Tolle: Writing – original draft. Valeria Tiranti: Writing – review and editing. Alessandro Prigione: Conceptualization; supervision; funding acquisition; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Acknowledgements
They acknowledge support from the Medical Faculty of Heinrich Heine University, Deutsche Forschungsgemeinschaft (DFG) (PR1527/5‐1 and PR1527/6‐1), European Joint Programme for Rare Diseases (EJPRD) and German Federal Ministry of Education and Research (BMBF) (01GM2002A), Leigh Syndrome International Consortium (LSIC), People Against Leigh syndrome (PALS), MitoHelp Foundation, and Cure Mito Foundation. This work was partially supported by the Italian Ministry of Health (RRC). VT is part of the Center for the Study of Mitochondrial Pediatric Diseases funded by the Mariani Foundation and is member of the European Reference Network for Rare Neuromuscular Diseases (ERN EURO‐NMD). Open Access funding enabled and organized by Projekt DEAL.
EMBO reports (2023) 24: e55678
References
- Agathocleous M, Harris WA (2013) Metabolism in physiological cell proliferation and differentiation. Trends Cell Biol 23: 484–492 [DOI] [PubMed] [Google Scholar]
- van den Ameele J, Li AYZ, Ma H, Chinnery PF (2020) Mitochondrial heteroplasmy beyond the oocyte bottleneck. Semin Cell Dev Biol 97: 156–166 [DOI] [PubMed] [Google Scholar]
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F et al (1981) Sequence and organization of the human mitochondrial genome. Nature 290: 457–465 [DOI] [PubMed] [Google Scholar]
- Antes A, Tappin I, Chung S, Lim R, Lu B, Parrott AM, Hill HZ, Suzuki CK, Lee C‐G (2010) Differential regulation of full‐length genome and a single‐stranded 7S DNA along the cell cycle in human mitochondria. Nucleic Acids Res 38: 6466–6476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzalone AV, Koblan LW, Liu DR (2020) Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol 38: 824–844 [DOI] [PubMed] [Google Scholar]
- Bacman SR, Williams SL, Duan D, Moraes CT (2012) Manipulation of mtDNA heteroplasmy in all striated muscles of newborn mice by AAV9‐mediated delivery of a mitochondria‐targeted restriction endonuclease. Gene Ther 19: 1101–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacman SR, Williams SL, Pinto M, Peralta S, Moraes CT (2013) Specific elimination of mutant mitochondrial genomes in patient‐derived cells by mitoTALENs. Nat Med 19: 1111–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baertling F, Rodenburg RJ, Schaper J, Smeitink JA, Koopman WJH, Mayatepek E, Morava E, Distelmaier F (2014) A guide to diagnosis and treatment of Leigh syndrome. J Neurol Neurosurg Psychiatry 85: 257–265 [DOI] [PubMed] [Google Scholar]
- Bannwarth S, Procaccio V, Lebre AS, Jardel C, Chaussenot A, Hoarau C, Maoulida H, Charrier N, Gai X, Xie HM et al (2013) Prevalence of rare mitochondrial DNA mutations in mitochondrial disorders. J Med Genet 50: 704–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Jehuda R, Shemer Y, Binah O (2018) Genome editing in induced pluripotent stem cells using CRISPR/Cas9. Stem Cell Rev Rep 14: 323–336 [DOI] [PubMed] [Google Scholar]
- Bottani E, Lamperti C, Prigione A, Tiranti V, Persico N, Brunetti D (2020) Therapeutic approaches to treat mitochondrial diseases: “one‐size‐fits‐all” and “precision medicine” strategies. Pharmaceutics 12: 1–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeron T, Chretien D, Rötig A, Munnich A, Rustin P (1993) Fate and expression of the deleted mitochondrial DNA differ between human heteroplasmic skin fibroblast and Epstein‐Barr virus‐transformed lymphocyte cultures. J Biol Chem 268: 19369–19376 [PubMed] [Google Scholar]
- Brinckmann A, Weiss C, Wilbert F, von Moers A, Zwirner A, Stoltenburg‐Didinger G, Wilichowski E, Schuelke M (2010) Regionalized pathology correlates with augmentation of mtDNA copy numbers in a patient with myoclonic epilepsy with ragged‐red fibers (MERRF‐syndrome). PLoS ONE 5: e13513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broomfield A, Sweeney MG, Woodward CE, Fratter C, Morris AM, Leonard JV, Abulhoul L, Grunewald S, Clayton PT, Hanna MG et al (2015) Paediatric single mitochondrial DNA deletion disorders: an overlapping spectrum of disease. J Inherit Metab Dis 38: 445–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunetti D, Dykstra W, Le S, Zink A, Prigione A (2021) Mitochondria in neurogenesis: implications for mitochondrial diseases. Stem Cells 39: 1289–1297 [DOI] [PubMed] [Google Scholar]
- Bugiardini E, Bottani E, Marchet S, Poole OV, Beninca C, Horga A, Woodward C, Lam A, Hargreaves I, Chalasani A et al (2020) Expanding the molecular and phenotypic spectrum of truncating MT‐ATP6 mutations. Neurol Genet 6: e381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporali L, Magri S, Legati A, Del Dotto V, Tagliavini F, Balistreri F, Nasca A, La Morgia C, Carbonelli M, Valentino ML et al (2020) ATPase domain AFG3L2 mutations Alter OPA1 processing and cause optic neuropathy. Ann Neurol 88: 18–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carelli V, Chan DC (2014) Mitochondrial DNA: impacting central and peripheral nervous systems. Neuron 84: 1126–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carelli V, Hirano M, Enríquez JA, Chinnery PF (2022) Implications of mitochondrial DNA mutations in human induced pluripotent stem cells. Nat Rev Genet 23: 69–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaliere A, Marchet S, Di Meo I, Tiranti V (2022) An In vitro approach to study mitochondrial dysfunction: a Cybrid model. J Vis Exp 10.3791/63452 [DOI] [PubMed] [Google Scholar]
- Chatre L, Ricchetti M (2013) Prevalent coordination of mitochondrial DNA transcription and initiation of replication with the cell cycle. Nucleic Acids Res 41: 3068–3078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Prosser R, Simonetti S, Sadlock J, Jagiello G, Schon EA (1995) Rearranged mitochondrial genomes are present in human oocytes. Am J Hum Genet 57: 239–247 [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Kirk K, Shurubor YI, Zhao D, Arreguin AJ, Shahi I, Valsecchi F, Primiano G, Calder EL, Carelli V et al (2018) Rewiring of glutamine metabolism is a bioenergetic adaptation of human cells with mitochondrial DNA mutations. Cell Metab 27: 1007–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Liang D, Guo J, Zhang J, Sun H, Zhang X, Jin J, Dai Y, Bao Q, Qian X et al (2022) DdCBE‐mediated mitochondrial base editing in human 3PN embryos. Cell Discov 8: 4–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry ABC, Gagne KE, McLoughlin EM, Baccei A, Gorman B, Hartung O, Miller JD, Zhang J, Zon RL, Ince TA et al (2013) Induced pluripotent stem cells with a mitochondrial DNA deletion. Stem Cells 31: 1287–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiaradia I, Lancaster MA (2020) Brain organoids for the study of human neurobiology at the interface of in vitro and in vivo. Nat Neurosci 23: 1496–1508 [DOI] [PubMed] [Google Scholar]
