

Summary
The main cause of high mortality in cancer patients is tumor metastasis. Exploring the underlying mechanism of tumor metastasis is of great significance for clinical treatments. Here, we identify the transcription factor Apt/FSBP is a suppressor for tumor metastasis. In Drosophila wing disc, knockdown of apt is able to trigger cell migration, whereas overexpression of apt hampers scrib-RNAi-induced tumor cell migration. Further studies show that loss of apt promotes cell migration through activating the JNK pathway. To investigate the role of FSBP, the homolog of Apt in mammals, we construct Fsbp liver-specific knockout mice. Knockout of Fsbp in liver does not cause any detectable physiological defects, but predisposes to tumorigenesis on DEN and CCl4 treatment. In addition, loss of Fsbp accelerates tumor metastasis from liver to diaphragm. Taken together, this study uncovers FSBP is a novel tumor suppressor, and provides it as a considerable drug target for tumor treatment.
Subject areas: Cancer, Cell biology, Cellular physiology
Graphical abstract
Highlights
-
•
Transcription factor Apt/FSBP is a suppressor for tumor metastasis
-
•
Loss of apt/Fsbp activates the JNK signaling pathway
-
•
Apt/FSBP is functional conserved in controlling cell migration from fly to mammals
Cancer; Cell biology; Cellular physiology
Introduction
Hepatocellular carcinoma (HCC) is one of the most common malignancies worldwide and the third leading cause of cancer-related deaths.1 The high mortality of HCC can be attributed to tumor metastasis.2 Tumor metastasis is a complex process involving activation of oncogenes and/or repression of tumor suppressors. Although the treatment of HCC has been greatly improved in recent years, there is still a lack of effective drugs to suppress tumor metastasis, mainly because the underlying mechanism of metastasis remains poorly understood.3,4 Therefore, it is imperative to dissect the mechanism of HCC metastasis.
The apontic (apt) gene is first identified to regulate tracheal system morphogenesis in Drosophila, and its deficiency leads to trachea defect.5 The following study show that apt encodes a putative bZIP (basic leucine zipper) transcription factor, which binds a consensus DNA sequence.6 In addition, Apt is involved in controlling Drosophila border cell migration through inhibiting JAK-STAT pathway.7 Despite its role as a transcription factor, the first direct transcriptional target gene of Apt has been identified until 2014.8 Apt governs cell cycle transition through activating cyclin E (cycE) transcription during Drosophila eye disc differentiation. Apt turns on cycE expression by directly binding its promoter.8 Phylogenetic analysis shows that Apt is evolutionarily conserved from Drosophila to mammals, especially its DNA-binding domain (bZIP). Besides, Apt is able to activate hedgehog (hh) expression to regulate wing development. Fibrinogen silencer binding protein (FSBP), the homolog of Apt, also positively regulates hh expression in cultured human cells,9 indicating that Apt/FSBP is functional conserved. FSBP expresses in several human tissues and is able to repress the transcription of the γ-chain of human fibrinogen gene.10 Fibrinogen is a soluble glycoprotein synthesized by hepatocytes in the liver, and its dysregulation has been implicated in several human diseases, including HCC.11 Previous studies have revealed that elevated fibrinogen tightly correlates with HCC stage and poor prognosis, suggesting its oncogenic role for HCC tumorigenesis.12 In addition, FSBP forms a complex with X11α to inhibit GSK3β transcription.10 Although our previous studies have clearly uncovered that Apt/FSBP promotes cell proliferation in Drosophila wing discs and human cell lines, the role of Apt/FSBP in metastasis is still unknown.
The JNK pathway is an important mechanism for tumor cell migration because it triggers the expression of several metalloproteinases, which hydrolyze the extracellular matrix and endow cells the ability to migrate.13 The JNK pathway is an evolutionarily conserved kinase cascade, which is strictly controlled by extrinsic and intrinsic stresses.14,15 In Drosophila, the tumor necrosis factor (TNF) homolog Eiger (Egr) binds to its receptor Grindelwald (Grnd)16 or Wengen (Wgn)17 to activate the downstream kinase cascade, culminating in JNK phosphorylation and activation.16,18 Activated JNK phosphorylates the putative transcriptional factors c-Jun and c-Fos to promote their nuclear accumulation.13 In mammals, MAPK kinases MKK4 and MKK7 activate the JNK signaling pathway by directly phosphorylating Thr183 and Tyr185 residues of JNK.19 The JNK pathway is involved in many cellular processes, such as apoptosis, proliferation, differentiation, cell migration and inflammation.15 Hyperactivation of the JNK signaling is sufficient to trigger tumor cell migration.20 Our previous study also shows that the deubiquitinase Usp8 promotes breast cancer metastasis through activating the JNK pathway.21 In HCC tissues, the JNK pathway aggravates tumorigenesis through activating the oncogenic targets, including c-Myc.22 Furthermore, conditional knockout of c-Jun in mouse hepatocytes dramatically hampers tumorigenesis with diethylnitrosamine (DEN) treatment.23 Albeit the important role of the JNK pathway in HCC tumorigenesis, its regulation remains unclear.
To examine the role of Apt/FSBP in tumor cell migration, we first constructed an in vivo Drosophila metastasis model using scrib RNAi and found that Apt suppressed metastasis. Next, FSBP was able to attenuate migration and invasion of HCC cell lines. To investigate the role of FSBP in liver cancer metastasis, we generated Fsbp liver condition knockout mice and induced tumorigenesis using DEN and CCl4 treatment. Consistently, knockout of Fsbp not only elevated liver tumor burden, but also promoted tumor migration. Mechanistically, activation of the JNK pathway is account for apt-RNAi-caused cell migration enhancement. Compared with wild type liver, RNA-seq results showed that Fsbp−/− liver exhibited upregulated JNK pathway activity. Thus, this study established an FSBP-JNK axis as a novel mechanism to regulate HCC metastasis.
