

Abstract
Background & Aims
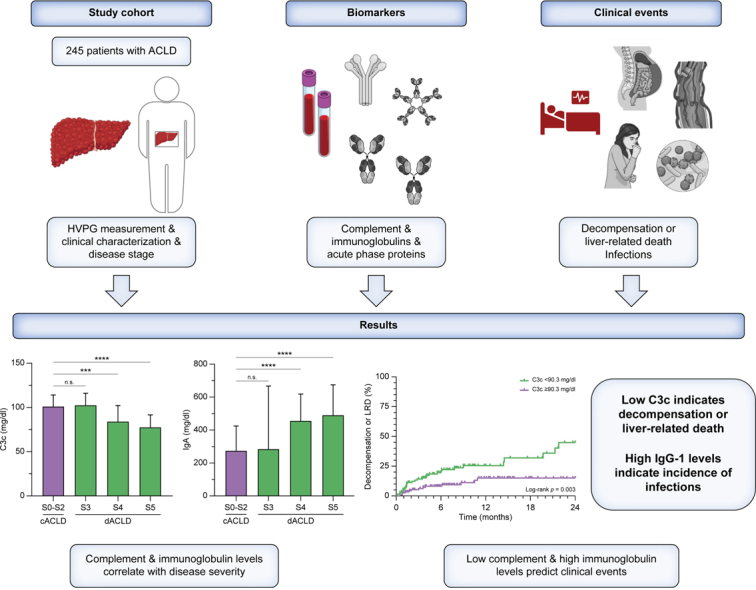
Cirrhosis-associated immune dysfunction (CAID) affects both innate and adaptive immunity. This study investigated the complement system, immunoglobulins, and acute-phase proteins and their prognostic relevance in patients with advanced chronic liver disease (ACLD).
Methods
Patients with ACLD (hepatic venous pressure gradient [HVPG] ≥6 mmHg) but without acute decompensation/infections were characterised by HVPG and by clinical EASL stages: compensated (cACLD; S0–2) vs. decompensated ACLD (dACLD) with previous variceal bleeding (S3), non-bleeding decompensation (S4), or further decompensation (S5). Complement factors (C3c, C4, CH50), immunoglobulins (IgA, IgM, IgG, IgG1–4), acute-phase proteins and systemic inflammation biomarkers (white blood cells, C-reactive protein, IL-6, procalcitonin) were measured.
Results
A total of 245 patients (median model for end-stage liver disease score: 11 [9–15], median HVPG: 17 [12–21] mmHg) were included with 150 (61%) presenting dACLD. Complement levels and activity significantly decreased in dACLD substages S4 and S5 (p <0.001). Total IgA/IgM/IgG and IgG1–4 subtype levels increased in patients with dACLD (all p <0.05). Complement and immunoglobulin levels correlated negatively and positively, respectively, with systemic inflammation (all p <0.05). High IgG-1 (adjusted hazard ratio per 100 mg/dl: 1.12, 1.04–1.19, p = 0.002) and IL-6 (adjusted hazard ratio: 1.03, 1.00–1.05, p = 0.023) levels predicted the development of infections during follow-up. High IgA (stratified by median; log-rank p <0.001), high IgG1 (log-rank p = 0.043) and low C3c (log-rank p = 0.003) indicated a higher risk of first/further decompensation or liver-related death (composite endpoint). Next to HVPG and IL-6, low C3c (adjusted hazard ratio per mg/dl: 0.99, 0.97–0.99, p = 0.040) remained independently associated with the composite endpoint on multivariate Cox regression analysis.
Conclusions
Complement levels and immunoglobulins may serve as surrogates of cirrhosis-associated immune dysfunction and associate with cirrhosis severity and systemic inflammation. Low complement C3c predicted decompensation and liver-related death, whereas high IgG-1 indicated an increased risk for infections.
Impact and Implications
Patients with cirrhosis are at increased risk for infections, which worsen their prognosis. We found a significant dysregulation of several essential components of the immune system that was linked to disease severity and indicated a risk for infections and other complications. Simple blood tests identify patients at particularly high risk, who may be candidates for preventive measures.
Clinical Trials Registration
This study is registered at ClinicalTrials.gov (NCT03267615).
Keywords: ACLD, Portal hypertension, Gut–liver axis, Immune dysfunction
Graphical abstract
Highlights
-
•
Complement factors, immunoglobulins, and acute-phase proteins are dysregulated in patients with advanced chronic liver disease (ACLD), correlate with disease severity, and indicate alterations of innate and adaptive immunity.
-
•
Low complement C3c levels independently predicted decompensation or liver-related death of patients with ACLD.
-
•
High IgG-1 levels independently predicted the incidence of infections in patients with ACLD.
Introduction
Cirrhosis-associated immune dysfunction (CAID) has been attributed a central pathophysiological role in advanced chronic liver disease (ACLD) and is characterised by systemic inflammation and impaired immunocompetence.1 The manifestation of CAID may range from increased systemic inflammation in patients without clinical evidence of infection as well as patients with acute-on-chronic liver failure (ACLF).1,2 In either situation, however, these immunological alterations seem to hold clinical significance, as previous studies from our and other centres demonstrated that systemic inflammation levels, as reflected by inflammatory cytokines and acute-phase proteins (APPs), predict disease progression and/or mortality in both clinically stable patients without overt infection3,4 as well as patients with acute hepatic decompensation and ACLF.5,6 Furthermore, the increased incidence of bacterial infections and their impact on prognosis in patients with ACLD underlines the profound clinical implications of CAID.7,8
Several mechanisms that are considered responsible for the development of CAID have been investigated in preclinical and clinical studies.1 For example, impairment of the gut–liver axis – involving intestinal dysbiosis and increased intestinal permeability – causes constant exposure of the immune system to bacterial pathogens and pathogen-associated molecular patterns (PAMPs).9 Furthermore, liver injury may promote inflammatory reactions in the absence of pathogenic triggers (‘sterile’ inflammation), that are often mediated by danger-associated molecular patterns (DAMPs).10 These conditions activate both innate and adaptive immune responses that might, on the one hand, promote systemic inflammation, and on the other hand, lead to exhaustion of the immune system.1
As mentioned above, inflammatory cytokines such as IL-6 and APPs such as C-reactive protein (CRP) were previously linked to disease severity and prognosis in ACLD.3,5,6 Previous studies have also demonstrated a relationship between complement factors (part of the innate immune system) and disease severity as well as the incidence of infections and mortality in patients with ACLD.11,12 Furthermore, circulating immunoglobulin levels (important elements of adaptive immunity) exhibited an association with the presence and/or severity of fibrosis/cirrhosis in patients with chronic liver disease.13,14 Nevertheless, the relationship between these immune components and hepatic dysfunction, portal hypertension, systemic inflammation, and complications of ACLD remains incompletely understood.