- Chichagova V, Hallam D, Collin J, Buskin A, Saretzki G, Armstrong L, Yu‐Wai‐Man P, Lako M, Steel DH (2017) Human iPSC disease modelling reveals functional and structural defects in retinal pigment epithelial cells harbouring the m.3243A>G mitochondrial DNA mutation. Sci Rep 7: 12320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnery PF, DiMauro S, Shanske S, Schon EA, Zeviani M, Mariotti C, Carrara F, Lombes A, Laforet P, Ogier H et al (2004) Risk of developing a mitochondrial DNA deletion disorder. Lancet 364: 592–596 [DOI] [PubMed] [Google Scholar]
- Chinnery PF, Craven L, Mitalipov S, Stewart JB, Herbert M, Turnbull DM (2014) The challenges of mitochondrial replacement. PLoS Genet 10: e1004315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S‐I, Lee S, Mok YG, Lim K, Lee J, Lee JM, Chung E, Kim J‐S (2022) Targeted A‐to‐G base editing in human mitochondrial DNA with programmable deaminases. Cell 185: 1764–1776 [DOI] [PubMed] [Google Scholar]
- Chou S‐J, Tseng W‐L, Chen C‐T, Lai Y‐F, Chien C‐S, Chang Y‐L, Lee H‐C, Wei Y‐H, Chiou S‐H (2016) Impaired ROS scavenging system in human induced pluripotent stem cells generated from patients with MERRF syndrome. Sci Rep 6: 23661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung S, Dzeja PP, Faustino RS, Perez‐Terzic C, Behfar A & Terzic A (2007) Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nat Clin Pract Cardiovasc Med 4 Suppl 1: S60‐7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciafaloni E, Santorelli FM, Shanske S, Deonna T, Roulet E, Janzer C, Pescia G, DiMauro S (1993) Maternally inherited Leigh syndrome. J Pediatr 122: 419–422 [DOI] [PubMed] [Google Scholar]
- Cliff TS, Dalton S (2017) Metabolic switching and cell fate decisions: implications for pluripotency, reprogramming and development. Curr Opin Genet Dev 46: 44–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortopassi GA, Arnheim N (1990) Detection of a specific mitochondrial DNA deletion in tissues of older humans. Nucleic Acids Res 18: 6927–6933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan CS, Renner M, De Gennaro M, Gross‐Scherf B, Goldblum D, Hou Y, Munz M, Rodrigues TM, Krol J, Szikra T et al (2020) Cell types of the human retina and its organoids at single‐cell resolution. Cell 182: 1623–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cree LM, Samuels DC, Chinnery PF (2009) The inheritance of pathogenic mitochondrial DNA mutations. Biochim Biophys Acta 1792: 1097–1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danese A, Patergnani S, Maresca A, Peron C, Raimondi A, Caporali L, Marchi S, La Morgia C, Del Dotto V, Zanna C et al (2022) Pathological mitophagy disrupts mitochondrial homeostasis in Leber's hereditary optic neuropathy. Cell Rep 40: 111124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Aurelio M, Vives‐Bauza C, Davidson MM, Manfredi G (2010) Mitochondrial DNA background modifies the bioenergetics of NARP/MILS ATP6 mutant cells. Hum Mol Genet 19: 374–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Dotto V, Ullah F, Di Meo I, Magini P, Gusic M, Maresca A, Caporali L, Palombo F, Tagliavini F, Baugh EH et al (2020) SSBP1 mutations cause mtDNA depletion underlying a complex optic atrophy disorder. J Clin Invest 130: 108–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Erchia AM, Atlante A, Gadaleta G, Pavesi G, Chiara M, De Virgilio C, Manzari C, Mastropasqua F, Prazzoli GM, Picardi E et al (2015) Tissue‐specific mtDNA abundance from exome data and its correlation with mitochondrial transcription, mass and respiratory activity. Mitochondrion 20: 13–21 [DOI] [PubMed] [Google Scholar]
- Deuse T, Hu X, Agbor‐Enoh S, Koch M, Spitzer MH, Gravina A, Alawi M, Marishta A, Peters B, Kosaloglu‐Yalcin Z et al (2019) De novo mutations in mitochondrial DNA of iPSCs produce immunogenic neoepitopes in mice and humans. Nat Biotechnol 37: 1137–1144 [DOI] [PubMed] [Google Scholar]
- Dunn DA, Pinkert CA (2012) Nuclear expression of a mitochondrial DNA gene: mitochondrial targeting of allotopically expressed mutant ATP6 in transgenic mice. J Biomed Biotechnol 2012: 541245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facucho‐Oliveira JM, St John JC (2009) The relationship between pluripotency and mitochondrial DNA proliferation during early embryo development and embryonic stem cell differentiation. Stem Cell Rev Rep 5: 140–158 [DOI] [PubMed] [Google Scholar]
- Falabella M, Minczuk M, Hanna MG, Viscomi C, Pitceathly RDS (2022) Gene therapy for primary mitochondrial diseases: experimental advances and clinical challenges. Nat Rev Neurol 18: 689–698 [DOI] [PubMed] [Google Scholar]
- Filograna R, Mennuni M, Alsina D, Larsson N‐G (2021) Mitochondrial DNA copy number in human disease: the more the better? FEBS Lett 595: 976–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folmes CDL, Nelson TJ, Martinez‐Fernandez A, Arrell DK, Lindor JZ, Dzeja PP, Ikeda Y, Perez‐Terzic C, Terzic A (2011) Somatic oxidative bioenergetics transitions into pluripotency‐dependent glycolysis to facilitate nuclear reprogramming. Cell Metab 14: 264–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folmes CDL, Martinez‐Fernandez A, Perales‐Clemente E, Li X, McDonald A, Oglesbee D, Hrstka SC, Perez‐Terzic C, Terzic A, Nelson TJ (2013) Disease‐causing mitochondrial Heteroplasmy segregated within induced pluripotent stem cell clones derived from a patient with MELAS. Stem Cells 31: 1298–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujikura J, Nakao K, Sone M, Noguchi M, Mori E, Naito M, Taura D, Harada‐Shiba M, Kishimoto I, Watanabe A et al (2012) Induced pluripotent stem cells generated from diabetic patients with mitochondrial DNA A3243G mutation. Diabetologia 55: 1689–1698 [DOI] [PubMed] [Google Scholar]
- Fuke S, Kubota‐Sakashita M, Kasahara T, Shigeyoshi Y, Kato T (2011) Regional variation in mitochondrial DNA copy number in mouse brain. Biochim Biophys Acta 1807: 270–274 [DOI] [PubMed] [Google Scholar]
- Galera‐Monge T, Zurita‐Díaz F, Canals I, Hansen MG, Rufián‐Vázquez L, Ehinger JK, Elmér E, Martin MA, Garesse R, Ahlenius H et al (2020) Mitochondrial Dysfunction and Calcium Dysregulation in Leigh Syndrome Induced Pluripotent Stem Cell Derived Neurons. Int J Mol Sci 21: 3191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammage PA, Rorbach J, Vincent AI, Rebar EJ, Minczuk M (2014) Mitochondrially targeted ZFNs for selective degradation of pathogenic mitochondrial genomes bearing large‐scale deletions or point mutations. EMBO Mol Med 6: 458–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammage PA, Moraes CT, Minczuk M (2018a) Mitochondrial genome engineering: the revolution may not Be CRISPR‐Ized. Trends Genet 34: 101–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammage PA, Viscomi C, Simard M‐L, Costa ASH, Gaude E, Powell CA, Van Haute L, McCann BJ, Rebelo‐Guiomar P, Cerutti R et al (2018b) Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat Med 24: 1691–1695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giang A‐H, Raymond T, Brookes P, de Mesy BK, Schwarz E, O'Keefe R, Eliseev R (2013) Mitochondrial dysfunction and permeability transition in osteosarcoma cells showing the Warburg effect. J Biol Chem 288: 33303–33311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein A, Falk MJ (1993) Mitochondrial DNA deletion syndromes. In GeneReviews, Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, LJH B, Gripp KW, Amemiya A (eds). Seattle, WA: University of Washington; [PubMed] [Google Scholar]
