

Summary
CRISPR-Cas9 cleavage efficacy and accuracy are the main challenges gene editing faces, and they are particularly affected by the optimal formation of the ribonucleoprotein (RNP) complex. We used nano differential scanning fluorimetry, a label and immobilization-free assay, to demonstrate that an equimolar ratio of Cas9 and guide RNA (gRNA) is optimal for RNP complex formation. We almost achieved 50% of green fluorescent protein (GFP) to blue fluorescent protein (BFP) conversion using a biallelic homozygous GFP human induced pluripotent stem cell line, when 0.4 μM of Cas9, equimolar Cas9/gRNA ratio and 2 μM of single-stranded oligonucleotide, were used and showed that increasing Cas9/gRNA ratio did not further improve KI efficiency. Additionally, excess gRNA decreased point mutation KI efficiency in rat embryos and drastically increased the occurrence of on-target large deletions. These findings highlight the importance of CRISPR/Cas9 stoichiometric optimization to ensure efficient and accurate KI generation, which will be applicable to other in vitro as well as in vivo models.
Subject areas: Biological sciences, Molecular biology, Molecular mechanism of gene regulation
Graphical abstract
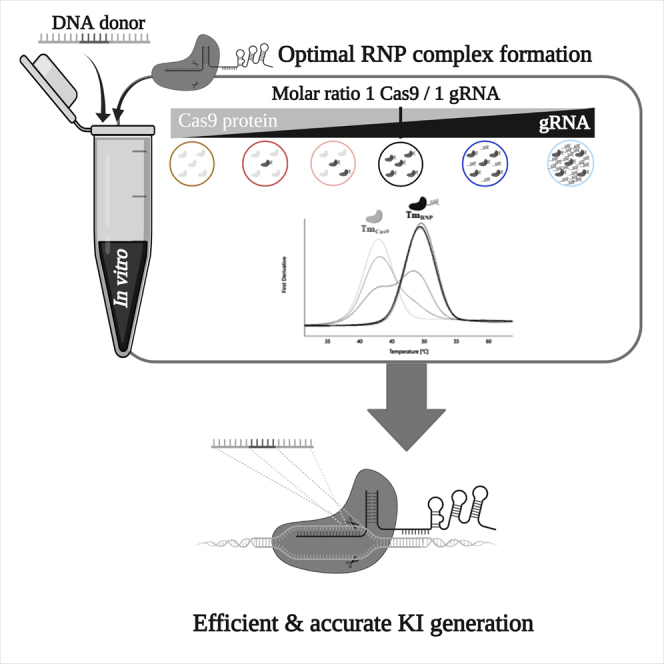
Highlights
-
•
NanoDSF quantitatively assessed the formation of the CRISPR-Cas9 RNP complex
-
•
An equimolar ratio of Cas9 and gRNA is optimal for efficient RNP complex formation
-
•
This ratio and excess of ssODN led to highly efficient knockin in hiPS and rats
-
•
Excess gRNA decreased knockin efficiency and increased on-target large deletions
Biological sciences; Molecular biology; Molecular mechanism of gene regulation
Introduction
Disease modeling relies on biological models, and our ability to perturb them. On one hand, animal models allow for the study of whole organisms and organ function. On the other hand, human induced pluripotent stem cells, reprogrammed from patient somatic cells, offer a human cellular model of choice. Both of these models (for a review in rat1 or human induced pluripotent stem cells2) can be perturbed genetically, especially by the CRISPR-Cas9 system.3
CRISPR-Cas9 mediated genetic manipulation enables the generation of a wide range of targeted knockout (KO) or knockin (KI) animal and cell models, such as single or multiplex KO, point mutation, conditional KO or KI of a reporter gene. Despite advances in our ability to manipulate CRISPR/Cas9 mediated editing, optimizing the efficiency and accuracy of this system remains poorly understood.
Targeted locus accessibility, quality of CRISPR-Cas9 reagents (expression, variants, structure …), delivery and cleavage efficiency and complexity of the generated model (multiple or large DNA donors and others)4 all influence genome editing efficiency to a greater or lesser extent. Control of unwanted editing by-products, such as unexpected DNA sequence modifications or large deletions, that are not always verified5 is highly desirable, and there is a need for in-depth analysis of editing parameters to fine-tune them in order to limit adverse genome editing.
A starting point is to more rigorously report experimental design. Indeed, in the literature, the Cas9/guide RNA (gRNA) molecular ratio used is often expressed in mass and span wide Cas9/gRNA ratio ranges (from 1/1 to 1/26) despite the fact that only one Cas9 can only load one gRNA.6 To the best of our knowledge, there is only one study of the formation of ribonucleoprotein (RNP) complex.7 To begin to address this, we decided to functionally link RNP complex formation and stoichiometry of gRNA. For better control of the delivered molecules throughout this study, we used RNP complexes prepared in vitro and expressed all stoichiometries in molar concentrations for Cas9, single-stranded oligonucleotide (ssODN) and gRNAs. We first demonstrated, in a GFP to BFP conversion hiPSC model,8 that an excess of gRNA did not improve KI efficiency while an excess of ssODN did. Indeed, we almost reached 50% of KI with 0.4 μM of Cas9, equimolar Cas9/gRNA and 2 μM of ssODN. Previous studies have shown increased editing efficiency with an excess of gRNA.9,10 Thus, we decided to assess RNP complex formation in vitro with equimolar Cas9/gRNA using a nano differential scanning fluorimetry (NanoDSF) assay. This label- and immobilization-free method enabled the detection of changes in Cas9 conformation including free Cas9 or complexed with a gRNA. We confirmed the optimal effect of an equimolar Cas9/gRNA ratio on a point mutation model on rat embryos11 and observed a deleterious effect of an excess of gRNA on KI efficiency in this model. Moreover, using a droplet digital PCR (ddPCR) approach, we also showed in rat embryos that equimolar Cas9/gRNA ratio drastically reduced the occurrence of on-site large deletions, compared to an excess of gRNA.
Our stoichiometric approach of KI generation brings a new technical perspective and helps to better control the CRISPR-Cas9 system. In particular, the development of an in vitro assay to assess RNP complex formation will facilitate the optimal delivery of the required molecules in vivo, limiting any side effects of free Cas9 or free gRNA on cellular repair mechanisms and integrity.
Results
Increasing ssODN concentration reduced KO and improved KI while increasing dgRNA did not affect KO or KI at an optimal Cas9 concentration of 0.4 μM
Biallelic homozygous GFP hiPSCs were converted into BFP expressing cells by CRISPR/Cas9 KI using a BFP ssODN (Figure 1A). Cells were phenotyped by flow cytometry 7 days post-electroporation to define different populations: GFP only expressing cells, non-fluorescent KO cells and KI BFP expressing cells (Figure 1B). Cells that have not been edited or for which editing has maintained GFP reading frame (GFP only expressing cells) could not be separated in our analysis and were not further studied. We analyzed cells that have lost GFP expression (non-fluorescent KO cells) and/or acquired BFP expression (KI BFP expressing cells) to define the stoichiometry for optimal KI generation of hiPSC (Figure 2).
Figure 1.

GFP to BFP conversion hiPSC model to study CRISPR/Cas9 genome editing and KI efficiency by flow cytometry
(A) Schematic representation of GFP to BFP conversion strategy using CRISPR/Cas9 and a BFP ssODN. PAM is written in bold, KI in blue and DraIII restriction site is underlined.
(B) Phenotyping strategy used to determine KI (BFP expressing cells) and KO (non-fluorescent cells) rates. The figures were created with https://www.biorender.com/ (Toronto, Canada). GFP, green fluorescent protein; BFP, blue fluorescent protein; dgRNA, dual guide RNA; ssODN, single-stranded oligonucleotide; hiPSCs, induced pluripotent stem cells; KI, knockin; KO, knockout.
