

Abstract
The increased production of derivatives of molecular oxygen and nitrogen in the form of reactive oxygen species (ROS) and reactive nitrogen species (RNS) lead to molecular damage called oxidative stress. Under normal physiological conditions, the ROS generation is tightly regulated in different cells and cellular compartments. Any disturbance in the balance between the cellular generation of ROS and antioxidant balance leads to oxidative stress. In this article, we discuss the sources of ROS (endogenous and exogenous) and antioxidant mechanisms. We also focus on the pathophysiological significance of oxidative stress in various cell types of the liver. Oxidative stress is implicated in the development and progression of various liver diseases. We narrate the master regulators of ROS-mediated signaling and their contribution to liver diseases. Non-alcoholic fatty liver diseases (NAFLD) are influenced by a “multiple parallel-hit model” in which oxidative stress plays a central role. We highlight the recent findings on the role of oxidative stress in the spectrum of NAFLD, including fibrosis and liver cancer. Finally, we provide a brief overview of oxidative stress biomarkers and their therapeutic applications in various liver-related disorders. Overall, the article sheds light on the significance of oxidative stress in the pathophysiology of the liver.
The Concept and Definition
In 1936, Hans Selye introduced the “stress concept” in understanding the response of animals to acute nonspecific nocuous agents such as cold, surgical injury, and drugs (349). He defined stress as a nonspecific response of the body to any demand (348). In the later 1970s, Paniker et al. showed that exposure of red blood cells to hydrogen peroxide (H2O2) induces “oxidative stress,” which is associated with glutathione reductase activity (299). In the 1980s, the term oxidative stress was introduced in redox and medicine biology in an introductory chapter (366). The review by Helmut Sies “Biochemistry of Oxidative Stress” described the biology of oxidative stress in biological systems, including the pro-oxidants, antioxidants, cause and effect, defense and repair mechanisms, and also control of oxidative stress. Oxidative stress is defined as the “state of condition wherein the cellular pro-oxidant and antioxidant balance is altered in favor of a pro-oxidant state.” In other words, oxidative stress occurs in response to increased production of reactive oxygen species and decreased production of antioxidants (135, 364, 365).
The concept of oxidative stress is associated with free radicals in biology and medicine (317). Free radicals are the molecules or atoms characterized by highly reactive unpaired electrons or atoms (39, 313, 412). All the free radicals are not equally toxic. The degree of reactivity or chemical nature of free radicals and their reactants depends on the extent of damage to the biological system. The reactivity of free radicals estimated by one-electron reduction potentially reflects the molecule’s affinity compared with hydrogen. Thus, most free radical products are assumed to be oxygen-based hydroxyl radicals and nitrogen-based peroxynitrite anion (241, 317).
Oxidative Stress-classification
In modern biology and medicine, several reactive species were identified. Among them, the most well-studied reactive species in the mammalian system include reactive oxygen species (ROS), reactive nitrogen species (RNS), reactive carbonyl species (RCS), and reactive sulfur species (RSS). Other biologically critical reactive species include selenium, chlorine, and bromine species.
Reactive oxygen species (ROS)
ROS comprise radical and nonradical oxygen species formed by the partial reduction of oxygen. ROS are majorly represented by superoxide anions (O2•−), hydroxyl radicals (HO•), and hydrogen peroxide (H2O2). Superoxide anions and hydroxyl radicals exist in a free-radical form characterized by highly unstable unpaired electrons (e.g., O2•− and HO•). While H2O2 is a nonradical, chemically stable, freely diffusible, and long-lived molecule (282, 365). ROS is formed through endogenous mechanisms such as mitochondrial electron transport chain (ETC), flavin-dependent oxidation, and microsomal oxidation (188, 280). ROS generation also occurs in response to exogenous pathways such as xenobiotic metabolism (e.g., antimycin and adriamycin) (285, 367). Furthermore, ROS could also be generated through exposure to nutrients, pollutants, and physical factors such as ultraviolet light, ultrasound, and X-rays (367). Auto-oxidation products such as flavins and hemoglobin also could lead to the formation of ROS (326, 347).
In the biological system, H2O2 is considered as the major ROS that plays a critical role in redox regulation (129, 324, 379). The production of H2O2 takes place in response to metabolic cues, cytokine, and chemokines (343). Under physiological levels, the redox signaling of H2O2 is mediated through oxidation of sulfur proteins, reversible methionine oxidation, selenoprotein, oxidation of metal centers, and lipids (408). However, the generation of higher amounts of H2O2 results in unspecific oxidation of proteins leading to cell growth arrest and death (128). The major endogenous enzymatic sources for H2O2 are NADPH oxidases and the mitochondrial ETC. Excess H2O2 is removed by peroxiredoxins and glutathione peroxidases, apart from catalases (141, 249). Besides, H2O2 levels are maintained by the exchange between organelle systems such as ER, mitochondria, and peroxisomes (453).
Reactive nitrogen species (RNS)
RNS is a family of nitrogen-associated molecules produced when nitrogen interacts with oxidants and reductants like superoxide and hydrogen peroxide, either endogenously or exogenously. The most common RNS includes nitric oxide (NO•) and peroxynitrite (ONOO−). Like ROS, RNS also exists in a free radical form (nitric oxide, nitrogen dioxide, and nitrite) or nonradical form (nitrous acid, peroxynitrite, and nitrosyl anions). The half-life, solubility, and biological reactivity of RNS depend on their precursors (1, 82, 297, 320).
Nitric oxide is the foremost and critical RNS known to play a significant role in redox biology. Nitric oxide is an easily diffusible free radical with a short half-life and is central to the formation of other RNS (320, 328). The generation of RNS begins with the synthesis of nitric oxide by the enzyme nitric oxide synthase (NOS), which exists in three forms; includes neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible NOS (iNOS). The eNOS and nNOS are constitutively expressed by the endothelial cells and neuronal cells, respectively, while iNOS expression is regulated at the transcriptional level in various cells (104, 105). In addition to nitric oxide, peroxynitrite is considered a major cellular nitrating agent, which derives from the reaction of NO with superoxide anion. Peroxynitrite is generated by the plasma membrane NADPH oxidases and mitochondrial respiratory chain. Although peroxynitrite is short-lived, it is a potent inducer of cell death (319, 320, 388).
Other species
In addition to ROS and RNS, several other highly reactive species regulate redox signaling. Among them, reactive sulfur species (RSS) comprises sulfur-containing reactive biomolecules that range from small molecules to proteins. The common RSS includes hydrogen sulfide (H2S), protein thiols, low-molecular-mass compounds such as glutathione, trypanothione, sulfenic acids (RSOH), and nitrosothiols (RSNO) (76, 122). Most of the RSS formed as a by-product of major thiols or oxidation of sulfite or sulfate molecules. Importantly, the per/polysulfides encompassing cysteine persulfide and polysulfide are the most abundant RSS identified in the mammalian and other biological systems (182, 203). Besides, several studies have shown that H2S is the most common short-lived RSS that has prolonged biological effects in the mammalian system (329). Recent studies have demonstrated a role for RSS in various diseases, including atherosclerosis, fatty liver, inflammation, and viral hepatitis (65, 189, 217). Further, a recent work by Zhang et al. shows that cellular polysulfides inhibit lipopolysaccharide-induced proinflammatory responses in the macrophages through toll-like receptor 4 (TLR4) signaling (462).
RCS are the biological compounds with one or more carbonyl groups that include metabolically generated aldehydes and electronically excited carbonyl molecules. Some of the RCS include acrolein, crotonaldehyde, glyoxal, acetone, and formaldehyde (350). RCSs are majorly generated through nonenzymatic processes such as lipid peroxidation, amino acid oxidation, and glycation (253, 369). Although RCSs exert beneficial effects, their overproduction, known as carbonyl stress, is the major contributing factor for aging, metabolic diseases, and other neurodegenerative diseases (95, 144). Mechanistically, RCS induce biological damage by forming α- and β-unsaturated aldehydes, dialdehydes, and keto-aldehydes, which are more reactive than saturated forms (5, 254). In aging, RCS accumulates in various tissues and peripheral blood in the form of advanced glycation end products (AGEs) (304, 400).
Physiological Significance of ROS
The cells generate ROS during oxidative metabolism by various chemical, environmental, and dietary cues, as described above (12). Maintaining a delicate balance between the oxidant-antioxidant mechanisms is modulated by its production, location, and inactivation. At physiological levels, ROS, considered as “redox biology,” plays a significant function in regulating signal transduction, gene expression, and cell proliferation (268, 292, 458). Recent studies have emphasized their role in blood pressure control (410, 418) and embryonic development (222, 437). Thus, ROS monitors cell fate indicating the existence of a “ROS rheostat” in the cells.
Under physiological conditions, mitochondrial reactive oxygen species (mROS) regulates biological functions such as autophagy, immunity, differentiation, longevity, and adaption to hypoxia (79, 351). As a defense mechanism, mitochondria are equipped with antioxidant enzyme machinery to minimize the risk of aberrant increases in ROS. For example, superoxide dismutase (SOD) families of enzymes such as SOD1, 2, and 3, peroxiredoxins, glutathione peroxidase (GPX), and catalases are localized in mitochondrial intermembrane space or matrix (172, 175, 421). Peroxiredoxins turn ROS signaling off (303), while GPXs buffers the excess ROS and bring them to normal levels (250).
The physiological significance of ROS is well-studied in mitophagy, a quality control process wherein damaged mitochondria are continuously removed from the cells (185). Impairment of mitophagy results in the accumulation of damaged mitochondria leading to a further increase in mROS/total ROS. Several studies have shown that mROS is required to induce mitophagy (92, 208). For instance, under starvation mROS drives the formation of autophagosome through the activation of PI3K pathway (342). Besides, mitophagy preserves mitochondrial bioenergetics, attenuates cell injury and progression of liver diseases by reducing oxidative stress (33, 243, 432). The other important physiological significance of mROS was demonstrated under hypoxic conditions. This concept has arrived from the data that the cells depleted of mitochondrial DNA do not stabilize HIFs under hypoxia (55). Thus, when the cells encounter low oxygen levels, they undergo an adaptive mechanism triggered by enhanced mROS generation. Similarly, several studies have shown that mROS regulate the immune system (106, 325), aging (211, 266, 332), and stem cell differentiation (161, 295). Thus, the level of ROS serves as an alarm to report the changing environment in the cell.
Liver and Oxidative Stress
The liver is the second-largest and key metabolic organ in the mammalian system (271). The liver consists of parenchymal (hepatocytes) and nonparenchymal cells. Hepatocytes are the major structural and functional units of the liver. The nonparenchymal cells include Kupffer cells, sinusoidal endothelial cells, stellate cells, periportal fibroblasts, and hepatic dendritic cells (404). The liver is the main site for synthesis, secretion, degradation, and coupled inter-conversion and biotransformation of amino acids, carbohydrates, and lipids. It is also involved in the storage and transport of micronutrients such as vitamins and minerals. In addition, the liver is the major site of detoxification of drugs, alcohol, and hormone metabolism and to some extent, filtration of blood. Collectively, it performs five essential functions, namely, (i) metabolism, (ii) storage, (iii) excretion and secretion, (iv) detoxification, and (v) blood filtration. These activities account for approximately 25% of the total metabolic rate. The liver is considered as the metabolic hub as it connects various organs in coordinating whole-body homeostasis of bio-molecules, hormones, macro-, and micronutrients (201, 314, 404).
Given its role in xenobiotic metabolism, the liver generates several oxygen (ROS) and nitrogen (RNS)-based free radical species. However, their persistent production results in oxidative stress leading to the dysregulation of liver homeostasis (69, 256). The initiation of ROS in the liver takes place due to increased mitochondrial respiration. mROS generated in the hepatocytes is responsible for the oxidative damage of lipids, proteins, and DNA (265, 280). Recent studies have reported that endoplasmic reticulum and peroxisomes also contribute to hepatic ROS levels (38, 207). Besides mitochondrial respiration, hepatic xanthine oxidase, which converts hypoxanthine to xanthine and xanthine to uric acid, generates superoxide anion and hydrogen peroxide (26).
Hepatocytes
Hepatocytes, which occupy nearly 80% of the liver volume, are the first cells that respond to the dietary contents after absorption and are prone to injuries from ingested toxins, alcohol, and other drugs. Hepatocytes prevent liver damage by storing free fatty acids in the form of lipid droplets. (14). Lipotoxicity impairs mitochondrial function and changes the redox state. Over time, dysfunctional mitochondria reduce ATP production resulting in increased ROS production and hepatocyte death (246). Thus, ROS released from the injured hepatocytes acts as a major stimulus for the progression of various liver diseases via activation of immune and hepatic stellate cells.
ROS generated in the hepatocytes also influences the function of the neighboring cells through various mechanisms. For example, ROS activates TGF-β and fibromodulin in the hepatocytes, which induces the migration and proliferation of HSCs leading to liver fibrosis (37). Apoptosis or necrosis of the hepatocytes also increases mROS releases and contributes to fibrogenesis (48, 416). Furthermore, increased hepatocyte ROS destroys the critical function of cellular macromolecules such as DNA, proteins, and lipids. For example, ROS oxidizes the protein kinase and phosphatases that regulate major signaling pathways such as mitogen-activated protein kinases (MAPKs) (153, 370). Hepatocytes also promote inflammation and oxidative stress by releasing hepatokines such as fetuin A, fetuin B, and IL-18. Further, hepatocytes store a large amount of iron and Fe-S containing mitochondria proteins, which are highly reactive toward O2− (389, 414). Thus, the excessive production of ROS generated from the hepatocytes plays a significant role in the development and progression of liver diseases (Figure 1).
Figure 1.
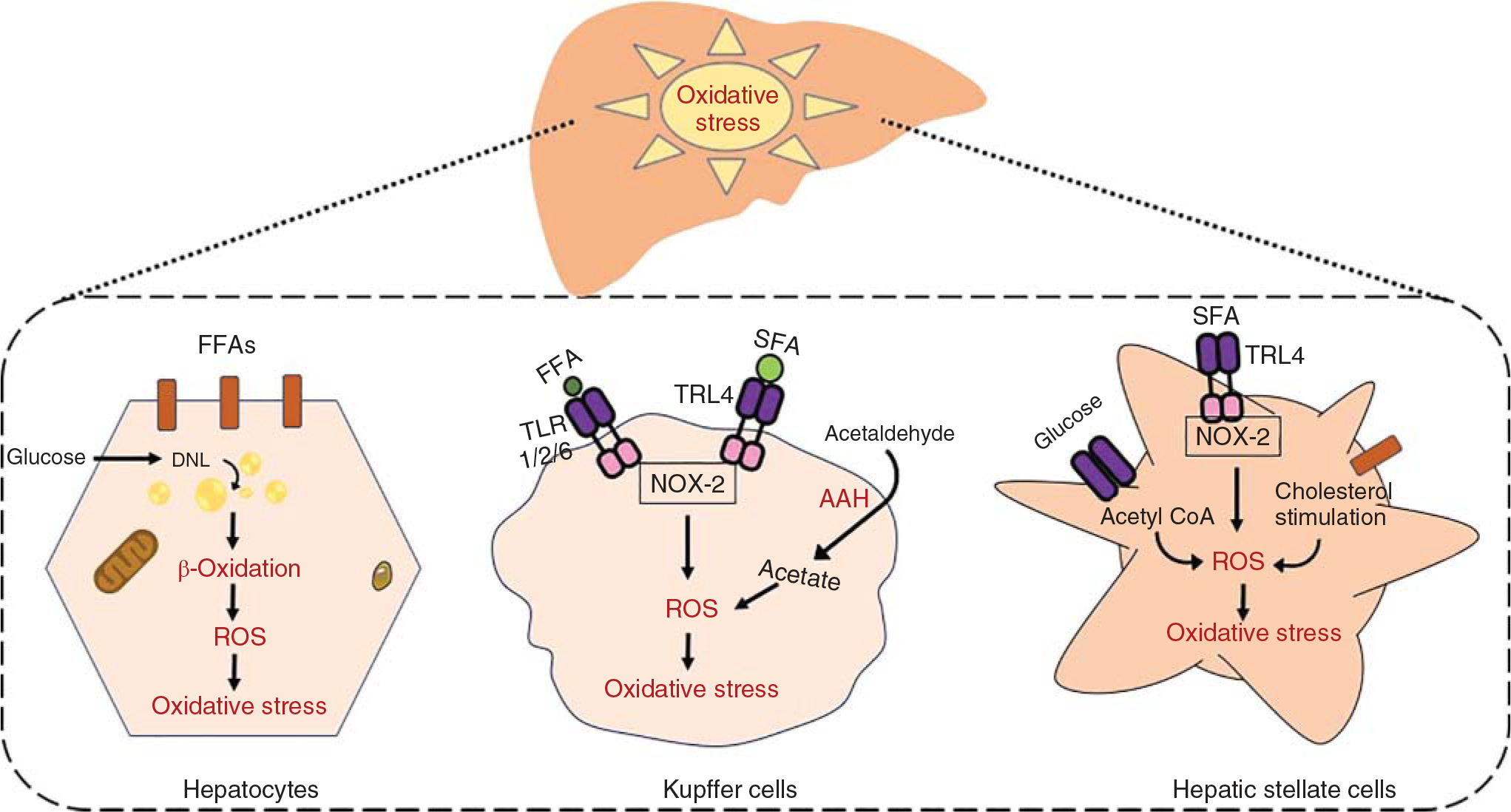
Mechanisms of generation of ROS-mediated oxidative stress in the liver. Increased lipid accumulation in hepatocytes via fatty acid uptake and de novo lipogenesis using several lipogenic enzymes augments mitochondrial β-oxidation leading to incessant generation of ROS. Hepatic nonparenchymal cells are also involved in ROS generation by activating the toll-like receptors (TLRs) on the Kupffer cells and HSCs. In Kupffer cells, TLR4 in response to SFA and TLR1/2/6 for FFA activation triggers NOX-2-mediated ROS generation, resulting in oxidative stress. In HSCs, accumulation of cholesterol and acetyl-CoA generates huge amount of ROS through glucose metabolism.
Kupffer cells
In the liver, Kupffer cells are the most abundant resident macrophages that eliminate invading microbes and their products. Kupffer cells reside in the hepatic sinusoidal cells, and their numbers are tightly regulated by various factors (91, 163). Although it seems that resident macrophages are essential to fight invading microbes, their activation plays a significant role in the initiation and progression of acute liver injuries and fatty liver disease (269). Recent evidence demonstrates that the recruitment and activation of Kupffer cells also occur in response to dietary and environmental factors (102, 391). The recruited macrophages initiate fatty liver and fibrosis in NAFLD (152, 406).