- Gorman GS, Chinnery PF, DiMauro S, Hirano M, Koga Y, McFarland R, Suomalainen A, Thorburn DR, Zeviani M, Turnbull DM (2016) Mitochondrial diseases. Nat Rev Dis Primers 2: 16080 [DOI] [PubMed] [Google Scholar]
- Goto Y, Nonaka I, Horai S (1990) A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 348: 651–653 [DOI] [PubMed] [Google Scholar]
- Granatiero V, Giorgio V, Calì T, Patron M, Brini M, Bernardi P, Tiranti V, Zeviani M, Pallafacchina G, De Stefani D et al (2016) Reduced mitochondrial Ca(2+) transients stimulate autophagy in human fibroblasts carrying the 13514A>G mutation of the ND5 subunit of NADH dehydrogenase. Cell Death Differ 23: 231–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield A, Braude P, Flinter F, Lovell‐Badge R, Ogilvie C, Perry ACF (2017) Assisted reproductive technologies to prevent human mitochondrial disease transmission. Nat Biotechnol 35: 1059–1068 [DOI] [PubMed] [Google Scholar]
- Gunnewiek TMK, Van Hugte EJH, Frega M, Morava E, Kasri NN, Foreman K, Panneman D, Mossink B, Linda K, Keller JM et al (2020) M.3243A>G‐induced mitochondrial dysfunction impairs human neuronal development and reduces neuronal network activity and synchronicity. Cell Rep 31: 107538 [DOI] [PubMed] [Google Scholar]
- Gunnewiek TMK, Verboven AHA, Pelgrim I, Hogeweg M, Schoenmaker C, Renkema H, Beyrath J, Smeitink J, De Vries BBA, Hoen PAC et al (2021) Sonlicromanol improves neuronal network dysfunction and transcriptome changes linked to m.3243A>G heteroplasmy in iPSC‐derived neurons. Stem Cell Reports 16: 2197–2212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Zhang X, Chen X, Sun H, Dai Y, Wang J, Qian X, Tan L, Lou X, Shen B (2021) Precision modeling of mitochondrial diseases in zebrafish via DdCBE‐mediated mtDNA base editing. Cell Discov 7: 4–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Chen X, Liu Z, Sun H, Zhou Y, Dai Y, Ma Y, He L, Qian X, Wang J et al (2022) DdCBE mediates efficient and inheritable modifications in mouse mitochondrial genome. Mol Ther Nucleic Acids 27: 73–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson CM, Falkenberg M, Larsson N‐G (2016) Maintenance and expression of mammalian mitochondrial DNA. Annu Rev Biochem 85: 133–160 [DOI] [PubMed] [Google Scholar]
- Guy J, Feuer WJ, Davis JL, Porciatti V, Gonzalez PJ, Koilkonda RD, Yuan H, Hauswirth WW, Lam BL (2017) Gene therapy for Leber hereditary optic neuropathy: low‐ and medium‐dose visual results. Ophthalmology 124: 1621–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagström E, Freyer C, Battersby BJ, Stewart JB, Larsson N‐G (2014) No recombination of mtDNA after heteroplasmy for 50 generations in the mouse maternal germline. Nucleic Acids Res 42: 1111–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn A, Zuryn S (2019) The cellular mitochondrial genome landscape in disease. Trends Cell Biol 29: 227–240 [DOI] [PubMed] [Google Scholar]
- Hämäläinen RH (2016) Mitochondrial DNA mutations in iPS cells: mtDNA integrity as standard iPSC selection criteria? EMBO J 35: 1960–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hämäläinen RH, Manninen T, Koivumäki H, Kislin M, Otonkoski T, Suomalainen A (2013) Tissue‐and cell‐type‐specific manifestations of heteroplasmic mtDNA 3243A>G mutation in human induced pluripotent stem cell‐derived disease model. Proc Natl Acad Sci USA 110: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hameed S, Tadi P (2022) Myoclonic epilepsy and ragged red fibers. Treasure Island, FL: StatPearls Publishing; [PubMed] [Google Scholar]
- Harding AE, Hammans SR (1992) Deletions of the mitochondrial genome. J Inherit Metab Dis 15: 480–486 [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Bacman SR, Peralta S, Falk MJ, Chomyn A, Chan DC, Williams SL, Moraes CT (2015) MitoTALEN: a general approach to reduce mutant mtDNA loads and restore oxidative phosphorylation function in mitochondrial diseases. Mol Ther 23: 1592–1599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama H, Katayama A, Komaki H, Nishino I, Goto YI (2015) Molecular pathomechanisms and cell‐type‐specific disease phenotypes of MELAS caused by mutant mitochondrial tRNA(Trp). Acta Neuropathol Commun 3: 52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heighton JN, Brady LI, Sadikovic B, Bulman DE, Tarnopolsky MA (2019) Genotypes of chronic progressive external ophthalmoplegia in a large adult‐onset cohort. Mitochondrion 49: 227–231 [DOI] [PubMed] [Google Scholar]
- Hernández‐Ainsa C, López‐Gallardo E, García‐Jiménez MC, Climent‐Alcalá FJ, Rodríguez‐Vigil C, Fernández G, de Villalta M, Artuch R, Montoya J, Ruiz‐Pesini E et al (2022) Development and characterization of cell models harbouring mtDNA deletions for in vitro study of Pearson syndrome. Dis Model Mech 15: dmm049083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt IJ, Harding AE, Petty RK, Morgan‐Hughes JA (1990) A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am J Hum Genet 46: 428–433 [PMC free article] [PubMed] [Google Scholar]
- Hudson G, Takeda Y, Herbert M (2019) Reversion after replacement of mitochondrial DNA. Nature 574: E8–E11 [DOI] [PubMed] [Google Scholar]
- Inak G, Lorenz C, Lisowski P, Zink A, Mlody B, Prigione A (2017) Concise review: induced pluripotent stem cell‐based drug discovery for mitochondrial disease. Stem Cells 35: 1655–1662 [DOI] [PubMed] [Google Scholar]
- Inak G, Rybak‐Wolf A, Lisowski P, Pentimalli TM, Jüttner R, Glažar P, Uppal K, Bottani E, Brunetti D, Secker C et al (2021) Defective metabolic programming impairs early neuronal morphogenesis in neural cultures and an organoid model of Leigh syndrome. Nat Commun 12: 1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Nakada K, Ogura A, Isobe K, Goto Y, Nonaka I, Hayashi JI (2000) Generation of mice with mitochondrial dysfunction by introducing mouse mtDNA carrying a deletion into zygotes. Nat Genet 26: 176–181 [DOI] [PubMed] [Google Scholar]
- Invernizzi F, D'Amato I, Jensen PB, Ravaglia S, Zeviani M, Tiranti V (2012) Microscale oxygraphy reveals OXPHOS impairment in MRC mutant cells. Mitochondrion 12: 328–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson CB, Turnbull DM, Minczuk M, Gammage PA (2020) Therapeutic manipulation of mtDNA Heteroplasmy: a shifting perspective. Trends Mol Med 26: 698–709 [DOI] [PubMed] [Google Scholar]
- Janssen MCH, Koene S, de Laat P, Hemelaar P, Pickkers P, Spaans E, Beukema R, Beyrath J, Groothuis J, Verhaak C et al (2019) The KHENERGY study: safety and efficacy of KH176 in mitochondrial m.3243A>G Spectrum disorders. Clin Pharmacol Ther 105: 101–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual‐RNA–guided DNA endonuclease in adaptive bacterial. Immunity 337: 816–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang E, Wang X, Tippner‐Hedges R, Ma H, Folmes CDL, Gutierrez NM, Lee Y, Van Dyken C, Ahmed R, Li Y et al (2016a) Age‐related accumulation of somatic mitochondrial DNA mutations in adult‐derived human iPSCs. Cell Stem Cell 18: 625–636 [DOI] [PubMed] [Google Scholar]
