

Abstract
Monoamine transporters (MATs) are targets of a wide range of compounds that have been developed as therapeutic treatments for various neuropsychiatric and neurodegenerative disorders such as depression, ADHD, neuropathic pain, anxiety disorders, stimulant use disorders, epilepsy, and Parkinson’s disease. The MAT family is comprised of three main members – the dopamine transporter (DAT), the norepinephrine transporter (NET), and the serotonin transporter (SERT). These transporters are through reuptake responsible for the clearance of their respective monoamine substrates from the extracellular space. The determination of X-ray crystal structures of MATs and their homologues bound with various substrates and ligands has resulted in a surge of structure-function-based studies of MATs to understand the molecular basis of transport function and the mechanism of various ligands that ultimately result in their behavioral effects. This review focusses on recent examples of ligand-based structure-activity relationship studies trying to overcome some of the challenges associated with previously developed MAT inhibitors. These studies have led to the discovery of unique and novel structurally diverse MAT ligands including allosteric modulators. These novel molecular scaffolds serve as leads for designing more effective therapeutic interventions by modulating the activities of MATs and ultimately their associated neurotransmission and behavioral effects.
Keywords: Dopamine, Serotonin, Norepinephrine, Antidepressants, Psychostimulants, Allosteric modulators
4.1. Introduction and Overview of Monoamine Transporters
The monoamine neurotransmitter transporters (MATs) are part of the solute carrier 6 (SLC6) family of transporters [1, 2] and are namely the dopamine transporter (DAT, SLC6A3), the norepinephrine transporter (NET, SLC6A2), and the serotonin transporter (SERT, SLC6A4) which mediate the reuptake of monoamine neurotransmitters dopamine (DA), norepinephrine (NE), and serotonin (5-HT), respectively, from the extracellular space into the intracellular presynaptic compartment. The MATs are membrane-bound transporters expressed in presynaptic neuronal terminals of their respective pathways within the CNS and mediate a rapid reuptake of neurotransmitters from the synaptic cleft into the presynaptic neurons, where the neurotransmitters are sequestered into synaptic vesicles (via vesicular monoamine transporters or VMAT) for recycling and release or are degraded by monoamine oxidase enzymes. The transport of substrates by the transporters is favored by the energy gradient produced by the movement of Na+ ions into the cell and therefore driven by the concentration gradient created by Na+/K+ ATPase. DAT and NET transport one dopamine or norepinephrine molecule along with 2 Na+ ions and one Cl− ion, whereas SERT cotransports one 5-HT molecule with one Na+ and one Cl− along with the counter-transport of one K+. Thus, the MATs are sometimes also referred to as Na+/Cl−-symporters [3].
4.2. Therapeutic Relevance of MATs
Since MATs control the signal amplitude and duration of monoaminergic neurotransmission, they remain a prominent therapeutic target of interest for the treatment of several neurological disorders [4]. In addition, the MATs are also the primary targets of action of a number of psychostimulants and recreational drugs of abuse such as cocaine, methamphetamine, 3,4-methylenedioxy-methamphetamine (“ecstasy” or MDMA), cathinones (or “bath salts”) which all either block or reverse the transport of neurotransmitters and as a result increase the synaptic neurotransmission leading to stimulatory effects.
Tricyclic antidepressants (e.g., clomipramine and amitriptyline) and the selective inhibitors of SERT (also known as selective serotoninreuptake inhibitors or SSRIs) such as fluoxetine, sertraline and paroxetine are commonly prescribed for depression, anxiety, and panic disorders. Bupropion, a DAT inhibitor, is an antidepressant and smoking cessation aid. Methylphenidate, another DAT-inhibitor is marketed for attention-deficit hyperactivity disorder (ADHD). Reboxetine is used in depression, panic disorder, and ADHD. Different from targeting the inhibition of MATs for use in mood- or cognition-related disorders, synthetic compounds that act as non-selective substrates of MATs and promote monoamine efflux have also found application for clinical use. For example, mixed amphetamine salts (racemic mixture of amphetamine isomers) are prescribed in low doses to treat ADHD and are commonly used off-label to improve cognition and to help in narcolepsy by promoting wakefulness. However, such drugs along with many other cognition-enhancing drugs that interact with MATs are strictly controlled and regulated because of their abuse liability.
4.3. Structural Insights and Transport Mechanism
Breakthrough discoveries such as crystal structures of homologous bacterial (Aquifex aeolicus) leucine transporter (LeuT) [5-7], Drosophila melanogaster dopamine transporter (dDAT) [8-10] and human serotonin transporter (hSERT) [11] have been pivotal in enhancing our understanding of the structural biology of the MATs and in guiding further studies to elucidate underlying molecular mechanisms of ligand transport and interaction.
The X-ray crystal structure of the bacterial leucine transporter (LeuT) from Aquifex aeolicus that shares 20–25% overall homology with the MATs [6] gave insights into the tertiary arrangement of the transporters and their functioning, especially of the core region which shares ~60% homology with the MATs. The co-crystal structures of Drosophila melanogaster DAT (dDAT) bound to the substrate DA, D-amphetamine, methamphetamine, cocaine, and many other ligands which has 50–55% homology with the human MATs [8-10] and the human SERT in complex with paroxetine and escitalopram [11-13] further enhanced our understanding of mechanism of transport and ligand induced structural conformations of MATs.
The structure of all MATs is comprised of 12 alpha-helical transmembrane spanning domains connected with flexible intracellular and extracellular loops. The N- and C-termini lie in the intracellular region. The high affinity primary substrate binding site, also referred to as the S1 or orthosteric site, lies at the core of the translocation pathway located between TM1 and TM6. The S1 site holds a substrate molecule along with one or two Na+ ions. The S1 pocket is comprised of a hydrophobic region, which holds the aromatic substituents of the substrates, and a hydrophilic or a polar region that contains a conserved aspartate residue (SERT: D98, DAT: D79, NET: D75) to form ionic interactions with the amine group of the substrates. The transporters translocate the substrate via a three-state “alternating access” mechanism through sequential binding and conformational changes [1]. According to the “alternating access” mechanism, the transporter adopts distinct conformations where the access to the central binding site is alternatingly sealed from the extracellular and the intracellular side facilitated by the opening and closing of the gating networks present above and below the S1 site. The substrate and the ions bind to the transporter when it adopts an outward-open conformation, which further triggers the closing of the transporter, and thereby occluding the substrate and the ions from either side of the membrane. Next, the occluded state is followed by the opening of the intracellular gate, leading to an inward-facing conformation, which releases the ions and substrate into the cytoplasm via diffusion facilitated by hydration of the substrate binding site. The recently reported SERT cryo-EM structure in complex with ibogaine provided further insights into the structural rearrangements that occur during the translocation cycle. Ibogaine is a hallucinogenic natural product with psychoactive and anti-addictive properties and a non-competitive inhibitor of transport. It is bound within the S1 site of SERT and the complexes were captured in outward-open, occluded, and inward-open conformations providing a snapshot of the neurotransmitter transport mechanism and inhibition [14].
4.4. Central Binding Site Versus Allosteric Binding Sites in MATs
LeuT crystal structures bound with leucine and tryptophan substrates within the S1 site are known in the literature. There are several known co-crystal structures of dDAT (shares 55% homology with human DAT at the amino acid level) in complex with dopamine, amphetamine, cocaine, tricyclic antidepressants, and several other inhibitors bound within the S1 site [10]. In addition, several crystal structures of LeuBAT (LeuT with key residues in the central binding site mutated to corresponding amino acids of human SERT) have been published with mazindol, TCAs, SSRIs, SNRIs, etc., all occupying the S1 site [15]. SERT crystal structures have recently been reported bound to the antidepressants sertraline, fluvoxamine, and paroxetine [12] occupying the central S1 site. Moreover, several homology models and computational studies, that have employed the known crystal structures as templates, have aided in the discovery and development of MAT ligands. Many novel MAT ligands have been identified as promising lead compounds through virtual screening studies using the computational models of MATs. Xue and colleagues [16] provide a comprehensive list of reported computational models known to date. Together, these structures have provided detailed insights into the molecular and structural details of the MAT central binding sites and have led to the advancement in the study of transporter interactions with their ligands and of structural differences among DAT, SERT, and NET [17]. Increasing structural and biochemical evidence have indicated the presence of additional ligand binding sites within the MATs other than the S1 central binding site. These sites most likely are present in the extracellular vestibular region of the transport channel above the S1 site. Several studies in the literature have endeavored to determine the exact molecular and structural features of ligand binding to this region of the transporters. One of the earliest evidences indicating the presence of ligand binding site in the extracellular region comes from the X-ray crystal structures of bacterial transporter LeuT in complex with several TCAs and SSRIs bound in the putative secondary binding site region [5-7, 18, 19]. More recently, hSERT crystal structure with two (S)-citalopram molecules simultaneously bound (one in the S1 site and the other in the extracellular vestibule region) [11] as well as the drastic increase in affinity of some bivalent ligands of SERT and DAT [20] compared to the common monovalent ligands strongly indicate the presence of distinct low-affinity ligand binding sites in the extracellular region within MATs other than the high-affinity primary binding site (i.e., S1). These results add to the earlier data on antagonist dissociation experiments of SERT that showed that there is a low-affinity allosteric site in SERT that slows the dissociation of inhibitors from a separate high-affinity site [21]. In this study, it was shown that S-citalopram or clomipramine impeded the dissociation of [3H]-citalopram, although with a relatively low potency of ~5 μM while it is known to bind to the central binding site with high potency. Later, Plenge et al. used simulations to show that ligand binding in the S1 propagates conformational changes in the S2 site. Recently, the same group reported cryo-EM structure of SERT bound with the antidepressant vilazodone in the extracellular vestibular [22]. In another study, molecular dynamics simulations and comparative genomics techniques identified another allosteric pocket [23, 24] in the extracellular vestibule of SERT and a virtual screening of a database against this site identified a few low potency ligands that specifically modulate the function of SERT ligands known to bind to the high-affinity S1 site. We employed a similar method of hybrid-based pharmacophore approach and virtual screening to identify a novel allosteric binding pocket within the extracellular region of DAT and discovered a screening hit, KM822, which modulates the activity of DAT in beneficial ways [25]. Thus, in addition to the primary binding site S1 observed in the crystal structures of LeuT and SERT, there is a possibility that multiple low affinity binding sites are present in regions distinct from the direct translocation pathway that could serve as allosteric sites for modulating conformational changes in the transporter and affect its function. The relevance of these allosteric sites as functional binding sites for monoamine substrates during translocation and for modulators is a topic of great interest currently.
4.5. Medicinal Chemistry of MAT Ligands
A wide range of MAT-interacting compounds have been developed to date as pharmacological and therapeutic tools to modulate and regulate neurotransmission. Different classes of non-selective or selective drugs that can inhibit, modulate, or promote the activity of the three transporters have been extensively developed via design and synthesis efforts to evaluate their efficacy in the treatment of many CNS-related diseases such as depression and the abuse of psychostimulants like cocaine and amphetamines. The following provides an overview of structural details of the most prominent ligands of MATs and summarizes the recent medicinal chemistry and structure-activity relationship studies of some of the MAT ligands that have been deemed “unique” and novel in terms of their mode of mechanism, site of action, neurochemical properties, and/or their behavioral effects.
4.5.1. Structure-Activity Relationship Studies of DAT Ligands
Cocaine (1, Fig. 4.1) is a highly addictive drug of abuse which binds to DAT and inhibits the reuptake of dopamine from the synapse into presynaptic neurons. This results in an increase in extracellular dopamine levels which is the primary mechanism of cocaine’s psychostimulant effects and abuse liability. Structurally, cocaine belongs to the tropane alkaloid class of drugs [26] and nonspecifically binds to DAT, NET, and SERT with similar potencies. Numerous substituted derivatives of cocaine with DAT selectivity have been evaluated as potential pharmacotherapies for psychostimulant abuse and other DAT-related disorders. In addition, SAR studies on cocaine have been of interest for many years to characterize the binding sites of cocaine and identify the potency and selectivity requirements of moieties within cocaine for its interaction with DAT. Carroll et al. reported several cocaine analogs through systematic structure activity relationship on cocaine’s structure [27, 28].
Fig. 4.1.