The present study aimed to simultaneously characterise circulating levels of complement and immunoglobulin levels and assess their dynamics across different ACLD stages, as well as their link to systemic inflammation (reflected by APPs) and disease prognosis in a well-characterised cohort of patients with ACLD undergoing hepatic venous pressure gradient (HVPG) measurement.
Patients and methods
Study design, patient selection, and clinical characterisation
Patients undergoing HVPG measurement were recruited into the Vienna Cirrhosis Study (VICIS; NCT03267615) between February 2019 and September 2021. The presence of ACLD was confirmed by an HVPG ≥6 mmHg that also defines the presence of sinusoidal portal hypertension. Patients with portal hypertension as a result of vascular liver disease, active malignancies (including hepatocellular carcinoma), previous liver transplantation, acute hepatic decompensation, or infection at the time point of HVPG measurement were excluded by review of prospectively collected clinical data. After exclusion of patients without information on laboratory parameters that are relevant to this study, the final study cohort comprised 245 patients with ACLD (Fig. S1). ACLD was staged according to the European Association for the Study of the Liver (EASL) guidelines15 and D’Amico et al.16 Patients with compensated ACLD (cACLD) were subsumed as stage 0–2 (S0–S2), whereas patients with decompensated ACLD (dACLD) were divided into patients with previous variceal bleeding (S3), patients with one non-bleeding decompensation event (S4), and patients with two or more decompensation events (S5).
Measurement of hepatic venous pressure gradient
Measurement of HVPG followed a standard operating procedure at the Vienna Hepatic Hemodynamic Lab, Medical University of Vienna, Austria, as described in previous publications.17,18 Briefly, after placing a catheter introducer sheath into the right internal jugular vein, the catheter tip was advanced into a large hepatic vein. After affirmation of the correct positioning of the catheter tip, at least three measurements of free and wedged hepatic vein pressures were performed. The mean of the differences between these pressures determined the HVPG.
Biomarker measurements
Biomarkers were measured in blood samples taken from the catheter introducer sheath at the time point of HVPG measurement. Measurements were performed without prior storage at the Department of Laboratory Medicine, Medical University of Vienna, Austria, according to standardised and ISO-certified procedures. Laboratory personnel were blinded to clinical and haemodynamic data. Quantitative determination of human complement factors C3c (normal range 90–180 mg/dl) and C4 (normal range 10–40 mg/dl), immunoglobulin subtypes IgG (normal range 700–1600 mg/dL), IgM (normal range 40–230 mg/dl), IgA (normal range 70–400 mg/dl), and IgG subclasses IgG1 (normal range 405–1011 mg/dl), IgG2 (normal range 169–786 mg/dl), IgG3 (normal range 11.0–85 mg/dl), and IgG4 (normal range 3.0–201 mg/dl), as well as the APPs alpha-1-acid glycoprotein (AGP; normal range 50–120 mg/dl), serum amyloid A (SAA; upper limit of normal 6.4 mg/L), and alpha-2-macroglobulin (A2M; normal range 130–300 mg/dl) were performed by nephelometry on a BNII System using N antisera (Siemens Healthcare Diagnostics, Vienna, Austria). Complement activity was assessed in serum by a CH50 in vitro liposome immunoassay (normal range 31.6–57.6 U/ml). Tissue transglutaminase IgA antibodies (TTG-IgA; upper limit of normal <10 U/ml) levels were determined in serum by an indirect ELISA (Orgentec Diagnostika, Mainz, Germany).
Statistical analysis
Statistical analyses were performed using IBM SPSS Statistics 27 (IBM, Armonk, NY, USA) or GraphPad Prism 9 (GraphPad Software, La Jolla, CA, USA). Continuous variables are presented as mean (± standard error of the mean) or median (interquartile range) depending on the normal distribution of variables, as assessed using D’Agostino & Pearson and Shapiro-Wilk tests. Comparison of continuous variables was performed using the Student t test, Mann–Whitney U test, Kruskal–Wallis test, or ANOVA, as appropriate. Tukey’s or Dunn’s multiple comparisons tests were applied. Categorical variables are presented as absolute and relative frequencies. Comparison of categorical variables was performed using the Χ2 test or Fisher’s Exact test. Pearson or Spearman correlation coefficients (95% CI) were determined to assess the correlation between continuous variables. Kaplan–Meier curves were used to illustrate the incidence of the composite endpoint ‘first/further decompensation or liver-related death’, as well as the incidence of infections, at 24 months in patients stratified by serum biomarker levels. The composite endpoint was defined as the incidence and/or worsening of ascites, variceal bleeding, hepatic encephalopathy, or liver-related death (LRD) during follow-up (see Supplementary material). The log-rank test was applied to compare the incidence of the respective endpoints between groups. Univariate and multivariate Cox proportional hazard models were used to determine the prognostic value of complement factors and immunoglobulin levels for these clinical events. The threshold for statistical significance was set at a two-sided p value <0.05 for all analyses.
Compliance with ethical standards
The study was conducted following the principles of the Declaration of Helsinki and its latest amendments and approved by the local ethics committee (EK 1262/2017). Patients gave written informed consent for liver vein catheterisation and participation in the VICIS study.
Results
Patient characteristics
Patients included in the study had a median age of 57 (50–66) years and displayed a median HVPG of 17 (12–21) mmHg, a median model for end-stage liver disease (MELD) score of 11 (9–15) points. Most patients (n = 213, 87%) had clinically significant portal hypertension (CSPH; HVPG ≥10 mmHg). Ninety-five patients (39%) had cACLD (S0–S2) and among patients with dACLD, 8 (3%) patients were in S3, 81 (33%) in S4, and 61 (25%) in S5. Alcohol-related liver disease (ALD) and viral hepatitis represented the main aetiologies of ACLD in the study cohort. Consistent with previous results,3 an increase in HVPG, MELD, systemic inflammation markers CRP and IL-6, as well as the prevalence of ALD were observed in patients with dACLD and the respective dACLD subgroups. Twenty-three (9%) patients reported intake of antibiotics as therapy for hepatic encephalopathy or prophylaxis for spontaneous bacterial peritonitis at the time point of HVPG measurement: 20 patients were on rifaximin, two on both rifaximin and norfloxacin, and one on norfloxacin. Patient characteristics are summarised in Table 1 and Table S1.
Table 1.
