

Abstract
Mechanisms underlying the depletion of NAD+ and accumulation of reactive oxygen species (ROS) in aging and age‐related disorders remain poorly defined. We show that reverse electron transfer (RET) at mitochondrial complex I, which causes increased ROS production and NAD+ to NADH conversion and thus lowered NAD+/NADH ratio, is active during aging. Genetic or pharmacological inhibition of RET decreases ROS production and increases NAD+/NADH ratio, extending the lifespan of normal flies. The lifespan‐extending effect of RET inhibition is dependent on NAD+‐dependent Sirtuin, highlighting the importance of NAD+/NADH rebalance, and on longevity‐associated Foxo and autophagy pathways. RET and RET‐induced ROS and NAD+/NADH ratio changes are prominent in human induced pluripotent stem cell (iPSC) model and fly models of Alzheimer's disease (AD). Genetic or pharmacological inhibition of RET prevents the accumulation of faulty translation products resulting from inadequate ribosome‐mediated quality control, rescues relevant disease phenotypes, and extends the lifespan of Drosophila and mouse AD models. Deregulated RET is therefore a conserved feature of aging, and inhibition of RET may open new therapeutic opportunities in the context of aging and age‐related diseases including AD.
Keywords: Alzheimer's disease, lifespan, mitochondrial complex I, NAD+/NADH ratio, reverse electron transport
Subject Categories: Metabolism, Molecular Biology of Disease
Reverse electron transport (RET) at mitochondrial complex I generates reactive oxygen species (ROS) and reduces NAD+/NADH ratio. Inhibition of RET genetically or pharmacologically extends animal lifespan and ameliorates Alzheimer's disease‐related phenotypes.
Introduction
A decline in mitochondrial activity and quality is associated with aging and a wide range of age‐related disorders (Wallace, 2005; Guarente, 2008; Cho et al, 2011; Sun et al, 2016). Along with loss of proteostasis, cellular senescence, deregulated nutrient sensing, stem cell exhaustion, epigenetic alteration, genome instability, telomere attrition, and altered intercellular communication, mitochondrial dysfunction is considered as one of the hallmarks of aging (Lopez‐Otin et al, 2013). These aging‐associated changes are not mutually exclusive, however. For example, mitochondrial dysfunction has been shown to contribute to proteostasis failure (Lu & Guo, 2020), cellular senescence (Campisi et al, 2019), and stem cell exhaustion (Zhang et al, 2018), highlighting the fundamental role of mitochondria in the aging process.
Mitochondria are the major source of ROS (Murphy, 2009), whose role in the aging process has been controversial. The free radical theory of aging proposed by Denham Harman dominated aging and longevity research for decades (Harman, 1956). However, this theory has been challenged by recent observations that while excessive ROS accumulation may be detrimental, moderately increased mitochondrial ROS can be beneficial during aging (Balaban et al, 2005), in part by triggering mitohormesis that elicits stress response and promotes mitochondrial repair and function (Ristow & Zarse, 2010; Yun & Finkel, 2014; Bar‐Ziv et al, 2020).
Mitochondria are also critical for NADH/NAD+ metabolism. During the tricarboxylic acid (TCA) cycle, NAD+ gains two electrons and a proton to be reduced to NADH. NADH is in turn oxidized to NAD+ upon donation of the electrons to NADH dehydrogenase (Complex I) of the electron transport chain (ETC). The central roles of NAD+ and NADH in TCA and ETC, respectively, underscore the importance of maintaining optimal NAD+/NADH ratio to mitochondrial function (Chini et al, 2021). Beyond this metabolic function, the biosynthesis, transport, and catabolism of NAD+ and its key intermediates critically regulate the activities of NAD+‐consuming enzymes, such as Sirtuins, poly‐ADP‐ribose polymerases (PARPs), and CD38/157 ectoenzymes, which are directly or indirectly involved in the aging process (Houtkooper et al, 2012; Imai & Guarente, 2014; Bonkowski & Sinclair, 2016; Covarrubias et al, 2021). NAD+/Sirtuin modulates longevity through the activation of mitochondrial stress responses such as the unfolded protein response (UPRmt) and the nuclear translocation and activation of Foxo (Mouchiroud et al, 2013), providing a molecular link between mitochondrial stress signaling and longevity. Although it is shown that NAD+ level declines with age (Yoshino et al, 2011; Gomes et al, 2013; Mouchiroud et al, 2013) and that replenishing NAD+ by supplementation of NAD+ precursor offers beneficial effects on aging and age‐related disorders (Rajman et al, 2018), the causes of NAD+ depletion during aging are not well understood. Deciphering the cellular mechanisms influencing NADH/NAD+ homeostasis may offer new ways to counter age‐related NAD+ decline.
During oxidative phosphorylation (OxPhos), NADH is oxidized to NAD+ at mitochondrial complex I (C‐I) and electrons are transferred from NADH to ubiquinone through several Fe‐S cluster‐containing proteins within C‐I (Hinchliffe & Sazanov, 2005; Hirst & Roessler, 2016). Under certain thermodynamic conditions, for example, when forward electron transport (FET) is blocked or when succinate accumulates to high level, RET can occur, moving electrons from ubiquinol to NAD+, producing a significant amount of ROS (RET‐ROS) in the process (Scialo et al, 2017; Onukwufor et al, 2019). Described many decades ago (Chance & Hollunger, 1961) and initially regarded as some sort of in vitro phenomenon, RET has been linked to physiological processes such as macrophage activation in response to bacterial infection (Mills et al, 2016). However, excessive RET‐ROS is implicated in ischemia–reperfusion injury (Chouchani et al, 2014; Brand et al, 2016). Another outcome of RET is the conversion of NAD+ to NADH, thereby decreasing NAD+ and lowering NAD+/NADH ratio, which has been shown to promote noncanonical Notch signaling in cancer stem cell growth and brain tumor settings (Ojha et al, 2022). Whether RET‐induced NAD+/NADH imbalance contributes to aging and age‐related neurodegenerative disease is not known.
Here, we show that RET is active during aging, contributing to age‐associated ROS accumulation and depressed NAD+/NADH ratio. Genetic or pharmacological inhibition of RET extended the lifespan of normal Drosophila. This lifespan‐extending effect of RET inhibition could be recapitulated by NAD+ precursor nicotinamide mononucleotide (NMN) supplementation and depended on the NAD+/Sirtuin, Foxo, and autophagy pathways, pathways intimated linked to aging (Riera et al, 2016), supporting that RET‐induced NAD+/NADH dyshomeostasis contributes to the normal aging process. We also found that genetic or pharmacological inhibition of RET effectively extended the lifespan and rescued disease phenotypes of Drosophila and mouse models of AD. Our results identify RET as a previously unrecognized mechanism by which mitochondrial dysfunction leads to hallmarks of aging and contributes to age‐related neurodegenerative disease and further suggest that rebalancing NAD+/NADH through inhibition of RET may be exploited for therapeutic purposes in the context of aging and age‐related diseases.
Results
RET is active during Drosophila aging
Increased ROS and decreased NAD+ levels are frequently observed in aging and age‐related diseases (Hur et al, 2014), but whether these phenomena are connected is not known. We examined brain samples of flies aged at 7 days (young), 30 days (middle aged), and 45 days (old), and observed significantly increased H2O2 as measured with the CM‐H2DCFDA dye in brain samples of aged flies, and a more moderate increase of H2O2 in middle‐aged flies, compared with young flies (Fig 1A and B). Staining with the mito‐SOX dye revealed increased mitochondrial ROS in aged brains (Fig EV1A and B). We also observed an age‐dependent gradual decrease in NAD+/NADH ratio (Fig 1C), consistent with age‐related decline of NAD+ observed in other species (Yoshino et al, 2011; Gomes et al, 2013; Mouchiroud et al, 2013).
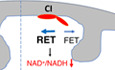
Figure 1. Active RET leads to increased ROS and decreased NAD+/NADH ratio in aged flies.
-
A, BCM‐H2DCFDA staining (A) and data quantification (B) showing H2O2 level in different aged fly brains with and without treatment with the indicated RET inhibitors (n = 5 per group).
-
CQuantification of NAD+/NADH ratio in different aged flies with and without CPT treatment (n = 3 groups, 20 flies per group).
-
DBN‐PAGE showing mitochondrial complex and supercomplex assembly in 10‐day, 35‐day and 45‐day‐old flies.
-
ETMRM staining assessing mitochondrial MMP in different aged flies and in old flies treated with CPT (n = 3).
-
FQuantification of mitochondrial succinate level in different aged flies and in aged flies treated with CPT.
-
G, HQuantification of RET‐ROS in mitochondria isolated from different aged flies and respiring under FET (G) or RET (H) condition (n = 4).
-
IDose‐dependent effect of CPT on RET‐ROS in mitochondria isolated from 45‐day‐old flies and respiring under RET condition (n = 4).
-
JQuantification of the effect of CPT on NAD+/NADH ratio in mitochondria isolated from 45‐day‐old flies and respiring under RET condition (n = 4).
-
KCM‐H2DCFDA staining showing H2O2 level in young flies treated with DES or DES + CPT (n = 5 per group).
-
LLive imaging with SoNar showing NAD+ (green) and NADH (blue) levels in different aged fly brains with or without treatment with the indicated chemicals.
Data information: Data are representative of at least three repeats. Data are shown as mean ± SEM. Asterisks indicate statistical significance (**P < 0.01, *P < 0.05) using single‐factor ANOVA with Scheffe's analysis as a post hoc test to compare two sets of data.
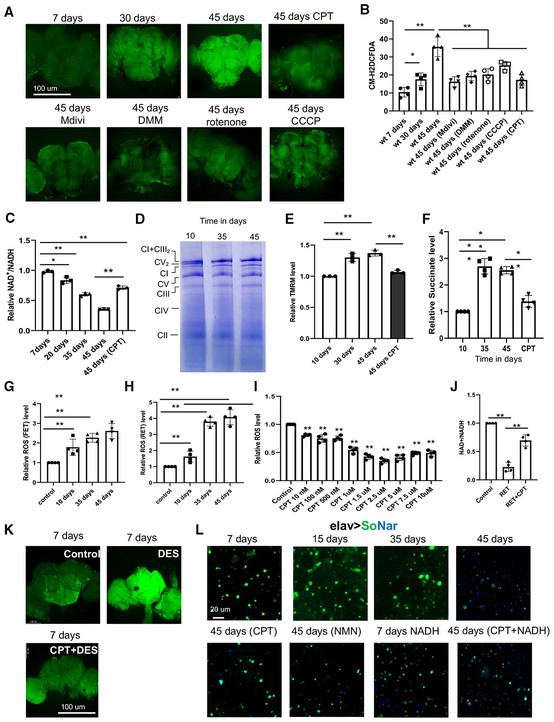
Figure EV1. Active RET leads to increased ROS and decreased NAD+/NADH ratio in aged flies.
-
A, BMito‐Sox staining (A) and data quantification (B) showing mitochondrial ROS level in different aged fly brains with or without treatment with the indicated RET inhibitors (n = 5 per group).
-
CQuantification of NAD+/NADH ratio in young flies, old flies, and old flies treated with the indicated chemicals.
-
DWestern blot analysis comparing NDUFS3 level in young and aged flies. **P < 0.01 in Student's t‐test.
-
EQuantification of FET‐ROS in isolated mitochondria purified from aged wild‐type flies and assayed under FET condition without or with treatment at the indicated concentrations of CPT (n = 4).
-
FQuantification of NAD+/NADH ratio in isolated mitochondria after induction of FET and with or without CPT (2.5 μM) treatment (n = 4).
-
GCo‐immunoprecipitation assay showing the effect of aging and CPT treatment on the various protein–protein interactions between C‐I proteins involved in RET.
-
HQuantification of ROS level from brain samples of DES and DES + CPT‐treated flies (n = 4).
-
IQuantification of NAD+/NADH ratio from brain samples of DES and DES + CPT‐treated flies (n = 5 sets, 20 flies per set).
-
JQuantification of NAD+ and NADH levels measured with SoNar in different aged fly brains with or without treatment with the indicated chemicals as shown in Fig 1D (n = 4 sets, 5 flies per set).
-
KQuantification of NAD+/NADH ratio in wild‐type flies treated with CPT or co‐treated with CPT and NADH (n = 4 sets, 20 flies per set).
-
LMitochondrial C‐I activity assay using mitochondria purified from different aged flies (n = 3).
-
MIn‐gel activity assays of respiratory complexes using mitochondria purified from different aged flies.
Data information: Data are representative of at least three repeats. Data are shown as mean ± SEM. Asterisks indicate statistical significance (**P < 0.01) in single‐factor ANOVA with Scheffe's analysis as a post hoc test.
We wondered whether RET might be the mechanism responsible for the observed ROS and NAD+/NADH ratio changes, as RET can simultaneously increase mito‐ROS and decrease NAD+/NADH ratio (Ojha et al, 2022). To test this possibility, we treated flies with the small molecule 6‐chloro‐3‐(2,4‐dichloro‐5‐methoxyphenyl)‐2‐mecapto‐7‐methoxyquinazolin‐4(3H)‐one (CPT2008 or CPT), which acts as an RET inhibitor by binding to complex I (C‐I) 30 kD subunit (C‐I30 or NDUFS3) and altering its interaction with other proteins in the soluble matrix arm of C‐I involved in electron transfer (Ojha et al, 2022). Both the increased ROS production and depressed NAD+/NADH ratio in aged flies were rescued by CPT treatment (Fig 1A and B), supporting that RET is a major mechanism accounting for the age‐related ROS and NAD+/NADH ratio changes. To further test the involvement of RET in conferring the aging‐associated ROS and NAD+/NADH ratio changes, we used mDivi‐1, a reversible C‐I inhibitor and RET inhibitor (Bordt et al, 2017), and rotenone, a C‐I inhibitor that preferentially inhibits RET at lower concentrations (Cino & Del Maestro, 1989; Liu et al, 2002). Both mDivi‐1 and rotenone mimicked CPT in rescuing age‐related ROS and NAD+/NADH ratio changes, albeit at lesser efficiencies than CPT (Fig 1A and B). We also treated flies with compounds that disrupt two of the driving forces of RET, mitochondrial membrane potential (MMP) and ubiquinol (CoQH2). CCCP was used to depolarize MMP, and dimethyl malonate (DMM), a competitive inhibitor of succinate dehydrogenase was used to lessen CoQH2 (Chouchani et al, 2014; Mills et al, 2016). Both CCCP and DMM rescued age‐related ROS (Fig 1A and B) and NAD+/NADH ratio (Fig EV1C) changes.