Chemical structures of DAT inhibitors
The 3-aryl analogues of cocaine were among the first series that provided highly potent and selective compounds against DAT. The position and type of substituents on the 3-aryl ring further improved the affinity and selectivity of these tropanes. WIN-35428 (1a, Fig. 4.1) emerged from the 3-phenyltropanes series with higher affinity and selectivity for DAT over NET and SERT as compared to cocaine. Although this series was explored to find analogs that could block the psychostimulant effects of cocaine by specifically binding to DAT and be useful as anti-addiction therapeutics, most of the analogs produced cocaine-like behavioral effects in animals. Nevertheless, WIN-35428 is extensively used to date as a pharmacological tool in DAT-binding assays and comparative studies as the removal of the phenyl ester group in cocaine’s structure imparted stability in aqueous solutions.
RTI-55 (1b) is another compound in this series which has selectivity for DAT and SERT over NET and is commonly used as a reference tool in pharmacological assays. Based on such results, it was hypothesized that cocaine analogs that inhibit DAT to block dopamine transport will have reinforcing effects similar to that of cocaine [29]. However, GBR 12909 (2), a piperazine-based selective DA uptake inhibitor which was in clinical evaluation for cocaine abuse, piqued interest in further discovery of compounds with a desirable behavioral profile [30, 31].
One of the most pivotal discoveries among the tropane class of DAT inhibitors was the compound benztropine (3), with a diphenylether substitution, similar to GBR 12909, at the 3α position and a lack of substituent at the 2-position. Interestingly, benztropine has high affinity and selectivity for DAT, but displayed “atypical” effects where it did not share cocaine’s stimulatory behavioral profile in animal models [32]. Although benztropine was a potent DAT ligand, its binding towards off-target sites confounded the interpretation of its behavioral effects in animal models of addiction. Further exploration of benztropine SAR studies to mitigate the off-target activity showed that the addition of halogen groups at 3- and 4-position of the phenyl rings and modification of the N-Methyl moiety could improve DAT affinity and selectivity of the benztropine analogs and minimize off-target engagement (Table 4.1) [33]. These efforts led to the discovery of JHW007 (3f), a potent and selective DAT inhibitor with a unique in vivo profile. JHW007, although inhibiting DAT with similar potency as cocaine, did not show cocaine-like stimulant or subjective effects. In addition, JHW007 is also shown to antagonize the behavioral effects of cocaine or methamphetamine [34-36]. Although JHW007 has never been evaluated clinically, benztropine did not produce a desirable response to acute cocaine administration in a clinical setting.
Table 4.1.
Binding data of tropane-based analogs at DAT, NET, and SERT
| Compound | DAT (Ki, nM) | NET (Ki, nM) | SERT (Ki, nM) |
|---|---|---|---|
| 1, cocaine | 187 ± 18.7 | 3300 ± 170 | 172 ± 15 |
| 1a, WIN35428 | 11 | NA | 160 |
| 1b, RTI-55 | 1.26 | NA | 4.21 |
| 2, GBR12935 | 12.0 ± 1.9 | 497 ± 17.0 | 105 ± 11.4 |
| 3, benztropine | 118 ± 18.7 | 1390 ± 134 | >10,000 |
| 3a | 36.8 ± 3.3 | 1010 ± 116 | 1320 ± 194 |
| 3b | 26.2 ± 2.1 | 508 ± 70.0 | 2100 ± 285 |
| 3c | 11.2 ± 1.2 | NA | NA |
| 3d | 11.8 ± 1.3 | 610 ± 81 | 3260 ± 110 |
| 3e | 399 ± 27.9 | 7660 ± 1240 | 3610 ± 214 |
| 3f, JHW007 | 24.6 ± 2.0 | 1730 ± 232 | 1330 ± 151 |
| 4, methylphenidate | 60 ± 0.01 | 100 ± 0.01 | 132.43 ± 10.71 |
Because of the desirable behavioral effects in the animal models, atypical DAT inhibitors have continued to be actively pursued and investigated to elucidate their mechanism and find their potential use as psychostimulant abuse medications. Their unique effects are suggestive of a different mode of action indicating a ligand-specific control of DAT function. Structure-function studies of typical and atypical DAT ligands have hypothesized that DAT ligands prefer different DAT conformations which subsequently dictate their behavioral effects. Continued efforts in the discovery of atypical ligands have shifted focus to a new generation of atypical DAT ligands comprised of modafinil and its analogs. Modafinil (5, Table 4.2), which is used as a wake-promoting agent for the treatment of narcolepsy and other sleep disorders [37], also has low affinity for DAT and low abuse potential according to the available clinical data [38, 39]. To discover modafinil analogs with improved pharmacological and behavioral profiles that could potentially be developed into addiction therapeutics [40], several SAR studies have been reported. Although modafinil is available as a racemic mixture, studies have suggested that R-(−)-modafinil is more metabolically stable, longer-acting than the (S)-enantiomer [41] and slightly more potent (threefold) than the S-(+) counterpart. In addition, although the racemic, R-, and S-enantiomers all stimulated locomotor activity in mice, they were much less effective and less potent than cocaine [37]. Nevertheless, in clinical trials, modafinil has demonstrated limited effectiveness in treating cocaine abuse, although there is some evidence of benefit in nonalcohol-dependent cocaine abusers [42]. Several attempts have been made to further increase the therapeutic efficacy for stimulant use disorders by the design, synthesis, and evaluation of several modafinil analogs to improve its water solubility and DAT affinity.
Table 4.2.
Binding affinities of Modafinil and its analogs against DAT, NET, and SERT
| Compound | X | Y | R | Z | DAT (Ki, nM) |
NET (Ki, nM) |
SERT (Ki, nM) |
|---|---|---|---|---|---|---|---|
| 5, modafinil | H | S=O | H | C=O | 2600 [2430–2780] | NA | NA |
| 5a | H | S | H | C=O | 12400 [10800–14300] | NA | NA |
| 5b | H | S=O | Methyl | C=O | 13100 [12600–13700] | NA | NA |
| 5c | 3,3′-di-F | S=O | H | C=O | 5930 [4990–7060] | NA | NA |
| 5d | 3,3′-di-Cl | S=O | H | C=O | 881 [763–1020] | NA | NA |
| 5e | 3,3′-di-Cl | S | H | C=O | 275 [257–295] | 45400 [39600–52000] | NA |
| 5f | 4′,4″-di-Br | S | Methyl | C=O | 3010 [2770–3260] | 11000 [9540–12600] | 5720 [5320–6150] |
| 5g | 4′,4″-di-Cl | S | Methyl | C=O | 4130 [3620–4710] | 9770 [9170–10400] | 10700 [7310–15700] |
| 5h | H | S | n-butyl | C=O | 23600 [20500–27100] | NA | NA |
| 5i | 4′,4″-di-Br | S | n-butyl | C=O | 722 [659–792] | 7580 [7210–7970] | 7090 [6990–8180] |
| 5j | 4′,4″-di-F | S | n-butyl | C=O | 6400 [5820–7050] | 56100 [53900–58500] | 25500 [23300–28000] |
| 5k | 4′,4″-di-CH3 | S=O | H | C=O | 12700 [12400–13100] | NA | NA |
| 5l | H | S | H | CH2 | 142 [131–155] | 980 [938–1020] | 221 [191–257] |
| 5m | H | S | Cyclopropylmethyl | CH2 | 435 [406–466] | 17300 [15400–19400] | 10000 [9570–10400] |
| 5n | H | S | n-butyl | CH2 | 310 [275–350] | 11500 [10700–12300] | 5700 [5040–6440] |
| 5o | H | S | 3-phenylpropyl | CH2 | 295 [268–325] | 5500 [5140–5880] | 927 [786–1090] |
| 5p (JJC8-016) | 4′,4″-di-F | S | 3-phenylpropyl | CH2 | 114 [97.4–132] | 3850 [3830–3870] | 354 [312–402] |