- Kang E, Wu J, Gutierrez NM, Koski A, Tippner‐Hedges R, Agaronyan K, Platero‐Luengo A, Martinez‐Redondo P, Ma H, Lee Y et al (2016b) Mitochondrial replacement in human oocytes carrying pathogenic mitochondrial DNA mutations. Nature 540: 270–275 [DOI] [PubMed] [Google Scholar]
- Kasahara A, Ishikawa K, Yamaoka M, Ito M, Watanabe N, Akimoto M, Sato A, Nakada K, Endo H, Suda Y et al (2006) Generation of trans‐mitochondrial mice carrying homoplasmic mtDNAs with a missense mutation in a structural gene using ES cells. Hum Mol Genet 15: 871–881 [DOI] [PubMed] [Google Scholar]
- Kazak L, Reyes A, Holt IJ (2012) Minimizing the damage: repair pathways keep mitochondrial DNA intact. Nat Rev Mol Cell Biol 13: 659–671 [DOI] [PubMed] [Google Scholar]
- Kelly RDW, Mahmud A, McKenzie M, Trounce IA, St John JC (2012) Mitochondrial DNA copy number is regulated in a tissue specific manner by DNA methylation of the nuclear‐encoded DNA polymerase gamma A. Nucleic Acids Res 40: 10124–10138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly RDW, Rodda AE, Dickinson A, Mahmud A, Nefzger CM, Lee W, Forsythe JS, Polo JM, Trounce IA, McKenzie M et al (2013) Mitochondrial DNA haplotypes define gene expression patterns in pluripotent and differentiating embryonic stem cells. Stem Cells 31: 703–716 [DOI] [PubMed] [Google Scholar]
- Kennedy SR, Salk JJ, Schmitt MW, Loeb LA (2013) Ultra‐sensitive sequencing reveals an age‐related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet 9: e1003794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keogh M, Chinnery PF (2013) Hereditary mtDNA heteroplasmy: a baseline for aging? Cell Metab 18: 463–464 [DOI] [PubMed] [Google Scholar]
- King MP, Attardi G (1989) Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 246: 500–503 [DOI] [PubMed] [Google Scholar]
- Klopstock T, Yu‐Wai‐Man P, Dimitriadis K, Rouleau J, Heck S, Bailie M, Atawan A, Chattopadhyay S, Schubert M, Garip A et al (2011) A randomized placebo‐controlled trial of idebenone in Leber's hereditary optic neuropathy. Brain 134: 2677–2686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodaira M, Hatakeyama H, Yuasa S, Seki T, Egashira T, Tohyama S, Kuroda Y, Tanaka A, Okata S, Hashimoto H et al (2015) Impaired respiratory function in MELAS‐induced pluripotent stem cells with high heteroplasmy levels. FEBS Open Bio 5: 219–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondoh H, Lleonart ME, Gil J, Beach D, Peters G (2005) Glycolysis and cellular immortalization. Drug Discov Today Dis Mech 2: 263–267 [Google Scholar]
- Kuijk E, Jager M, van der Roest B, Locati MD, Van Hoeck A, Korzelius J, Janssen R, Besselink N, Boymans S, van Boxtel R et al (2020) The mutational impact of culturing human pluripotent and adult stem cells. Nat Commun 11: 2493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA et al (2005) Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309: 481–484 [DOI] [PubMed] [Google Scholar]
- Lancaster MA, Renner M, Martin C‐A, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA (2013) Cerebral organoids model human brain development and microcephaly. Nature 501: 373–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange C, Turrero Garcia M, Decimo I, Bifari F, Eelen G, Quaegebeur A, Boon R, Zhao H, Boeckx B, Chang J et al (2016) Relief of hypoxia by angiogenesis promotes neural stem cell differentiation by targeting glycolysis. EMBO J 35: 924–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson NG, Holme E, Kristiansson B, Oldfors A, Tulinius M (1990) Progressive increase of the mutated mitochondrial DNA fraction in Kearns‐Sayre syndrome. Pediatr Res 28: 131–136 [DOI] [PubMed] [Google Scholar]
- Latorre‐Pellicer A, Moreno‐Loshuertos R, Lechuga‐Vieco AV, Sánchez‐Cabo F, Torroja C, Acín‐Pérez R, Calvo E, Aix E, González‐Guerra A, Logan A et al (2016) Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature 535: 561–565 [DOI] [PubMed] [Google Scholar]
- Le S, Petersilie L, Inak G, Menacho‐Pando C, Kafitz KW, Rybak‐Wolf A, Rajewsky N, Rose CR, Prigione A (2021) Generation of human brain organoids for mitochondrial disease modeling. J Vis Exp 10.3791/62756 [DOI] [PubMed] [Google Scholar]
- Lee H, Lee S, Baek G, Kim A, Kang BC, Seo H, Kim JS (2021) Mitochondrial DNA editing in mice with DddA‐TALE fusion deaminases. Nat Commun 12: 10–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Lee H, Baek G, Kim J‐S (2022a) Precision mitochondrial DNA editing with high‐fidelity DddA‐derived base editors. Nat Biotechnol 10.1038/s41587-022-01486-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Lee H, Baek G, Namgung E, Park JM, Kim S, Hong S, Kim J‐S (2022b) Enhanced mitochondrial DNA editing in mice using nuclear‐exported TALE‐linked deaminases and nucleases. Genome Biol 23: 211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Z, Meng H, Liu L, Zhao H, Rao X, Yan Y, Wu H, Liu M, He A, Yi C (2022) Mitochondrial base editor induces substantial nuclear off‐target mutations. Nature 606: 804–811 [DOI] [PubMed] [Google Scholar]
- Lertrit P, Noer AS, Byrne E, Marzuki S (1992) Tissue segregation of a heteroplasmic mtDNA mutation in MERRF (myoclonic epilepsy with ragged red fibers) encephalomyopathy. Hum Genet 90: 251–254 [DOI] [PubMed] [Google Scholar]
- Lester Sequiera G, Srivastava A, Alagarsamy KN, Rockman‐Greenberg C, Dhingra S (2021) Generation and evaluation of isogenic iPSC as a source of cell replacement therapies in patients with Kearns Sayre syndrome. Cell 10: 568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis‐Israeli YR, Wasserman AH, Gabalski MA, Volmert BD, Ming Y, Ball KA, Yang W, Zou J, Ni G, Pajares N et al (2021) Self‐assembling human heart organoids for the modeling of cardiac development and congenital heart disease. Nat Commun 12: 5142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim AZ, Ng YS, Blain A, Jiminez‐Moreno C, Alston CL, Nesbitt V, Simmons L, Santra S, Wassmer E, Blakely EL et al (2022a) Natural history of Leigh syndrome: a study of disease burden and progression. Ann Neurol 91: 117–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim K, Cho SI, Kim JS (2022b) Nuclear and mitochondrial DNA editing in human cells with zinc finger deaminases. Nat Commun 13: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CS, Sharpley MS, Fan W, Waymire KG, Sadun AA, Carelli V, Ross‐Cisneros FN, Baciu P, Sung E, McManus MJ et al (2012) Mouse mtDNA mutant model of Leber hereditary optic neuropathy. Proc Natl Acad Sci USA 109: 20065–20070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D‐S, Huang Y‐W, Ho C‐S, Hung P‐L, Hsu M‐H, Wang T‐J, Wu T‐Y, Lee T‐H, Huang Z‐D, Chang P‐C et al (2019) Oxidative insults and mitochondrial DNA mutation promote enhanced autophagy and Mitophagy compromising cell viability in pluripotent cell model of mitochondrial disease. Cell 8: 65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisowski P, Kannan P, Mlody B, Prigione A (2018) Mitochondria and the dynamic control of stem cell homeostasis. EMBO Rep 19: e45432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López‐Gallardo E, López‐Pérez MJ, Montoya J, Ruiz‐Pesini E (2009) CPEO and KSS differ in the percentage and location of the mtDNA deletion. Mitochondrion 9: 314–317 [DOI] [PubMed] [Google Scholar]