Patient characteristics.
| Compensated ACLD (n = 95) |
Decompensated ACLD (n = 150) |
p value | |||
|---|---|---|---|---|---|
| Stage 0-2 | Stage 3Bleeding (n = 8) | Stage 4Non-bleeding decompensation (n = 81) | Stage 5Further decompensation (n = 61) | ||
| Age (years) | 58 (47–66) | 58 (54–61) | 56 (49–66) | 58 (48–68) | 0.863 |
| Sex (M, %) | 64 (67) | 6 (75) | 52 (64) | 39 (64) | 0.899 |
| Aetiology (n, %) | |||||
| ALD | 30 (32) | 3 (38) | 51 (63) | 40 (66) | <0.001 |
| Viral | 24 (25) | 4 (50) | 4 (5) | 3 (5) | |
| ALD + Viral | 3 (3) | 0 (0) | 6 (7) | 8 (13) | |
| NASH | 17 (18) | 0 (0) | 0 (0) | 1 (2) | |
| Cholestatic | 12 (13) | 0 (0) | 5 (6) | 2 (3) | |
| Other | 9 (10) | 1 (12) | 15 (19) | 7 (12) | |
| HVPG (mmHg) | 12 (9–18) | 18 (12–23) | 19 (15–22) | 19 (15–23) | <0.001 |
| MELD (points) | 9 (8–11) | 10 (9–12) | 12 (10–15) | 14 (11–17) | <0.001 |
| Bilirubin (mg/dl) | 0.87 (0.67–1.35) | 1.25 (0.81–1.39) | 1.55 (0.97–2.39) | 1.59 (0.92–2.34) | <0.001 |
| Albumin (g/L) | 39.9 (36.7–42.4) | 39.4 (37.8–42.1) | 35.2 (31.1–38.4) | 34.4 (31.3–37.0) | <0.001 |
| AST (U/L) | 38 (27–56) | 34 (23–49) | 44 (31–62) | 42 (29–54) | 0.275 |
| CRP (mg/dl) | 0.16 (0.07–0.36) | 0.21 (0.09–0.98) | 0.42 (0.17–0.91) | 0.45 (0.24–1.16) | <0.001 |
| IL-6 (pg/ml) | 5.45 (3.34–9.08) | 5.72 (4.82–13.8) | 11.2 (6.69–18.2) | 15.3 (9.77–27.8) | <0.001 |
| WBCs (G/L) | 4.82 (3.31–6.17) | 3.26 (2.64–4.64) | 4.86 (3.57–6.32) | 4.69 (3.75–6.10) | 0.134 |
| PCT (ng/ml) | 0.07 (0.05–0.11) | 0.06 (0.04–0.18) | 0.11 (0.07–0.17) | 0.13 (0.07–0.19) | <0.001 |
| LBP (μg/ml) | 6.45 (4.91–8.48) | 7.60 (6.18–11.2) | 6.66 (4.83–9.39) | 6.31 (5.22–8.23) | 0.521 |
| C3c (mg/dl) | 101 (86.9–114) | 103 (88.8–116) | 84.0 (67.7–102) | 77.5 (59.8–91.6) | <0.001 |
| C4 (mg/dl) | 14.8 (10.3–19.9) | 16.1 (10.6–17.2) | 13.7 (10.3–17.0) | 10.3 (8.00–14.3) | <0.001 |
| CH50 (U/ml) | 57.9 (48.9–60.0) | 58.1 (44.8–60.0) | 51.1 (36.5–59.7) | 44.5 (27.7–54.4) | <0.001 |
| IgA (mg/dl) | 274 (199–424) | 284 (242–667) | 456 (300–618) | 490 (298–674) | <0.001 |
| IgM (mg/dl) | 118 (68.2–175) | 151 (62.6–196) | 159 (92.3–237) | 145 (87.9–230) | 0.036 |
| IgG (mg/dl) | 1340 (1060–1670) | 1510 (1360–1705) | 1560 (1275–1970) | 1510 (1220–1845) | 0.006 |
| IgG1 (mg/dl) | 882 (684–1100) | 1145 (775–1318) | 1050 (786–1380) | 976 (776–1225) | 0.062 |
| IgG2 (mg/dl) | 302 (220–412) | 282 (172–534) | 341 (262–536) | 354 (247–478) | 0.094 |
| IgG3 (mg/dl) | 47.8 (31.7–72.1) | 51.2 (35.4–67.8) | 56.2 (37.7–86.1) | 59.2 (37.2–88.9) | 0.066 |
| IgG4 (mg/dl) | 44.8 (21.6–94.4) | 65.7 (37.2–118) | 61.5 (26.4–143) | 57.0 (33.1–108) | 0.174 |
| AGP (mg/dl) | 51.3 (40.7–62.3) | 52.3 (39.3–58.5) | 47.0 (32.3–62.0) | 46.9 (34.1–61.1) | 0.349 |
| SAA (mg/L) | 4.16 (4.16–7.01) | 4.16 (4.16–4.63) | 4.16 (4.16–7.27) | 4.16 (4.16–8.79) | 0.818 |
| A2M (mg/dl) | 286 (228–331) | 360 (285–379) | 231 (198–263) | 205 (159–260) | <0.001 |
Statistical analysis: Kruskal-Wallis test was applied to compare continuous variables between groups. Values of p <0.05 are indicated in bold.
A2M, alpha-2-macroglobulin; AGP, alpha-1-acid glycoprotein; ALD, alcohol-related liver disease; AST, aspartate aminotransferase; c/dACLD, compensated/decompensated advanced chronic liver disease; C3c, complement C3 component; CRP, C-reactive protein; HVPG, hepatic venous pressure gradient; LBP, lipopolysaccharide binding protein; M, male; MELD, model for end-stage liver disease; NASH, non-alcoholic steatohepatitis; PCT, procalcitonin; SAA, serum amyloid A.
Complement levels and complement activity
Complement component C3c and C4 levels decreased in disease stages S4 and S5 as compared with patients with S0–S2: C3c levels were 101 (86.9–114) in S0–S2, 84.0 (67.7–102) mg/dl in S4, and 77.5 (59.8–91.6) in S5. C4 levels were 14.8 (10.3–19.9) mg/dl in S0–S2, 13.7 (10.3–17.0) mg/dl in S4, and 10.3 (8.00–14.3) mg/dl in S5 (all p <0.001). Concordantly, CH50 activity – reflecting the haemolytic activity of the complement system in vitro – decreased in S4 and S5 as compared with S0–S2: CH50 was 57.9 (48.9–60.0) mg/dl in S0–S2, 51.1 (36.5–59.7) mg/dl in S4, and 44.5 (27.7–54.4) mg/dl in S5 (p <0.001). The decrease of complement (activity) levels was even more pronounced in S5 as compared with S4, indicating that these markers are closely associated with further decompensation (Fig. 1). Concordantly, complement markers exhibited a significant negative association with HVPG and MELD (Fig. S2).
Fig. 1.

Complement and immunoglobulin serum levels across different stages of advanced chronic liver disease. Statistical analysis: Kruskal-Wallis test with Dunn’s multiple comparisons test was applied to compare continuous variables between groups. n.s., not significant; ∗p <0.05; ∗∗p <0.01; ∗∗∗p <0.001; ∗∗∗∗p <0.0001. C3c, complement C3 component; C4, complement C4; c/dACLD, compensated/decompensated advanced chronic liver disease; IgA/G/M, immunoglobulin A/G/M.