In one of the earlier SARs explorations of modafinil, it was found that reducing the sulfoxide with sulfide decreased DAT affinity and improved SERT binding if the diphenyl rings are unsubstituted (see Table 4.2) [43]. Addition of alkyl groups on terminal amide nitrogen reduced DAT affinity. Binding affinity at DAT increased with halogen substitutions at para position of the aryl rings in the order of Br > Cl > F ≥ H. In addition, this halogen substitution order was also followed by halogens at meta-positions of the diphenyl rings [43]. Compound 5e displayed remarkable selectivity towards DAT as compared to SERT and NET with 3,3′-di-Cl substitution with thioacetamide scaffold. 4,4′-dimethyl substitution on the diphenyl rings (5k) decreased DAT affinity by fivefold but did not affect the DAT selectivity of the compound. For those analogues that contained unsubstituted diphenyl rings, it was generally observed that for the thioethanamine scaffold, DAT affinity increased with more bulky substituents on terminal amine nitrogen whereas in thioacetamides, DAT affinity decreased with an increase in bulk at the terminal amine nitrogen [43]. Compound 5l with the thioethanamine scaffold and unsubstituted diphenyl rings was one of the most potent compound towards DAT, however, lost significant DAT selectivity. This indicated that the replacement of amide with amine to increase water solubility was well tolerated. The affinity of compound 5o, with N-propylphenylamine substituent, for DAT increased by tenfold as compared to modafinil and was found to be more selective towards DAT than NET and SERT [41]. The DAT affinity further increased 2.5-fold with 4,4′-difluoro substituted diphenyl-moiety (5p, JJC8-016). In addition, N-cyclopropylmethyl and N-n-butyl substituted analogs (compounds 5m and 5n) were also among the most DAT-selective compounds in this series. Further functionalization of the terminal nitrogen was attempted by incorporating the piperazine ring in the main scaffold (Table 4.3) [42].
Table 4.3.
Binding affinities of piperazine-based modafinil analogs
| Compound | X | Y | Z | R | DAT (Ki, nM) |
NET (Ki, nM) | SERT (Ki, nM) |
|---|---|---|---|---|---|---|---|
| 5q | H | S=O | C=O | 3-phenylpropyl | 33.0 ± 2.83 | 54300 ± 3210 | 15200 ± 1100 |
| 5r | 4,4′-diF | S=O | C=O | 3-phenylpropane | 37.6 ± 1.86 | 12000 ± 1430 | 1320 ± 152 |
| 5s | 4,4′-diF | S=O | C=O | 2-OH-propyl | 752 ± 87.4 | NA | 32800 ± 4430 |
| 5t | 3,3′-diCl | S=O | C=O | 2-OH-propyl | 1380 ± 191 | >50 uM | NA |
| 5u | H | S | C | 2-OH-propyl | 49.6 ± 4.31 | 44500 ± 2400 | 26700 ± 2630 |
| 5v (JJC8-091) | 4,4′-diF | S | C | 2-OH-propyl | 16.7 ± 1.22 | 17800 ± 885 | 1770 ± 234 |
| 5w (JJC8-088) | 4,4′-diF | S | C | 2-OH, 3-phenylpropyl | 6.72 ± 0.977 | 1950 ± 227 | 213 ± 13.2 |
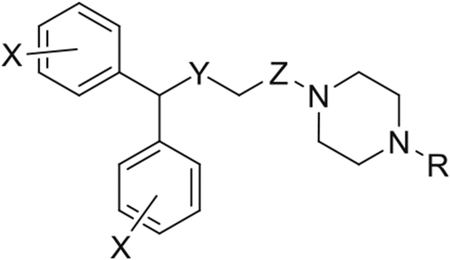
Compound 5q with a piperazine amide substitution retaining the 3-phenylpropyl moiety of compound 5p displayed 100-fold higher potency than modafinil and high selectivity for DAT as compared to SERT and NET, with additional 4,4′-diF groups imparting a modest change in affinity (5r). This series was further diversified by hydroxylating the N-bearing propyl chain. Compounds 5s and 5t with 2-OH-propyl side chain lost potency for DAT but retained high DAT selectivity and were almost inactive at SERT and NET. The removal of amide carbonyl C=O to get compounds like 5u and 5v was well tolerated as well [42]. Comparison of compounds 2-hydroxypropyl 5v (JJC8-091) and 2-hydroxyphenylpropyl 5w (JJC8-088) revealed that phenyl moiety imparted greater affinity to the compound with DAT affinity threefold higher for 5w than 5v with similar selectivity profile for DAT versus SERT. Compounds JJC8-016, JJC8-088, and JJC8-091 emerged as the most promising compounds with superior DAT affinity compared to modafinil and better aqueous solubility [44]. These compounds were evaluated for their effects on the neurochemistry via brain microdialysis and fast-scan cyclic voltammetry and behavior through ambulatory activity of male Swiss-Webster mice. The results of these studies are summarized in the literature [44-46]. Briefly, compound JJC8-016 reduced the psychostimulant effects in various animal models of addiction. For example, cocaine enhanced locomotion, cocaine self-administration, and cocaine-induced reinstatement of drug seeking behavior were dose-dependently reduced by JJC8-016 pretreatment [47] although there is evidence that it might possess cardiotoxic adverse effects similar to that seen with its analog, GBR12909 [48]. Compound JJC8-088 displayed cocaine-like effects and preferred open DAT conformation similar to that of cocaine. Compound JJC8-091 showed promising results in behavioral testing in a murine locomotor assay where it was effective in reducing stimulant effects in rats exposed to long-access (6 h) methamphetamine [48, 49]. Furthermore, JJC8-091 reduced cocaine self-administration and cocaine-primed reinstatement of cocaine seeking [49]. Hence, JJC8-091 emerged as a promising lead compound and structural modifications of this scaffold continue to be reported that have better profile in behavioral models of cocaine abuse and are more metabolically stable [50]. Giancola and colleagues have recently reported a new series of modafinil where they have replaced the metabolically susceptible piperazine ring of compounds like JJC8-016, JJC8-088, and JJC8-091 with more stable bioisosteric moieties aminopiperidine and piperidine amine (Table 4.4). The importance of the tertiary amine was explored through compounds with either a tertiary amine or amide, and the oxidation state of the sulfide was varied. Of note, the bis(4-F-phenyl)methyl moiety of the previous lead compounds (JJC8-091, JJC8-088, and JJC8-089) was retained in these new series [51]. Several compounds were designed and synthesized in this series with terminally attached alkyl-phenyl and para-substituted phenyl moieties.
Table 4.4.
Binding affinities of aminopiperidine and piperidine amine-containing modafinil analogs
| Compound | Scaffold | Y | Z | R1 | R2 | DAT | SERT | σ1 |
|---|---|---|---|---|---|---|---|---|
| Ki ± SEM (nM) | ||||||||
| 5aa | A | S=O | C=O | Phenyl | H | 79.1 ± 20.6 | 7780 ± 734 | 585 ± 21.5 |
| 5bb | A | S=O | C=O | 4-fluorophenyl | H | 77.2 ± 4.54 | 4640 ± 381 | 1440 ± 131 |
| 5cc | A | S=O | CH2 | 4-fluorophenyl | H | 50.6 ± 11.2 | 373 ± 23.8 | 26.5 ± 3.88 |
| 5dd | A | S | C=O | 2,4-difluorophenyl | H | 30.0 ± 8.25 | 296 ± 35.3 | 20.6 ± 2.38 |
| 5ee | A | S | CH2 | Phenyl | H | 32.9 ± 5.86 | 409 ± 58.2 | 3.81 ± 0.639 |
| 5ff | A | S=O | CH2 | 1-OH-2-phenylethyl | H | 91.8 ± 21.3 | 599 ± 54.4 | 351 ± 25.9 |
| 5gg | A | S | CH2 | 1-OH-2-phenylethyl | H | 31.4 ± 9.64 | 129 ± 33.4 | 309 ± 38.3 |
| 5hh | B | S | CH2 | Phenyl | H | 108 ± 17.5 | 331 ± 34.6 | 60.9 ± 8.90 |
| 5ii | B | S | CH2 | 4-fluorophenyl | H | 55.9 ± 6.08 | 268 ± 9.33 | 41.4 ± 10.9 |
| 5jj | B | S | CH2 | 1-OH-2-phenylethyl | H | 47.7 ± 2.62 | 66.8 ± 2.82 | 88 ± 4.02 |
| 5kk | A | S | C=O | 2,4-difluorophenyl | H | 30.0 ± 8.25 | 296 ± 35.3 | 20.6 ± 2.38 |

The aminopiperidines were slightly more potent than the piperidine amine analogs; however, the trend was opposite with 2-OH-propylphenyl-substituted analogs. Reduced sulfides were slightly more potent than their sulfoxide counterparts with a moderate loss in DAT selectivity compared to SERT (5ff versus 5gg). Overall, all compounds resulted in moderate binding affinity for DAT and relatively improved potency towards SERT. This series of compounds was also explored for activity against σ1 receptors due to preclinical precedence related to the therapeutic value of dual DAT/σ1 inhibitors [52]. All compounds were inactive towards NET. The most promising among this series of compounds were 5bb, 5cc, and 5dd, with highest stability, which were carried forward for further in vitro and in vivo evaluation. None of these analogues showed a cocaine-like profile as systemic administration produced only minimal stimulation of ambulatory activity. Kalaba et al. [53] have reported another series where the carboxamide of modafinil is replaced with a number of five- and six-membered aromatic heterocycles to improve activity and selectivity and in vitro/in vivo profiles (Table 4.5).
Table 4.5.
Inhibition of DAT, NET, and SERT-mediated uptake of respective radioligands by thiazole-containing modafinil analogues
| Compound | Scaffold | X | Z | n | A | B | DAT (IC50 ± SD, μM) |
NET (IC50 ± SD, μM) |
SERT (IC50 ± SD, μM) |
|---|---|---|---|---|---|---|---|---|---|
| 5ll | A | H | – | – | H | H | 14.73 | 420.1 | NA |
| 5 mm | B | H | S=O | 1 | H | CH3 | 3.25 | 174 | NA |
| 5nn | B | H | S=O | 1 | H | Cyclopropyl | 4.1 ± 0.8 | 687.2 ± 152.8 | 436.1 ± 129 |
| 5oo | B | H | S=O | 1 | H | n-propyl | 28.43 ± 1.93 | NA | NA |
| 5pp | B | H | S=O | 1 | H | n-butyl | 13.50 ± 2.76 | 164 ± 56.88 | NA |
| 5qq | B | CH3 | S=O | 1 | H | H | 7.32 ± 0.04 | NA | NA |
| 5rr | B | F | S=O | 1 | H | H | 6.76 ± 0.67 | NA | NA |
| 5ss | C | CH3 | – | – | – | – | 10.57 ± 0.80 | 215.50 ± 66.01 | NA |
| 5tt | C | OCH3 | – | – | – | – | 71.59 ± 8.28 | NA | NA |
| 5uu | – | – | – | – | – | – | 0.65 ± 0.07 | 73.25 ± 9.15 | NA |