Figure 2.

Increasing ssODN concentration reduced KO and improved KI while increasing dgRNA did not affect KO or KI at an optimal Cas9 concentration of 0.4 μM
(A and B) Effect of increased concentrations of Cas9 protein (from 0.2 to 0.5 μM) with Cas9/gRNA/ssODN molar ratios 1/4/4 on KI or B. KO efficiency.
(C and D) Improvement of KI but not D. KO efficiency by increasing Cas9/dgRNA/ssODN molar ratios with 0.4 μM of Cas9.
(E and F) Similar KI and F. KO rates with increased dgRNA concentrations and fixed 1.6 μM of ssODN (1/4 Cas9/ssODN molar ratio).
(G and H) Improved percentage of KI cells and H. reduced percentage of KO cells with increased concentrations of ssODN and fixed 0.4 μM or 1.6 μM dgRNA (1/1 or 1/4 Cas9/dgRNA molar ratio, respectively). Data are represented as individual replicates with mean ± SEM. Replicates from conditions with Cas9/dgRNA/ssODN ratio 1/4/4 with 0.2 μM (N = 3), 0.3 μM (N = 6), 0.4 μM (N = 6) and 0.5 μM (N = 2) of Cas9 (Figures 2A and 2B) and conditions with 0.4 μM of Cas9 with Cas9/dgRNA/ssODN ratios 1/1/1 (N = 3), 1/2/2 (N = 2), 1/3/3 (N = 3) and 1/4/4 (N = 6) (Figures 2C and 2D) were electroporated on different days. Conditions 0.4 μM of Cas9 with Cas9/dgRNA/ssODN ratios 1/2/4 (N = 3), 1/3/4 (N = 3) and 1/4/4 (N = 6) were electroporated on 3, 2 and 6 different days, respectively (Figures 2E and 2F). With 0.4 μM of Cas9, replicates with Cas9/dgRNA/ssODN 1/1/1 (N = 3) and 1/4/4 (N = 6) were electroporated on different days, while ratios 1/1/5 (N = 8), 1/4/2 (N = 3) and 1/4/3 (N = 3) were electroporated on 7, 2 and 2 different days, respectively (Figures 2G and 2H). Nonparametric unpaired Mann-Whitney t-tests have been run between all conditions on each figure. Asterisks highlight the statistical differences between conditions (∗p ≤ 0.05, ∗∗p ≤ 0.01). KI, knockin; KO, knockout; dgRNA, dual guide RNA; ssODN, single-stranded oligonucleotide; hiPSCs, human induced pluripotent stem cells.
Skarnes et al. electroporated 800,000 hiPSCs using Amaxa 4D nucleofector pulse code CA-137 in 100 μL with 20 μg of Cas9 (1.2 μM), 16 μg (5 μM) of gRNA and 200 pmol of ssODN (2 μM).12 That is to say, Cas9/gRNA/ssODN 1/4.2/1.7 M ratio. We electroporated hiPSCs with similar parameters, after scaling down to 200,000 cells in 20 μL, in strips. First, we defined the optimal Cas9 concentration. Two days post electroporation, cells electroporated with 1 μM of Cas9 were scarce (less than 5% of confluency, data not shown) suggesting cell toxicity, thus we chose a concentration range from 0.2 μM to 0.5 μM for KI (Figure 2A) and KO (Figure 2B) analysis. An excess of dgRNA and ssODN (Cas9/gRNA/ssODN molar ratio 1/4/4) was used to ensure Cas9 concentration would be the limiting factor in this analysis. The percentage of KI cells was significantly improved when increasing Cas9 concentrations from 0.2 to 0.3 μM or to 0.4 μM (24.3%, 36.6% and 38.8%, respectively) (Figure 2A). The percentage of KO cells also improved with an increase in Cas9 concentration (Figure 2B). For both KI and KO cells analysis, 0.5 μM of Cas9 tended to reduce efficiency (Figures 2A and 2B), thus 0.4 μM of Cas9 seemed to be the optimal concentration for efficient editing on hiPSC in our electroporation conditions. We then analyzed whether both dgRNA and ssODN concentrations can impact KI (Figure 2C) and KO (Figure 2D) efficiencies with 0.4 μM of Cas9. We observed a trend for KI improvement with an increased Cas9/dgRNA/ssODN ratio (Figure 2C), especially from 1/1/1 to 1/4/4 ratios (15.3% and 38.8%, respectively) while a trend for KO reduction was observed (Figure 2D). To figure out whether this increase was mediated by an excess of dgRNA or ssODN, we analyzed both parameters separately. With a constant concentration of 0.4 μM Cas9 and a Cas9/ssODN molar ratio of 1/4, increasing dgRNA concentration induced similar percentages of KI (Figure 2E) and KO (Figure 2F) cells. Interestingly, with a constant concentration of 0.4 μM Cas9 and Cas9/dgRNA molar ratio of 1/1 or 1/4, increasing ssODN concentration improved KI rate (Figure 2G) and reduced KO rate (Figure 2H). In particular, increasing ssODN concentration from 0.4 μM (1/1/1 Cas9/dgRNA/ssODN molar ratio) to 2 μM (1/1/5) or 1.6 μM (1/4/4) significantly improved KI efficiency (15.3%, 40.2% and 38.8%, respectively) (Figure 2G). In contrast, the percentage of KO cells was significantly reduced from 0.4 μM (1/1/1 Cas9/dgRNA/ssODN molar ratio) to 2 μM (1/1/5) of ssODN (63.3% and 27.8%, respectively) suggesting a switch in repair mechanism in favor of KI (Figure 2H). This switch from high rate of KO cells to high rate of KI cells with ssODN concentration increase was not observed with Cas9/dgRNA molar ratio 1/4. Indeed, 1/4/2, 1/4/3 or 1/4/4 Cas9/dgRNA/ssODN molar ratios displayed similar KI rates than 1/1/5 (Figure 2G). KO rate was significantly increased from Cas9/dgRNA/ssODN molar ratio 1/1/5 condition to 1/4/2, 1/4/3 or 1/4/4 conditions (27.8%, 51.2%, 40.6% and 44.7%, respectively) (Figure 2H). Overall, the condition 1/1/1 produced the greatest KO rate (63.3%) and the lowest KI rate (15.3%) but increasing ssODN concentration to 2 μM (1/1/5) led to the highest KI rate (40.2%). These results suggest that dgRNA molar concentration should be similar to Cas9 concentration to limit free dgRNA negative impact and ssODN concentration high to favor DNA template-mediated repair.
Characterization of cell subsets generated by genome editing of hiPSC with 0.4 μM of Cas9, equimolar Cas9/dgRNA molar ratio and 2 μM of ssODN
Our results support that 0.4 μM of Cas9 with equimolar Cas9/dgRNA ratio induced the most efficient cleavage of the RNP complex and increasing ssODN concentration to 1.6 or 2 μM led to almost 50% of KI efficiency. We tried to increase further the ssODN concentration but 3 and 4 μM of ssODN resulted in elevated cell toxicity (less than 10% confluency observed by microscopy after 48h) (Figure S1). Thus, we further characterized the 0.4 μM of Cas9 with equimolar Cas9/dgRNA ratio and 2 μM of ssODN (1/1/5 Cas9/dgRNA/ssODN molar ratio) by cell sorting and genotyping (Figures 3 and S2). Flow cytometry analysis distinguished GFP/BFP expressing cells (GFP+/BFP+), cells with lower BFP expression (GFP-/BFP low) and cells with higher BFP expression (GFP-/BFP high) (Figure 3A). Capillary electrophoresis of PCR amplified GFP locus (genotyping primers Up: CGTAAACGGCCACAAGTTCA and Lo: CTTGTAGTTGCCGTCGTCCT) targeted by the dgRNA and digested by DraIII enzyme showed presence of DraIII restriction sites in GFP-/BFP high cells, GFP-/BFP low cells and GFP+/BFP+, confirming the KI status observed by phenotyping (Figure 3B). Only GFP-/BFP high cells display full DraIII digestion, suggesting the presence of two KI alleles per cell. Sanger sequencing was performed on each of these subsets to precisely determine their genotype and confirmed homozygous KI status of GFP- BFP high cells (Figure 3C). As expected, GFP-/BFP- cells appear to be KO/KO, GFP+/BFP+ to be WT/KI, GFP-/BFP low KO/KI and GFP-/BFP high KI/KI (Figure 3D).