- Lorenz C, Prigione A (2016) Aging vs. rejuvenation: reprogramming to iPSCs does not turn back the clock for somatic mitochondrial DNA mutations. Stem Cell Investig 3: 43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz C, Lesimple P, Bukowiecki R, Zink A, Inak G, Mlody B, Singh M, Semtner M, Mah N, Auré K et al (2017) Human iPSC‐derived neural progenitors are an effective drug discovery model for neurological mtDNA disorders. Cell Stem Cell 20: 659–674 [DOI] [PubMed] [Google Scholar]
- Luo S, Valencia CA, Zhang J, Lee N‐C, Slone J, Gui B, Wang X, Li Z, Dell S, Brown J et al (2018) Biparental inheritance of mitochondrial DNA in humans. Proc Natl Acad Sci USA 115: 13039–13044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Folmes CDL, Wu J, Morey R, Mora‐Castilla S, Ocampo A, Ma L, Poulton J, Wang X, Ahmed R et al (2015) Metabolic rescue in pluripotent cells from patients with mtDNA disease. Nature 524: 234–238 [DOI] [PubMed] [Google Scholar]
- Magnusson J, Orth M, Lestienne P, Taanman J‐W (2003) Replication of mitochondrial DNA occurs throughout the mitochondria of cultured human cells. Exp Cell Res 289: 133–142 [DOI] [PubMed] [Google Scholar]
- Maitra A, Arking DE, Shivapurkar N, Ikeda M, Stastny V, Kassauei K, Sui G, Cutler DJ, Liu Y, Brimble SN et al (2005) Genomic alterations in cultured human embryonic stem cells. Nat Genet 37: 1099–1103 [DOI] [PubMed] [Google Scholar]
- McCann BJ, Cox A, Gammage PA, Stewart JB, Zernicka‐Goetz M, Minczuk M (2018) Delivery of mtZFNs into early mouse embryos. Methods Mol Biol 1867: 215–228 [DOI] [PubMed] [Google Scholar]
- McCauley HA, Wells JM (2017) Pluripotent stem cell‐derived organoids: using principles of developmental biology to grow human tissues in a dish. Development 144: 958–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnight CL, Low YC, Elliott DA, Thorburn DR, Frazier AE (2021) Modelling mitochondrial disease in human pluripotent stem cells: what have we learned? Int J Mol Sci 22: 7730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minczuk M, Papworth MA, Miller JC, Murphy MP, Klug A (2008) Development of a single‐chain, quasi‐dimeric zinc‐finger nuclease for the selective degradation of mutated human mitochondrial DNA. Nucleic Acids Res 36: 3926–3938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura Y, Li M‐Y, Revah O, Yoon S‐J, Narazaki G, Pașca SP (2022) Engineering brain assembloids to interrogate human neural circuits. Nat Protoc 17: 15–35 [DOI] [PubMed] [Google Scholar]
- Mok BY, de Moraes MH, Zeng J, Bosch DE, Kotrys AV, Raguram A, Hsu FS, Radey MC, Peterson SB, Mootha VK et al (2020) A bacterial cytidine deaminase toxin enables CRISPR‐free mitochondrial base editing. Nature 583: 631–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok BY, Kotrys AV, Raguram A, Huang TP, Mootha VK, Liu DR (2022a) CRISPR‐free base editors with enhanced activity and expanded targeting scope in mitochondrial and nuclear DNA. Nat Biotechnol 40: 1378–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok YG, Lee JM, Chung E, Lee J, Lim K, Cho SI, Kim JS (2022b) Base editing in human cells with monomeric DddA‐TALE fusion deaminases. Nat Commun 13: 4038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraes CT, Ciacci F, Silvestri G, Shanske S, Sciacco M, Hirano M, Schon EA, Bonilla E, DiMauro S (1993) Atypical clinical presentations associated with the MELAS mutation at position 3243 of human mitochondrial DNA. Neuromuscul Disord 3: 43–50 [DOI] [PubMed] [Google Scholar]
- Nakada K, Inoue K, Ono T, Isobe K, Ogura A, Goto YI, Nonaka I, Hayashi JI (2001) Inter‐mitochondrial complementation: mitochondria‐specific system preventing mice from expression of disease phenotypes by mutant mtDNA. Nat Med 7: 934–940 [DOI] [PubMed] [Google Scholar]
- Nakano T, Ando S, Takata N, Kawada M, Muguruma K, Sekiguchi K, Saito K, Yonemura S, Eiraku M, Sasai Y (2012) Self‐formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 10: 771–785 [DOI] [PubMed] [Google Scholar]
- Nesbitt V, Pitceathly RDS, Turnbull DM, Taylor RW, Sweeney MG, Mudanohwo EE, Rahman S, Hanna MG, McFarland R (2013) The UK MRC mitochondrial disease patient cohort study: clinical phenotypes associated with the m.3243A>G mutation‐‐implications for diagnosis and management. J Neurol Neurosurg Psychiatry 84: 936–938 [DOI] [PubMed] [Google Scholar]
- Nilsen H, Otterlei M, Haug T, Solum K, Nagelhus TA, Skorpen F, Krokan HE (1997) Nuclear and mitochondrial uracil‐DNA glycosylases are generated by alternative splicing and transcription from different positions in the UNG gene. Nucleic Acids Res 25: 750–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissanka N, Bacman SR, Plastini MJ, Moraes CT (2018) The mitochondrial DNA polymerase gamma degrades linear DNA fragments precluding the formation of deletions. Nat Commun 9: 2491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong S‐E, Walford GA, Sugiana C, Boneh A, Chen WK et al (2008) A mitochondrial protein compendium elucidates complex I disease biology. Cell 134: 112–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palombo F, Peron C, Caporali L, Iannielli A, Maresca A, Di Meo I, Fiorini C, Segnali A, Sciacca FL, Rizzo A et al (2021) The relevance of mitochondrial DNA variants fluctuation during reprogramming and neuronal differentiation of human iPSCs. Stem Cell Reports 16: 1953–1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palozzi JM, Jeedigunta SP, Minenkova AV, Monteiro VL, Thompson ZS, Lieber T, Hurd TR (2022) Mitochondrial DNA quality control in the female germline requires a unique programmed mitophagy. Cell Metab 34: 1809–1823 [DOI] [PubMed] [Google Scholar]
- Pașca SP, Arlotta P, Bateup HS, Camp JG, Cappello S, Gage FH, Knoblich JA, Kriegstein AR, Lancaster MA, Ming G‐L et al (2022) A nomenclature consensus for nervous system organoids and assembloids. Nature 609: 907–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlakis SG, Phillips PC, DiMauro S, De Vivo DC, Rowland LP (1984) Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: a distinctive clinical syndrome. Ann Neurol 16: 481–488 [DOI] [PubMed] [Google Scholar]
- Payne BAI, Wilson IJ, Hateley CA, Horvath R, Santibanez‐Koref M, Samuels DC, Price DA, Chinnery PF (2011) Mitochondrial aging is accelerated by anti‐retroviral therapy through the clonal expansion of mtDNA mutations. Nat Genet 43: 806–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne BAI, Wilson IJ, Yu‐Wai‐Man P, Coxhead J, Deehan D, Horvath R, Taylor RW, Samuels DC, Santibanez‐Koref M, Chinnery PF (2013) Universal heteroplasmy of human mitochondrial DNA. Hum Mol Genet 22: 384–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeva V, Blei D, Trombly G, Corsi S, Szukszto MJ, Rebelo‐Guiomar P, Gammage PA, Kudin AP, Becker C, Altmüller J et al (2018) Linear mitochondrial DNA is rapidly degraded by components of the replication machinery. Nat Commun 9: 1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pek NMQ, Phua QH, Ho BX, Pang JKS, Hor J‐H, An O, Yang HH, Yu Y, Fan Y, Ng S‐Y et al (2019) Mitochondrial 3243A>G mutation confers pro‐atherogenic and pro‐inflammatory properties in MELAS iPS derived endothelial cells. Cell Death Dis 10: 802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perales‐Clemente E, Fernández‐Silva P, Acín‐Pérez R, Pérez‐Martos A, Enríquez JA (2011) Allotopic expression of mitochondrial‐encoded genes in mammals: achieved goal, undemonstrated mechanism or impossible task? Nucleic Acids Res 39: 225–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perales‐Clemente E, Cook AN, Evans JM, Roellinger S, Secreto F, Emmanuele V, Oglesbee D, Mootha VK, Hirano M, Schon EA et al (2016) Natural underlying mt DNA heteroplasmy as a potential source of intra‐person hi PSC variability. EMBO J 35: 1979–1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peron C, Mauceri R, Iannielli A, Cavaliere A, Legati A, Rizzo A, Sciacca FL, Broccoli V, Tiranti V (2021) Generation of two human iPSC lines, FINCBi002‐a and FINCBi003‐a, carrying heteroplasmic macrodeletion of mitochondrial DNA causing Pearson's syndrome. Stem Cell Res 50: 102151 [DOI] [PubMed] [Google Scholar]