Under pathological conditions, Kupffer cells are activated by various inflammatory cells, chemokines, and other growth-modulating factors (93, 215). Also, the accumulation of ROS activates Kupffer cells (110). Further, activated Kupffer cells could also contribute to ROS generation under various acute and chronic liver injuries. In support of this notion, the treatment of rats and mice with carbon tetrachloride (CCl4) results in the activation of Kupffer cells, thereby increasing the reactive oxygen intermediates (9, 176, 327). In addition to resident macrophages, recruited macrophages also produce ROS through various mechanisms, including mitochondrial damage, ER stress, and increased NADPH oxidases (NOXs) (83, 115, 132). The iNOS expressed by the Kupffer cells generate RNS and its redox derivatives. NOX-derived ROS involves the production of proinflammatory cells and other chemokines in response to LPS and other fattyacids (Figure1) (142, 181, 228, 311). Recent studies show that the phenotypic switch in the resident macrophages from classically activated inflammatory macrophages (M1) to alternatively activated anti-inflammatory macrophages (M2) is strongly associated with increased generation of ROS in the liver (169, 290).
Hepatic stellate cells (HSCs)
HSCs located in the space of Disse occupy nearly 8% to 12% of the total liver cell population. HSCs play a significant role in maintaining hepatic architecture and blood flow by regulating the synthesis/degradation of the extracellular matrix. HSCs also exhibit immune function by secreting various cytokines, chemokines, and growth factors. HSCs store retinoids including vitamin A and its isoforms. Because of these characteristics, HSCs regulate the functions of hepatocytes and other liver cells through a paracrine and juxtracrine mechanism (Figure 1) (340, 353, 354). ROS generated by the Kupffer cells and leukocytes could activate HSCs. A positive correlation between ROS and HSCs activation was demonstrated using various models such Fas the CCl4-induced and diet-induced liver injury in rodent animals. Although CCL4 does not affect the HSCs directly, substances released from the injured hepatocytes, including ROS activates HSCs (27, 272). Activation of HSCs leads to excessive synthesis and deposition of extracellular matrix proteins resulting in liver fibrosis (457). Although the exact mechanisms of HSC activation are unclear, several studies demonstrated that inflammatory cytokines and chemokines drive the activation and proliferation of HSCs (6, 216).
Endothelial cells
The liver sinusoidal endothelial cells (LSEC) are highly specialized and distinctive micro-vascular cell types that play a key role in maintaining the liver microenvironment. LSECs are considered as the first defense barrier and contribute to metabolite transport, inflammation, and angiogenesis by interacting with various neighboring cell types in the liver (42). LSECs also govern the regenerative process in response to liver injury (84, 309, 417). Thus, the dysfunction of LSECs leads to the initiation and progression of various liver-related diseases, including liver fibrosis and cirrhosis (137, 257). Free fatty acids, triglycerides, ethanol, and HCV core protein could trigger LSEC dysfunction (18, 140, 263). Inflammatory cells and activated HSCs also drive liver injury by altering LSEC response to injury (174, 360). It has been shown that ROS selectively targets LSEC during prolonged liver injury. Importantly, LSECs are highly sensitive to ROS compared to other liver cells due to higher expression of NADPH oxidase (NOX) 2 and 4, the major source of ROS via NADPH oxidases, and their reduced capacity to enzymatically detoxify H2O2 (259, 267, 358). Moreover, impairment in autophagy in LSEC results in an improper response to oxidative damage and leading to increased ROS generation. This further helps LSEC recruit and activate macrophages and modulating the expression of proinflammatory cytokines and, thereby disease progression (136, 330).
Immune cells
The liver is considered as “the organ of the immune system.” The liver immune cells are distributed and localized strategically in various compartments and circulate in the sinusoids. The liver immune cells majorly include dendritic cells, neutrophils, lymphocytes (B & T), and natural killer cells (74, 284). The hepatic immune cell population is established as early as early embryonic life and creates a niche for blood surveillance. Although liver immune cells are prerequisites for proper host immune responses, excessive accumulation or recruitment of immune cells could lead to several liver-related diseases. For example, neutrophils infiltrated at the site of injury recruit blood monocytes and other immune cells, which further activates dendritic cells and macrophages (160, 431, 447).
One of the hallmarks of inflammation is the generation of large quantities of superoxide radicals via NADPH oxidase complex in the liver immune cells. In addition, most of the oxidants, except superoxide and hydrogen peroxide, are produced by the enzyme myeloperoxidase (MPO) in the immune cells, especially neutrophils (197). ROS generated in the immune cells induces tissue damage and disease progression. Indeed, studies have shown that neutrophils were the primary cell that displays NADPH oxidase activity and generates a whole spectrum of both radicals and nonradical relevant to ROS (382, 447). T cells also generate ROS via NADPH oxidase and act as the primary source for mROS (352). This concept is tested in several studies wherein pharmacologic treatment of primary T cells with antioxidants attenuated the proliferation of T cells (168, 277). Similarly, continuous generation of ROS in primary B cells in response to B cell antigen receptor stimulation results in proliferation and activation of B cells (430). Likewise, oxidative stress in the liver also enhances the dendritic cell response leading to T cell activation (289, 296).
Cellular Organelles and ROS
In the cells, ROS is generated in various cellular compartments, including the cytoplasm, mitochondria, ER, lysosomes, and peroxisomes.
Mitochondria
Mitochondria play an essential role in the generation of ATP through the oxidation of metabolic intermediates through the ETC. Mitochondria account for nearly 90% of oxygen consumption and therefore acts as the most redox-active compartment in the mammalian cells (46, 464). However, the accumulation of excess fatty acids creates an imbalance between the delivery and outflow of electrons to the respiratory chain in the mitochondria leading to the production of ROS (287, 377). In general, the mitochondrial ROS (mROS) in ETC occurs in the form of superoxide anion radical and its dismutation product H2O2 (40, 210). Although mitochondria are occupied with several complexes in generating energy, complex I and III are the major sites of electron transfer to O2 to generate O2•− (62, 195). Studies have shown that ROS could also be generated from pyruvate dehydrogenase (44), α-ketoglutarate dehydrogenase (378), glycerol-3-phosphate dehydrogenase (293), and monoamine oxidase (375). Under pathological conditions, several molecular mechanisms drive the overproduction of mROS. For example, mitochondria in the apoptotic cells produce superoxide in response to the release of cytochrome c (130, 166). Thus, mitochondria are considered the main source of ROS.
Given its role in ROS production, mitochondria are well equipped with antioxidant and scavenging mechanisms such as manganese superoxide dismutase (Mn-SOD), copper zinc-superoxide dismutase (CuZn-SOD), mitochondrial glutathione (mGSH), glutathione peroxidase, and catalase. In addition, the mitochondrial matrix is equipped with two antioxidant enzymatic systems: GSH-dependent glutathione peroxidase and NADPH-dependent thioredoxin-2 systems (338, 351). These enzymatic reactions help to maintain the balance between the pro and antioxidants. However, under pathological conditions, mitochondria generate huge amounts of ROS due to dysregulation in one or several of the aforementioned oxidant and antioxidant mechanisms. The well-studied liver pathologies that involve increased mitochondrial ROS include alcoholic-fatty liver disease (AFLD), nonalcoholic fatty liver disease (NAFLD), cirrhosis, viral hepatitis, and hepatocellular carcinoma (HCC). For example, chronic alcohol consumption leads to enhanced ROS production in the mitochondria partly due to the accumulation of cholesterol in the inner mitochondrial membrane and further disturbing the mGSH import from the cytosol. The significance of the mitochondrial import of GSH was demonstrated by restoring the membrane fluidity by administering the antioxidant N-acetylcysteine. Moreover, alcohol-induced ROS causes oxidative damage to the mitochondrial DNA, thereby increasing the risk of double-strand breaks and somatic mutations (32, 53, 118, 138). Thus, several studies have demonstrated targeting oxidative stress as a potential therapeutic avenue for liver-related diseases such as AFLD and NAFLD.
Endoplasmic reticulum
The endoplasmic reticulum (ER) is the major site for protein synthesis, folding, modification, and trafficking (41). ER is also involved in the biosynthesis of steroids, lipids, and carbohydrates (154). Oxidative protein folding in the ER generates ROS, and in fact, the overall estimate is that nearly 25% of ROS generated in the cell is contributed by the ER. An oxidizing environment needs to be maintained in the ER lumen to introduce the disulfide bonds during protein folding and trafficking (346). Interestingly, ER is impermeable to GSSH; therefore, it relies on its capacity to generate GSSH. The electron transport required to generate GSSH in the ER membrane is regulated by the protein disulfide isomerase (PDI) and ER oxidoreductin 1(ERO1) (36, 312). Under pathological conditions, overactivation of unfolded protein response results in the generation of ROS and is strongly associated with the progression of NASH and cirrhosis (207, 374). Furthermore, ER stress results in the accumulation of calcium in the mitochondria, which promotes exacerbated mitochondrial ROS production leading to disease progression (126, 134). ER is also the home for the enzyme cytochrome 2E1 (CYP2E1), the enzyme responsible for ethanol catabolism. Thus, the ER plays a central role in ethanol-induced ROS production in the hepatocytes (265). Moreover, in HCV infections, viral replication and their gene products induce ROS production from ER (133, 307).
Peroxisomes
Peroxisomes are the oxidation sites of very long-chain and branched-chain fatty acids that cannot directly enter into the mitochondria. The dynamic nature of peroxisomes to enlarge, elongate, and proliferate confers its ability to involve in oxidative and detoxification reactions (205). In other words, peroxisomes majorly serve two functions to protect against liver diseases: (i) by the degradation of very long-chain fatty acids and (ii) disposing of excess ROS. Peroxisomes produce large amounts of H2O2 from the continuous oxidation of fatty acids (107, 165, 288). As a protective mechanism, peroxisomes are equipped with high detoxifying enzymes such as catalase, GPx, Mn-SOD, and CuZn-SOD (288, 398). Thus, structural and functional disturbances in peroxisomes of hepatocytes are sufficient to induce spontaneous hepatic steatosis through excessive generation and release of ROS (158).
Lysosomes
Lysosomes are the major nutrient-sensitive organelle involved in autophagy, including the removal of damaged mitochondria through a process called mitophagy (204). Recent studies have demonstrated a reciprocal relationship between ROS and autophagy (219). For example, Atg4, a cysteine protease, was identified as a direct target of ROS (342). Further, starvation concomitantly increases autophagy and mitochondrial ROS production through the inhibition of mTOR pathway (359). This concept was supported by a study wherein nutrient deprivation in hepatocytes results in the accumulation of defective mitochondria and increased oxidative stress (180). Moreover, ROS-mediated mitophagy is known to play a critical role in the first hit (lipid accumulation) and second hit (oxidative stress and inflammation) of NASH pathogenesis (212). The other fascinating role of lysosomes in oxidative stress arises from their ability to accumulate iron, wherein it catalyzes the Fenton reaction with H2O2 in various liver disease models (227, 399). Thus, lysosomal disorders associated with several chronic diseases are mainly related to increased generation of mitochondrial ROS.
External Mediators of Oxidative Stress
In addition to intracellular sources, several external factors trigger ROS generation. They include alcohol, drugs, nutrients, toxicants, pollutants, and physical stressors (UV, X-ray, and ionizing radiation). All these factors promote the initiation and progression of various liver-related diseases by altering the redox signaling.
Alcohol
Alcohol metabolism occurs in the liver with the help of enzymes alcohol dehydrogenase and aldehyde dehydrogenase resulting in the generation of one molecule of NADH. During this process, the respiratory activity is increased, resulting in increased oxygen consumption and the generation of greater amounts of ROS. When alcohol consumption is excessive, other enzymes such as NADH-dependent cytochrome C reductase, aldehyde oxidase, and xanthine oxidase drive ROS generation (52, 455). Alcohol also increases ROS generation by noncanonical mechanisms. For example, alcohol induces oxidative stress by increasing intestinal iron absorption leading to iron overload in the liver (157). Several studies have shown that the accumulation of iron in the liver is strongly associated with oxidative stress and the development of several liver-associated disorders (58, 99, 405, 411). Further, alcohol increases cytochrome P2E1 (CYP2E1) activity, an important player in the metabolism of alcohol. The induction of CYP2E1 strongly correlates with the generation of hydroxyethyl radicals and other lipid radicals (109, 276). For example, the exposure of HepG2 cells expressing human CYP2E1 to alcohol results in ROS generation, mitochondrial damage, and cell toxicity (435). Studies have also established that oxidative stress is the primary cause of alcohol-induced hepatocyte toxicity (54, 202). In support of this notion, alcohol-induced hepatocyte injury and cell death are mitigated by the administration of antioxidants (60, 231, 424).
Lipids
In the mammalian system, lipids act not only as the source of energy but also provide the structural components of the cell membranes. Lipids can also act as signaling molecules by forming a permeability barrier of cells and lipid bilayer in subcellular organelles. The fluidity of the membrane is highly dependent on the composition of fatty acids (i.e., degree of saturation: unsaturation fatty acids) in the lipid bilayer (384, 438). Thus, it becomes essential to maintain not only the lipid content but also the ratio of saturated to unsaturated fatty acids. Lipids are more prone to damage from several exogenous stimuli. Most importantly, excessive accumulation of ROS in subcellular organelle could directly damage the lipids (449). The most common forms of the ROS that affect the lipids are hydroxyl radical and hydroperoxyl. ROS damages the lipids through the process of lipid peroxidation, wherein they attack the lipids containing carbon-carbon double bond(s), especially the polyunsaturated fatty acids (PUFAs), which consists of two or more double bonds belonging to omega-3 (n−3) and omega-6 (n−6) fatty acids (315, 448). In addition, a wealth of literature has shown that cholesterol, membrane phospholipids, and glycolipids also target lipid peroxidation (51, 310). Lipid peroxidation generates a variety of lipid hydroperoxides such as MDA, 4-HNE, propanal, and hexanal. Among them, MDA is considered as the most mutagenic product of lipid peroxidation, whereas 4-HNE is the most toxic form. Further, MDA and 4-HNE are used as bioactive markers of lipid peroxidation, which could signal and regulate various transcriptional factors (16, 23).
Diet/Nutrition
The diet has a profound effect on redox biology by regulating the balance between the pro- and antioxidant mechanisms. The major dietary micro- and macronutrients that play a critical role in the production of ROS include proteins, lipids, carbohydrates, vitamins, and minerals (127, 390). For example, excessive consumption of carbohydrates or high-fat diet increases mitochondrial respiration, subsequently producing high levels of superoxides and free radicals. In particular, the consumption of high fructose diet results in lipid peroxidation, cytokine secretion, and ROS production. These diets induce hepatic mitochondrial dysfunction by supplying electrons continuously to the ETC by upregulating the TCA cycle and thereby impair mitochondrial complex IV activity. Thus, a higher release of ROS favors the development and progression of fructose-induced NAFLD (50, 397). Further, Mohanty et al. have shown that increased lipid and protein intake is strongly associated with the ROS generation in polymorphonuclear leukocytes and mononuclear cells (270).
Vitamins and minerals essentially prevent the generation of excess ROS by upregulating the antioxidant enzyme activities. Among them, vitamins E (α-tocopherol), C, and B12 are widely studied for their antioxidant properties (49, 334). For example, vitamin E deficiency reduces antioxidant enzymes such as liver GSH peroxidase, glutathione reductase, and catalase resulting in increased lipid peroxidation (403). Further, vitamin E and C have been shown to improve the clinical symptoms of NAFLD, enhance glucose metabolism, and lower liver injury (94, 291). Similarly, minerals such as selenium, magnesium, manganese, and copper prevent mitochondrial dysfunction and free radical-induced liver damage (429). For example, selenium acts as a cofactor for several enzymes, including glutathione peroxidase and selenoprotein P (402). Several studies have linked mineral deficiency to oxidative stress and increased susceptibility to lipid peroxidation (220, 441, 465).
Aging
Aging is a natural process that involves the loss of tissue and organ function over time. During aging, macromolecules (lipids, DNA, and proteins) undergo various structural and functional changes due to the accumulation of ROS and RNS from endogenous and exogenous sources (229). Although the exact mechanism(s) of ROS-induced aging is unclear, several lines of studies have elucidated a role for ROS in cellular senescence. Oxidative stress induces cellular senescence through multiple mechanisms. For example, the accumulation of ROS and RNS increases the proinflammatory cytokines, chemokines, and other growth factors that are central to the progression of liver diseases (20, 103). ROS also increases the expression of several MMPs such as MMP-1, 2, 7, and 9, which affects the mechanical properties of the extracellular matrix leading to the development of senescence (80, 108, 387). Further, ROS and RNS decrease the expression of forehead box protein (FOXO), sirtuins, and sarco-endoplasmic reticulum Ca2+-ATPase activities, which are involved in various pathways associated with age-related diseases (100, 186, 279, 368).
It is also well-established that mitochondrial integrity and function decline with age due to mitochondrial DNA damage. The increased sensitivity of mitochondrial DNA to oxidative stress led to the concept of a “vicious cycle” wherein ROS-induced impairment of mitochondrial function leads to a further increase in oxidant production (308, 362). It is also accepted that old mitochondria show impaired mitochondrial morphology and function due to significant impairment in electron transport( 260). Furthermore, studies with genetically modified animals such as SOD1, SOD2, and p66shc-deficient animals show that mitochondrial dysfunction could trigger premature aging (294, 413, 426). The epigenetic and DNA methylation modifications induced by ROS also play a role in the aging mechanisms (117, 131, 322).
Pathophysiological Sources of ROS
Inflammation
Oxidative stress and inflammation are tightly interrelated and often present simultaneously, making it difficult to discern the causal role of oxidative stress in inflammatory diseases such as NASH. Oxidative stress plays a dual role in tissue damage by triggering innate immune response and infiltration of inflammatory cells (neutrophils, monocytes, and lymphocytes). For example, oxidative stress due to impaired mitophagy triggers an innate immune response by activating the NOD-like receptor protein 3 (NLRP3), a pattern recognition receptor (461). NLRP3 regulates genes involved in cytoprotective and antioxidant protection. A recent study found that the NLRP3 inflammasome accentuated oxidative stress by suppressing the nuclear factor (erythroid-derived 2; Nrf2), a basic leucine zipper (bZIP) transcription factor (149). On the other hand, sustained NRF2 activation protects mice from NASH progression by inhibiting oxidative stress and inflammation (440).