Compound 5ll from this SAR studies was one prototype with comparable DAT potency as modafinil, and negligible activity towards SERT and NET. Compounds 5mm and 5nn were almost fivefold more potent than modafinil against DAT and also were inactive against SERT and NET. Larger, bulkier substituents in place of methyl or cyclopropyl of 5mm and 5nn resulted in two- to threefold loss in potency (5oo to 5pp). However, a chloro substituent was well tolerated (5oo). Para-substitutions at the diphenyl rings (dimethyl in 5qq and difluoro in 5rr) continued to provide highly potent and selective compounds in this series. Other analogs with thiazole ring linked to the main scaffold via 2- and 5-position led to reduced potency. The authors have also reported results of chiral separation of several analogs with promising potency and selectivity. One of the chiral HPLC separation products (5uu, Table 4.5) displayed highest DAT activity (IC50 = 0.65 ± 0.07 μM) and 100-fold less active on NET (IC50 = 73 ± 9 μM) and inactive for SERT. Hence, this series of compounds also represent potential new leads for developing psychostimulant therapeutics. Overall, the SAR studies of modafinil analogs provide promising new leads to develop drugs with an atypical DAT inhibitor profile that could have utility as anti-addiction therapies. Discovery of such atypical DAT inhibitors that lack “cocaine-like” profile piqued further interest in determining their molecular interactions with DAT. It is suggested that these ligands bind and stabilize a DAT conformation which is disparate from that of cocaine-bound DAT conformation resulting in contrasting psychostimulant effects. Experimental evidence suggest that cocaine stabilizes outward-open DAT conformation whereas JHW-007, GBR-12909, modafinil, and its analogs tend to be less affected by conformational changes and most likely favor a more inward-facing occluded conformation [54]. This conformational “preference” could attribute to the lack of addiction potential of atypical inhibitors [55]. Another reason for reduced addiction liability of atypical DAT inhibitors could be their slow rate of onset of action in the brain or their off-target effects that might also contribute to their reduced behavioral reinforcing effects [55]. Hence, further studies related to specific structural basis of interaction of DAT ligands are needed to promote the discovery and development of improved medications for psychostimulant addiction.
With the growing interest in the possible therapeutic potential of allosteric modulators of DAT, a library of diphenylmethyl (benzhydryl)-containing compounds were screened for their allosteric activity against the MATs (Fig. 4.2) [55, 56]. Among these, SRI-9804 (6a) was reported to inhibit both uptake and efflux of [3H]-dopamine, but only with partial efficacy (40–60%). Similar to allosteric activity of S-citalopram in SERT, SRI-9804 reduced the dissociation of a radioligand prebound in the orthosteric site of DAT. Two other compounds, SRI-20040 (6b) and SRI-20041 (6c), were eventually reported to partially inhibit [125I]RTI-55 binding and [3H]-DA uptake slow the dissociation rate of [125I]RTI-55 from the DAT, and allosterically modulate d-amphetamine-induced, DAT-mediated DA release. Subsequently, a series of SRI-20041 analogs were designed, synthesized, and evaluated in order to increase the affinity for DAT. This resulted in SRI-29574 (6d), one of the high-affinity and selective DAT inhibitors in this series, showing similar pharmacological features to its parent compound. SRI-29574 partially inhibited DAT uptake (IC50 = 2.3 ± 0.4 nM) while being inactive in inhibiting DAT binding. The binding site of these compounds is still unknown, but regardless of their potential clinical application, their unconventional transporter pharmacology makes them potentially interesting candidates to further explore DAT allosteric modulators.
Fig. 4.2.

Chemical structures of DAT allosteric ligands
Our group recently reported a novel allosteric inhibitor of DAT known as KM822 (7, Fig. 4.2) [25, 57]. KM822 was identified using in silico screening against the putative allosteric site present in the extracellular region of the DA translocation pathway. Biochemical investigation revealed it to be a noncompetitive inhibitor of DAT with relatively good selectivity. It was also shown to reduce psychostimulant-mediated stimulatory effects in various ex vivo and in vivo models of addiction and thus represent a possible candidate for exploring another novel class of drugs to treat addiction.
4.5.2. Structure-Activity Relationship Studies of SERT Ligands
Several classes of antidepressant drugs that target SERT have been developed and have been classified as tricyclic antidepressants (TCAs), selective serotonin reuptake inhibitors (SSRIs), and serotonin-norepinephrine reuptake inhibitors (SNRIs) (Fig. 4.3). The first-generation antidepressants included the TCAs like imipramine (8), amitriptyline (9), desipramine (10), and clomipramine (11). However, due to their nonspecific effect on cholinergic, histaminergic, and α-adrenergic pathways and their low therapeutic index, their use now is limited to treatment-resistant patients who have failed to respond to newer generations of antidepressants [58]. In the search for highly efficient antidepressant drugs with an improved safety and tolerability profile, the considerable effort of the last five decades led to the development of the selective monoamine reuptake inhibitors, such as SSRIs. SSRIs are currently the most prescribed antidepressant pharmacotherapy and the most common SSRIs used till date are fluvoxamine (12), fluoxetine (13), citalopram (16), paroxetine (14), and sertraline (15). SSRIs have a diverse chemical structure, and, in many instances, they do not share common structural motifs. The structural explanation of how these diverse ligands bind to SERT have been summarized by Coleman and Gouaux [12] by obtaining and comparing X-ray crystal structures of SERT in complex with paroxetine, citalopram, sertraline, and fluvoxamine. The diversity of SSRI chemical structures, in turn, results in compounds with varied affinities towards SERT and substantial pharmacological differences. The most studied SSRI to date is citalopram. It is the most SERT-selective SSRI. The S-enantiomer (16a) is responsible for the high selectivity and affinity for SERT and is ~30-fold more potent than R-citalopram (16b). In the past, several SAR studies on citalopram have been reported [59-61]. These SAR studies employing the analogues of citalopram have been pivotal in characterizing the S1 site for SERT [62, 63, 65, 76, 61]. More recently, the interest in achieving higher drug target selectivity to minimize side effects have led to an impetus in the study and development of SERT inhibitors that target the allosteric sites. The SSRIs like sertraline, paroxetine, clomipramine, and citalopram have been shown to possess allosteric activity as they can impair dissociation of a pre-bound high affinity radioligand to the transporter [74]. However, the allosteric potencies of these compounds are all in the micromolar range, while they bind to the orthosteric site with low-nanomolar affinity [74]. The co-crystal structure of S-citalopram bound in the allosteric region of SERT has made it feasible to further explore and understand the allosteric regulation mechanism of MATs [11].
Fig. 4.3.

Chemical structures of clinically-approved SERT ligands

Banala and colleagues [64] were the initial ones to explore the SAR of citalopram analogs for binding at the S2 site by measuring the ability to decrease the dissociation rate of [3H]-S-citalopram from the S1 site via allosteric modulation at S2 (Table 4.6). All compounds were initially assessed for SERT-S1 potency by competitive radioligand binding displacement of [3H]-S-citalopram. The replacement of 5-CN substituent with amide, amino, or any other aliphatic heterocyclic groups (compounds 16c to 16g) resulted in a decrease in the SERT S1 binding affinity compared to citalopram but only marginally changed for the aldehyde substitution (16d). With exception of the amide (16c), all other replacements retained high SERT selectivity versus NET. Substitution with bulkier heterocycles or substituted aryl rings also reduced affinity with a few exceptions. Substitutions 4-nitrophenyl (16h) and 4-aminophenyl (16i) reduced SERT-S1 binding almost 20-fold. However, these two compounds displayed allosteric site S1 binding by prolonging the dissociation of the S1 radioligand. For the 4′-fluoro phenyl moiety, replacing 4′-fluoro with bulky substituted aryls (e.g., 16j) decreased the SERT-S1 binding affinity, no binding at SERT-S2 site. Replacement of a methyl group of dimethyl amine moiety with bulky long chain aryl or heterocycles (compounds 16k to 16m) reduced both SERT-S1 affinity and selectivity; however, some of these compounds showed SERT-S2 binding.
Table 4.6.
Citalopram analogues assessed by radioligand binding displacement of [3H]-escitalopram (for SERT), tritiated-nisoxetine (for NET), and potency of 30 μM compound in inhibiting dissociation of [3H]-escitalopram from SERT
| Compound | R1 | R2 | R3 | SERT (Ki ± SEM, nM) |
NET (Ki ± SEM, nM) |
[3H] escitalopram dissoc. t1/2 (min) |
|---|---|---|---|---|---|---|
| Citalopram (16) | CN | F | CH3 | 1.94 ± 0.198 | 5950 ± 77.4 | 16.1 ± 1.0 |
| S-citalopram (16a) | CN | F | CH3 | 0.89 ± 0.132 | 10500 ± 893 | 68 ± 12 |
| 16c | CONH2 | F | CH3 | 17.7 ± 1.80 | 123 ± 17.7 | NA |
| 16d | CHO | F | CH3 | 4.3 ± 0.096 | NA | NA |
| 16e | CH2NH2 | F | CH3 | 41.9 ± 5.73 | NA | 17.4 ± 0.8 |
| 16f |