Results
Loss of Apt promotes tumor cell migration through activating the JNK pathway
Increasing studies have revealed that the transcription factor Apt is involved in multiple physiological processes, including cell proliferation, but its role in tumor cell migration is still unclear. To explore whether Apt modulates tumor cell migration, we first silenced apt using patched-gal4 (ptc-gal4), which expresses along the anterior/posterior (A/P) boundary in the wing disc. Compared with the control (Figures 1A and 1C), knockdown of apt triggered cell migration (Figures 1B and 1C). In Drosophila wing discs, knockdown of the cell polarity gene scribbled (scrib) confers cell migration, which has been used as an ideal model to study tumor cell migration.24 We employed this model to test whether Apt regulates scrib-RNAi-induced tumor cell migration. Compared with scrib-RNAi control (Figures 1E and 1H), random knockdown of apt using FLP-out technique aggravated cell migration (Figures 1G and 1H), whereas overexpression of apt blocked cell migration (Figures 1F and 1H). Taken together, Apt is possible a suppressor for tumor cell migration.
Figure 1.

Apt knockdown promotes cell migration through JNK pathway
(A) The third-instar larvae wing disc of ptc-gal4 UAS-GFP was used as a control.
(B) Knockdown of apt induced cell migration from A/P boundary to posterior.
(C) Quantification analyses of migrating cell numbers in A, B (N = 15), data are represented as means ± SD.
(D) Clone formed under normal conditions.
(E) Scrib-RNAi induced peripheral migration of the cloned cells.
(F) Overexpression of apt inhibited the migration of clone cells induced by scrib-RNAi.
(G) apt-RNAi promoted the migration of clone cells induced by scrib-RNAi.
(H) Quantification analyses of migrating cell numbers in D-G (N = 15), data are represented as means ± SD. The t-test was used for statistical analyses, ∗∗∗∗p < 0.0001.
(I) A control third-instar larvae wing disc expressing GFP under ptc-gal4 driver was stained to show GFP (green) and Mmp1 (red).
(J) Knockdown of apt increased Mmp1 level.
(K) A control third-instar larvae wing disc expressing GFP under ptc-gal4 driver was stained to show GFP (green) and puc-lacZ (red).
(L) Knockdown of apt increased puc-lacZ level.
(M) A control wing disc expressing GFP was stained with GFP (green) and p-JNK (red).
(N) Knockdown of apt in the wing disc by ptc-gal4 elevated p-JNK level.
(O) Knockdown of apt alone caused cell migration and increased puc-LacZ level.
(P) Knockdown of jnk could rescue the phenotype induced by apt-RNAi.
(Q and R) Overexpression of FSBP inhibited the cell migration induced by apt-RNAi in the wing disc. Scale bars: 50 μm for all disc images.
It is well known that the JNK pathway is a key driver for tumor cell migration because of its target gene matrix metalloproteinase (mmp1), which encodes a proteinase to hydrolyze extracellular matrix. Thus, it is necessary to test whether Apt modulates the JNK pathway. Compared with the control disc (Figure 1I), knockdown of apt substantially upregulated Mmp1 (Figure 1J). Furthermore, knockdown of apt was able to increase puc-lacZ and pJNK (Figures 1K-1N), which serve as well-documented readouts of the JNK pathway. Overall, these results have demonstrated that knockdown of apt activates the JNK pathway.
Having established that knockdown of apt activates the JNK pathway, we next examined whether the JNK pathway activation is required for apt-RNAi-induced tumor cell migration. Compared with apt-RNAi alone (Figure 1O), knockdown of jnk effectively inhibited apt-RNAi-induced cell migration and puc-lacZ upregulation (Figure 1P). Of interest, ectopic expression of FSBP, the homolog of Apt in human, was able to restore apt-RNAi-induced cell migration and Mmp1 upregulation (Figures 1Q and 1R), indicating the conserved role of Apt/FSBP in controlling tumor cell migration.
FSBP inhibits HCC cell metastasis
Albeit the above results reveal that FSBP is able to replace Apt to regulate tumor cell migration in Drosophila wing discs, we needed to investigate its role in human tumor cell lines. We chose hepatocellular carcinoma (HCC) cell lines for following study since the high mortality of HCC. First, we checked the expression levels of FSBP in six HCC cell lines and found that FSBP showed high expression in SK-Hep1 cells and low in SMMC-7721 cells (Figures 2A and 2B). Thus, we selected SK-Hep1 cells for FSBP knockdown assays and SMMC-7721 cells for FSBP overexpression assays. Overexpression of FSBP apparently decreased the ability of migration and invasion in SMMC-7721 cells (Figures 2C and 2D). In addition, FSBP was able to attenuate wound healing in SMMC-7721 cells (Figure 2G), together suggesting that FSBP is sufficient to suppress HCC cell metastasis. Conversely, knockdown of FSBP promoted migration and invasion of SK-Hep1 cells (Figure 2F). The western blot result showed that FSBP siRNA effectively silenced endogenous FSBP (Figure 2E). In line with this, knockdown of FSBP also quickened wound healing of SK-Hep1 cells (Figure 2H), indicating that FSBP is a suppressor for HCC cell metastasis. Taken together, these results uncover that FSBP is sufficient and necessary to inhibit HCC cell metastasis.
Figure 2.

FSBP inhibits the metastasis of hepatocellular carcinoma cells
(A and B) The expression of FSBP in liver cancer cell lines.
(C) The expression of Fg-FSBP in SMMC-7721 cells determined by western blot. The SMMC-7721 cells invasion and migration abilities were analyzed by Transwell assay, invasion assay (D) and wound-healing assay (G), respectively. Quantification analysis was shown on the right.
(E) The knockdown efficiency of FSBP in SK-Hep1 cells was determined by western blot. The SK-Hep1 cell invasion and migration abilities were analyzed by Transwell assay, invasion assay (F) and wound-healing assay (H), respectively. Quantification analysis was shown on the right, data are represented as means ± SD. The t-test was used for statistical analyses, ∗p < 0.05, ∗∗∗p < 0.001. Scale bars: 100 μm.