Figure 3.

Characterization of cell subsets generated by genome editing of hiPSC with 0.4 μM of Cas9, equimolar Cas9/dgRNA molar ratio and 2 μM of ssODN
(A) Cell sorting of KO and KI subsets on hiPSC GFP and BFP expressing cells. See also Figure S2 for cell subsets sorting purity.
(B) Genotyping of GFP-/BFP high, GFP-/BFP low, GFP+/BFP+, and GFP-/BFP- subsets by PCR amplification and DraIII digestion on capillary electrophoresis. Capillary Electrophoresis gel has been cropped up and down to focus on bands between 100 and 300bp.
(C) Sanger sequencing results details on each sorted subset.
(D) Subsets genotypes summarized on phenotyping graph. The figure was created with BioRender.com (Toronto, Canada).hiPSC, human induced pluripotent stem cells; GFP, Green Fluorescent Protein; BFP, Blue Fluorescent Protein; ssODN, single-stranded oligonucleotide; WT, wild-type; KI, knockin; KO, knockout; dgRNA, dual guide RNA.
Highly efficient GFP to BFP conversion with equimolar Cas9/dgRNA molar ratio, 0.4 μM of Cas9 and 2 μM of ssODN
We next investigated the impact of gRNA stoichiometry in the context of 0.4 μM of Cas9 and 2 μM ssODN molar ratio 1/1/5 and compared to a higher dgRNA concentration (1/2/5) (Figure 4A). The proportion of KO/KO cells for 1/1/5 and 1/2/5 conditions were still similarly high (27.8% and 30.9%). KI BFP expressing cells subsets also display similar proportions, KO/KI and KI/KI cells being the most represented ones for the 1/1/5 condition (20.3% and 14.0%, respectively) and the 1/2/5 condition 21.8% and 18.4%, respectively) and WT/KI the less represented one (5.7% and 3.5% for 1/1/5 and 1/2/5 conditions, respectively). WT/KI subset was the result of KI on one allele and perfect repair or no cleavage on the other allele, that is why this subset is rare, in particular with efficient cleavage conditions. Overall, the total KI BFP expressing cells percentage is similarly high between conditions 1/1/5 and 1/2/5 (40.2% and 44.1%, respectively) (Figure 4B). These results were consistent with those observed with 1/2/4, 1/3/4 and 1/4/4 Cas9/dgRNA/ssODN (Figures 2E and 2F).
Figure 4.

Highly efficient GFP to BFP conversion with equimolar Cas9/dgRNA molar ratio, 0.4 μM of Cas9 and 2 μM of ssODN
(A) The percentage of KO/KO, WT/KI, KO/KI and KI/KI are not significantly different after genome editing with equimolar or 1/2 Cas9/dgRNA molar ratio, 0.4 μM of Cas9 and 2 μM of ssODN.
(B) The percentages of KI BFP expressing cells are similar between Cas9/dgRNA/ssODN molar ratio 1/1/5 and 1/2/5. Data are represented as individual replicates with mean ± SEM. Conditions with 0.4 μM of Cas9 with Cas9/dgRNA/ssODN molar ratio 1/1/5 (N = 8) and 1/2/5 (N = 6) were electroporated on 7 and 5 different days, respectively. Nonparametric unpaired Mann-Whitney t-tests have been run between all conditions. GFP, green fluorescent protein; BFP, blue fluorescent protein; dgRNA, dual guide RNA; ssODN, single-stranded oligonucleotide; hiPSCs, human induced pluripotent stem cells; WT, wild-type; KI, knockin; KO, knockout.
Overall, our studies on hiPSC genome editing indicated that 0.4 μM of Cas9, equimolar Cas9/gRNA ratio and 2 μM of ssODN is optimal to achieve efficient GFP to BFP conversion in hiPSCs.
NanoDSF assay showed efficient RNP complex formation with equimolar Cas9/dgRNA GFP ratio
Our results are contradicting previous studies showing that excess gRNA could improve on-target cleavage.9,10 To support our conclusions, we then set up an in vitro assay to assess RNP complex formation efficiency, using NanoDSF. NanoDSF is a label-free technology that detects fluorescence of tryptophan and tyrosine of proteins during an increasing ramp of temperature. The shift in emission at 350 and 330 nm is plotted versus time, automatically fitted and allows the determination of thermal unfolding transition points. NanoDSF detects conformational changes in a protein that lead to a difference in thermal stability, defined by the melting temperature (Tm). Since Cas9 is an allosteric enzyme (for a review13) containing tryptophan and tyrosine residues (Figure S3), NanoDSF may be helpful for the detection of RNP complex formation that induces major rearrangements of Cas9.14 We confirmed that it was the case when 0.75 μM of Cas9 was used. To define the efficacy of RNP complex formation with the dgRNA used in hiPSC to target the GFP locus (dgRNA GFP), we obtained the denaturation curve induced by increased temperature with a range of dgRNA GFP concentration, expressed in Cas9/dgRNA molar ratio (Figure 5A). Using the first derivative, we determined the Tm of Cas9 alone (TmCas9) or with a range of Cas9/dgRNA molar ratios (TmRNP) from 1/1 to 1/5 (Figure 5B). Increased amounts of dgRNA GFP used at 1/1 (TmRNP = 49.6°C), 1/2 (TmRNP = 50.0°C), 1/3 (TmRNP = 49.9°C) or 1/5 (TmRNP = 49.8°C) Cas9/dgRNA molar ratio induced a drastic Tm increase (Figure 2C), but the Tm of these molar ratios were not significantly different, compared to that of Cas9 alone (TmCas9 = 43.2°C). This reflects RNP complex formation. There is no free Cas9 detected by this method using equimolar Cas9/dgRNA ratio. Thus, our RNP complex formation is optimal. We then used a point mutation model on rat embryos to verify if equimolar Cas9/dgRNA leads to optimal KI rate and if excess of dgRNA has an impact on KI efficiency.
Figure 5.

NanoDSF assay showed efficient RNP complex formation with equimolar Cas9/dgRNA GFP ratio
(A) Schematic representation of the nanoDSF assay using a constant concentration of Cas9 (0.75 μM) and increased concentrations of dgRNA, expressed as Cas9/dgRNA molar ratios. One Cas9 is only able to load one dgRNA, leading to free molecules for other ratios than equimolar Cas9/dgRNA ratio. The figures were created with https://www.biorender.com/ (Toronto, Canada).
(B) NanoDSF curves and derivative curves used to determine Tm of Cas9 (TmCas9) and RNP complex (TmRNP) for each condition tested with dgRNA GFP.
(C) Comparison of RNP complex formation efficiency with increasing concentrations of dgRNA in the same conditions described in A. and B. by analysis of TmCas9 and TmRNP with dgRNA GFP. Data are represented as individual replicates with mean ± SEM. No significant difference was noted when nonparametric unpaired Mann-Whitney t-tests (N = 3) were done. dgRNA, dual guide RNA; RNP, ribonucleoprotein complex; Tm, melting temperature; GFP, green fluorescent protein.