Further, cytokines, chemokines, and nitric oxide released by the activated inflammatory cells induce tissue damage, thereby augment oxidative stress. Thus, a vicious feed-forward regulation of inflammation by oxidative stress and vice versa plays a critical role in the initiation and progression of liver diseases. Therefore, antioxidant and anti-inflammatory therapy is beneficial in the management of liver diseases. Consistently, enhancing the scavenging of ROS by SIRT1 deacetylases (stem cell therapy) decreases oxidative stress and inflammation in NASH (221).
In NASH patients, ROS induces necroptosis of hepatocytes by upregulating IL-1β, IL-8, and TNF-α. It is also possible that the hypomethylated CpG islands and formyl peptides released from the damaged mitochondria stimulate innate immune response (281). For example, glutathionylated peroxiredoxin 2 and thioredoxin released from the activated macrophages induce oxidative stress in a TNFα-dependent manner (335). Sometimes, the release of mitochondrial DNA into the cytosol induces cGAS-Stimulator of Interferon (STING) pathway, further aggravating liver injury (61, 415). In a mouse model of NAFLD, activation of STING in macrophages aggravates hepatic inflammation and fibrosis (420). Exposure of formyl peptides also stimulates the innate immune system in a CXC chemokine receptor 2 (CXCR2)-dependent recruitment of neutrophils in acute liver injury models (218).
Insulin resistance
Insulin regulates glucose homeostasis by promoting peripheral glucose uptake and utilization in various tissues. When insulin fails to promote glucose metabolism, a condition known as insulin resistance (IR), blood glucose is chronically elevated. Insulin secretion from pancreatic islets is increased as compensation to IR (173). Thus, chronic hyperinsulinemia is the hallmark of IR. However, sustained hyperglycemia eventually leads to failure of the pancreatic islets leading to type 2 diabetes. The polygenic nature of IR makes it challenging to elucidate its origin.
Nonetheless, over three decades of research indicate ROS as an integral part of insulin signaling. High levels of ROS are associated with IR. For example, the thiol-dependent enzymes, namely protein tyrosine phosphatase (PTP) regulated by the cellular redox state, play a crucial role in inhibiting insulin signaling (409). Moreover, insulin increases H2O2 generation by inducing NOX4, and low levels of H2O2 are essential for insulin-mediated translocation of glucose transporter 4 (Glut4) via PI3K/PLC (72). While physiological levels of ROS promote insulin sensitivity by promoting Glut4 translocation, an overload of ROS suppresses Glut1 translocation to the plasma membrane by downregulating AKT phosphorylation (101).
Oxidative stress also reduces insulin packaging and impairs glucose-stimulated insulin secretion (101, 113). Furthermore, oxidative stress inhibits β-cell differentiation via its effect on transcription factors such as pancreatic and duodenal homeobox 1 (Pdx-1), homeobox protein Nkx6.1, neurogenin-3 (Ngn.3), FOXO, and MafA (121). Higher concentrations of free radicals inhibit insulin gene expression at the transcriptional level by repressing Pdx-1 (insulin promoter factor 1) and MafA (a transcription factor) (143). However, a recent study in adipocytes demonstrated that ROS-mediated transcriptional response is insufficient to cause IR. Nonetheless, overexpression of PRDX3 and MnSOD decreased blood glucose levels and improved insulin sensitivity in HFD-induced obesity (59). The contribution of mitochondrial ROS to IR is further evidenced by a rapid reversal of IR by inhibitors of mitochondrial respiratory complexes and uncouplers of oxidative phosphorylation (7). Patients with NASH present with high levels of nonesterified fatty acids sufficient to induce oxidative stress and IR through activation of JNK/p38MAPK pathway (116). These results suggest that approaches that prevent ROS generation provide excellent protection against IR.
Oxidative Stress and Cellular Dysfunction
Mitochondrial dysfunction
Mitochondrial dysfunction could arise from impaired mitochondrial biogenesis and clearance of damaged mitochondria (273, 274). Several transcription factors are essential for mitochondrial biogenesis and function, including PGC-1α and TFAM that are decreased in fatty liver disease (3, 283). Moreover, the mitochondria appear swollen with disrupted architecture in NASH patients (337). In addition to aberrant mitochondrial structures, the expression and activity of mitochondrial respiration enzymes are decreased in both alcoholic and nonalcoholic patients. In rat models, alcohol-containing diets severely impair the expression and activity of mitochondrial respiratory complexes (almost all I, III, IV, and V) (71, 118). Although the efficacy is affected by the route of administration (intragastric ingestion vs. oral feeding), the ultimate effect of alcohol on mitochondrial dysfunction does not vary significantly (138). Similarly, high-fat diet-fed mice display reduced cytochrome b-complex, ATP complex subunits, and citrate synthase enzyme expression and activity (119). Moreover, the mitochondrial defects attribute to the decreased half-life of mitochondrial respiratory proteins. Due to reduced mitochondrial ATP production, these animals exhibit impaired mitophagy, enhancing ROS generation.
The mitochondrial respiratory chain produces a substantial amount of ROS as it consumes molecular oxygen during oxidative phosphorylation. Efficient scavenging of ROS in the NASH models improves mitochondrial integrity and ameliorates NASH. A recent study demonstrated that sirtuin (SIRT) 3 improves mitochondrial integrity and protects hepatocytes from oxidative injury by enhancing ROS scavenging (232). Moreover, overexpression of SIRT1 or SIRT3 increased antioxidants in chronic hepatocyte injury models (Figure 2) (232).
Figure 2.
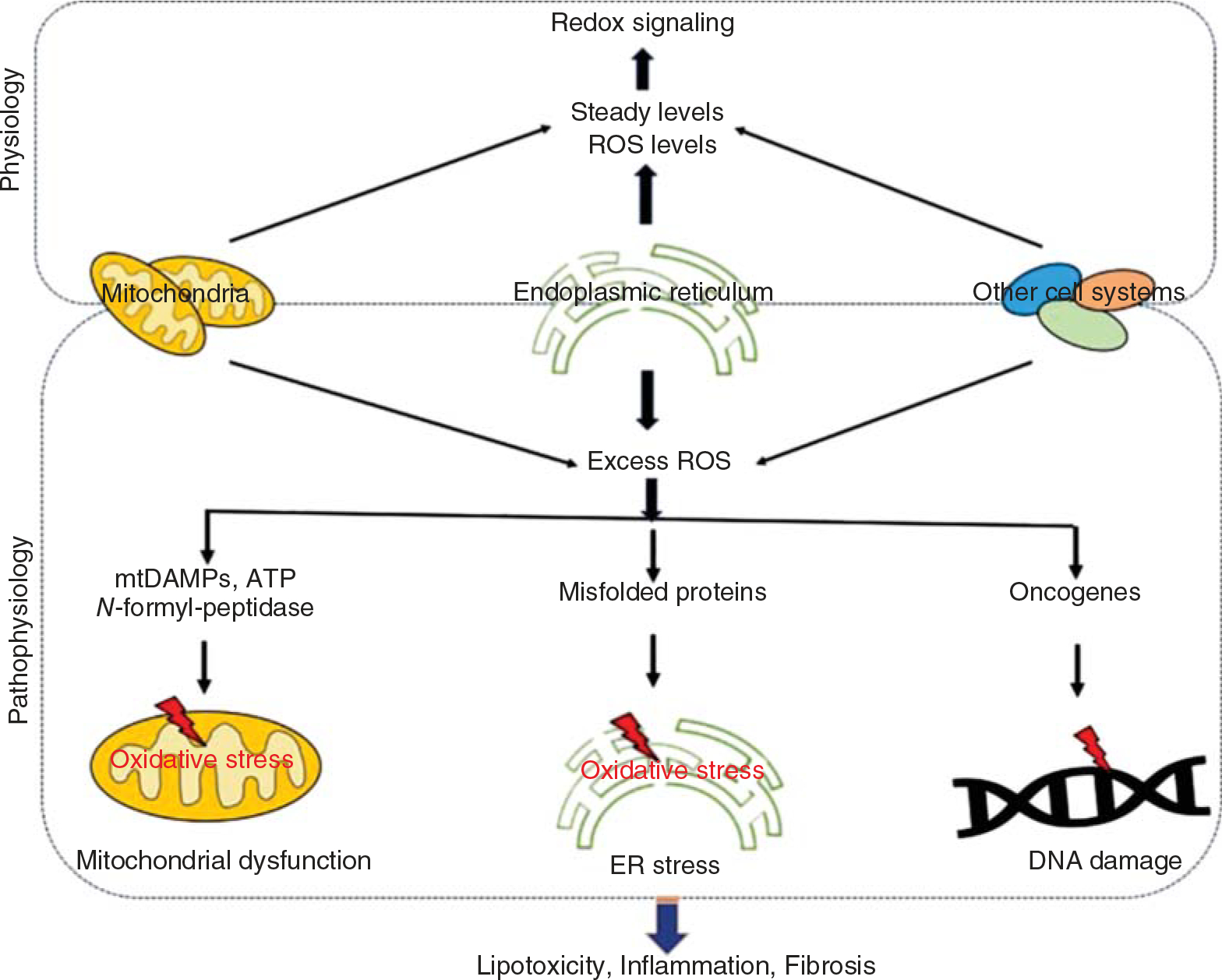
Putative sources of ROS and their contribution to pathologies of liver diseases. Under physiological conditions, organelle systems generate steady-state ROS levels that promote redox signaling. On the other hand, under pathological conditions, excess ROS produced various danger signals such as DAMPS and ATP from the mitochondria and unfolded/misfolded proteins from the ER and oncogenes that promote mitochondrial dysfunction, ER stress, and DNA damage. All these factors lead to the development of liver pathologies such as inflammation, fibrosis, and even cancer.
Endoplasmic reticulum (ER) stress
ER is the site of protein synthesis, folding, and maturation. Misfolded or unfolded proteins undergo either refolding or degradation. Aberrations in protein folding or clearance of misfolded proteins induce an unfolded protein response (UPR) mediated by protein kinase RNA-like ER kinase (PERK), activating transcription factors (ATFs), and inositol-requiring signaling protein 1 (IRE1). PERK temporarily halts the global protein translation by phosphorylating the eukaryotic translation initiation factor 2a (eIF2a). Activated eIF2a phosphorylates nuclear factor erythroid 2-related factor 2 (Nrf2), leading to the dissociation of the Nrf2 from Kelch-like ECH-associated protein 1 (Keap1) complex, which eventually induces the expression of antioxidant enzymes, including heme oxygenase-1 (HO-1) (41, 209). ATF4 induces CCAAT/enhancer-binding protein homologous protein (CHOP) to activate an antioxidant response. IRE1 and ATF6 co-operate to upregulate several UPR target genes, heat-shock protein 70 (Hsp70), and other chaperones to restore the ER redox state. ATF6 also induces X-box binding protein-1 (XBP1), which stimulates an inflammatory response through NFκB and C-Jun N-terminal kinase (JNK) signaling pathways. Thus, UPR helps the cells to recover from ER stress and restore ER equilibrium. If UPR persists, IRE1 increases tumor necrosis factor (TNF) receptor-associated factor 2 (TRAF2)-induced JNK activation, which then triggers cell death by inducing the expression of proapoptotic BH3-only proteins including p53 upregulated modulator of apoptosis (PUMA), Bcl-2 like protein (BIM), and BH3-interacting-domain death agonist (BID).
Similar to mitochondria, the redox state is critical for the metabolic homeostasis of ER. In NASH patients and model organisms, ER stress occurs due to oxidative damage (445). Chronic ER stress increases ROS production by upregulating CHOP. HO-1 induced by Nrf2 helps to mitigate oxidative stress. Figure 2 with genetic deletion of Nrf2 mice are highly susceptible to oxidative stress and show aggravated NASH phenotype (264).
Overexpression of HO-1 protects against oxidative stress due to increased expression of antioxidant chaperones, enzymes, and anti-inflammatory cytokines such as IL-22 (57). Paradoxically, PERK inactivates Nrf2 signaling during ER stress; however, its physiological significance is unclear (373). In alcohol-induced liver injury, ER stress is strongly associated with oxidative stress. Alcohol feeding increases the expression of ER stress-associated proteins such as Hsp70, binding immunoglobulin protein (BiP), and Grp 94, CHOP as well as caspase 12 as early as 2-weeks (139). Exaggerated hepatocyte apoptosis with no difference in hepatic steatosis in alcohol-fed CHOP null mice suggests that ER stress plays a significant role in injury-mediated apoptosis (162). Further, hepatic overexpression of the ER chaperones, such as ORP150/HYOU1 and GRP78/BiP improved insulin sensitivity and hepatic steatosis (451). This suggests that ER stress response plays a vital role in maintaining hepatic homeostasis during metabolic insults. Interestingly, hepatocyte-specific IRE1 knockout mice exhibit steatosis when exposed to tunicamycin, while deletion of IRE1 in the hepatic stellate cells attenuates HCC progression (70, 460). Thus, a considerable difference in the effect of ER stress and UPR response between various cell types in the liver poses a severe limitation in therapeutic targeting ER stress and UPR pathways for the treatment of NASH.
DNA damage
Several endogenous and exogenous genotoxic insults induce spontaneous mutations in the DNA. Therefore, precise DNA repair mechanisms are critical in maintaining genome integrity. Genomic instabilities are the characteristic feature of HCC due to erroneous DNA repair mechanisms. ROS, UV light, radiation, and environmental mutagen are the major DNA damage inducers. Oxidative stress and ROS co-operate with the mutagens to drive the progression of NASH to HCC. DNA damage activates p53-mediated cell apoptosis (81, 376). A recent review has elaborated on how dysregulation of p53 function leads to metabolic disorders, including NASH and HCC (196). However, the mechanisms involved in p53-mediated metabolic dysregulation are not fully understood. Studies show that p53-binding protein 1 (p53BP1), a DNA damage response protein, forms nuclear foci in response to double-strand DNA breaks. The number of p53BP1 foci increases in NASH patients implying significant DNA double-strand breaks (4). ROS and p53 have a versatile partnership. ROS can act upstream and downstream to p53 to regulate cellular processes. ROS modifies DNA by forming stable covalent bonds leading to base modifications, including thymidine glycol, 5-hydroxylmethyluracil, and 8-OHdG. Thus, hepatitis viral infections that generate 8-OHdG often lead to liver cancer. An elevated level of 8-OHdG is used as a novel prognostic marker in HCC and is often associated with poor patient survival (225). 8-OHdG also serves as a marker in hemochromatosis, Wilson’s disease, chronic hepatitis, hepatoblastoma, and primary biliary cirrhosis (73). ROS also induces double-stranded DNA breaks, thereby increasing mutations or chromosomal aberrations resulting in tumorigenesis (Figure 2) (22).
Advanced glycation end products (AGEs)
In obesity and NASH, AGEs levels are significantly elevated (63). AGEs that include methylglyoxal, glyoxal, and HNE, reduce sarcoplasmic (ER) reticulum Ca2+ ATPase (SERCA) activity (428). AGE products also possess highly reactive moieties and interact with a wide range of biomolecules, including proteins. AGE could also signal through several receptors called receptors for AGE (RAGE), whose identity is revealed in recent investigations. For example, galectin 3a and Oligosaccharyltransferase-48 (OST48) are characterized as AGE binding proteins. OST48 is an integral part of the ER membrane that acts as a clearance receptor for AGE. Overexpression of OST48 is associated with a high risk of liver fibrosis and liver failure (468). RAGE/TLR4 signaling also induces ER stress-mediated liver fibrosis. For example, a recent study shows that high mobility group protein (HMGB1) released from the injured hepatocyte induces ER stress in hepatic stellate cells via RAGE/TLR4 signaling leading to liver fibrosis (146). The significance of these mechanisms in NASH or ASH patient needs further investigation. Uncoupling protein (UCP) 2, an important regulator of ER stress in a wide range of tissues, intersect with AGE/RAGE signaling. For instance, UCP2 deficiency decreases hepatic glyoxalase-1 resulting in a reciprocal increase in methylglyoxal associated with high mortality in young mice (194).
Molecular Master Regulators of Oxidative Stress
The oxidative stress is regulated by numerous transcription factors, which helps to minimize tissue damage. These transcription factors either promote or suppress oxidative stress depending on their expression levels.
Nuclear factor erythroid 2-related factor 2 (Nrf2)
The transcription factor, Nrf2 regulates numerous antioxidant genes by binding to the antioxidant response elements (ARE) on the target gene promoters. The transcriptional activity of Nrf2 is regulated by p62 and Keap1. Oxidative stress increases the binding of NRF2 to p62, thereby limiting the interaction with Keap1, a cullin-3-type ubiquitin ligase that degrades Nrf2 (190). Tank binding kinase 1 (TBK1) further acts as an upstream regulator of Nrf2 by stabilizing p62(67). Recent studies demonstrate that noncoding RNAs and micro-RNAs are also involved in stabilizing Nrf2 during oxidative stress (425).
Nrf2 expression is a cardinal feature of acute or chronic oxidative stress. Targets of Nrf2 include HO-1, SOD1, catalase, and glutathione S-transferase (GST). Therefore, Nrf2 could potentially control the oxidative stress response as it targets an array of genes that possess stress-responsive elements similar to GST. Mice with hepatocyte-specific Keap1 deletion, display enhanced Nrf2 levels leading to elevated expression of enzymes involved in glutathione synthesis, including GST-peroxidase and glutamate-cysteine ligase (233). Initially, Nrf2 expression is thought to be protective against almost all liver injury models ranging from acute hepatitis to cholangitis and NASH to liver cancer. However, a growing body of evidence suggests that its expression is vital in the progression of liver diseases (183, 392). Although the hepatocyte-specific Keap1 knockout mice enhanced Nrf2 expression and showed a protective phenotype from cadmium-induced acute liver injury (233), Nrf2 knockout mice are highly susceptible to ethanol-induced liver fibrosis and steatosis (199). Wang et al. found that genetic deletion of Nrf2 improved liver phenotype by suppressing the expression of very-low-density lipoprotein receptors in alcohol-fed mice(422). The latter observation was further supported by clinical studies showing that Nrf2 activation increases with the incidence of HCC (156). Moreover, Nrf2 knockout mice display severe NASH symptoms when fed on a methionine choline-deficient diet (199). Nevertheless, these observations indicate that NRF2 could be a sensitive player to define the state of the liver phenotype in various experimental settings of liver diseases (Figure 3).
Figure 3.