Immunoglobulins and immunoglobulin subtypes
Circulating levels of the immunoglobulin subtypes IgA, IgM, and IgG, as well as the IgG isoforms IgG 1–4 and TTG-IgA antibody levels, increased in patients with dACLD (S3–S5) as compared with cACLD (S0–S2; all p <0.05; Table S1; Fig. S3), and correlated with the severity of portal hypertension (reflected by HVPG) and liver dysfunction (reflected by MELD; all p <0.05; Fig. S4). When differentiating patients by different decompensated ACLD stages, IgA levels exhibited a stepwise increase from S0–S2 (274 [199–424] mg/dl, p <0.001; Fig. 1) to patients with S4 (456 [300–618] mg/dl) and S5 (490 [298–674] mg/dl). The IgA isoform TTG-IgA – a potential marker of mucosal immunity, as described for other mucosa-associated antibodies in the context of cirrhosis19 – exhibited a similar increase in S4 (2.10 [1.25–3.15] U/ml) and S5 (2.10 [1.20–3.00] U/ml; S0–S2: 1.20 [1.00–2.20] U/ml; p <0.001), indicating that TTG-IgA levels are also linked to disease severity in ACLD. Of note, all patients displayed TTG-IgA levels below the diagnostic threshold for coeliac disease. IgA and TTG-IgA serum levels exhibited a strong correlation (Spearman’s rs = 0.797, 0.75–0.84, p <0.001; Fig. S5). As mentioned above, IgG and IgM levels increased in patients with dACLD upon comparison with cACLD. When stratifying patients by different dACLD stages, however, rather weak statistical trends were observed, which might be related to the smaller sample size and adjustment for multiple testing (Table 1; Fig. 1; Fig. S6). Nevertheless, these results may suggest that the increase of IgA in patients with S4 and S5 represents the most pronounced change among circulating immunoglobulin subtypes in dACLD.
Acute-phase reaction and systemic inflammation
Systemic inflammation parameters CRP, IL-6, and procalcitonin (PCT) increased across disease stages, whereas lipopolysaccharide binding protein (LBP) and white blood cells (WBCs) did not display significant dynamics across patient groups (Table 1). To assess the acute-phase reaction in more detail, we quantified the circulating levels of AGP, SAA, A2M – being regarded as APPs and modulators of inflammation and immunity – across different stages of ACLD severity. AGP and SAA levels were, however, similar across ACLD stages (p = 0.349 and p = 0.818, respectively; Table 1). Conversely, A2M exhibited a decrease in patients with S4 (231 [198–263] mg/dl) and S5 (205 [159–260], p <0.001) as compared with S0–S2 (286 [228–331] mg/dl; p <0.001). All markers displayed a weak negative association with MELD, indicating that the synthesis of these proteins may be impaired as liver dysfunction progresses (Figs. S7 and S8).
The link between innate and adaptive immunity, acute-phase, and systemic inflammation
To explore whether circulating components of innate and adaptive immunity align with markers of systemic inflammation and bacterial translocation, their correlation with CRP, IL-6, PCT, and LBP levels was assessed (Fig. 2). IL-6 (rather than CRP or PCT) exhibited significant associations with complement factors (negative correlation) and immunoglobulin levels (positive correlation) in the study cohort. Among studied parameters, IL-6 displayed the strongest link to IgA (rs = 0.501, 0.40–0.59, p <0.001), TTG-IgA (rs = 0.382, 0.27–0.49, p <0.001) and A2M (rs = -0.436, -0.53 to -0.33, p <0.001) levels. LBP levels had no meaningful correlation with immunoglobulin levels in the circulation, however, exhibited positive correlations with complement factors, AGP, and SAA levels. A correlation matrix of all studied parameters (including all immunoglobulin subtypes and complement levels) is displayed in Fig. S9.
Fig. 2.

Correlation between biomarkers of systemic inflammation and circulating components of innate and adaptive immunity. Statistical analysis: Spearman’s correlation coefficients were calculated to assess the association between continuous variables. ∗p <0.05; ∗∗p <0.01; ∗∗∗p <0.001. A2M, alpha-2-macroglobulin; AGP, alpha-1-acid glycoprotein; C3c, complement C3 component; c/dACLD, compensated/decompensated advanced chronic liver disease; CRP, C-reactive protein; LBP, lipopolysaccharide binding protein; PCT, procalcitonin; SAA, serum amyloid A.
Furthermore, we explored whether the link between systemic inflammation and circulating immune components was restricted to patients with either cACLD or dACLD. The link between IL-6 and IgA, TTG-IgA, and IgG levels was observed in both cACLD and dACLD (Fig. 2). Other immunoglobulin subtypes and complement factors exhibited only weak correlations with inflammation parameters in the overall cohort and displayed similar results (i.e. non-significant or weak correlations) in patients with cACLD and dACLD, respectively.
Prediction of decompensation and liver-related mortality
Considering that CAID is believed to impact on the course of ACLD, we assessed whether serum levels of innate and adaptive immunity components in the systemic circulation were indicative of disease progression – as reflected by the composite endpoint of first/further decompensation or LRD at 24 months. The median transplant-free follow-up was 6.90 (3.30–12.9) months. Eight (3%) patients had no follow-up, and thus, were excluded for outcome analyses. Forty-three (18%) patients developed decompensation or LRD during follow-up.
Patients were stratified into groups above and below the median values of immunoglobulins and complement factors investigated in this study. The incidence of the composite endpoint was significantly higher in patients with IgA >365 mg/dl (log-rank hazard ratio [HR]: 3.61, 1.98–6.58, p <0.001), total IgG >1,470 mg/dl (log-rank HR: 1.96, 1.08–3.57, p = 0.032) and IgG1 >973 mg/dl (log-rank HR: 1.89, 1.04–3.44, p = 0.043), as well as reduced complement levels, particularly C3c (log-rank HR: 2.58, 1.42–4.70, p = 0.003) (Fig. 3; Fig. S10; Table S2).
Fig. 3.

Incidence of decompensation/liver-related death in patients stratified by complement and immunoglobulin levels. Statistical analysis: patients were stratified by median complement and immunoglobulin levels. The incidence of events in different patient groups was compared with the log-rank test. C3c, complement C3 component; LRD, liver-related death.
To determine parameters that were independent predictors of disease progression, multivariate Cox regression models were performed. Besides established parameters such as HVPG, MELD, and IL-6, complement and most immunoglobulin levels were associated with decompensation or LRD on univariate Cox regression. On multivariate analysis that was also adjusted for the presence of dACLD, C3c (adjusted HR [aHR] per mg/dl: 0.98, 0.97–0.99, p <0.001), and IgG-1 (aHR per 100 mg/dl: 1.05, 0.99–1.12, p = 0.103) – next to HVPG and IL-6 – displayed an independent association with the composite endpoint (Table 2).
Table 2.
