

Abstract
We report the identification of a cell population that shares pericyte, stromal and stemness features, does not harbor the Kras G12D mutation and drives tumoral growth in vitro and in vivo. We term these cells pericyte stem cells (PeSCs) and define them as CD45−EPCAM−CD29+CD106+CD24+CD44+ cells. We perform studies with p48‐Cre;Kras G12D (KC), pdx1‐Cre;Kras G12D ;Ink4a/Arf fl/fl (KIC) and pdx1‐Cre;Kras G12D ;p53 R172H (KPC) and tumor tissues from PDAC and chronic pancreatitis patients. We also perform single‐cell RNAseq analysis and reveal a unique signature of PeSC. Under steady‐state conditions, PeSCs are barely detectable in the pancreas but present in the neoplastic microenvironment both in humans and mice. The coinjection of PeSCs and tumor epithelial cells leads to increased tumor growth, differentiation of Ly6G+ myeloid‐derived suppressor cells, and a decreased amount of F4/80+ macrophages and CD11c+ dendritic cells. This population induces resistance to anti‐PD‐1 immunotherapy when coinjected with epithelial tumor cells. Our data reveal the existence of a cell population that instructs immunosuppressive myeloid cell responses to bypass PD‐1 targeting and thus suggest potential new approaches for overcoming resistance to immunotherapy in clinical settings.
Keywords: myeloid‐derived suppressor cells, pancreatic cancer, PD‐1 therapy, pericyte stem cells
Subject Categories: Cancer, Immunology, Stem Cells & Regenerative Medicine
Pericyte stem cells (PeSCs) represent a newly identified cell population that shares pericyte, stromal and stemness features and drives tumoral growth in pancreatic cancer. This study reports on the role of these cells within the tumor microenvironment.
Introduction
The pancreatic tumor stroma consists of a variety of components, including fibroblasts, myofibroblasts, blood vessels, inflammatory and immune cells, extracellular matrix (ECM) and matricellular proteins (Neesse et al, 2011; Gore & Korc, 2014). High stromal activity, as assessed by α‐smooth muscle actin (α‐SMA) expression, is associated with poor prognosis in patients with pancreatic cancer (Erkan et al, 2008). Nevertheless, the depletion of α‐SMA‐expressing cells (Ozdemir et al, 2014) also resulted in undifferentiated PDA tumors and decreased survival in mice, suggesting that solely this parameter is not defining the whole complexity of the stromal activity. Fibrous proteins, such as collagens, laminin, and fibronectin, and noncollagenous proteins, such as glycoproteins, proteoglycans, and glycosaminoglycans, together make up the ECM that constitutes the stromal component. The cellular component of the stroma includes immune cells such as lymphocytes, macrophages, mast cells, and myeloid‐derived suppressor cells (MDSCs) along with vascular and neural elements (endothelial cells and neurons, respectively).
Accumulating evidence indicates the presence of a close and complex interplay of paracrine interactions between tumor cells and the stroma that facilitates cancer progression (Gore & Korc, 2014). Because the stromal compartment evolves rapidly starting from the beginning of the neoplastic reaction, we investigated the existence and role of stromal stem cells within the neoplastic microenvironment that are capable of driving the stromal reaction. Since the discovery of cancer stem cells (CSCs) in 1997 by Bonnet and Dick in leukemia (Bonnet & Dick, 1997), these cells have been found in several other types of solid tumors, including colon, breast, brain, skin, and pancreatic tumors. Here, we describe the identification of a cell population that shares pericyte, stem, and stromal and properties within the neoplastic microenvironment of pancreatic cancer. We have named these cells pericyte stem cells (PeSCs). We generated primary cell lines of PeSCs from mouse pancreatic neoplasms that stably express a fluorescent tag to assess the impact of the stromally elicited signals on tumor growth and the response to immunotherapy in pancreatic cancer.
Results
The CD106 + population is uniquely localized in pancreatic injury and precancerous lesions
CD106 has been proposed as a specific marker for defining mesenchymal stem cells in tissue repair and regeneration (Dominici et al, 2006; Acharya et al, 2013). Therefore, we performed IHC staining for CD106 in human PDAC samples (stages II and III). We found that CD106 positive cells were exclusively localized in the tumor adjacent stroma rather than in the tumor core (Fig 1A and Appendix Fig S1). We also observed CD106 positive cells in the intraductal papillary mucinous tumor (IPMT). No staining was detected in the normal pancreas or in the tumor core of PanIN III and IPMT (Fig EV1A and B). By identifying the tumor core versus the adjacent region of paired surgical samples, we verified that CD106 positive cells were labeled in all the adjacent regions of Pancreatic Intraepithelial Neoplasia (PanIN) I to PanIN III and of IPMT (Fig EV1B). Immunohistology staining of human tumor microarray (TMA) for CD106 further indicated that these cells were uniquely present in pancreatitis and precancerous early lesions PanINs rather than at the PDAC stage (Fig EV1C and Appendix Table S1), which is similar to the results obtained from our patient samples (Figs 1A and EV1B, and Appendix Fig S1). Next, we took advantage of the p48‐Cre;Kras G12D (KC) mouse model, which develops PanINs from the age of 1.5 months (Hingorani et al, 2003). We found that the CD106+ population was in close contact with αSMA+ cells (Fig 1B left) and absent in the CK19+ nascent duct region (Fig 1B right). To obtain further insights into their precise phenotype and frequencies, we used FACS staining to determine their presence in the pancreas of KC mice compared with that of WT mice. In our analysis, we excluded CD45+ cells (hematopoietic cells) and CD31+ cells (endothelial cells). The gated CD106+ cells represented 1.5% of the CD45−CD31− population and 0.15% of the total population of pancreatic cells but were not detected in the WT mice (Fig 1C). Furthermore, CD45−CD31−CD106+CD29+ cells also exhibited positive CD44 and CD24 expression and expressed no EPCAM and low levels of PDGRFα (which define duct cells and cancer‐associated fibroblasts, respectively; Fig 1C). Previous studies noted the expression of CD24, CD44, and EPCAM at the cell surface and identified CSCs in the pancreas (Li et al, 2007). This suggests that the CD106+ population might carry both stemness and stromal properties. Therefore, we next perform double immunofluorescence staining for CD24 and CD44 and we identified a population of cells located within the stromal compartment of the pancreas of KC mice that displayed a spindle‐shaped morphology (Fig 1D, left). This population was diminished in mice with PDAC, that is, pdx1‐Cre;Kras G12D ;Ink4a/Arf fl/fl (KIC; Unpublished observations, Wu), and pdx1‐Cre;Kras G12D ;p53 R172H (KPC) mice (Fig 1D, right), which develop adenocarcinoma beginning at 5 and 16 weeks of age, respectively (Aguirre et al, 2003; Hingorani et al, 2005).
Figure 1. The CD106+ population is uniquely localized in pancreatic injury and precancerous lesions.
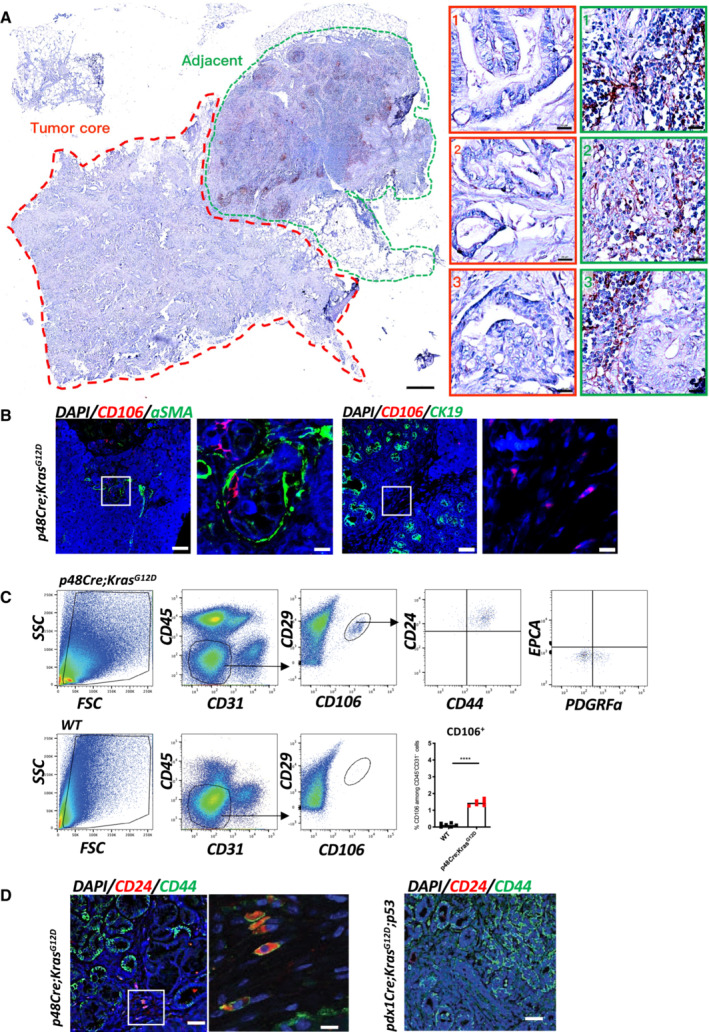
- Representative IHC staining of tumor core and adjacent region in PDAC patient (stage II) for CD106+. The corresponding magnification insets of the tumor core (red dotted line) or adjacent region (green dotted line) are displayed on the right. Scale bar, 500 μm in gross, 20 μm in magnification.
- Immunofluorescence staining of the pancreas obtained from a 2.5‐month‐old KC mouse for CD106 (red), α‐SMA (green) or CK19 (green), and DAPI (blue). Scale bar, 50 μm, 10 μm.
- Gating strategy for the identification of the CD106+CD24+CD44+ population in KC and WT mice. FACS analysis of the percentage of CD106+ among CD45−CD31− cells; each dot represents one mouse. The bars are representing the mean. The P‐values were calculated using Student's t‐test.
- Immunofluorescence staining of pancreases obtained from a KC mouse (left) and a 2.5‐month‐old KPC mouse (right) for CD44 (green), CD24 (red), and DAPI (blue). Scale bar, 50, 10 μm.
Source data are available online for this figure.
Figure EV1. The CD106+ population is uniquely localized to pancreatic injury and precancerous lesions.
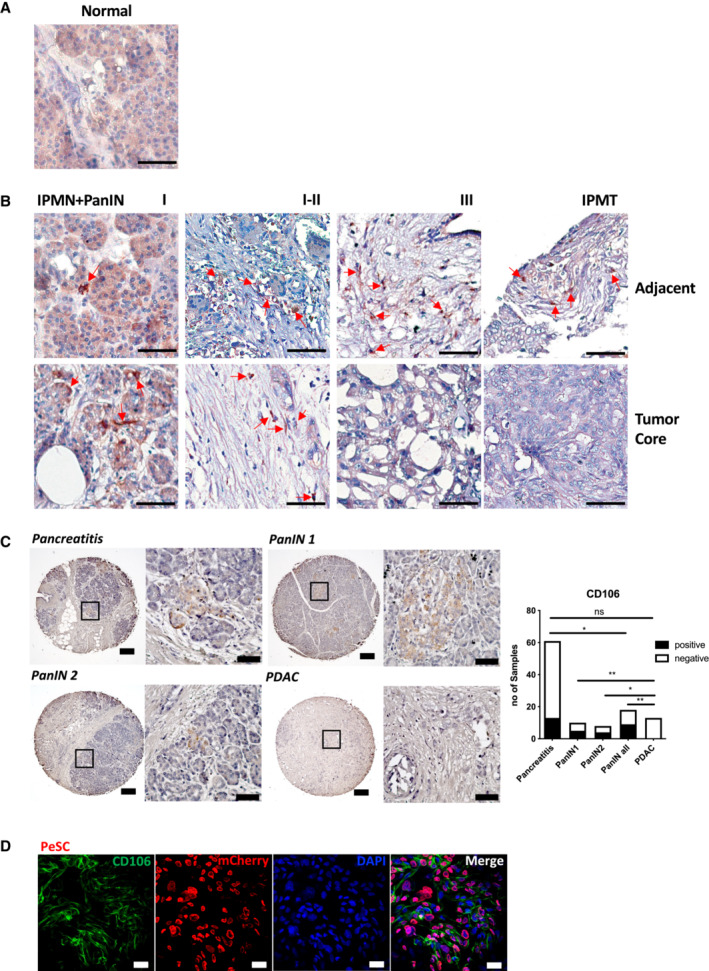
- Representative IHC staining for CD106+ in normal pancreas. Scale bar, 50 μm.
- Representative IHC staining for CD106+ in IPMN+PanIN I‐III and IPMT human samples. The adjacent region (above) and tumor core (below) were paired from the same patient respectively, corresponding to the diagnosis. The red arrow indicated the CD106+ spindle‐like cells. Scale bar, 50 μm.
- Representative IHC staining of pancreatic lesions of human TMA for CD106+. The magnification region indicates representative CD106+ cells. Scale bar, 200, 50 μm. Quantification of CD106 expression in pancreatitis, PanIN and PDAC lesions according to patients' pathological references of TMA (Appendix Table S1). *P < 0.05 and **P < 0.01. ****P < 0.0001. The P‐values were calculated using Student's t‐test.
- Representative IF staining of the PeSC cell line on a coverslip for CD106 (green), mCherry (red), and DAPI (blue). Scale bar, 50 μm.
To get further insight into the phenotype of this population, we then performed RNAseq single‐cell analysis. We have performed first a FACS sorting to obtain a fibroblast‐enriched fraction and a tumor duct forming PanINs. Pancreatic cells from 5 KC (2 months of age) mice were dissociated into single cells. From the pooled cells we isolated two fractions of cells by FACS: (i) a viable cell population of fibroblasts‐enriched population and (ii) a viable cell population of PanIN‐enriched population (Fig 2A and Appendix Fig S2). Sorted cells were subjected to the same droplet‐based protocol for single‐cell capture and library preparation. Since the identified CD106+ population was associated with PanINs, we analyzed the sequencing data from the viable cell PanIN enriched population (2,000 cells) isolated from the pooled KC pancreases were clustered in 10 clusters (Fig 2B). We analyzed the most expressed genes of each cluster (Fig 2C) and performed Wiki pathway identification_2019 and identified a unique population (cluster 7) that have stem properties, focal adhesion, and inflammatory response pathways (Fig 2D).
Figure 2. Single‐cell RNAseq analysis reveals a cell population distinct from the PanIN signature population.
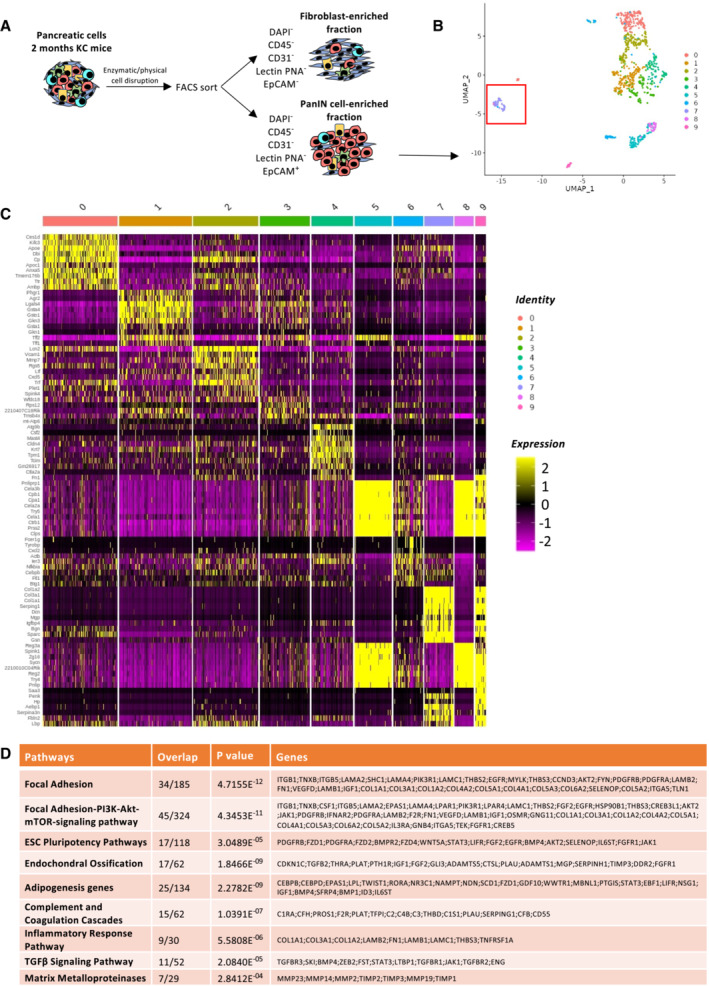
- Graphical scheme describing the workflow. Murine 2‐month KC pancreases were dissociated into single cells. Two fractions of cells were collected by FACS from each sample: (i) viable cell fraction (DAPI−) fibroblast‐enriched fraction (DAPI−, CD45−, CD31−, Lectin PNA,− and EPCAM−) and (ii) viable cell fraction (DAPI−) PanIN‐enriched fraction (DAPI−, CD45−, CD31−, Lectin PNA−, and EPCAM+). The sorted cells from each fraction were subjected to single‐cell capture, barcoding, and reverse transcription using the 10X Genomics platform.