We further studied mechanisms underlying aging‐related RET changes. We first characterized complex I assembly using blue native polyacrylamide gel electrophoresis (BN‐PAGE) on 10‐day‐, 35‐day‐, and 45‐day‐old flies. We found that supercomplex (C‐I+C‐III2) assembly observed in young flies was reduced with old age (Fig 1D). Correspondingly, holo‐complex I level was increased in old animals (Fig 1D), which was also confirmed by examining the level of C‐I protein Ndufs3 (Fig EV1D), although C‐V level was not significantly changed. Not that our assignment of the identity of C‐I and supercomplex bands on BN‐PAGE is consistent with that previously reported for fly mitochondrial complexes (Murari et al, 2020). As supercomplex formation limits ROS production from C‐I (Maranzana et al, 2013), presumably because supercomplex is not conducive to RET due to the lack of C‐II, this offered one mechanism of age‐associated RET change. We further examined other factors that could affect RET and found significantly increased MMP in aged fly samples (Fig 1E). Moreover, mitochondrial succinate level was also increased in old flies (Fig 1F). Both were rescued by CPT treatment. Although the mechanisms of MMP and succinate change remain to be determined, because both can serve as drivers of RET, their changes offered another mechanism of age‐associated RET increase. To more directly measure age‐related RET change, we used mitochondrial purified from fly tissues in in vitro assays by supplying mitochondrial complex I with RET (succinate) and FET (malate + glutamate) substrates. We found that while FET‐ROS was modestly increased with age, RET‐ROS was increased dramatically in old age (Fig 1G and H). Using this in vitro assay, we analyzed the effect of CPT on RET and FET. We found that CPT decreased RET‐ROS in a dose‐dependent manner (Fig 1I), with 2.5 μM CPT being an optimal dose to decrease RET‐ROS in purified mitochondrial samples (Fig 1I), while no such effect was observed for FET‐ROS (Fig EV1E), supporting that CPT preferentially inhibits RET. The mild reduction of FET‐ROS by CPT could be due to an antioxidant property of the ‐SH group in CPT. The amelioration of RET‐induced NAD+/NADH ratio change by CPT in purified mitochondria (Fig 1J), and the lack of effect of CPT on NAD+/NADH ratio under FET condition (Fig EV1F), also supported the RET inhibitor activity of CPT. Our previous study demonstrated that RET is associated with altered protein–protein interactions within C‐I (Ojha et al, 2022). We found that NDUFV1 interaction with NDUFS3 was decreased with age while NDUFV2 interaction with NDUFS3 was increased. Treatment with CPT lessened NDUFS3 interaction with NDUFV2 and Notch but strengthened NDUFS3 interaction with NDUFV1 (Fig EV1G). Finally, we tested whether we could induce age‐related changes by activating RET. Treatment of young flies with diethylsuccinate (DES), a cell‐permeable form of the complex II substrate succinate that can drive RET (Mills et al, 2016; Ojha et al, 2022), efficiently induced ROS and decreased NAD+/NADH, effects that were inhibited by CPT (Figs 1K and EV1H and I). Together, these data demonstrate that multiple mechanisms can contribute to age‐related RET increase and that CPT serves as a specific RET inhibitor to block aging‐associated RET‐ROS and NAD+/NADH changes.
To further examine the effect of aging on NAD+/NADH levels, we used flies expressing a genetically encoded NAD+/NADH reporter SoNar (Bonnay et al, 2020). SoNar fluorescence is intrinsically ratiometric with two excitation wavelengths having opposing responses to NAD+ and NADH, allowing quantitative determination of NAD+ or NADH ratiometrically (Zhao et al, 2015). With this approach, we observed a gradual drop in NAD+/NADH ratio with age, and CPT treatment significantly restored NAD+/NADH ratio in aged flies (Figs 1L and EV1J). As controls, we showed that feeding flies with the NAD+ precursor nicotinamide mononucleotide (NMN) had similar effect as CPT in restoring NAD+/NADH ratio (Figs 1L and EV1J), and the effect of CPT on the SoNar reporter was dampened by NADH co‐treatment (Figs 1L and EV1K). We also measured C‐I activities using two approaches: (i) In vitro C‐I activity assay using malate and glutamate as substrates (Fig EV1L) and (ii) In‐gel C‐I activity assay (Fig EV1M). In both assays, we did not find a significant change in C‐I activity during fly aging. In‐gel activity assays of C‐II, C‐III, and C‐IV also did not reveal significant changes (Fig EV1M). These data strongly support that RET is active during aging and contributes to age‐related ROS increase and NAD+/NADH ratio decrease.
Inhibition of RET from midlife onwards extends Drosophila lifespan
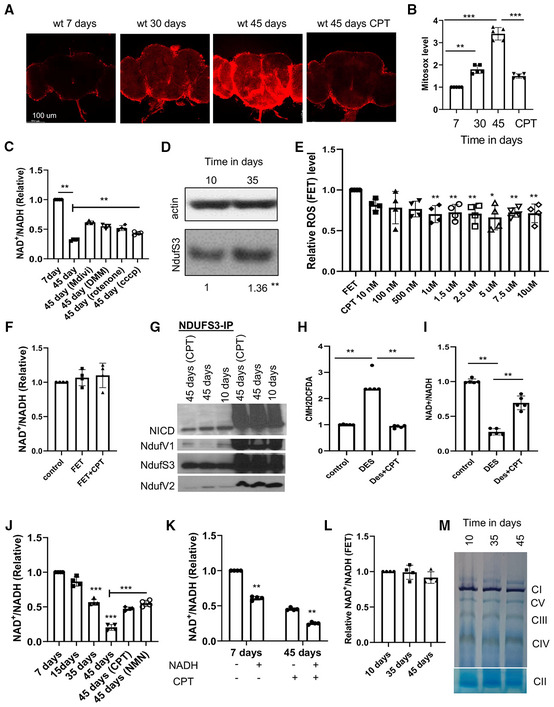
Given the effect of CPT on ROS and NAD+/NADH metabolism, two key parameters associated with aging, we tested the effect of CPT treatment on fly lifespan. Consistent with previous finding that genetic manipulations of mitochondria‐related functions throughout the adulthood might not have obvious impact on longevity, whereas initiating the same manipulations from midlife onwards could significantly extend lifespan (Rana et al, 2017), we found that continuous feeding of newly eclosed flies with CPT did not have obvious effect on lifespan (Fig EV2A), whereas treating flies with CPT starting from midlife (days 20, 35, and 45) onwards resulted in significant extension of median and maximal lifespans in both males (Fig 2A–D) and females (Fig EV2B and C), except day 45 females (Fig EV2D). We also found that 10 or 20 μM CPT produced robust lifespan extension, whereas 5 or 40 μM CPT failed to show obvious effect (Figs 2E–H and EV2E). As diet restriction could extend lifespan, we tested whether CPT might affect food intake. Using food mixed with the Brilliant Blue dye to follow intake, we found CPT did not affect feeding (Fig EV2F). Because midlife 10 μM CPT treatment resulted in pronounced lifespan extension while consuming less drug, this regime was used for further studies.
Figure EV2. Inhibition of RET by CPT treatment extends Drosophila lifespan.
-
ASurvival curves comparing control flies and flies fed with food containing 10 μM CPT from day 1 onwards.
-
B–DSurvival curves comparing overall survival (by logrank test) and maximum lifespan (by Wang–Allison test) of control flies and flies fed with food containing 10 μM CPT from 20 days (**P < 0.001 logrank, **P < 0.001 Wang–Allison) (B), 35 days (**P < 0.001 logrank, **P < 0.001 Wang–Allison) (C), and 45 days (ns) (D) onwards (n = 3 groups, 20–25 flies per group).
-
ESurvival curves comparing control flies and flies fed with food containing 40 μM CPT from 35 days onwards (ns; n = 3 groups, 20–25 flies per group).
-
FQuantification of food consumption in control and CPT‐fed flies (n = 4 groups, 20 per group).
Data information: Data are representative of at least three repeats. Data are shown as mean ± SEM in (F). Survival curves were analyzed with logrank test along with Wang–Allison test for maximum lifespan. Dashed lines mark time points of 50% survival. Asterisks indicate statistical significance (**P < 0.01, ns: not significant) in single‐factor ANOVA with Scheffe's analysis as a post hoc test.
Figure 2. Inhibition of RET by CPT treatment extends Drosophila lifespan.
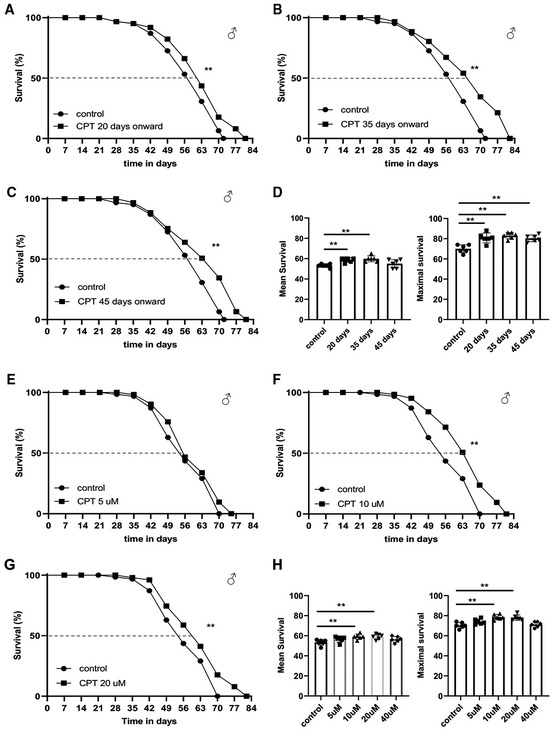
-
A–DSurvival curves comparing overall survival (by logrank test) and maximum lifespan (by Wang–Allison test) of control flies and flies fed with food containing 10 μM CPT from 20 days (**P < 0.01 logrank, **P < 0.01 Wang–Allison) (A), 30 days (**P < 0.01 logrank, **P < 0.01 Wang–Allison) (B), or 45 days (**P < 0.01 logrank, **P < 0.01 Wang–Allison) (C) onwards. Bar graphs (D) show the quantification of mean and maximal lifespan (n = 3 groups, 20–25 flies per group).
-
E–HSurvival curves comparing control flies and flies fed with food containing 5 μM CPT (E), 10 μM CPT (**P < 0.01 logrank, **P < 0.01 Wang–Allison) (F), or 20 μM CPT (**P < 0.01 logrank, **P < 0.01 Wang–Allison) (G) from 35 days onwards. Bar graphs (H) show the quantification of mean and maximal lifespan (n = 3 groups, 20–25 flies per group), using single‐factor ANOVA with Scheffe's analysis as a post hoc test.
Data information: Data are representative of at least three repeats. (D, H) Data are shown as mean ± SEM. Survival curves were analyzed with logrank test along with Wang–Allison test for maximum lifespan. Asterisks indicate statistical significance (**P < 0.01, *P < 0.05). Dashed lines mark time points of 50% survival.
RET inhibition maintains proteostasis, improves mitochondrial function, and extends healthspan in aged flies
To better understand the effect of CPT on organismal health, we examined parameters of health span. We observed age‐related decline in locomotor activity as measured with the climbing assay, and this deficit was significantly rescued by CPT (Fig 3A). Loss of intestinal barrier function is a hallmark of aging (Rera et al, 2011). To determine the impact of CPT on intestinal barrier integrity, we used the “Smurf” assay that monitors the presence of nonabsorbed dyes outside of the digestive tract after feeding (Rera et al, 2011). This assay revealed improved intestinal barrier integrity after 10 days of CPT treatment (Fig 3B; Appendix Fig S1A).
Figure 3. Effect of RET inhibition by CPT on healthspan in Drosophila .
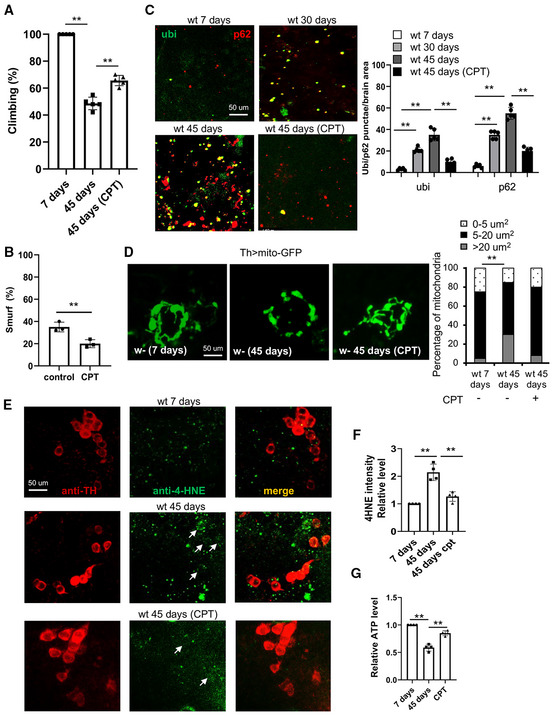
-
AClimbing assay of young flies, old flies, and old flies treated with CPT (n = 3 groups, 20 per group).
-
BSmurf assay of intestinal integrity in aged flies with or without CPT treatment (n = 3 groups, 20 per group).
-
CImages and data quantification showing accumulation of protein aggregates in the brains of aged flies with or without CPT treatment (n = 5).
-
DImages and data quantification showing DA neuron mitochondrial morphology in the brains of aged flies with or without CPT treatment (n = 5).
-
E, FImages (E) and data quantification (F) showing 4‐HNE levels within or near TH‐positive DA neurons in young flies, old flies, and old flies treated with CPT (n = 4 sets, 5 samples per set). Arrows point to HNE signals co‐labeled with TH‐ positive DA neurons.
-
GQuantification of ATP levels in young flies, old flies, and old flies treated with CPT.
Data information: Data are representative of at least three repeats. Data are shown as mean ± SEM. Asterisks indicate statistical significance (**P < 0.01) in single‐factor ANOVA with Scheffe's analysis as a post hoc test.
Proteostasis failure and mitochondrial dysfunction are cellular hallmarks of aging. To test whether CPT treatment affected these features, we examined brain and indirect flight muscle tissues of control and CPT‐treated flies. Aged fly brains accumulated ubiquitin‐positive protein aggregates, indicating proteostasis failure (Fig 3C). Treatment with CPT from midlife onwards remarkably reduced the size and number of ubiquitin‐positive protein aggregates (Fig 3C). Similar result was seen in flight muscle (Appendix Fig S1B). However, we did not observe a significant effect of CPT treatment on global ubiquitination level (Appendix Fig S1C), suggesting that CPT may affect proteostasis through other mechanisms, or that the ubiquitination of only a small subset of proteins is affected that is beyond our current detection.