Excess of dgRNA reduced KI efficiency and drastically increased on-target large deletion in a point mutation rat animal model
Initially, we tested RNP complex formation efficiency using NanoDSF assay regarding excess of Cas9, equimolar Cas9/dgRNA ratio or excess of dgRNA in rat embryos (Figure 6A). In all conditions, addition of dgRNA targeted epoxide hydrolase 2 gene (rEphx2) (hereinafter defined as dgRNA rEphx2), induced a drastic and significant Tm increase compared to Cas9 alone suggesting RNP complex formation (Figure 6B). For RNP (Cas9/dgRNA) molar ratios rEphx2 in favor of Cas9 (RNP 5/1 and 2/1 ratios) we observed a denaturation curve with two transitions represented as TmCas9 and TmRNP. Compared to Cas9 alone (TmCas9 Cas9 = 43.2°C), TmCas9 for those two RNP ratios were not significantly different (TmCas9 RNP 5/1 = 43.0°C; TmCas9 RNP 2/1 = 43.3°C, p = 0.4809 and 0.9353, respectively) while TmRNP were (TmRNP RNP 5/1 = 48.0°C; TmRNP RNP 2/1 = 48.9°C, p = 0.029 for both). These results indicate that RNP complex has been formed but free Cas9 remains in these conditions. With an equimolar ratio (RNP 1/1) or excess of dgRNA rEphx2 (RNP 1/2, 1/3 and 1/5), we observed only one transition (TmRNP RNP 1/1 = 49.7°C; TmRNP RNP 1/2 = 49.9°C; TmRNP RNP 1/3 = 49.8°C; TmRNP RNP 1/5 = 49.7°C). These Tm were not significantly different from each other. Moreover, TmRNP for RNP 1/1 ratio with dgRNA rEphx2 is significantly different from that for RNP 2/1 ratio (TmRNP RNP 2/1 = 48.9°C; TmRNP RNP 1/1 = 49.7°C, p = 0.0206) and Cas9 alone (TmCas9 Cas9 = 43.2°C; TmRNP RNP 1/1 = 49.7°C, p < 0.0001). This indicates that most Cas9 have formed an RNP complex with RNP 1/1 and excess of dgRNA. Thus, our RNP complex formation is efficient and an equimolar Cas9/gRNA ratio limits free Cas9 and, probably also free dgRNA.
Figure 6.

Excess of dgRNA reduced KI efficiency and drastically increased on-target large deletion in a point mutation rat animal model
(A) Schematic representation of nanoDSF assay and associated derivative curves used to determine Tm of Cas9 (TmCas9) and RNP complex (TmRNP) for each concentration of dgRNA rEphx2 with a constant concentration of Cas9 (0.75 μM).
(B) Comparison of RNP complex formation efficiency with increasing concentrations of dgRNA in the same conditions described in A. by analysis of TmCas9 (gray) and TmRNP (black) with dgRNA rEphx2. Data are represented as individual replicates with mean ± SEM. Asterisks highlight the statistical differences between conditions using unpaired nonparametric Mann-Whitney t-tests (p > 0.05 (ns), ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001 and ∗∗∗∗p ≤ 0.0001).
(C) Schematic representation of the point mutation generated by KI with a XbaI ssODN on the rat Ephx2 locus.
(D) Comparison of KI (blue squares), total editing (gray bars) and large deletion (red dots) rates with an equimolar (1/1) Cas9/dgRNA ratio or excess of dgRNA (1/2) and a constant dose of Cas9 (0.2 μM) and ssODN (2 μM). Conditions with Cas9/dgRNA ratio 1/1 (N = 30) and 1/2 (N = 24) were electroporated on 3 different days.
(E) Distribution of KI copy number among KI embryos with the same conditions as described in D. using a boxplot combined with a violin plot. Points represent each embryo carrying KI allele. No significant difference was noted when Kruskal-Wallis test was done.
(F) Allele copy number analysis around Cas9 cleavage site for each condition by ddPCR on rat embryos presenting no large deletion (black), large deletions with (blue) or without KI allele (red). Each curve represents one embryo and each point a ddPCR assay. The figures were created with https://www.biorender.com/ (Toronto, Canada). dgRNA, dual guide RNA; RNP, ribonucleoprotein complex; Tm, melting temperature; ns, non-significant, ssODN, single-stranded oligonucleotide; KI, knockin; bp, base pair; nt, nucleotide.
We tested the effect of an excess of dgRNA (Cas9/dgRNA 1/2) on a point mutation model on rat embryos using dgRNA rEphx2 and a ssODN containing an XbaI restriction site (Figure 6C).11 One-cell rat embryos were electroporated with NEPA21 electroporator (NEPA GENE Co. Ltd, Sonidel, Dublin, Ireland) with large cuvette and implanted into foster mothers. Embryos were harvested at embryonic day 14. Notably, we confirmed from our previous experiments that 0.2 μM of Cas9 and 2 μM of ssODN were the optimal doses (data not shown). Each embryo was genotyped by PCR (genotyping primers Fwd: GGCAGGGTTTCTAGTTCTTGG and Rev: TCTTGTAAACTGAGGCGGGTA) followed by a heteroduplex mobility assay using capillary electrophoresis (HMA-CE)15 with XbaI digestion and Sanger sequencing. Overall genotyping results are summed-up in Table S1. If genome editing occurred at the zygote stage, only one cell was edited. Thus, WT/WT, WT/indel, indel/indel, indel/KI, WT/KI and KI/KI genotypes can be detected among the harvested embryos 14 days after transfer of electroporated zygotes into recipient females. For one cell-embryos, WT or KI copy numbers should be near 0%, 50% or 100%. Genome editing can also take place when the embryo has started to divide. This situation frequently leads to generation of mosaic animals displaying more than two different alleles but also unwanted DNA repair events.16 In this case, WT or KI copy number would be between 0 and 50% or 50% and 100%. Indeed, a first attempt of copy counting by qPCR using SYBR Green,17 with a highly specific 16 bases primer for the KI or the WT allele, revealed high mosaicism (Table S1). We then further analyzed our samples with ddPCR, a more suitable method for rare allele detection and quantification.18 Primers and probes were designed to confidently count copies of WT and KI alleles but also to count copies of genomic DNA flanking dgRNA target site (maximum set to 2 because embryos are diploid). Assays were thus designed at 0.4, 0.8, 1.4 and 2.1 kb on both side of cleavage site. From these assays, we defined large deletions as a loss of copies (copy number below 2) at least 400 bp upstream and/or downstream of cleavage site.
KI, genome editing (cumulated indels and KI) and large deletions (more than 400bp deleted in accordance with ddPCR assays at 5′ 400bp and 3′ 400bp locations) were quantified for each condition (Figure 6D and Table S2). Highly efficient KI was observed with equimolar Cas9/dgRNA ratio (50%) while excess of dgRNA decreased it to 37.5%. Surprisingly, we observed a very high percentage of large deletions in rat embryos when they were electroporated with an excess of dgRNA (75.0%) compared to equimolar Cas9/dgRNA ratio (16.7%).
Interestingly, among KI embryos, the equimolar Cas9/dgRNA condition showed 3 homozygous KI embryos and 1 with more than 1.75 copies while Cas9/dgRNA 1/2 condition only showed 1 embryo with 1.75 copies (Figure 6E). These results suggest an interference of the excess of gRNA with repair mechanisms. Through ddPCR analysis, we showed that large deletions occur both on 5′- and 3′- sides of the cleavage site or cover it. These results allowed us to design personalized PCR reactions to decipher large deletion size and extremities by HMA-CE and Sanger sequencing. All of these three possibilities of large deletions location were similarly represented in each electroporated condition studies (Figure 6F and Table S3). Sequences on the extremities of these large deletions were also studied and showed a prevalence of microhomologies but no repeated patterns (Table S3). With this strategy, we were able to fully characterize most of the embryos in this study. Nevertheless, a few deletions were too large (length >4 kb) to be define under the designed assays (called "MegaDel" in Table S3).