- Unsupervised clustering UMAP representation of the PanIN enriched fraction. The red square is indicating cluster 7.
- Heat map showing scaled normalized expression of discriminative marker genes between the nine subclusters, with cells as columns and genes as rows.
- Wiki pathway identification_2019 analysis reveals nine most up‐regulated pathways. The number of overlapped genes of the pathways are indicated, as well as the P‐value and the names of the genes detected.
Source data are available online for this figure.
The CD106 + population shares pericyte, stem, and stromal features
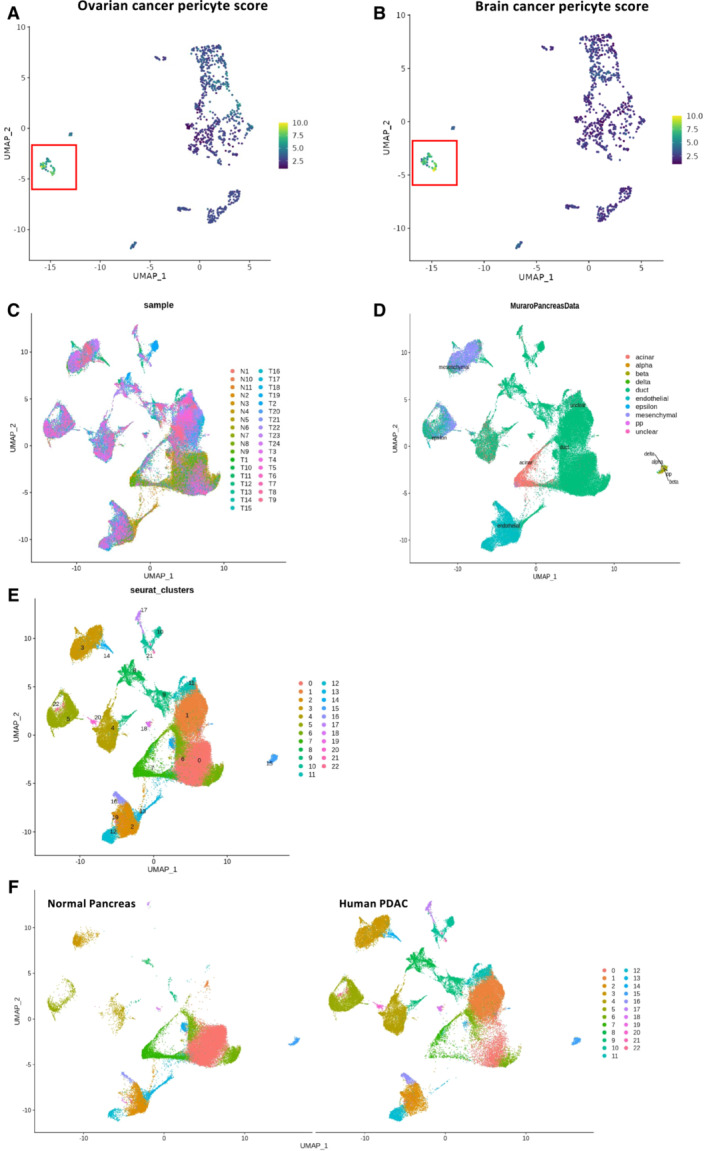
Because the frequency of the CD106+ cells was low in the neoplastic pancreas, we thought to amplify the cells by serum deprivation as previously described (Ferro et al, 2019). KC pancreases from 2.5‐month‐old mice were recovered and digested with collagenase, and a single‐cell suspension was cultured for 14 days in the absence of serum. After 14 days of serum starvation, the CD106+ population represented approximately 4% of the CD45−EPCAM−CD31− population (Appendix Fig S3A). We obtained similar results from two different pancreata of KC mice (Unpublished observations, Wu). To further characterize their properties, we decided to generate a primary cell line via several successive passages (up to passage 20) and infected the cells to stably express the mCherry tag (Figs 3A and EV1D, and Appendix Fig S3A and B). During passaging from P0 to P20, the cells underwent spontaneous dynamic selection of the CD106+ population from 15 to 90% (Figs 3A and EV1D, and Appendix Fig S3A–C). We defined these cells as PeSCs. We also generated an epithelial tumoral PanIN cell line (defined as Epi) and a lung metastasis cell line (defined as Meta) from the pancreas and lung of 2‐month‐old KIC mice, respectively, that express an eGFP tag (Appendix Fig S3D and E). We tested the presence of the floxed KrasG12D allele in the three cell lines and showed that the PeSCs, in contrast to the Epi and Meta cell lines, did not carry the recombined KrasG12D gene, which confirmed that the PeSCs were not of tumoral origin (Fig 3B). We also confirmed that PeSC does not express the KrasG12D mutated protein by Western Blot approach using an anti‐KrasG12D‐specific Ab (Fig 3C). A FACS phenotypic analysis confirmed that the PeSCs were CD44+CD24low, whereas the Epi and Meta cells were CD44+CD24high and CD44+CD24−, respectively, and clustered differently on a CD24 CD44 and CD24 CD106 dot plot (Fig 3D and Appendix Fig S3F). We further investigated their transcriptomic profile by RNA sequencing. The principal component analysis (PCA) and gene expression profile of the RNA‐seq data showed that the PeSCs exhibited an upregulated stemness and interferon gene signature compared with the Epi and Meta cells (Fig 3E and F). Furthermore, the PeSCs presented downregulated cell cycle genes compared with the Epi and Meta cells and did not exhibit epithelial characteristics (Fig 3F). We confirmed the RNAseq data through a qPCR analysis of several upregulated genes detected in the transcriptomic analysis (Fig 3G). We confirmed that Nanog and Oct3/4 were specifically upregulated in PeSCs and not in Epi and Meta cells. Furthermore, we detected an upregulation of genes associated with the pericyte signature in the PeSCs (Armulik et al, 2011). The transcriptomic analysis revealed that the RNA expression levels of NKX3.2, Anpep (encoding Anpep [alanyl (membrane) aminopeptidase]), Cd248, Eng (encoding endoglin), Pdgfrb (encoding PDGFRβ), and Rgs5 (encoding RGS5 [regulator of G‐protein signaling 5]) were higher in the PeSCs than in the Epi and Meta cells and we then confirmed their expression by qPCR and FACS staining (Fig 3G and Appendix Fig S3C). To define if the cells obtained were reflecting the population observed ex vivo, we have generated a PeSC score from the RNAseq data of the PeSC cell line and applied the score to the single‐cell RNAseq analysis. As shown in Fig 3H, the highest PeSC score is displayed by cluster 7 (Fig 2B). Cluster 7 stands for the minority population in this PanIN enriched fraction and their distribution indeed diverges from the other major clusters (cluster 1–6, 8, 9, Fig 2B and C). The detailed genes of the PeSC score are depicted in plots in Appendix Fig S4. We further checked the expression of EPCAM in the PeSC fraction and found that cluster 7 (PeSC fraction) had no expression of EPCAM (Appendix Fig S3G). As PeSC is in close contact with the PanIN lesions according to the staining on the tissue section (Figs 1B and EV1B), these cells are sorted with the ductal enriched fraction as contamination. Moreover, we show now that, in addition to our PeSC score, cluster 7 is enriched in pericyte scores independently (Fig EV2A and B) obtained from the literature (ovarian cancer pericytes; Sinha et al, 2016) as well as brain pericytes (Oudenaarden et al, 2022). More interestingly, we checked the distribution of this PeSC in the human PDAC context by applying the PeSC score to the public data of human PDAC single‐cell RNAseq. The database contains 35 samples (24 PDAC and 11 nontumor) and altogether over 80 k cells (Chen et al, 2021; Fig EV2C). The general identities of cell type were annotated based on Muraro's single‐cell dataset as a reference (Muraro et al, 2016; Fig EV2D). Unsupervised clustering showed 23 different clusters of cells in the integration of whole samples (Fig EV2E), as well as in grouping samples into normal pancreas or PDAC, respectively (Fig EV2F). Consistent with our findings in GEMMs, the PeSC population is significantly clustering in human PDAC with the highest PeSC scores among the mesenchymal populations (Fig EV2D) compared to the normal pancreas (Fig 3I). Altogether these data point out the existence of a new defined population both in PDAC GEMMs and human PDAC, distinct from PanIN epithelial cells and from fibroblast enriched fractions with unique stem properties. To determine their origin, we performed bone marrow chimeras. For this purpose, 3 weeks KC mice were irradiated and iv injected with bone marrow cells from a Tomato expressing mouse and sacrificed at day 60 (Fig EV3A). We found that the efficiency of reconstitution in the generated chimeras was around 50% of donor bone marrow in the periphery (Fig EV3B) and around 30% in the pancreas (Fig EV3B). We further analyzed the % CD106+ in the pancreas. We gated on CD45−CD106+ cells, and then quantified % of the Tomato+ cells. The % of Tomato+ among CD106+ cells was around 20–30% (Fig EV3C) in the pancreas, which is consistent with the % of the above‐mentioned reconstituted Tomato+ cells in the pancreas of the chimeras (Fig EV3B), suggesting that the majority of CD106 were of bone marrow origin. We verified by immunofluorescence staining that CD106 was coexpressed with Tomato in the stroma around PanIN lesions demonstrating the bone marrow origin of this population (Fig EV3D). Generally, stem cells are continuously losing their stemness during conventional ex vivo 2‐D culture (Banerjee & Bhonde, 2006). Hanging drop spheroid assay has been described to verify cell stemness in vitro (Banerjee & Bhonde, 2006), indicating that only stem cells are capable of forming spheroids. Furthermore, by seeding these stem cells trypsinized from spheroids in the culture plates, their proliferation to confluency indicates retention of their self‐duplication capacity. We performed 2‐D and 3‐D hanging drop combination assay using PeSC cell lines in both early and late passages. We showed that either PeSC early passages (P1) or late passages (P20) were able to form spheroids by hanging drop assay (Fig 4A and B), while the Epi cells did not (Fig 4B). Moreover, with the notion that PeSC shares stem cell characteristics in the gene expression profile, we tested their ability to differentiate into multiple cell lineages. We cultured the cells in specific chondrogenic and osteogenic media and determined that PeSCs had the potential to differentiate into cartilage and bone cells (Fig 4C). We then questioned whether the PeSCs influenced the proliferation of tumor cells in vitro. For this purpose, we cocultured PeSCs with the Epi cell line in vitro at different ratios to determine the impact of PeSCs on tumor epithelial cell growth. We observed that PeSCs significantly potentiated Epi cell proliferation when the cells were cocultured at Epi cells:PeSC ratios as low as 10:1 or 100:1 (Figs 4D and EV4A). These IncuCyte analyses were confirmed by Ki67 intracellular staining and FACS analysis (Fig EV4B). In contrast, we found that the Epi cell line had no impact on PeSC proliferation under the same conditions (Fig EV4C and D). Because pericytes are mural cells capable of sustaining vessel formation, we performed an in vitro analysis of vessel formation using C57BL/6 mouse primary pancreatic microvascular endothelial cells under 3D conditions (Fig 4E). PeSCs had the potential to sustain vessel formation in 3D cell culture conditions. Moreover, in the presence of Epi cells, PeSC was able to structure the vessel in structures containing the three cell types (Fig 4E inset Endo‐Epi‐PeSC). A large variety of anti‐ and pro‐angiogenic growth factors, extracellular matrix proteins, proteases, and adhesion molecules interacting with multiple cells and tissues are required for angiogenesis. It has been described that the vasculature was reinforced and the endothelial cells were protected by maintaining a balance between the endothelial cells and pericytes controlled by the PDGF‐BB signaling pathway operating in a paracrine manner to keep pericytes in the quiescent state. Pericytes turned into an activated state to migrate by the cause of PDGF‐BB expression disruption, dissociating pericyte‐endothelial connections in injury (Wong et al, 2015). To further study the impact of PDGF‐BB on PeSCs, we treated PeSCs with PDGF‐BB + TGF‐β for 7 days and perform q‐PCR together with FACS analysis. We found that the expression of stroma/CAF‐related genes and markers, that is, PDGFRα, PDGFRβ, α‐SMA, Desmin and CD61 were significantly diminished after the treatment at both RNA (Fig 4F) and protein level (Fig 4G). Furthermore, several EMT‐related genes were also down‐regulated by the treatment (Fig EV4E). This indicated that PeSCs could be transformed to the quiescent state to sustain the vasculature by PDGF‐BB as described in the literature.
Figure 3. The CD106+ population shares pericyte, stem cell and stromal features.
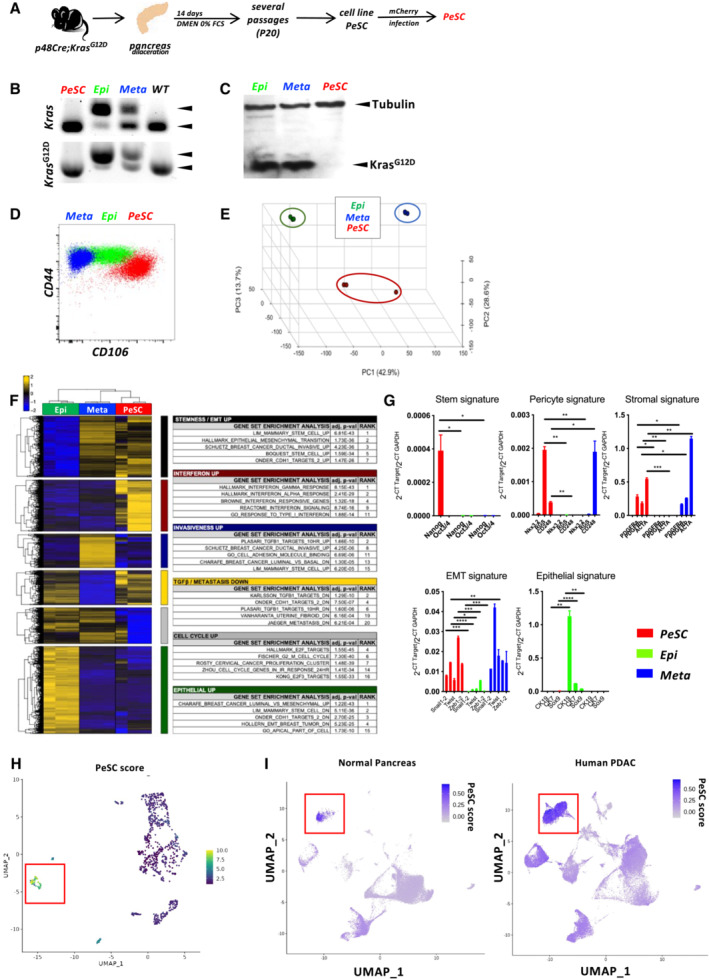
- Schematic representation of the experimental setting for PeSC line generation.