- Pfeffer G, Horvath R, Klopstock T, Mootha VK, Suomalainen A, Koene S, Hirano M, Zeviani M, Bindoff LA, Yu‐Wai‐Man P et al (2013) New treatments for mitochondrial disease‐no time to drop our standards. Nat Rev Neurol 9: 474–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips AF, Millet AR, Tigano M, Dubois SM, Crimmins H, Babin L, Charpentier M, Piganeau M, Brunet E, Sfeir A (2017) Single‐molecule analysis of mtDNA replication uncovers the basis of the common deletion. Mol Cell 65: 527–538 [DOI] [PubMed] [Google Scholar]
- Pica‐Mattoccia L, Attardi G (1972) Expression of the mitochondrial genome in HeLa cells. IX. Replication of mitochondrial DNA in relationship to cell cycle in HeLa cells. J Mol Biol 64: 465–484 [DOI] [PubMed] [Google Scholar]
- Picard M (2021) Blood mitochondrial DNA copy number: what are we counting? Mitochondrion 60: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M, Shirihai OS (2022) Mitochondrial signal transduction. Cell Metab 34: 1620–1653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M, Zhang J, Hancock S, Derbeneva O, Golhar R, Golik P, O'Hearn S, Levy S, Potluri P, Lvova M et al (2014) Progressive increase in mtDNA 3243A>G heteroplasmy causes abrupt transcriptional reprogramming. Proc Natl Acad Sci USA 111: E4033–E4042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickett SJ, Grady JP, Ng YS, Gorman GS, Schaefer AM, Wilson IJ, Cordell HJ, Turnbull DM, Taylor RW, McFarland R (2018) Phenotypic heterogeneity in m.3243A>G mitochondrial disease: the role of nuclear factors. Ann Clin Transl Neurol 5: 333–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitceathly RDS, Rahman S, Hanna MG (2012) Single deletions in mitochondrial DNA‐‐molecular mechanisms and disease phenotypes in clinical practice. Neuromuscul Disord 22: 577–586 [DOI] [PubMed] [Google Scholar]
- Prigione A, Fauler B, Lurz R, Lehrach H, Adjaye J (2010) The senescence‐related mitochondrial/oxidative stress pathway is repressed in human induced pluripotent stem cells. Stem Cells 28: 721–733 [DOI] [PubMed] [Google Scholar]
- Prigione A, Lichtner B, Kuhl H, Struys EA, Wamelink M, Lehrach H, Ralser M, Timmermann B, Adjaye J (2011) Human induced pluripotent stem cells harbor homoplasmic and heteroplasmic mitochondrial DNA mutations while maintaining human embryonic stem cell‐like metabolic reprogramming. Stem Cells 29: 1338–1348 [DOI] [PubMed] [Google Scholar]
- Qi X, Chen X, Guo J, Zhang X, Sun H, Wang J, Qian X, Li B, Tan L, Yu L et al (2021) Precision modeling of mitochondrial disease in rats via DdCBE‐mediated mtDNA editing. Cell Discov 7: 95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath S, Sharma R, Gupta R, Ast T, Chan C, Durham TJ, Goodman RP, Grabarek Z, Haas ME, Hung WHW et al (2021) MitoCarta3.0: an updated mitochondrial proteome now with sub‐organelle localization and pathway annotations. Nucleic Acids Res 49: D1541–D1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausser S, Trumpff C, McGill MA, Junker A, Wang W, Ho S‐H, Mitchell A, Karan KR, Monk C, Segerstrom SC et al (2021) Mitochondrial phenotypes in purified human immune cell subtypes and cell mixtures. Elife 10: e70899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy P, Ocampo A, Suzuki K, Luo J, Bacman SR, Williams SL, Sugawara A, Okamura D, Tsunekawa Y, Wu J et al (2015) Selective elimination of mitochondrial mutations in the germline by genome editing. Cell 161: 459–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero‐Morales AI, Robertson GL, Rastogi A, Rasmussen ML, Temuri H, McElroy GS, Chakrabarty RP, Hsu L, Almonacid PM, Millis BA et al (2022) Human iPSC‐derived cerebral organoids model features of Leigh syndrome and reveal abnormal corticogenesis. Development 149: dev199914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi A, Lickfett S, Martins S, Prigione A (2022) A call for consensus guidelines on monitoring the integrity of nuclear and mitochondrial genomes in human pluripotent stem cells. Stem Cell Reports 17: 707–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotig A, Colonna M, Bonnefont JP, Blanche S, Fischer A, Saudubray JM, Munnich A (1989) Mitochondrial DNA deletion in Pearson's marrow/pancreas syndrome. Lancet 1: 902–903 [DOI] [PubMed] [Google Scholar]
- Rowe RG, Daley GQ (2019) Induced pluripotent stem cells in disease modelling and drug discovery. Nat Rev Genet 20: 377–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell OM, Fruh I, Rai PK, Marcellin D, Doll T, Reeve A, Germain M, Bastien J, Rygiel KA, Cerino R et al (2018) Preferential amplification of a human mitochondrial DNA deletion in vitro and in vivo. Sci Rep 8: 1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell OM, Gorman GS, Lightowlers RN, Turnbull DM (2020) Mitochondrial diseases: hope for the future. Cell 181: 168–188 [DOI] [PubMed] [Google Scholar]
- Saada A (2014) Mitochondria: mitochondrial OXPHOS (dys) function ex vivo—the use of primary fibroblasts. Int J Biochem Cell Biol 48: 60–65 [DOI] [PubMed] [Google Scholar]
- Sabharwal A, Kar B, Restrepo‐Castillo S, Holmberg SR, Mathew ND, Kendall BL, Cotter RP, Warejoncas Z, Seiler C, Nakamaru‐Ogiso E et al (2021) The FusX TALE base editor (FusXTBE) for rapid mitochondrial DNA programming of human cells in vitro and zebrafish disease models in vivo. CRISPR J 4: 799–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer PM, Scherer Alves L, Lvova M, Huang J, Rathi K, Janssen K, Butic A, Yardeni T, Morrow R, Lott M et al (2022) Combination of common mtDNA variants results in mitochondrial dysfunction and a connective tissue dysregulation. Proc Natl Acad Sci USA 119: e2212417119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholle LM, Zierz S, Mawrin C, Wickenhauser C, Urban DL (2020) Heteroplasmy and copy number in the common m.3243A>G mutation‐a post‐mortem genotype‐phenotype analysis. Genes (Basel) 11: 212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sercel AJ, Carlson NM, Patananan AN, Teitell MA (2021) Mitochondrial DNA dynamics in reprogramming to pluripotency. Trends Cell Biol 31: 311–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakiba N, Fahmy A, Jayakumaran G, McGibbon S, David L, Trcka D, Elbaz J, Puri MC, Nagy A, van der Kooy D et al (2019) Cell competition during reprogramming gives rise to dominant clones. Science 364: eaan0925 [DOI] [PubMed] [Google Scholar]