Fsbp deficiency promotes hepatocarcinogenesis and metastasis
To further investigate the function of Fsbp for hepatocarcinogenesis in vivo, Fsbp was specifically knocked out in mouse liver and the status of tumorigenesis was checked. We generated transgenic Fsbpfl/fl mouse, which two loxP sequences were inserted into the encoding region of Fsbp (Figure S1A). Fsbpfl/fl mouse was then crossed with Alb-Cre mouse to knock out Fsbp (Figure S1A). The PCR result revealed that the corresponding encoding sequence was indeed excised in the liver (Figures S1A and S1B). To investigate the role of Fsbp in HCC initiation and progress, we established a mouse liver tumor model by single injection of diethylnitrosamine (DEN) and multiple injections of carbon tetrachloride (CCl4) into Fsbp−/− and Fsbpfl/fl mice. Both DEN and CCl4 are toxic carcinogens that can cause liver cell death, DNA damage, fibrosis, and ultimately liver cancer. The mice were sacrificed at 26 weeks for following study (Figure 3A). We collected mouse livers, measured the number and size of tumors, and calculated the ratios of liver weight to body weight. After calculation, we observed that the tumor number, tumor size larger than 5 mm, and liver weight to body weight ratios were higher in the liver of Fsbp−/− mice than in Fsbpfl/fl mice (Figures 3A and 3B). Hematoxylin-eosin (HE) staining showed that Fsbp−/− mice had larger tumor area compared with Fsbpfl/fl mice (Figure 3C). Immunohistochemistry (IHC) results showed that the number of Ki67-positive cells in the liver of Fsbp−/− mice was increased (Figure 3D). In addition, we found many tumor nodules in the diaphragm of Fsbp−/− mice but not Fsbpfl/fl mice (Figure 3E). According to pathological analysis and Ki67 staining, diaphragmatic nodules were metastatic tumors (Figures 3F and 3G). Overall, these data have shown that Fsbp deletion promotes liver cancer initiation and metastasis in mice.
Figure 3.

FSBP deficiency promotes hepatocarcinogenesis and metastasis in vivo
(A) Schematic of the experimental design. Fsbp−/− (n = 12) and Fsbpfl/fl (n = 12) mice were injected with DEN (20 mg/kg i.p.) at day 14 postpartum, followed by twice of week injections of CCl4 (0.5 mL/kg i.p., 1:4 dissolved in corn oil) for 12 weeks starting at 4 weeks after birth. The mice were sacrificed at 24 weeks after DEN injection.
(B) Tumor numbers, liver-weight-to-body-weight ratios, and number of tumors larger than 5 mm in diameter of the Fsbp−/− and Fsbpfl/fl mice were analyzed and compared.
(C) HE staining of the liver tissues from Fsbp−/− and Fsbpfl/fl mice was shown. T indicated tumor.
(D) Immunohistochemical analysis of Ki67 expression in mice liver tumors.
(E) Tumor metastasis loci in diaphragm from Fsbp−/− and Fsbpfl/fl mice were compared.
(F) HE staining of the diaphragmatic tumor metastases from Fsbp−/− and Fsbpfl/fl mice were showed.
(G) Immunohistochemical analysis of Ki67 expression in mice diaphragm. The t-test was used for statistical analyses, data are represented as means ± SD, ∗p < 0.05, ∗∗p < 0.01. Scale bars: 100 μm.
FSBP suppresses EMT in mouse liver and human HCC cell lines
Although the above in vivo data suggest that deletion of Fsbp promotes liver cancer metastasis, we needed to confirm this result at molecular level. We examined EMT marker, such as N-Cadherin and Vimentin, to reflect cell metastasis. First, we found that overexpression of FSBP reduced mesenchymal markers, including N-Cadherin, β-Catenin, Vimentin, FAK and Snail, although increased epithelial marker E-Cadherin in SK-Hep1 cells and SMMC-7721 (Figure 4A), suggesting that cell migration is inhibited. In contrast, knockdown of FSBP upregulated mesenchymal markers, but downregulated epithelial marker (Figure 4B), indicating EMT occurs in cells. On the other hand, we further examined EMT markers in liver tissues of Fsbp knockout mice, and found Fsbp−/− mice showed increased mesenchymal markers and decreased epithelial marker compared to Fsbpfl/fl mice (Figure 4C). Consistently, IHC staining also showed that knockout of Fsbp elevated EMT signals in liver tissues (Figure 4D). Altogether, these results suggest that FSBP suppresses EMT in mouse liver and human HCC cells.
Figure 4.

FSBP suppresses EMT in mouse liver and human HCC
(A and B) Western blot analysis of the expression of EMT markers (E-Cadherin, N-cadherin, β-Catenin, Vimentin, FAK and Snail) in SK-Hep1 cells and SMMC-7721 cells with overexpression (A) or silencing (B) of FSBP. Actin acts as a loading control.
(C) Western blot analysis of the expression of EMT markers in mouse liver tumor with knocking out Fsbp. Actin acts as a loading control.
(D) Immunohistochemical analysis of EMT markers (E-Cadherin, β-Catenin, Vimentin, FAK and Snail) expression in Fsbp−/− and Fsbpfl/fl mice. The t-test was used for statistical analyses, data are represented as means ± SD, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Scale bars: 50 μm.
Loss of Fsbp activates the JNK pathway in mouse livers
Our above results have demonstrated that knockdown of apt promotes tumor cell migration through activating the JNK pathway in Drosophila wing discs. Because the JNK pathway is evolutionarily conserved from insects to human,15 we sought to test whether FSBP inhibits HCC metastasis via controlling the JNK pathway. At first, we performed RNA-seq analysis of livers from Fsbpfl/fl and Fsbp−/− mice (n = 3). KEGG pathway enrichment analysis showed that the differential genes in MAPK signaling pathway were more significant (Figure 5A). Hierarchical clustering revealed that multiple genes (c-Jun, c-Myc, Fos, etc.) downstream of the JNK signaling pathway were up-regulated in Fsbp−/− livers compared with Fsbpfl/fl (Figure 5B and Tables S2 and S3). As expected, the protein levels of p-JNK, p-c-Jun, c-Myc and MMP9 (the ortholog of Mmp1 in human) in hepatoma cells of Fsbp−/− mice were higher than those of Fsbpfl/fl mice (Figure 5C). In consistent, RT-qPCR results showed that the mRNA levels of c-Jun, c-Myc, MMP9 also upregulated in Fsbp−/− livers compared to Fsbpfl/fl livers (Figure 5D). Therefore, knockout of Fsbp in mouse liver is able to activate the JNK pathway.
Figure 5.