- PCR for genomic Kras and KrasG12D.
- Western Blot analysis for tubulin and mutated KrasG12D proteins in total proteins extracts of PeSC, Epi, and Meta cells.
- Representative dot plot of the CD44 and CD24 staining of PeSCs and Epi and Meta cells.
- Principal component analysis (PCA) of RNA‐seq data from the indicated cell populations (n = 3).
- Pathway enrichment of a list of genes.
- qPCR analysis of several genes identified in the RNAseq analysis. Treatment was performed in biological triplicates and qPCR analysis was performed by technical duplicates. The bars are representing the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. The P‐values were calculated using Student's t‐test.
- Definition of the PeSC score (bulk RNAseq) applied to the single‐cell RNAseq analysis and identification of cluster 7.
- Application of PeSC score to the human PDAC public single‐cell RNAseq database and identification of a cluster with the highest PeSC (red square) among the mesenchymal population (Fig EV2D) in either Normal Pancreas (left) or Human PDAC (right).
Source data are available online for this figure.
Figure EV2. PeSC signature in human cancer single‐cell data.
-
A, BApplication of ovarian cancer pericyte (A) and brain cancer pericyte score (B) to our mouse single‐cell RNAseq analysis showing enrichment in the unique cluster of PeSC.
-
CPooled single‐cell RNAseq data to UMAP analysis from 35 samples (11 normal pancreas and 24 PDAC patients), including a total number of over 80,000 cells. The UMAP shows the distribution of each cell cluster.
-
DCell types of each cluster were identified based on Muraro's single‐cell dataset as a reference.
-
E, FUnsupervised clustering UMAP analysis distinguishes 23 subclusters in the integration of whole 35 samples (E), pooled from 11 normal pancreas samples and 24 PDAC samples (F).
Figure EV3. Chimera approach identifies the origin of PeSC.
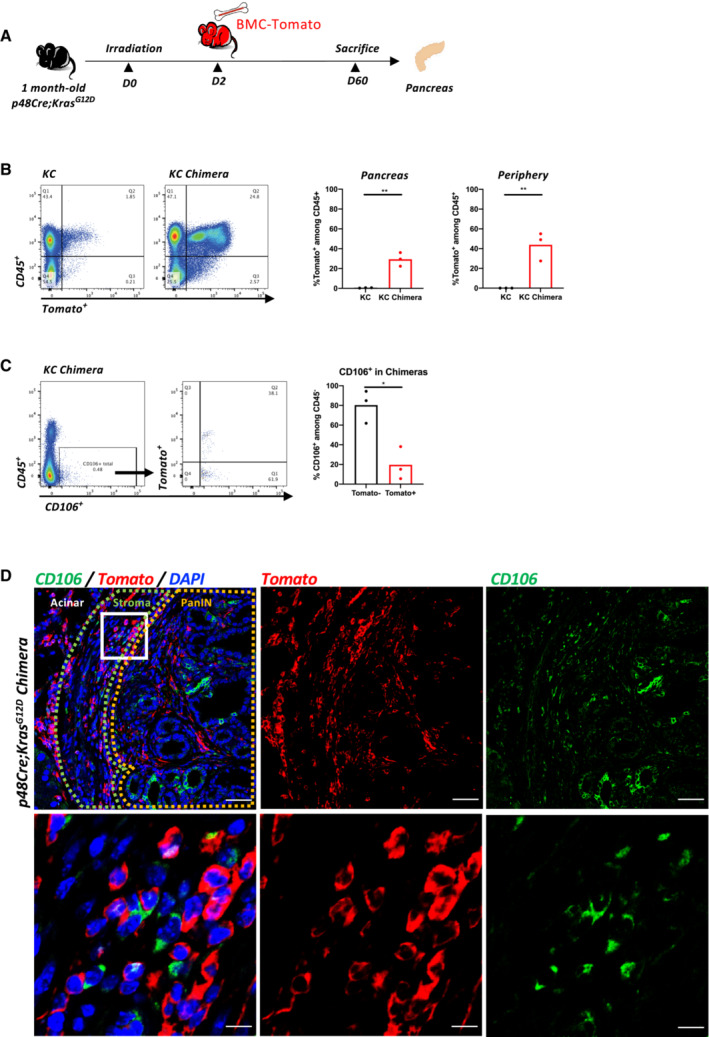
- Experimental protocol for bone marrow chimeras. One month old KC mice were sublethally irradiated and reconstituted with bone marrow cells from a Tomato expressing mouse. After reconstitution, the chimeras were sacrificed on day 60.
- Representative dot plots of reconstituted bone marrow cells (Tomato+) in KC chimeras compared to the nonreconstituted KC mice as control by FACS analysis. Quantification of reconstituted cells (Tomato+) among CD45+ immune cells in either pancreas or periphery of KC chimeras compared to the control. Three mice in each condition.
- Representative dot plots and gating strategy of distinguishing the reconstituted CD106+ cells (Tomato+) and nonreconstituted CD106+ cells (Tomato−) in KC chimeras. Three mice in the group, each dot represents one mouse.
- Representative IF staining for CD106 (green) and Tomato (red) in the pancreas of chimera mice. Inset shows colocalization of CD106‐ and Tomato‐stained cells. Scale bar, 50 μm (upper), 10 μm (lower).
Data information: ns not significant, *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001. The P‐values were calculated using Student's t‐test.
Figure 4. Stem properties of PeSCs and their impact on tumor cell proliferation on vessel formation.
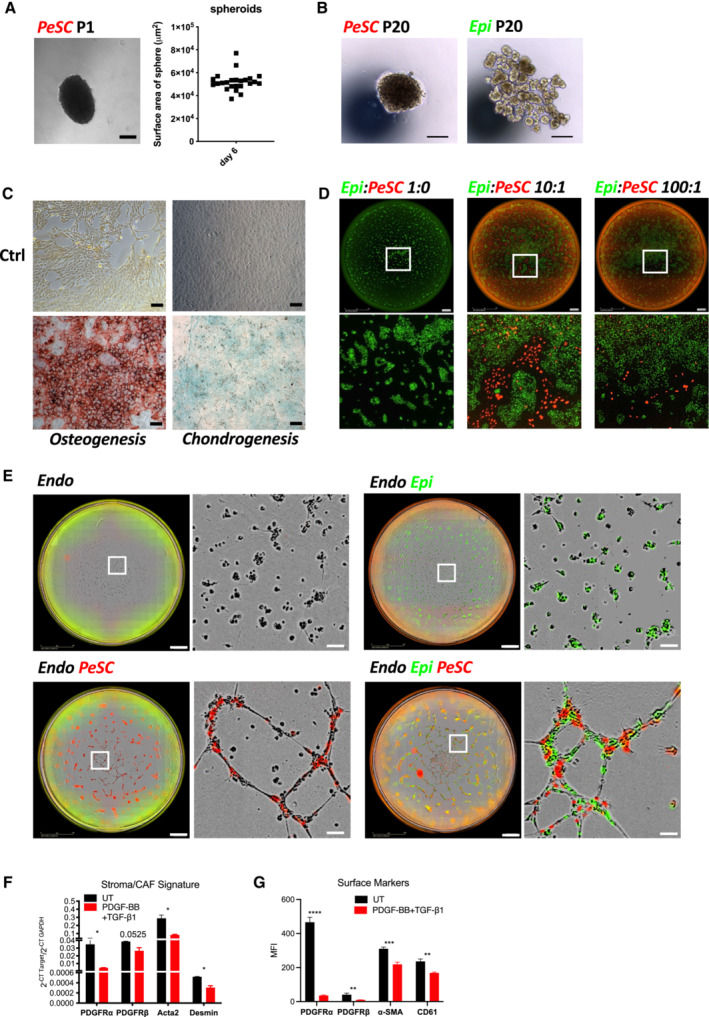
- Representative image of PeSC spheroid in early passages (P1) performed by hanging drop approach after 6 days. Scale bar, 100 μm. Quantification of PeSC spheroids surface area. Unit, μm2.
- Representative images of spheroid formed by PeSCs and cell clusters formed by Epi cells, respectively, in late passages (P20). Scale bar, 100 μm.
- Representative Alizarin Red S staining (left) and Alcian blue staining (right) of PeSCs in a 12‐well culture plate observed with a 10× optical lens. Scale bar, 200 μm.
- Representative full‐well view (upper Scale bar, 1.4 mm) and 5× magnification (lower) of cocultures of Epi cells + PeSCs at the indicated ratio in 96‐well plates observed by IncuCyte after 48 h.
- Pericyte property of PeSCs impact tube vessel formation. Representative 3D culture illustration of the cell lines in the indicated condition: primary pancreatic endothelial cells (Endo), Endo + Epi, Endo + PeSC, and Endo + Epi + PeSC. The tube forming test has been performed twice. Scale bar, 500 μm, insets 1,500 μm.
- qPCR analysis of stroma/CAF‐related genes expressed by PeSCs after the treatment of PDGF‐BB + TGF‐β. Treatment was performed in biological triplicates and qPCR analysis was performed by technical duplicates. The bars are representing the mean ± SD.
- FACS analysis of stroma/CAF‐related surface markers expressed by PeSC after the treatment of PDGF‐BB + TGF‐β. Treatment and FACS analysis were both performed by biological triplicates. The bars are representing the mean ± SD. *P < 0.05, ***P < 0.001, and ****P < 0.0001. The P‐values were calculated using Student's t‐test.
Source data are available online for this figure.
Figure EV4. Stem properties of PeSCs and their impact on tumor cells.
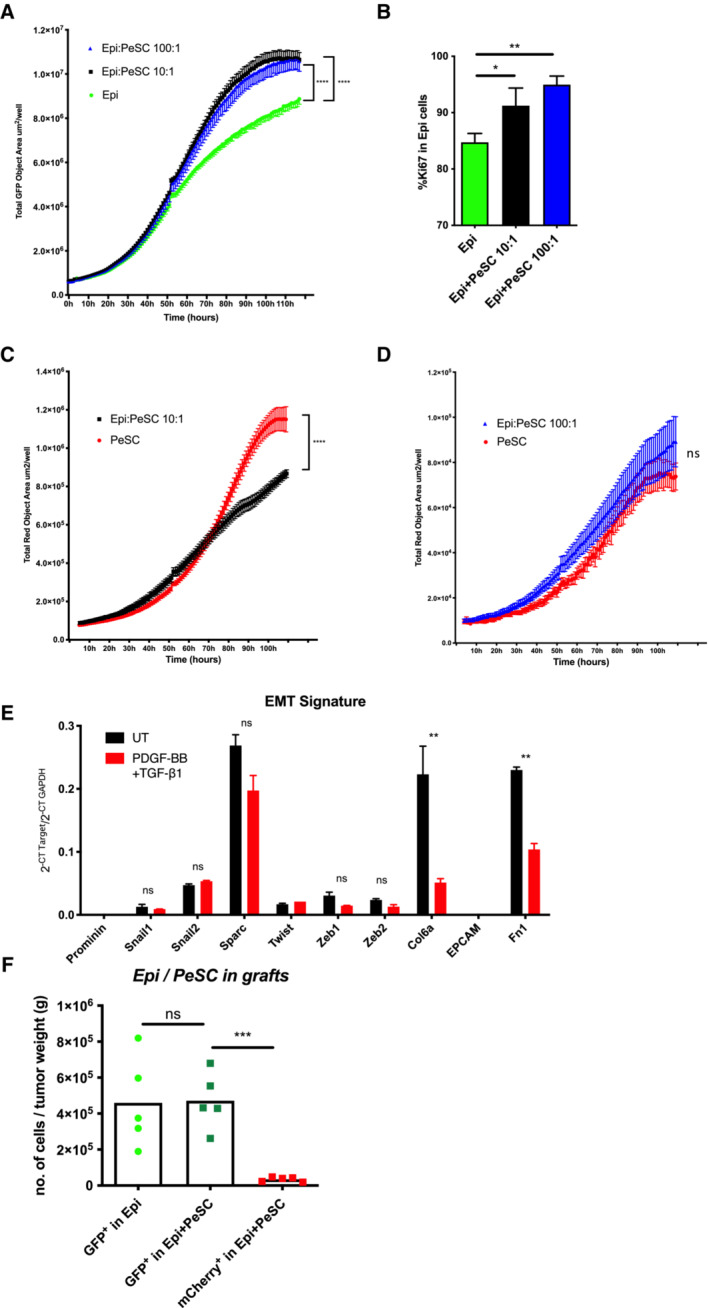
-
AProliferation curves of Epi cells in the indicated culture conditions based on the analysis of total green (GFP) object area in each well observed by IncuCyte. The data of each curve are summarized from six biological replicates of each condition. The bars are representing the mean ± SD.
-
BFACS analysis of the percentage of Ki67+ Epi cells in each culture condition at 48 h. FACS analysis was performed by biological triplicates corresponding to the indicated culture condition.
-
C, DThe bars are representing the mean ± SD (C, D) Proliferation curves of PeSCs in the indicated culture conditions based on the analysis of the total red (mCherry) object area in each well observed by IncuCyte. The data of each curve are summarized from six biological replicates of each condition. The bars are representing the mean ± SD.
-
EqPCR analysis of EMT‐related genes expressed by PeSCs after the treatment of PDGF‐BB and TGF‐β compared to the untreated ones (UT). Treatment was performed in biological triplicates and qPCR analysis was performed by technical duplicates. The bars are representing the mean ± SD.
-
FQuantification of Epi (GFP+) or PeSC (mCherry+) cells in the s.c. tumor grafts in the indicated conditions based on FACS analysis. Five mice in each group.
Data information: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. The P‐values were calculated using Student's t‐test.
Altogether, these data suggest the identification of a pericyte stromal cell with stemness, self‐renewal, asymmetric division and differentiation character and the ability to increase epithelial tumor cell proliferation in vitro.