Cox proportional hazard regression model assessing predictors of decompensation/liver-related death.
| Parameter | Univariable |
Multivariable (last step) |
||||
|---|---|---|---|---|---|---|
| HR | 95% CI | p value | HR | 95% CI | p value | |
| Age (per year) | 1.01 | 0.99–1.04 | 0.299 | |||
| Sex (male) | 0.66 | 0.36–1.21 | 0.177 | |||
| MELD (per point) | 1.15 | 1.07–1.24 | <0.001 | 0.98 | 0.87–1.10 | 0.724 |
| HVPG (per mmHg) | 1.13 | 1.07–1.18 | <0.001 | 1.09 | 1.03–1.15 | 0.002 |
| IL-6 (per pg/ml) | 1.03 | 1.02–1.05 | <0.001 | 1.02 | 1.01–1.04 | 0.009 |
| dACLD (yes) | 2.46 | 1.18–5.13 | 0.017 | 0.94 | 0.41–2.15 | 0.888 |
| C3c (per mg/dl) | 0.98 | 0.97–0.99 | <0.001 | 0.99 | 0.97–0.99 | 0.040 |
| IgA (per 25 mg/dl) | 1.04 | 1.02–1.06 | <0.001 | 1.00 | 0.98–1.03 | 0.725 |
| IgM (per 100 mg/dl) | 1.20 | 1.05–1.37 | 0.009 | 1.07 | 0.91–1.25 | 0.405 |
| IgG-1 (per 100 mg/dl) | 1.10 | 1.05–1.17 | <0.001 | 1.05 | 0.99–1.12 | 0.092 |
| IgG-2 (per 100 mg/dl) | 1.04 | 0.90–1.21 | 0.589 | |||
| IgG-3 (per 25 mg/dl) | 1.04 | 0.96–1.13 | 0.333 | |||
| IgG-4 (per 25 mg/dl) | 1.05 | 1.01–1.09 | 0.023 | 1.03 | 0.99–1.07 | 0.195 |
Statistical analysis: multivariable analysis was performed using a backward stepwise Cox proportional hazards regression model. p values in bold denotes statistical significance.
C3c, complement C3 component; dACLD, decompensated advanced chronic liver disease; HVPG, hepatic venous pressure gradient; MELD, model for end-stage liver disease.
Prediction of infections
Furthermore, we explored whether the susceptibility to infections – indicating a potential clinical manifestation of CAID – was related to immunoglobulin or complement levels. Infections during follow-up were recorded in 28 (11%) patients at 24 months (cACLD: n = 11/95; 12%; dACLD: n = 17/150, 11%): n = 6 had spontaneous bacterial peritonitis, n = 6 urinary tract infection, n = 5 respiratory infection (n = 1 COVID-19), n = 2 sepsis, n = 2 gastrointestinal infection, n = 1 cholangitis, n = 1 superinfected pancreatitis, n = 2 other infections (n = 1 incarcerated hernia with secondary peritonitis and sepsis, n = 1 rectal abscess), and in n = 3 cases antibiotic treatment was recorded as a result of clinical and laboratory signs of infection/inflammation, although no clear focus was identified.
Again, patients were initially stratified by median immunoglobulin and complement levels. The incidence of infections was significantly higher in patients with low C3c (log-rank HR: 2.34, 1.12–4.92, p = 0.030), and tended to correlate with high IgG1 levels (log-rank HR: 2.14, 1.02–4.48, p = 0.054; Figs S11 and S12; Table S3). Importantly, IL-6, IgG-1 (all p <0.05), and IgG-4 (p = 0.076) were associated with the development of infections on univariate Cox regression analysis. Multivariate analysis indicated that IL-6 (aHR per pg/ml: 1.03, 1.00–1.05, p = 0.023) and IgG-1 (aHR per 100 mg/dl: 1.12, 1.04–1.19, p = 0.002) were independently linked to the development of infections in patients with ACLD (Table 3).
Table 3.
Cox proportional hazard regression model assessing predictors of infections during follow-up.
| Parameter | Univariable |
Multivariable (last step) |
||||
|---|---|---|---|---|---|---|
| HR | 95% CI | p value | HR | 95% CI | p value | |
| Age (per year) | 1.02 | 0.99–1.05 | 0.246 | |||
| Sex (male) | 0.82 | 0.38–1.74 | 0.596 | |||
| MELD (per point) | 1.05 | 0.95–1.16 | 0.346 | |||
| HVPG (per mmHg) | 1.05 | 0.99–1.11 | 0.116 | |||
| IL-6 (per pg/ml) | 1.03 | 1.01–1.05 | 0.007 | 1.03 | 1.00–1.05 | 0.023 |
| dACLD (yes) | 0.95 | 0.44–2.02 | 0.887 | |||
| C3c (per mg/dl) | 0.99 | 0.98–1.01 | 0.191 | |||
| IgA (per 25 mg/dl) | 1.02 | 0.99–1.05 | 0.191 | |||
| IgM (per 100 mg/dl) | 1.05 | 0.83–1.32 | 0.719 | |||
| IgG-1 (per 100 mg/dl) | 1.13 | 1.06–1.22 | <0.001 | 1.12 | 1.04–1.19 | 0.002 |
| IgG-2 (per 100 mg/dl) | 0.91 | 0.74–1.11 | 0.354 | |||
| IgG-3 (per 25 mg/dl) | 1.04 | 0.93–1.15 | 0.521 | |||
| IgG-4 (per 25 mg/dl) | 1.04 | 1.00–1.09 | 0.076 | 1.03 | 0.98–1.07 | 0.282 |
| TTG-IgA (per U/ml) | 1.09 | 0.88–1.35 | 0.442 | |||
Statistical analysis: multivariable analysis was performed using a backward stepwise Cox proportional hazards regression model. p values in bold denotes statistical significance.
C3c, complement C3 component; dACLD, decompensated advanced chronic liver disease; HVPG, hepatic venous pressure gradient; MELD, model for end-stage liver disease; TTG, tissue transglutaminase.
Discussion
In the present study, we demonstrate a dysregulation of innate and adaptive immunity in 245 well-characterised patients with ACLD without infections or acute decompensation. Importantly, systemic C3c and IgG1 levels seem to be valuable biomarkers indicative of CAID1 in patients with ACLD. CAID is considered to progress across the spectrum of ACLD and to exert a decisive impact on the progression of disease.1,20 A dysfunctional gut–liver axis and bacterial translocation are believed to promote hepatic and systemic inflammatory responses, and thus, the development of CAID.1,21,22 Concordantly, previous studies reported dysfunction of neutrophils and monocytes,23 B cells,24 T cells,25 as well as increased systemic inflammation3,5,6 in different patient populations of ACLD.