Hepatic Fsbp deficiency activates the JNK signaling pathway
(A) KEGG pathway analysis of the number of differential genes and (B) Hierarchical clustering analysis of up and downregulated genes in livers from Fsbp−/− and Fsbpfl/fl mice.
(C) Knockout of Fsbp increased p-JNK, p-c-Jun, c-Myc and MMP9 protein levels in Fsbp−/− and Fsbpfl/fl mice.
(D) Relative mRNA expression of Fsbp, JNK, c-Jun, c-Myc and MMP9 in livers of Fsbp−/− and Fsbpfl/fl mice. The t-test was used for statistical analyses, data are represented as means ± SD, ns (not significant), ∗p < 0.05, ∗∗∗p < 0.001.
FSBP inhibits HCC cell metastasis via the JNK pathway
Having established that loss of Fsbp triggers the JNK pathway in mouse livers, it is necessary to test whether FSBP regulates HCC metastasis through controlling the JNK pathway. At first, we validated the regulation of FSBP on the JNK pathway in several HCC cell lines. Overexpression of FSBP apparently decreased p-JNK, p-c-Jun, c-Myc and MMP9 protein levels in SK-Hep1 cells and SMMC-7721 cells (Figure 6A), reflecting the inhibition of the JNK pathway. In contrast, knockdown of FSBP activated the JNK signaling in SK-Hep1 cells and SMMC-7721 cells (Figure 6B). In addition, RT-qPCR analyses confirmed that FSBP inhibited the expression of JNK pathway target genes (Figures 6C and S2A), whereas FSBP siRNA exerts an opposite effect (Figures 6D and S2B).
Figure 6.

FSBP-deficiency leads to activation of the JNK signaling pathway in vitro
(A) Overexpression of FSBP reduced p-JNK, p-c-Jun, c-Myc and MMP9 protein levels in SK-Hep1 cells and SMMC-7721 cells.
(B) Knockdown of FSBP increased p-JNK, p-c-Jun, c-Myc and MMP9 protein levels in SK-Hep1 cells and SMMC-7721 cells.
(C and D) Relative mRNA expression of FSBP, JNK, c-Jun, JunD, c-Myc and MMP9 in HCC cells.
(E–G) SK-Hep1 cells and SMMC-7721 cells migration and p-JNK expression were examined by western blot and transwell assay in the presence or absence of JNK inhibitors. (E and G) Showed p-JNK protein expression in SK-Hep1 cells and SMMC-7721 cells. (F and H) SK-Hep1 cells and SMMC-7721 cells migration were shown. Quantification analysis was shown on the right. The t-test was used for statistical analyses, data are represented as means ± SD, ns (not significant), ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Scale bars: 100 μm.
In fact, several JNK pathway inhibitors are under preclinical trial. Given that knockdown of FSBP promotes HCC cell metastasis via activating the JNK pathway, we next investigated whether the JNK pathway inhibitor is able to suppress FSBP siRNA-induced cell migration. The JNK inhibitor (SP600125) treatment indeed decreased p-JNK under FSBP knockdown in SK-Hep1 cells (Figure 6E), suggesting that the activation of the JNK pathway is blocked. Consistently, SP600125 treatment effectively suppressed FSBP siRNA-induced cell metastasis enhancement of SK-Hep1 cells (Figure 6F). We validated these results in SMMC-7721 cells. Taken together, loss of FSBP promotes HCC cell metastasis via activating the JNK pathway, thus JNK inhibitor is a possible drug for FSBP-related HCC.
HCC samples show decreased FSBP
The above data have demonstrated that FSBP acts as a suppressor for HCC metastasis in cell lines and mouse model. We next examined the expression of FSBP in human HCC samples. Compared with the corresponding non-cancerous liver tissues, FSBP were apparently decreased in HCC samples (Figure 7A). We analyzed 30 pairs of human liver cancer patients and found that HCC samples showed decreased expression of FSBP, indicating the downregulation of FSBP is possible a cause of HCC.
Figure 7.

HCC samples show decreased FSBP
(A) Western blot was performed to detect the relative protein levels of FSBP in paired HCC samples. The relative band densities from all the detected patients were analyzed by ImageJ software and normalized by β-Actin (right panel). The t-test was used for statistical analyses, data are represented as means ± SD, ∗∗p < 0.01.
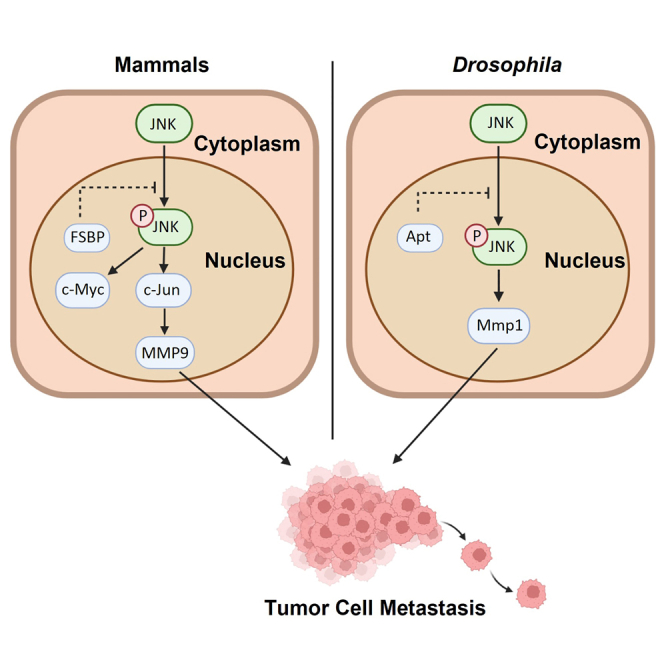
(B) The proposed mechanism of FSBP-mediated regulation of HCC.