PeSCs lead to increased tumor growth and the accumulation of Ly6G + myeloid‐derived suppressor cells in vivo
Because PeSCs potentiated the in vitro proliferation of Epi cells, we subsequently sought to investigate the impact of PeSCs on epithelial tumor growth in vivo. To explore this issue, we injected s.c. Epi cells alone (one flank) or in the presence of PeSCs (the other flank) in Matrigel plugs (Fig 5A) at a 1:1 ratio in Rag2KO recipient mice. We sacrificed the mice 10 days after the injection and found that the coinjection of Epi with PeSCs induced an increased tumor weight compared with the injection of Epi cells alone (Fig 5B). By quantifying the number of grafted cells on day 10, we found that although we injected the equivalent number of cells of both Epi and PeSC, the amount of PeSC is about 1% of the Epi cells, which is similar to the observed level of PeSC in KC pancreas (Figs EV4F and 1C). This confirmed that the increase in graft weight is predominantly due to the proliferation of Epi cells in both Epi alone and Epi + PeSC condition rather than the proliferative contribution by PeSCs. The percentage of recruited CD45+ hematopoietic cells obtained with the coinjection of Epi cells PeSCs were higher than those obtained with the injection of Epi cells alone (Fig 5C), and among the CD45+ recruited cells, a markedly higher proportion of Ly6G+ MDSCs (defined as CD11b+Ly6G+) was obtained with the coinjection of Epi cells and PeSCs (Fig 5D). As described in the literature, PDAC is characterized by abundant myeloid cell infiltrates that predominantly include monocytes, granulocytes, and macrophages (Mitchem et al, 2013). These infiltrates are associated with immunosuppression, fibrosis, T‐cell dysfunction, and poor prognosis in patients with PDAC (DeNardo et al, 2011; Nywening et al, 2018). The F4/80+ macrophage and CD11c+ dendritic cell populations were strongly reduced (Fig 5E and F), and the expression of the costimulatory molecules CD80 and CD86 on the surface of macrophages and dendritic cells were also markedly decreased (Fig 5G). The effect on the Ly6G MDSC, F4/80, and CD11c populations was observed even with very low numbers of injected PeSCs (i.e., Epi cell:PeSC ratio of 100:1, Unpublished observations, Wu). Furthermore, we performed immunofluorescence staining with anti‐CK19 Ab to label epithelial PanIN duct cells and anti‐mCherry and anti‐GFP Abs to identify implanted cells (PeSCs and Epi) on graft sections (Fig 5H). We observed that Epi cells formed PanIN structures similar to those found on the pancreas of KC mice (Figs 5H and 1B right). In the presence of PeSCs, PanIN structures displayed a loss of nuclear polarity, which suggested the development of more advanced lesions (Fig 5H). We confirmed the change in PanIN structures by immunofluorescence using ZO‐1 and E‐cad staining. As previously described (Polette et al, 2007), we found that ZO‐1 and Ecad expression shifted from tight junctions to the cytoplasm of duct cells (Fig 5I). Altogether, these results indicate that PeSCs influence tumor progression with a concomitant loss of nuclear polarization in epithelial tumor cells in vivo by inducing reprogramming of the innate immune cell microenvironment, and this reprogramming was characterized by increases in Gr‐MDSCs and reductions in F4/80+ macrophages and mature CD11c+ dendritic cells. Because these three populations share CD11b expression, we hypothesized that PeSCs might impact CD11b differentiation upon recruitment to the tumor microenvironment. To test this hypothesis, we sought to determine whether PeSCs promote the differentiation of monocytes into Gr‐MDSCs expressing Ly6G+ rather than macrophages and dendritic cells. For this purpose, we isolated and cultured mouse bone marrow precursors (Fig 6A) and tested the effects of coculturing PeSCs, Epi cells or Epi cells + PeSCs with bone marrow precursors. After the coculture of bone marrow cells in the presence of PeSCs or Epi cells alone, approximately 15–20% of the bone marrow cells were Ly6G+CD11b+, whereas this population was strongly increased in the coculture with Epi cells + PeSCs (Fig 6B and C). Extended analysis of the cytokine/chemokine production by LegendPlex assay pointed out an increased production of CCL2 and CCL5 but also CCL20, CXCL1, CXCL5, and CXCL10 (Appendix Fig S5A–H).
Figure 5. In vivo injection of PeSCs in the context of epithelial tumors induce Ly6G+ MDSCs in the microenvironment.
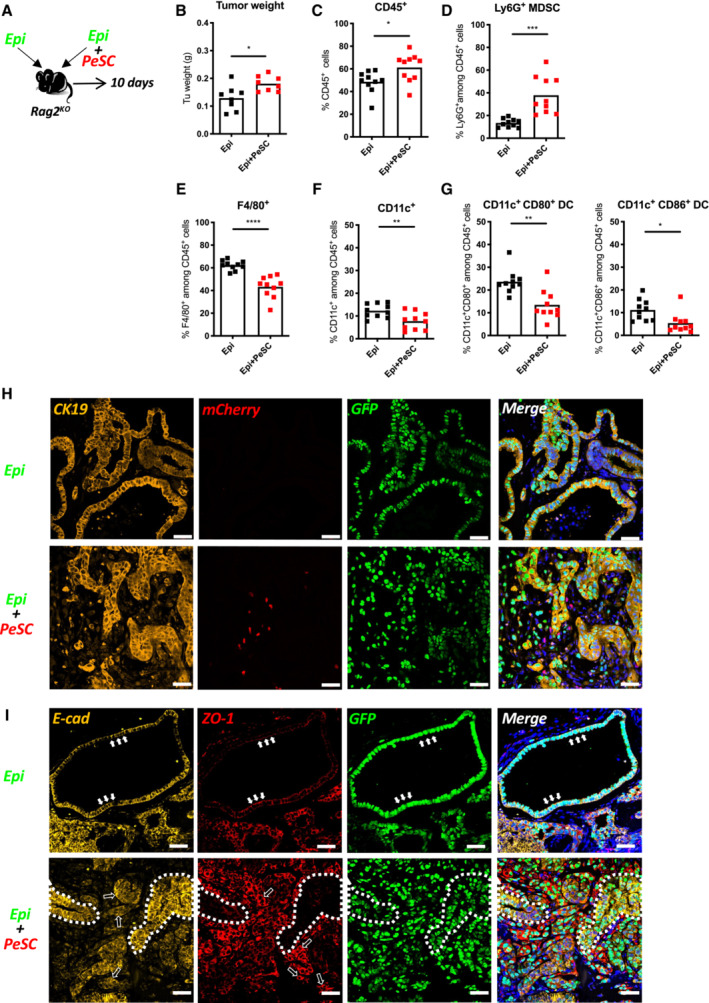
-
AExperimental setting.
-
B–G(B) Tumor weight. FACS analysis of the percentages of CD45+ cells (C), Ly6G+CD45+ MDSCs (D), F4/80+CD45+ Macrophages (E), CD11c+CD45+ DCs (F), and CD11c+CD80+/CD11c+CD86+ DCs (G). The results shown are cumulative from three independent experiments (each dot represents one mouse, 12–15 mice per group).
-
H, IRepresentative immunofluorescence staining of implanted tumors in Rag2KO mice for CK19, mCherry, GFP and DAPI (H) and E‐cadherin, ZO‐1 and GFP (I). Solid white arrows indicated the representative cells which expressed E‐cadherin and ZO‐1 in the cell junctions. Hollow white arrows indicated the representative cells whose E‐cadherin or ZO‐1 expression was shifting to the cytoplasm. The white dotted line indicated two representative areas where E‐cadherin and ZO‐1 were reversely expressed. Scale bar, 50 mm. *P < 0.05, **P < 0.01 and ***P < 0.001. The P‐values were calculated using Student's t‐test.
Source data are available online for this figure.
Figure 6. Tumor‐PeSCs crosstalk induces Ly6G+ MDSC differentiation.
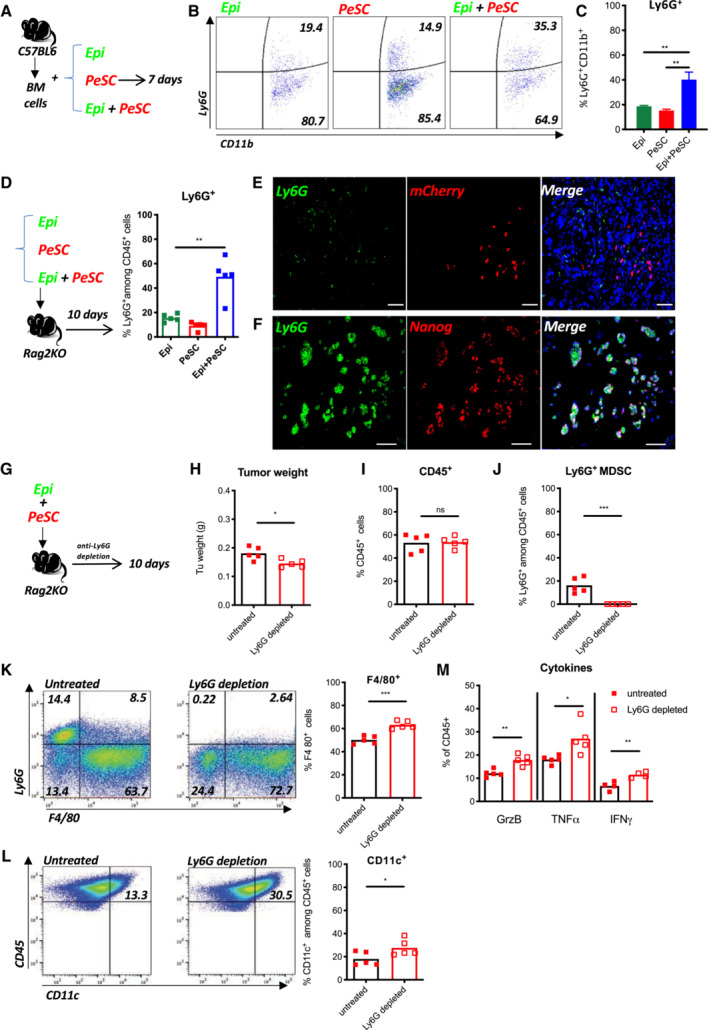
-
AExperimental setting of bone marrow differentiation.
-
BRepresentative dot plots of CD11b and Ly6G staining in bone marrow cells cultured in the presence of Epi cells, PeSCs or both Epi cells, and PeSCs.
-
CQuantification of the percentage of Ly6G+ cells among CD11b+ cells (cumulative from three independent experiments). The bars are representing the mean ± SD.
-
DExperimental setting for in vivo injection. FACS analysis of the percentages of Ly6G+CD45+ cells (each dot represents one mouse per group).
-
E, FRepresentative immunofluorescence staining of coimplanted Epi cells + PeSCs for Ly6G, mCherry, Nanog, and DAPI. Scale bar, 50 mm.
-
G–MIn vivo depletion of Ly6G+ MDSCs diminishes tumor growth. (G) Experimental setting. (H) Tumor weight. FACS analysis of the percentage of CD45+ cells (I), and Ly6G+ MDSCs (J) derived from the tumor. (K) Representative dot plots of Ly6G‐ and F4/80‐stained CD45+ cells. FACS analysis of the percentage of F4/80+ cells. (L) Representative dot plots of CD45‐ and CD11c‐stained cells. FACS analysis of the percentage of CD11c+ cells among CD45+ cells. (M) FACS analysis of GrzB+, TNFα+, and IFNγ+ cells among CD45+ cells. Eight mice in each group, five for FACS analysis and three for histological analysis. Each dot represents one mouse.
Data information: *P < 0.05, **P < 0.01, and ***P < 0.001. The P‐values were calculated using Student's t‐test.
Source data are available online for this figure.
Furthermore, we analyze the accumulation of the Ly6G+CD11b+ population in three s.c. grafting condition, respectively, that is, Epi, PeSC, or Epi + PeSC, in the Rag2KO mice. The result revealed that the increased amount of Ly6G+CD11b+ was only obtained in the Epi + PeSC condition (Fig 6D), and these Ly6G+ cells were in close contact with the mCherry+ PeSCs observed by IF staining of the graft section (Fig 6E) and express Nanog (Fig 6F). Furthermore, the use of anti‐CCL5‐depleting Ab did not affect Ly6G+CD11b+ differentiation but did affect macrophage differentiation, which expressed less CD103+ in vitro and in vivo (Appendix Fig S5I and J), and these findings suggest a role for CCL5 in the local instruction of macrophages and residency. The use of anti‐CD106‐depleting Ab did not affect Ly6G+CD11b+ differentiation or macrophage differentiation but reduced the tumor weight and number of PeSC and Epi GFP tumor cells in the tumor graft (Fig EV5A and B). Altogether, these data demonstrate that the presence of PeSCs increased the tumor weight and favored the accumulation of Ly6G+CD11b+ within the tumor microenvironment. To determine whether Ly6G+ MDSCs are required for the in vivo effects of PeSCs, we coinjected Epi cells and PeSCs subcutaneously and treated the mice with an anti‐Ly6G‐depleting Ab (Fig 6G). We observed a significant decrease in the tumor weight (Fig 6H) with a similar PeSC amount (Appendix Fig S6D). The depletion was effective because we detected no Ly6G+ cells in the tumor grafts (Fig 6J). Similar percentages of CD45+ cells were observed in both conditions (Fig 6I). In contrast, we detected increased populations of F4/80+ macrophages and CD11c+ dendritic cells (Fig 6K and L). We also detected increases in the populations of NKp46+ and Ly6C+ cells, whereas the percentage of CD206+ among F4/80 cells were decreased (Appendix Fig S6A–C). More importantly, we detected increased antitumoral cytokine production, including TNFα, IFNγ, and cytotoxic GrzB (Fig 6M). Altogether, these results indicate that Ly6G+ MDSCs play an important role in the immunosuppressive role of PeSCs.
Figure EV5. Tumor‐PeSC crosstalk affects the immune system.
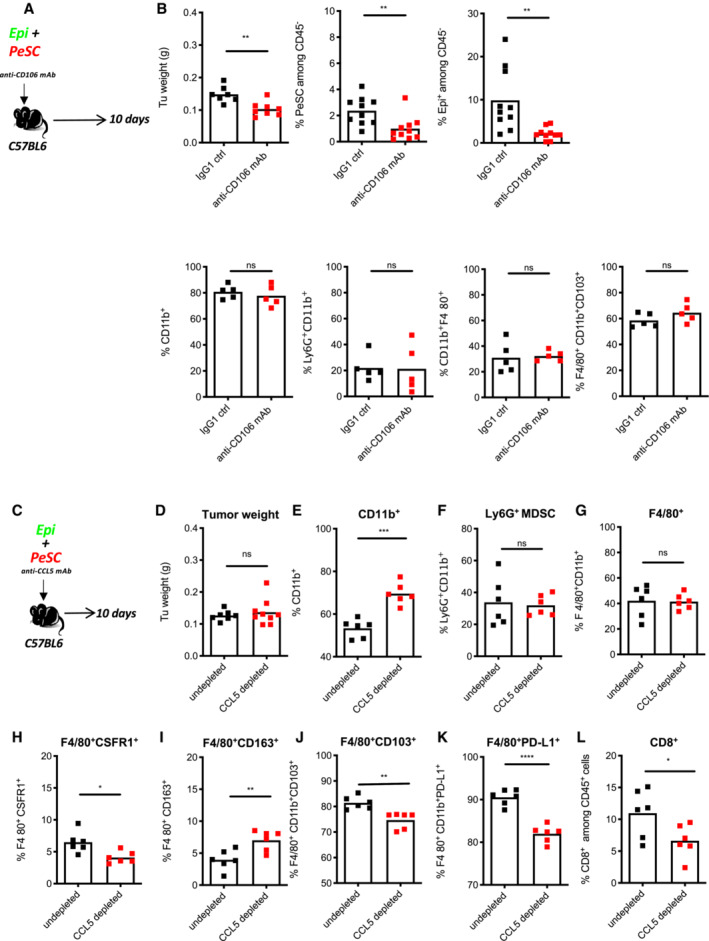
-
A, BIn vivo neutralization of CD106: (A) Experiment setting. (B) Tumor weight, FACS analysis of % PeSC+ among CD45− cells, % Epi+ among CD45− cells, % CD11b+ among CD45+ cells, % Ly6G+CD11b+ among CD45+ cells, % CD11b+F4/80+ among CD45+ cells, % CD11b+F4/80+CD103+ among CD45+ cells (eight mice in each group, five for FACS analysis, and three for histological analysis. Quantification of PeSCs and Epi cells were cumulative from two FACS panels as technical duplicates).
-
C–LIn vivo depletion of CCL5 impact the macrophages differentiation: (C) Experimental setting. (D) Tumor weight of the grafts formed by Epi + PeSC in CCL5‐depleted conditions compared with undepleted conditions. FACS analysis of the percentage of CD11b+ cells (E), Ly6G+CD11b+ (F), F4/80+CD11b+ (G), F4/80+CSFR1+ (H), F4/80+CD163+ (I), F4/80+CD11b+CD103+ (J), F4/80+CD11b+PD‐L1+ (K) and CD8+ cells (L) among CD45+ cells derived from the tumor grafts. Ten mice in each group, six for FACS analysis and four for histological analysis.
Data information: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. The P‐values were calculated using Student's t‐test.