Mitochondrial function is intimately linked to morphological dynamics regulated by fission and fusion. Previous studies in fly muscle revealed decreased fission/increased fusion with age and that upregulation of fission from midlife onwards was sufficient to extend fly lifespan (Rana et al, 2017). Consistent with this observation, we found that mitochondria labeled with a mito‐GFP reporter in dopaminergic neurons (Fig 3D), or immunostained with anti‐ATP5α in flight muscle (Appendix Fig S1D), were more fused and aggregated in aged flies and that CPT treatment resulted in smaller and less aggregated mitochondria, suggesting that CPT promoted fission. No significant change in mitochondrial mass was observed (Appendix Fig S1H). As we observed increased RET‐ROS with age, and ROS‐induced oxidative damage is another hallmark of aging, we assessed the effect of CPT on ROS‐induced oxidative damage to lipids (lipid peroxidation) using the anti‐4‐Hydroxynonenal (HNE) antibody. Treatment with 10 μM CPT for 10 days significantly reduced age‐associated lipid peroxidation in the brain (Fig 3E and F). Finally, consistent with mitochondrial function being improved by CPT, the decline of ATP production in aged fly muscle was ameliorated by CPT treatment (Fig 3G), supporting that CPT does not significantly inhibit FET or OxPhos.
Knockdown of NDUFS3 or supplementation of NAD + precursor mimics the effect of pharmacological RET inhibition on lifespan extension
To further support the idea that inhibition of RET is the mechanism responsible for lifespan extension by CPT, we sought a genetic approach. Previous studies identified NDUFS3 (C‐I30) and NDUFV1 of C‐I as potential binding targets of CPT (Ojha et al, 2022). We tested whether inhibition of RET by RNAi of NDUFS3, NDUFV1, or a closely associated NDUFS2, might mimic the effect of CPT. We performed tissue‐specific knockdown of NDUFS2, NDUFS3, and NDUFV1 in muscle or neuronal tissues. We observed significant extension of lifespan when NDUFS3 and NDUFS2 were knocked down in muscle using the Mhc‐GAL4 (Fig 4A and B), with the effect of NDUFS3 seemingly stronger than NDUFS2. However, the effect was not as prominent with neuronal knockdown using the elav‐GAL4 (Fig EV3A and B). By contrast, reduced lifespan was observed when NDUFV1 was knocked down (Fig 4C). Correlating with the lifespan effects, both NDUFS3‐ and NDUFS2‐RNAi attenuated age‐related ROS elevation (Fig 4D) and NAD+/NADH decline (Fig 4E), indicating that they inhibited RET and thus mimicked the CPT effect. Although ROS level reduction could be caused by a reduction of mitochondrial mass, no obvious change in mitochondrial mass by NDUFS3‐ or NDUFS2‐RNAi was observed (Appendix Fig S1E). On the contrary, NDUFV1‐RNAi further reduced NAD+/NADH ratio (Fig 4E). Given that NDUFV1 is the only subunit in C‐I that accepts NADH and initiates FET, its knockdown might have reduced NADH entry into the ETC, resulting in NADH accumulation and NAD+ shortage and explaining its lifespan effect. We also performed qRT–PCR analysis to assess knockdown efficiency. NDUFS2 and NDUFS3 mRNA levels were reduced by 55 and 59%, respectively, whereas NDUFV1 was reduced by 85% (Appendix Fig S1F). NDUFS3 protein knockdown was confirmed with antibody that is available (Appendix Fig S1G). Thus, another possibility is that mild knockdown of C‐I subunits involved in RET is beneficial, whereas excessive knockdown as in NDUFV1 RNAi is detrimental. Further studies are needed to test this possibility.
Figure 4. Inhibition of RET by genetic manipulation of C‐I subunits extends fly lifespan.
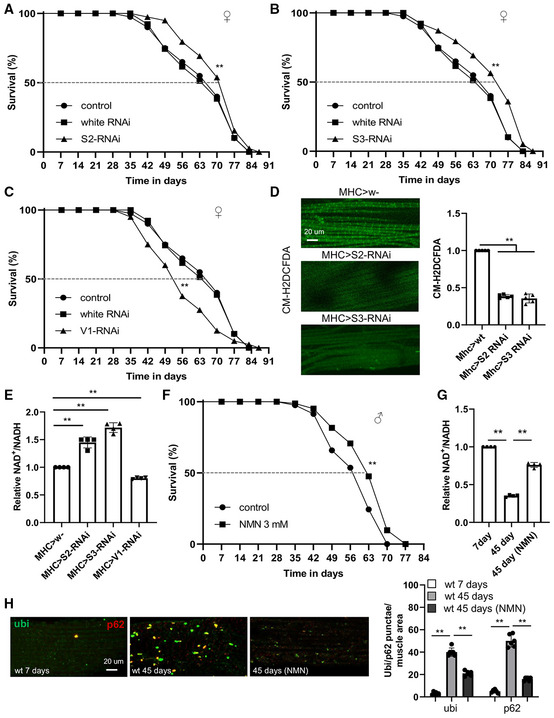
-
A–CSurvival curves of control flies and flies with muscle‐specific knockdown of NDUFS2 (**P < 0.01 logrank) (A), NDUFS3 (**P < 0.01 logrank, **P < 0.01 Wang–Allison) (B), or NDUFV1 (**P < 0.01 logrank) (C) (n = 3 groups, 20–25 flies per group).
-
DCM‐H2DCFDA staining of H2O2 and quantification of signal intensity in the muscle tissues of 50‐day‐old control flies and flies with NDUFS2 or NDUFS3 knockdown (n = 5 per group).
-
EQuantification of NAD+/NADH in 50‐day‐old control flies and flies with NDUFS2, NDUFS3, or NDUFV1 knocked down by RNAi.
-
FSurvival curves of wild‐type flies with or without NMN treatment from 35 days onwards (**P < 0.01 logrank, **P < 0.01 Wang–Allison) (n = 3 groups, 20–25 flies per group).
-
GQuantification of NAD+/NADH in young flies, old flies, and old flies treated with CPT.
-
HImages and data quantification showing accumulation of protein aggregates in the muscle of young flies, old flies, and old flies treated with CPT (n = 5 per group).
Data information: Data are representative of at least three repeats. (D, E, G, H) Data are shown as mean ± SEM. Survival curves were analyzed with logrank test along with Wang–Allison test for maximum lifespan. Asterisks indicate statistical significance (**P < 0.01) in single‐factor ANOVA with Scheffe's analysis as a post hoc test. Dashed lines mark time points of 50% survival.
Figure EV3. Inhibition of RET by genetic manipulation of C‐I subunits extends fly lifespan.
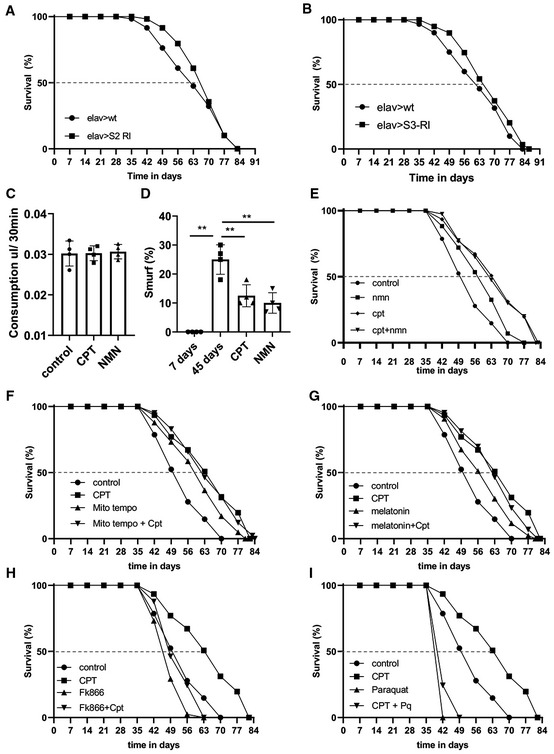
-
A, BSurvival curves of control flies and flies with NDUFS2 (A), or NDUFS3 (B) knocked down in neurons (n = 3 groups, 20–25 flies per group).
-
CQuantification of food consumption in flies treated with CPT or NMN.
-
DQuantification of Smurf assay data in young flies, old flies, and old flies treated with CPT and NMN (n = 3 groups, 20 flies per group).
-
E–ISurvival curves showing the lifespan effect of CPT in combination with NMN (E) (control vs NMN: **P < 0.01 logrank, **P < 0.01 Wang Alisson; CPT vs CPT + NMN, ns), mito‐Tempo (F) (control vs mito‐Tempo: **P < 0.01 logrank, **P < 0.01 Wang Alisson; CPT vs CPT + mito‐Tempo: ns), melatonin (G) (control vs melatonin: **P < 0.01 logrank, **P < 0.01 Wang Alisson; CPT vs CPT+ melatonin: ns), FK866 (H) (control vs FK866: **P < 0.01 logrank, **P < 0.01 Wang Alisson; FK866 vs FK866 + CPT: ns), and paraquat (I) (control vs paraquat: **P < 0.01 logrank, **P < 0.01 Wang Alisson; paraquat vs paraquat+CPT: ns) (n = 3 groups, 20 flies per group).
Data information: Data are representative of at least three repeats. Data are shown as mean ± SEM in (C, D). Survival curves were analyzed with logrank test along with Wang–Allison test for maximum lifespan. Dashed lines mark time points of 50% survival. Asterisks indicate statistical significance (**P < 0.01) in single‐factor ANOVA with Scheffe's analysis as a post hoc test.
To further test the idea that inhibition of RET‐induced NAD+/NADH ratio change is the mechanistic basis of the observed lifespan extension by CPT, we treated flies with the NAD+ precursor NMN. Wild‐type flies treated with 3 mM NMN from 35 days onwards extended their lifespan to similar extent as with CPT (Fig 4F), without obvious effect on food intake (Fig EV3C). This was correlated with the restoration of NAD+/NADH ratio reduced by aging (Fig 4G). Moreover, similar as CPT, NMN also restored proteostasis in aged fly muscle (Fig 4H) and maintained intestinal integrity (Fig EV3D). These data support that RET‐induced NAD+/NADH ratio change is a major contributor to aging which is antagonized by CPT.
We further tested the contribution of increased NAD+/NADH ratio and reduced mito‐ROS to the antiaging effect of CPT by evaluating CPT behavior in combination with other drugs, such as oxidants/antioxidant and NMN. When flies were fed both CPT and NMN, their lifespan was similar as that provided by CPT (Fig EV3E), suggesting that CPT and NMN act through similar mechanisms to extend lifespan. Interestingly, flies fed with mito‐Tempo, a mitochondrial‐targeted antioxidant (Jiang et al, 2009), exhibited increases lifespan, suggesting that mito‐ROS is generally detrimental to fly lifespan. However, mito‐Tempo and CPT co‐treatment did not provide additional lifespan benefit than CPT alone (Fig EV3F). Similarly, we found that the antioxidant melatonin also provided lifespan benefit. However, in combination with CPT it did not provide additional benefit (Fig EV3G). On the contrary, treatment with FK866, which inhibits NAD+ synthesis and blocks the anticancer effect of CPT (Ojha et al, 2022), abolished the lifespan benefit conferred by CPT (Fig EV3H), and CPT failed to provide protection against the lifespan‐shortening effect of the oxidant paraquat (Fig EV3I). Together, these data support that the effect of CPT on NAD+/NADH critically mediates its lifespan effect, although its effect on mito‐ROS may also contribute, given the detrimental effect of mito‐ROS on lifespan as revealed by mito‐Tempo.
Lifespan extension by RET inhibition is dependent on Sirtuin, Foxo, and autophagy pathways
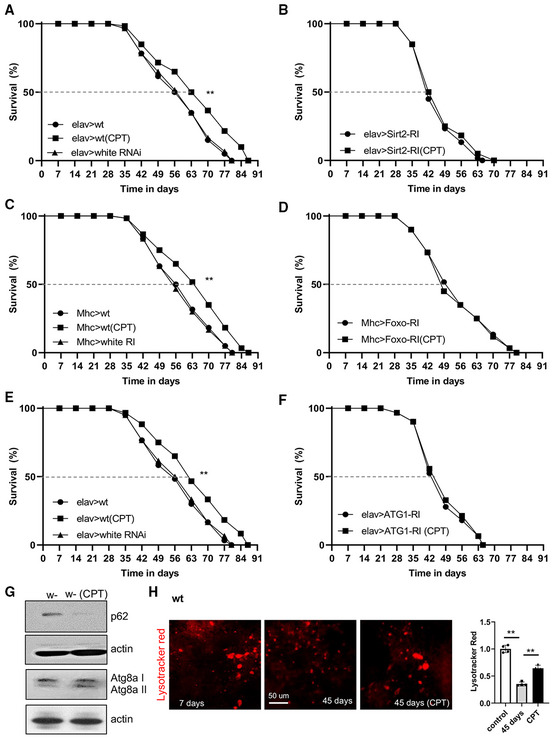
We further probed the signaling mechanisms by which inhibition of RET extends lifespan. Our results so far strongly supported the role of RET‐associated NAD+/NADH homeostasis in mediating the CPT effect. Given the intricate relationship between NAD+ and Sirtuins, with Sirtuins being NAD+‐consuming enzymes and NAD+ a coenzyme needed for Sirtuin activity, we examined whether NAD+/Sirtuin signaling might mediate the CPT effect on lifespan. The Drosophila genome encodes 5 Sirtuins (dSirt1, 2, 4, 6, 7). Although dSirt1, which is homologous to Sir2 in yeast, has been most extensively studied, its role in lifespan regulation is still controversial (Newman et al, 2002; Wood et al, 2004; Burnett et al, 2011). Previous studies indicated that dSirt2 might be involved in longevity, as its knockdown decreased fly lifespan (Kusama et al, 2006). We wondered whether dSirt2 might mediate the CPT effect on lifespan. Neuronal knockdown of dSirt2 shortened lifespan (Fig 5A and B), consistent with the previously reported role of dSirt2 (Kusama et al, 2006). Importantly, the lifespan‐extension effect of CPT seen in WT flies was diminished by dSirt2‐RNAi (Fig 5A and B), suggesting that dSIRT2 is required for CPT to extend lifespan. However, we cannot exclude potential roles of other Sirtuins in mediating the effect of CPT.
Figure 5. Lifespan effect of RET inhibition by CPT is mediated by Sirtuin, Foxo, and autophagy pathways.
-
A, BSurvival curves showing the effect of neuronal dSirt2 knockdown on fly lifespan under normal and CPT treatment conditions (n = 3 groups, 20–25 flies per group).
-
C, DSurvival curves showing the effect of muscle dFoxo knockdown on fly lifespan under normal and CPT treatment conditions (n = 3 groups, 20–25 flies per group).
-
E, FSurvival curves showing the effect of neuronal ATG1 knockdown on fly lifespan under normal and CPT treatment conditions (n = 3 groups, 20–25 flies per group).
-
GImmunoblots showing the effect of CPT on p62, ATG8a‐I, and ATG8a‐II levels in fly muscle.
-
HImages and data quantification in single‐factor ANOVA with Scheffe's analysis as a post hoc test showing lysotracker staining in the muscle of young flies, old flies, and old flies treated with CPT (n = 5 per group).
Data information: Data are representative of at least three repeats. Data are shown as mean ± SEM (H). Survival curves were analyzed with logrank test along with Wang–Allison test for maximum lifespan. Asterisks indicate statistical significance (**P < 0.01). Dashed lines mark time points of 50% survival.