Discussion
Although metastasis is a key cause for HCC-induced death, its underlying mechanism remains unclear. In this study, we provide that the transcription factor Apt/FSBP is a suppressor for tumor cell metastasis in Drosophila wing discs and HCC cell lines. First, we used a well-known Drosophila tumor model to reveal that knockdown of apt promotes tumor cell migration, whereas overexpression of apt exerts an opposite effect. Mechanistically, loss of apt activates the JNK pathway. Blockade of the JNK pathway is able to suppress apt-RNAi-induced tumor cell migration. Moreover, we demonstrate that FSBP can substitute Apt to control cell migration. Next, we construct Fsbp liver-specific knockout mouse and find that it predisposes to tumorigenesis on DEN and CCl4 treatment. In addition, deletion of Fsbp promotes liver tumor metastasis. We used several human HCC cells to validate that FSBP negatively regulates EMT. Through RNA-seq, we find that loss of Fsbp activates the JNK pathway in mouse liver. The JNK pathway inhibitor is capable of suppressing EMT induced by FSBP knockdown, providing JNK pathway inhibitor as putative drug. At last, we reveal that human HCC samples show decreased FSBP expression. Thus, our study uncovers an evolutionarily conserved mechanism to suppress tumor metastasis, in which Apt/FSBP suppresses the oncogenic JNK pathway (Figure 7B).
Our previous study has illustrated that Apt/FSBP is able to turn on the Hh pathway via activating hh transcription in Drosophila and mammalian cells.9 The Hh pathway is well-documented oncogenic, especially in lung cancers and medulloblastoma. From this respective, Apt/FSBP is an oncogene. Thus, we propose the possible hypothesis that Apt/FSBP plays an oncogenic or anti-tumor role in a context-dependent manner. As a transcription factor, it is acceptable that Apt/FSBP controls the expression of numerous genes, which serve as oncogenes or tumor suppressors. Thus, Apt/FSBP possibly plays multiple roles in different tissues through activating distinct targets. In the future, it will be interesting to examine the exact roles of FSBP in other types of cancers.
Although we provide enough evidence to support that Apt/FSBP negatively regulates the JNK pathway, its direct transcriptional target responsible for JNK pathway inactivation is still unknown. As a matter of fact, we make great efforts to identify the target of Apt/FSBP in the JNK pathway, but fail. Increasing numbers of JNK pathway regulators are reported in recent years. Thus, it is possible that Apt/FSBP inhibits the JNK pathway via activating a non-canonical negative modulator upstream JNK.
In addition to promoting tumor cell migration, the JNK pathway also regulates physiological cell migration during embryogenesis.25 Whether Apt/FSBP is involved in modulating embryonic cell migration through JNK pathway is unclear. In fact, null allele mutation of Apt leads to embryonic death of Drosophila.26 Whether this death is because of a defect in cell migration needs to be further determined. Furthermore, cell migration plays an important role in the tissue repair after damage.27 Whether Apt/FSBP participates in the regulation of repair also needs to be further explored. This study will have certain significance for regenerative medicine.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-Actin | Genscript | Cat#A00702 (RRID: AB_914102) |
| Mouse anti-Fg | Sigma-Aldrich | Cat#F3165 (RRID: AB_259529) |
| Mouse anti-Myc | Santa Cruz Biotechnology | Cat#SC-40 (RRID: AB_627268) |
| Rabbit anti-FSBP | Atlas antibodies | Cat#HPA026509 (RRID: AB_10602307) |
| Rabbit anti-E-Cadherin | ABclonal | Cat#A16811 (RRID: AB_2768812) |
| Rabbit anti-N-Cadherin | ABclonal | Cat#A3045 (RRID: AB_2863024) |
| Rabbit anti-β-Catenin | Proteintech | Cat#17565-1-AP (RRID: AB_2088102) |
| Rabbit anti-FAK | ABclonal | Cat#A11195 (RRID: AB_2758455) |
| Rabbit anti-Vimentin | Proteintech | Cat#60330-1-Ig (RRID: AB_2881439) |
| Rabbit anti-Snail | ABclonal | Cat#A5243 (RRID: AB_2766076) |
| Rabbit anti-MMP9 | ABclonal | Cat#A0289 (RRID: AB_2757101) |
| Rabbit anti-pJNK | ABclonal | Cat#AP0276 (RRID: AB_2771315) |
| Rabbit anti-phospho-c-Jun | ABclonal | Cat#AP0105 (RRID: AB_2863804) |
| Rabbit anti-c-Myc | ABclonal | Cat#A1309 (RRID: AB_2759938) |
| Rabbit anti-Ki67 | ABclonal | Cat#A2094 (RRID: AB_2764114) |
| Goat anti-rabbit IgG | Jackson ImmunoResearch | Cat#111-035-144 (RRID: AB_2307391) |
| Goat anti-mouse IgG | Jackson ImmunoResearch | Cat#115-035-146 (RRID: AB_2307392) |
| Rabbit anti-β-Galactosidase | MBL International | Cat#PM049 (RRID: AB_1520806) |
| Mouse anti-Mmp1 | Development Studies Hybridoma Bank | Cat#14A3D2 (RRID: AB_579782) |
| Rabbit anti-pJNK (Thr183/Tyr185) | Cell Signaling Technology | Cat#9251 (RRID: AB_331659) |
| Chemicals, peptides, and recombinant proteins | ||
| Diethylnitrosamine | Sigma-Aldrich | CAS: 55-18-5 |
| CCl4 | Aladdin | C131583, CAS: 56-23-5 |
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat#11668019 |
| 3, 3′-diaminobenzidine | TIANGEN | Cat#PA110 |
| Protease inhibitor cocktail | Glpbio | Cat#GK10014 |
| ECL Plus | Millipore | Cat#WBKLS0500 |
| Trizol | Thermo Fisher Scientific | Cat#15596026 |
| SP600125 | Beyotime | Cat#S1876 |
| ChamQ Universal SYBR qPCR Master Mix | Vazyme | Cat#Q711 |
| HiScript III RT SuperMix for qPCR | Vazyme | Cat#R323 |
| DAPI | Sigma-Aldrich | Cat#D9564 |
| Software and algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| GraphPad Prism 5 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| DNAMAN | Lynnon BioSoft | https://www.lynnon.com/dnaman.html |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to Qingxin Liu (liuqingxin@sdau.edu.cn).
Materials availability
Requests for materials should be directed to the corresponding authors.