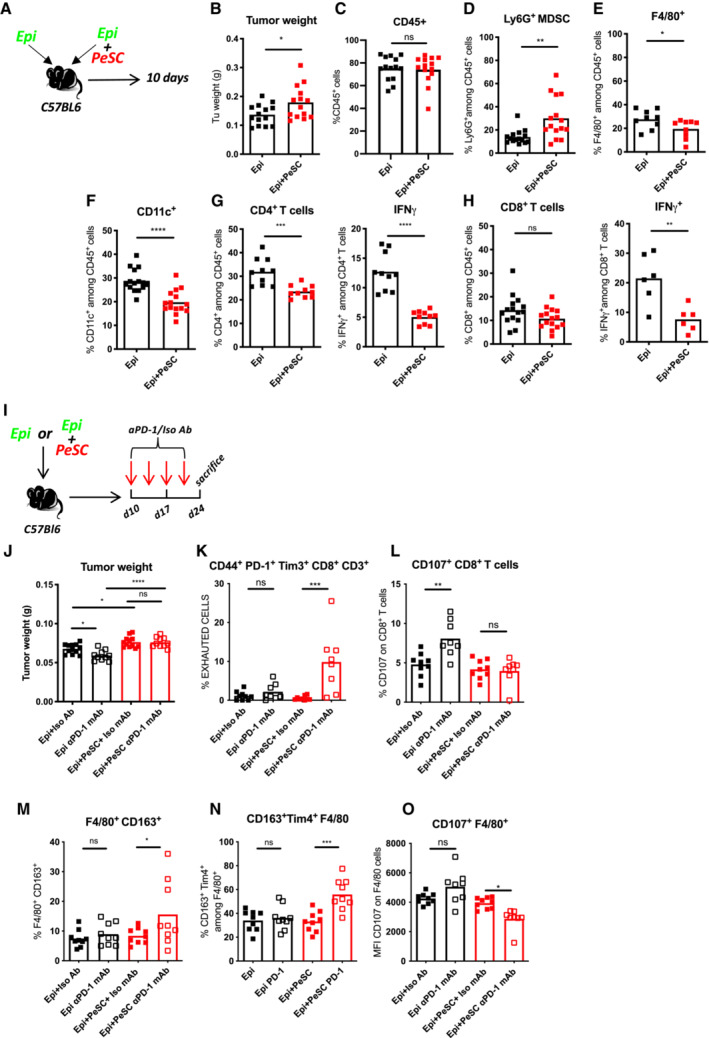
Previous studies have shown that the targeted depletion of MDSCs unmasks pancreatic ductal adenocarcinoma to adaptive immunity (Stromnes et al, 2014). We then questioned whether PeSCs impact the innate immune response in fully immune‐competent mice. To explore this hypothesis, we injected s.c. Epi cells alone (one flank) or in the presence of PeSCs (the other flank) in Matrigel plugs (Fig 7A) at a 1:1 ratio in C57BL/6 immunocompetent recipient mice. Similar to the results obtained with Rag2KO recipient mice, we found that the coinjection of Epi cells with PeSCs induced an increase in the tumor weight compared with that obtained with the injection of Epi cells alone (Fig 7B). Although the percentage of CD45+ hematopoietic cells was not increased with the injection of Epi cells + PeSC as previously found in Rag2KO mice (Figs 7C and 5C), the percentage of Ly6G+ MDSC population was unanimously increased (Fig 7D). Moreover, as previously described, the populations of F4/80+ macrophages and CD11c+ dendritic cells expressing the costimulatory molecule CD86 were strongly reduced (Fig 7E and F). Furthermore, a markedly lower percentage of CD4+ T cells and a lower production of IFNγ were obtained with the injection of Epi cells + PeSCs compared with the injection of Epi cells alone (Fig 7G). The CD8+ T cell compartment was also affected but to a lesser extent (nonsignificant reduction in the percentage of CD8+ T cells), and a significant reduction in the percentage of IFNγ among CD8+ T cells was detected (Fig 7H). This effect was partially due to the local accumulation of the CCL5 chemokine because the depletion of this chemokine in vivo diminishes PD‐L1 expression in F4/80 cells and decreases CD8+ T cells without affecting the Ly6G+CD11b+ compartment (Fig EV5C–L). Altogether, these results suggest that the presence of PeSCs in the tumor microenvironment reprogram CD11b+ monocyte differentiation to result in diminished T cell priming and activation.
Figure 7. PeSCs drive PD‐1 resistance in the tumor microenvironment.
-
AExperimental setting.
-
B–H(B) Tumor weight. FACS analysis of the percentage of CD45+ cells derived from the tumor (C), Ly6G+ MDSCs (D), F4/80+CD45+ cells (E), CD11c+CD45+ cells (F), CD4+CD45+ and IFNγ+CD4+ (G) and CD8+CD45+ and IFNγ+CD8+ (H). The results shown are cumulative from three independent experiments (each dot represents one mouse, 12–15 mice per group).
-
IExperimental setting.
-
J–O(J) Tumor weight. FACS analysis of CD44+PD‐1+Tim3+CD8+CD3+ T cells (K), CD107+CD8+ T cells (L), F4/80+CD163+ Macrophages (M), CD163+ Tim4+ Macrophages (N) and CD107+ Macrophages (O). The results shown are cumulative from three independent experiments (each dot represents one mouse, 9–12 mice per group).
Data information: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. The P‐values were calculated using Student's t‐test.
Source data are available online for this figure.
PeSCs drive resistance to PD‐1 treatment in pancreatic cancer
Extensive immunosuppressive myeloid cell infiltration in PDAC (Bayne et al, 2012; Pylayeva‐Gupta et al, 2012; Stromnes et al, 2014) tissues has been shown to be associated with resistance to immunotherapy (Blair et al, 2019). Strategies that target monocyte or granulocyte trafficking or macrophage survival (Zhu et al, 2014) in combination with checkpoint immunotherapies have shown promise in preclinical studies, and these studies have transitioned into ongoing clinical trials for the treatment of pancreatic and other cancer types. Because the presence of PeSCs in the tumor microenvironment suppressed the T cell response, the next step was to determine whether PeSCs influence the response to immune therapy, that is, anti‐PD‐1 Ab treatment.
To test this hypothesis, we injected s.c. Epi cells alone or in the presence of PeSCs at a ratio of 1:1 into the same C57BL/6 recipient mice. Ten days later, we injected anti‐PD1 or ctrl isotype Ab twice a week for 2 weeks and sacrificed the mice 14 days later (Fig 7I).
We found that mice injected with Epi cells alone responded to anti‐PD‐1 therapy because they displayed a diminished tumor weight compared to isotype ctrl‐treated animals in contrast to the coinjection of Epi + PeSC with led to equivalent tumor weight (Fig 7J). The analysis of the immune compartment in each condition showed that CD8+ T cells displayed an exhausted phenotype characterized by concomitant expression of CD44, PD‐1, and Tim‐3 (Fig 7K). Furthermore, CD8+ T cells have an increased ability to produce CD107a upon anti‐PD1 treatment solely in Epi conditions but not in combo conditions (Fig 7L). With respect to the macrophage response, we observed increased percentages of F4/80 expressing CD163 as well as both CD163 and Tim4 in anti‐PD‐1 treated combo condition (Fig 7M and N). These macrophages were expressing less CD107a (Fig 7O). Altogether these data suggest that PeSC drives PD‐1 resistance by affecting the monocyte differentiation in situ.
Discussion
The role of the stromal compartment in regulating antitumoral immune responses and tumoral growth is indisputable (Hanahan & Weinberg, 2011), but this “ménage‐à‐trois” tumor–stromal–immune cell interaction is a difficult process to fine‐tune with the aim of achieving a positive therapeutic outcome. Here, we describe the existence of a cell population that emerges very early during the process of transformation, shares features of pericyte, stromal, and stem cells and is able to modulate and pervert the interaction between tumor cells and the innate immune system from the beginning of neoplasia. This population is defined by the expression of CD106, CD29, CD24, and CD44 and the exclusion of CD45 and EPCAM. This population, which harbors neoplastic lesions in mice and humans, has the capacity to instruct recruited monocytes to become granulocytic MDSCs “in situ” and skew the outcome of the antitumoral T cell response by reducing their inflammatory cytokine production and therefore sustaining exhaustion and ultimately driving resistance to conventional anti‐PD‐1 therapy.
Pericytes of perivascular cells are important cells that have been previously described to regulate the T cell response in different locations (in the brain and lung; Balabanov et al, 1996) and pathophysiological contexts (acute respiratory distress syndrome). However, their role in the tumoral progression is not well understood. In the context of glioblastoma, pericytes are able to support tumor growth through immune suppression (Valdor et al, 2017; Guerra et al, 2018; Sena et al, 2018). Recent studies revealed that the cancer burden is controlled by mural cells expressing integrin β3 in the context of an implanted B16 melanoma model (Wong et al, 2020). The researchers showed that CCL2 secretion is important for MEK1 and ROCK2 activity in tumor cells. In our context, even though both CCL2 and CCL5 were secreted by PeSCs in the proximity of tumor cells, CCL5 appears to play a more important local effect because the neutralization of CCL5 led to decreased CD103 and PD‐L1 expression on F4/80 macrophages. CCL20, CXCL1, CXCL5, and CXCL10 were also found increased suggesting a complex orchestration of chemokines that needs further investigation. Our data showed that PeSCs were able to structure the vessels in vitro similarly to the pericyte functions in tissue repair related to pancreatic injury in our model. These CD106+ cells likely play an important role in the context of neoplastic transformation because we detected them in a human pancreas TMA and in the GEMM mice in the PanIN areas. One limitation of the study comes from the fact that we coinject Epi cells with PeSC at a ratio of 1:1 which does not reflect the biological cellular relationships within the tissue. Nevertheless, the percentage of PeSC in the grafts represented about 1% of the Epi cells amount at day 10 when the mice are sacrificed. This level is similar to the in vivo PeSC observation in the KC pancreas. Because tumors evolve as heterogeneous in vivo, our preliminary IHC staining on human samples verified that the majority of CD106+ cells are indeed distributed in the tumor adjacent regions, indicating their contribution greatly correlated to tumor initiation from the early stage.
One important feature of the identified PeSC population is their “stem” cell property, which allows these cells to adapt to the local signals in the tumor microenvironment. We demonstrated that these cells were able to directly increase the proliferation of tumor cells, whereas the tumor cells did not have feedback on their proliferation status, at least in vitro. Furthermore, the cells were able to differentiate into chondrocytes and osteoblasts, and the choice of the cell was highly related to their location in the pancreas (Van Noorden et al, 1995; Pilarsky et al, 2008).
Because these cells are present at the beginning of the transformation in the pancreas, the next question was related to the crosstalk of PeSCs with the early actors of the immune response, that is, the innate immune response. The early bone marrow recruitment of monocyte precursors in the context of neoplasia is a well‐documented process (Movahedi et al, 2010; Franklin et al, 2014). The plasticity of PeSCs cells in the tumor microenvironment represents a double‐edged sword mechanism. Recent data demonstrate that the reprogramming of the innate immune compartment by CD11b agonism can render tumors more sensitive to checkpoint blockade (Panni et al, 2019), which indicates the importance of early events in the kinetics of the immune response. CD11b+Ly6G+ MDSCs are associated with immune suppression in several cancer types, and the GM‐CSF cytokine plays an important role as a driver mechanism (Pylayeva‐Gupta et al, 2012; Stromnes et al, 2014). IL6 production also plays an important role in the observed effect (Weber et al, 2020). Our data revealed no differences in the production of those cytokines from PeSCs in the TME. The suppressive function of MDSCs has previously been linked to their immature state (Gabrilovich et al, 2012). In our model, we found that a vast majority of the Ly6G+ cells are Nanog+, which sustains their immature phenotype (Fig 6F). The depletion of the Ly6G+ population through an Ab approach led to increased antitumoral cytokine production and a decrease in tumor weight. Furthermore, this depletion was accompanied by restoration of the F4/80+ and CD11c+ percentages in the TME, which suggests that the CD11b+ precursors of these three populations play a key role in mediating the effect of PeSCs on the modulation of the tumor microenvironment.
Last but not least, we discovered that this initial interaction between PeSCs and innate CD11b+ precursors has an impact on the adaptive immune response by impacting the quantity and quality of the T cell immune response. We found a drastic reduction in the percentage of CD4+ T cells and the production of IFNγ by CD8+ T cells in the presence of PeSCs in the tumor microenvironment. This reduction was associated with an increase in the Ly6G+CD11b+ population in the tumor microenvironment and led to anti‐PD1 resistance. One important finding is that Epi tumor cells respond to anti‐PD1 treatment, which suggests that PD‐1 resistance in pancreatic cancer is not driven by the paucity of tumor antigen presentation but rather local immune suppressive signals, which include MDSCs (Stromnes et al, 2014) and stromal exclusion (Jiang et al, 2016, 2020).
Our study provides the first demonstration that CD106+ PeSCs are able to target the innate CD11b compartment to drive the differentiation of Ly6G+ MDSCs and inhibit the antitumoral differentiation of F4/80 macrophages and CD11c+ dendritic cells, which ultimately leads to PD‐1 immunotherapy resistance (Appendix Fig S7). Further studies should identify strategies for targeting this population in the tumor microenvironment to unleash the potential of existing immunotherapy for pancreatic cancer.