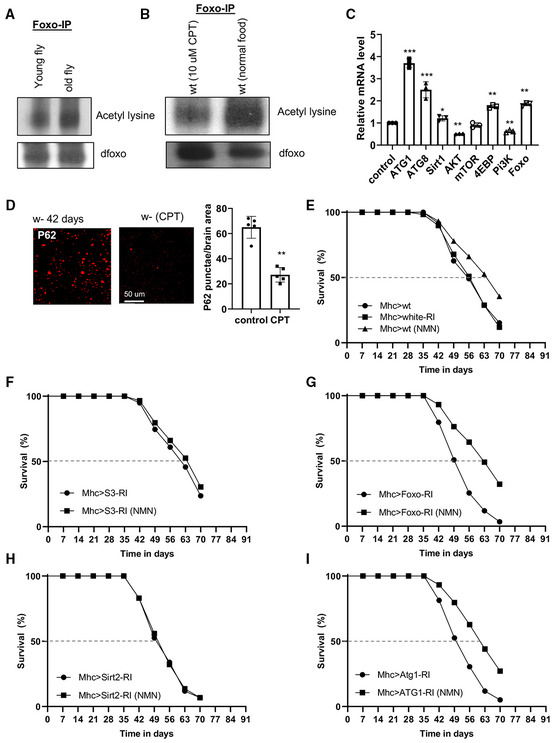
Sirtuins regulate cellular metabolism, stress response, and longevity, likely through specific substrates. The Foxo transcription factor is a substrate of Sirtuin (Brunet et al, 2004) and a mediator of the lifespan effect of NAD+ supplementation (Mouchiroud et al, 2013). We found that dFoxo‐RNAi diminished the lifespan extension by CPT in WT flies (Fig 5C and D). At the biochemical level, Sirtuin activity as measured with dFoxo deacetylation was reduced in aged flies (Fig EV4A), and CPT significantly decreased levels of deacetylated dFoxo (Fig EV4B), consistent with CPT boosting Sirtuin activity toward Foxo, presumably through NAD+ regulation.
Figure EV4. Lifespan effect of RET inhibition by CPT is mediated by Sirtuin, Foxo, and autophagy pathways.
-
A, BImmunoblots showing the effect of aging on dFoxo acetylation (A), and effect of CPT treatment on dFoxo acetylation (B). dFoxo was immunoprecipitated from whole fly extracts and detected with anti‐acetyl‐K antibody.
-
CRT–PCR analysis of levels of the indicated transcripts in aged flies relative to young flies.
-
DImages and data quantification showing p62 staining in the brain of old flies and old flies treated with CPT (n = 5 per group).
-
E–ISurvival curve showing the effect of NMN on lifespan in control (Mhc > wt) flies (**P < 0.01, logrank) (E), and NDUFS3‐RNAi (F), Foxo‐RNAi (**P < 0.01, logrank) (G), Sirt‐2‐RNAi (H), and ATG1‐RNAi (**P < 0.01, logrank) (I) flies (n = 3 groups, 20 flies per group).
Data information: Data are representative of at least three repeats. Data are shown as mean ± SEM in (C, D). Survival curves were analyzed with logrank test along with Wang–Allison test for maximum lifespan. Dashed lines mark time points of 50% survival. Asterisks indicate statistical significance (***P < 0.001, **P < 0.01, *P < 0.05) in single‐factor ANOVA with Scheffe's analysis as a post hoc test.
We further investigated signaling mechanisms downstream of Sirtuins and Foxo that mediate the effect of CPT on lifespan. Sirtuins and Foxo can activate the autophagy pathway (Hariharan et al, 2010; Ng & Tang, 2013), an aging‐relevant process in which defective proteins or organelles are engulfed into vesicles which fuse with lysosomes to form autophagosomes, leading to cargo degradation. Our RT–PCR analysis showed that autophagy genes ATG1 and ATG8 were upregulated by CPT. dSirt1, dFoxo, and 4EBP, a transcriptional target of dFoxo, were also upregulated, whereas AKT and PI3K were downregulated (Fig EV4C). We next tested whether autophagy was required for the lifespan extension by CPT. Intriguingly, knockdown of the key autophagy regulator ATG1 blocked the CPT effect on lifespan extension (Fig 5E and F). The knockdown efficiency of ATG1, dSirt2, and Foxo by the transgenes used in this study was 72, 70, and 76%, respectively (Appendix Fig S1F). ATG1 and Foxo protein knockdown was confirmed with antibodies that are available (Appendix Fig S1G).
Autophagy receptors play important roles in the autophagy process by linking autophagy cargos with the autophagy machinery through LC3. Drosophila has only one autophagy receptor p62 and one LC3 homolog ATG8a. Prominent p62‐positive protein aggregates were observed in aged fly brains, presumably reflecting a defect in autophagic flux, as p62 itself is normally turned over by the lysosome during active autophagy. CPT treatment dramatically reduced p62‐positive aggregates (Fig EV4D), which was further validated by examining p62 protein level using western blot (Fig 5G). Moreover, the increased conversion of ATG8a‐I to ATG8a‐II, a lipidated and active form of ATG8a, also indicated the activation of autophagy by CPT (Fig 5G). We further validated these findings by detecting autophagosome‐lysosome fusion using lysotracker and observed decreased autolysosome formation in aged brains, which was effectively restored by CPT treatment (Fig 5H). Thus, the autophagy pathway is compromised during aging, and RET inhibition by CPT can restore autophagy activity.
We also tested whether the lifespan benefit provided by NMN is dependent on Sirtuin, ATG1, or Foxo. We found that dSirt2‐RNAi sufficiently blocked the NMN effect in lifespan extension, whereas ATG1‐RNAi and Foxo‐RNAi were less effective (Figs EV4E and G–I). Moreover, NMN did not offer additional lifespan benefit in NDUFS3‐RNAi flies (Fig EV4F). Thus, although dSirt2‐RNAi was effective in blocking the lifespan effects of CPT and NMN, ATG1‐RNAi and Foxo‐RNAi were effective in blocking the effect of CPT but not NMN, suggesting that ATG1 and Foxo may mediate other effects of CPT than NAD+/NADH, such as its antioxidant activity.
Inhibition of RET extends lifespan and rescues disease phenotypes in fly AD models
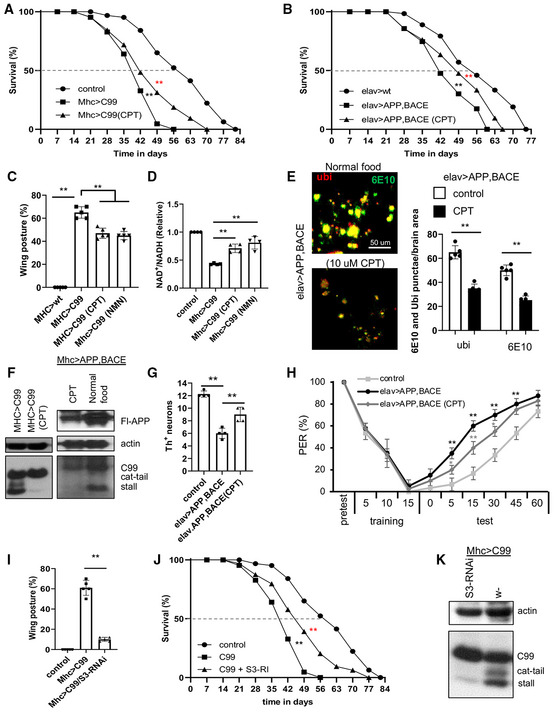
Aging is a major risk factor for chronic diseases. It is assumed that aging and age‐related diseases may share some basic biological mechanisms, but this has not been rigorously demonstrated. AD is an exemplary age‐related neurodegenerative disease. We were interested in testing whether RET, implicated in aging by our current study, is also involved in AD pathogenesis. We first tested CPT in a transgenic model expressing the C‐terminal fragment of amyloid precursor protein (APP.C99). APP.C99 is emerging as an etiological driver in AD pathogenesis (Lauritzen et al, 2019). Expression of APP.C99 in fly muscle using the Mhc‐GAL4 driver resulted in significantly shortened lifespan (Fig 6A). Treating Mhc > APP.C99 flies with CPT from day 35 onwards significantly extended their median as well as maximal lifespans (Fig 6A). Co‐expression of full‐length APP with beta‐secretase (BACE), a combination that results in efficient APP processing to APP.C99, also resulted in shortened lifespan when transgenes were driven by the pan‐neuronal driver elav‐GAL4 (Fig 6B). CPT treatment of these flies from day 35 onwards significantly extended their lifespan (Fig 6B).
Figure 6. Inhibition of RET rescues disease phenotypes of fly AD models.
-
A, BSurvival curves showing the effect of CPT treatment on the lifespan of Mhc > APP.C99 flies (**P < 0.01 logrank, **P < 0.01 Wang–Allison) (A) and elav > APP.BACE flies (**P < 0.01 logrank, **P < 0.01 Wang–Allison) (B) (n = 3 groups, 20–25 flies per group).
-
C, DQuantification of the effect of CPT and NMN treatment on wing posture (C) and NAD+/NADH ratio (D) in Mhc > APP.C99 flies.
-
EImmunofluorescence images and data quantification showing the effect of CPT on the formation of ubiquitin‐ and 6E10‐positive amyloid aggregates in the brain of elav > APP.BACE flies (n = 5).
-
FImmunoblots showing the effect of CPT on aberrant APP.C99 translation products in Mhc > APP.C99 and Mhc > APP/BACE flies.
-
GQuantification of the effect of CPT on DA neuron number in the PPL1 cluster of elav > APP/BACE fly brain (n = 4 sets, 5 brains per set).
-
HTaste memory assay showing the effect of CPT on learning and memory in elav > APP/BACE flies (n = 3 groups, 10 flies per group).
-
IEffect of NDUFS3‐RNAi on the wing posture defect of Mhc > APP.C99 flies (n = 3 groups, 20–25 flies per group).
-
JSurvival curves showing the effect of NDUFS3‐RNAi on the lifespan of Mhc > APP.C99 flies (**P < 0.01 logrank, **P < 0.01 Wang–Allison) (n = 3 groups, 20 flies per group).
-
KImmunoblots showing the effect of NDUFS3‐RNAi on aberrant APP.C99 translation products in Mhc > APP.C99 flies.
Data information: Data are representative of at least three repeats. Data are shown as mean ± SEM (C–E, G). Survival curves were analyzed with logrank test along with Wang–Allison test for maximum lifespan. Asterisks indicate statistical significance (**P < 0.01) in single‐factor ANOVA with Scheffe's analysis as a post hoc test (C–E, I). Dashed lines mark time points of 50% survival.
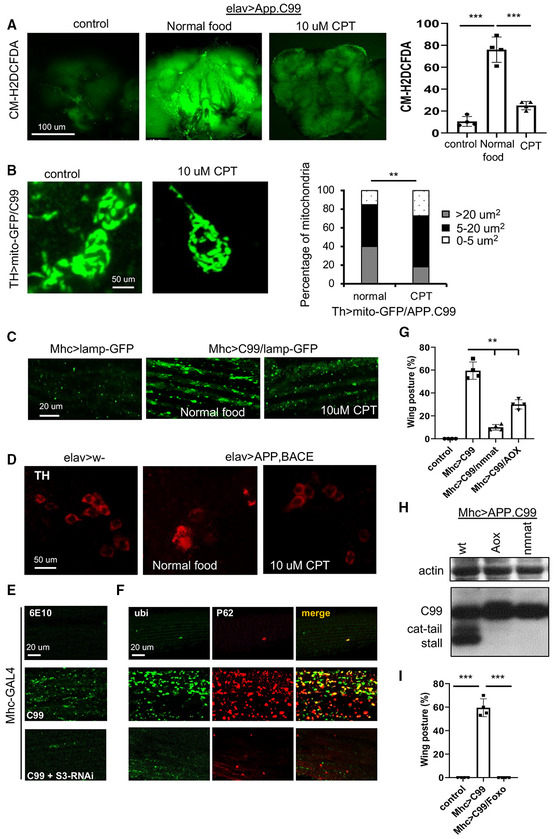
Overexpression of APP.C99 in fly muscle disrupts indirect flight muscle function and structural integrity (Rimal et al, 2021), manifested as abnormal wing postures. CPT significantly rescued APP.C99‐induced wing posture defect (Fig 6C). Similarly, NMN also rescued APP.C99‐induced abnormal wing posture (Fig 6C). These data support that CPT may exert its protective effect through NAD+. We found that expression of APP.C99 resulted in decreased NAD+/NADH ratio (Fig 6D), increased ROS level (Fig EV5A), and altered mitochondrial morphology (Fig EV5B). These phenotypes were effectively rescued by CPT (Figs 6D and EV5A and B). These results suggest that RET is active in the APP.C99 AD model and contributes to disease pathogenesis and lifespan reduction.
Figure EV5. Inhibition of RET rescues disease phenotypes of fly AD models.
-
AStaining images and data quantification showing the effect of CPT on ROS level in the brain of elav > APP.BACE flies (n = 3 set, 5 brains per set).
-
BImmunofluorescence images and data quantification showing the effect of CPT on mitochondrial morphology in DA neurons of TH > APP.C99 flies co‐expressing a mito‐GFP reporter (n = 5 brains per group).
-
CImmunofluorescence images showing the effect of CPT treatment on lysosomal morphology in the muscle of Mhc > APP.C99 flies.
-
DImmunofluorescence images showing the effect of CPT treatment on DA neuron number in the PPL1 cluster of elav > APP.BACE fly brain.
-
E, FImmunostaining showing the effect of NDUFS3‐RNAi on 6E10‐positive amyloid (E) and ubiquitin‐ and p62‐positive protein aggregates (F) in the muscle of Mhc > APP.C99 flies.
-
GQuantification of the effect of AOX or NMNAT overexpression on wing posture in Mhc > APP.C99 flies (n = 4 groups, 20 flies per group).
-
HImmunoblots showing the effect of AOX or NMNAT overexpression on aberrant APP.C99 translation products in Mhc > APP.C99 flies.
-
IQuantification of the effect of dFoxo overexpression on wing posture in Mhc > APP.C99 flies (n = 4 groups, 20 flies per group).
Data information: Data are representative of at least three repeats. Data are shown as mean ± SEM. Asterisks indicate statistical significance (***P < 0.001,**P < 0.01) using single=factor ANOVA with Scheffe's analysis as a post hoc test.
In the fly models we studied, APP.C99 induces proteostasis failure and endolysosomal defects (Rimal et al, 2021), which are regarded as cellular hallmarks of AD (Nixon, 2017). CPT reduced the accumulation of ubiquitinated protein aggregates in elav > APP/BACE fly brain, suggesting that it helped to maintain proteostasis (Fig 6E). Furthermore, autophagic flux defect manifesting as enlarged lysosomes detected with the lysosome marker Lamp1‐GFP in Mhc > APP.C99 fly muscle was ameliorated by CPT treatment (Fig EV5C).