|
F | CH3 | 3.24 ± 0.328 | 11100 ± 1490 | 16.4 ± 0.3 |
| 16g |

|
F | CH3 | 22.7 ± 2.62 | NA | 24.8 ± 1.1 |
| 16h | 4-nitrophenyl | F | CH3 | 18.6 ± 1.65 | 8280 ± 825 | 40.2 ± 7.0 |
| 16i | 4-aminophenyl | F | CH3 | 29.2 ± 4.11 | ND | 52.6 ± 4.1 |
| 16j | CN | 3-cyanophenyl | CH3 | 51.5 ± 6.63 | 2800 ± 353 | NA |
| 16k | CN | F | C4H9 | 61.6 ± 6.19 | 276 ± 41.1 | NA |
| 16l | CN | F |

|
22.8 ± 2.29 | ND | 59.6 ± 1.0 |
| 16m | CN | F |

|
98.8 ± 13.3 | 272 ± 48.5 | 60.1 ± 1.0 |
NA not active

Larsen et al. [68] performed a systematic SAR study on citalopram (which is >1000-fold selective for S1 than S2) to identify S2 selective compounds by exploring all possible combinations of substituents between citalopram and talopram (Table 4.7, 16n, a close analog of citalopram and only possess low SERT-S1 affinity) [63]. The authors measured the IC50 for inhibition of [3H]-S-citalopram dissociation by determining the [3H]-S-citalopram dissociation rates in the presence of increasing concentrations of allosteric inhibitor [21]. The authors concluded that – CN group (absent in talopram) is absolutely necessary for S2 activity. Compounds without –CN displayed a fivefold reduction in S2 potency compared to their –CN congeners. The presence of para-fluoro was also important for S2 activity but its removal did not affect the S1 activity much. The addition of phthalane dimethyl groups in ##3 as compared to citalopram markedly reduced S1 affinity with a significant increase in the allosteric potency. Changing the dimethyl amine to monosubstitution showed almost no change in S2 activity but a reduction in S1 activity. The compound with the highest allosteric potency and lowest orthosteric affinity reported was dimethyl citalopram (16p), with a twofold increase in the allosteric potency compared to the orthosteric affinity [68]. The tested compounds were all racemic mixtures. To date, investigation of the enantioselectivity of these compounds has not been reported.
Table 4.7.
Systematic SAR exploration of citalopram and talopram analogs
| Compound | X | Y | Z | R | SERT S1 binding IC50 (μM) |
T1/2 for [3H]S-CIT dissociation (min) |
SERT allosteric potency IC50 (μM) |
|---|---|---|---|---|---|---|---|
| Escitalopram (16a) | CN | F | H | CH3 | 0.010 [0.0008; 0.013] | 790 ± 160 | 5.8 [5.4; 6.3] |
| Talopram (16n) | H | H | CH3 | H | 0.718 | 100 ± 3.8 | ND |
| 16o | CN | F | H | H | 0.041 [0.032; 0.051] | 380 ± 26 | 10.1 [10.0; 10.2] |
| 16p | CN | F | CH3 | CH3 | 6.4 [4.7; 8.8] | 1090 ± 130 | 3.6 [3.3; 3.8] |
| 16q | CN | H | CH3 | CH3 | 10 [9.2; 12] | 440 ± 30 | 12 [10; 13] |
ND not determined
A recent in silico screening of a library of citalopram analogs and subsequent in vitro evaluation for their allosteric potency led to the discovery of Lu AF60097 (17, Fig. 4.4) with an allosteric potency of 31 nM (measure of imipramine dissociation inhibition at S1), which is more than a 150-fold increase compared with citalopram [74]. Lu AF60097 also possess a S1 binding component with SERT S1 binding potency of ~265 nM and hence could be another template for future SAR studies to isolate S2 selective compounds. Another potential allosteric modulator of recent interest is vilazodone (18, Fig. 4.4). Vilazodone is an approved anti-depressant with partial agonist activity at 5-HT1A receptor. Recent molecular pharmacology and cryo-EM structural elucidation have revealed that vilazodone’s primary binding site is the allosteric site S2 [22], as opposed to previously reported results of MD simulations that had suggested that vilazodone binds to the S1 site similarly to all other TCAs and SSRIs [80]. Vilazodone inhibits [3H]5-HT transport with an apparent Ki of 1.1 nM and causes a decrease in the VMAX of [3H]5-HT without having any significant effect on KM suggesting a noncompetitive binding with 5-HT. Vilazodone also inhibits [3H]-imipramine dissociation with an allosteric potency of 14 nM and [3H]S-citalopram dissociation with an allosteric potency of 250 nM. The high-affinity profile of such allosteric compounds along with the elucidation of the molecular binding structures opens the possibility of developing compounds with beneficial therapeutic profiles.
Fig. 4.4.

Structures of SERT allosteric ligands
4.5.3. Structure-Activity Relationship Studies of NET Ligands
Ligands of NET are commonly classified into: selective norepinephrine reuptake inhibitors (sNRIs), serotonin-norepinephrine reuptake inhibitors (SNRIs), norepinephrine-dopamine reuptake inhibitors, and triple reuptake inhibitors (TRIs). Selective NRIs (Fig. 4.5) such as nisoxetine (19), reboxetine (20), and atomoxetine (21) are useful in the treatment of depression and ADHD. Nisoxetine is a selective, potent NRI with (R)-nisoxetine much more potent than (S)-enantiomer. Reboxetine is marketed as a racemic mixture, with (+)-(S,S)-enantiomer is much more potent for NET inhibition as compared to SERT, with (−)-(R,R)-enantiomer gaining potency for SERT. Substitutions at the aryloxy ether group affected the selectivity of compounds between NET and SERT, and in general, the (S,S)-enantiomers were more potent for NET than SERT. Several SAR studies have been reported for reboxetine where the aryloxy ether group is replaced by a number of substituted arylthiolether functionalities which in general retain the potency against NET. Atomoxetine was developed and launched by Eli Lilly as a treatment for ADHD and is a potent, selective inhibitor of NET. It is an (−)-isomer of an ortho-methylphenoxy analog of nisoxetine, and is a derivative of phenoxypropylamine. Its mechanism of action in the treatment of ADHD is unclear but is thought to be related to its selective inhibition of presynaptic norepinephrine reuptake in the prefrontal cortex, resulting in increased noradrenergic transmission, important for attention, learning, memory, and adaptive response [66]. The X-ray crystal structure of dDAT with nisoxetine and reboxetine exhibit outward-open conformations with inhibitors bound in the central S1 pocket provide insights into the pharmacophores that dictate selectivity towards hNET [9]. The new generation of antidepressants has seen a paradigm shift where a mixed action on both SERT and NET is desirable to achieve additional therapeutic benefits such as improved antidepressant efficacy and faster onset of action. Approved SNRIs such as duloxetine (22), milnacipran (23), and venlafaxine (25) may have improved antidepressant efficacy. In another study, X-ray crystal structures of dDAT with NE as well as SNRIs duloxetine and milnacipran were identified. Bupropion (24) belongs to the NDRI class of dual reuptake inhibitors and has been approved to treat major depressive disorder (MDD) and for smoking cessation [70]. Despite the approval of several antidepressants, they still lack satisfactory therapeutic effect due to the low remission rates and delayed therapeutic onset, especially in the case of major depressive disorder (MDD) [67]. TRIs which inhibit all three transporters (DAT, NET, and SERT) are currently of great interest and several compounds have advanced into clinical trials (Fig. 4.6). Such polypharmacological profile of TRIs has led to their better efficacy, safety, and less tolerance in clinical evaluation for MDD, ADHD, binge eating disorder, and cocaine addiction [77].
Fig. 4.5.

Structures of NET ligands
Fig. 4.6.

Structures of TRIs currently under clinical investigation
One of the earliest TRIs developed was compound 26a that reached clinical trials for pain treatment [79]. Extensive SAR analysis of 26a was conducted by industrial groups to develop compounds with superior potency at all three transporters. Compounds 26b, 26c, and 26d that emerged from the SAR studies on 26a are also in the clinical development stage for ADHD and depression. Amitifadine (26c) is currently in a Phase 3 trial for MDD, and centanafadine (26d) is in a Phase 3 trial for MDD and ADHD. NS2359, another promising TRI currently in a Phase 2 clinical trial for cocaine addiction, MDD, and ADHD, was developed through structural modifications on naturally occurring TRI, cocaine. Dasotraline, 27 was discovered as part of SAR development around cis-sertraline. Tu et al. [78] have recently compared the binding modes of dasotraline (27), NS2359 (28), and centanafadine (26d) by docking them into the central binding sites of the crystal structure of SERT, and the homology models of DAT and NET, and created a pharmacophore model for TRI binding to understand their polypharmacological profile which could be useful in discovering new and improved TRIs.