Materials and Methods
Reagents and Tools table
| Reagent/Resource | Reference or Source | Identifier or Catalog Number |
|---|---|---|
| Experimental Models | ||
| Pericyte stem cells | This paper | N/A |
| Tumoral epithelial cells (Epi) | Goehrig et al (2019) | Goehrig et al (2019) |
| Lung metastatic cells (Meta) | This paper | N/A |
| C57BL/6 mouse primary pancreatic microvascular endothelial cells | Cell Biologics | Cat#C57‐6206 |
| C57BL/6 mice (wild type) | Envigo | C57bL/6JOlaHsd |
| Rag2KO mice | Charles Rivers | |
| P48+/Cre;KrasG12D (KC) mice | Hingorani et al (2003) | See reference |
| pdx1+/Cre;KrasG12D;p53R172H (KPC) mice | Hingorani et al (2005) | See reference |
| pdx1+/Cre;KrasG12D;Ink4a/Arffl/fl (KIC) mice | Aguirre et al (2003) | See reference |
| Recombinant DNA | ||
| H2B‐GFP | Addgene | Cat#25999, RRID:Addgene_25999 |
| H2B‐mCherry | Addgene | Cat#20972, RRID:Addgene_20972 |
| Antibodies | ||
| Mouse monoclonal anti‐CD24 antibody (clone SN3b) | Invitrogen/Thermo Fisher Scientific | Cat# MA5‐11833, RRID:AB_10985938 |
| Rabbit polyclonal anti‐CD44 antibody | Abcam | Cat# ab157107, RRID:AB_2847859 |
| Rabbit monoclonal recombinant anti‐VCAM1 (CD106) antibody (clone EPR5047) | Abcam | Cat# ab134047, RRID:AB_2721053 |
| Goat polyclonal anti‐alpha smooth muscle actin antibody, N‐term | GeneTex | Cat# GTX89701, RRID:AB_10721877 |
| Rat monoclonal anti‐mouse cytokeratin 19 antibody, unconjugated | DSHB | Cat# TROMA‐III, RRID:AB_2133570 |
| Rabbit monoclonal recombinant anti‐ZO1 tight junction protein antibody (clone EPR19945‐296) | Abcam | Cat# ab221547, RRID:AB_2892660 |
| Rat monoclonal anti‐E‐cadherin antibody (clone ECCD‐2) | Invitrogen/Thermo Fisher Scientific | Cat# 13‐1900, RRID:AB_2533005 |
| Rabbit polyclonal anti‐mCherry antibody | Abcam | Cat# ab167453, RRID:AB_2571870 |
| Goat polyclonal anti‐GFP antibody | Abcam | Cat# ab6673, RRID:AB_305643 |
| Rat monoclonal purified anti‐mouse Ly‐6G antibody (clone 1A8) | BioLegend | Cat# 127602, RRID:AB_1089180 |
| Rabbit polyclonal StemAb™ anti‐mouse Nanog antibody | ReproCell Incorporated | Cat# RCAB002P‐F |
| Horse anti‐goat IgG antibody (H+L), biotinylated | Vector Laboratories/Eurobio Scientific | Cat# BA‐9500, RRID:AB_2336123 |
| Goat anti‐mouse IgG antibody (H+L), biotinylated | Vector Laboratories/Eurobio Scientific | Cat# BA‐9200, RRID:AB_2336171 |
| Horse anti‐rabbit IgG antibody (H+L), biotinylated | Vector Laboratories/Eurobio Scientific | Cat# BA‐1100, RRID:AB_2336201 |
| Goat anti‐rat IgG antibody, mouse adsorbed (H+L), biotinylated | Vector Laboratories/Eurobio Scientific | Cat# BA‐9401, RRID:AB_2336208 |
| Chicken polyclonal anti‐Rat IgG (H+L) Cross‐Adsorbed Secondary Antibody, Alexa Fluor 647 | Invitrogen/Thermo Fisher Scientific | Cat# A‐21472, RRID:AB_2535875 |
| Donkey polyclonal anti‐Rat IgG (H+L) Highly Cross‐Adsorbed Secondary Antibody, Alexa Fluor 488 | Invitrogen/Thermo Fisher Scientific | Cat# A‐21208, RRID:AB_2535794 |
| Donkey polyclonal anti‐Rabbit IgG (H+L) Highly Cross‐Adsorbed Secondary Antibody, Alexa Fluor 555 | Invitrogen/Thermo Fisher Scientific | Cat# A‐31572, RRID:AB_162543 |
| Donkey polyclonal anti‐Rabbit IgG (H+L) Highly Cross‐Adsorbed Secondary Antibody, Alexa Fluor 488 | Invitrogen/Thermo Fisher Scientific | Cat# A‐21206, RRID:AB_2535792 |
| Donkey polyclonal anti‐Goat IgG (H+L) Cross‐Adsorbed Secondary Antibody, Alexa Fluor 488 | Invitrogen/Thermo Fisher Scientific | Cat# A‐11055, RRID:AB_2534102 |
| Donkey polyclonal anti‐Mouse IgG (H+L) Highly Cross‐Adsorbed Secondary Antibody, Alexa Fluor 555 | Invitrogen/Thermo Fisher Scientific | Cat# A‐31570, RRID:AB_2536180 |
| Rat monoclonal anti‐mouse CD45 (clone 30‐F11), APC/Cyanine7 conjugated | BioLegend | Cat# 103116, RRID:AB_312981 |
| Rat monoclonal anti‐mouse CD45 (clone 30‐F11), APC conjugated, eBioscience | Invitrogen/Thermo Fisher Scientific | Cat# 17‐0451‐82, RRID:AB_469392 |
| Rat monoclonal anti‐mouse CD31 (clone 390), Brilliant Violet 421 conjugated | BioLegend | Cat# 102423, RRID:AB_2562186 |
| Rat monoclonal anti‐mouse/rat CD29 (clone HMβ1‐1), PerCP/Cyanine5.5 conjugated | BioLegend | Cat# 102228, RRID:AB_2572079 |
| Rat monoclonal anti‐mouse CD106 (clone 429 [MVCAM.A]), Alexa Fluor 647 conjugated | BioLegend | Cat# 105712, RRID:AB_493429 |
| Rat monoclonal anti‐mouse CD24 (clone M1/69), PE/Cyanine7 conjugated | BioLegend | Cat# 101822, RRID:AB_756048 |
| Rat monoclonal anti‐mouse/human CD44 (clone IM7), Alexa Fluor 700 conjugated | BioLegend | Cat# 103026, RRID:AB_493713 |
| Rat monoclonal anti‐mouse PDGFRα (clone APA5), Super Bright 600 conjugated, eBioscience | Invitrogen/Thermo Fisher Scientific | Cat# 63‐1401‐82, RRID:AB_2734880 |
| Rat monoclonal anti‐mouse CD326 (Ep‐CAM) (clone G8.8), Brilliant Violet 510 conjugated | BioLegend | Cat# 118231, RRID:AB_2632774 |
| Rat monoclonal anti‐mouse PDGFRβ (clone APB5), Super Bright 780 conjugated | Invitrogen/Thermo Fisher Scientific | Cat# 78‐1402‐80, RRID:AB_2784899 |
| Armenian hamster monoclonal anti‐mouse CD34 (clone HM34), PE conjugated | BioLegend | Cat# 128610, RRID:AB_2074601 |
| Rat monoclonal anti‐mouse CD90.2 (clone 30‐H12), Brilliant Violet 570 conjugated | BioLegend | Cat# 105329, RRID:AB_10917055 |
| Rat monoclonal anti‐mouse CD105 (clone MJ7/18), Pacific Blue conjugated | BioLegend | Cat# 120412, RRID:AB_2098890 |
| Rat monoclonal anti‐mouse CD146 (clone ME‐9F1), PerCP/Cyanine5.5 conjugated | BioLegend | Cat# 134710, RRID:AB_11203708 |
| Mouse monoclonal anti‐mouse/human/rat αSMA (clone 1A4), eFluor 570 conjugated, eBioscience | Invitrogen/Thermo Fisher Scientific | Cat# 41‐9760‐82, RRID:AB_2573631 |
| Armenian hamster monoclonal anti‐mouse/rat CD61 (clone 2C9.G2 [HMβ3‐1]), PE/Cyanine7 conjugated | BioLegend | Cat# 104318, RRID:AB_2687361 |
| Rat monoclonal anti‐mouse Ly‐6G (clone 1A8), Brilliant Violet 785 conjugated | BioLegend | Cat# 127645, RRID:AB_2566317 |
| Rat monoclonal anti‐mouse Ly‐6C (clone HK1.4), PE conjugated | BioLegend | Cat# 128008, RRID:AB_1186132 |
| Rat monoclonal anti‐mouse F4/80 (clone BM8), PE/Cyanine7 conjugated | BioLegend | Cat# 123114, RRID:AB_893478 |
| Rat monoclonal anti‐mouse F4/80 (clone BM8), PerCP/Cyanine5.5 conjugated | BioLegend | Cat# 123128, RRID:AB_893484 |
| Armenian hamster monoclonal anti‐mouse CD11c (clone N418), Alexa Fluor 700 conjugated, eBioscience | Invitrogen/Thermo Fisher Scientific | Cat# 56‐0114‐82, RRID:AB_493992 |
| Armenian hamster monoclonal anti‐mouse CD80 (clone 16‐10A1), Brilliant Violet 605 conjugated | BioLegend | Cat# 104729, RRID:AB_11126141 |
| Rat monoclonal anti‐mouse CD86 (clone GL‐1), APC/Cyanine7 conjugated | BioLegend | Cat# 105030, RRID:AB_2244452 |
| Rat monoclonal anti‐mouse/human CD11b (clone M1/70), Brilliant Violet 421 conjugated | BioLegend | Cat# 101251, RRID: AB_2562904 |
| Human/mouse monoclonal anti‐human/mouse Granzyme B (clone GB11), Alexa Fluor 647 conjugated | BioLegend | Cat#515406, RRID:AB 2566333 |
| Rat monoclonal anti‐mouse Granzyme B (clone 16G6), Biotin, eBioscience | Invitrogen/Thermo Fisher Scientific | Cat# 13‐8822‐80, RRID:AB_466954 |
| Rat monoclonal anti‐mouse TNF‐α (clone MP6‐XT22), Brilliant Violet 605 conjugated | BioLegend | Cat# 506329, RRID:AB_11123912 |
| Rat monoclonal anti‐mouse IFN‐γ (clone XMG1.2), Brillant Violet 650 conjugated | BioLegend | Cat#505831, RRID:AB_11142685 |
| Rat monoclonal anti‐mouse CD4 (clone RM4‐5), Brilliant Violet 510 conjugated | BioLegend | Cat# 100553, RRID:AB_2561388 |
| Rat monoclonal anti‐mouse CD8a (clone 53‐6.7), Brilliant Violet 421 conjugated | BioLegend | Cat# 100738, RRID:AB_11204079 |
| Rat monoclonal anti‐mouse CD279 (PD‐1) (clone 29F.1A12), PE conjugated | BioLegend | Cat# 135206, RRID:AB_1877231 |
| Rat monoclonal anti‐mouse CD366 (Tim‐3) (clone B8.2C12), APC conjugated | BioLegend | Cat# 134008, RRID:AB_2562998 |
| Rat monoclonal anti‐mouse CD3 (clone 17A2), Brilliant Violet 711 conjugated | BioLegend | Cat# 100241, RRID:AB_2563945 |
| Rat monoclonal anti‐mouse CD107a (LAMP‐1) (clone 1D4B), PE/Cyanine7 conjugated | BioLegend | Cat# 121619, RRID:AB_2562146 |
| Rat monoclonal anti‐mouse CD163 (clone TNKUPJ), PE conjugated, eBioscience | Invitrogen/Thermo Fisher Scientific | Cat# 12‐1631‐82, RRID:AB_2716924 |
| Rat monoclonal anti‐mouse Tim‐4 (clone RMT4‐54), Alexa Fluor 647 conjugated | BioLegend | Cat# 130008, RRID:AB_2271648 |
| Armenian Hamster monoclonal anti‐mouse CD103 (clone 2E7), Brilliant Violet 510 conjugated | BioLegend | Cat# 121423, RRID:AB_2562713 |
| Rat monoclonal anti‐mouse CD335 (NKp46) (clone 29A1.4), Brilliant Violet 421 conjugated | BioLegend | Cat# 137611, RRID:AB_10915472 |
| Rat monoclonal anti‐mouse CD206 (MMR) (clone C068C2), Brilliant Violet 650 conjugated | BioLegend | Cat# 141723, RRID:AB_2562445 |
| Rat monoclonal anti‐mouse CD115 (CSF‐1R) (clone AFS98), PerCP/Cyanine5.5 conjugated | BioLegend | Cat# 135526, RRID:AB_2566462 |
| Rat monoclonal anti‐mouse CD274 (B7‐H1, PD‐L1) (clone 10F.9G2), PerCP/Cyanine5.5 conjugated | BioLegend | Cat# 124334, RRID:AB_2629832 |
| Lectin PNA from arachis hypogaea (peanut), Alexa Fluor 647 conjugate | Invitrogen/Thermo Fisher Scientific | Cat# L32460 |
| InVivoMAb mouse IgG1 isotype control (clone MOPC‐21) | Bio X cell | Cat# BE0083, RRID:AB_1107784 |
| InVivoMab anti‐mouse CD106 (VCAM‐1) | Bio X Cell | Cat# BE0027, RRID:AB_1107572 |
| InVivoPlus anti‐mouse PD‐1 (CD279) | Bio X Cell | Cat# BE0146, RRID:AB_10949053 |
| InVivoPlus anti‐mouse Ly6G antibody | Bio X Cell | Cat# BE0075‐1, RRID:AB_1107721 |
| Mouse IgG2A isotype control (clone 20102) | R&D systems | Cat# MAB003, RRID:AB_357345 |
| Mouse CCL5/RANTES Antibody | R&D Systems | Cat# MAB478, RRID:AB_2290968 |
| Ras (G12D Mutant Specific) (D8H7) Rabbit mAb | Cell Signaling | Cat# 14429S, RRID:AB_2728748 |
| Peroxidase‐AffiniPure donkey anti‐rabbit IgG (H+L) antibody | Jackson ImmunoResearch | Cat# 711‐035‐152, RRID:AB_10015282 |
| Oligonucleotides and other sequence‐based reagents | ||
| Primers for Nanog: Forward: 5′‐TCTCTCAGGCCCAGCTGTGT‐3′ and Reverse: 5′‐GCTTGCACTTCATCCTTTG‐3′ | Intergrated DNA Technologies | N/A |
| Primers for Oct3/4: Forward: 5′‐GCCCTGCAGAAGGAGCTAGAAC‐3′ and Reverse: 5′‐GGAATACTCAATACTTGATCT‐3′ | Intergrated DNA Technologies | N/A |
| Primers for Nkx3.2: Forward: 5′‐AGATGTCAGCCAGCGTTTC‐3′ and Reverse: 5′‐AGGGCTAACGCTGTCATCCT‐3′ | Intergrated DNA Technologies | N/A |
| Primers for RGS: Forward: 5′‐GCTTTGACTTGGCCCAGAAA‐3′ and Reverse: 5′‐CCTGACCAGATGACTACTTGATTAGCT‐3′ | Intergrated DNA Technologies | N/A |
| Primers for PDGFRα: Forward: 5′‐TCTGTGACTTTTAAGGATGCTTCA‐3′ and Reverse: 5′‐GATGCCCACATAGCCTTCATTC‐3′ | Intergrated DNA Technologies | N/A |
| Primers for PDGFRβ: Forward: 5′‐TCATGAAGCCAGCAAGAGTG‐3′ and Reverse: 5′‐GTGGTAATCCCGTCAGCATC‐3′ | Intergrated DNA Technologies | N/A |
| Primers for ACTA2: Forward: 5′‐GCCAGTCGCTGTCAGGAACCC‐3′ and Reverse: 5′‐AGCCGGCCTTACAGAGCCCA‐3′ | Intergrated DNA Technologies | N/A |
| Primers for Prominin1: Forward: 5′‐GCCCAAGCTGGAAGAATATG‐3′ and Reverse: 5′‐CAGCAGAAAGCAGACAATCAA‐3′ | Intergrated DNA Technologies | N/A |
| Primers for Snail1: Forward: 5′‐CTTGTGTCTGCACGACCTGT‐3′ and Reverse: 5′‐CAGGAGAATGGCTTCTCACC‐3′ | Intergrated DNA Technologies | N/A |
| Primers for Snail2: Forward: 5′‐CATTGCCTTGTGTCTGCAAG‐3′ and Reverse: 5′‐AGAAAGGCTTTTCCCCAGTG‐3′ | Intergrated DNA Technologies | N/A |
| Primers for Sparc: Forward: 5′‐AGAGGAAACGGTCGAGGAG‐3′ and Reverse: 5′‐CTCACACACCTTGCCATGTT‐3′ | Intergrated DNA Technologies | N/A |
| Primers for Twist: Forward: 5′‐AGCTACGCCTTCTCCGTCT‐3′ and Reverse: 5′‐TCCTTCTCTGGAAACAATGACA‐3′ | Intergrated DNA Technologies | N/A |
| Primers for Zeb2: Forward: 5′‐CCAGAGGAAACAAGGATTTCAG‐3′ and Reverse: 5′‐AGGCCTGACATGTAGTCTTGTG‐3′ | Intergrated DNA Technologies | N/A |
| Primers for Zeb1: Forward: 5′‐TGAGCACACAGGTAAGAGGCC‐3′ and Reverse: 5′‐GGCTTTTCCCCAGAGTGCA‐3′ | Intergrated DNA Technologies | N/A |
| Primers for Col6a: Forward: 5′‐GCAAGGATGAGCTGGTCAA‐3′ and Reverse: 5′‐GTCCACGTGCTCTTGCATC‐3′ | Intergrated DNA Technologies | N/A |
| Primers for Epcam: Forward: 5′‐GATTCTGCACGTGAGACCTG‐3′ and Reverse: 5′‐GATACCAAGTCAAACCGAGAACTT‐3′ | Intergrated DNA Technologies | N/A |
| Primers for Fn1: Forward: 5′‐CGGAGAGAGTGCCCCTACTA‐3′ and Reverse: 5′‐CGATATTGGTGAATCGCAGA‐3′ | Intergrated DNA Technologies | N/A |
| Primers for Krt19: Forward: 5′‐TCCCAGCTCAGCATGAAAGCT‐3′ and Reverse: 5′‐AAAACCGCTGATCACGCTCTG‐3′ | Intergrated DNA Technologies | N/A |
| Primers for Krt7: Forward: 5′‐CACGAACAAGGTGGAGTTGGA‐3′ and Reverse: 5′‐TGTCTGAGATCTGCGACTGCA‐3′ | Intergrated DNA Technologies | N/A |
| Primers for Sox9: Forward: 5′‐CAAGACTCTGGGCAAGCTCTG‐3′ and Reverse: 5′‐TCCGCTTGTCCGTTCTTCAC‐3′ | Intergrated DNA Technologies | N/A |
| Primers for GAPDH: Forward: 5′‐CAACGACCCCTTCATTGACC‐3′ and Reverse: 5′‐GGTCTCGCTCCTGGAAGATG‐3′ | Intergrated DNA Technologies | N/A |
| PrimePCR™ SYBR® Green Assay CD248 for Mouse | Bio‐Rad | Cat#10025636 |
| KrasG12D conditional primers for PCR: con1: 5′‐GTC TTTCCCCAGCACAGTGC‐3′; con2: 5′‐CTCTTGCCTACGCCACCAGCT C‐3′; con3: 5′‐AGCTAGCCACCATGGCTTGAGTAAGTCTGCA‐3′ | Intergrated DNA Technologies | https://jacks‐lab.mit.edu/protocols/genotyping/kras_cond |
| Chemicals, Enzymes and other reagents | ||
| Complete mouse endothelial cell medium | Cell Biologics | Cat# M1168 |
| Phosphate‐buffered saline (PBS) 10× | Gibco/Thermo Fisher Scientific | Cat# 14190‐144 |
| Dulbecco's Modified Eagle Medium (DMEM) | Gibco/Thermo Fisher Scientific | Cat# 61965059 |
| Fetal calf serum (FCS) | Eurobio Scientific | Cat# CVFSVF00‐01 |
| Penicillin‐Streptomycin (P/S) | Gibco/Thermo Fisher Scientific | Cat# 15140‐122 |