Recent studies implicated stalled translation and ribosome collision as previously unrecognized mechanisms in generating toxic protein species that perturb proteostasis and cause neurodegenerative disease (Wu et al, 2019; Li et al, 2020b; Rimal et al, 2021). In the case of APP.C99, translation stalling and ribosome collision‐induced aberrant APP.C99 species with possible C‐terminal Ala and Thr additions (CAT‐tails) could accumulate due to inadequacy of ribosome‐associated quality control (RQC), causing proteostasis failure and defective endolysosomal and autophagy systems (Rimal et al, 2021). CPT strongly decreased the stalled and CAT‐tailed APP.C99 species, without affecting the full‐length APP.C99 products which are produced by ribosomes that successfully pass the stall (Fig 6F). CPT also reduced the aberrant APP.C99 species derived from full‐length APP in Mhc > APP/BACE flies (Fig 6F). These results reveal a potential link between RET and RQC, although the exact molecular mechanism remains to be elucidated.
We next examined the effect of RET inhibition by CPT on the neurodegeneration and behavioral deficits in elav > APP/BACE flies. APP/BACE co‐expression in the brain led to dopaminergic neuron loss (Rimal et al, 2021), which was rescued by CPT (Figs 6G and EV5D). For learning and memory testing, we used the taste memory assay (Masek et al, 2015). Applying sucrose solution (appetitive tastant) to the tarsi (feet) of a starved fly induced robust feeding behavior as measured by the proboscis extension reflex (PER). After rounds of paired application of sucrose to the tarsi and quinine (aversive tastant) to the proboscis, normal flies learned from experience and showed attenuated PER response to subsequent application of sucrose alone. The elav > APP/BACE flies exhibited compromised memory of the aversive taste they experienced. This behavioral deficit was rescued by CPT (Fig 6H).
To complement the pharmacological approach, we applied a genetic approach to test the idea that RET and RET‐induced NAD+/NADH dyshomeostasis are involved in APP.C99‐associated AD pathogenesis. As described earlier, NDUFS3 is a C‐I protein participating in RET and a target of CPT (Ojha et al, 2022). Knockdown of NDUFS3 ameliorated indirect flight muscle deterioration, as indicated by its rescue of the abnormal wing posture seen in Mhc > APP.C99 flies (Fig 6I), and extended their lifespan (Fig 6J). This was correlated with a reduction of 6E10‐positive, APP.C99‐derived amyloid aggregates (Fig EV5E). Moreover, ubiquitin‐ and p62‐positive protein aggregates were also dramatically reduced by NDUFS3‐RNAi in Mhc > APP.C99 flies (Fig EV5F), suggesting the restoration of proteostasis. At the molecular level, the aberrant APP.C99 species resulting from stalled translation and defective RQC were also effectively removed by NDUFS3‐RNAi (Fig 6K).
To further test the importance of RET and RET‐induced NAD+/NADH imbalance in APP.C99 pathogenesis, we overexpressed alternative oxidase (AOX) and nicotinamide mononucleotide acetyltransferase (NMNAT) in Mhc > APP.C99 background. AOX is an enzyme from Ciona intestinalis that consumes CoQH2, and NMNAT is a rate‐limiting enzyme in NAD+ biosynthesis. APP.C99 flies overexpressing AOX or NMNAT showed normal wing posture (Fig EV5G) and removal of translationally stalled aberrant APP.C99 species (Fig EV5H). Moreover, overexpression of dFoxo effectively rescued the wing posture defect in Mhc > APP.C99 flies (Fig EV5I). This result, together with our previous finding of rescue of APP.C99 toxicity by the activation of autophagy through ATG1 overexpression (Rimal et al, 2021), supports that the Foxo and autophagy pathways known to mediate the effect of NAD+/Sirtuin on aging are also relevant to AD. Thus, RET and RET‐induced NAD+/NADH imbalance impinge on the basic molecular and cellular mechanisms underlying aging and AD, and inhibition of RET pharmacologically or genetically is beneficial in AD settings.
Inhibition of RET rescues disease phenotypes and extends lifespan in mammalian models of AD
We next sought to validate the role of RET in mammalian models of AD. We first used patient iPSC‐derived neuronal models. Using a method that facilitates the differentiation of iPSCs into cortical neurons through the introduction of the transcription factor Ngn2 (Zhang et al, 2013), we obtained human neurons (Appendix Fig S2A) from iPSCs carrying a familial AD (FAD) mutation involving APP duplication (Israel et al, 2012), or from iPSCs derived from Down syndrome (DS) patients who develop AD due to an extra copy of APP gene on Chr. 21 (Weick et al, 2013). Compared with control neurons, the FAD and DS iPSC‐derived neurons exhibited higher ROS level (Fig 7A; Appendix Fig S2B) and lower NAD+/NADH ratio (Fig 7B). These defects were rescued by CPT treatment (Fig 7A and B; Appendix Fig S2B), suggesting that patient iPSC‐derived neurons undergo active RET. Microglial proliferation and activation in the brain is a prominent feature of AD. Various studies have revealed multiple states of microglial activation in disease settings, which likely underlie the disparate roles of microglia in the development and progression of AD pathology (Deczkowska et al, 2018). In the murine microglial BV‐2 cell line, we found that treatment of these cells with DES, an inducer of RET as seen in ischemic stroke, inflammatory immune response, and cancer settings (Chouchani et al, 2014; Mills et al, 2016; Ojha et al, 2022), there was a significant increase in RET‐generated ROS. CPT effectively reduced DES‐induced ROS (Appendix Fig S2C and D). CPT treatment also significantly increased NAD+/NADH ratio in BV‐2 cells (Appendix Fig S2E), indicating microglial cells are adept at pathological RET.
Figure 7. RET inhibition by CPT rescues disease phenotypes in mammalian AD models.
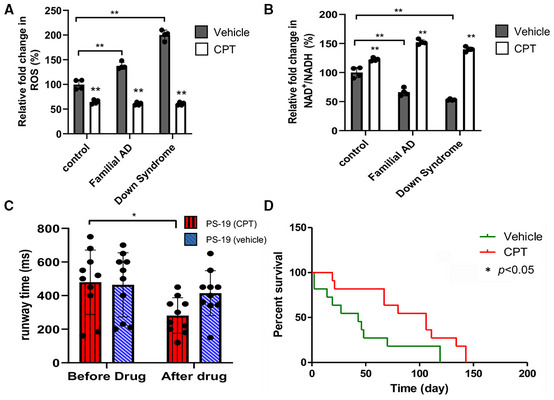
-
A, BQuantification of effect of CPT on ROS level as detected by staining with the H2O2 dye CM‐H2DCFDA (A), and on NAD+/NADH ratio (B) in control, FAD, and DS iPSC‐derived neurons.
-
CQuantification showing the effect of CPT on locomotor activity in PS19 tauopathy mice (n = 10 per group).
-
DSurvival curves showing the effect of CPT on lifespan of PS19 tauopathy mice (*P < 0.05 logrank) (n = 9 in vehicle group, 8 in CPT group).
Data information: Data are shown as mean ± SEM (C). Survival curves were analyzed with logrank test along with Wang–Allison test for maximum lifespan. Asterisks indicate statistical significance (**P < 0.01, *P < 0.05) in single‐factor ANOVA with Scheffe's analysis as a post hoc test (A–C).
The defining neuropathological features of AD include the senile plaques and neurofibrillary tangles (NFTs). NFTs are mainly composed of aggregated hyperphosphorylated tau (Grundke‐Iqbal et al, 1986). They are observed in AD and a host of neurodegenerative diseases known as tauopathies. There is considerable evidence that tau abnormality contributes to AD, and tau is increasingly being recognized as a potential therapeutic target for AD (Iqbal et al, 2018). The PS19 transgenic model expressing the P301S mutant form of human tau associated with frontotemporal dementia is a well‐established mouse model for studying neurodegenerative tauopathy and AD‐related changes (Yoshiyama et al, 2007). PS19 tauopathy mice exhibited decline of locomotor activity at around 8 months of age. Treatment of PS19 mice starting at 8 months of age with CPT for 1 month significantly increased their locomotor activity as measured by their running speed in a runway assay (Fig 7C). Importantly, treatment of PS19 mice with CPT starting at a symptomatic stage at 10 months of age significantly extended their lifespan (Fig 7D). Together, these results indicate that active RET contributes to age‐associated disease pathogenesis in multiple models of AD.
Discussion
NAD+ serves as a central regulator of metabolism and longevity by acting as a coenzyme in redox reactions that link glycolysis and the TCA cycle to OxPhos, and as a cosubstrate for key enzymes regulating longevity, including Sirtuins and PARPs. The realization that NAD+ levels decline with age has spurred intense efforts to restore NAD+ homeostasis using NAD+ precursors or PARP inhibitors. A major unsettled question is why NAD+ levels decline in the first place. Previous studies have shown that CD38, a membrane‐bound NAD+ hydrolyase, is upregulated during aging. CD38 knockout mice better preserve NAD+ and mitochondrial function during aging, suggesting that destruction by CD38 is one of the mechanisms of age‐related NAD+ decline (Camacho‐Pereira et al, 2016). Our results implicate RET as a previously unrecognized mechanism that reduces steady‐state NAD+ levels by converting NAD+ to NADH at the point of C‐I where NADH normally enters the ETC during FET. Supporting its clinical relevance, increased RET is observed in aging human muscle, although the functional significance is unclear (Capel et al, 2005). Since genetic or pharmacological manipulation of RET had strong effect on NAD+/NADH ratio when we used total tissue samples for NAD+ and NADH measurements, mitochondrial RET appears to be a major mechanism influencing whole cell NAD+/NADH homeostasis during aging. A major distinction between NAD+/NADH modulation by RET inhibition versus precursor supplementation is that the former changes NAD+/NADH ratio without changing total NAD(H) content, whereas the latter increases total NAD(H) content, the long‐term effect of which remains to be determined in preclinical animals and humans.
Mito‐ROS is the main source of free radicals in most cells. ROS plays multifaceted roles, with physiological levels of ROS exerting signaling functions whereas pathological levels causing oxidative damage and contributing to aging and diseases (Schieber & Chandel, 2014). RET is the main contributor of mito‐ROS, and elevated RET‐ROS has been linked to disease (Murphy, 2009; Brand et al, 2016; Scialo et al, 2017). Intriguingly, a previous study suggested that physiological level of RET‐ROS regulates stress adaptation and extends lifespan in flies (Scialo et al, 2016), although the study was based mainly on overexpression of yeast NDI1, which in addition to increasing RET‐ROS through CoQH2 also increases NAD+/NADH ratio (Ojha et al, 2022). If the pro‐longevity effect of physiological RET‐ROS holds true, the lifespan‐extending effect observed with RET inhibitors might have underestimated the effect of their rebalancing of NAD+/NADH on lifespan, as these inhibitors presumably also reduced beneficial RET‐ROS. However, our observation that mito‐Tempo can extend fly lifespan suggests that the roles of mito‐ROS and RET‐ROS in lifespan regulation are likely more complex than currently appreciated and require further investigation. On the contrary, the facts that NAD+ supplementation had similar effect as CPT on lifespan, that co‐treatment with CPT and NAD+ supplementation did not offer further lifespan benefit than individual treatment, and that CPT or NAD+ supplementation provided generally stronger lifespan effect than antioxidants, strongly supported our conclusion that the effect on NAD+/NADH ratio is a major contributor to the lifespan benefit of CPT. This is consistent with findings in cancer settings, where CPT exerted anticancer effects that antioxidants were not able to provide (Ojha et al, 2022).
Our results also show that RET inhibition with CPT is beneficial in Drosophila and mouse models of AD. This is correlated with RET being active in the brain tissue of AD fly models and in human AD iPSC‐derived neurons, accounting for the reduced NAD+/NADH ratio in the AD models shown here, and in other AD mouse models (Hou et al, 2021). Although ROS is generally considered the causative agent of cellular damages in neurodegenerative settings, ROS‐directed therapeutic agents have yielded disappointing results (Carvalho et al, 2017), and the specific role of RET‐ROS in these diseases remains undefined. Nevertheless, given that NAD+ supplementation was recently shown to reduce inflammation, DNA damage, apoptosis and cellular senescence, induce mitophagy, and improve cognitive and synaptic function in APP/PS1 AD mice (Hou et al, 2021), the beneficial effect of RET inhibition in our studies is at least partially mediated by its effect on NAD+/NADH ratio.
One important finding of our studies is that RET inhibition directly affects the mechanism underlying the generation of aberrant translation products of APP.C99, which cause proteostasis failure and impair the endolysosomal and autophagy systems, thus supporting a causal role of RET, and by extension mitochondrial dysfunction, in AD pathogenesis. Mitochondrial dysfunction and AD pathogenesis has previously been linked (Swerdlow, 2020). How mitochondrial RET is mechanistically linked to the RQC of translational stalling and ribosome collision during APP/APP.C99 protein synthesis, which occurs at the ER translocon (Rimal et al, 2021), is an intriguing question that warrants further investigation. Nevertheless, our findings of a tight correlation between the beneficial effect of RET manipulations in the AD model, either genetically or pharmacologically, and the removal of proteostasis‐perturbing aberrant APP.C99 species resulting from inadequate RQC of translation stalling and ribosome collision, offer further support for the hypothesis that translation stalling and ribosome collision is central to AD pathogenesis (Lu, 2022).
Sirtuins are NAD+‐dependent enzymes involved in metabolism, stress response, and longevity (Imai & Guarente, 2014). Mammals express a family of seven Sirtuins (SIRT1‐7), three of which are localized to mitochondria (Sirt3, Sirt4, and Sirt5), highlighting the importance of Sirtuins to mitochondrial homeostasis and metabolism. The Drosophila genome encodes 5 Sirtuins (dSirt1, 2, 4, 6, 7) named after their closest mammalian homologs, although in the case of dSirt2, it is homologous to both human Sirt2 and Sirt3. Genetic studies have implicated dSirt1, dSirt2, dSirt4, and dSirt6 in lifespan regulation (Wood et al, 2004, 2018; Kusama et al, 2006; Taylor et al, 2022), although the role of dSirt1 in aging is questioned (Burnett et al, 2011). We found that dSirt2‐RNAi and Foxo‐RNAi blocked the lifespan‐extending effect of CPT. Although it is possible that dSirt2 may feedback regulate C‐I to influence RET or FET, this feedback regulation is unlikely to be the main mechanism of Sirt2 action on lifespan; Otherwise, CPT would be able to rescue the shortened lifespan of dSirt2‐RNAi flies. Instead, we observed that dSirt2‐RNAi blocked the CPT effect on lifespan. Our results support the idea that the NAD+/Sirtuin‐Foxo pathway, which is involved in lifespan extension by NAD+ supplementation in worms (Mouchiroud et al, 2013), mediates the effect of RET‐related NAD+/NADH change on fly lifespan. Moreover, our results identified the autophagy pathway as a mediator of the CPT effect on lifespan, presumably downstream of NAD+/Sirtuin‐Foxo signaling. Previous studies also implicated the activation of UPRmito in mediating the lifespan‐extending effect of NAD+ supplementation (Mouchiroud et al, 2013). Future studies will test if the lifespan‐extending effect and AD‐protective effect of CPT may also involve the activation of UPRmito.