Recently, a number of 4-benzylpiperidine carboxamides were explored as TRIs (Table 4.8) [72]. The potency was compared to venlafaxine, the control drug for SERT and NET activity, and GBR12909 to compare DAT potency. In general, changing 3-carbon linker to 2-carbon drastically improved DAT inhibition with no change in NET and SERT inhibition (example, compound 29c versus 29d). The authors found that they could modulate the inhibition of DAT, NET, and SERT by changing the R substituents. Although most R substituents provided high potency at NET, 4-biphenyl substitution (with 3-carbon linker) provided exceptionally high inhibitory potency against NET and SERT (29a). Another novel compound, LPM580098 (toludesvenlafaxine, Fig. 4.5, compound 25b), was designed and synthesized based on the structure of venlafaxine and was reported to show promising preclinical results in neuropathic pain models [69]. It is a prodrug that can quickly convert to the SNRI desvenlafaxine (25a) under the hydrolysis of ubiquitous esterase in vivo [81]. Another series of TRIs reported recently contain the asymmetric pyran scaffold (Table 4.9) [75].
Table 4.8.
SAR exploration of 4-benzylpiperidine carboxamide containing TRIs
| Compound | N | R | SERT | NET | DAT |
|---|---|---|---|---|---|
| IC50 (in μM) are shown as 95% confidence intervals | |||||
| 29a | 3 | 4-biphenyl | 0.04–0.07 | 0.01–2.12 | >10 |
| 29b | 2 | 4-biphenyl | 0.986 | 0.17–1.28 | >10 |
| 29c | 3 | 2-naphthyl | 0.06–0.18 | 0.15–0.76 | >10 |
| 29d | 2 | 2-naphthyl | 0.39–1.31 | 0.28–0.57 | 0.71–3.70 |
| Venlafaxine | – | – | 0.07–0.52 | 2.16–3.01 | – |
| GBR12909 | – | – | – | – | 0.02–0.08 |
Table 4.9.
Systematic SAR exploration of pyran-based TRIs
| Compound | DAT (Ki, nM) | NET (Ki, nM) | SERT (Ki, nM) |
|---|---|---|---|
| 30a | 13.3 ± 2.0 | 13.2 ± 3.5 | 46.7 ± 17.0 |
| 30b | 16.2 ± 1.5 | 3.23 ± 0.99 | 16.2 ± 1.5 |
| 30c | 29.8 ± 4.1 | 524 ± 132 | 259 ± 77 |
| 30d | 7.94 ± 0.66 | 14.6 (6) ± 2.9 | 367 ± 52 |
| 30e | 182 ± 50 | 27.9 ± 4.9 | 1540 ± 299 |
| 30f | 24.5 ± 1.2 | 3.92 (5) ± 0.71 | 339 ± 39 |
| 30g | 29.6 ± 6.9 | 2.14 ± 0.58 | 1.72 ± 0.48 |
| 30h | 56.3 ± 1.1 | 20.8 ± 4.6 | 13.6 ± 1.6 |
| 30i | 6.29 ± 1.52 | 6.74 ± 1.80 | 30.1 ± 5.3 |

The authors used extensive SAR studies to map out the structural requirements for interaction of the pyran derivatives with MATs to design superior TRIs (Table 4.9). The group had previously reported compounds 30a and 30b as orally active TRIs with high efficacy in the rat animal model of depression. Molecular docking studies have revealed that 30b makes strong, conserved H-bonds and hydrophobic interactions within the S1 site of all three transporters. Further modification of the aryl ring of N-benzyl moieties of 30a and 30b provided compounds with varying degrees of potency against the three transporters. Subtle changes in the aryl ring substitution, e.g., comparing 30c and 30d, and drastic changes in potency are observed within the two positional isomers, with DAT and SERT potencies only moderately affected. The para-fluoro substitutions on the biphenyl rings were important for TRI activity as their removal (30e versus 30f) resulted in tenfold reduction in NET potency, sevenfold reduction in DAT activity, and fivefold decrease in SERT inhibition. The importance of (S)-hydroxyl was evident when it was removed from 30g to get 30h resulting in tenfold reduction in NET and SERT activity, but only a moderate decrease in DAT potency. Inverting the stereochemistry of hydroxyl from axial position in 30a to equatorial position to get 30i slightly improved the TRI activity as compared to 30a.
Paudel et al. have designed, synthesized, and analyzed the SAR of another promising class of TRIs with the 1,5-disubstituted tetrazole scaffold [71, 73]. The prototype compound 31 (Fig. 4.7) in this series showed potent inhibitory activity against the three reuptake transporters (IC50; 158.7 nM for 5-HT; 99 nM for NE; 97.5 nM for DA).
Fig. 4.7.