Discussion
Effectiveness and accuracy of CRISPR-Cas9-based genome editing are variable among targeted loci, cells and types of genetic modifications (such as short or large KO or KI). Undoubtedly, efficiency and accuracy rely on an optimal formation of the RNP complex. Nevertheless, because of a lack of appropriate tools, this issue has never been assessed. Recently, a size exclusion chromatography method has been described to characterize aggregation in RNP complex formation by Camperi et al.,7 who highlighted the importance of optimal conformation of the gRNA, but did not analyze RNP complex formation efficiency. Indeed, Camperi et al.7 used 1/3 Cas9/gRNA molar ratio in one of their experiments where free Cas9 was no longer detected, but gRNA clearly remained. This technique can thus detect Cas9, gRNA and RNP complex, but may not be able to detect subtle conformational changes in Cas9 protein. Differential Scanning Fluorimetry (also called Thermal Shift Assay) has previously been used by two teams to qualitatively characterize Cas9 thermostability in the presence of truncated or unfolded gRNA19 or in the presence of different DNA target binding conditions.20 Furthermore, this method requires quadruplicate measurements which highlight the low reproducibility of dye labeling of Cas9 (data not shown). Here, we describe a NanoDSF-based assay, which is a label- and immobilization-free technique used to evaluate Cas9 thermostability. We used this technique to assess RNP complex formation efficiency and limit free Cas9 delivery to cells or embryos. This assay relies on Cas9 conformation and is very sensitive and reproducible. In our experimental conditions, we showed that the use of an equimolar Cas9/gRNA ratio resulted in optimal RNP complex formation, but an excess of Cas9 protein was sub-optimal for RNP complex formation. An excess of gRNA did not further improve Cas9 thermostability and thus did not ameliorate RNP complex formation. NanoDSF assays may be useful for quality controls of RNP complex formation, for instance with different formats of gRNA (single or dual gRNA) or in different buffers. This technique could also be used to characterize subtle changes in Cas9 conformations that could be, for instance, induced by the presence of a DNA donor or a DNA target or magnesium activation in a much easier way than with crystallography in a first attempt. NanoDSF is thus a promising technique complementary to existing tools that will help improving gene editing but also help fully decipher Cas9 mechanism of action.
Several studies showed an increase in genome editing efficiency when excess amounts of gRNA were used in vivo, but they failed to check the in vitro RNP complex formation efficiency as we did in our study.9,10 A sub-optimal RNP complex formation in vitro could explain the difference in genome editing efficiency between our studies and others. Cas9 protein formulation and storage can cause aggregation9 or degradation and may affect the complex formation. We have shown that the Tm of a free Cas9 is of about 43°C in our buffer and that at 37°C denaturation has already started through our NanoDSF study, highlighting the thermosensitivity of this protein that should be taken in consideration when preparing RNP complex. The excess amount of gRNA may also be responsible for poor RNP complex formation, probably due to unexpected secondary structures21,22 or degradation. One Cas9 protein can only load one gRNA,6 RNP (Cas9/gRNA) ratio should then be close to 1/1 for efficient conditions of preparation of RNP complex in vitro as we have demonstrated in this study.
This rational molecular approach facilitated the generation of KI models using iPSCs and rat embryos and demonstrated the superiority of KI when equimolar Cas9/gRNA ratio was employed. We achieved about 50% of KI for both models. Moreover, we demonstrated that an excess of gRNA does not impact KI efficiency nor proportions of homozygous or heterozygous KI subsets in a hiPSC GFP to BFP conversion model with optimized RNP complex formation. We even showed a negative effect of an excess of gRNA on KI rate and reduced number of homozygous KI embryos in a rat point mutation model. This could probably be explained by a surprisingly high large deletion rate occurring in this condition. The remaining free gRNA might be able to interfere with repair mechanisms, thus preventing KI to occur. We have also observed rather few large deletions on our sorted subsets of hiPSC after editing with 0.4 μM of Cas9 with RNP1/1 and 2 μM of ssODN (data not shown).
CRISPR-Cas9 is known for uncontrollable damage generation in on- and off-targets.16 We confirmed the importance of this by analyzing allele-specific copy numbers by ddPCR or qPCR to detect these very frequent on-target damages. Indeed, we believe that copy counting of deleted segments should be part of the quality control required for validating genetically modified models. For genome editing of embryos, high mosaicism is often observed and complicates their full characterization. ddPCR provides a snapshot of the detected locus at a specific time point and drives large deletion characterization by personalized primer design to facilitate genotyping. The use of Next Generation Sequencing (NGS), as exemplified by Oxford Nanopore Technology, is also an option which has been described for KI allele sequence quality control.23 Nevertheless, this is still very expensive, especially for low throughput analysis, and more robust pipelines for analysis are required. New techniques are emerging to identify and quantify chromosomal aberrations and off-targets effects24,25 and will facilitate complete analysis of side effects associated with genome editing.
In this study, an excess amount of gRNA delivered into a cell or embryo decreased KI efficiency. Therefore, the minimal amount of gRNA may be required for inducing an efficient KI event. We used a high amount of ssODN as a repair template to increase the chance for precise HDR at the cleavage site. To limit the delivery of unnecessary DNA donor and have a better control on repair outcome, the DNA donor can be attached to any component of the RNP complex.1 Only one molecule of genomic DNA can be targeted by one RNP complex and double-stranded break repair may require only one molecule of DNA donor. As for RNP complex formation, a stochiometric analysis can be performed on DNA donor/DNA target ratio but also RNP/DNA target ratios. Nevertheless, in the literature, concentrations are often expressed in mass, limiting the molar ratio extrapolation, especially when key information may be missing. The expression of conditions with molar concentrations will facilitate the rational design of stochiometric studies in the future. By using lower amount of RNP complex, we reduced the imbalanced ratio of RNP/large DNA donor, increasing the chance of DNA donor availability at the Cas9 cut site. The present study will help to define conditions for more efficient KI with long DNA donor.
In conclusion, the strategy presented in this study is easy to use and applicable to all models. For hiPSCs, it will lower the number of clones generated during KI campaigns, alleviating the tissue culture burden. For animal models, it will reduce the number of required embryos and enables relevant studies to be conducted more in line with the 3R’s objectives.
Limitations of the study
Rat embryos usage is framed by ethical issues and hiPSC projects are very demanding. These two aspects limited the number of studied loci. Larger studies on multiple loci and species will help to understand the impact of excess of gRNA on repair mechanisms.