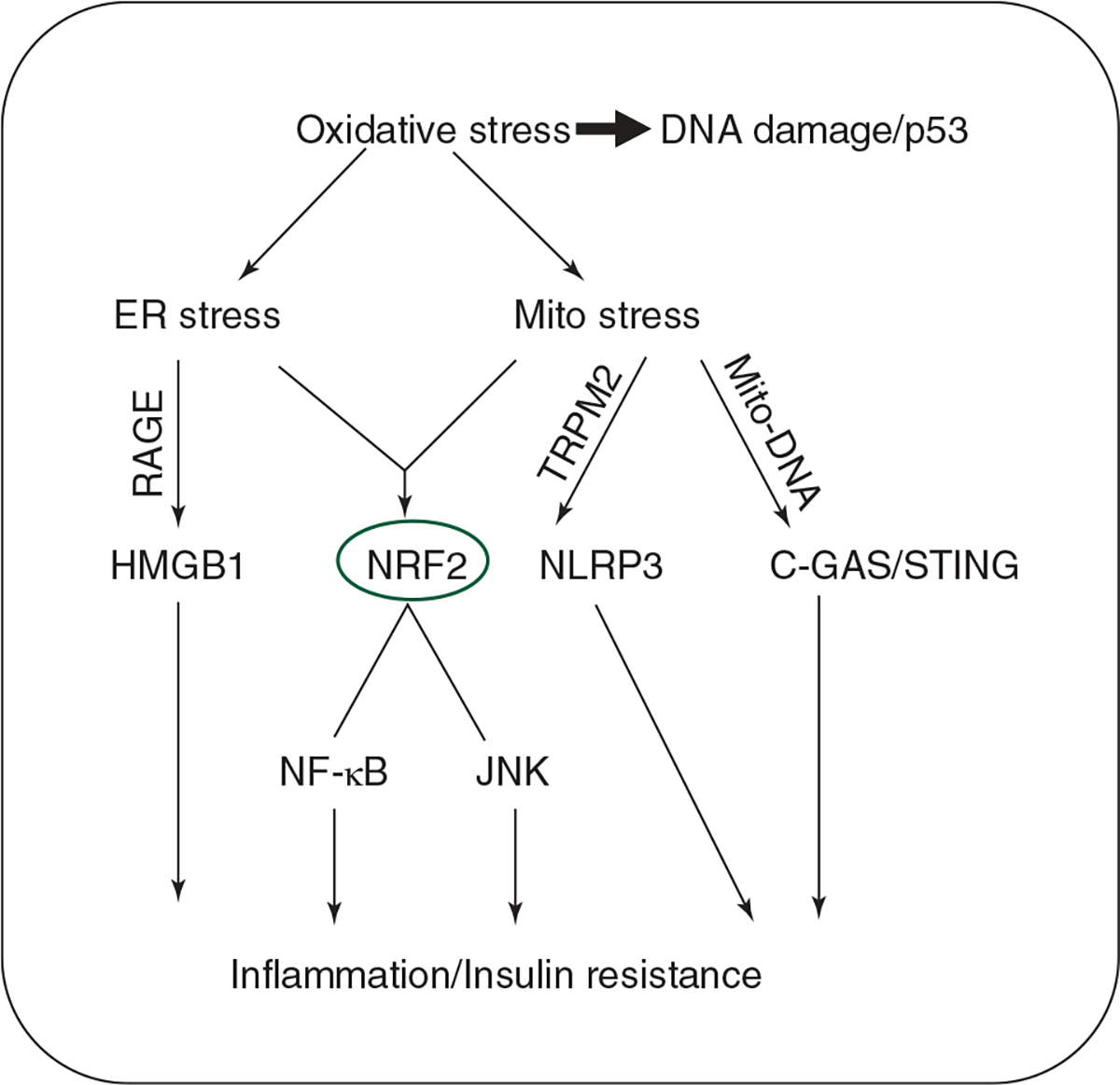
Master regulators of oxidative stress response leading to inflammation and insulin resistance: Oxidative stress causes severe DNA damage sensed by p53 accumulation. Oxidative stress exerts its effects through the ER and mitochondrial stress (complete events mentioned in the review). NRF2 is the cumulative stress response marker induced by the ER and mitochondrial stress. ER stress-induced inflammation is mediated by the recently discovered HMGB1 transcription factor, which further intersects with RAGE signaling. C-GAS/STING pathway is the intracellular DNA-sensing pathway activated by the mitochondrial DNA leaked into the cytosol. NLRP3 is either directly activated by TRPM2 or could be upstream of C-GAS/STING pathway. Thus, NLRP3 co-operates with C-GAS/STING pathway to promote inflammation and insulin resistance.
Nuclear factor-kappa B (NF-κB)
Canonical and noncanonical pathways regulate the activation of NF-κB. In the canonical mechanism, phosphorylation of inhibitory kappa Bα (IκBα) by IκB kinase (IKK) induces degradation of the IκBα from NF-κB:IκBα complex, resulting in the nuclear translocation of NF-κB. In contrast, the noncanonical mechanism involves NF-κB-inducing kinase (NIK)-mediated processing of NF-κB2 p100 into NF-κB2 p52, which complex with RelB to induce the expression of NF-κB target genes (177, 363).
NF-κB plays a dual role in liver injury, where its deletion in hepatocytes is deleterious but protective against liver injury in other cell types. A moderate activation of NF-κB signaling in hepatocytes is protective in states of low-grade lipogenesis in the absence of inflammation (240, 380). Therefore, it appears that NF-κB-induced by oxidative stress helps to prevent hepatocyte death and promote a compensatory proliferation of hepatocytes. In contrast, the deletion of IKK2α in unstressed hepatocytes does not affect oxidative stress markers (237). Thus, NF-κB serves as the master regulator of inflammation and cell death. The role of NF-κB in inducing cell death appears to be dependent on its ability to activate its downstream target JNK/p38 MAPK (discussed below).
NF-κB activation in acute or chronic liver disease is partly mediated by the inflammatory response (442). NF-κB transcriptionally regulates several antioxidants, and pro-oxidant gene expression in a spatial-temporal manner, and their details are reviewed elsewhere (275). ROS stimulates NF-κB activation in the cytoplasm but inhibits NF-κB DNA binding activity in the nucleus (164). ROS could also regulate DNA binding activity modifying specific residues in NF-κB (184).
Forkhead box protein O (FOXO)
Forkhead box protein O (FOXO) transcription factors are highly conserved in higher organisms with an essential role in hepatic glucose and lipid metabolism. There are four FOXO proteins expressed in mammals, among which FOXO1, 3, and 4 express ubiquitously, while FOXO6 expression is specific to neuronal cells. In mouse models, the deletion of FOXO1 and 3 induced spontaneous hepatic steatosis and decreased glucose metabolism (459). Mouse with triple knockouts for FOXO1, 3, and 4 also show similar phenotypes in hepatic glucose and lipid metabolism (393). FOXO-dependent suppression of fatty acid synthase and nicotinamide phosphoribosyl transferase (NMPT) regulates hepatic steatosis. Emerging evidence shows a crucial role for FOXO transcription factors in oxidative stress. For example, FOXO can control oxidative stress via NAD-dependent deacetylase SIRT1, as NMPT is essential for the synthesis of NAD (393). A recent study showed that FOXO1 expression is associated with fatty liver, whereas pharmacological inhibition of FOXO1 improved hepatic steatosis (89). This contradicting data is due to high levels of ER stress and hepatocyte necroptosis in these models (89). An independent study using triple knockout mice (FOXO1, 3, and 4) further established the significance of FOXO signaling in suppressing oxidative stress (10). Thus, the controversial data suggest a complex yet significant role of FOXOs in regulating redox biology in the liver.
JNK
c-JUN N-terminal kinases (JNK) and p38 are the principal members of MAPK family, which phosphorylate and activate several transcription factors, including c-Jun, ATF-2, and TCF/Elk-1 (87). Vertebrates express three isoforms of JNKs, where JNK1 and 2 are ubiquitously expressed, and JNK3 is expressed only in the heart, brain, and testicles. Although JNK1/2 are important mediators of liver injury, there appears to be a specific role for each JNK isoform in liver disease depending on their binding partners and downstream signaling mediators (167, 187, 371). For example, JNK promotes TNF-α-induced cell death when NF-κB is not active in hepatocytes (333). JNK then phosphorylates and activates its downstream targets such as c-JUN, JUN-B, and JUN-D. The targets of JNK pathway involve apoptosis-related genes such as BCL-2 and Bax (433). A recent study shows that JNK activation induces Bim in hepatocytes, enhancing oxidative stress leading to IR and steatosis (230). Thus, Bim knockout mice exhibit improved mitochondrial function, reduced oxidative stress, and are protected against diet-induced obesity and IR (230).
MAP3K, MLK3, and ASK1 are implicated in the regulation of hepatic JNK signaling in NASH models. Mice with the knockout of MLK3 alone or double knockout for MLK3 and MLK2 are protected from diet-induced obesity due to attenuated JNK activation (112, 170). ASK1 inhibitors (GS-4997, GS-444217) are currently on phase 2 clinical trials for liver fibrosis in humans (235). Whether ASK1 inhibitors ameliorate hepatic fibrosis through inhibition of JNK-oxidant signaling needs further investigation (235).
Sirtuins
Sirtuins are a class of NAD+-dependent deacetylases, which confers protection against a wide range of metabolic disorders, including obesity and age-associated diseases. Accumulating evidence suggests that sirtuins regulate autophagy in response to physiological and environmental stresses (8, 90). Humans express seven sirtuins that include SIRT1 to SIRT7. Spatiotemporal expression of sirtuins mediates their specific role from gene expression to cell cycle regulation. Among the sirtuins, SIRT3, SIRT4, and SIRT5 are exclusively localized to the mitochondria and involve in posttranslational modifications of mitochondrial proteins. In response to oxidative stress, mitochondrial sirtuins deacetylate a network of proteins involved in the TCA cycle and oxidative phosphorylation complexes (56).
SIRT1 expression protects against liver injury by maintaining mitochondria health. Insults such as hypoxia, ischemia, and pro-oxidants that are highly prevalent in fatty liver deplete SIRT1 levels and impair autophagy (407). Thus, the ectopic expression of SIRT1 restores autophagy dramatically. Surprisingly, SIRT1 interacts with mitofusin-2, a mitochondrial fusion protein, suggesting that SIRT1 is a linker between the nucleus and mitochondria under stress conditions (34). Moreover, SIRT1 overexpression restores autophagy in MFN2 deficient cells indicating that SIRT1 plays a role in alleviating mitochondrial dysfunction, thereby decreasing oxidative stress. Small molecules for SIRT1 such as SRT2104 and SRT1720 show a profound effect on lipid peroxidation and carbonylation of proteins in the liver (43). SIRT1 also regulates p53 activity by acetylation and stabilization of p53 in damaged or stress-induced cells (155). SIRT1 is also an essential regulator of insulin signaling and is downregulated in conditions such as obesity and T2DM. Inhibition of SIRT1 recapitulates IR phenotype, while overexpression of SIRT1 improved insulin sensitivity, mainly via repressing PTP1B (381). These studies indicate that SIRT1 has an essential role in depleting oxidative stress developed during disease progression. However, studies with liver and hepatocyte-specific knockdown of SIRT3 show a negative role for SIRT3 on the regulation of autophagy, lipotoxicity, and susceptibility to chronic alcohol consumption (223, 244).
NOD-like receptor protein 3 (NLRP3)
The inflammasome nucleotide-binding oligomerization domain-like receptor family pyrin domain containing 3 (NLRP3) is often investigated in liver diseases. NLRP3 belongs to the innate immune response complex consisting of NLRP3, pathogen/danger associated recognition receptors, apoptosis-associated speck-like protein (ASC), and pro-caspase-1. NLRP3 activation converts pro-caspase-1 into active caspase-1 leading to the induction of mediators of local inflammation and pyroptosis such as IL-1β, IL-17, and IL-18 (198, 238). Studies with NLRP-3 knockout mice show that alcohol-induced liver inflammation and fibrosis were attenuated in the absence of NLRP3 (86, 278). Global and myeloid cell-specific Nlrp3 overexpression mice display hyperactive NLRP3, which induces inflammation, hepatocyte pyroptosis, and fibrosis in the liver (434). It has been demonstrated that mROS is required for the activation of NLRP3 and TRPM2, a calcium channel required for this pathway (466). However, it warrants future studies to investigate the role of mROS in NLRP3 signaling and its implications in NASH. Hepatitis-B virus toxin induces NLPR3-mediated inflammation and hepatocyte pyroptosis in ROS (H2O2)-dependent manner (439). In diabetic conditions, increased NLRP3 activation from elevated oxidative stress induces hepatocyte pyroptosis during ischemic reperfusion injury (361). Studies using NLRP3, ASC, and caspase 1 deletions also suggest the role of NLRP3 inflammasome in the pathogenesis of NAFLD in obesity (148).
Oxidative Stress and Pathophysiology of Liver Disease
Oxidative stress and NAFLD/NASH
The presence of cytoplasmic triglyceride lipid droplets in more than 5% of hepatocytes or triglyceride content accounting for more than 5% of the liver weight is defined as hepatic steatosis/fatty liver. Hepatic steatosis is classified as microvesicular (smaller lipid droplets) and macrovesicular (large lipid droplets). Macrovesicular steatosis results from an imbalance in triglyceride synthesis, whereas microvesicular steatosis develops due to mitochondrial dysfunction (defective fatty acid oxidation) (206, 372). Simple hepatic steatosis is often self-limiting; however, it can progress to nonalcoholic steatohepatitis (NASH) characterized by hepatocyte injury (hepatocyte ballooning and cell death) and infiltration of immune cells. NASH is followed by induction of liver fibrosis leading to cirrhosis culminating in hepatocellular carcinoma (HCC) (11, 258). The prevalence of NAFLD is estimated to be around 20% to 30% in Western countries and 5% to 18% in Asia (31, 341).
The pathogenesis of NAFLD is explained by “multiple parallel-hit hypothesis” wherein the “first hit” involves the accumulation of free fatty acids (FFAs) leading to lipotoxicity and oxidative stress denoted as “second hit.” The accumulation of FFAs in the liver alters mitochondrial function and increases ROS generation (28, 47). Therefore, the development and progression of NAFLD are strongly associated with the continuous generation of ROS/RNS and oxidative damage to the organelles. This is further accentuated by dysfunction in the counteractive antioxidant mechanisms. The mitochondrial abnormalities range from lipid-related changes to mitochondrial DNA damage to sirtuin imbalance (214, 301, 383). For example, mitochondrial DNA damage and alterations in genes encoding mitochondrial proteins result in a rapid increase in oxidative stress and, thereby, trigger the progression of simple steatosis to fibrosis (251). The mitochondrial dysfunction mostly alters fatty acid oxidation leading to the accumulation of fat in the hepatocytes, exacerbating steatosis (88). Oxidative stress also affects lipid metabolism in hepatocytes. For example, Seo et al. demonstrated that treatment of Huh7 and AML12 cell with H2O2 increased mRNA expression levels of genes involved in lipid (lipin) and cholesterol metabolism (SREBP-2) and further, the lipid overload in the hepatocytes increases inner mitochondrial membrane permeability, loss of membrane potential, and ATP synthesis capacity (355). Further, hepatocyte-derived ROS releases highly unstable reactive aldehydic derivatives that impair mitochondrial respiration and drive disease progression (78).
NAFLD is strongly associated with an increase in nonesterified fatty acids (NEFA) and free cholesterol (FC). Further, the NEFA and FC undergo oxidation with the help of lipoxygenases, cyclogeneses and cytochromes P450 family and, thereby, produce lipid peroxides such as malondialdehyde (MDA) and 4-hydroxynonenal (HNE) (30). Several studies have a positive correlation between lipid peroxidation and NAFLD. For example, plasma levels 8-isoprostane, a product of lipid peroxidation was significantly increased in patients of NAFLD (191). Recent studies show that lipid peroxidation is increased in pediatric NAFLD; however, no change with liver CYP 2E1 expression was observed (29). In addition, lipid peroxides such as MDA and 4-HNE generated in NAFLD act as biomarkers of NAFLD and NASH (450). Further, 4-HNE activates and increases ROS production in HSCs (456). Thus, alteration of lipid metabolism during NAFLD leads to accumulation of toxic lipids and oxidative stress and helps to the progression to NASH (Figure 4).
Figure 4.
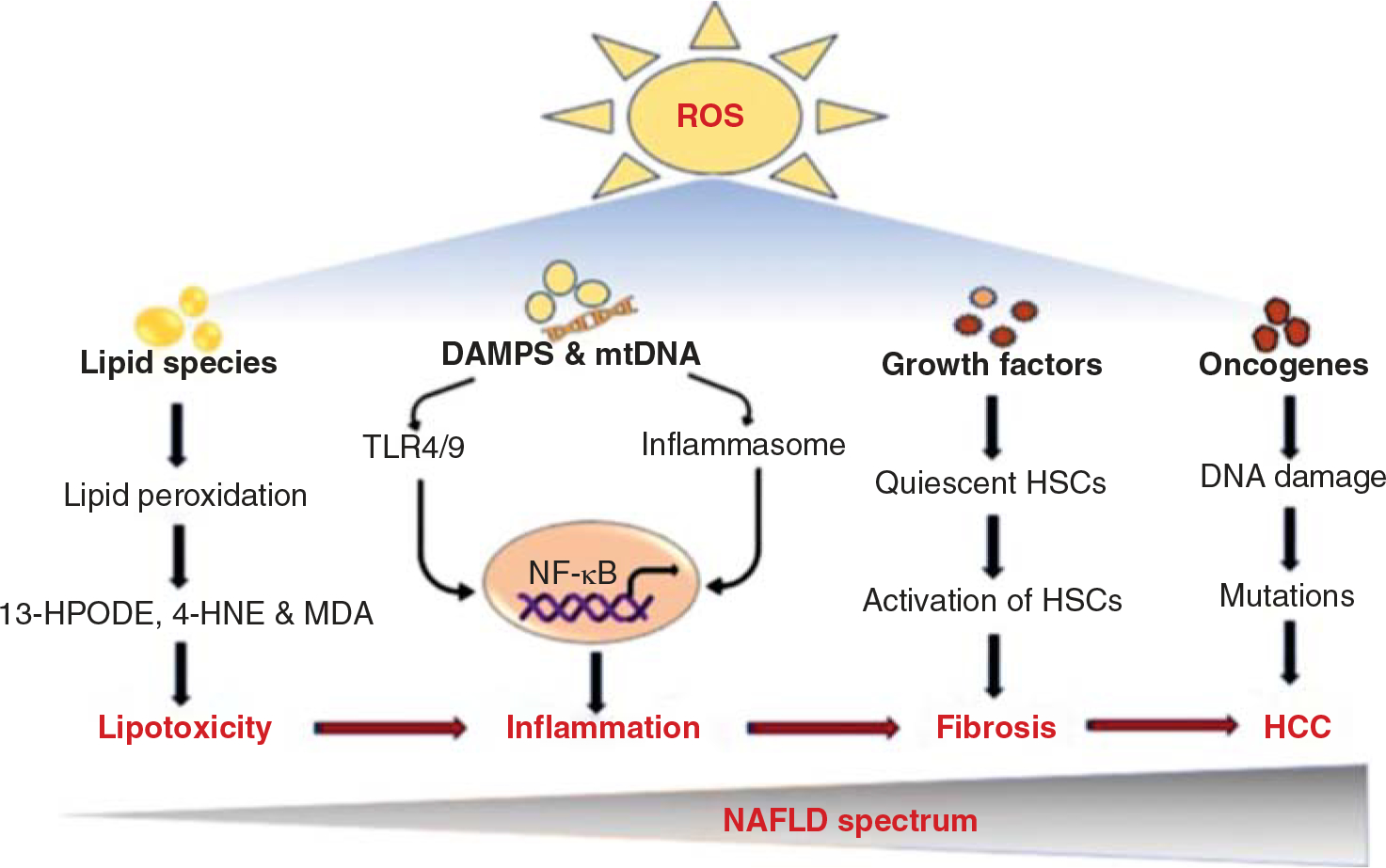
The mechanisms of ROS-induced oxidative stress in the pathogenesis of NAFLD. ROS can oxidize stored lipids through the process of lipid peroxidation, releasing lipid peroxidation reactive aldehydes, which result in lipotoxicity. Lipotoxicity involves in the production of several hepatic inflammatory mediators. ROS also increases the production of danger signals and mtDNA stimulating the innate immune system and inflammatory cytokines to promote liver inflammation. ROS-associated lipid peroxidation and cytokines contribute to the inflammatory cell infiltrate. On the other hand, ROS-mediated oxidative stress is a feature of liver fibrosis that activates HSCs by releasing several profibrotic stimuli and growth factors such as TGF-β, leptin, AGEs, and PDGF. Further, ROS induces DNA damage, resulting in cancer cell transformation.