Complement proteins are primarily synthesised by hepatocytes and are involved in the removal of pathogens through opsonisation, mediation of inflammatory processes, and cytotoxicity.26,27 Systemic inflammation may upregulate the hepatic synthesis of complement factors in physiological conditions.27 However, decreased complement levels have been reported in patients with cirrhosis – presumably caused by hepatic dysfunction – and linked to an increased risk for infections and a worse prognosis.11,12,28 In the present study, we were able to confirm the link between complement levels and liver dysfunction as well as portal hypertension in a large cohort of thoroughly characterised patients with ACLD. Importantly, we demonstrate a pronounced decrease in complement levels, as well as the in vitro complement activity assay CH50, particularly in patients with S4 (non-bleeding decompensation) and S5 (further decompensation). The decrease in complement levels/activity in ACLD is likely related to both impaired synthetic function and increased complement consumption (e.g. in response to bacterial translocation). Such et al.29 observed that intestinal decontamination increased C3 levels in serum and ascitic fluid in a small study on patients with cirrhosis and ascites. Another study in patients with ascites reported increased activation of the complement system in the presence of bacterial antigens.30 We acknowledge that our study cannot provide further mechanistic evidence on the source of reduced complement levels in patients with dACLD. Nevertheless, the observed negative correlation between IL-6 and complement levels, and the prognostic value of complement levels towards disease progression, underline the clinical significance of reduced complement levels in ACLD.
Furthermore, the production of immunoglobulins by B cells is an important part of the adaptive immune system. Previous studies have suggested B-cell dysfunction in cirrhosis,24,31 however, various changes in the lymphocyte compartment have been proposed to occur in ACLD.1,32 Elevated immunoglobulin levels were previously shown in patients with chronic liver disease and correlated with the severity of fibrosis.13,14 Importantly, immunoglobulin levels (IgA, IgM, IgG) decreased in patients with alcohol-related cirrhosis after regeneration of hepatic function following liver transplantation.33 Our study demonstrates that the immunoglobulin subtypes IgA, IgG, and IgM increased in patients with dACLD and were linked to portal hypertension and liver dysfunction. Interestingly, IgA serum levels exhibited a pronounced increase in patients with S4 and S5 and exhibited a significant correlation with systemic inflammation markers. Considering the impaired intestinal barrier in cirrhosis and the resulting exposure of gut-associated lymphoid tissue to pathogens from the intestinal lumen,9,21 it is tempting to speculate that the observed elevation of IgA in patients with dACLD (S5 > S4) reflect bacterial translocation in ACLD. Concordantly, Massonnet et al.34 suggested that the upregulation of IgA production in cirrhosis is related to activation of Toll-like receptor pathways. Furthermore, the strong correlation between IgA and ‘subclinical’ TTG-IgA levels (i.e. below the threshold indicative of coeliac disease) also suggests a potential link to mucosal immunity in cirrhosis. To this end, also anti-gliadin antibodies were previously associated with portal hypertension severity and intestinal permeability in patients with ACLD.19
Although IgA levels showed strong dynamics across ACLD stages, only the IgG isoform IgG1 (next to HVPG, IL-6, and C3c) displayed an independent predictive value for first/further decompensation or liver-related death on multivariate analysis. The prognostic value of IL-6 and IgG1 likely underline the clinical significance of a proinflammatory state in ACLD. Interestingly, the finding of increased IgG-4 levels in dACLD – which is considered as a mediator of anti-inflammatory effects or even of immunotolerance in other contexts35 support the concept that both inflammation and immune paralysis are relevant concomitant features of CAID.1,36
CAID is believed to ultimately predispose patients with ACLD to infections and decompensation. When we analysed the association of our biomarkers with the development of infections during follow-up, IgG1 and IL-6 were independently predictive for infections in our cohort of ACLD patients. These results further emphasise the potential role of circulating biomarkers of immune response to bacterial translocation as surrogates for CAID, particularly because patients developing infections were at increased risk of liver-related complications in previous studies.8,37 Although it can only be speculated why complement factors were not indicative towards developing infections, it is possible that the local defence against the entry of pathogens might not be primarily related to the amount of circulating complement factors and their activity. Furthermore, patients with dACLD exhibited a similar infection rate in our study as compared with patients with cACLD, which is surprising, for example when considering the high number of infections reported in patients with acute decompensation included in the PREDICT study.38 One could speculate that the selection of patients without acute decompensation, infections, or non-elective hospitalisation at the time point of HVPG measurement may be the key difference to other studies focusing on patients with dACLD. For example, patients allocated to the ‘stable decompensated cirrhosis’ group in the PREDICT study exhibited the most infection events ‘at’ or shortly ‘before and after’ study inclusion (i.e. the time point of acute decompensation).38 Notably, the relatively short follow-up interval of our cohort needs to be considered when interpreting the predictive value of these biomarkers towards infections, indicating that this question should be addressed in future studies with long-term follow-up of patients with dACLD.
Finally, our study investigated whether certain APPs were related to ACLD stage or to systemic inflammation: AGP exerts numerous immunomodulatory functions that include adhesion, pathogen binding, and regulation of leucocyte functions,39 whereas SAA reportedly functions as a soluble pattern recognition receptor.40 A2M functions as a protease inhibitor and may scavenge cytokines and other proinflammatory mediators.41 Interestingly, A2M decreased in patients with dACLD (S4 and S5), displayed a negative correlation with CRP or IL-6, and was also linked to clinical events during follow-up. These results emphasise that the mechanistic implications caused by dysregulation of certain APPs such as A2M may further contribute to immune dysfunction and thus, to CAID in patients with ACLD.41
In summary, our study demonstrates that the dysregulation of complement factors, immunoglobulins, and APPs indicate the presence of CAID and are closely linked to ACLD severity and prognosis. Low C3c was an independent predictor of the composite endpoint ‘decompensation or liver-related death’, whereas high IgG1 was an independent predictor of infections. Further mechanistic studies are warranted to decipher the underlying mechanistic effects as potential therapeutic targets and to explore the effects of therapies targeting the gut–liver axis on complement factors and immunoglobulins as biomarkers of CAID.
Financial support
No funding was received for this study.
Authors’ contributions
Study concept and design: BeSi, MM, TR. Data acquisition: all authors. Data analysis: BeSi, MM, TR. Data interpretation: all authors. Drafted the manuscript: BeSi, TR. Critical revision of manuscript: LH, MJ, DJMB, BeSc, BSH, AFS, RM, MT, MM.
Data availability statement
Data are available at reasonable request to the corresponding author.