Cultured cells are heterogeneous in terms of DNA content mainly because of cell phase differences, making it difficult to confidently assess copy numbers.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Alt-R® S.p. Cas9 Nuclease V3 | Integrated DNA Technologies | 1081058 |
| Alt-R® CRISPR-Cas9 tracrRNA | Integrated DNA Technologies | 1072532 |
| Alt-R® CRISPR-Cas9 crRNA, see Table S4 | Integrated DNA Technologies | custom |
| DraIII-HF®restriction enzyme | New England Biolabs | R3510 |
| Fixable Viability Stain 440UV | BD Bioscience | 566332 |
| PMSG | Med'Vet | SYNCRO-PART PMSG 500 UI |
| hCG | Med'Vet | CHORULON® 1500 UI |
| XbaI restriction enzyme | New England Biolabs | R0145 |
| Critical Commercial Assays | ||
| P3 Primary Cell Nucleofector™ Solution | Lonza | V4XP-3032 |
| Herculase II fusion Enzyme with dNTP Combo | Agilent Technologies | 600677 |
| ddPCR supermix for probes (no dUTP) | Bio-Rad | 1863024 |
| DNA 1K/12K/Hi Sensitivity Assay LabChip | Perkin Elmer | 760517 |
| DNA High Sensitivity Reagent Kit | Perkin Elmer | CLS760672 |
| Master Mix Fast SYBR™ Green | Applied Biosystems | 4385612 |
| Experimental Models: Cell Lines | ||
| LON71-019 | (Gaignerie et al.,26 2018) | N/A |
| Experimental Models: Organisms/Strains | ||
| Sprague-Dawley rats | Janvier Labs | RjHan:SD |
| Oligonucleotides | ||
| ssODN, see Table S4 | Integrated DNA Technologies | Ultramer® DNA Oligo, standard desalting |
| HMA-CE and qPCR primers, see Table S4 | Eurofins | custom |
| ddPCR-VIC labeled reference Sdha assay, see Table S4 | ThermoFisher Scientific | custom |
| ddPCR FAM-labelled custom rat rEphx2 assays, see Table S4 | Integrated DNA Technologies | custom |
| Software and Algorithms | ||
| PR.Therm Control Software | NanoTemper | https://nanotempertech.com/prometheus-pr-thermcontrol-software/ |
| FlowJo | FlowJo software | https://www.flowjo.com/ |
| QuantaSoft Analysis Pro™ software | Bio-Rad | https://www.bio-rad.com/fr-fr/life-science/digital-pcr/qx200-droplet-digital-pcr-system/quantasoft-software-regulatory-edition |
| GraphPad Prism 7.0 | phPad Software, Inc | http://www.graphpad.com/ |
| Rstudio 2022.07.2 | Posit | https://posit.co/download/rstudio-desktop/ |
| LabChip GX | Perkin Elmer | https://www.perkinelmer.com/fr/lab-products-and-services/resources/labchip-and-optimizer-software-downloads.html |
| Other | ||
| nanoDSF grade high sensitivity capillaries | NanoTemper Technologies | PR-C006 |
| Matrigel® Matrix | Corning | 354277 |
| mTeSR1 medium | Stem Cell Technologies | Catalog #85850 |
| StemMACSTM | Miltenyi Biotec | 130-104-368 |
| TrypLE™ Express Enzyme | Thermofisher Scientific | 12605010 |
| Rock inhibitor Y 27632 dihydrochloride | Axon Medchem | 1683 |
| StemFlex | Thermofisher Scientific | A3349401 |
| M16 medium | Sigma-Aldrich | M7292 |
| Droplet generation oil for probes | Bio-Rad | 1863005 |
| Droplet generator DG8 Gasket | Bio-Rad | 1863009 |
| DG8 Cartridges for QX100/QX200 droplet generator | Bio-Rad | 1864008 |
| Automated droplet generation oil for probes | Bio-Rad | 1864110 |
| DG32 cartridge for automated droplet generator | Bio-Rad | 1864108 |
| Pierceable foil heat seal | Bio-Rad | 1814040 |
| ddPCR droplet reader oil | Bio-Rad | 1863004 |
| Twin-tec PCR plate 96, semi-skirted, red | Eppendorf | 30128613 |
Resource availability
Lead contact
Requests for resources and reagents or contact for further information should be addressed to Ignacio Anegon (ianegon@nantes.inserm.fr).
Materials availability
Homozygous GFP hiPSC are available from the iPSC core facility (BioCore, Nantes Université, France), through MTA.
Rat embryos were harvested at embryonic day 14, they are thus not available.
Experimental models and subject details
hiPSCs
Culture of hiPSCs and generation of GFP iPSC lines
iPSCs (mâle cells) were cultured at 37°C, 5% CO2 in feeder-free culture conditions using stem cell-qualified Matrigel-coated plates (0.1 mg/mL; BD Bioscience, San Jose, USA) with mTeSR1 medium (Stem Cell Technologies, Vancouver, Canada) and passaged using the Passaging Solution XF (StemMACSTM, Miltenyi Biotec, Bergisch Gladbach, Germany). When using single cells suspension, cells were detached with TrypLE Express Enzyme (Thermofisher Scientific, Waltham, USA) and culture medium was supplemented with 10 μM Rock inhibitor (Sigma-Aldrich, Saint-Louis, USA).
For the GFP expressing lines, 1 000 000 hiPSCs “LON71-019”26 were transfected with 3 μg TALEN Right (addgene #35442) + 3 μg TALEN Left (addgene #35431) + 4.8 μg AAV-CAGGS-GFP DNA donor (addgene #22212, kindly provided by L. Daheron, Harvard Stem Cells Institute iPSC core facility) using Amaxa Nucleofector II (P3 kit, B-16 program) to obtain KI in the AAV locus. Clones were manually selected and first maintained on matrigel (Matrigel Matrix, Corning, Corning, USA)-coated plates with Stemflex (Thermofisher Scientific, Waltham, USA), then switched to mTeSR1 medium (Stem Cell Technologies, Vancouver, Canada). Genomic PCR of the AAV locus allowed to select cells that were biallelically edited.
Animal models
Sprague-Dawley (SD) rats were the only strain used and sourced from Janvier Labs (Le Genest-Saint-Isle, France). Prepubescent females were 3–4 weeks old (between 75 and 100 g), pseudo-pregnant females and vasectomized or fertile males were 3–6 months old. All the animal care and procedures performed in this study were approved by the Animal Experimentation Ethics Committee of the Pays de la Loire region, France, in accordance with the guidelines from the French National Research Council for the Care and Use of Laboratory Animals (Permit Numbers: CEEA-PdL-2020-26567).
Method details
CRISPR-Cas9 components and RNP formation
Cas9 protein, crRNA and tracrRNA (Alt-R CRISPR Cas9 system) were purchased from IDT Integrated DNA technologies (Coralville, USA) as well as ultramer ssODN modified on both extremities with phosphorothioate bonds. Sequences are specified in Table S4.
DgRNA is formed by equimolar incubation of crRNA and tracrRNA in Nuclease-Free Duplex Buffer (30 mM HEPES, pH 7.5; 100 mM potassium acetate) from IDT Integrated DNA technologies (Coralville, USA). It is denatured for 5 min at 95°C and incubated for 5 min at 25°C to ensure correct folding of gRNA. RNP complex is formed by incubation of Cas9 protein and gRNA, at concentrations specified for each experiment, 10 min at 25°C in a buffer (HEPES 20 mM, KCl 150 mM, pH 7,5). All these steps were made in a thermal cycler to ensure the reproducibility of the experiments. ssODN is finally added at concentrations specified for each experiment after RNP formation.
NanoDSF
Ten μL of RNP complex prepared with 0.75 μM of Cas9 were loaded into nanoDSF grade high sensitivity capillaries (NanoTemper Technologies, Munich, Germany) and installed on capillary array on the Prometheus NT.48 nanoDSF instrument (NanoTemper Technologies, Munich, Germany). The temperature was increased from 20°C to 95°C with a linear thermal ramp (at a rate of 1°C/min). Changes in fluorescence of tryptophan at 330 and 350 nm were recorded at a rate of 10 datapoints per minute. The 350/330 nm ratio of the two wavelengths were plotted against temperature and the first derivative analysis allowed the determination of Tm using the PR.Therm Control Software (NanoTemper Technologies, Munich, Germany).
hiPSC GFP to BFP editing
Gentle TryplE (Gibco, Carlsbad, USA) enzymatic digestion was performed to obtain single cells suspension. 200 000 cells, resuspended in P3 Primary Cell Nucleofector™ Solution (Lonza, Basel, Switzerland), were mixed with RNP complex and the ssODN that contains the sequence that will edit GFP to BFP sequences as well as a new DraIII restriction site8, and were electroporated in 16-well Nucleocuvette Strips (Lonza, Basel, Switzerland) using CA-137 program. Seven days after transfection, cells were dissociated using TryplE. Dead cells were stained with Fixable Viability Stain 440UV (BD Bioscience, San Jose, USA). Cells were analyzed using BD FACSCelesta (GFP: laser: 488 nm, filter: 530/30; BFP: 405 nm laser: 450/40, Viability: laser Ultraviolet, filter: 379/28) and sorted with BD FACSAria (GFP: laser: 488 nm, filter: 530/30; BFP: 405 nm laser: 450/40).