Oxidative stress and liver inflammation
The accumulation of toxic lipids and lipid peroxides in the liver is one of the common manifestations of inflammation in both ALD and NAFLD/NASH. Inflammation is the physiological responses to tissue injury and infection represented by various inflammatory cells, including cytokines and other lipid mediators such as eicosanoids. Numerous studies have shown that the persistence of inflammatory cells and inflammatory mediators in the liver cells play a significant role in the pathogenies of ALD and NAFLD (345, 394). It is believed that liver inflammatory cells rapidly recruit and activate liver macrophages, neutrophils, and other immune cells that promote hepatic injury and thereby NASH progression (114). Further, liver inflammation is triggered by extrahepatic tissues such as the adipose tissue and the gut by releasing various lipid mediators and innate immune response mediators (68).
Most importantly, the common causative factor for the sustained activation of inflammatory cells during the NAFLD/NASH is oxidative stress. The lipid peroxide-induced oxidative stress plays a critical role in the NASH progression through inflammatory signaling. For instance, inflammation and oxidative stress are inter-linked with several liver diseases (69, 323). Under physiological levels, ROS helps to kill the pathogens and modulate signaling events through redox regulation (298). However, during hepatocyte injury, ROS from leukocytes triggers the release of damage-associated molecular patterns (DAMPs), including heat shock proteins, DNA, and RNA (19, 193). Studies have shown that DAMPs and their associated complexes activate the liver resident Kupffer cells and neutrophils via toll-like receptors (TLRs) (Figure 4) (261, 306). For example, TLR4 activates the superoxide-producing enzymes such as NADPH oxidase and MPO in the neutrophils and thereby promotes inflammation (300). Further, MPO is also shown to be involved in the production of hypochlorous acid and contributes to the formation of chlorotyrosine protein and oxidative stress protein adducts (35). Thus, all these changes result in increased production of ROS in various liver cells and help in the NASH progression.
The oxidative stress or reactive species in NAFLD/NASH also involves in the activation of various inflammatory mediators such as TNF-α, IL-1, IL-6, IL-18, and MCP-1 and some of the inflammatory pathways and among them, the most common is NF-κB activation. The ROS is known to activate the NF-κB through several inflammatory cytokines and oxidized lipids and also LPS (124). For example, H2O2 is shown to activate the NF-κB even at micromolar concentrations in human neutrophils and is further rescued by treatment with antioxidant N-acetylcysteine (344). The ROS-induced NF-κB activation takes place in various liver cells in NASH or injury models influencing the survival of hepatocytes, Kupffer cells, and HSCs (171). Thus, given its broad roles, NF-κB acts as the central player in various chronic liver diseases.
Oxidative stress and liver fibrosis/cirrhosis
Liver fibrosis is a complex phenomenon wherein continuous production and accumulation of extracellular matrix (ECM) in the liver occurs due to repeated injury and inflammation. Hepatic steatosis, inflammation, alcohol consumption, viral hepatitis, cholestasis, and iron overload are some of the major causative factors of liver fibrosis (25, 192, 396). The generation of ROS in the liver plays a critical role in the initiation and progression of fibrogenesis through its effect on hepatocytes, Kupffer cells, and HSCs (179, 239). The oxidative products and lipid peroxidation augment the production and release of other pro-fibrogenic factors such as inflammatory cytokines (17, 245). Mechanistically, ROS-driven hepatocyte injury increases the secretion of several pro-fibrotic mediators such as TNF-α and transforming growth factor (TGF-β), which further aggravate the inflammatory and fibrotic responses (Figure 4) (234). Oxidative stress in hepatocytes could also increase fibrogenesis through indirect mechanisms via suppression of antioxidant enzymes (GSH) in a NF-κB-dependent manner (236). In addition to the hepatocytes, Kupffer cells and resident macrophages produce a significant amount of ROS upon activation by TGF-β (150, 151). A study has shown that the treatment of macrophages with 4-HNE results in TGF-β-mediated myofibroblast activation and, thereby, fibrosis (213). Further, activation of Kupffer cells with profibrogenic toxins such as iron, copper, and dichlorobenzene generates abundant oxidative stress in the liver (213, 336). Hydrogen peroxide produced by the Kupffer cells increases collagen type 1 from HSCs (120). Similarly, HSCs co-cultured with stimulated neutrophils show a significant increase in the procollagen α1 expression due to the activation of HSCs from ROS released from the neutrophils (467). HSCs themselves express Nox1 and 2 involved in liver fibrosis in association with TLRs (200, 228). Thus, oxidative stress drives liver fibrosis through a complex mechanism that involves the activation of HSCs in the liver.
Oxidative stress and liver cancer
Cirrhosis often progresses to hepatocellular carcinoma (HCC), the leading cause of cancer-related deaths worldwide. Hepatitis B virus, hepatitis C virus, and metabolic diseases are some of the risk factors associated with the development of HCC (21, 395, 452). Oxidative stress plays a pivotal role in every stage of HCC (302, 357, 423). For example, in NASH, inflammation induced-ROS and RNS cause DNA damage and significantly impair the DNA repair mechanisms resulting in mutations leading to HCC (Figure 4) (81, 446). Recent studies have shown that oxidative stress directly regulates cancer cell proliferation, survival, invasion, and/or metastasis of HCC (2, 145, 427).
The role of oxidative stress in HCC was demonstrated with the decreased levels of antioxidant enzymes, including the SODs, glutathione reductase, and glutathione peroxidase. For example, under chronic HCV-induced oxidative stress, the glutathione levels were significantly reduced, and the ratio between the oxidized and reduced forms of glutathione was increased (64, 385). A recent study has shown that thioredoxin reductase and glutathione reductase-null mice are more susceptible to chemical-induced liver cancer through elevated DNA damage (262). Oxidative stress also represses the Nrf2-Keap1 complex and increases the expression of thioredoxin reductase and glutathione reductase above basal levels (386). Further, oxidative stress also enhances Nrf2 and 8-OHdG levels in HCC cell lines, and these effects could be rescued through antioxidant mechanisms (255). Oxidative stress also dysregulates autophagy mechanisms and thereby, activates Nrf2 leading to the proliferation and survival of HCC (24, 190). Although the Nrf2 is considered as an antioxidant, persistent activation of oxidative stress-induced Nrf2 is one of the critical mechanisms that control the development of HCC.
HCV infections also increase the expression of the NADPH oxidase family, thereby causing DNA damage and tissue remodeling (75, 96, 436). The interaction of core proteins of HCV with the inner mitochondrial enzymes results in ROS generation through the oxidation of glutathione and reduction of NADPH content in the liver (226, 419). The HBV-mediated ROS also alters DNA methylation and suppresses SOCS3 expression, which results in the proliferation of the cancer cells (454). The other important environmental factor associated with increased ROS production in the HCC patients is iron-related Fenton reactions. Several studies have demonstrated a strong link between iron toxicity and HCC via ROS generation (15, 125, 252).
Oxidative Stress-biomarkers
Estimation of serum alanine transaminase (ALT) and aspartate transaminase (AST) levels is the gold standard to evaluate liver injury. However, AST levels increase in conditions such as muscle injury, celiac disease, and even pregnancy. Moreover, 20% of patients with cirrhosis may have normal ALT levels. Therefore, several studies aimed at identifying a reliable biomarker for liver diseases. The estimation of oxidative stress and antioxidants have a significant impact on the clinical management of liver injuries. Direct measurement of ROS is highly variable depending on their species, origin, and hyperreactivity. Therefore, instead of considering actual free radical species, considering their reactive products such as lipids, DNA, amino acids, and glycated proteins could yield a better and reliable estimation of ROS in redox state (286). The by-products of ROS reaction with lipids are MDA, 4-HNE, and 8-isoprostane. Nucleic acid-derived oxidative products are 8-hydroxy-2′-deoxyguanosine (8-OH-2dG) and 8-hydroxyguanine (8-OH-G). Proteins or amino acid-derived oxidative products are hydroxyproline, 3-nitrotyrosine, and 2-oxohistidine. AGE products also serve as a valuable biomarker of oxidative stress. Different methodologies estimate these markers depending on the disease context. For example, thiobarbituric acid reactive substances (TBARS), MDA, and 4-HNE are commonly used to assess lipid peroxidation in the serum of NASH patients. Among these, MDA is the most indicative of oxidative stress (111). Oxidative stress markers can also be identified in the liver or serum by simple ELISA and fluorescence-based detection methods. Mass spectrometry could be the most powerful methodology to identify the species of ROS (45). The determination of ROS levels combined with the standard liver function tests will be highly beneficial in predicting the clinical outcome of liver diseases (13).
Oxidative Stress as a Therapeutic Approach
The generation or maintenance of a minimal amount of ROS is essential for proper liver function. It is clear from the literature that antioxidants play a significant role in balancing ROS levels. Antioxidants have a high affinity for reducing the reactivity of ROS-generated free radicals. The most commonly used antioxidants include curcumin, resveratrol, coffee, flavonoids, and silymarin. These antioxidants reduce the risk of liver injury by activating various signaling cascades (49, 247, 248, 334). For example, plant-derived flavonoids protect the liver from ROS-mediated damages such as inflammation, liver fibrosis, and cancer (224, 463). One of the most studied flavonoids as hepatoprotectant is silymarin, a complex of seven flavonolignans present in the milk of thistle extracts (98, 123). Similarly, the antioxidant potential of flavonolignans silybin is tested in a spectrum of NAFLD, including liver inflammation, fibrosis, and cirrhosis (97). Besides, silybin also improves mitochondrial function and thereby attenuates the progression of NAFLD to end-stage liver disease (356). In addition, the polyphenolic compound curcumin ameliorates oxidative stress in the liver by increasing the expression of Nrf2 and HO-1 and also various antioxidants such as glutamate-cysteine ligase, activating transcription factor (ATF), peroxiredoxin 3, SOD, and catalase. (77, 331, 443). Resveratrol, also a polyphenolic compound that has been shown to improve mitochondrial function by increasing AMPK and SIRT1 activity in the liver (147, 401). In comparison, quercetin reduces the risk of progression of liver fibrosis by attenuating the expression of TGF-β, CTGF, and collagen-1α (321, 444). Most importantly, vitamins, including vitamin E and C and superoxide scavengers such as N-acetylcysteine (NAC) and tempol are well known for their antioxidant properties. For example, vitamin E reduces the incidence of NAFLD and its progressive forms by limiting lipid peroxidation, inflammation, and hepatic fibrosis (159, 305, 316, 339). Recently, a study has shown that superoxide dismutase mimetics, NAC, and tempol attenuated the development of liver pathologies such as cell necrosis, fibrosis, and ER stress through suppressing mTORC1 signaling (66). Currently, vitamin E, D, C, and NAC are very effective in treating chronic liver cancer patients who do not respond well with interferon and chemotherapy-based treatments (85, 178, 242, 318). Thus, targeting oxidative stress in NAFLD patients provides immediate and effective therapy.
Conclusion
In modern redox biology and medicine, ROS signaling is one of the comprehensive and unified central homeostatic mechanisms at molecular, organellar, cellular, and tissue level. Several studies have emphasized its significance in health and diseases. Since the liver acts as the hub of various metabolic and detoxification processes, all most all the liver cells are involved in redox homeostasis. Any perturbation in hepatic redox homeostasis dysregulates the fundamental cellular processes such as insulin signaling, mitophagy, cell proliferation, and hypoxic signaling leading to the initiation and progression of liver diseases. Especially in NASH, oxidative stress plays a critical role in the pathogenesis, independent of heterogeneous cellular damage. Furthermore, oxidative stress is the hallmark of liver cirrhosis/cancer. However, targeting oxidative stress in liver-related diseases is partially successful because of its diverse role in regulating physiological processes such as mitophagy and hypoxic signaling. Thus, by considering all these traits, there has been continued interest in the therapeutic targeting of redox medicine/biology to treat liver-related diseases.
Didactic Synopsis.
Major teaching points
In biology and medicine, oxidative stress is defined as a “state of condition wherein the cellular pro-oxidant and antioxidant balance is altered in favor of a pro-oxidant state.”
The most common free radicals are (a) reactive oxygen species (ROS), such as superoxide anions (O2•−), hydroxyl radicals (HO•), and hydrogen peroxide (H2O2); and (b) reactive nitrogen species (RNS), such as nitric oxide (NO•) and peroxynitrite (ONOO−). In addition, several other reactive species, such as reactive sulfur species (RSS) and reactive carbonyl species (RCS), have been characterized.
ROS generation is driven by endogenous (mitochondrial electron transport chain and transmembrane NADPH oxidases) and exogenous mechanisms (nutrients, drugs, toxicants, and physical stressors).
The liver acts as the metabolic hub and the detoxication center in our body and, therefore, generates a significant amount of ROS.
All the cell types in the liver are equipped with a battery of enzymatic (superoxide anion dismutase, catalase, glutathione peroxidases) and nonenzymatic antioxidants (vitamins A, C, GSH) to defend against oxidative stress.
At physiological levels, ROS plays a significant role in diverse cellular mechanisms, including signal transduction, gene expression, mitophagy, cell proliferation, and immune regulation.
The key molecular mediators/regulators of Redox signaling are nuclear factor erythroid 2-related factor 2 (Nrf2), NF-κB, sirtuins, and forkhead box protein O (FOXO).
Imbalance in the liver Redox biology is one of the critical components of “multiple parallel-hit hypotheses” responsible for the initiation and progression of liver diseases, including nonacholic fatty liver diseases (NAFLD), liver cirrhosis, and HCC.
Studies have identified several biomarkers of oxidative stress such as protein carbonyls, AGEs, 4-hydroxynonenal, and malondialdehyde (MDA), which could be detected by ELISA, fluorescence-based methods, and mass spectrophotometry.
Redox-modulation by natural or pharmacological antioxidants appears to be an attractive and viable therapeutic option in several liver-related diseases.