Experimental model and subject details
Fly stocks
The ptc-gal4, Ay-gal4, UAS-GFP, apt-RNAi (V4289), scrib-RNAi and UAS-apt had been described in our previous studies.9,21,28 The puc-lacZ (#109029, KDSC) and bsk-RNAi (TH04355.N, THFC) flies were purchased from Kyoto Drosophila Stock Center (KDSC) or Tsinghua Fly Center (THFC). The UAS-FSBP transgenic fly was generated by injection of UAS-attB-FSBP construct into the Drosophila embryos through the phiC31 integration system. All fly stocks were reared on cornmeal and agar medium at 25°C. For all fly cross experiments, unmated females were crossed with male flies of corresponding RNAi or transgenes at 29°C.
Cell lines and cell culture
Two human HCC cell lines (SK-Hep-1, SMMC-7721) were purchased from the ATCC and routinely checked for mycoplasma contamination using the Vazyme Mycoplasma detection kit. SK-Hep-1 cells were cultured in DMEM medium supplemented with 10% fetal bovine serum, and SMMC-7721 cells were cultured in RPMI1640 medium supplemented with 10% fetal bovine serum, cells were maintained at 37°C in a 5% CO2 incubator.
Mice and animal models
Fsbpfl/fl and Alb-Cre mice in C57BL/6 background were generated by Cyagen Biosciences Inc. Fsbp-/- mice were specifically knocked out of the Fsbp gene in the liver, and Fsbpfl/fl mice were used as control mice. Fsbp-/- (n=12) and Fsbpfl/fl (n=12) male mice were injected with DEN (20mg/kg, i.p.) at day 14 postpartum, followed by twice of week injections of CCl4 (0.5 ml/kg i.p., 1:4 dissolved in corn oil) for 12 weeks starting at 4 weeks after birth. The mice were sacrificed at 26 weeks after the induction for experiments. Mice were maintained in a sterile individually ventilated cage (IVC) system at 22-26°C under a standard 12:12-h light/dark cycle, with adequate food and water. All procedures followed the National Institutes of Health guidelines for the care and use of laboratory animals.
Method details
Small interference RNA
To knockdown FSBP in human HCC cells, the siRNAs were synthesized by GenePharma (Shanghai, China), and the sequences were as follows: mock-siRNA: 5ʹ-UUC UCC GAA CGU GUC ACG UdTdT-3ʹ; FSBP-siRNA: 5′-GCU AGA GGA AGA GCU ACU AdTdT-3′. siRNAs were transfected at a final concentration of 100nM via lipo2000 (Invitrogen) according to the manufacturer’s instructions. Cells were harvested for WB or real-time PCR after cultured for 72h with serum containing medium and detection of knockdown efficiency.
Transwell assays
Transwell assays were performed according to our previous published paper.29 Briefly, the SK-Hep-1 or SMMC-7721 cells (1 × 105 cells) were plated into the upper chamber with serum-free medium and with 10% FBS medium in the lower chamber of 24-well plate (Corning) after transfection for 48h. After additional 24h (SK-Hep-1 cells) or 48h (SMMC-7721 cells), migrating cells or invading cells were fixed with 20% methanol for 20min and stained with 0.1% crystal violet (Sangon Biotech). The numbers of migrating cell and invading cells per well were counted under a light microscope in nine random fields. Data are presented as means ± SD of these nine fields.
Wound healing assays
Wound healing assays were carried out as previously described.29 In brief, after transfection for 48h, the SK-Hep-1 or SMMC-7721 cells (2 × 105 cells/well) were plated into 6-well plates and cultured overnight to form a confluent monolayer. Then make a gap of constant width with a sterile micropipette, the detached cells were washed away with PBS. Cells were cultured for 24h (SK-Hep-1 cells) or 48h (SMMC-7721 cells) in a 37°C incubator, images of wounds were taken under a light microscope and the width of gaps were counted by the Image J software.
Immunohistochemical staining
The 4% paraformaldehyde-fixed embedded paraffin sections were deparaffinized in xylene and hydrated in different gradients of ethanol. Endogenous peroxidase was blocked with 3% H2O2 for 15 min, followed by microwave antigen retrieval. Then sections were blocked with 10% goat serum at room temperature for half an hour, incubated the primary antibody overnight at 4°C. Primary antibodies were showed as following: rabbit anti-Ki-67 (1:100; ABclonal), rabbit anti-E-Cadherin (1:1000; ABclonal), rabbit anti-β-Catenin (1:200; Proteintech), rabbit anti-FAK (1:200; ABclonal), rabbit anti-Vimentin (1:1000; Proteintech), rabbit anti-Snail (1:200; ABclonal). After incubated with HRP-conjugated anti-rabbit antibody at room temperature for 60 minutes, the visualization signal was developed with 3, 3′-diaminobenzidine tetrachloride.
Immunostaining
Immunostaining of imaginal discs were performed with previous protocols.30 In brief, third-instar larvae were dissected in phosphate buffered solution (PBS) and fixed in freshly made 4% formaldehyde in PBS at room temperature for 20 min, then washed three times in PBT (PBS plus 0.1% Triton X-100). The dissected larvae were incubated overnight with needed primary antibodies in PBT at 4°C, then washed with PBT for three times and incubated with corresponding fluorophore-conjugated secondary antibody 2h at room temperature. After washing for three times in PBT, imaginal discs were dissected and mounted in 40% glycerol. Images were captured with Zeiss confocal microscope. Primary antibodies were showed as following: rabbit anti-β-Galactosidase (1:500, MBL), mouse anti-Mmp1 (1:10; DSHB), rabbit anti-pJNK (1:200; CST) and DAPI (1:1000; Sigma). Secondary antibodies used in this study were bought from Jackson ImmunoResearch, and were diluted at 1:500.