Conflicts of interest
BeSi has received travel support from AbbVie and Gilead and was supported by an International Research scholar by Gilead Sciences awarded to TR. DB has received travel support from AbbVie and Gilead, speaker fees from AbbVie and Siemens, as well as grant support from Philips, Siemens, and Gilead. BeSc received travel support from AbbVie, Ipsen, and Gilead. MT received grant support from Albireo, Alnylam, Cymabay, Falk, Gilead, Intercept, MSD, Takeda and Ultragenyx, honoraria for consulting from Albireo, Boehringer Ingelheim, BiomX, Boehringer Ingelheim, Falk, Genfit, Gilead, Intercept, Janssen, Merck, MSD, Novartis, Phenex, Regulus and Shire, speaker fees from BMS, Falk, Gilead, Intercept, and MSD, as well as travel support from Abbvie, Falk, Gilead, and Intercept. MM served as a speaker and/or consultant and/or advisory board member for AbbVie, Gilead, Collective Acumen, and W. L. Gore & Associates and received travel support from AbbVie and Gilead. TR received grant support from Abbvie, Boehringer-Ingelheim, Gilead, MSD, Philips Healthcare, Gore; speaking honoraria from Abbvie, Gilead, Gore, Intercept, Roche, MSD; consulting/advisory board fee from Abbvie, Bayer, Boehringer-Ingelheim, Gilead, Intercept, MSD, Siemens; and travel support from Abbvie, Boehringer-Ingelheim, Gilead, and Roche. RP, AFS, BH, LH, MJ and RM declare no conflicts of interest.
Please refer to the accompanying ICMJE disclosure forms for further details.
Footnotes
Author names in bold designate shared co-first authorship.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jhepr.2023.100712.
Supplementary data
The following are the supplementary data to this article:
References
- 1.Albillos A., Martin-Mateos R., Van der Merwe S., Wiest R., Jalan R., Álvarez-Mon M. Cirrhosis-associated immune dysfunction. Nat Rev Gastroenterol Hepatol. 2022;19:112–134. doi: 10.1038/s41575-021-00520-7. [DOI] [PubMed] [Google Scholar]
- 2.Martin-Mateos R., Alvarez-Mon M., Albillos A. Dysfunctional immune response in acute-on-chronic liver failure: it takes two to tango. Front Immunol. 2019;10:973. doi: 10.3389/fimmu.2019.00973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Costa D., Simbrunner B., Jachs M., Hartl L., Bauer D., Paternostro R., et al. Systemic inflammation increases across distinct stages of advanced chronic liver disease and correlates with decompensation and mortality. J Hepatol. 2021;74:819–828. doi: 10.1016/j.jhep.2020.10.004. [DOI] [PubMed] [Google Scholar]
- 4.Mandorfer M., Schwabl P., Paternostro R., Pomej K., Bauer D., Thaler J., et al. Von Willebrand factor indicates bacterial translocation, inflammation, and procoagulant imbalance and predicts complications independently of portal hypertension severity. Aliment Pharmacol Ther. 2018;47:980–988. doi: 10.1111/apt.14522. [DOI] [PubMed] [Google Scholar]
- 5.Trebicka J., Amoros A., Pitarch C., Titos E., Alcaraz-Quiles J., Schierwagen R., et al. Addressing profiles of systemic inflammation across the different clinical phenotypes of acutely decompensated cirrhosis. Front Immunol. 2019;10:476. doi: 10.3389/fimmu.2019.00476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Claria J., Stauber R.E., Coenraad M.J., Moreau R., Jalan R., Pavesi M., et al. Systemic inflammation in decompensated cirrhosis: characterization and role in acute-on-chronic liver failure. Hepatology. 2016;64:1249–1264. doi: 10.1002/hep.28740. [DOI] [PubMed] [Google Scholar]
- 7.Jalan R., Fernandez J., Wiest R., Schnabl B., Moreau R., Angeli P., et al. Bacterial infections in cirrhosis: a position statement based on the EASL Special Conference 2013. J Hepatol. 2014;60:1310–1324. doi: 10.1016/j.jhep.2014.01.024. [DOI] [PubMed] [Google Scholar]
- 8.Arvaniti V., D'Amico G., Fede G., Manousou P., Tsochatzis E., Pleguezuelo M., et al. Infections in patients with cirrhosis increase mortality four-fold and should be used in determining prognosis. Gastroenterology. 2010;139:1246–1256. doi: 10.1053/j.gastro.2010.06.019. [DOI] [PubMed] [Google Scholar]
- 9.Simbrunner B., Mandorfer M., Trauner M., Reiberger T. Gut-liver axis signaling in portal hypertension. World J Gastroenterol. 2019;25:5897–5917. doi: 10.3748/wjg.v25.i39.5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kubes P., Mehal W.Z. Sterile inflammation in the liver. Gastroenterology. 2012;143:1158–1172. doi: 10.1053/j.gastro.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 11.Homann C., Varming K., Høgåsen K., Mollnes T.E., Graudal N., Thomsen A.C., et al. Acquired C3 deficiency in patients with alcoholic cirrhosis predisposes to infection and increased mortality. Gut. 1997;40:544–549. doi: 10.1136/gut.40.4.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baumann M., Witzke O., Canbay A., Patschan S., Treichel U., Gerken G., et al. Serum C3 complement concentrations correlate with liver function in patients with liver cirrhosis. Hepatogastroenterology. 2004;51:1451–1453. [PubMed] [Google Scholar]
- 13.Lin S., Sun Q., Mao W., Chen Y. Serum immunoglobulin A (IgA) level is a potential biomarker indicating cirrhosis during chronic hepatitis B infection. Gastroenterol Res Pract. 2016;2016 doi: 10.1155/2016/2495073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Watt K., Uhanova J., Gong Y., Kaita K., Doucette K., Pettigrew N., et al. Serum immunoglobulins predict the extent of hepatic fibrosis in patients with chronic hepatitis C virus infection. J Viral Hepat. 2004;11:251–256. doi: 10.1111/j.1365-2893.2004.00507.x. [DOI] [PubMed] [Google Scholar]
- 15.EASL Clinical Practice Guidelines for the management of patients with decompensated cirrhosis. J Hepatol. 2018;69:406–460. doi: 10.1016/j.jhep.2018.03.024. [DOI] [PubMed] [Google Scholar]
- 16.D'Amico G., Morabito A., D’Amico M., Pasta L., Malizia G., Rebora P., et al. Clinical states of cirrhosis and competing risks. J Hepatol. 2018;68:563–576. doi: 10.1016/j.jhep.2017.10.020. [DOI] [PubMed] [Google Scholar]
- 17.Reiberger T., Schwabl P., Trauner M., Peck-Radosavljevic M., Mandorfer M. Measurement of the hepatic venous pressure gradient and transjugular liver biopsy. J Vis Exp. 2020;160 doi: 10.3791/58819. [DOI] [PubMed] [Google Scholar]