Acknowledgment
This study was funded by the National Institute of Diabetes and Digestive and Kidney Diseases (DK110537) and Pittsburgh Liver Research Center Pilot and Feasibility grant (P30DK120531) to S.K.R.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Adams L, Franco MC, Estevez AG. Reactive nitrogen species in cellular signaling. Exp Biol Med (Maywood) 240: 711–717, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aggarwal V, Tuli HS, Varol A, Thakral F, Yerer MB, Sak K, Varol M, Jain A, Khan MA, Sethi G. Role of reactive oxygen species in cancer progression: Molecular mechanisms and recent advancements. Biomolecules 9: 735, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aharoni-Simon M, Hann-Obercyger M, Pen S, Madar Z, Tirosh O. Fatty liver is associated with impaired activity of PPARgamma-coactivator 1alpha (PGC1alpha) and mitochondrial biogenesis in mice. Lab Invest 91: 1018–1028, 2011. [DOI] [PubMed] [Google Scholar]
- 4.Akazawa Y, Nakashima R, Matsuda K, Okamaoto K, Hirano R, Kawasaki H, Miuma S, Miyaaki H, Malhi H, Abiru S, Itoh M, Kondo H, Fukuoka J, Nakao K, Nakashima M. Detection of DNA damage response in nonalcoholic fatty liver disease via p53-binding protein 1 nuclear expression. Mod Pathol 32: 997–1007, 2019. [DOI] [PubMed] [Google Scholar]
- 5.Aldini G, Orioli M, Carini M. Alpha, beta-unsaturated aldehydes adducts to actin and albumin as potential biomarkers of carbonylation damage. Redox Rep 12: 20–25, 2007. [DOI] [PubMed] [Google Scholar]
- 6.Aleffi S, Petrai I, Bertolani C, Parola M, Colombatto S, Novo E, Vizzutti F, Anania FA, Milani S, Rombouts K, Laffi G, Pinzani M, Marra F. Upregulation of proinflammatory and proangiogenic cytokines by leptin in human hepatic stellate cells. Hepatology 42: 1339–1348, 2005. [DOI] [PubMed] [Google Scholar]
- 7.Alexopoulos SJ, Chen SY, Brandon AE, Salamoun JM, Byrne FL, Garcia CJ, Beretta M, Olzomer EM, Shah DP, Philp AM, Hargett SR, Lawrence RT, Lee B, Sligar J, Carrive P, Tucker SP, Philp A, Lackner C, Turner N, Cooney GJ, Santos WL, Hoehn KL. Mitochondrial uncoupler BAM15 reverses diet-induced obesity and insulin resistance in mice. Nat Commun 11: 2397, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allaire M, Rautou PE, Codogno P, Lotersztajn S. Autophagy in liver diseases: Time for translation? J Hepatol 70: 985–998, 2019. [DOI] [PubMed] [Google Scholar]
- 9.Alric L, Orfila C, Carrere N, Beraud M, Carrera G, Lepert JC, Duffaut M, Pipy B, Vinel JP. Reactive oxygen intermediates and eicosanoid production by kupffer cells and infiltrated macrophages in acute and chronic liver injury induced in rats by CCl4. Inflamm Res 49: 700–707, 2000. [DOI] [PubMed] [Google Scholar]
- 10.Ambrogini E, Almeida M, Martin-Millan M, Paik JH, Depinho RA, Han L, Goellner J, Weinstein RS, Jilka RL, O’Brien CA, Manolagas SC. FoxO-mediated defense against oxidative stress in osteoblasts is indispensable for skeletal homeostasis in mice. Cell Metab 11: 136–146, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M. From NASH to HCC: Current concepts and future challenges. Nat Rev Gastroenterol Hepatol 16: 411–428, 2019. [DOI] [PubMed] [Google Scholar]
- 12.Apel K, Hirt H. Reactive oxygen species: Metabolism, oxidative stress, and signal transduction. Annu Rev Plant Biol 55: 373–399, 2004. [DOI] [PubMed] [Google Scholar]
- 13.Arauz J, Ramos-Tovar E, Muriel P. Redox state and methods to evaluate oxidative stress in liver damage: From bench to bedside. Ann Hepatol 15: 160–173, 2016. [DOI] [PubMed] [Google Scholar]
- 14.Araya J, Rodrigo R, Videla LA, Thielemann L, Orellana M, Pettinelli P, Poniachik J. Increase in long-chain polyunsaturated fatty acid n - 6/n - 3 ratio in relation to hepatic steatosis in patients with non-alcoholic fatty liver disease. Clin Sci (Lond) 106: 635–643, 2004. [DOI] [PubMed] [Google Scholar]
- 15.Asare GA, Mossanda KS, Kew MC, Paterson AC, Kahler-Venter CP, Siziba K. Hepatocellular carcinoma caused by iron overload: A possible mechanism of direct hepatocarcinogenicity. Toxicology 219:41–52, 2006. [DOI] [PubMed] [Google Scholar]
- 16.Ayala A, Munoz MF, Arguelles S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev 2014: 360438, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aziz IA, Yacoub M, Rashid L, Solieman A. Malondialdehyde; Lipid peroxidation plasma biomarker correlated with hepatic fibrosis in human Schistosoma mansoni infection. Acta Parasitol 60: 735–742, 2015. [DOI] [PubMed] [Google Scholar]
- 18.Baiocchini A, Del Nonno F, Taibi C, Visco-Comandini U, D’Offizi G, Piacentini M, Falasca L. Liver sinusoidal endothelial cells (LSECs) modifications in patients with chronic hepatitis C. Sci Rep 9: 8760, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bajwa E, Pointer CB, Klegeris A. The role of mitochondrial damage-associated molecular patterns in chronic neuroinflammation. Mediators Inflamm 2019: 4050796, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bakunina N, Pariante CM, Zunszain PA. Immune mechanisms linked to depression via oxidative stress and neuroprogression. Immunology 144: 365–373, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balogh J, Victor D 3rd, Asham EH, Burroughs SG, Boktour M, Saharia A, Li X, Ghobrial RM, Monsour HP Jr. Hepatocellular carcinoma: A review. J Hepatocell Carcinoma 3: 41–53, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bankoglu EE, Tschopp O, Schmitt J, Burkard P, Jahn D, Geier A, Stopper H. Role of PTEN in oxidative stress and DNA damage in the liver of whole-body pten haplodeficient mice. PLoS One 11: e0166956, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barrera G, Pizzimenti S, Daga M, Dianzani C, Arcaro A, Cetrangolo GP, Giordano G, Cucci MA, Graf M, Gentile F. Lipid peroxidation-derived aldehydes, 4-hydroxynonenal and malondialdehyde in aging-related disorders. Antioxidants (Basel) 7: 102, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bartolini D, Dallaglio K, Torquato P, Piroddi M, Galli F. Nrf2-p62 autophagy pathway and its response to oxidative stress in hepatocellular carcinoma. Transl Res 193: 54–71, 2018. [DOI] [PubMed] [Google Scholar]
- 25.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest 115: 209–218, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Battelli MG, Polito L, Bortolotti M, Bolognesi A. Xanthine oxidoreductase-derived reactive species: Physiological and pathological effects. Oxid Med Cell Longev 2016: 3527579, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bedossa P, Houglum K, Trautwein C, Holstege A, Chojkier M. Stimulation of collagen alpha 1(I) gene expression is associated with lipid peroxidation in hepatocellular injury: A link to tissue fibrosis? Hepatology 19: 1262–1271, 1994. [PubMed] [Google Scholar]
- 28.Begriche K, Massart J, Robin MA, Bonnet F, Fromenty B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 58: 1497–1507, 2013. [DOI] [PubMed] [Google Scholar]
- 29.Bell LN, Molleston JP, Morton MJ, Klipsch A, Saxena R, Vuppalanchi R, Chalasani N. Hepatic lipid peroxidation and cytochrome P-450 2E1 in pediatric nonalcoholic fatty liver disease and its subtypes. J Clin Gastroenterol 45: 800–807, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bellanti F, Villani R, Facciorusso A, VendemialeG, ServiddioG.Lipid oxidation products in the pathogenesis of non-alcoholic steatohepatitis. Free Radic Biol Med 111: 173–185, 2017. [DOI] [PubMed] [Google Scholar]
- 31.Benedict M, Zhang X. Non-alcoholic fatty liver disease: An expanded review. World J Hepatol 9: 715–732, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bernstein JD, Penniall R. Effects of chronic ethanol treatment upon rat liver mitochondria. Biochem Pharmacol 27: 2337–2342, 1978. [DOI] [PubMed] [Google Scholar]
- 33.Bhogal RH, Weston CJ, Velduis S, GDL H, Reynolds GM, Davies S, Nyguet-Thin L, Alfaifi M, Shepard EL, Boteon Y, Wallace L, Oo YH, Adams DH, Mirza DF, Mergental H, Muirhead G, Stephenson BTF, Afford SC. The reactive oxygen species-mitophagy signaling pathway regulates liver endothelial cell survival during ischemia/reperfusion injury. Liver Transpl 24: 1437–1452, 2018. [DOI] [PubMed] [Google Scholar]
- 34.Biel TG, Lee S, Flores-Toro JA, Dean JW, Go KL, Lee MH, Law BK, Law ME, Dunn WA Jr, Zendejas I, Behrns KE, Kim JS. Sirtuin 1 suppresses mitochondrial dysfunction of ischemic mouse livers in a mitofusin 2-dependent manner. Cell Death Differ 23: 279–290, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Binder V, Ljubojevic S, Haybaeck J, Holzer M, El-Gamal D, Schicho R, Pieske B, Heinemann A, Marsche G. The myeloperoxidase product hypochlorous acid generates irreversible high-density lipoprotein receptor inhibitors. Arterioscler Thromb Vasc Biol 33: 1020–1027, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Birk J, Meyer M, Aller I, Hansen HG, Odermatt A, Dick TP, Meyer AJ, Appenzeller-Herzog C. Endoplasmic reticulum: Reduced and oxidized glutathione revisited. J Cell Sci 126: 1604–1617, 2013. [DOI] [PubMed] [Google Scholar]
- 37.Black D, Lyman S, Qian T, Lemasters JJ, Rippe RA, Nitta T, Kim JS, Behrns KE. Transforming growth factor beta mediates hepatocyte apoptosis through Smad3 generation of reactive oxygen species. Biochimie 89: 1464–1473, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bonekamp NA, Volkl A, Fahimi HD, Schrader M. Reactive oxygen species and peroxisomes: Struggling for balance. Biofactors 35: 346–355, 2009. [DOI] [PubMed] [Google Scholar]
- 39.Boveris A Biochemistry of free radicals: From electrons to tissues. Medicina (B Aires) 58: 350–356, 1998. [PubMed] [Google Scholar]
- 40.Boveris A, Oshino N, Chance B. The cellular production of hydrogen peroxide. Biochem J 128: 617–630, 1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Braakman I, Hebert DN. Protein folding in the endoplasmic reticulum. Cold Spring Harb Perspect Biol 5: a013201, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Braet F, Wisse E. Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: A review. Comp Hepatol 1: 1, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303: 2011–2015, 2004. [DOI] [PubMed] [Google Scholar]
- 44.Bunik VI, Sievers C. Inactivation of the 2-oxo acid dehydrogenase complexes upon generation of intrinsic radical species. Eur J Biochem 269: 5004–5015, 2002. [DOI] [PubMed] [Google Scholar]
- 45.Butterfield DA, Gu L, Di Domenico F, Robinson RA. Mass spectrometry and redox proteomics: Applications in disease. Mass Spectrom Rev 33: 277–301, 2014. [DOI] [PubMed] [Google Scholar]
- 46.Cadenas S Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim Biophys Acta Bioenerg 1859: 940–950, 2018. [DOI] [PubMed] [Google Scholar]
- 47.Caldwell SH, Swerdlow RH, Khan EM, Iezzoni JC, Hespenheide EE, Parks JK, Parker WD Jr. Mitochondrial abnormalities in non-alcoholic steatohepatitis. J Hepatol 31: 430–434, 1999. [DOI] [PubMed] [Google Scholar]
- 48.Cao L, Quan XB, Zeng WJ, Yang XO, Wang MJ. Mechanism of hepatocyte apoptosis. J Cell Death 9: 19–29, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Casas-Grajales S, Muriel P. Antioxidants in liver health. World J Gastrointest Pharmacol Ther 6: 59–72, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Castro MC, Francini F, Gagliardino JJ, Massa ML. Lipoic acid prevents fructose-induced changes in liver carbohydrate metabolism: Role of oxidative stress. Biochim Biophys Acta 1840: 1145–1151, 2014. [DOI] [PubMed] [Google Scholar]
- 51.Catala A, Diaz M. Editorial: Impact of lipid peroxidation on the physiology and pathophysiology of cell membranes. Front Physiol 7: 423, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cederbaum AI. Alcohol metabolism. Clin Liver Dis 16: 667–685, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cederbaum AI, Lieber CS, Rubin E. Effects of chronic ethanol treatment of mitochondrial functions damage to coupling site I. Arch Biochem Biophys 165: 560–569, 1974. [DOI] [PubMed] [Google Scholar]
- 54.Cederbaum AI, Lu Y, Wu D. Role of oxidative stress in alcohol-induced liver injury. Arch Toxicol 83: 519–548, 2009. [DOI] [PubMed] [Google Scholar]
- 55.Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A 95: 11715–11720, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chang HC, Guarente L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol Metab 25: 138–145, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chang M, Xue J, Sharma V, Habtezion A. Protective role of hemeoxygenase-1 in gastrointestinal diseases. Cell Mol Life Sci 72: 1161–1173, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chapman RW, Morgan MY, Laulicht M, Hoffbrand AV, Sherlock S. Hepatic iron stores and markers of iron overload in alcoholics and patients with idiopathic hemochromatosis. Dig Dis Sci 27: 909–916, 1982. [DOI] [PubMed] [Google Scholar]
- 59.Chen L, Na R, Gu M, Salmon AB, Liu Y, Liang H, Qi W, Van Remmen H, Richardson A, Ran Q. Reduction of mitochondrial H2O2 by overexpressing peroxiredoxin 3 improves glucose tolerance in mice. Aging Cell 7: 866–878, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen MF, Huang CC, Liu PS, Chen CH, Shiu LY. Saikosaponin a and saikosaponin d inhibit proliferation and migratory activity of rat HSC-T6 cells. J Med Food 16: 793–800, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, Jacob L, Patwa R, Shah H, Xu K, Cross JR, Massague J. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 533: 493–498, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: Central role of complex III. J Biol Chem 278: 36027–36031, 2003. [DOI] [PubMed] [Google Scholar]
- 63.Chen Z, Tian R, She Z, Cai J, Li H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic Biol Med 152: 116–141, 2020. [DOI] [PubMed] [Google Scholar]
- 64.Cheng SB, Liu HT, Chen SY, Lin PT, Lai CY, Huang YC. Changes of oxidative stress, glutathione, and its dependent antioxidant enzyme activities in patients with hepatocellular carcinoma before and after tumor resection. PLoS One 12: e0170016, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cheung SH, Kwok WK, To KF, Lau JY. Anti-atherogenic effect of hydrogen sulfide by over-expression of cystathionine gamma-lyase (CSE) gene. PLoS One 9: e113038, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cho CS, Kowalsky AH, Namkoong S, Park SR, Wu S, Kim B, James A, Gu B, Semple IA, Tohamy MA, Solanki S, Cho US, Greenson JK, Shah YM, Kim M, Lee JH. Concurrent activation of growth factor and nutrient arms of mTORC1 induces oxidative liver injury. Cell Discov 5: 60, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cho CS, Park HW, Ho A, Semple IA, Kim B, Jang I, Park H, Reilly S, Saltiel AR, Lee JH. Lipotoxicity induces hepatic protein inclusions through TANK binding kinase 1-mediated p62/sequestosome 1 phosphorylation. Hepatology 68: 1331–1346, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Choi S, Diehl AM. Role of inflammation in nonalcoholic steatohepatitis. Curr Opin Gastroenterol 21: 702–707, 2005. [DOI] [PubMed] [Google Scholar]
- 69.Cichoz-Lach H, Michalak A. Oxidative stress as a crucial factor in liver diseases. World J Gastroenterol 20: 8082–8091, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cinaroglu A, Gao C, Imrie D, Sadler KC. Activating transcription factor 6 plays protective and pathological roles in steatosis due to endoplasmic reticulum stress in zebrafish. Hepatology 54: 495–508, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Clugston RD, Jiang H, Lee MX, Piantedosi R, Yuen JJ, Ramakrishnan R, Lewis MJ, Gottesman ME, Huang LS, Goldberg IJ, Berk PD, Blaner WS. Altered hepatic lipid metabolism in C57BL/6 mice fed alcohol: A targeted lipidomic and gene expression study. J Lipid Res 52: 2021–2031, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Contreras-Ferrat A, Llanos P, Vasquez C, Espinosa A, Osorio-Fuentealba C, Arias-Calderon M, Lavandero S, Klip A, Hidalgo C, Jaimovich E. Insulin elicits a ROS-activated and an IP(3)-dependent Ca(2)(+) release, which both impinge on GLUT4 translocation. J Cell Sci 127: 1911–1923, 2014. [DOI] [PubMed] [Google Scholar]
- 73.Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J 17: 1195–1214, 2003. [DOI] [PubMed] [Google Scholar]
- 74.Crispe IN. The liver as a lymphoid organ. Annu Rev Immunol 27: 147–163, 2009. [DOI] [PubMed] [Google Scholar]
- 75.Crosas-Molist E, Bertran E, Sancho P, Lopez-Luque J, Fernando J, Sanchez A, Fernandez M, Navarro E, Fabregat I. The NADPH oxidase NOX4 inhibits hepatocyte proliferation and liver cancer progression. Free Radic Biol Med 69: 338–347, 2014. [DOI] [PubMed] [Google Scholar]
- 76.Cuevasanta E, Moller MN, Alvarez B. Biological chemistry of hydrogen sulfide and persulfides. Arch Biochem Biophys 617: 9–25, 2017. [DOI] [PubMed] [Google Scholar]
- 77.Cunningham RP, Moore MP, Moore AN, Healy JC, Roberts MD, Rector RS, Martin JS. Curcumin supplementation mitigates NASH development and progression in female Wistar rats. Physiol Rep 6: e13789, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cyr A, Chambers L, Waltz PK, Whelan SP, Kohut L, Carchman E, Dyer M, Luciano J, Kautza B, Gomez HD, Otterbein LE, Rosengart MR, Shiva S, Zuckerbraun BS. Endotoxin engages mitochondrial quality control via an iNOS-reactive oxygen species signaling pathway in hepatocytes. Oxid Med Cell Longev 2019: 4745067, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dan Dunn J, Alvarez LA, Zhang X, Soldati T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol 6: 472–485, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dasgupta J, Kar S, Liu R, Joseph J, Kalyanaraman B, Remington SJ, Chen C, Melendez JA. Reactive oxygen species control senescence-associated matrix metalloproteinase-1 through c-Jun-N-terminal kinase. J Cell Physiol 225: 52–62, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Davalli P, Marverti G, Lauriola A, D’Arca D. Targeting oxidatively induced dna damage response in cancer: Opportunities for novel cancer therapies. Oxid Med Cell Longev 2018: 2389523, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Del Rio LA. ROS and RNS in plant physiology: An overview. J Exp Bot 66: 2827–2837, 2015. [DOI] [PubMed] [Google Scholar]
- 83.dela Pena A, Leclercq IA, Williams J, Farrell GC. NADPH oxidase is not an essential mediator of oxidative stress or liver injury in murine MCD diet-induced steatohepatitis. J Hepatol 46: 304–313, 2007. [DOI] [PubMed] [Google Scholar]