Western blotting
Mice tissues or cells were lysed with lysis buffer add with protease inhibitor cocktail (#GK10014, Glpbio). Equal amounts of protein from treated tissues or cells were separated by SDS-PAGE and transferred onto a PVDF membrane. The membrane was incubated with specific primary antibodies along with HRP-conjugated secondary antibodies. The protein signals were detected using ECL Plus (#WBKLS0500, Millipore). Primary antibodies were showed as following: mouse anti-Actin (1:5000; Genscript), mouse anti-Fg (1:2000; Sigma), mouse anti-Myc (1:2000; Santa Cruz), rabbit anti-FSBP (1:1000; Atlas antibodies), rabbit anti-E-Cadherin (1:2000; ABclonal), rabbit anti-N-Cadherin (1:1000; ABclonal), rabbit anti-β-Catenin (1:5000; Proteintech), rabbit anti-FAK (1:2000; ABclonal), rabbit anti-Vimentin (1:2000; Proteintech), rabbit anti-Snail (1:2000; ABclonal), rabbit anti-MMP9 (1:1000; ABclonal), rabbit anti-pJNK (1:1000; ABclonal), rabbit anti-phospho-c-Jun (1:1000; ABclonal), rabbit anti-c-Myc (1:1000; ABclonal). secondary antibodies were showed as following: goat anti-rabbit (1:10000; Jackson ImmunoResearch), goat anti-mouse (1:10000; Jackson ImmunoResearch).
Total RNA extraction and RT-qPCR
Total RNA was isolated from tissues or cells by Trizol (#15596026, Invitrogen) and reverse transcribed into cDNA using a transcription kit (#R323, Vazyme). Real-time PCR was performed at 95°C for 30s, followed by 40 cycles of 95°C for 10s and 60°C for 30s with SYBR Green I as a fluorescent dye (#Q711, Vazyme). The data were analyzed with the 2−ΔΔCt method using Actin as a loading control. The primers used in the study were listed in the Table S1.
RNA-seq analysis
Samples of liver tissues (100 mg) were removed from each of Fsbpfl/fl and Fsbp-/- mice (n=3), and total RNA was isolated. RNA was subjected to RNA-Seq analysis on BGISEQ-500 system by Beijing Genomics Institute (BGI), China. In addition, the RNA is reverse transcribed to obtain cDNA for library construction for library construction. High-quality reads were aligned to the mouse reference genome (GRCm38). Significant differential expression was set if a gene with > 1.2-fold expression difference versus the control with adjusted p-value of < 0.05. Gene expressions were normalized to fragments per kilobase of exon model per million (FPKM) mapped reads from RNA-Seq by Expectation Maximization (RSEM). Gene Ontology (GO) and signaling pathway annotation and enrichment analyses were based on the Gene Ontology Database (http://www.geneontology.org/) and KEGG pathway database (http://www.genome.jp/kegg/), respectively.
Quantification and statistical analysis
All statistical analyses were performed using GraphPad Prism software. Data shown in Figures are representative of at least three independent experiments and were analyzed by one-way Student's t-test, with p < 0.05 considered statistically significant. Statistical insignificance is shown as ns (not significant), ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 and ∗∗∗∗p < 0.0001.
Acknowledgments
We sincerely thank Dr. Susumu Hirose (National Institute of Genetics, Japan) and Yasushi Hiromi (National Institute of Genetics, Japan) for discussions and comments on the manuscript. We also appreciate National Institute of Genetics of Japan (NIG), Kyoto Drosophila Stock Center (KDSC) or Tsinghua Fly Center (THFC) and Developmental Studies Hybridoma Bank (DSHB) for providing fly stocks and reagents. This work was supported by grants from the National Natural Science Foundation of China (32272945, 31922011, 31872971, 81760724), the National Key Research and Development Program of China (2017YFE0129800) and the Natural Science Foundation of Jiangxi (No. 20181BAB205026).
Author contributions
Q.L. and Y.Z. conceived the study. Q.L., Z.Z., Y.Z., and X.Y. designed the study. F.S., W.Z., X.L., and X.C. performed experiments. M.J. provided HCC samples. F.S., W.Z., and Y.Z analyzed the data and made Figures. F.S., W.Z., Y.Z., Q.L., and Z.Z. wrote the manuscript. All authors approved the final submission.
Declaration of interests
The authors declare no competing interests.
Published: March 17, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106440.
Contributor Information
Yunhe Zhao, Email: yhzhao@sdau.edu.cn.
Qingxin Liu, Email: liuqingxin@sdau.edu.cn.
Zizhang Zhou, Email: zhouzz@sdau.edu.cn.
Supplemental information
Data and code availability
-
•
Data reported in this paper are available from the lead contact upon reasonable request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Couri T., Pillai A. Goals and targets for personalized therapy for HCC. Hepatol. Int. 2019;13:125–137. doi: 10.1007/s12072-018-9919-1. [DOI] [PubMed] [Google Scholar]
- 2.Ge X., Yao Y., Li J., Li Z., Han X. Role of LncRNAs in the epithelial-mesenchymal transition in hepatocellular carcinoma. Front. Oncol. 2021;11:690800. doi: 10.3389/fonc.2021.690800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oura K., Morishita A., Tani J., Masaki T. Tumor immune microenvironment and immunosuppressive therapy in hepatocellular carcinoma: a review. Int. J. Mol. Sci. 2021;22:5801. doi: 10.3390/ijms22115801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Valastyan S., Weinberg R.A. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147:275–292. doi: 10.1016/j.cell.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gellon G., Harding K.W., McGinnis N., Martin M.M., McGinnis W. A genetic screen for modifiers of Deformed homeotic function identifies novel genes required for head development. Development. 1997;124:3321–3331. doi: 10.1242/dev.124.17.3321. [DOI] [PubMed] [Google Scholar]
- 6.Liu Q.X., Jindra M., Ueda H., Hiromi Y., Hirose S. Drosophila MBF1 is a co-activator for Tracheae Defective and contributes to the formation of tracheal and nervous systems. Development. 2003;130:719–728. doi: 10.1242/dev.00297. [DOI] [PubMed] [Google Scholar]