| Collagenase P | Roche/Sigma‐Aldrich | Cat# 11213865001 |
| 16% Formaldehyde solution (PFA) | Termo Fisher | Cat# 28908 |
| 10% Neutral buffered formalin | Diapath S.p.A. | REF: F0043 |
| Triton X‐100 | Sigma‐Aldrich | Cat# T8787 |
| Antigen Unmasking Solution, Citrate‐Based | Vector Laboratories/Eurobio Scientific | Cat# H‐3300 |
| DAB Substrate Kit, Peroxidase (HRP), with Nickel, (3,3'‐diaminobenzidine) | Vector Laboratories/Eurobio Scientific | Cat# SK‐4100 |
| Mayer Hematoxylin | Diapath S.p.A. | REF: C0303 |
| Antibody diluent Reagent | Life Techno/Thermo Fisher | Cat# 003218 |
| Vectashield mounting medium with DAPI | Vector Laboratories/Eurobio Scientific | Cat# H‐1200 |
| NuPAGE 4‐12% Bis‐Tris polyacrylamide gel | Invitrogen/Thermo fischer | Cat# NP0335BOX |
| Immune‐Blot PVDF membrane | BIO‐RAD | Cat# 1620177 |
| Tris buffered saline (TBS) | Euromedex | Cat# ET220 |
| Tween 20 | VWR | Cat# 28829.296 |
| Bovine serum albumin (BSA) | Sigma | Cat# A2153‐100G |
| RIPA buffer | Pierce/Thermo Fisher | Cat# 89900 |
| Protease complete EDTA‐free | Roche/Sigma‐Aldrich | Cat# 04 693 159 001 |
| Phosphatase phoSTOP | Roche/Sigma‐Aldrich | Cat# 04 906 837 001 |
| Cell Activation Cocktail (w/o Brefeldin A) | BioLegend | Cat# 423301 |
| Brefeldin A solution | BioLegend | Cat# 420601 |
| Corning® Matrigel® Basement Membrane Matrix High Concentration | Corning | Cat# 354248 |
| Corning® Matrigel® Growth Factor Reduced (GFR) Basement Membrane Matrix, Phenol Red‐free, LDEV‐free | Corning | Cat# 356231 |
| Recombinant Murine PDGF‐BB | PeproTech/Thermo Fisher Scientific | Cat# 315‐18 |
| TGF‐β1 | Biotechne | Cat#7666‐MB |
| IncuCyte® Zoom live‐cell analysis system | Essen Bioscience | |
| Other | ||
| PA485: Pancreatitis and matched pancreatic cancer tissue array | US Bioomax, Inc. | https://www.biomax.us/tissue‐arrays/pancreas/PA485 |
| BIC14011a: Pancreas intraepithelial neoplasia, pancreatitis and cancer tissue array | US Bioomax, Inc. | https://www.biomax.us/tissue‐arrays/pancreas/BIC14011a |
| StemPro® Osteogenesis Differentiation Kit | Gibco/Thermo Fisher Scientific | Cat#10072‐01 |
| StemPro® Chondrogenesis Differentiation Kit | Gibco/Thermo Fisher Scientific | Cat#10071‐01 |
| Alizarin Red S Staining Kit | ScienCell Research Laboratories | Cat#0223 |
| Alcian Blue 8GX | Sigma‐Aldrich | Cat#A3157‐10G |
| True‐Nuclear™ Transcription Factor Buffer Set | BioLegend | Cat#424401 |
| NucleoSpin® RNA Plus kit | MACHEREY‐NAGEL | REF 740984 |
| CellTrace™ CFSE Cell Proliferation Kit | Invitrogen/Thermo Fisher Scientific | Cat#C34554 |
| LEGENDplex™ Custom Mouse 5‐plex Panel | BioLegend | Cat# B285861 |
| ECL Western Blotting Substrate | Pierce/Thermo Fisher Scientific | Cat# 32106 |
Methods and Protocols
Mice
The Ptf1a/p48‐Cre;Kras G12D (KC), Pdx1‐Cre;Kras G12D;Ink4a/Arf fl/fl (KIC), and Pdx1‐Cre;Kras G12D;Tp53 R172H (KPC) mice have been previously described (Aguirre et al, 2003; Ascher et al, 2003; Hingorani et al, 2005). Rag2KO and C57BL/6 mice were obtained from The Charles River Laboratories and Envigo (France) and used as hosts in the s.c. tumor implantation experiments. All mice were kept under specific pathogen‐free conditions at the Animale en Cancérologie (AniCan) platform at the Cancer Research Center of Lyon (CRCL). All animal procedures and experiments were performed in compliance with the ethical guidelines of the CRCL Animal Care and Use Committee, with approval of the Experimental Animal Ethics Committee of the Rhône‐Alpes region (CECCAPP; CECCAPP_CLB_2019_002).
Mouse primary cell lines
The isolation and culture of cells were performed using a protocol adapted from a previously published protocol (Bayne et al, 2012; Goehrig et al, 2019). PeSCs were obtained from the pancreas of 2.5‐month‐old neoplastic KC mice and dissociated. Tumor primary cell lines (Epi) and metastatic lung cell lines (Meta) were obtained from the pancreas of 2‐month‐old KIC mice respectively, using the same protocol. The cells were then plated in six‐well plates with serum‐free DMEN (Gibco), and after 2 weeks, the media was changed to DMEN‐complete. The KrasG12D mutation was detected by PCR and WB. After several passages as indicated in Fig 3A and Appendix Fig S3D, the cells were infected with a lentivector expressing H2B‐mCherry (PeSC) or H2B GFP (Epi or Meta cells) as previously described (Deygas et al, 2018). C57BL/6 mouse primary pancreatic microvascular endothelial cells were purchased from Cell Biologics.
Matrigel tube formation assay
The assay of tube formation performed with endothelial cells based on Matrigel embodiment has been previously described (Sorrell et al, 2009; Zhang et al, 2015; Kutikhin et al, 2020). Briefly, Matrigel (Growth Factor‐Reduced Basement Membrane Matrix, Phenol Red‐Free, LDEV‐free) was melted on ice and then paved onto a 96‐well plate. Subsequently, 20 ml of Matrigel was distributed into each well, and the plate was then centrifuged at 500 g for 1 min to ensure uniform coverage. Afterward, the plate was immediately placed in an incubator for 30 min for gel formation. C57BL/6 mouse primary pancreatic microvascular endothelial cells (Endo) were trypsinized and then seeded on the polymerized gel in a complete endothelial culture medium (Cell Biologics) at a density of 10,000 cells/cm2. For the coculture with the Endo cells, Epi cells and PeSCs were seeded at the same condition and above‐mentioned density. A monolayer culture with no Matrigel polymer was used as a control. Each condition was performed in triplicate. The cultures were monitored using IncuCyte Zoom®, and photographs were captured every 2 h after initiation of the assay. Endothelial cells were observed under phase contrast, whereas combined phase contrast with green and red fluorescence optics was used for the analysis of the Epi cells and/or PeSCs in coculture conditions.
Cell culture, proliferation and differentiation
Cells were cultured in DMEM supplemented with 10% FCS and 1% penicillin/streptomycin for proliferation. PeSCs were cocultured with Epi cells at the indicated ratios, and these cell lines were then observed through the red and green channels, respectively, using IncuCyte Zoom®. The proliferation curves of the Epi cells were depicted based on the total green object area count (μm2/well) analyzed and exported from the IncuCyte® system. For the PeSC differentiation experiments, the cells were plated using StemPro® differentiation kits (Gibco) for osteogenesis and chondrogenesis in accordance with the provided instructions. Alizarin Red S staining for osteogenic lineages and Alcian Blue staining for chondrogenic lineages were also performed according to the protocols recommended by the above‐mentioned commercial kits. For the PDGF‐BB treatment assay, PeSCs were stimulated by PDGF‐BB (10 ng/ml) and TGF‐β1 (2 ng/ml). PDGF‐BB and TGF‐β1 were replenished every 2 days for 7 days. After 7 days, the cells were recovered for FACS analysis and mRNA is subject to RT‐qPCR for stemness, epithelial, fibroblast, and mesenchymal‐related genes.
Short‐term PeSC and tumor cell implantation studies
PeSC or Epi cells (1 × 105) were embedded as plugs in a 1:1 Matrigel‐PBS mix (Matrigel® Basement Membrane Matrix High Concentration, HC) into the flanks of C57BL/6 or Rag2 KO mice. The mice were then monitored and sacrificed at the indicated time points. The tumor grafts were weighed, measured, and processed for staining prior to flow cytometry analysis. Ly6G+ cells were depleted in vivo by administering two consecutive i.p. injections of anti‐Ly6G mAbs to the mice (8.5 mg/kg) at the indicated time points. The anti‐PD‐1 strategy involved the i.p. injection of anti‐PD‐1 into the mice (5 mg/kg) twice a week at the indicated time points for 2 weeks. CCL5 depletion was administrated by pre‐mixing anti‐CCL5 mAbs (5 μg/kg or 0.5 μg/ml) in the Matrigel® (HC) plugs before implanting. Control mice were treated with anti‐IgG2A (5 μg/kg or 0.5 μg/ml).
Reverse transcription and qPCR
RNA from pelleted islets was extracted using a NucleoSpin® RNA Plus kit according to the manufacturer's instructions. The RNA concentrations were measured using a Nanodrop spectrophotometer. Reverse transcription (RT) was performed using equivalent quantities of extracted RNAs (greater than 300 ng), and cDNA was used for quantitative polymerase chain reaction (qPCR) analyses with SsoAdvanced Universal SYBR® Green Supermix (Bio‐Rad). The following primers were used: Nanog, Oct3/4, Nkx3.2, RGS, PDGFRα, PDGFRβ, Acta2, Desmin, Prominin, Snail1, Snail2, Sparc, Twist, Zeb1, Zeb2, Col6a, EpCAM, Fn‐1, Krt19, Krt, Sox9, and PrimePCR™ SYBR® Green Assay CD248 for Mouse (Bio‐Rad).
RNAseq analysis
Raw sequencing data quality controls were performed with FastQC (v 0.11.5). Salmon (0.10.0) was used for the quantification of gene expression from the raw sequencing reads, and the reference mouse genome GRCm38 and the annotations of protein‐coding genes from gencode vM20 were utilized as an index. Unless otherwise specified, the analyses were performed using R (v3.5.1). Starting from the Salmon transcript quantification, we used the R packages tximport (Soneson et al, 2015; v1.10.1) and DESeq2 (Love et al, 2014; v1.22.2) to perform the differential expression analyses (Wald test and P‐value correction with the Benjamini–Hochberg method). We used the R packages clusterProfiler (Yu et al, 2012; v 3.10.1) and msgidbr (v 6.2.1) to test the pathway enrichment of a list of genes. We tested the list of genes against pathways from the msigdb hallmark, C2 and C5 gene sets. Overrepresentation P‐values were corrected with the Benjamini–Hochberg method.
Single cell RNA sequencing (scRNAseq)
CAFs and ductal tumor cells were obtained from a pool of 5 KC mice, as described above.
After tissue dissection and dissociation, FACS purified suspended cells were partitioned into nanoliter‐scale Gel Bead‐In‐Emulsions (GEMs) with the Chromium Single Cell Controller (10XGenomics) at the Single Cell Platform (CLB/CRCL). After cell encapsulation and barcoding, library preparation followed the standard 10XGenomics 3′scRNAseq protocol comprising reverse transcription, amplification and indexing. Sequencing was performed using a NovaSeq Illumina device (Illumina). Illumina bcl files were basecalled, demultiplexed and aligned to the mouse mm10 genome using the cellranger software (10XGenomics). All downstream analyses were performed using R/Bioconductor/CRAN packages, R version 4.0.3 (2020‐10‐10)—“Bunny‐Wunnies Freak Out” [https://cran.r‐project.org/; http://www.bioconductor.org/; https://cran.r‐project.org/] on a linux platform (x86_64‐pc‐linux‐gnu [64‐bit]). Filtered barcoded matrices were used to create a Seurat object (Stuart et al, 2019) for CAFs and Ducts cells that were subsequently merged. A total of 4,949 cells (4,072 CAFs and 877 Ducts) remained after filtering for quality parameters (number of features per cell < 6,000, fraction of mitochondrial genes < 10%). SCTransform was used to simultaneously normalize, identify variable features, and scale the data. Following dimension reduction with PCA, the first 20 dimensions were used to construct a shared nearest neighbor (SNN) graph using the FindNeighbours function. Clusters were identified with a resolution of 0.5 and projected in two‐dimensional plots using Uniform Manifold Approximation and Projection (UMAP; arXiv:1802.03426v3).
Differentially expressed genes for each cluster were identified using the FindMarkers function. In addition, a list of PeSC markers obtained from bulk RNAseq data was used to calculate a PeSC score. This permitted the identification of a common cluster between CAFs and Ducts with a high PeSC score. This putative PeSC cluster was isolated and pathway analysis was performed in its full list of unique markers using EnrichR R (Kuleshov et al, 2016) and pathfindR (Ulgen et al, 2019) CRAN packages. Significant markers and pathways were selected with a threshold of adjusted P‐value (FDR) below 0.05.
Immunohistochemistry and immunofluorescence
BIC14011a and PA485 human tissue microarrays (TMAs) were obtained from a commercial source (US Biomax, Inc.). BIC14011a is a pancreas array that contains 22 pancreatitis cases, 18 pancreatic intraepithelial neoplasia cases, and eight pancreatic adenocarcinoma cases. PA485 is pancreatitis and matching pancreatic adenocarcinoma array containing 43 cases of pancreatitis and five matched pancreatic adenocarcinomas. Mouse tumor grafts were harvested, fixed overnight in 10% neutral buffered formalin, embedded in paraffin, and sectioned at 4 μm. All immunohistochemical (IHC) staining procedures were performed following heat‐induced epitope retrieval (Antigen‐unmasking solution), and the sections were incubated with the primary antibodies overnight at 4°C. IHC staining was revealed using 3,3′‐diaminobenzidine (DAB kit), and the sections were counterstained with hematoxylin. The CD106 positivity was visually quantified by the identification of at least one positive cell within each individual TMA spot. IF staining was performed using a standard protocol, and the sections were counterstained with VECTASHIELD® Antifade Mounting Medium with DAPI for nuclear counterstaining. The primary antibodies used were the following: anti‐CD24, anti‐CD44, anti‐CD106 (VCAM‐1), anti‐αSMA, anti‐CK19 (Troma III), anti‐ZO‐1, anti‐E‐cadherin, anti‐Nanog, anti‐Ly6G, anti‐mCherry, and anti‐GFP. Specific anti‐F(ab')2‐Alexa Fluor 647, anti‐F(ab')2‐Alexa Fluor 555 and anti‐F(ab')2‐Alexa Fluor 488 antibodies were used as secondary antibodies.