The molecular players involved in RET functioning and regulation are just beginning to be revealed. Previous study showed that CPT inhibits RET by interacting with subunits of the soluble matrix arm of the L‐shaped C‐I (Ojha et al, 2022). These C‐I subunits are positioned at the NADH entry point (or the beginning of FET) and the end point of RET, with CPT altering the dynamic protein–protein interactions among them. The age‐related change in protein–protein interactions among C‐I subunits is unlikely to cause C‐I disassembly, otherwise we would have detected age‐related C‐I activity changes in our assays. The subtle protein–protein interaction change likely alters the direction or kinetics of electron flow, as some of these proteins contain the electron‐transporting Fe‐S clusters. Our data show that genetic interference by RNAi of two of these subunits, NDUFS2 and NDUFS3, mimicked the effect of CPT treatment. Importantly, the C‐I proteins shown to interact with CPT or participate in RET, especially NDUFS3, are reported to be downregulated in long‐lived mammals (Miwa et al, 2014), and their knockdown in worms (Dillin et al, 2002; Lee et al, 2003) or flies (Copeland et al, 2009; Owusu‐Ansah et al, 2013) extended lifespan, supporting the importance of C‐I in aging (Hur et al, 2014). We propose that these subunits participate in a conserved mechanism of lifespan regulation, whereby modulation of RET and RET‐associated NAD+/NADH homeostasis and/or ROS signaling underpin their longevity effects. RET is well suited to detect a range of physiological changes that require metabolic adaptation and is likely subjected to intricate regulation. In this regard, Notch has been shown to interact with C‐I subunits involved in RET and this interaction stimulates RET and promotes cancer stem cell proliferation (Ojha et al, 2022). Notch has previously been linked to aging (Conboy et al, 2005) and AD (Ho et al, 2020). Our results show that protein–protein interactions among C‐I subunits involved in RET and Notch are altered during aging and that CPT restores some of these interactions. Further studies of RET regulators such as Notch in aging and age‐related diseases will undoubtedly shed the much‐needed light on the cause of RET deregulation during aging, expand our knowledge on the mechanisms of lifespan regulation, and offer new targets for therapeutic intervention.
Materials and Methods
Fly genetics
The following flies were obtained from the Bloomington Drosophila Stock Center: elav‐GAL4 (8765), UAS‐APP.C99 (33783), UAS‐APP (6700), UAS‐APP;UAS‐BACE (33798), UAS‐LAMP1‐GFP (42714), UAS‐Foxo (9575), UAS‐NMNAT (39702), UAS‐ND75 (NDUFS1) RNAi (33911), UAS‐ND30 (NDUFS3) RNAi (44535), UAS‐ND49 (NDUFS2) RNAi (28573), UAS‐ND‐51 (NDUFV1) RNAi (36701), and UAS‐ND‐24 (NDUFV2) RNAi (51855). We received the following flies from Vienna Drosophila Stock Center: UAS‐ATG1‐RNAi (16133), UAS‐Sirt2‐RNAi (103790). The sources of the other fly stocks are as follows: Dr. T. Littleton (MHC‐GAL4), Dr. J. Knoblich (UAS‐SoNar), Dr. S. Birman (TH‐GAL4), Dr. H Jacobs (UAS‐AOX), and Dr. William Saxton (UAS‐mito‐GFP). The indicated UAS RNAi and OE fly lines were crossed to Mhc‐Gal4 or elav‐Gal4 driver lines for muscle or pan‐neuronal expression, respectively. Fly culture and crosses were performed according to standard procedures. Adult flies were generally raised at 25°C and with 12/12 h dark/light cycles. Fly food was prepared with a standard receipt (Water, 17 L; Agar, 93 g; Cornmeal, 1,716 g; Brewer's yeast extract, 310 g; Sucrose, 517 g; Dextrose, 1,033 g).
Fly lifespan analysis
Flies were reared in vials containing cornmeal medium. Flies were anesthetized using CO2 and collected at a density of 20 flies/vial. Initial experiments were done on both males and females. As no difference in lifespan effect was observed between sexes, later studies were done mostly in males. All flies were kept at humidified, 12 h on/off light cycle at 25°C. Flies were flipped into fresh vial every 3 days, and the number of dead animals was recorded. Each set of experiment was carried out ≥ 4 times.
Drug treatments
CPT was obtained from Cerepeut Inc. under a Materials Transfer Agreement between Cerepeut Inc. and Stanford University. The compound was synthesized for Cerepeut Inc. by the Chemistry Branch of Medicilon. Details of chemical synthesis are available upon request. Flies were treated with 5–20 μM CPT, 3 mM NMN (Sigma), 10 mM DMM (Sigma), 10 μM CCCP (Sigma), 10 μM mdivi‐1 (Enzo Life Sciences), 250 nM rotenone (Sigma), 10 mM DES (Sigma), 4 mM Paraquat (Sigma), 0.5 mM melatonin (Sigma), 100 nM FK866 (Cayman chemicals), 10 μM mito‐Tempo (Sigma), and 10 μM NADH (Sigma) for the indicated periods of time. Drug‐containing fly food was prepared by adding drugs to microwave‐melted fly food kept at warm temperature (~ 50°C). After thorough mixing, the fly food was left at room temperature to harden and kept at 4°C. Flies were changed to fresh drug‐containing food every 2 days.
Aversive taste memory assay
Taste memory assay was performed as described previously (Masek et al, 2015), with slight modifications. Briefly, one‐week‐old flies were starved for 12–18 h in an empty vial on wet Kimwipe paper. Flies were anesthetized using ice and fixed on a glass slide by applying nail polish to their wings. A total of 10–15 flies were used for each set of experiment. Flies were then incubated in a humid chamber for 2 h to allow recovery from the procedure. In the pretest phase, flies were presented with 500 mM sucrose stimuli (attractive tastant) to their legs using Kimwipe wick. Flies that showed positive proboscis extension to the stimulus were used for the next phases. In the training phase, the flies were presented with 500 mM sucrose stimuli at their legs while being simultaneously punished with 10 mM quinine (aversive tastant) applied to their extended proboscis. Training was repeated 15 times for each fly. The last phase is the test phase where the flies were given 500 mM sucrose at their legs at different time intervals (0, 5, 15, 30, 45, and 60 min), and proboscis extension was recorded. Each experiment was carried out ≥ 4 times.
NAD +/NADH measurement
NAD+/NADH was measured essentially as described previously (Ojha et al, 2022), using an NAD+/NADH quantification colorimetric kit according to the manufacturer's instructions (AAT Bioquest #15273). Briefly, 10 whole flies or thoraces were taken for one set of experiment and lysed using the lysis buffer for 15 min at 4°C and lysates were collected after centrifugation at 12,000 g for 15 min. For the measurement of total NAD+ amount, NAD extraction solution into the lysates was added and incubated at 37°C for 15 min; thereafter, neutralization solution was added to neutralize the NAD extracts. Absorbance was monitored at 460 nm after adding NAD/NADH working solution and 1 h incubation at room temperature with protection from light. To measure total NAD+ and NADH amount, control extraction solution was added into the lysates and incubated at 37°C for 15 min; thereafter, again control extraction solution was added. Absorbance was monitored at 460 nm after adding NAD/NADH working solution and 1 h incubation at room temperature with protection from light. The ratio of NAD+ /NADH was determined by the following equation: . Each experiment was performed ≥ 4 times.
We used a genetically encoded NAD+/NADH sensor SoNar, to measure NAD+/NADH by live imaging. Briefly, the UAS‐Gal4 system was used to express the UAS‐SoNar transgene (Bonnay et al, 2020) in neurons using the pan‐neuronal elav‐Gal4 driver. Flies with or without drug treatment were dissected at the indicated ages, and brain samples were kept in Schneider's media and immediately visualized on a Leica SP8 confocal microscope with laser 405 nm and 488 nm to detect NADH or NAD+ signals. Since the SoNar‐NADH signal is sensitive to photobleaching, we first detected NADH signal using laser 405. The signal intensity was measured by using the NIH Image J software.
H2O2 measurement by CM‐H2DCFDA staining
CM‐H2DCFDA (5 μM) was added to freshly dissected muscle of brain tissue and allowed to incubate for 30 min. Tissue samples were immediately imaged on a Leica SP8 confocal microscope with an excitation laser of 492–495 nm and detection set for 517–527 nm using a 40× oil‐objective lens.
Western blot and co‐immunoprecipitation analyses
Around 5 fly thoraces were homogenized in 75 μl of either regular lysis buffer (50 mM Tris–HCl, 150 mM NaCl, 1% Triton X100, protease inhibitors) or Urea lysis buffer (6 M Urea, 50 mM Tris–HCl, 150 mM NaCl, 0.1% Triton X100, protease inhibitors) on ice. Samples were homogenized using a hand‐held mechanical homogenizer for 30 s. The homogenized samples were incubated on ice for 30 min before centrifuging at 21,130 g for 20 min at 4°C. 30 μl of supernatant was mixed with 10 μl of 4× Lammaelli buffer (BioRad #161–0747) and boiled for 5 min at 100°C. The protein lysate was cooled, centrifuged and loaded onto 4–12% Bis‐Tris gel (Invitrogen #NP0321) or 16% Tricine gel (Invitrogen #EC66955) with 1× MES (Invitrogen #NP0002) as running buffer as previously described (Wu et al, 2019; Rimal et al, 2021). Co‐immunoprecipitation assays for detecting the effect of aging and drug treatment on protein–protein interaction between C‐I proteins were performed essentially following a protocol described before (Rimal et al, 2021).
Immunohistochemistry
Immunostaining of adult fly muscle was performed as described previously (Li et al, 2020b). Briefly, fly thoraxes were dissected and fixed with 4% paraformaldehyde (Electron Microscopy Sciences, cat. no. 15710) in phosphate‐buffered saline and 0.3% Triton X‐100 (PBS‐T). The tissues were then washed three times with PBS‐T. Samples were incubated for 30 min at room temperature in blocking buffer: 0.5% goat serum in PBS‐T. The following primary antibodies were added and samples were incubated overnight at 4°C: anti‐Ubiquitin, Abcam ab140601, 1:1,000; anti‐P62, Abcam ab178440, 1:1,000; 6E10, Bio legend 803001, 1:1,000; and anti‐LAMP1, DSHB 1D4B, 1:100. The samples were washed three times with PBS‐T and subsequently incubated with the following secondary antibodies for 4 h at 4°C: Alexa Flour 488 (A32723), Alexa flour 594 (A11036), Invitrogen, both at 1:200. The tissues were washed three times with PBS‐T and mounted in slow fade gold buffer (Invitrogen).
Immunostaining of adult brains was performed as described previously (Rimal et al, 2021). Briefly, brain tissues of adult flies were dissected and fixed on ice for 30–45 min in fixing buffer (940 μl of 1% PBS‐T and 60 μl of 37% formaldehyde). Tissues were washed three times in 0.1% PBS‐T and blocked overnight at 4°C in blocking buffer (1 ml 1× PBS, 0.1% Triton X, 5 mg/ml BSA). This was followed by incubating for 16 h at 4°C with the following primary antibodies: anti‐TH, Pel‐Freez P40101‐150, 1:1,000; anti‐4‐HNE, R&D system MAB3249, 1:1,000; anti‐Ubiquitin, Abcam ab140601, 1:1,000; 6E10, Bio legend 803001, 1:1,000; anti‐p62, Abcam ab178440, 1:1,000. Tissues were washed three times with 0.1% PBS‐T and subsequently incubated with the following secondary antibodies for 4 h at 4°C: Alexa Flour 488 (A32723), Alexa flour 594 (A11036), Invitrogen, both at 1:200. Samples were finally mounted in slow fade gold buffer (Invitrogen) and viewed using a Leica SP8 confocal microscope.
ATP measurement
ATP measurements were performed according as described previously (Li et al, 2020b), using a luciferase‐based bioluminescence assay (ATP Bioluminescence Assay Kit HS II, Roche Applied Science). For each test, three thoraxes were taken by removing legs, wings, head, and abdomen and quickly homogenized in 100 μl lysis buffer (provided by kit). Subsequently, the tissue lysates were boiled for 5 min at 100°C and cleared by centrifugation at 20,000 g for 2 min. The supernatant was transferred to a new tube and kept on ice. 2.5 μl of cleared tissue lysate was mixed with 187.5 μl dilution buffer and 10 μl luciferase reagent. The luminescence signal was measured using a Lumat LB 9507 tube luminometer (Berthold Technologies). Each group was analyzed at least three times.
Smurf assay
We performed Smurf assay to analyze intestinal barrier function in flies in aged flies as described previously (Rera et al, 2011). Briefly, flies at the indicated ages were allowed to feed 2.5% Brilliant blue dye mixed with normal food for 16 h. Flies were counted as Smurf when dye coloration was observed outside digestive tract. Each experiment was performed ≥ 4 times.
Climbing activity and wing posture assays
Climbing assay was performed as described previously (Rimal et al, 2021). Briefly, around 10–20 male flies were transferred to a clean plastic vial. The flies were allowed to get accustomed to the new environment for 3–4 min and subsequently measured for bang‐induced vertical climbing distance. The performance was scored as percentage of flies crossing the 8 cm mark within 12 s. Each experiment was performed ≥ 4 times. To assay wing posture, cohorts of flies raised at 25 degrees at the indicated ages were visually inspected for straight, held‐up, or droopy wing postures. The number of flies with normal (straight) or abnormal (held‐up or droopy) wing postures was counted and quantified as the percentage of the total number of flies.
Feeding quantification
The feeding quantification assay was carried out as previously described with slight modification. Briefly, 3‐ to 6‐day‐old flies were allowed to feed on fly food containing 0.1% (w/v) Brilliant Blue FCF food coloring and with or without 10 μM CPT or 3 mM NMN for 30 min without starvation. After feeding control diet or drug‐containing diet, flies were sacrificed by freezing at −20°C. Six male flies were transferred into individual 1.5 ml Eppendorf tubes filled with 200 μl PBS‐T (1× PBS with 0.2% Triton X‐100). The flies were completely ground, and subsequently, 800 μl of PBS‐T was added. The tube was centrifuged for 5 min at 15,871 g at 4°C. Supernatant was loaded in cuvettes for spectrophotometry by measuring absorbance at 630 nm. PBS‐T was used as blank control. The ingestion index (I.I.) was calculated based on the difference in optical density (O.D.) between control‐diet and chemical‐diet groups. I.I is defined as (O.D. chemical diet − O.D. control diet)/O.D. control diet. Each experiment was carried out six times.