- 7.Starz-Gaiano M., Melani M., Wang X., Meinhardt H., Montell D.J. Feedback inhibition of Jak/STAT signaling by apontic is required to limit an invasive cell population. Dev. Cell. 2008;14:726–738. doi: 10.1016/j.devcel.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 8.Liu Q.X., Wang X.F., Ikeo K., Hirose S., Gehring W.J., Gojobori T. Evolutionarily conserved transcription factor Apontic controls the G1/S progression by inducing cyclin E during eye development. Proc. Natl. Acad. Sci. USA. 2014;111:9497–9502. doi: 10.1073/pnas.1407145111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang X.F., Shen Y., Cheng Q., Fu C.L., Zhou Z.Z., Hirose S., Liu Q.X. Apontic directly activates hedgehog and cyclin E for proper organ growth and patterning. Sci. Rep. 2017;7:12470. doi: 10.1038/s41598-017-12766-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lau K.F., Perkinton M.S., Rodriguez L., McLoughlin D.M., Miller C.C.J. An X11α/FSBP complex represses transcription of the GSK3β gene promoter. Neuroreport. 2010;21:761–766. doi: 10.1097/WNR.0b013e32833bfca0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davalos D., Akassoglou K. Fibrinogen as a key regulator of inflammation in disease. Semin. Immunopathol. 2012;34:43–62. doi: 10.1007/s00281-011-0290-8. [DOI] [PubMed] [Google Scholar]
- 12.Zhang X., Long Q. Elevated serum plasma fibrinogen is associated with advanced tumor stage and poor survival in hepatocellular carcinoma patients. Medicine. 2017;96:e6694. doi: 10.1097/MD.0000000000006694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.La Marca J.E., Richardson H.E. Two-faced: roles of JNK signalling during tumourigenesis in the Drosophila model. Front. Cell Dev. Biol. 2020;8:42. doi: 10.3389/fcell.2020.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weston C.R., Davis R.J. The JNK signal transduction pathway. Curr. Opin. Cell Biol. 2007;19:142–149. doi: 10.1016/j.ceb.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Davis R.J. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 16.Andersen D.S., Colombani J., Palmerini V., Chakrabandhu K., Boone E., Röthlisberger M., Toggweiler J., Basler K., Mapelli M., Hueber A.O., Léopold P. The Drosophila TNF receptor Grindelwald couples loss of cell polarity and neoplastic growth. Nature. 2015;522:482–486. doi: 10.1038/nature14298. [DOI] [PubMed] [Google Scholar]
- 17.Kanda H., Igaki T., Kanuka H., Yagi T., Miura M. Wengen, a member of the Drosophila tumor necrosis factor receptor superfamily, is required for Eiger signaling. J. Biol. Chem. 2002;277:28372–28375. doi: 10.1074/jbc.C200324200. [DOI] [PubMed] [Google Scholar]
- 18.Igaki T., Kanda H., Yamamoto-Goto Y., Kanuka H., Kuranaga E., Aigaki T., Miura M. Eiger a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J. 2002;21:3009–3018. doi: 10.1093/emboj/cdf306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gkouveris I., Nikitakis N.G. Role of JNK signaling in oral cancer: a mini review. Tumour Biol. 2017;39 doi: 10.1177/1010428317711659. 1010428317711659. [DOI] [PubMed] [Google Scholar]
- 20.Chan K.K.S., Leung C.O.N., Wong C.C.L., Ho D.W.H., Chok K.S.H., Lai C.L., Ng I.O.L., Lo R.C.L. Secretory Stanniocalcin 1 promotes metastasis of hepatocellular carcinoma through activation of JNK signaling pathway. Cancer Lett. 2017;403:330–338. doi: 10.1016/j.canlet.2017.06.034. [DOI] [PubMed] [Google Scholar]
- 21.Zhao Y., Peng D., Liu Y., Zhang Q., Liu B., Deng Y., Ding W., Zhou Z., Liu Q. Usp8 promotes tumor cell migration through activating the JNK pathway. Cell Death Dis. 2022;13:286. doi: 10.1038/s41419-022-04749-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Udden S.N., Kwak Y.T., Godfrey V., Khan M.A.W., Khan S., Loof N., Peng L., Zhu H., Zaki H. NLRP12 suppresses hepatocellular carcinoma via downregulation of cJun N-terminal kinase activation in the hepatocyte. Elife. 2019;8:e40396. doi: 10.7554/eLife.40396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eferl R., Ricci R., Kenner L., Zenz R., David J.P., Rath M., Wagner E.F. Liver tumor development: c-Jun antagonizes the proapoptotic activity of p53. Cell. 2003;112:181–192. doi: 10.1016/s0092-8674(03)00042-4. [DOI] [PubMed] [Google Scholar]
- 24.Sun X., Ding Y., Zhan M., Li Y., Gao D., Wang G., Gao Y., Li Y., Wu S., Lu L., et al. Usp7 regulates Hippo pathway through deubiquitinating the transcriptional coactivator Yorkie. Nat. Commun. 2019;10:411. doi: 10.1038/s41467-019-08334-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deng Y., Peng D., Xiao J., Zhao Y., Ding W., Yuan S., Sun L., Ding J., Zhou Z., Zhan M. Inhibition of the transcription factor ZNF281 by SUFU to suppress tumor cell migration. Cell Death Differ. 2023;30:702–715. doi: 10.1038/s41418-41022-01073-41411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou Z., Yao X., Li S., Xiong Y., Dong X., Zhao Y., Jiang J., Zhang Q. Deubiquitination of Ci/Gli by Usp7/HAUSP regulates hedgehog signaling. Dev. Cell. 2015;34:58–72. doi: 10.1016/j.devcel.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ding Y., Wang G., Zhan M., Sun X., Deng Y., Zhao Y., Liu B., Liu Q., Wu S., Zhou Z. Hippo signaling suppresses tumor cell metastasis via a Yki-Src42A positive feedback loop. Cell Death Dis. 2021;12:1126. doi: 10.1038/s41419-021-04423-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kushnir T., Mezuman S., Bar-Cohen S., Lange R., Paroush Z., Helman A. Novel interplay between JNK and Egfr signaling in Drosophila dorsal closure. PLoS Genet. 2017;13:e1006860. doi: 10.1371/journal.pgen.1006860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takasu-Ishikawa E., Yoshihara M., Ueda A., Rheuben M.B., Hotta Y., Kidokoro Y. Screening for synaptic defects revealed a locus involved in presynapticand postsynaptic functions in Drosophila embryos. J. Neurobiol. 2001;48:101–119. [PubMed] [Google Scholar]
- 30.Sahu S., Sridhar D., Abnave P., Kosaka N., Dattani A., Thompson J.M., Hill M.A., Aboobaker A. Ongoing repair of migration-coupled DNA damage allows planarian adult stem cells to reach wound sites. Elife. 2021;10:e63779. doi: 10.7554/eLife.63779. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data reported in this paper are available from the lead contact upon reasonable request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.