Western blot
WB is performed among Epi, Meta and PeSC cells for Kras G12D detection. Cells were collected by scratching in 10 ml of PBS. After centrifugation, the proteins were isolated using RIPA buffer supplemented with protease and phosphatase inhibitors. Twenty microliters of the obtained lysate were separated by NuPAGE 4–12% Bis‐Tris Gel electrophoresis system and transferred to an Immune‐Blot PVDF membrane. After transfer, the immune blots were blocked by incubating with 5% BSA in Tris‐buffered saline (TBS) containing 0.05% Tween 20. The blots were then probed overnight with Ras (G12D) mAb at 1:1,000. After washing in TBS‐Tween 0.05%, the membranes were revealed with secondary antibodies for 1 h at room temperature. After washing, the blots were developed using the ECL chemiluminescence method according to the manufacturer's protocol (Pierce™ ECL Western Blotting Substrate). HRP‐conjugated anti‐rabbit Ab was used for primary antibody binding.
FACS analysis
Cells were collected by trypsinzing from the culture plate and then distributed into the 96‐well plate for FACS staining. In the case of tumor implantation, the tumor grafts were excised, weighed, and measured. The tumor grafts were then digested in 1% PBS supplemented with collagenase at 1 mg/ml for 20 min at 37°C. The digested tissue was homogenized by passage through a 100 μm cell strainer. Single‐cell suspensions derived from tumor grafts were stained with fluorescently labeled antibodies for 20 min at 4°C. For cytokine analysis and intracellular staining, single‐cell suspensions were incubated with a complete culture medium containing a cell activation cocktail (PMA + ionomycin + Brefeldin A) for 2–4 h at 37°C prior to surface staining to retain intracellular proteins. After surface staining, cells were fixed and permeabilized using the True‐Nuclear™ Transcription Factor Buffer Set according to the manufacturer's protocol. Intracellular staining was performed for 30 min at 4°C. Fluorescently labeled cells were acquired on a BD Fortessa Flow Cytometer (BD; Franklin Lakes/NJ/USA) and analyzed using FlowJo. The monoclonal Abs used in flow cytometry were summarized in the key resources table.
Bone marrow chimera approach
Briefly, recipient mice were sublethally irradiated with 7 Gy via a cesium γ source 2 days prior to transplantation. Bone marrow cells were harvested from the femurs and tibias of donor mice in RPMI‐1640. Recipient mice were administered approximately 2 × 106 bone marrow cells in 0.2 ml medium via iv injection. Five weeks after transplantation, recipient mice were subjected to whole blood sampling to determine the degree of chimerism by flow cytometry determination of Tomato expression.
Quantification and statistical analysis
The P‐values were calculated using Student's t‐test with GraphPad Prism as indicated in the figure legends: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. One‐way ANOVA with Tukey's post hoc test was used for multiple comparisons. The contingency analysis was performed using Fisher's exact test.
Author contributions
Zhichong Wu: Investigation; writing – original draft; writing – review and editing. Kevin Thierry: Investigation; writing – review and editing. Sophie Bachy: Investigation. Xinyi Zhang: Investigation. Pia Gamradt: Investigation. Hector Hernandez‐Vargas: Formal analysis. Ivan Mikaelian: Investigation. Laurie Tonon: Software; formal analysis. Roxanne Pommier: Data curation; software; formal analysis. Yajie Zhao: Investigation. Philippe Bertolino: Writing – original draft. Ana Hennino: Conceptualization; supervision; funding acquisition; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
The authors thank the staff at ANICAN (Cancer Research Center of Lyon) staff for the maintenance of the mouse strains and Christophe Vanbelle for the helpful assistance with the confocal microscopy experiments. This study was supported by grants from La Ligue Contre le Cancer (AH and PB), Bristol Meyers Squibb Foundation (AH), INSERM Transfert (AH), Sanofi iAward (AH), Institut National du cancer (INCA) AAP 2019, and Fondation de France (AH and PG), ZCW and YJZ were both supported by a Chinese Scholarship Council (CSC) Fellowship. ZCW was supported by Science and Technology Commission of Shanghai Municipality (Shanghai Administration of Foreign Experts Affairs), no. 21430711900.
EMBO reports (2023) 24: e56524
Data availability
The scRNAseq data from this publication have been deposited to the Gene Expression Omnibus (GEO) with the identifier GSE220687 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE220687).
References
- Acharya C, Cline RA, Jaligama D, Noel P, Delany JP, Bae K, Furlan A, Baty CJ, Karlsson JM, Rosario BL et al (2013) Fibrosis reduces severity of acute‐on‐chronic pancreatitis in humans. Gastroenterology 145: 466–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, Redston MS, DePinho RA (2003) Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev 17: 3112–3126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armulik A, Genove G, Betsholtz C (2011) Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell 21: 193–215 [DOI] [PubMed] [Google Scholar]
- Ascher E, Markevich N, Schutzer RW, Kallakuri S, Jacob T, Hingorani AP (2003) Cerebral hyperperfusion syndrome after carotid endarterectomy: predictive factors and hemodynamic changes. J Vasc Surg 37: 769–777 [DOI] [PubMed] [Google Scholar]
- Balabanov R, Washington R, Wagnerova J, Dore‐Duffy P (1996) CNS microvascular pericytes express macrophage‐like function, cell surface integrin alpha M, and macrophage marker ED‐2. Microvasc Res 52: 127–142 [DOI] [PubMed] [Google Scholar]
- Banerjee M, Bhonde RR (2006) Application of hanging drop technique for stem cell differentiation and cytotoxicity studies. Cytotechnology 51: 1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, Vonderheide RH (2012) Tumor‐derived granulocyte‐macrophage colony‐stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 21: 822–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair AB, Kim VM, Muth ST, Saung MT, Lokker N, Blouw B, Armstrong TD, Jaffee EM, Tsujikawa T, Coussens LM et al (2019) Dissecting the stromal signaling and regulation of myeloid cells and memory effector T cells in pancreatic cancer. Clin Cancer Res 25: 5351–5363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet D, Dick JE (1997) Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 3: 730–737 [DOI] [PubMed] [Google Scholar]
- Chen K, Wang Q, Li M, Guo H, Liu W, Wang F, Tian X, Yang Y (2021) Single‐cell RNA‐seq reveals dynamic change in tumor microenvironment during pancreatic ductal adenocarcinoma malignant progression. EBioMedicine 66: 103315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SA et al (2011) Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov 1: 54–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deygas M, Gadet R, Gillet G, Rimokh R, Gonzalo P, Mikaelian I (2018) Redox regulation of EGFR steers migration of hypoxic mammary cells towards oxygen. Nat Commun 9: 4545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominici M, Le Blanc K, Mueller I, Slaper‐Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop D, Horwitz E (2006) Minimal criteria for defining multipotent mesenchymal stromal cells. The international society for cellular therapy position statement. Cytotherapy 8: 315–317 [DOI] [PubMed] [Google Scholar]
- Erkan M, Michalski CW, Rieder S, Reiser‐Erkan C, Abiatari I, Kolb A, Giese NA, Esposito I, Friess H, Kleeff J (2008) The activated stroma index is a novel and independent prognostic marker in pancreatic ductal adenocarcinoma. Clin Gastroenterol Hepatol 6: 1155–1161 [DOI] [PubMed] [Google Scholar]
- Ferro F, Spelat R, Shaw G, Duffy N, Islam MN, O'Shea PM, O'Toole D, Howard L, Murphy JM (2019) Survival/adaptation of bone marrow‐derived mesenchymal stem cells after long‐term starvation through selective processes. Stem Cells 37: 813–827 [DOI] [PubMed] [Google Scholar]
- Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, Pamer EG, Li MO (2014) The cellular and molecular origin of tumor‐associated macrophages. Science 344: 921–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich DI, Ostrand‐Rosenberg S, Bronte V (2012) Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 12: 253–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goehrig D, Nigri J, Samain R, Wu Z, Cappello P, Gabiane G, Zhang X, Zhao Y, Kim IS, Chanal M et al (2019) Stromal protein betaig‐h3 reprogrammes tumour microenvironment in pancreatic cancer. Gut 68: 693–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore J, Korc M (2014) Pancreatic cancer stroma: friend or foe? Cancer Cell 25: 711–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra DAP, Paiva AE, Sena IFG, Azevedo PO, Silva WN, Mintz A, Birbrair A (2018) Targeting glioblastoma‐derived pericytes improves chemotherapeutic outcome. Angiogenesis 21: 667–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646–674 [DOI] [PubMed] [Google Scholar]
- Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA et al (2003) Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4: 437–450 [DOI] [PubMed] [Google Scholar]
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA (2005) Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7: 469–483 [DOI] [PubMed] [Google Scholar]
- Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, Nywening TM, Hawkins WG, Shapiro IM, Weaver DT et al (2016) Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med 22: 851–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Liu X, Knolhoff BL, Hegde S, Lee KB, Jiang H, Fields RC, Pachter JA, Lim KH, DeNardo DG (2020) Development of resistance to FAK inhibition in pancreatic cancer is linked to stromal depletion. Gut 69: 122–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A et al (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44: W90–W97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutikhin AG, Tupikin AE, Matveeva VG, Shishkova DK, Antonova LV, Kabilov MR, Velikanova EA (2020) Human peripheral blood‐derived endothelial Colony‐forming cells are highly similar to mature vascular endothelial cells yet demonstrate a transitional transcriptomic signature. Cell 9: 876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF, Simeone DM (2007) Identification of pancreatic cancer stem cells. Cancer Res 67: 1030–1037 [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica‐Worms D et al (2013) Targeting tumor‐infiltrating macrophages decreases tumor‐initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res 73: 1128–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Movahedi K, Laoui D, Gysemans C, Baeten M, Stange G, Van den Bossche J, Mack M, Pipeleers D, In't Veld P, De Baetselier P et al (2010) Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res 70: 5728–5739 [DOI] [PubMed] [Google Scholar]
- Muraro MJ, Dharmadhikari G, Grun D, Groen N, Dielen T, Jansen E, van Gurp L, Engelse MA, Carlotti F, de Koning EJ et al (2016) A single‐cell transcriptome atlas of the human pancreas. Cell Syst 3: 385–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neesse A, Michl P, Frese KK, Feig C, Cook N, Jacobetz MA, Lolkema MP, Buchholz M, Olive KP, Gress TM et al (2011) Stromal biology and therapy in pancreatic cancer. Gut 60: 861–868 [DOI] [PubMed] [Google Scholar]
- Nywening TM, Belt BA, Cullinan DR, Panni RZ, Han BJ, Sanford DE, Jacobs RC, Ye J, Patel AA, Gillanders WE et al (2018) Targeting both tumour‐associated CXCR2(+) neutrophils and CCR2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 67: 1112–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oudenaarden C, Sjolund J, Pietras K (2022) Upregulated functional gene expression programmes in tumour pericytes mark progression in patients with low‐grade glioma. Mol Oncol 16: 405–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdemir BC, Pentcheva‐Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV et al (2014) Depletion of carcinoma‐associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25: 719–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panni RZ, Herndon JM, Zuo C, Hegde S, Hogg GD, Knolhoff BL, Breden MA, Li X, Krisnawan VE, Khan SQ et al (2019) Agonism of CD11b reprograms innate immunity to sensitize pancreatic cancer to immunotherapies. Sci Transl Med 11: eaau9240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilarsky C, Ammerpohl O, Sipos B, Dahl E, Hartmann A, Wellmann A, Braunschweig T, Lohr M, Jesenofsky R, Friess H et al (2008) Activation of Wnt signalling in stroma from pancreatic cancer identified by gene expression profiling. J Cell Mol Med 12: 2823–2835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polette M, Mestdagt M, Bindels S, Nawrocki‐Raby B, Hunziker W, Foidart JM, Birembaut P, Gilles C (2007) Beta‐catenin and ZO‐1: shuttle molecules involved in tumor invasion‐associated epithelial‐mesenchymal transition processes. Cells Tissues Organs 185: 61–65 [DOI] [PubMed] [Google Scholar]
- Pylayeva‐Gupta Y, Lee KE, Hajdu CH, Miller G, Bar‐Sagi D (2012) Oncogenic Kras‐induced GM‐CSF production promotes the development of pancreatic neoplasia. Cancer Cell 21: 836–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sena IFG, Paiva AE, Prazeres P, Azevedo PO, Lousado L, Bhutia SK, Salmina AB, Mintz A, Birbrair A (2018) Glioblastoma‐activated pericytes support tumor growth via immunosuppression. Cancer Med 7: 1232–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha D, Chong L, George J, Schluter H, Monchgesang S, Mills S, Li J, Parish C, Bowtell D, Kaur P et al (2016) Pericytes promote malignant ovarian cancer progression in mice and predict poor prognosis in serous ovarian cancer patients. Clin Cancer Res 22: 1813–1824 [DOI] [PubMed] [Google Scholar]
- Soneson C, Love MI, Robinson MD (2015) Differential analyses for RNA‐seq: transcript‐level estimates improve gene‐level inferences. F1000Res 4: 1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrell JM, Baber MA, Caplan AI (2009) Influence of adult mesenchymal stem cells on in vitro vascular formation. Tissue Eng Part A 15: 1751–1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromnes IM, Brockenbrough JS, Izeradjene K, Carlson MA, Cuevas C, Simmons RM, Greenberg PD, Hingorani SR (2014) Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut 63: 1769–1781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, Satija R (2019) Comprehensive integration of single‐cell data. Cell 177: 1888–1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulgen E, Ozisik O, Sezerman OU (2019) pathfindR: an R package for comprehensive identification of enriched pathways in omics data through active subnetworks. Front Genet 10: 858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdor R, Garcia‐Bernal D, Bueno C, Rodenas M, Moraleda JM, Macian F, Martinez S (2017) Glioblastoma progression is assisted by induction of immunosuppressive function of pericytes through interaction with tumor cells. Oncotarget 8: 68614–68626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Noorden CJ, Jonges GN, Vogels IM, Hoeben KA, Van Urk B, Everts V (1995) Ectopic mineralized cartilage formation in human undifferentiated pancreatic adenocarcinoma explants grown in nude mice. Calcif Tissue Int 56: 145–153 [DOI] [PubMed] [Google Scholar]
- Weber R, Riester Z, Huser L, Sticht C, Siebenmorgen A, Groth C, Hu X, Altevogt P, Utikal JS, Umansky V (2020) IL‐6 regulates CCR5 expression and immunosuppressive capacity of MDSC in murine melanoma. J Immunother Cancer 8: e000949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SP, Rowley JE, Redpath AN, Tilman JD, Fellous TG, Johnson JR (2015) Pericytes, mesenchymal stem cells and their contributions to tissue repair. Pharmacol Ther 151: 107–120 [DOI] [PubMed] [Google Scholar]
- Wong PP, Munoz‐Felix JM, Hijazi M, Kim H, Robinson SD, De Luxan‐Delgado B, Rodriguez‐Hernandez I, Maiques O, Meng YM, Meng Q et al (2020) Cancer burden is controlled by mural cell‐β3‐integrin regulated crosstalk with tumor cells. Cell 181: 1346–1363 [DOI] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, He QY (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16: 284–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Huang Q, Tang M, Zhang J, Fan W (2015) Complement factor H expressed by retinal pigment epithelium cells can suppress neovascularization of human umbilical vein endothelial cells: an in vitro study. PLoS ONE 10: e0129945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, Wang‐Gillam A, Goedegebuure SP, Linehan DC, DeNardo DG (2014) CSF1/CSF1R blockade reprograms tumor‐infiltrating macrophages and improves response to T‐cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res 74: 5057–5069 [DOI] [PMC free article] [PubMed] [Google Scholar]