Analysis of gene expression
We used TRIzol (Invitrogen) to extract mRNA from dissected fly thorax and the iscript cDNA synthesis kit (BioRad) to synthesize cDNA. Real‐time quantitative PCR (RT–qPCR) was performed using SYBR Green. Sequence of RT–qPCR primers are as follows:
AKT forward: GCTATGACGCCATCTGAAC,
AKT Reverse: CGCCGCTGCTATTACAAG,
ATG1 forward: GCCAGCTCCATCGAAAATAACC,
ATG1 Reverse: GCGGCGCAGCAGGCACAG,
ATG8a Forward: CCAATACAAGGAGGAGCAC,
ATG8a Reverse: AGGAAGTAGAACTGACCGA,
mTOR Forward: ACCGATGACGAGGAGAATG,
mTOR Reverse: AGCGAAGATACTGTTCAATGG
4 E‐BP Forward: CCAGGAAGGTTGTCATGTG
4 E‐BP Reverse: GGAGTGGTGGAGTAGAGG
PI3K Forward: CCTAATCTGCCTGTTGCCCA
PI3K Reverse: ACTGAGTCGCTTCGTTTCGT
Sirt 1 Forward: ACGAGGAAATTCGCCTGGGCC
Sirt 1 Reverse: GTGGCCATGACCGTGTGGTACT
NDUFV1 Forward: ACCAACCTCTATGGACGAC
NDUFV1 Reverse: GATTCCCTCTCGGCATGGCGT
NDUFV2 Forward: GCCAACATCCGGGCAATTGCCA
NDUFV2 Reverse: TAGTCATCGTTGATCGCCACC
NDUFS2 Forward: CCGCATATTGGGCTGCTGCA
NDUFS2 Reverse: GCCAGATCCACGCAACATAACT
NDUFS3 Forward: CTCGCATCTCTCCGATTTCGG
NDUFS3 Reverse: AACTGTTCCCAAGGCGCCGAC
Sirtuin 2 Forward: GCTGGCATTCCGGATTTCAGA
Sirtuin 2 Reverse: CCCTATCGCCCGAAGTCATCT
Foxo Forward: TTCGAGCCACAGACCCGG
Foxo Reverse: TTCGAGCCACAGACCCGG
BN‐PAGE and in‐gel activity assays
Mitochondria were isolated from the whole flies as described previously (Li et al, 2020a). Isolated mitochondrial pellets were suspended in native PAGE sample buffer (Invitrogen BN20032) along with 1% digitonin and protease inhibitors. Following 15 min incubation on ice, samples were centrifuged at 20,000 g for 30 min. Supernatant was collected and mixed with the G‐250 sample additive and loaded onto 4–16% precast native PAGE gels. The NativeMark protein standard (Life Technologies) was run along with samples to estimate molecular weight. Electrophoreses were conducted using Native PAGE running buffer (Invitrogen) as anode and Native PAGE running buffer containing Cathode buffer additive (Invitrogen) as cathode. Gels were stained with Coomassie blue staining reagent to visualize protein bands.
In‐gel activity assays were performed as described previously (Jha et al, 2016). Complex I activity assay was performed by incubating BN‐PAGE gel with 0.1 mg/ml NADH, 2.5 mg/ml Nitrotetrazolium blue chloride, and 5 mM Tris–HCl (at pH 7.4) at room temperature for 15–20 min. Complex II activity was measured by incubating BN‐PAGE gel with 2.5 mg/ml Nitrotetrazolium blur chloride, 0.2 mM of Phenazine Methosulphate, 20 mM Sodium Succinate, and 5 mM Tris–HCl at room temperature for 40 min. Complex III and IV activities were measured by incubating BN‐PAGE gel with 50 mM Sodium phosphate (at pH 7.2), 0.05% Diaminobenzidine, 50 mM cytochrome c at room temperature for 30–40 min.
Mitochondrial reactive oxygen species (mtROS), membrane potential (MMP), and succinate assays
Mitochondrial ROS assay was performed as described previously (McManus et al, 2019). Briefly, mitochondrial ROS was determined by monitoring the oxidation of Amplex Red by H2O2 in presence of horseradish peroxidase. Isolated mitochondria were incubated in an assay medium (125 mM KCL, 20 mM Hepes, 2 mM K2HPO4, 1 mM Mgcl2, 0.1 mM EGTA, 0.025% BSA (pH)). To induce forward electron transport, 2.5 mM malate and 2.5 mM glutamate were supplemented as a substrate. Similarly, to induce reverse electron transport 5 mM succinate and 1 μg/ml oligomycin were supplemented into the mitochondrial samples. 1 μM Amplex Red and 5 U. ml‐1 horseradish peroxidase were added into the reaction media followed by the addition of CPT. Finally, fluorescence was recorded at excitation 560 and emission 590 nm. MMP was examined using 100 mM TMRM, and fluorescence was recorded at 535 nm excitation and 600 nm emission. Mitochondria succinate level was examined with the succinate assay kit from abcam (ab204718), following the manufacturer's protocol.
Neuronal and BV‐2 cell culture
Transformed murine microglial cell line BV‐2 was obtained from Dr. Creed Michael Stary at Department of Anesthesiology, Stanford University School of Medicine. Cells were cultured in DMEM high‐glucose media (11965084, Gibco) supplemented with 10% FBS (100‐602, Gemini Bio) and Penicillin–Streptomycin (15140‐122, Gibco; 1:100). Cells were maintained under standard tissue culture conditions (5% CO2, 37°C). Cells were seeded at 0.5 × 106 cells/ml. Cells were treated with DES (5 mM), CPT (20 μM), or DES (5 mM) plus CPT (20 μM) for 30 min to 4 h, and thereafter, cells were trypsinized and processed for staining with CM‐H2DCFDA. Cells were incubated with CM‐H2DCFDA (5 μM) for 15 min in the dark at 37°C. After incubation, cells were washed and resuspended in PBS. Samples were immediately observed using an LSR II flow cytometer, and data were analyzed using the FlowJo software.
iPSC‐derived neuronal culture
AD iPSC cell lines (UCSD239i‐APP2‐1 and UCSD234i‐SAD2‐3) were obtained from WiCell Research Resources (Wicell, WI). Normal human ESCs (H1) were gifts from Dr. Marius Wernig at Department of Pathology, Stanford University School of Medicine. H1 ESCs and AD iPSCs were differentiated according to a protocol previously described (Zhang et al, 2013). Briefly, ESCs and iPSCs were treated with Accutase and plated as dissociated cells on Matrigel‐coated plates using mTeSR1 supplemented with ROCK inhibitor. The next day, cells were transfected with TetO inducible NGN2 lentivirus using polybrene (8 μg/ml, Sigma). The following day, culture medium was replaced with N2 containing Doxycycline (2 μg/ml) with subsequent puromycin selection (2 μg/ml). After puromycin selection, ESCs and iPSCs were allowed to differentiate into neurons by retaining them in N2 medium with change in medium every day for 10 days. Differentiated neurons were subsequently treated with CPT (2 μM) for 1 h and processed for ROS (CM‐H2DCFDA) measurements or treated with CPT (2 μM) for 12 h and processed for NAD+/NADH measurements.
Mouse strains and treatments
B6;C3‐Tg (Prnp‐MAPT*P301S) PS19Vle/J (Strain #008169; PS19) mice were ordered from Jackson Laboratory. After mice were received from the vendor, they were kept in the animal facility of Stanford University School of Medicine and bred to wild‐type mice to obtain sufficient number of animals for the planned experiments. Animals were housed in groups of up to 3 per cage and maintained as colonies at the Stanford University Animal Facility. Food and water were available ad libitum, and the cage contained shredded paper bedding as standard housing. Animals were kept on a 12‐h light/dark cycle (lights on at 6:00 h), and the room temperature was controlled via a thermostat set at 21°C. No obvious differences between male and female mice were observed, and we used both male and female mice for the present studies. All animal experiments were performed in accordance with the protocols approved by the Administrative Panel on Laboratory Animal Care (APLAC) at Stanford University and comply with all regulations for ethical conduct of animal research. For the CPT treatment of PS19 mice, 8‐month‐old animals were treated with vehicle (DMSO:PEG300:20%cyclodextrin = 5:45:50) or CPT dissolved in vehicle at a dose of 50 mg/kg body weight, by daily i.p injection for 2 months for the behavioral studies. For the lifespan study, 10‐month‐old animals were treated with vehicle (DMSO:PEG300:20% cyclodextrin = 5:45:50) or CPT dissolved in vehicle at a dose of 50 mg/kg body weight, by daily i.p injection until all animals reach the terminal stage of life.
Mouse brain frozen section immunofluorescence staining
Animals were sacrificed by cervical dislocation, and subsequently, half of the brain tissues were harvested followed by storing in 4% formalin (Sigma) for 24 h. Tissues were then moved into 10% sucrose 24 h, 20% sucrose 24 h, respectively. After processing, tissues were embedded with OCT. Ten‐micrometer sections were fixed in cold acetone for 10 min then air dried at room temperature for 1 h. To remove OCT, slides were placed in 1XPBS wash buffer for 10 min followed by processing with 0.3% PBS‐T. After washing with 1XPBS, excess wash buffer was wiped off from slide without drying sections. Tissues were blocked in 5% goat serum for 1 h at room temperature followed by incubating with primary antibodies for 6E10, IBA‐1, and GFAP overnight at 4°C. Samples were rinsed in wash buffer for 5 min. After wiping off excess buffer, we incubated samples with secondary antibody (conjugated to fluorophores) in the dark for 2 h at room temperature. After washing with PBS for 3 times, samples were ready for mounting with mounting media. Images were taken by Leica SP8 microscope.
Runway test
We devised the runway test to measure the locomotor activity of PS19 mice. Animals were toe dipped in black ink and placed on a 20 cm x 100 cm white paper (the runway) placed on a flat surface, and with the home cage at one end of the paper. Animals were initially restrained at the tail, placed at one end of the runway, and allowed to run to their home cage after releasing the tail restraint. Animal movement was tracked by video recording. The time it took for the animals to complete the runway was measured based on footprint analysis. The runway time was represented in milliseconds.
Mouse lifespan assay
Ten‐month‐old PS19 mice with obvious disease symptoms as indicated by lack of activity and signs of a hunched back and paralysis were treated with vehicle or CPT at 50 mg/kg by daily i.p injection. Mice were inspected once daily. If mice appeared to be too weak to obtain food, a mush of ground pellets and water was placed on the cage bottom to prevent dying from starvation. Moribund mice were killed if they appeared severely ill and were judged not able to survive another 48 h. A mouse was considered severely moribund if it exhibited more than one of the following clinical signs: inability to eat or drink; abnormally low body temperature; severe lethargy; severe balance or gait disturbance; and rapid weight loss for a week or more. The age at which a moribund mouse was killed was used to estimate its natural lifespan.
Quantification and statistical analysis
Statistical analysis was performed using GraphPad Prism 8 (Windows version 8, GraphPad Software, San Diego, CA, USA). Student's t‐test, logrank, Wang–Allison test, and one‐way ANOVA test with Scheffe's analysis as a post hoc test for comparing two sets of data were used in statistical analyses. All data are represented as mean ± SEM, with P < 0.05 being considered statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001. ns, not significant.
Study approval
All experiments using mice were performed in accordance with the protocols approved by the Administrative Panel on Laboratory Animal Care (APLAC) at Stanford # University (Protocol#APLAC‐22400). Experiments using human iPSCs were performed in accordance with protocols approved by Stem Cell Research Oversight (SCRO) at Stanford University (Protocol SCRO‐493).
Author contributions
Bingwei Lu: Conceptualization; resources; supervision; funding acquisition; methodology; writing—original draft; project administration; writing—review and editing. Suman Rimal: Conceptualization; data curation; formal analysis; visualization; methodology; writing—review and editing. Ishaq Tantray: Data curation; formal analysis. Yu Li: Data curation; formal analysis. Tejinder Khaket: Data curation; formal analysis. Yanping Li: Data curation; formal analysis. Sunil Bhurtel: Data curation. Wen Li: Data curation. Cici Zeng: Data curation.
Disclosure and competing interests statement
Bingwei Lu is a co‐founder and serves on the Scientific Advisory Board of Cerepeut Inc.
Supporting information
Appendix
Expanded View Figures PDF
PDF+
Acknowledgments
We are grateful to Drs. J. Knoblich, S. Birman, T. Littleton, W. Saxton, the Vienna Drosophila RNAi Center, and the Bloomington Drosophila Stock Center for fly stocks; Drs. Marius Wernig and Bo Zhou for help with iPSC culture and neuronal induction; Stanford PAN facility for primer synthesis; Dr. Nay Saw and Stanford Behavioral and Functional Neuroscience Laboratory for help with mouse behavioral assays; The Axelrod, Bogyo, Lipsick, and Svensson Laboratories in the Department of Pathology, Stanford University School of Medicine, for sharing reagents and equipment. Special thanks go to J. Gaunce and W. Jiao for maintaining fly stocks and providing technical supports, and members of the Lu laboratory for discussions. This work was supported by the NIH (R01NS084412, R21AG07414, and R01AR074875 to B.L.).
EMBO reports (2023) 24: e55548
Data availability
There are no data described in this study that requires deposition.