Electroporation of rat intact zygotes and embryo transfer
The electroporation procedure and embryo transfer were performed as described below. Briefly, fertilized 1-cell stage embryos were collected from super-ovulated prepubescent females27 (30 IU PMSG at day-2 before breeding and 20 IU hCG the day of breeding; INTERVET, Beaucouzé, France) and kept in M16 medium (Sigma-Aldrich, Saint-Louis, USA) at 37°C under 5% CO2 until electroporation.
Electroporation of 30–50 embryos with 50 μL of gRNA/Cas9 RNP complex with ssODN was performed using the NEPA21 system (NEPA GENE Co. Ltd, Sonidel, Dublin, Ireland) with 4 poring pulses of 300 V 0.5 ms in length, 50 ms interval and 5 transfer pulses of 20V, 50 ms in length, 50 ms interval, polarity +/− to limit toxicity. Surviving embryos were maintained under 5% CO2 at 37°C until implantation the same day in the oviduct of pseudo-pregnant females (0.5 days postcoitum). Developed embryos were harvested at embryonic day 14 (E14).
Genotyping
Briefly, hiPSCs and E14 embryos were digested overnight at 56°C in 1 mL of tissue digestion buffer (0.1 mol/L Tris-HCl, pH 8.3, 5 mmol/L EDTA, 0.2% SDS, 0.2 mol/L NaCl, 100 μg/mL proteinase K). The CRISPRs nuclease targeted regions were PCR-amplified from diluted lysed samples (1/20) with a high-fidelity polymerase (Herculase II fusion polymerase, Agilent Technologies, Santa Clara, USA). To detect gene editing, locus specific primers GFP and rEphx2 (Table S4) were used and mutations were analyzed using an HMA-CE assay15 using a DNA 1K/12K/Hi Sensitivity Assay LabChip (Perkin Elmer Waltham, USA) with DNA High Sensitivity Reagent Kit (Perkin Elmer, Waltham, USA) followed by PCR amplicon direct digestion by XbaI (New England Biolabs, Ipswich, USA) and sequencing. Capillary electrophoresis results were analyzed using LabChip GX software (Perkin Elmer, Waltham, USA). qPCR was done with master Mix Fast SYBR Green (Applied Biosystem, Waltham, USA) and primers described in Table S4 to quantify both WT and KI alleles.
Allele copy counting by ddPCR
DdPCR was used to determine copy number variation28 in genome-edited rat embryos. Experiments were performed as duplex reactions, where the sequence employed as a donor was amplified using a fluorescein amidite (FAM)-labelled assay. Custom assays were ordered from IDT Integrated DNA technologies (Coralville, USA). Custom assays were used in parallel with 2′-chloro-7′phenyl-1,4-dichloro-6-carboxy-fluorescein derived from Aequorea victoria (VIC)-labelled reference gene assay (Table S4) set at two copies on the Bio-Rad QX200 ddPCR System (Bio-Rad, Hercules, USA). Reaction mixes (22 μL) contained 2 μL crude DNA lysate or 50 ng of phenol/chloroform purified genomic DNA, 1× ddPCR Supermix for probes (Bio-Rad, Hercules, USA), 225 nM of each primer (two primers per assay) and 50 nM of each probe (one VIC-labelled probe for the reference gene assay and one FAM-labelled for the target genomic region assays). These reaction mixes were loaded in plate format into the Bio-Rad QX200 AutoDG and the droplets generated as per the manufacturer’s instructions. Post droplet generation, the oil/reagent emulsion was transferred to a 96-well semi-skirted plate (Eppendorf, Hamburg, Germany), and the samples were amplified on a Bio-Rad T100 thermocycler (95°C for 10 min, followed by 40 cycles of 94°C for 30 s and 60°C for 60 s, with a final elongation step of 98°C for 10 min, where all temperature ramping was set to 3°C/s). The plate containing the droplet amplicons was subsequently loaded into the QX200 Droplet Reader (Bio-Rad, Hercules, USA). Standard reagents and consumables supplied by Bio-Rad (Hercules, USA) were used, including cartridges, droplet generation oil and droplet reader oil. Copy numbers were assessed using the QuantaSoft Analysis Pro software (Bio-Rad, Hercules, USA) using at least 10 000 accepted droplets per sample. The copy numbers were calculated by applying Poisson statistics to the fraction of end-point positive reactions.
Quantification and statistical analysis
Graphs and statistical analyses were performed using nonparametric unpaired Mann-Whitney tests with GraphPad Prism (GraphPad Software, Inc, San Diego, USA) v5.03 excepted for Figures 6E and 6F which were done with Rstudio (Posit, Inc, Boston, USA). Kruskal-Wallis and Dunn tests were performed for KI copy numbers distribution analysis among KI embryos (Figure 6E). Detailed are provided in each figure legend.
Acknowledgments
We thank the TRIP, iPSC and P2R core facilities (Biogenouest and IBiSA endorsed), CR2TI and genOway for their support. We thank in particular Claire Usal and Séverine Ménoret (TRIP) for their animal expertise, Laure-Hélène Ouisse, Ghenima Ahmil (TRIP), Séverine Bézie and Jenny Greig (CR2TI) for their flow cytometry and cell sorting expertise, Aude Guiffes (TRIP) for her work on rat genotyping, Anne Gaignerie (iPSC BioCore) for hiPSC culture support and Sarah Duponchel (genOway) for her wise advices. NanoDSF studies was supported by funding from the Biogenouest network and from the “Pays de la Loire” region. This work was also supported by a Medical Research Council grant (A410 to LTeboul).
Author contributions
VC participated in all experiments design and analysis. IL and LD designed hiPSC experiments and IL performed experiments and analysis. SR and IL phenotyped hiPSCs by flow cytometry. VC performed RNP complex formation for rat embryo electroporation and nanoDSF studies. SR handled rat embryos and electroporation of RNP complex. LTesson set up and performed hiPSCs and rat embryos genotyping. AF and KB performed nanoDSF experiments and analysis. AA, DA, AC and LTeboul participated in the design set-up, performance and analysis of ddPCR experiments. YC, LTeboul, LD and IA secured funding, defined experiments as well as interpreted and analyzed data. VC wrote this manuscript with all authors intellectual contribution.
Declaration of interests
YC and VC were genOway employees. The other authors declare no conflict of interest.
Published: March 14, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106399.
Supplemental information
MegaDel means no amplification with primer situated at 3.4 kb 5′ side or 2.4 Kb 3’side, size and sequence ND; ND, not determined; microhomologies are written in red related to Figure 6
∗indicates bases with phosphorothioate modification. reaction; GFP, Green Fluorescent Protein; BFP, Blue Fluorescent Protein; hiPS, human induced pluripotent stem cells. ssODN, single-stranded oligonucleotide; KI, knockin, qPCR, HMA-CE, heteroduplex mobility assay by capillary electrophoresis; quantitative polymerase chain related to STAR Methods
Data availability
Any additional information on the data generated in this paper is available from the lead contact upon request.