Prototype of 1,5-disubstituted tetrazole series of TRIs
In conclusion, several SAR studies are currently in place to characterize novel TRI compounds that can be further utilized for developing better therapeutic agents against neurological disorders.
4.6. Conclusion
DAT, NET, and SERT represent important therapeutic targets for a number of CNS-related pathophysiological conditions. The discovery of crystal structures of transporter homologs bound with substrate and inhibitors and corresponding homology-based computational studies have substantially progressed our understanding of structure-function profiles of MATs and has aided in several medicinal chemistry-based drug discovery efforts. Over the years, a multitude of drug classes have been explored mainly via ligand-based drug design and discovery. These compounds target the MATs with unique and interesting behavioral profiles and have been explored as a promising starting point for developing therapeutics against various neurological disorders. Because the vastness of the literature available on SAR studies of MATs is substantial and beyond the scope of this review, we have attempted to summarize the most recent developments that have been made targeting each of the transporters with a particular focus on drug developments that have resulted in unique and superior behavioral effects.
References
- 1.Kristensen AS, Andersen J, Jorgensen TN, Sorensen L, Eriksen J, Loland CJ, Stromgaard K, Gether U. SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol Rev. 2011;63(3):585–640. 10.1124/pr.108.000869. [DOI] [PubMed] [Google Scholar]
- 2.Rudnick G, Kramer R, Blakely RD, Murphy DL, Verrey F. The SLC6 transporters: perspectives on structure, functions, regulation, and models for transporter dysfunction. Pflugers Arch. 2014;466(1):25–42. 10.1007/s00424-013-1410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aggarwal S, Mortensen OV. Overview of monoamine transporters. Curr Protoc Pharmacol. 2017;79:12.16.11–7. 10.1002/cpph.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Howell LL, Negus SS. Monoamine transporter inhibitors and substrates as treatments for stimulant abuse. Adv Pharmacol. 2014;69:129–76. 10.1016/B978-0-12-420118-7.00004-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singh SK, Yamashita A, Gouaux E. Antidepressant binding site in a bacterial homologue of neurotransmitter transporters. Nature. 2007;448(7156):952–6. 10.1038/nature06038. [DOI] [PubMed] [Google Scholar]
- 6.Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl--dependent neurotransmitter transporters. Nature. 2005;437(7056):215–23. 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 7.Zhou Z, Zhen J, Karpowich NK, Goetz RM, Law CJ, Reith ME, Wang DN. LeuT-desipramine structure reveals how antidepressants block neurotransmitter reuptake. Science. 2007;317(5843):1390–3. 10.1126/science.1147614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Penmatsa A, Wang KH, Gouaux E. X-ray structure of dopamine transporter elucidates anti-depressant mechanism. Nature. 2013;503(7474):85–90. 10.1038/nature12533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Penmatsa A, Wang KH, Gouaux E. X-ray structures of drosophila dopamine transporter in complex with nisoxetine and reboxetine. Nat Struct Mol Biol. 2015;22(6):506–8. 10.1038/nsmb.3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang KH, Penmatsa A, Gouaux E. Neurotransmitter and psychostimulant recognition by the dopamine transporter. Nature. 2015;521(7552):322–7. 10.1038/nature14431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coleman JA, Green EM, Gouaux E. X-ray structures and mechanism of the human serotonin transporter. Nature. 2016;532(7599):334–9. 10.1038/nature17629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coleman JA, Gouaux E. Structural basis for recognition of diverse antidepressants by the human serotonin transporter. Nat Struct Mol Biol. 2018;25(2):170–5. 10.1038/s41594-018-0026-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coleman JA, Navratna V, Antermite D, Yang D, Bull JA, Gouaux E. Chemical and structural investigation of the paroxetine-human serotonin transporter complex. elife. 2020;9:e56427. 10.7554/eLife.56427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coleman JA, Yang D, Zhao Z, Wen PC, Yoshioka C, Tajkhorshid E, Gouaux E. Serotonin transporter-ibogaine complexes illuminate mechanisms of inhibition and transport. Nature. 2019;569(7754):141–5. 10.1038/s41586-019-1135-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang H, Goehring A, Wang KH, Penmatsa A, Ressler R, Gouaux E. Structural basis for action by diverse antidepressants on biogenic amine transporters. Nature. 2013;503(7474):141–5. 10.1038/nature12648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xue W, Fu T, Zheng G, Tu G, Zhang Y, Yang F, Tao L, Yao L, Zhu F. Recent advances and challenges of the drugs acting on monoamine transporters. Curr Med Chem. 2020;27(23):3830–76. 10.2174/0929867325666181009123218. [DOI] [PubMed] [Google Scholar]
- 17.Ortore G, Orlandini E, Betti L, Giannaccini G, Mazzoni MR, Camodeca C, Nencetti S. Focus on human monoamine transporter selectivity. New human DAT and NET models, experimental validation, and SERT affinity exploration. ACS Chem Neurosci. 2020;11(20):3214–32. 10.1021/acschemneuro.0c00304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loland CJ. The use of LeuT as a model in elucidating binding sites for substrates and inhibitors in neurotransmitter transporters. Biochim Biophys Acta. 2015;1850(3):500–10. 10.1016/j.bbagen.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 19.Singh SK, Piscitelli CL, Yamashita A, Gouaux E. A competitive inhibitor traps LeuT in an open-to-out conformation. Science. 2008;322(5908):1655–61. 10.1126/science.1166777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andersen J, Ladefoged LK, Kristensen TN, Munro L, Grouleff J, Stuhr-Hansen N, Kristensen AS, Schiott B, Stromgaard K. Interrogating the molecular basis for substrate recognition in serotonin and dopamine transporters with high-affinity substrate-based bivalent ligands. ACS Chem Neurosci. 2016;7(10):1406–17. 10.1021/acschemneuro.6b00164. [DOI] [PubMed] [Google Scholar]
- 21.Plenge P, Shi L, Beuming T, Te J, Newman AH, Weinstein H, Gether U, Loland CJ. Steric hindrance mutagenesis in the conserved extracellular vestibule impedes allosteric binding of antidepressants to the serotonin transporter. J Biol Chem. 2012;287(47):39316–26. 10.1074/jbc.M112.371765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Plenge P, Yang D, Salomon K, Laursen L, Kalenderoglou IE, Newman AH, Gouaux E, Coleman JA, Loland CJ. The antidepressant drug vilazodone is an allosteric inhibitor of the serotonin transporter. Nat Commun. 2021;12(1):5063. 10.1038/s41467-021-25363-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kortagere S, Fontana AC, Rose DR, Mortensen OV. Identification of an allosteric modulator of the serotonin transporter with novel mechanism of action. Neuropharmacology. 2013;72:282–90. 10.1016/j.neuropharm.2013.04.026. [DOI] [PubMed] [Google Scholar]
- 24.Mortensen OV, Kortagere S. Designing modulators of monoamine transporters using virtual screening techniques. Front Pharmacol. 2015;6:223. 10.3389/fphar.2015.00223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aggarwal S, Liu X, Rice C, Menell P, Clark PJ, Paparoidamis N, Xiao YC, Salvino JM, Fontana ACK, Espana RA, Kortagere S, Mortensen OV. Identification of a novel allosteric modulator of the human dopamine transporter. ACS Chem Neurosci. 2019;10(8):3718–30. 10.1021/acschemneuro.9b00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kohnen-Johannsen KL, Kayser O. Tropane alkaloids: chemistry, pharmacology, biosynthesis and production. Molecules. 2019;24(4):796. 10.3390/molecules24040796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carroll FI, Gao Y, Abraham P, Lewin AH, Lew R, Patel A, Boja JW, Kuhar MJ. Probes for the cocaine receptor. Potentially irreversible ligands for the dopamine transporter. J Med Chem. 1992;35(10):1813–7. 10.1021/jm00088a017. [DOI] [PubMed] [Google Scholar]
- 28.Cline EJ, Scheffel U, Boja JW, Carroll FI, Katz JL, Kuhar MJ. Behavioral effects of novel cocaine analogs: a comparison with in vivo receptor binding potency. J Pharmacol Exp Ther. 1992;260(3):1174–9. [PubMed] [Google Scholar]
- 29.Kuhar MJ, Ritz MC, Boja JW. The dopamine hypothesis of the reinforcing properties of cocaine. Trends Neurosci. 1991;14(7):299–302. 10.1016/0166-2236(91)90141-G. [DOI] [PubMed] [Google Scholar]
- 30.Lewis D, Zhang Y, Prisinzano T, Dersch CM, Rothman RB, Jacobson AE, Rice KC. Further exploration of 1-[2-[bis-(4-fluorophenyl)methoxy]ethyl]piperazine (GBR 12909): role of N-aromatic, N-heteroaromatic, and 3-oxygenated N-phenylpropyl substituents on affinity for the dopamine and serotonin transporter. Bioorg Med Chem Lett. 2003;13(7):1385–9. 10.1016/s0960-894x(03)00108-2. [DOI] [PubMed] [Google Scholar]
- 31.Prisinzano T, Greiner E, Johnson EM 2nd, Dersch CM, Marcus J, Partilla JS, Rothman RB, Jacobson AE, Rice KC. Piperidine analogues of 1-[2-[bis(4-fluorophenyl)methoxy]ethyl]-4-(3-phenylpropyl)piperazine (GBR 12909): high affinity ligands for the dopamine transporter. J Med Chem. 2002;45(19):4371–4. 10.1021/jm020264r. [DOI] [PubMed] [Google Scholar]
- 32.Newman AH, Agoston GE. Novel benztropine [3a-(diphenylmethoxy)tropane] analogs as probes for the dopamine transporter. Curr Med Chem. 1998;5(4):305–19. [PubMed] [Google Scholar]
- 33.Newman AH, Kulkarni S. Probes for the dopamine transporter: new leads toward a cocaineabuse therapeutic – A focus on analogues of benztropine and rimcazole. Med Res Rev. 2002;22(5):429–64. 10.1002/med.10014. [DOI] [PubMed] [Google Scholar]
- 34.Dassanayake AF, Canales JJ. Replacement treatment during extinction training with the atypical dopamine uptake inhibitor, JHW-007, reduces relapse to methamphetamine seeking. Neurosci Lett. 2018;671:88–92. 10.1016/j.neulet.2018.02.025. [DOI] [PubMed] [Google Scholar]
- 35.Ferragud A, Velazquez-Sanchez C, Canales JJ. Modulation of methamphetamine's locomotor stimulation and self-administration by JHW 007, an atypical dopamine reuptake blocker. Eur J Pharmacol. 2014;731:73–9. 10.1016/j.ejphar.2014.03.015. [DOI] [PubMed] [Google Scholar]
- 36.Velazquez-Sanchez C, Ferragud A, Murga J, Carda M, Canales JJ. The high affinity dopamine uptake inhibitor, JHW 007, blocks cocaine-induced reward, locomotor stimulation and sensitization. Eur Neuropsychopharmacol. 2010;20(7):501–8. 10.1016/j.euroneuro.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 37.Loland CJ, Mereu M, Okunola OM, Cao J, Prisinzano TE, Mazier S, Kopajtic T, Shi L, Katz JL, Tanda G, Newman AH. R-modafinil (armodafinil): a unique dopamine uptake inhibitor and potential medication for psychostimulant abuse. Biol Psychiatry. 2012;72(5):405–13. 10.1016/j.biopsych.2012.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Myrick H, Malcolm R, Taylor B, LaRowe S. Modafinil: preclinical, clinical, and post-marketing surveillance – a review of abuse liability issues. Ann Clin Psychiatry. 2004;16(2):101–9. 10.1080/10401230490453743. [DOI] [PubMed] [Google Scholar]