- 84.DeLeve LD. Liver sinusoidal endothelial cells and liver regeneration. J Clin Invest 123: 1861–1866, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Deng J, Liu AD, Hou GQ, Zhang X, Ren K, Chen XZ, Li SSC, Wu YS, Cao X. N-acetylcysteine decreases malignant characteristics of glioblastoma cells by inhibiting Notch2 signaling. J Exp Clin Cancer Res 38: 2, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.DeSantis DA, Ko CW, Liu Y, Liu X, Hise AG, Nunez G, Croniger CM. Alcohol-induced liver injury is modulated by Nlrp3 and Nlrc4 inflammasomes in mice. Mediators Inflamm 2013: 751374, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dhanasekaran DN, Reddy EP. JNK-signaling: A multiplexing hub in programmed cell death. Genes Cancer 8: 682–694, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Diao L, Auger C, Konoeda H, Sadri AR, Amini-Nik S, Jeschke MG. Hepatic steatosis associated with decreased beta-oxidation and mitochondrial function contributes to cell damage in obese mice after thermal injury. Cell Death Dis 9: 530, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ding HR, Tang ZT, Tang N, Zhu ZY, Liu HY, Pan CY, Hu AY, Lin YZ, Gou P, Yuan XW, Cai JH, Dong CL, Wang JL, Ren HZ. Protective properties of FOXO1 inhibition in a murine model of non-alcoholic fatty liver disease are associated with attenuation of ER stress and necroptosis. Front Physiol 11: 177, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ding RB, Bao J, Deng CX. Emerging roles of SIRT1 in fatty liver diseases. Int J Biol Sci 13: 852–867, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dixon LJ, Barnes M, Tang H, Pritchard MT, Nagy LE. Kupffer cells in the liver. Compr Physiol 3: 785–797, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dodson M, Darley-Usmar V, Zhang J. Cellular metabolic and autophagic pathways: Traffic control by redox signaling. Free Radic Biol Med 63: 207–221, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Duarte N, Coelho IC, Patarrao RS, Almeida JI, Penha-Goncalves C, Macedo MP. How inflammation impinges on NAFLD: A role for Kupffer cells. Biomed Res Int 2015: 984578, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.El Hadi H, Vettor R, Rossato M. Vitamin E as a treatment for nonalcoholic fatty liver disease: Reality or myth? Antioxidants (Basel) 7: 12, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ellis EM. Reactive carbonyls and oxidative stress: Potential for therapeutic intervention. Pharmacol Ther 115: 13–24, 2007. [DOI] [PubMed] [Google Scholar]
- 96.Eun HS, Cho SY, Joo JS, Kang SH, Moon HS, Lee ES, Kim SH, Lee BS. Gene expression of NOX family members and their clinical significance in hepatocellular carcinoma. Sci Rep 7: 11060, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Federico A, Dallio M, Loguercio C. Silymarin/silybin and chronic liver disease: A marriage of many years. Molecules 22: 191, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Feher J, Lengyel G. Silymarin in the prevention and treatment of liver diseases and primary liver cancer. Curr Pharm Biotechnol 13: 210–217, 2012. [DOI] [PubMed] [Google Scholar]
- 99.Feierman DE, Winston GW, Cederbaum AI. Ethanol oxidation by hydroxyl radicals: Role of iron chelates, superoxide, and hydrogen peroxide. Alcohol Clin Exp Res 9: 95–102, 1985. [DOI] [PubMed] [Google Scholar]
- 100.Ferber EC, Peck B, Delpuech O, Bell GP, East P, Schulze A. FOXO3a regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression. Cell Death Differ 19: 968–979, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fernandes R, Hosoya K, Pereira P. Reactive oxygen species downregulate glucose transport system in retinal endothelial cells. Am J Physiol Cell Physiol 300: C927–C936, 2011. [DOI] [PubMed] [Google Scholar]
- 102.Ferrere G, Leroux A, Wrzosek L, Puchois V, Gaudin F, Ciocan D, Renoud ML, Naveau S, Perlemuter G, Cassard AM. Activation of Kupffer Cells Is Associated with a Specific Dysbiosis Induced by Fructose or High Fat Diet in Mice. PLoS One 11: e0146177, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res 122: 877–902, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Forstermann U, Closs EI, Pollock JS, Nakane M, Schwarz P, Gath I, Kleinert H. Nitric oxide synthase isozymes. Characterization, purification, molecular cloning, and functions. Hypertension 23: 1121–1131, 1994. [DOI] [PubMed] [Google Scholar]
- 105.Forstermann U, Sessa WC. Nitric oxide synthases: Regulation and function. Eur Heart J 33: 829–837, 837a-837d, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Franchina DG, Dostert C, Brenner D. Reactive oxygen species: Involvement in T cell signaling and metabolism. Trends Immunol 39: 489–502, 2018. [DOI] [PubMed] [Google Scholar]
- 107.Fransen M, Nordgren M, Wang B, Apanasets O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochim Biophys Acta 1822: 1363–1373, 2012. [DOI] [PubMed] [Google Scholar]
- 108.Freitas-Rodriguez S, Folgueras AR, Lopez-Otin C. The role of matrix metalloproteinases in aging: Tissue remodeling and beyond. Biochim Biophys Acta Mol Cell Res 1864: 2015–2025, 2017. [DOI] [PubMed] [Google Scholar]
- 109.French SW, Morimoto M, Reitz RC, Koop D, Klopfenstein B, Estes K, Clot P, Ingelman-Sundberg M, Albano E. Lipid peroxidation, CYP2E1 and arachidonic acid metabolism in alcoholic liver disease in rats. J Nutr 127: 907S–911S, 1997. [DOI] [PubMed] [Google Scholar]
- 110.Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem 275: 2247–2250, 2000. [DOI] [PubMed] [Google Scholar]
- 111.Frijhoff J, Winyard PG, Zarkovic N, Davies SS, Stocker R, Cheng D, Knight AR, Taylor EL, Oettrich J, Ruskovska T, Gasparovic AC, Cuadrado A, Weber D, Poulsen HE, Grune T, Schmidt HH, Ghezzi P. Clinical relevance of biomarkers of oxidative stress. Antioxid Redox Signal 23: 1144–1170, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gadang V, Kohli R, Myronovych A, Hui DY, Perez-Tilve D, Jaeschke A. MLK3 promotes metabolic dysfunction induced by saturated fatty acid-enriched diet. Am J Physiol Endocrinol Metab 305: E549–E556, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gallagher PE, Brent TP. Human 3-methyladenine-DNA glycosylase: Demonstration of a stimulatory factor. Biochem Biophys Res Commun 101: 956–962, 1981. [DOI] [PubMed] [Google Scholar]
- 114.Gao B, Tsukamoto H. Inflammation in alcoholic and nonalcoholic fatty liver disease: friend or foe? Gastroenterology 150: 1704–1709, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gao J, Jiang Z, Wang S, Zhou Y, Shi X, Feng M. Endoplasmic reticulum stress of Kupffer cells involved in the conversion of natural regulatory T cells to Th17 cells in liver ischemia-reperfusion injury. J Gastroenterol Hepatol 31: 883–889, 2016. [DOI] [PubMed] [Google Scholar]
- 116.Gao W, Du X, Lei L, Wang H, Zhang M, Wang Z, Li X, Liu G, Li X. NEFA-induced ROS impaired insulin signalling through the JNK and p38MAPK pathways in non-alcoholic steatohepatitis. J Cell Mol Med 22: 3408–3422, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gao X, Zhang Y, Burwinkel B, Xuan Y, Holleczek B, Brenner H, Schottker B. The associations of DNA methylation alterations in oxidative stress-related genes with cancer incidence and mortality outcomes: A population-based cohort study. Clin Epigenetics 11: 14, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Garcia-Ruiz C, Fernandez-Checa JC. Mitochondrial oxidative stress and antioxidants balance in fatty liver disease. Hepatol Commun 2: 1425–1439, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Garcia-Ruiz I, Solis-Munoz P, Fernandez-Moreira D, Grau M, Colina F, Munoz-Yague T, Solis-Herruzo JA. High-fat diet decreases activity of the oxidative phosphorylation complexes and causes nonalcoholic steatohepatitis in mice. Dis Model Mech 7: 1287–1296, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Garcia-Trevijano ER, Iraburu MJ, Fontana L, Dominguez-Rosales JA, Auster A, Covarrubias-Pinedo A, Rojkind M. Transforming growth factor beta1 induces the expression of alpha1(I) procollagen mRNA by a hydrogen peroxide-C/EBPbeta-dependent mechanism in rat hepatic stellate cells. Hepatology 29: 960–970, 1999. [DOI] [PubMed] [Google Scholar]
- 121.Gerber PA, Rutter GA. The role of oxidative stress and hypoxia in pancreatic beta-cell dysfunction in diabetes mellitus. Antioxid Redox Signal 26: 501–518, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Giles GI, Jacob C. Reactive sulfur species: An emerging concept in oxidative stress. Biol Chem 383: 375–388, 2002. [DOI] [PubMed] [Google Scholar]
- 123.Gillessen A, Schmidt HH. Silymarin as supportive treatment in liver diseases: A narrative review. Adv Ther 37: 1279–1301, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gloire G, Legrand-Poels S, Piette J. NF-kappaB activation by reactive oxygen species: Fifteen years later. Biochem Pharmacol 72: 1493–1505, 2006. [DOI] [PubMed] [Google Scholar]
- 125.Gordeuk VR, McLaren CE, MacPhail AP, Deichsel G, Bothwell TH. Associations of iron overload in Africa with hepatocellular carcinoma and tuberculosis: Strachan’s 1929 thesis revisited. Blood 87: 3470–3476, 1996. [PubMed] [Google Scholar]
- 126.Gorlach A, Bertram K, Hudecova S, Krizanova O. Calcium and ROS: A mutual interplay. Redox Biol 6: 260–271, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gorlach A, Dimova EY, Petry A, Martinez-Ruiz A, Hernansanz-Agustin P, Rolo AP, Palmeira CM, Kietzmann T. Reactive oxygen species, nutrition, hypoxia and diseases: Problems solved? Redox Biol 6: 372–385, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gough DR, Cotter TG. Hydrogen peroxide: A Jekyll and Hyde signalling molecule. Cell Death Dis 2: e213, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Grisham MB. Methods to detect hydrogen peroxide in living cells: Possibilities and pitfalls. Comp Biochem Physiol A Mol Integr Physiol 165: 429–438, 2013. [DOI] [PubMed] [Google Scholar]
- 130.Guerra-Castellano A, Diaz-Quintana A, Perez-Mejias G, Elena-Real CA, Gonzalez-Arzola K, Garcia-Maurino SM, De la Rosa MA, Diaz-Moreno I. Oxidative stress is tightly regulated by cytochrome c phosphorylation and respirasome factors in mitochondria. Proc Natl Acad Sci U S A 115: 7955–7960, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Guillaumet-Adkins A, Yanez Y, Peris-Diaz MD, Calabria I, Palanca-Ballester C, Sandoval J. Epigenetics and oxidative stress in aging. Oxid Med Cell Longev 2017: 9175806, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Guo C, Sun L, Chen X, Zhang D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen Res 8: 2003–2014, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Guo X, Liu WL, Yang D, Shen ZQ, Qiu ZG, Jin M, Li JW. Hepatitis C virus infection induces endoplasmic reticulum stress and apoptosis in human fetal liver stem cells. J Pathol 248: 155–163, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gyorke S, Carnes C. Dysregulated sarcoplasmic reticulum calcium release: Potential pharmacological target in cardiac disease. Pharmacol Ther 119: 340–354, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Halliwell B Biochemistry of oxidative stress. Biochem Soc Trans 35: 1147–1150, 2007. [DOI] [PubMed] [Google Scholar]
- 136.Hammoutene A, Biquard L, Lasselin J, Kheloufi M, Tanguy M, Vion AC, Merian J, Colnot N, Loyer X, Tedgui A, Codogno P, Lotersztajn S, Paradis V, Boulanger CM, Rautou PE. A defect in endothelial autophagy occurs in patients with non-alcoholic steatohepatitis and promotes inflammation and fibrosis. J Hepatol 72: 528–538, 2020. [DOI] [PubMed] [Google Scholar]
- 137.Hammoutene A, Rautou PE. Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J Hepatol 70: 1278–1291, 2019. [DOI] [PubMed] [Google Scholar]
- 138.Han D, Johnson HS, Rao MP, Martin G, Sancheti H, Silkwood KH, Decker CW, Nguyen KT, Casian JG, Cadenas E, Kaplowitz N. Mitochondrial remodeling in the liver following chronic alcohol feeding to rats. Free Radic Biol Med 102: 100–110, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Han H, Hu J, Lau MY, Feng M, Petrovic LM, Ji C. Altered methylation andexpressionofER-associateddegradationfactorsinlong-termalcohol and constitutive ER stress-induced murine hepatic tumors. Front Genet 4: 224, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hang TC, Lauffenburger DA, Griffith LG, Stolz DB. Lipids promote survival, proliferation, and maintenance of differentiation of rat liver sinusoidal endothelial cells in vitro. Am J Physiol Gastrointest Liver Physiol 302: G375–G388, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hanschmann EM, Godoy JR, Berndt C, Hudemann C, Lillig CH. Thioredoxins, glutaredoxins, and peroxiredoxins--molecular mechanisms and health significance: From cofactors to antioxidants to redox signaling. Antioxid Redox Signal 19: 1539–1605, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Harbrecht BG, Billiar TR. The role of nitric oxide in Kupffer cell-hepatocyte interactions. Shock 3: 79–87, 1995. [PubMed] [Google Scholar]
- 143.Harmon JS, Bogdani M, Parazzoli SD, Mak SS, Oseid EA, Berghmans M, Leboeuf RC, Robertson RP. beta-Cell-specific overexpression of glutathione peroxidase preserves intranuclear MafA and reverses diabetes in db/db mice. Endocrinology 150: 4855–4862, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Haus JM, Thyfault JP. Therapeutic potential of carbonyl-scavenging carnosine derivative in metabolic disorders. J Clin Invest 128: 5198–5200, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.He JD, Wang Z, Li SP, Xu YJ, Yu Y, Ding YJ, Yu WL, Zhang RX, Zhang HM, Du HY. Vitexin suppresses autophagy to induce apoptosis in hepatocellular carcinoma via activation of the JNK signaling pathway. Oncotarget 7: 84520–84532, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.He Q, Fu Y, Ding X, Li D, Wang Z, Tian D, Yan W. High-mobility group box 1 induces endoplasmic reticulum stress and activates hepatic stellate cells. Lab Invest 98: 1200–1210, 2018. [DOI] [PubMed] [Google Scholar]
- 147.Heeboll S, Thomsen KL, Clouston A, Sundelin EI, Radko Y, Christensen LP, Ramezani-Moghadam M, Kreutzfeldt M, Pedersen SB, Jessen N, Hebbard L, George J, Gronbaek H. Effect of resveratrol on experimental non-alcoholic steatohepatitis. Pharmacol Res 95–96: 34–41, 2015. [DOI] [PubMed] [Google Scholar]
- 148.Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, Camporez JP, Shulman GI, Gordon JI, Hoffman HM, Flavell RA. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482: 179–185, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hennig P, Garstkiewicz M, Grossi S, Di Filippo M, French LE, Beer HD. The crosstalk between Nrf2 and Inflammasomes. Int J Mol Sci 19: 562, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Herrera B, Alvarez AM, Sanchez A, Fernandez M, Roncero C, Benito M, Fabregat I. Reactive oxygen species (ROS) mediates the mitochondrial-dependent apoptosis induced by transforming growth factor (beta) in fetal hepatocytes. FASEB J 15: 741–751, 2001. [DOI] [PubMed] [Google Scholar]
- 151.Herrera B, Murillo MM, Alvarez-Barrientos A, Beltran J, Fernandez M, Fabregat I. Source of early reactive oxygen species in the apoptosis induced by transforming growth factor-beta in fetal rat hepatocytes. Free Radic Biol Med 36: 16–26, 2004. [DOI] [PubMed] [Google Scholar]
- 152.Heymann F, Hammerich L, Storch D, Bartneck M, Huss S, Russeler V, Gassler N, Lira SA, Luedde T, Trautwein C, Tacke F. Hepatic macrophage migration and differentiation critical for liver fibrosis is mediated by the chemokine receptor C-C motif chemokine receptor 8 in mice. Hepatology 55: 898–909, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Holmstrom KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol 15: 411–421, 2014. [DOI] [PubMed] [Google Scholar]
- 154.Hu J, Zhang Z, Shen WJ, Azhar S. Cellular cholesterol delivery, intracellular processing and utilization for biosynthesis of steroid hormones. Nutr Metab (Lond) 7: 47, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hussain SP, Amstad P, He P, Robles A, Lupold S, Kaneko I, Ichimiya M, Sengupta S, Mechanic L, Okamura S, Hofseth LJ, Moake M, Nagashima M, Forrester KS, Harris CC. p53-induced up-regulation of MnSOD and GPx but not catalase increases oxidative stress and apoptosis. Cancer Res 64: 2350–2356, 2004. [DOI] [PubMed] [Google Scholar]
- 156.Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O, Watanabe S, Ando J, Iwadate M, Yamamoto M, Lee MS, Tanaka K, Komatsu M. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol 193: 275–284, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ioannou GN, Dominitz JA, Weiss NS, Heagerty PJ, Kowdley KV. The effect of alcohol consumption on the prevalence of iron overload, iron deficiency, and iron deficiency anemia. Gastroenterology 126: 1293–1301, 2004. [DOI] [PubMed] [Google Scholar]
- 158.Islam SMT, Won J, Khan M, Chavin KD, Singh I. Peroxisomal foot-print in the pathogenesis of nonalcoholic steatohepatitis. Ann Hepatol 19 (5): 466–471, 2020. [DOI] [PubMed] [Google Scholar]
- 159.Ivancovsky-Wajcman D, Fliss-Isakov N, Salomone F, Webb M, Shibolet O, Kariv R, Zelber-Sagi S. Dietary vitamin E and C intake is inversely associated with the severity of nonalcoholic fatty liver disease. Dig Liver Dis 51: 1698–1705, 2019. [DOI] [PubMed] [Google Scholar]
- 160.Jaeschke H, Hasegawa T. Role of neutrophils in acute inflammatory liver injury. Liver Int 26: 912–919, 2006. [DOI] [PubMed] [Google Scholar]
- 161.Ji AR, Ku SY, Cho MS, Kim YY, Kim YJ, Oh SK, Kim SH, Moon SY, Choi YM. Reactive oxygen species enhance differentiation of human embryonic stem cells into mesendodermal lineage. Exp Mol Med 42: 175–186, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ji C, Mehrian-Shai R, Chan C, Hsu YH, Kaplowitz N. Role of CHOP in hepatic apoptosis in the murine model of intragastric ethanol feeding. Alcohol Clin Exp Res 29: 1496–1503, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ju C, Tacke F. Hepatic macrophages in homeostasis and liver diseases: From pathogenesis to novel therapeutic strategies. Cell Mol Immunol 13: 316–327, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kabe Y, Ando K, Hirao S, Yoshida M, Handa H. Redox regulation of NF-kappaB activation: Distinct redox regulation between the cytoplasm and the nucleus. Antioxid Redox Signal 7: 395–403, 2005. [DOI] [PubMed] [Google Scholar]
- 165.Kakimoto PA, Tamaki FK, Cardoso AR, Marana SR, Kowaltowski AJ. H2O2 release from the very long chain acyl-CoA dehydrogenase. Redox Biol 4: 375–380, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kalpage HA, Wan J, Morse PT, Zurek MP, Turner AA, Khobeir A, Yazdi N, Hakim L, Liu J, Vaishnav A, Sanderson TH, Recanati MA, Grossman LI, Lee I, Edwards BFP, Huttemann M. Cytochrome c phosphorylation: Control of mitochondrial electron transport chain flux and apoptosis. Int J Biochem Cell Biol 121: 105704, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 120: 649–661, 2005. [DOI] [PubMed] [Google Scholar]