References
- 1.Chenouard V., Remy S., Tesson L., Ménoret S., Ouisse L.-H., Cherifi Y., Anegon I. Advances in genome editing and application to the generation of genetically modified rat models. Front. Genet. 2021;12:615491. doi: 10.3389/fgene.2021.615491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karagiannis P., Takahashi K., Saito M., Yoshida Y., Okita K., Watanabe A., Inoue H., Yamashita J.K., Todani M., Nakagawa M., et al. Induced pluripotent stem cells and their use in human models of disease and development. Physiol. Rev. 2019;99:79–114. doi: 10.1152/physrev.00039.2017. [DOI] [PubMed] [Google Scholar]
- 3.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Javaid N., Choi S. CRISPR/Cas system and factors affecting its precision and efficiency. Front. Cell Dev. Biol. 2021;9 doi: 10.3389/fcell.2021.761709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burgio G., Teboul L. Anticipating and identifying collateral damage in genome editing. Trends Genet. 2020;36:905–914. doi: 10.1016/j.tig.2020.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nishimasu H., Ran F.A., Hsu P.D., Konermann S., Shehata S.I., Dohmae N., Ishitani R., Zhang F., Nureki O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156:935–949. doi: 10.1016/j.cell.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Camperi J., Moshref M., Dai L., Lee H.Y. Physicochemical and functional characterization of differential CRISPR-cas9 ribonucleoprotein complexes. Anal. Chem. 2022;94:1432–1440. doi: 10.1021/acs.analchem.1c04795. [DOI] [PubMed] [Google Scholar]
- 8.Glaser A., McColl B., Vadolas J. GFP to BFP conversion: a versatile assay for the quantification of CRISPR/cas9-mediated genome editing. Mol. Ther. Nucleic Acids. 2016;5:e334. doi: 10.1038/mtna.2016.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nguyen D.N., Roth T.L., Li P.J., Chen P.A., Apathy R., Mamedov M.R., Vo L.T., Tobin V.R., Goodman D., Shifrut E., et al. Polymer-stabilized Cas9 nanoparticles and modified repair templates increase genome editing efficiency. Nat. Biotechnol. 2020;38:44–49. doi: 10.1038/s41587-019-0325-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wei T., Cheng Q., Min Y.-L., Olson E.N., Siegwart D.J. Systemic nanoparticle delivery of CRISPR-Cas9 ribonucleoproteins for effective tissue specific genome editing. Nat. Commun. 2020;11:3232. doi: 10.1038/s41467-020-17029-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Remy S., Chenouard V., Tesson L., Usal C., Ménoret S., Brusselle L., Heslan J.-M., Nguyen T.H., Bellien J., Merot J., et al. Generation of gene-edited rats by delivery of CRISPR/Cas9 protein and donor DNA into intact zygotes using electroporation. Sci. Rep. 2017;7 doi: 10.1038/s41598-017-16328-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Skarnes W.C., Pellegrino E., McDonough J.A. Improving homology-directed repair efficiency in human stem cells. Methods. 2019;165:18–28. doi: 10.1016/j.ymeth.2019.06.016. [DOI] [PubMed] [Google Scholar]
- 13.Zuo Z., Liu J. Allosteric regulation of CRISPR-Cas9 for DNA-targeting and cleavage. Curr. Opin. Struct. Biol. 2020;62:166–174. doi: 10.1016/j.sbi.2020.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jinek M., Jiang F., Taylor D.W., Sternberg S.H., Kaya E., Ma E., Anders C., Hauer M., Zhou K., Lin S., et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science. 2014;343 doi: 10.1126/science.1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chenouard V., Brusselle L., Heslan J.-M., Remy S., Ménoret S., Usal C., Ouisse L.-H., NGuyen T.H., Anegon I., Tesson L. A rapid and cost-effective method for genotyping genome-edited animals: a heteroduplex mobility assay using microfluidic capillary electrophoresis. J Genet Genomics. 2016;43:341–348. doi: 10.1016/j.jgg.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 16.Bunton-Stasyshyn R.K., Codner G.F., Teboul L. Screening and validation of genome-edited animals. Lab. Anim. 2022;56:69–82. doi: 10.1177/00236772211016922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tesson L., Rémy S., Ménoret S., Usal C., Anegon I. Analysis by quantitative PCR of zygosity in genetically modified organisms. Methods Mol. Biol. 2010;597:277–285. doi: 10.1007/978-1-60327-389-3_19. [DOI] [PubMed] [Google Scholar]
- 18.Owens D.D.G., Caulder A., Frontera V., Harman J.R., Allan A.J., Bucakci A., Greder L., Codner G.F., Hublitz P., McHugh P.J., et al. Microhomologies are prevalent at Cas9-induced larger deletions. Nucleic Acids Res. 2019;47:7402–7417. doi: 10.1093/nar/gkz459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tillotson E.E., Harmange G., Selleck W., Jayaram H. editas MEDICINE; 2017. Assessing Cas9-gRNA Ribonucleoprotein Complex Formation for Development of Ex-Vivo Therapeutics.https://www.editasmedicine.com/newsroom/ [Google Scholar]
- 20.Jiang F., Taylor D.W., Chen J.S., Kornfeld J.E., Zhou K., Thompson A.J., Nogales E., Doudna J.A. Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science. 2016;351:867–871. doi: 10.1126/science.aad8282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thyme S.B., Akhmetova L., Montague T.G., Valen E., Schier A.F. Internal guide RNA interactions interfere with Cas9-mediated cleavage. Nat. Commun. 2016;7 doi: 10.1038/ncomms11750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Riesenberg S., Helmbrecht N., Kanis P., Maricic T., Pääbo S. Improved gRNA secondary structures allow editing of target sites resistant to CRISPR-Cas9 cleavage. Nat. Commun. 2022;13:489. doi: 10.1038/s41467-022-28137-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCabe C.V., Codner G.F., Allan A.J., Caulder A., Christou S., Loeffler J., Mackenzie M., Malzer E., Mianné J., Pike F.J., et al. Application of long-read sequencing for robust identification of correct alleles in genome edited animals. bioRxiv. 2019 doi: 10.1101/838193. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turchiano G., Andrieux G., Klermund J., Blattner G., Pennucci V., El Gaz M., Monaco G., Poddar S., Mussolino C., Cornu T.I., et al. Quantitative evaluation of chromosomal rearrangements in gene-edited human stem cells by CAST-Seq. Cell Stem Cell. 2021;28:1136–1147.e5. doi: 10.1016/j.stem.2021.02.002. [DOI] [PubMed] [Google Scholar]
- 25.Wienert B., Wyman S.K., Richardson C.D., Yeh C.D., Akcakaya P., Porritt M.J., Morlock M., Vu J.T., Kazane K.R., Watry H.L., et al. Unbiased detection of CRISPR off-targets in vivo using DISCOVER-Seq. Science. 2019;364:286–289. doi: 10.1126/science.aav9023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gaignerie A., Lefort N., Rousselle M., Forest-Choquet V., Flippe L., Francois–Campion V., Girardeau A., Caillaud A., Chariau C., Francheteau Q., et al. Urine-derived cells provide a readily accessible cell type for feeder-free mRNA reprogramming. Sci. Rep. 2018;8 doi: 10.1038/s41598-018-32645-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ménoret S., Remy S., Usal C., Tesson L., Anegon I. Generation of transgenic rats by microinjection of short DNA fragments. Methods Mol. Biol. 2010;597:81–92. doi: 10.1007/978-1-60327-389-3_6. [DOI] [PubMed] [Google Scholar]
- 28.Codner G.F., Lindner L., Caulder A., Wattenhofer-Donzé M., Radage A., Mertz A., Eisenmann B., Mianné J., Evans E.P., Beechey C.V., et al. Aneuploidy screening of embryonic stem cell clones by metaphase karyotyping and droplet digital polymerase chain reaction. BMC Cell Biol. 2016;17:13–30. doi: 10.1186/s12860-016-0108-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MegaDel means no amplification with primer situated at 3.4 kb 5′ side or 2.4 Kb 3’side, size and sequence ND; ND, not determined; microhomologies are written in red related to Figure 6
∗indicates bases with phosphorothioate modification. reaction; GFP, Green Fluorescent Protein; BFP, Blue Fluorescent Protein; hiPS, human induced pluripotent stem cells. ssODN, single-stranded oligonucleotide; KI, knockin, qPCR, HMA-CE, heteroduplex mobility assay by capillary electrophoresis; quantitative polymerase chain related to STAR Methods
Data Availability Statement
Any additional information on the data generated in this paper is available from the lead contact upon request.