- 39.Vosburg SK, Hart CL, Haney M, Rubin E, Foltin RW. Modafinil does not serve as a reinforcer in cocaine abusers. Drug Alcohol Depend. 2010;106(2–3):233–6. 10.1016/j.drugalcdep.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hersey M, Bacon AK, Bailey LG, Coggiano MA, Newman AH, Leggio L, Tanda G. Psychostimulant use disorder, an unmet therapeutic goal: can modafinil narrow the gap? Front Neurosci. 2021;15:656475. 10.3389/fnins.2021.656475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cao J, Prisinzano TE, Okunola OM, Kopajtic T, Shook M, Katz JL, Newman AH. Structure-activity relationships at the monoamine transporters for a novel series of modafinil (2-[(diphenylmethyl)sulfinyl]acetamide) analogues. ACS Med Chem Lett. 2010;2(1):48–52. 10.1021/ml1002025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cao J, Slack RD, Bakare OM, Burzynski C, Rais R, Slusher BS, Kopajtic T, Bonifazi A, Ellenberger MP, Yano H, He Y, Bi GH, Xi ZX, Loland CJ, Newman AH. Novel and high affinity 2-[(Diphenylmethyl)sulfinyl]acetamide (modafinil) analogues as atypical dopamine transporter inhibitors. J Med Chem. 2016;59(23):10676–91. 10.1021/acs.jmedchem.6b01373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okunola-Bakare OM, Cao J, Kopajtic T, Katz JL, Loland CJ, Shi L, Newman AH. Elucidation of structural elements for selectivity across monoamine transporters: novel 2-[(diphenylmethyl)sulfinyl]acetamide (modafinil) analogues. J Med Chem. 2014;57(3):1000–13. 10.1021/jm401754x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tanda G, Hersey M, Hempel B, Xi ZX, Newman AH. Modafinil and its structural analogs as atypical dopamine uptake inhibitors and potential medications for psychostimulant use disorder. Curr Opin Pharmacol. 2021;56:13–21. 10.1016/j.coph.2020.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Keighron JD, Giancola JB, Shaffer RJ, DeMarco EM, Coggiano MA, Slack RD, Newman AH, Tanda G. Distinct effects of (R)-modafinil and its (R)- and (S)-fluoro-analogs on mesolimbic extracellular dopamine assessed by voltammetry and microdialysis in rats. Eur J Neurosci. 2019;50(3):2045–53. 10.1111/ejn.14256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keighron JD, Quarterman JC, Cao J, DeMarco EM, Coggiano MA, Gleaves A, Slack RD, Zanettini C, Newman AH, Tanda G. Effects of (R)-modafinil and modafinil analogues on dopamine dynamics assessed by voltammetry and microdialysis in the mouse nucleus accumbens shell. ACS Chem Neurosci. 2019;10(4):2012–21. 10.1021/acschemneuro.8b00340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang HY, Bi GH, Yang HJ, He Y, Xue G, Cao J, Tanda G, Gardner EL, Newman AH, Xi ZX. The novel modafinil analog, JJC8-016, as a potential cocaine abuse pharmacotherapeutic. Neuropsychopharmacology. 2017;42(9):1871–83. 10.1038/npp.2017.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tunstall BJ, Ho CP, Cao J, Vendruscolo JCM, Schmeichel BE, Slack RD, Tanda G, Gadiano AJ, Rais R, Slusher BS, Koob GF, Newman AH, Vendruscolo LF. Atypical dopamine transporter inhibitors attenuate compulsive-like methamphetamine self-administration in rats. Neuropharmacology. 2018;131:96–103. 10.1016/j.neuropharm.2017.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Newman AH, Cao J, Keighron JD, Jordan CJ, Bi GH, Liang Y, Abramyan AM, Avelar AJ, Tschumi CW, Beckstead MJ, Shi L, Tanda G, Xi ZX. Translating the atypical dopamine uptake inhibitor hypothesis toward therapeutics for treatment of psychostimulant use disorders. Neuropsychopharmacology. 2019;44(8):1435–44. 10.1038/s41386-019-0366-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Slack RD, Ku TC, Cao J, Giancola JB, Bonifazi A, Loland CJ, Gadiano A, Lam J, Rais R, Slusher BS, Coggiano M, Tanda G, Newman AH. Structure-activity relationships for a series of (bis(4-fluorophenyl)methyl)sulfinyl alkyl alicyclic amines at the dopamine transporter: functionalizing the terminal nitrogen affects affinity, selectivity, and metabolic stability. J Med Chem. 2020;63(5):2343–57. 10.1021/acs.jmedchem.9b01188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Giancola JB, Bonifazi A, Cao J, Ku T, Haraczy AJ, Lam J, Rais R, Coggiano MA, Tanda G, Newman AH. Structure-activity relationships for a series of (bis(4-fluorophenyl)methyl) sulfinylethyl-aminopiperidines and -piperidine amines at the dopamine transporter: bio-isosteric replacement of the piperazine improves metabolic stability. Eur J Med Chem. 2020;208:112674. 10.1016/j.ejmech.2020.112674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hiranita T, Hong WC, Kopajtic T, Katz JL. Sigma receptor effects of N-substituted benztropine analogs: implications for antagonism of cocaine self-administration. J Pharmacol Exp Ther. 2017;362(1):2–13. 10.1124/jpet.117.241109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kalaba P, Ilic M, Aher NY, Dragacevic V, Wieder M, Zehl M, Wackerlig J, Beyl S, Sartori SB, Ebner K, Roller A, Lukic N, Beryozkina T, Gonzalez ERP, Neill P, Khan JA, Bakulev V, Leban JJ, Hering S, Pifl C, Singewald N, Lubec J, Urban E, Sitte HH, Langer T, Lubec G. Structure-activity relationships of novel thiazole-based modafinil analogues acting at monoamine transporters. J Med Chem. 2020;63(1):391–417. 10.1021/acs.jmedchem.9b01938. [DOI] [PubMed] [Google Scholar]
- 54.Schmitt KC, Rothman RB, Reith ME. Nonclassical pharmacology of the dopamine transporter: atypical inhibitors, allosteric modulators, and partial substrates. J Pharmacol Exp Ther. 2013;346(1):2–10. 10.1124/jpet.111.191056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reith ME, Blough BE, Hong WC, Jones KT, Schmitt KC, Baumann MH, Partilla JS, Rothman RB, Katz JL. Behavioral, biological, and chemical perspectives on atypical agents targeting the dopamine transporter. Drug Alcohol Depend. 2015;147:1–19. 10.1016/j.drugalcdep.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rothman RB, Ananthan S, Partilla JS, Saini SK, Moukha-Chafiq O, Pathak V, Baumann MH. Studies of the biogenic amine transporters 15. Identification of novel allosteric dopamine transporter ligands with nanomolar potency. J Pharmacol Exp Ther. 2015;353(3):529–38. 10.1124/jpet.114.222299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Refai O, Aggarwal S, Cheng MH, Gichi Z, Salvino JM, Bahar I, Blakely RD, Mortensen OV. Allosteric Modulator KM822 Attenuates Behavioral Actions of Amphetamine in Caenorhabditis elegans through interactions with the dopamine transporter DAT-1. Mol Pharmacol. 2022;101(3):123–31. 10.1124/molpharm.121.000400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Little A. Treatment-resistant depression. Am Fam Physician. 2009;80(2):167–72. [PubMed] [Google Scholar]
- 59.Bigler AJ, Bøgesø K, et al. Quantitative structure-activity relationship in a series of selective 5-HT uptake inhibitors. Eur J Med Chem. 1977;12:289–95. [Google Scholar]
- 60.Eildal JNN, Andersen J, Kristensen AS, Jørgensen AM, Bang-Andersen B, Jørgensen M, Strømgaard K. From the selective serotonin transporter inhibitor citalopram to the selective norepinephrine transporter inhibitor talopram: synthesis and structure–activity relationship studies. J Med Chem. 2008;51(10):3045–8. 10.1021/jm701602g. [DOI] [PubMed] [Google Scholar]
- 61.Zhang P, Cyriac G, Kopajtic T, Zhao Y, Javitch JA, Katz JL, Newman AH. Structure–activity relationships for a novel series of citalopram (1-(3-(dimethylamino)propyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-carbonitrile) analogues at monoamine transporters. J Med Chem. 2010;53(16):6112–21. 10.1021/jm1005034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Andersen J, Olsen L, Hansen KB, Taboureau O, Jørgensen FS, Jørgensen AM, Bang-Andersen B, Egebjerg J, Strømgaard K, Kristensen AS. Mutational mapping and modeling of the binding site for (S)-citalopram in the human serotonin transporter. J Biol Chem. 2010;285(3):2051–63. 10.1074/jbc.M109.072587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Andersen J, Stuhr-Hansen N, Zachariassen L, Toubro S, Hansen SM, Eildal JN, Bond AD, Bogeso KP, Bang-Andersen B, Kristensen AS, Stromgaard K. Molecular determinants for selective recognition of antidepressants in the human serotonin and norepinephrine transporters. Proc Natl Acad Sci U S A. 2011;108(29):12137–42. 10.1073/pnas.1103060108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Banala AK, Zhang P, Plenge P, Cyriac G, Kopajtic T, Katz JL, Loland CJ, Newman AH. Design and synthesis of 1-(3-(dimethylamino)propyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-carboni trile (citalopram) analogues as novel probes for the serotonin transporter S1 and S2 binding sites. J Med Chem. 2013;56(23):9709–24. 10.1021/jm4014136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Coleman JA, Green EM, Gouaux E. Thermostabilization, expression, purification, and crystallization of the human serotonin transporter bound to S-citalopram. J Vis Exp. 2016;(117):54792. 10.3791/54792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Garnock-Jones KP, Keating GM. Atomoxetine: a review of its use in attention-deficit hyperactivity disorder in children and adolescents. Pediatr Drugs. 2009;11(3):203–26. [DOI] [PubMed] [Google Scholar]
- 67.Lane RM. Antidepressant drug development: focus on triple monoamine reuptake inhibition. J Psychopharmacol. 2015;29(5):526–44. 10.1177/0269881114553252. [DOI] [PubMed] [Google Scholar]
- 68.Larsen MA, Plenge P, Andersen J, Eildal JN, Kristensen AS, Bøgesø KP, Gether U, Strømgaard K, Bang-Andersen B, Loland CJ. Structure-activity relationship studies of citalopram derivatives: examining substituents conferring selectivity for the allosteric site in the 5-HT transporter. Br J Pharmacol. 2016;173(5):925–36. 10.1111/bph.13411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li N, Li C, Han R, Wang Y, Yang M, Wang H, Tian J. LPM580098, a novel triple reuptake inhibitor of serotonin, noradrenaline, and dopamine, attenuates neuropathic pain. Front Pharmacol. 2019;10:53. 10.3389/fphar.2019.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Patel K, Allen S, Haque MN, Angelescu I, Baumeister D, Tracy DK. Bupropion: a systematic review and meta-analysis of effectiveness as an antidepressant. Ther Adv Psychopharmacol. 2016;6(2):99–144. 10.1177/2045125316629071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Paudel S, Acharya S, Yoon G, Kim KM, Cheon SH. Design, synthesis and in vitro activity of 1,4-disubstituted piperazines and piperidines as triple reuptake inhibitors. Bioorg Med Chem. 2017;25(7):2266–76. 10.1016/j.bmc.2017.02.051. [DOI] [PubMed] [Google Scholar]
- 72.Paudel S, Kim E, Zhu A, Acharya S, Min X, Cheon SH, Kim KM. Structural requirements for modulating 4-benzylpiperidine carboxamides from serotonin/norepinephrine reuptake inhibitors to triple reuptake inhibitors. Biomol Ther (Seoul). 2021;29(4):392–8. 10.4062/biomolther.2020.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Paudel S, Min X, Acharya S, Khadka DB, Yoon G, Kim KM, Cheon SH. Triple reuptake inhibitors: design, synthesis and structure-activity relationship of benzylpiperidine-tetrazoles. Bioorg Med Chem. 2017;25(20):5278–89. 10.1016/j.bmc.2017.07.046. [DOI] [PubMed] [Google Scholar]
- 74.Plenge P, Abramyan AM, Sørensen G, Mørk A, Weikop P, Gether U, Bang-Andersen B, Shi L, Loland CJ. The mechanism of a high-affinity allosteric inhibitor of the serotonin transporter. Nat Commun. 2020;11(1):1491. 10.1038/s41467-020-15292-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Santra S, Kortagere S, Vedachalam S, Gogoi S, Antonio T, Reith MEA, Dutta AK. Novel potent dopamine–norepinephrine and triple reuptake uptake inhibitors based on asymmetric pyran template and their molecular interactions with monoamine transporters. ACS Chem Neurosci. 2021;12(8):1406–18. 10.1021/acschemneuro.1c00078. [DOI] [PubMed] [Google Scholar]
- 76.Sorensen L, Andersen J, Thomsen M, Hansen SM, Zhao X, Sandelin A, Stromgaard K, Kristensen AS. Interaction of antidepressants with the serotonin and norepinephrine transporters: mutational studies of the S1 substrate binding pocket. J Biol Chem. 2012;287(52):43694–707. 10.1074/jbc.M112.342212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Subbaiah MAM. Triple reuptake inhibitors as potential therapeutics for depression and other disorders: design paradigm and developmental challenges. J Med Chem. 2018;61(6):2133–65. 10.1021/acs.jmedchem.6b01827. [DOI] [PubMed] [Google Scholar]
- 78.Tu G, Fu T, Yang F, Yang J, Zhang Z, Yao X, Xue W, Zhu F. Understanding the polypharmacological profiles of triple reuptake inhibitors by molecular simulation. ACS Chem Neurosci. 2021;12(11):2013–26. 10.1021/acschemneuro.1c00127. [DOI] [PubMed] [Google Scholar]
- 79.Zhang M, Jovic F, Vickers T, Dyck B, Tamiya J, Grey J, Tran JA, Fleck BA, Pick R, Foster AC, Chen C. Studies on the structure–activity relationship of bicifadine analogs as monoamine transporter inhibitors. Bioorg Med Chem Lett. 2008;18(13):3682–6. 10.1016/j.bmcl.2008.05.077. [DOI] [PubMed] [Google Scholar]
- 80.Zhang Y, Zheng G, Fu T, Hong J, Li F, Yao X, Xue W, Zhu F. The binding mode of vilazodone in the human serotonin transporter elucidated by ligand docking and molecular dynamics simulations. Phys Chem Chem Phys. 2020;22(9):5132–44. 10.1039/c9cp05764a. [DOI] [PubMed] [Google Scholar]
- 81.Zhu H, Wang W, Sha C, Guo W, Li C, Zhao F, Wang H, Jiang W, Tian J. Pharmacological characterization of Toludesvenlafaxine as a triple reuptake inhibitor. Front Pharmacol. 2021;12:741794–4. 10.3389/fphar.2021.741794. [DOI] [PMC free article] [PubMed] [Google Scholar]