- 18.Simbrunner B., Semmler G., Stadlmann A., Scheiner B., Schwabl P., Paternostro R., et al. Vitamin A levels reflect disease severity and portal hypertension in patients with cirrhosis. Hepatol Int. 2020;14:1093–1103. doi: 10.1007/s12072-020-10112-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reiberger T., Ferlitsch A., Payer B.A., Mandorfer M., Heinisch B.B., Hayden H., et al. Non-selective betablocker therapy decreases intestinal permeability and serum levels of LBP and IL-6 in patients with cirrhosis. J Hepatol. 2013;58:911–921. doi: 10.1016/j.jhep.2012.12.011. [DOI] [PubMed] [Google Scholar]
- 20.Arroyo V., Angeli P., Moreau R., Jalan R., Clària J., Trebicka J., et al. The systemic inflammation hypothesis: towards a new paradigm of acute decompensation and multiorgan failure in cirrhosis. J Hepatol. 2021;74:670–685. doi: 10.1016/j.jhep.2020.11.048. [DOI] [PubMed] [Google Scholar]
- 21.Tranah T.H., Edwards L.A., Schnabl B., Shawcross D.L. Targeting the gut-liver-immune axis to treat cirrhosis. Gut. 2020;70:982–994. doi: 10.1136/gutjnl-2020-320786. [DOI] [PubMed] [Google Scholar]
- 22.Simbrunner B., Trauner M., Reiberger T. Therapeutic aspects of bile acid signalling in the gut-liver axis. Aliment Pharmacol Ther. 2021;54:1243–1262. doi: 10.1111/apt.16602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tritto G., Bechlis Z., Stadlbauer V., Davies N., Francés R., Shah N., et al. Evidence of neutrophil functional defect despite inflammation in stable cirrhosis. J Hepatol. 2011;55:574–581. doi: 10.1016/j.jhep.2010.11.034. [DOI] [PubMed] [Google Scholar]
- 24.Doi H., Iyer T.K., Carpenter E., Li H., Chang K.-M., Vonderheide R.H., et al. Dysfunctional B-cell activation in cirrhosis resulting from hepatitis C infection associated with disappearance of CD27-positive B-cell population. Hepatology. 2012;55:709–719. doi: 10.1002/hep.24689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lebossé F., Gudd C., Tunc E., Singanayagam A., Nathwani R., Triantafyllou E., et al. CD8(+)T cells from patients with cirrhosis display a phenotype that may contribute to cirrhosis-associated immune dysfunction. EBioMedicine. 2019;49:258–268. doi: 10.1016/j.ebiom.2019.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qin X., Gao B. The complement system in liver diseases. Cell Mol Immunol. 2006;3:333–340. [PubMed] [Google Scholar]
- 27.Gao B., Jeong W.I., Tian Z. Liver: an organ with predominant innate immunity. Hepatology. 2008;47:729–736. doi: 10.1002/hep.22034. [DOI] [PubMed] [Google Scholar]
- 28.Glargaard S., Boysen T., Pilely K., Garred P., Ytting H. Prognostic value of lectin pathway molecules and complement proteins in ascitic fluid and blood in patients with liver cirrhosis. Scand J Gastroenterol. 2018;53:64–69. doi: 10.1080/00365521.2017.1386710. [DOI] [PubMed] [Google Scholar]
- 29.Such J., Guarner C., Soriano G., Teixidó M., Barrios J., Tena F., et al. Selective intestinal decontamination increases serum and ascitic fluid C3 levels in cirrhosis. Hepatology. 1990;12:1175–1178. doi: 10.1002/hep.1840120516. [DOI] [PubMed] [Google Scholar]
- 30.Francés R., González-Navajas J.M., Zapater P., Muñoz C., Caño R., Pascual S., et al. Bacterial DNA induces the complement system activation in serum and ascitic fluid from patients with advanced cirrhosis. J Clin Immunol. 2007;27:438–444. doi: 10.1007/s10875-007-9090-2. [DOI] [PubMed] [Google Scholar]
- 31.Girón J.A., Alvarez-Mon M., Menéndez-Caro J.L., Abreu L., Albillos A., Manzano L., et al. Increased spontaneous and lymphokine-conditioned IgA and IgG synthesis by B cells from alcoholic cirrhotic patients. Hepatology. 1992;16:664–670. doi: 10.1002/hep.1840160309. [DOI] [PubMed] [Google Scholar]
- 32.Wiest R., Lawson M., Geuking M. Pathological bacterial translocation in liver cirrhosis. J Hepatol. 2014;60:197–209. doi: 10.1016/j.jhep.2013.07.044. [DOI] [PubMed] [Google Scholar]
- 33.González-Quintela A., López-Ben S., Pérez L.F., Graña B., Varela M., Tomé S., et al. Time-course changes of serum immunoglobulins (IgA, IgG, IgM) after liver transplantation for alcoholic cirrhosis. Transpl Immunol. 2003;11:73–77. doi: 10.1016/S0966-3274(02)00084-9. [DOI] [PubMed] [Google Scholar]
- 34.Massonnet B., Delwail A., Ayrault J.M., Chagneau-Derrode C., Lecron J.C., Silvain C. Increased immunoglobulin A in alcoholic liver cirrhosis: exploring the response of B cells to Toll-like receptor 9 activation. Clin Exp Immunol. 2009;158:115–124. doi: 10.1111/j.1365-2249.2009.04004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crescioli S., Correa I., Karagiannis P., Davies A.M., Sutton B.J., Nestle F.O., et al. IgG4 characteristics and functions in cancer immunity. Curr Allergy Asthma Rep. 2016;16:7. doi: 10.1007/s11882-015-0580-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Márquez M., Fernández-Gutiérrez C., Montes-de-Oca M., Blanco M.J., Brun F., Rodríguez-Ramos C., et al. Chronic antigenic stimuli as a possible explanation for the immunodepression caused by liver cirrhosis. Clin Exp Immunol. 2009;158:219–229. doi: 10.1111/j.1365-2249.2009.04005.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Villanueva C., Albillos A., Genescà J., Garcia-Pagan J.C., Brujats A., Calleja J.L., et al. Bacterial infections adversely influence the risk of decompensation and survival in compensated cirrhosis. J Hepatol. 2021;75:589–599. doi: 10.1016/j.jhep.2021.04.022. [DOI] [PubMed] [Google Scholar]
- 38.Trebicka J., Fernandez J., Papp M., Caraceni P., Laleman W., Gambino C., et al. The PREDICT study uncovers three clinical courses of acutely decompensated cirrhosis that have distinct pathophysiology. J Hepatol. 2020;73:842–854. doi: 10.1016/j.jhep.2020.06.013. [DOI] [PubMed] [Google Scholar]
- 39.Ceciliani F., Lecchi C. The immune functions of α(1) acid glycoprotein. Curr Protein Pept Sci. 2019;20:505–524. doi: 10.2174/1389203720666190405101138. [DOI] [PubMed] [Google Scholar]
- 40.Smole U., Gour N., Phelan J., Hofer G., Köhler C., Kratzer B., et al. Serum amyloid A is a soluble pattern recognition receptor that drives type 2 immunity. Nat Immunol. 2020;21:756–765. doi: 10.1038/s41590-020-0698-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vandooren J., Itoh Y. Alpha-2-macroglobulin in inflammation, immunity and infections. Front Immunol. 2021;12 doi: 10.3389/fimmu.2021.803244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available at reasonable request to the corresponding author.