- Shi Y, Inoue H, Wu JC, Yamanaka S (2017) Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov 16: 115–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu A, Mito T, Hayashi C, Ogasawara E, Koba R, Negishi I, Takenaga K, Nakada K, Hayashi J‐I (2014) Transmitochondrial mice as models for primary prevention of diseases caused by mutation in the tRNA(Lys) gene. Proc Natl Acad Sci USA 111: 3104–3109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu A, Mito T, Hashizume O, Yonekawa H, Ishikawa K, Nakada K, Hayashi J‐I (2015) G7731A mutation in mouse mitochondrial tRNALys regulates late‐onset disorders in transmitochondrial mice. Biochem Biophys Res Commun 459: 66–70 [DOI] [PubMed] [Google Scholar]
- Shoffner JM, Wallace DC (1992) Mitochondrial genetics: principles and practice. Am J Hum Genet 51: 1179–1186 [PMC free article] [PubMed] [Google Scholar]
- Silva‐Pinheiro P, Minczuk M (2022) The potential of mitochondrial genome engineering. Nat Rev Genet 23: 199–214 [DOI] [PubMed] [Google Scholar]
- Silva‐Pinheiro P, Mutti CD, Van Haute L, Powell CA, Nash PA, Turner K, Minczuk M (2022a) A library of base editors for the precise ablation of all protein‐coding genes in the mouse mitochondrial genome. Nat Biomed Eng 10.1038/s41551-022-00968-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva‐Pinheiro P, Nash PA, Van Haute L, Mutti CD, Turner K, Minczuk M (2022b) In vivo mitochondrial base editing via adeno‐associated viral delivery to mouse post‐mitotic tissue. Nat Commun 13: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Moraes CT (2001) Manipulating mitochondrial DNA heteroplasmy by a mitochondrially targeted restriction endonuclease. Hum Mol Genet 10: 3093–3099 [DOI] [PubMed] [Google Scholar]
- St John JC (2016) Mitochondrial DNA copy number and replication in reprogramming and differentiation. Semin Cell Dev Biol 52: 93–101 [DOI] [PubMed] [Google Scholar]
- Stenton SL, Zou Y, Cheng H, Liu Z, Wang J, Shen D, Jin H, Ding C, Tang X, Sun S et al (2022) Leigh syndrome: a study of 209 patients at the Beijing Children's hospital. Ann Neurol 91: 466–482 [DOI] [PubMed] [Google Scholar]
- Stewart JB (2021) Current progress with mammalian models of mitochondrial DNA disease. J Inherit Metab Dis 44: 325–342 [DOI] [PubMed] [Google Scholar]
- Stewart JB, Chinnery PF (2015) The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat Rev Genet 16: 530–542 [DOI] [PubMed] [Google Scholar]
- Stewart JB, Chinnery PF (2021) Extreme heterogeneity of human mitochondrial DNA from organelles to populations. Nat Rev Genet 22: 106–118 [DOI] [PubMed] [Google Scholar]
- Stewart JB, Freyer C, Elson JL, Wredenberg A, Cansu Z, Trifunovic A, Larsson N‐G (2008) Strong purifying selection in transmission of mammalian mitochondrial DNA. PLoS Biol 6: e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Amato P, Sparman M, Woodward J, Sanchis DM, Ma H, Gutierrez NM, Tippner‐Hedges R, Kang E, Lee H‐S et al (2013) Towards germline gene therapy of inherited mitochondrial diseases. Nature 493: 627–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi A, Kaku Y, Ohmori T, Sharmin S, Ogawa M, Sasaki H, Nishinakamura R (2014) Redefining the in vivo origin of metanephric nephron progenitors enables generation of complex kidney structures from pluripotent stem cells. Cell Stem Cell 14: 53–67 [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126: 663–676 [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131: 861–872 [DOI] [PubMed] [Google Scholar]
- Takebe T, Sekine K, Enomura M, Koike H, Kimura M, Ogaeri T, Zhang R‐R, Ueno Y, Zheng Y‐W, Koike N et al (2013) Vascularized and functional human liver from an iPSC‐derived organ bud transplant. Nature 499: 481–484 [DOI] [PubMed] [Google Scholar]
- Tanaka M, Borgeld H‐J, Zhang J, Muramatsu S, Gong J‐S, Yoneda M, Maruyama W, Naoi M, Ibi T, Sahashi K et al (2002) Gene therapy for mitochondrial disease by delivering restriction endonuclease SmaI into mitochondria. J Biomed Sci 9: 534–541 [DOI] [PubMed] [Google Scholar]
- Tani H, Ishikawa K, Tamashiro H, Ogasawara E, Yasukawa T, Matsuda S, Shimizu A, Kang D, Hayashi J‐I, Wei F‐Y et al (2022) Aberrant RNA processing contributes to the pathogenesis of mitochondrial diseases in trans‐mitochondrial mouse model carrying mitochondrial tRNALeu(UUR) with a pathogenic A2748G mutation. Nucleic Acids Res 50: 9382–9396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanosaki S, Tohyama S, Kishino Y, Fujita J, Fukuda K (2021) Metabolism of human pluripotent stem cells and differentiated cells for regenerative therapy: a focus on cardiomyocytes. Inflamm Regen 41: 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RW, Schaefer AM, McFarland R, Maddison P, Turnbull DM (2002) A novel mitochondrial DNA tRNA(Ile) (A4267G) mutation in a sporadic patient with mitochondrial myopathy. Neuromuscul Disord 12: 659–664 [DOI] [PubMed] [Google Scholar]
- Thorburn DR, Rahman J, Rahman S (1993) Mitochondrial DNA‐associated Leigh syndrome and NARP. In GeneReviews, Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, LJH B, Gripp KW, Amemiya A (eds). Seattle, WA: University of Washington; [PubMed] [Google Scholar]
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly‐Y M, Gidlöf S, Oldfors A, Wibom R et al (2004) Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429: 417–423 [DOI] [PubMed] [Google Scholar]
- Tsang SH, Aycinena ARP, Sharma T (2018) Mitochondrial disorder: Kearns‐Sayre syndrome. Adv Exp Med Biol 1085: 161–162 [DOI] [PubMed] [Google Scholar]
- Tyynismaa H, Mjosund KP, Wanrooij S, Lappalainen I, Ylikallio E, Jalanko A, Spelbrink JN, Paetau A, Suomalainen A (2005) Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late‐onset mitochondrial disease in mice. Proc Natl Acad Sci USA 102: 17687–17692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vafai SB, Mootha VK (2012) Mitochondrial disorders as windows into an ancient organelle. Nature 491: 374–383 [DOI] [PubMed] [Google Scholar]
- Van Haute L, Spits C, Geens M, Seneca S, Sermon K (2013) Human embryonic stem cells commonly display large mitochondrial DNA deletions. Nat Biotechnol 31: 20–23 [DOI] [PubMed] [Google Scholar]
- Venco P, Bonora M, Giorgi C, Papaleo E, Iuso A, Prokisch H, Pinton P, Tiranti V (2015) Mutations of C19orf12, coding for a transmembrane glycine zipper containing mitochondrial protein, cause mis‐localization of the protein, inability to respond to oxidative stress and increased mitochondrial Ca2+ . Front Genet 6: 185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva‐Paz M, Povea‐Cabello S, Villalón‐García I, Suárez‐Rivero JM, Álvarez‐Córdoba M, de la Mata M, Talaverón‐Rey M, Jackson S, Sánchez‐Alcázar JA (2019) Pathophysiological characterization of MERRF patient‐specific induced neurons generated by direct reprogramming. Biochim Biophys Acta Mol Cell Res 1866: 861–881 [DOI] [PubMed] [Google Scholar]
- Vincent AE, Picard M (2018) Multilevel heterogeneity of mitochondrial respiratory chain deficiency. J Pathol 246: 261–265 [DOI] [PubMed] [Google Scholar]