References
- Balaban RS, Nemoto S, Finkel T (2005) Mitochondria, oxidants, and aging. Cell 120: 483–495 [DOI] [PubMed] [Google Scholar]
- Bar‐Ziv R, Bolas T, Dillin A (2020) Systemic effects of mitochondrial stress. EMBO Rep 21: e50094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonkowski MS, Sinclair DA (2016) Slowing ageing by design: the rise of NAD(+) and sirtuin‐activating compounds. Nat Rev Mol Cell Biol 17: 679–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnay F, Veloso A, Steinmann V, Kocher T, Abdusselamoglu MD, Bajaj S, Rivelles E, Landskron L, Esterbauer H, Zinzen RP et al (2020) Oxidative metabolism drives immortalization of neural stem cells during tumorigenesis. Cell 182: 1490–1507.e19 [DOI] [PubMed] [Google Scholar]
- Bordt EA, Clerc P, Roelofs BA, Saladino AJ, Tretter L, Adam‐Vizi V, Cherok E, Khalil A, Yadava N, Ge SX et al (2017) The putative Drp1 inhibitor mdivi‐1 is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Dev Cell 40: 583–594.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD, Goncalves RL, Orr AL, Vargas L, Gerencser AA, Borch Jensen M, Wang YT, Melov S, Turk CN, Matzen JT et al (2016) Suppressors of superoxide‐H2O2 production at site IQ of mitochondrial complex I protect against stem cell hyperplasia and ischemia‐reperfusion injury. Cell Metab 24: 582–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY et al (2004) Stress‐dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303: 2011–2015 [DOI] [PubMed] [Google Scholar]
- Burnett C, Valentini S, Cabreiro F, Goss M, Somogyvari M, Piper MD, Hoddinott M, Sutphin GL, Leko V, McElwee JJ et al (2011) Absence of effects of Sir2 overexpression on lifespan in C. elegans and Drosophila . Nature 477: 482–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho‐Pereira J, Tarrago MG, Chini CCS, Nin V, Escande C, Warner GM, Puranik AS, Schoon RA, Reid JM, Galina A et al (2016) CD38 dictates age‐related NAD decline and mitochondrial dysfunction through an SIRT3‐dependent mechanism. Cell Metab 23: 1127–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J, Kapahi P, Lithgow GJ, Melov S, Newman JC, Verdin E (2019) From discoveries in ageing research to therapeutics for healthy ageing. Nature 571: 183–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capel F, Rimbert V, Lioger D, Diot A, Rousset P, Mirand PP, Boirie Y, Morio B, Mosoni L (2005) Due to reverse electron transfer, mitochondrial H2O2 release increases with age in human vastus lateralis muscle although oxidative capacity is preserved. Mech Ageing Dev 126: 505–511 [DOI] [PubMed] [Google Scholar]
- Carvalho AN, Firuzi O, Gama MJ, Horssen JV, Saso L (2017) Oxidative stress and antioxidants in neurological diseases: is there still hope? Curr Drug Targets 18: 705–718 [DOI] [PubMed] [Google Scholar]
- Chance B, Hollunger G (1961) The interaction of energy and electron transfer reactions in mitochondria. I. General properties and nature of the products of succinate‐linked reduction of pyridine nucleotide. J Biol Chem 236: 1534–1543 [PubMed] [Google Scholar]
- Chini CCS, Zeidler JD, Kashyap S, Warner G, Chini EN (2021) Evolving concepts in NAD(+) metabolism. Cell Metab 33: 1076–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho J, Hur JH, Walker DW (2011) The role of mitochondria in drosophila aging. Exp Gerontol 46: 331–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC et al (2014) Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515: 431–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cino M, Del Maestro RF (1989) Generation of hydrogen peroxide by brain mitochondria: the effect of reoxygenation following postdecapitative ischemia. Arch Biochem Biophys 269: 623–638 [DOI] [PubMed] [Google Scholar]
- Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA (2005) Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature 433: 760–764 [DOI] [PubMed] [Google Scholar]
- Copeland JM, Cho J, Lo T Jr, Hur JH, Bahadorani S, Arabyan T, Rabie J, Soh J, Walker DW (2009) Extension of drosophila life span by RNAi of the mitochondrial respiratory chain. Curr Biol 19: 1591–1598 [DOI] [PubMed] [Google Scholar]
- Covarrubias AJ, Perrone R, Grozio A, Verdin E (2021) NAD(+) metabolism and its roles in cellular processes during ageing. Nat Rev Mol Cell Biol 22: 119–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deczkowska A, Keren‐Shaul H, Weiner A, Colonna M, Schwartz M, Amit I (2018) Disease‐associated microglia: a universal immune sensor of neurodegeneration. Cell 173: 1073–1081 [DOI] [PubMed] [Google Scholar]
- Dillin A, Hsu AL, Arantes‐Oliveira N, Lehrer‐Graiwer J, Hsin H, Fraser AG, Kamath RS, Ahringer J, Kenyon C (2002) Rates of behavior and aging specified by mitochondrial function during development. Science 298: 2398–2401 [DOI] [PubMed] [Google Scholar]
- Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP et al (2013) Declining NAD(+) induces a pseudohypoxic state disrupting nuclear‐mitochondrial communication during aging. Cell 155: 1624–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundke‐Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM (1986) Microtubule‐associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem 261: 6084–6089 [PubMed] [Google Scholar]
- Guarente L (2008) Mitochondria—a nexus for aging, calorie restriction, and sirtuins? Cell 132: 171–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hariharan N, Maejima Y, Nakae J, Paik J, DePinho RA, Sadoshima J (2010) Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation‐induced autophagy in cardiac myocytes. Circ Res 107: 1470–1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman D (1956) Aging: a theory based on free radical and radiation chemistry. J Gerontol 11: 298–300 [DOI] [PubMed] [Google Scholar]
- Hinchliffe P, Sazanov LA (2005) Organization of iron‐sulfur clusters in respiratory complex I. Science 309: 771–774 [DOI] [PubMed] [Google Scholar]
- Hirst J, Roessler MM (2016) Energy conversion, redox catalysis and generation of reactive oxygen species by respiratory complex I. Biochim Biophys Acta 1857: 872–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho DM, Artavanis‐Tsakonas S, Louvi A (2020) The notch pathway in CNS homeostasis and neurodegeneration. Wiley Interdiscip Rev Dev Biol 9: e358 [DOI] [PubMed] [Google Scholar]
- Hou Y, Wei Y, Lautrup S, Yang B, Wang Y, Cordonnier S, Mattson MP, Croteau DL, Bohr VA (2021) NAD(+) supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer's disease via cGAS‐STING. Proc Natl Acad Sci U S A 118: e2011226118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Pirinen E, Auwerx J (2012) Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol 13: 225–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur JH, Stork DA, Walker DW (2014) Complex‐I‐ty in aging. J Bioenerg Biomembr 46: 329–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Guarente L (2014) NAD+ and sirtuins in aging and disease. Trends Cell Biol 24: 464–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K, Liu F, Gong CX (2018) Recent developments with tau‐based drug discovery. Expert Opin Drug Discov 13: 399–410 [DOI] [PubMed] [Google Scholar]
- Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS et al (2012) Probing sporadic and familial Alzheimer's disease using induced pluripotent stem cells. Nature 482: 216–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha P, Wang X, Auwerx J (2016) Analysis of mitochondrial respiratory chain Supercomplexes using blue native polyacrylamide gel electrophoresis (BN‐PAGE). Curr Protoc Mouse Biol 6: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Stoyanovsky DA, Belikova NA, Tyurina YY, Zhao Q, Tungekar MA, Kapralova V, Huang Z, Mintz AH, Greenberger JS et al (2009) A mitochondria‐targeted triphenylphosphonium‐conjugated nitroxide functions as a radioprotector/mitigator. Radiat Res 172: 706–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusama S, Ueda R, Suda T, Nishihara S, Matsuura ET (2006) Involvement of drosophila Sir2‐like genes in the regulation of life span. Genes Genet Syst 81: 341–348 [DOI] [PubMed] [Google Scholar]
- Lauritzen I, Pardossi‐Piquard R, Bourgeois A, Becot A, Checler F (2019) Does intraneuronal accumulation of carboxyl‐terminal fragments of the amyloid precursor protein trigger early neurotoxicity in Alzheimer's disease? Curr Alzheimer Res 16: 453–457 [DOI] [PubMed] [Google Scholar]
- Lee SS, Lee RY, Fraser AG, Kamath RS, Ahringer J, Ruvkun G (2003) A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet 33: 40–48 [DOI] [PubMed] [Google Scholar]
- Li S, Wu Z, Li Y, Tantray I, De Stefani D, Mattarei A, Krishnan G, Gao FB, Vogel H, Lu B (2020a) Altered MICOS morphology and mitochondrial ion homeostasis contribute to poly(GR) toxicity associated with C9‐ALS/FTD. Cell Rep 32: 107989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Wu Z, Tantray I, Li Y, Chen S, Dong J, Glynn S, Vogel H, Snyder M, Lu B (2020b) Quality‐control mechanisms targeting translationally stalled and C‐terminally extended poly(GR) associated with ALS/FTD. Proc Natl Acad Sci U S A 117: 25104–25115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Fiskum G, Schubert D (2002) Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem 80: 780–787 [DOI] [PubMed] [Google Scholar]
- Lopez‐Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013) The hallmarks of aging. Cell 153: 1194–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B (2022) Translational regulation by ribosome‐associated quality control in neurodegenerative disease, cancer, and viral infection. Front Cell Dev Biol 10: 970654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Guo S (2020) Mechanisms linking mitochondrial dysfunction and Proteostasis failure. Trends Cell Biol 30: 317–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maranzana E, Barbero G, Falasca AI, Lenaz G, Genova ML (2013) Mitochondrial respiratory supercomplex association limits production of reactive oxygen species from complex I. Antioxid Redox Signal 19: 1469–1480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masek P, Worden K, Aso Y, Rubin GM, Keene AC (2015) A dopamine‐modulated neural circuit regulating aversive taste memory in drosophila. Curr Biol 25: 1535–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus MJ, Picard M, Chen HW, De Haas HJ, Potluri P, Leipzig J, Towheed A, Angelin A, Sengupta P, Morrow RM et al (2019) Mitochondrial DNA variation dictates expressivity and progression of nuclear DNA mutations causing cardiomyopathy. Cell Metab 29: 78–90.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, Tourlomousis P, Dabritz JHM, Gottlieb E, Latorre I et al (2016) Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167: 457–470.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwa S, Jow H, Baty K, Johnson A, Czapiewski R, Saretzki G, Treumann A, von Zglinicki T (2014) Low abundance of the matrix arm of complex I in mitochondria predicts longevity in mice. Nat Commun 5: 3837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Canto C, Mottis A, Jo YS, Viswanathan M, Schoonjans K et al (2013) The NAD(+)/Sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 154: 430–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murari A, Rhooms SK, Goparaju NS, Villanueva M, Owusu‐Ansah E (2020) An antibody toolbox to track complex I assembly defines AIF's mitochondrial function. J Cell Biol 219: e202001071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J 417: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman BL, Lundblad JR, Chen Y, Smolik SM (2002) A drosophila homologue of Sir2 modifies position‐effect variegation but does not affect life span. Genetics 162: 1675–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng F, Tang BL (2013) Sirtuins' modulation of autophagy. J Cell Physiol 228: 2262–2270 [DOI] [PubMed] [Google Scholar]
- Nixon RA (2017) Amyloid precursor protein and endosomal‐lysosomal dysfunction in Alzheimer's disease: inseparable partners in a multifactorial disease. FASEB J 31: 2729–2743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojha R, Tantray I, Rimal S, Mitra S, Cheshier S, Lu B (2022) Regulation of reverse electron transfer at mitochondrial complex I by unconventional notch action in cancer stem cells. Dev Cell 57: 260–276.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onukwufor JO, Berry BJ, Wojtovich AP (2019) Physiologic implications of reactive oxygen species production by mitochondrial complex I reverse electron transport. Antioxidants 8: 285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owusu‐Ansah E, Song W, Perrimon N (2013) Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell 155: 699–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajman L, Chwalek K, Sinclair DA (2018) Therapeutic potential of NAD‐boosting molecules: the in vivo evidence. Cell Metab 27: 529–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana A, Oliveira MP, Khamoui AV, Aparicio R, Rera M, Rossiter HB, Walker DW (2017) Promoting Drp1‐mediated mitochondrial fission in midlife prolongs healthy lifespan of Drosophila melanogaster. Nat Commun 8: 448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rera M, Bahadorani S, Cho J, Koehler CL, Ulgherait M, Hur JH, Ansari WS, Lo T Jr, Jones DL, Walker DW (2011) Modulation of longevity and tissue homeostasis by the Drosophila PGC‐1 homolog. Cell Metab 14: 623–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riera CE, Merkwirth C, De Magalhaes Filho CD, Dillin A (2016) Signaling networks determining life span. Annu Rev Biochem 85: 35–64 [DOI] [PubMed] [Google Scholar]
- Rimal S, Li Y, Vartak R, Geng J, Tantray I, Li S, Huh S, Vogel H, Glabe C, Grinberg LT (2021) Inefficient quality control of ribosome stalling during APP synthesis generates CAT‐tailed species that precipitate hallmarks of Alzheimer's disease. Acta Neuropathol Commun 9: 169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristow M, Zarse K (2010) How increased oxidative stress promotes longevity and metabolic health: the concept of mitochondrial hormesis (mitohormesis). Exp Gerontol 45: 410–418 [DOI] [PubMed] [Google Scholar]
- Schieber M, Chandel NS (2014) ROS function in redox signaling and oxidative stress. Curr Biol 24: R453–R462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scialo F, Sriram A, Fernandez‐Ayala D, Gubina N, Lohmus M, Nelson G, Logan A, Cooper HM, Navas P, Enriquez JA et al (2016) Mitochondrial ROS produced via reverse electron transport extend animal lifespan. Cell Metab 23: 725–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scialo F, Fernandez‐Ayala DJ, Sanz A (2017) Role of mitochondrial reverse electron transport in ROS signaling: potential roles in health and disease. Front Physiol 8: 428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N, Youle RJ, Finkel T (2016) The mitochondrial basis of aging. Mol Cell 61: 654–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow RH (2020) The mitochondrial hypothesis: dysfunction, bioenergetic defects, and the metabolic link to Alzheimer's disease. Int Rev Neurobiol 154: 207–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JR, Wood JG, Mizerak E, Hinthorn S, Liu J, Finn M, Gordon S, Zingas L, Chang C, Klein MA et al (2022) Sirt6 regulates lifespan in Drosophila melanogaster . Proc Natl Acad Sci U S A 119: e2111176119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC (2005) A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet 39: 359–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weick JP, Held DL, Bonadurer GF 3rd, Doers ME, Liu Y, Maguire C, Clark A, Knackert JA, Molinarolo K, Musser M et al (2013) Deficits in human trisomy 21 iPSCs and neurons. Proc Natl Acad Sci U S A 110: 9962–9967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, Sinclair D (2004) Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature 430: 686–689 [DOI] [PubMed] [Google Scholar]
- Wood JG, Schwer B, Wickremesinghe PC, Hartnett DA, Burhenn L, Garcia M, Li M, Verdin E, Helfand SL (2018) Sirt4 is a mitochondrial regulator of metabolism and lifespan in Drosophila melanogaster . Proc Natl Acad Sci U S A 115: 1564–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Tantray I, Lim J, Chen S, Li Y, Davis Z, Sitron C, Dong J, Gispert S, Auburger G et al (2019) MISTERMINATE mechanistically links mitochondrial dysfunction with proteostasis failure. Mol Cell 75: 835–848.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino J, Mills KF, Yoon MJ, Imai S (2011) Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet‐ and age‐induced diabetes in mice. Cell Metab 14: 528–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM (2007) Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53: 337–351 [DOI] [PubMed] [Google Scholar]
- Yun J, Finkel T (2014) Mitohormesis. Cell Metab 19: 757–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, Marro S, Patzke C, Acuna C, Covy J et al (2013) Rapid single‐step induction of functional neurons from human pluripotent stem cells. Neuron 78: 785–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Menzies KJ, Auwerx J (2018) The role of mitochondria in stem cell fate and aging. Development 145: dev143420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Hu Q, Cheng F, Su N, Wang A, Zou Y, Hu H, Chen X, Zhou HM, Huang X et al (2015) SoNar, a highly responsive NAD+/NADH sensor, allows high‐throughput metabolic screening of anti‐tumor agents. Cell Metab 21: 777–789 [DOI] [PMC free article] [PubMed] [Google Scholar]