- Wai T, Ao A, Zhang X, Cyr D, Dufort D, Shoubridge EA (2010) The role of mitochondrial DNA copy number in mammalian fertility. Biol Reprod 83: 52–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC (1999) Mitochondrial diseases in man and mouse. Science 283: 1482–1488 [DOI] [PubMed] [Google Scholar]
- Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, Elsas LJ 2nd, Nikoskelainen EK (1988) Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science 242: 1427–1430 [DOI] [PubMed] [Google Scholar]
- Wei W, Chinnery PF (2020) Inheritance of mitochondrial DNA in humans: implications for rare and common diseases. J Intern Med 287: 634–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Gaffney DJ, Chinnery PF (2021) Cell reprogramming shapes the mitochondrial DNA landscape. Nat Commun 12: 5241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Schon KR, Elgar G, Orioli A, Tanguy M, Giess A, Tischkowitz M, Caulfield MJ, Chinnery PF (2022a) Nuclear‐embedded mitochondrial DNA sequences in 66,083 human genomes. Nature 611: 105–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Li Z, Xu K, Feng H, Xie L, Li D, Zuo Z, Zhang M, Xu C, Yang H et al (2022b) Mitochondrial base editor DdCBE causes substantial DNA off‐target editing in nuclear genome of embryos. Cell Discov 8: 391–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Xu C, Feng H, Xu K, Li Z, Hu J, Zhou L, Wei Y, Zuo Z, Zuo E et al (2022c) Human cleaving embryos enable efficient mitochondrial base‐editing with DdCBE. Cell Discov 8: 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissig V (2020) Drug development for the therapy of mitochondrial diseases. Trends Mol Med 26: 40–57 [DOI] [PubMed] [Google Scholar]
- Wilkins HM, Carl SM, Swerdlow RH (2014) Cytoplasmic hybrid (cybrid) cell lines as a practical model for mitochondriopathies. Redox Biol 2: 619–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmer RA, Leopoldi A, Aichinger M, Wick N, Hantusch B, Novatchkova M, Taubenschmid J, Hämmerle M, Esk C, Bagley JA et al (2019) Human blood vessel organoids as a model of diabetic vasculopathy. Nature 565: 505–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winanto KZJ, Soh BS, Fan Y, Ng SY (2020) Organoid cultures of MELAS neural cells reveal hyperactive notch signaling that impacts neurodevelopment. Cell Death Dis 11: 182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman MJ, Mahe MM, Trisno S, Poling HM, Watson CL, Sundaram N, Chang C‐F, Schiesser J, Aubert P, Stanley EG et al (2017) Engineered human pluripotent‐stem‐cell‐derived intestinal tissues with a functional enteric nervous system. Nat Med 23: 49–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YR, Wang AG, Chen YT, Yarmishyn AA, Buddhakosai W, Yang TC, Hwang DK, Yang YP, Shen CN, Lee HC et al (2018) Bioactivity and gene expression profiles of hiPSC‐generated retinal ganglion cells in MT‐ND4 mutated Leber's hereditary optic neuropathy. Exp Cell Res 363: 299–309 [DOI] [PubMed] [Google Scholar]
- Yahata N, Matsumoto Y, Omi M, Yamamoto N, Hata R (2017) TALEN‐mediated shift of mitochondrial DNA heteroplasmy in MELAS‐iPSCs with m . 13513G > a mutation. Sci Rep 7: 15557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahata N, Boda H, Hata R (2021) Elimination of mutant mtDNA by an optimized mpTALEN restores differentiation capacities of Heteroplasmic MELAS‐iPSCs. Mol Ther Methods Clin Dev 20: 54–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Wu H, Kang X, Liang Y, Lan T, Li T, Tan T, Peng J (2018) Targeted elimination of mutant mitochondrial DNA in MELAS‐iPSCs by mitoTALENs. Protein Cell 9: 283–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y‐P, Nguyen PNN, Lin T‐C, Yarmishyn AA, Chen W‐S, Hwang D‐K, Chiou G‐Y, Lin T‐W, Chien C‐S, Tsai C‐Y et al (2019) Glutamate stimulation dysregulates AMPA receptors‐induced signal transduction pathway in Leber's inherited optic neuropathy patient‐specific hiPSC‐derived retinal ganglion cells. Cell 8: 625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T‐C, Yarmishyn AA, Yang Y‐P, Lu P‐C, Chou S‐J, Wang M‐L, Lin T‐C, Hwang D‐K, Chou Y‐B, Chen S‐J et al (2020) Mitochondrial transport mediates survival of retinal ganglion cells in affected LHON patients. Hum Mol Genet 29: 1454–1464 [DOI] [PubMed] [Google Scholar]
- Yokota M, Shitara H, Hashizume O, Ishikawa K, Nakada K, Ishii R, Taya C, Takenaga K, Yonekawa H, Hayashi J‐I (2010) Generation of trans‐mitochondrial Mito‐mice by the introduction of a pathogenic G13997A mtDNA from highly metastatic lung carcinoma cells. FEBS Lett 584: 3943–3948 [DOI] [PubMed] [Google Scholar]
- Yoneda M, Chomyn A, Martinuzzi A, Hurko O, Attardi G (1992) Marked replicative advantage of human mtDNA carrying a point mutation that causes the MELAS encephalomyopathy. Proc Natl Acad Sci USA 89: 11164–11168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MA, Larson DE, Sun C‐W, George DR, Ding L, Miller CA, Lin L, Pawlik KM, Chen K, Fan X et al (2012) Background mutations in parental cells account for most of the genetic heterogeneity of induced pluripotent stem cells. Cell Stem Cell 10: 570–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu‐Wai‐Man P, Turnbull DM, Chinnery PF (2002) Leber hereditary optic neuropathy. J Med Genet 39: 162–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu‐Wai‐Man P, Newman NJ, Carelli V, Moster ML, Biousse V, Sadun AA, Klopstock T, Vignal‐Clermont C, Sergott RC, Rudolph G et al (2020) Bilateral visual improvement with unilateral gene therapy injection for Leber hereditary optic neuropathy. Sci Transl Med 12: eaaz7423 [DOI] [PubMed] [Google Scholar]
- Zaidi AA, Wilton PR, Su MS‐W, Paul IM, Arbeithuber B, Anthony K, Nekrutenko A, Nielsen R, Makova KD (2019) Bottleneck and selection in the germline and maternal age influence transmission of mitochondrial DNA in human pedigrees. Proc Natl Acad Sci USA 116: 25172–25178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambelli F, Mertens J, Dziedzicka D, Sterckx J, Markouli C, Keller A, Tropel P, Jung L, Viville S, Van de Velde H et al (2018) Random mutagenesis, clonal events, and embryonic or somatic origin determine the mtDNA variant type and load in human pluripotent stem cells. Stem Cell Reports 11: 102–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zekonyte U, Bacman SR, Smith J, Shoop W, Pereira CV, Tomberlin G, Stewart J, Jantz D, Moraes CT (2021) Mitochondrial targeted meganuclease as a platform to eliminate mutant mtDNA in vivo. Nat Commun 12: 3210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Liu H, Luo S, Lu Z, Chávez‐Badiola A, Liu Z, Yang M, Merhi Z, Silber SJ, Munné S et al (2017) Live birth derived from oocyte spindle transfer to prevent mitochondrial disease. Reprod Biomed Online 34: 361–368 [DOI] [PubMed] [Google Scholar]
- Zheng X, Boyer L, Jin M, Kim Y, Fan W, Bardy C, Berggren T, Evans RM, Gage FH, Hunter T (2016a) Alleviation of neuronal energy deficiency by mTOR inhibition as a treatment for mitochondria‐related neurodegeneration. Elife 5: e13378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Boyer L, Jin M, Mertens J, Kim Y, Ma L, Ma L, Hamm M, Gage FH, Hunter T (2016b) Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. Elife 5: e13374 [DOI] [PMC free article] [PubMed] [Google Scholar]