- 168.Kaminski M, Kiessling M, Suss D, Krammer PH, Gulow K. Novel role for mitochondria: Protein kinase Ctheta-dependent oxidative signaling organelles in activation-induced T-cell death. Mol Cell Biol 27: 3625–3639, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kang K, Reilly SM, Karabacak V, Gangl MR, Fitzgerald K, Hatano B, Lee CH. Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab 7: 485–495, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kant S, Barrett T, Vertii A, Noh YH, Jung DY, Kim JK, Davis RJ. Role of the mixed-lineage protein kinase pathway in the metabolic stress response to obesity. Cell Rep 4: 681–688, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Karin M NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol 1: a000141, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Karnati S, Luers G, Pfreimer S, Baumgart-Vogt E. Mammalian SOD2 is exclusively located in mitochondria and not present in peroxisomes. Histochem Cell Biol 140: 105–117, 2013. [DOI] [PubMed] [Google Scholar]
- 173.Kartchner MM. Stroke prevention. West J Med 130: 254–255, 1979. [PMC free article] [PubMed] [Google Scholar]
- 174.Kaur S, Tripathi D, Dongre K, Garg V, Rooge S, Mukopadhyay A, Sakhuja P, Sarin SK. Increased number and function of endothelial progenitor cells stimulate angiogenesis by resident liver sinusoidal endothelial cells (SECs) in cirrhosis through paracrine factors. J Hepatol 57: 1193–1198, 2012. [DOI] [PubMed] [Google Scholar]
- 175.Kawamata H, Manfredi G. Import, maturation, and function of SOD1 and its copper chaperone CCS in the mitochondrial intermembrane space. Antioxid Redox Signal 13: 1375–1384, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kawashima R, Mochida S, Matsui A, YouLuTu ZY, Ishikawa K, Toshima K, Yamanobe F, Inao M, Ikeda H, Ohno A, Nagoshi S, Uede T, Fujiwara K. Expression of osteopontin in Kupffer cells and hepatic macrophages and Stellate cells in rat liver after carbon tetrachloride intoxication: A possible factor for macrophage migration into hepatic necrotic areas. Biochem Biophys Res Commun 256: 527–531, 1999. [DOI] [PubMed] [Google Scholar]
- 177.Kendellen MF, Bradford JW, Lawrence CL, Clark KS, Baldwin AS. Canonical and non-canonical NF-kappaB signaling promotes breast cancer tumor-initiating cells. Oncogene 33: 1297–1305, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Khan S, Ali A, Khan S, Bakillah A, Damanhouri G, Khan A, Makki A, AlAnsari I, Banu N. Current therapies in alleviating liver disorders and cancers with a special focus on the potential of vitamin D. Nutr Metab (Lond) 15: 13, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Khomich O, Ivanov AV, Bartosch B. Metabolic hallmarks of hepatic stellate cells in liver fibrosis. Cells 9: 24, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kim I, Lemasters JJ. Mitochondrial degradation by autophagy (mitophagy) in GFP-LC3 transgenic hepatocytes during nutrient deprivation. Am J Physiol Cell Physiol 300: C308–C317, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kim SY, Jeong JM, Kim SJ, Seo W, Kim MH, Choi WM, Yoo W, Lee JH, Shim YR, Yi HS, Lee YS, Eun HS, Lee BS, Chun K, Kang SJ, Kim SC, Gao B, Kunos G, Kim HM, Jeong WI. Pro-inflammatory hepatic macrophages generate ROS through NADPH oxidase 2 via endocytosis of monomeric TLR4-MD2 complex. Nat Commun 8: 2247, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kimura H Physiological roles of hydrogen sulfide and polysulfides. Handb Exp Pharmacol 230: 61–81, 2015. [DOI] [PubMed] [Google Scholar]
- 183.Klaassen CD, Reisman SA. Nrf2 the rescue: Effects of the antioxidative/electrophilic response on the liver. Toxicol Appl Pharmacol 244: 57–65, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Klatt P, Lamas S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur J Biochem 267: 4928–4944, 2000. [DOI] [PubMed] [Google Scholar]
- 185.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 290: 1717–1721, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Klotz LO, Sanchez-Ramos C, Prieto-Arroyo I, Urbanek P, Steinbrenner H, Monsalve M. Redox regulation of FoxO transcription factors. Redox Biol 6: 51–72, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kluwe J, Pradere JP, Gwak GY, Mencin A, De Minicis S, Osterreicher CH, Colmenero J, Bataller R, Schwabe RF. Modulation of hepatic fibrosis by c-Jun-N-terminal kinase inhibition. Gastroenterology 138: 347–359, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Knock GA. NADPH oxidase in the vasculature: Expression, regulation and signalling pathways; role in normal cardiovascular physiology and its dysregulation in hypertension. Free Radic Biol Med 145: 385–427, 2019. [DOI] [PubMed] [Google Scholar]
- 189.Kolluru GK, Shen X, Kevil CG. Reactive sulfur species: A new redox player in cardiovascular pathophysiology. Arterioscler Thromb Vasc Biol 40: 874–884, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, Kim M, Nishito Y, Iemura S, Natsume T, Ueno T, Kominami E, Motohashi H, Tanaka K, Yamamoto M. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 12: 213–223, 2010. [DOI] [PubMed] [Google Scholar]
- 191.Konishi M, Iwasa M, Araki J, Kobayashi Y, Katsuki A, Sumida Y, Nakagawa N, Kojima Y, Watanabe S, Adachi Y, Kaito M. Increased lipid peroxidation in patients with non-alcoholic fatty liver disease and chronic hepatitis C as measured by the plasma level of 8-isoprostane. J Gastroenterol Hepatol 21: 1821–1825, 2006. [DOI] [PubMed] [Google Scholar]
- 192.Koyama Y, Brenner DA. Liver inflammation and fibrosis. J Clin Invest 127: 55–64, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, Lambrecht BN, Vandenabeele P. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol 32: 157–164, 2011. [DOI] [PubMed] [Google Scholar]
- 194.Kuhla A, Trieglaff C, Vollmar B. Role of age and uncoupling protein-2 in oxidative stress, RAGE/AGE interaction and inflammatory liver injury. Exp Gerontol 46: 868–876, 2011. [DOI] [PubMed] [Google Scholar]
- 195.Kushnareva Y, Murphy AN, Andreyev A. Complex I-mediated reactive oxygen species generation: Modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem J 368: 545–553, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Lacroix M, Riscal R, Arena G, Linares LK, Le Cam L. Metabolic functions of the tumor suppressor p53: Implications in normal physiology, metabolic disorders, and cancer. Mol Metab 33: 2–22, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol 4: 181–189, 2004. [DOI] [PubMed] [Google Scholar]
- 198.Lamkanfi M, Vande Walle L, Kanneganti TD. Deregulated inflammasome signaling in disease. Immunol Rev 243: 163–173, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Lamle J, Marhenke S, Borlak J, von Wasielewski R, Eriksson CJ, Geffers R, Manns MP, Yamamoto M, Vogel A. Nuclear factor-eythroid 2-related factor 2 prevents alcohol-induced fulminant liver injury. Gastroenterology 134: 1159–1168, 2008. [DOI] [PubMed] [Google Scholar]
- 200.Lan T, Kisseleva T, Brenner DA. Deficiency of NOX1 or NOX4 Prevents Liver Inflammation and Fibrosis in Mice through Inhibition of Hepatic Stellate Cell Activation. PLoS One 10: e0129743, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Langhans W Role of the liver in the control of glucose-lipid utilization and body weight. Curr Opin Clin Nutr Metab Care 6: 449–455, 2003. [DOI] [PubMed] [Google Scholar]
- 202.Larosche I, Choumar A, Fromenty B, Letteron P, Abbey-Toby A, Van Remmen H, Epstein CJ, Richardson A, Feldmann G, Pessayre D, Mansouri A. Prolonged ethanol administration depletes mitochondrial DNA in MnSOD-overexpressing transgenic mice, but not in their wild type littermates. Toxicol Appl Pharmacol 234: 326–338, 2009. [DOI] [PubMed] [Google Scholar]
- 203.Lau N, Pluth MD. Reactive sulfur species (RSS): Persulfides, polysulfides, potential, and problems. Curr Opin Chem Biol 49: 1–8, 2019. [DOI] [PubMed] [Google Scholar]
- 204.Lawrence RE, Zoncu R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat Cell Biol 21: 133–142, 2019. [DOI] [PubMed] [Google Scholar]
- 205.Lazarow PB. The role of peroxisomes in mammalian cellular metabolism. J Inherit Metab Dis 10 (Suppl 1): 11–22, 1987. [DOI] [PubMed] [Google Scholar]
- 206.Lazarus JV, Colombo M, Cortez-Pinto H, Huang TT, Miller V, Ninburg M, Schattenberg JM, Seim L, Wong VWS, Zelber-Sagi S. NAFLD - sounding the alarm on a silent epidemic. Nat Rev Gastroenterol Hepatol 17: 377–379, 2020. [DOI] [PubMed] [Google Scholar]
- 207.Lebeaupin C, Vallee D, Hazari Y, Hetz C, Chevet E, Bailly-Maitre B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J Hepatol 69: 927–947, 2018. [DOI] [PubMed] [Google Scholar]
- 208.Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem J 441: 523–540, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Lee S, Kim S, Hwang S, Cherrington NJ, Ryu DY. Dysregulated expression of proteins associated with ER stress, autophagy and apoptosis in tissues from nonalcoholic fatty liver disease. Oncotarget 8: 63370–63381, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Lee S, Tak E, Lee J, Rashid MA, Murphy MP, Ha J, Kim SS. Mitochondrial H2O2 generated from electron transport chain complex I stimulates muscle differentiation. Cell Res 21: 817–834, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Lee SJ, Hwang AB, Kenyon C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr Biol 20: 2131–2136, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Lemasters JJ, Zhong Z. Mitophagy in hepatocytes: Types, initiators and role in adaptive ethanol metabolism. Liver Res 2: 125–132, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Leonarduzzi G, Scavazza A, Biasi F, Chiarpotto E, Camandola S, Vogel S, Dargel R, Poli G. The lipid peroxidation end product 4-hydroxy-2,3-nonenal up-regulates transforming growth factor beta1 expression in the macrophage lineage: A link between oxidative injury and fibrosclerosis. FASEB J 11: 851–857, 1997. [DOI] [PubMed] [Google Scholar]
- 214.Leveille M, Estall JL. Mitochondrial dysfunction in the transition from NASH to HCC. Metabolites 9: 233, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Li H, Zhou Y, Wang H, Zhang M, Qiu P, Zhang M, Zhang R, Zhao Q, Liu J. Crosstalk between liver macrophages and surrounding cells in nonalcoholic steatohepatitis. Front Immunol 11: 1169, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Li J, Zhao YR, Tian Z. Roles of hepatic stellate cells in acute liver failure: From the perspective of inflammation and fibrosis. World J Hepatol 11: 412–420, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Li JJ, Li Q, Du HP, Wang YL, You SJ, Wang F, Xu XS, Cheng J, Cao YJ, Liu CF, Hu LF. Homocysteine triggers inflammatory responses in macrophages through inhibiting CSE-H2S signaling via DNA hypermethylation of CSE promoter. Int J Mol Sci 16: 12560–12577, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Li L, Chen K, Xiang Y, Yoshimura T, Su S, Zhu J, Bian XW, Wang JM. New development in studies of formyl-peptide receptors: Critical roles in host defense. J Leukoc Biol 99: 425–435, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Li L, Tan J, Miao Y, Lei P, Zhang Q. ROS and autophagy: Interactions and molecular regulatory mechanisms. Cell Mol Neurobiol 35: 615–621, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Li L, Yang X. The essential element manganese, oxidative stress, and metabolic diseases: Links and interactions. Oxid Med Cell Longev 2018: 7580707, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Li M, Guo K, Vanella L, Taketani S, Adachi Y, Ikehara S. Stem cell transplantation upregulates Sirt1 and antioxidant expression, ameliorating fatty liver in type 2 diabetic mice. Int J Biol Sci 11: 472–481, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Li Q, Zhao Z. Influence of N-acetyl-L-cysteine against bisphenol a on the maturation of mouse oocytes and embryo development: In vitro study. BMC Pharmacol Toxicol 20: 43, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Li S, Dou X, Ning H, Song Q, Wei W, Zhang X, Shen C, Li J, Sun C, Song Z. Sirtuin 3 acts as a negative regulator of autophagy dictating hepatocyte susceptibility to lipotoxicity. Hepatology 66: 936–952, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Li S, Tan HY, Wang N, Cheung F, Hong M, Feng Y. The potential and action mechanism of polyphenols in the treatment of liver diseases. Oxid Med Cell Longev 2018: 8394818, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Li S, Wang X, Wu Y, Zhang H, Zhang L, Wang C, Zhang R, Guo Z. 8-Hydroxy-2’-deoxyguanosine expression predicts hepatocellular carcinoma outcome. Oncol Lett 3: 338–342, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Li XD, Sun L, Seth RB, Pineda G, Chen ZJ. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci U S A 102: 17717–17722, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Li Y, Chen M, Xu Y, Yu X, Xiong T, Du M, Sun J, Liu L, Tang Y, Yao P. Iron-mediated lysosomal membrane permeabilization in ethano-linduced hepatic oxidative damage and apoptosis: Protective effects of quercetin. Oxid Med Cell Longev 2016: 4147610, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Liang S, Kisseleva T, Brenner DA. The role of NADPH oxidases (NOXs) in liver fibrosis and the activation of myofibroblasts. Front Physiol 7: 17, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Liguori I, Russo G, Curcio F, Bulli G, Aran L, Della-Morte D, Gargiulo G, Testa G, Cacciatore F, Bonaduce D, Abete P. Oxidative stress, aging, and diseases. Clin Interv Aging 13: 757–772, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Litwak SA, Pang L, Galic S, Igoillo-Esteve M, Stanley WJ, Turatsinze JV, Loh K, Thomas HE, Sharma A, Trepo E, Moreno C, Gough DJ, Eizirik DL, de Haan JB, Gurzov EN. JNK activation of BIM promotes hepatic oxidative stress, steatosis, and insulin resistance in obesity. Diabetes 66: 2973–2986, 2017. [DOI] [PubMed] [Google Scholar]
- 231.Liu H, Qi X, Cao S, Li P. Protective effect of flavonoid extract from Chinese bayberry (Myrica rubra Sieb. et Zucc.) fruit on alcoholic liver oxidative injury in mice. J Nat Med 68: 521–529, 2014. [DOI] [PubMed] [Google Scholar]
- 232.Liu J, Li D, Zhang T, Tong Q, Ye RD, Lin L. SIRT3 protects hepatocytes from oxidative injury by enhancing ROS scavenging And mitochondrial integrity. Cell Death Dis 8: e3158, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Liu J, Wu KC, Lu YF, Ekuase E, Klaassen CD. Nrf2 protection against liver injury produced by various hepatotoxicants. Oxid Med Cell Longev 2013: 305861, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Liu RM, Desai LP. Reciprocal regulation of TGF-beta and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol 6: 565–577, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Loomba R, Lawitz E, Mantry PS, Jayakumar S, Caldwell SH, Arnold H, Diehl AM, Djedjos CS, Han L, Myers RP, Subramanian GM, McHutchison JG, Goodman ZD, Afdhal NH, Charlton MR, Investigators G-U. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology 67: 549–559, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Lou H, Kaplowitz N. Glutathione depletion down-regulates tumor necrosis factor alpha-induced NF-kappaB activity via IkappaB kinase-dependent and -independent mechanisms. J Biol Chem 282: 29470–29481, 2007. [DOI] [PubMed] [Google Scholar]
- 237.Lu H, Lei X, Zhang Q. Moderate activation of IKK2-NF-kB in unstressed adult mouse liver induces cytoprotective genes and lipogenesis without apparent signs of inflammation or fibrosis. BMC Gastroenterol 15: 94, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Luan J, Ju D. Inflammasome: A double-edged sword in liver diseases. Front Immunol 9: 2201, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Luangmonkong T, Suriguga S, Mutsaers HAM, Groothuis GMM, Olinga P, Boersema M. Targeting oxidative stress for the treatment of liver fibrosis. Rev Physiol Biochem Pharmacol 175: 71–102, 2018. [DOI] [PubMed] [Google Scholar]
- 240.Luedde T, Schwabe RF. NF-kappaB in the liver--linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol 8: 108–118, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Lushchak VI. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem Biol Interact 224: 164–175, 2014. [DOI] [PubMed] [Google Scholar]
- 242.Lv H, Wang C, Fang T, Li T, Lv G, Han Q, Yang W, Wang H. Vitamin C preferentially kills cancer stem cells in hepatocellular carcinoma via SVCT-2. NPJ Precis Oncol 2: 1, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Ma X, McKeen T, Zhang J, Ding WX. Role and mechanisms of mitophagy in liver diseases. Cells 9: 837, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Ma Y, Chai H, Ding Q, Qian Q, Yan Z, Ding B, Dou X, Li S. Hepatic SIRT3 upregulation in response to chronic alcohol consumption contributes to alcoholic liver disease in mice. Front Physiol 10: 1042, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.MacDonald GA, Bridle KR, Ward PJ, Walker NI, Houglum K, George DK, Smith JL, Powell LW, Crawford DH, Ramm GA. Lipid peroxidation in hepatic steatosis in humans is associated with hepatic fibrosis and occurs predominately in acinar zone 3. J Gastroenterol Hepatol 16: 599–606, 2001. [DOI] [PubMed] [Google Scholar]
- 246.Machado MV, Diehl AM. Pathogenesis of nonalcoholic steatohepatitis. Gastroenterology 150: 1769–1777, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Mahesh M, Bharathi M, Raja Gopal Reddy M, Pappu P, Putcha UK, Vajreswari A, Jeyakumar SM. Carrot juice ingestion attenuates high fructose-induced circulatory pro-inflammatory mediators in weanling Wistar rats. J Sci Food Agric 97: 1582–1591, 2017. [DOI] [PubMed] [Google Scholar]
- 248.Mahesh M, Bharathi M, Reddy MR, Kumar MS, Putcha UK, Vajreswari A, Jeyakumar SM. Carrot juice administration decreases liver stearoyl-CoA desaturase 1 and improves docosahexaenoic acid levels, but not steatosis in high fructose diet-fed weanling Wistar rats. Prev Nutr Food Sci 21: 171–180, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Mailloux RJ. Mitochondrial antioxidants and the maintenance of cellular hydrogen peroxide levels. Oxid Med Cell Longev 2018: 7857251, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Mailloux RJ, McBride SL, Harper ME. Unearthing the secrets of mitochondrial ROS and glutathione in bioenergetics. Trends Biochem Sci 38: 592–602, 2013. [DOI] [PubMed] [Google Scholar]
- 251.Malik AN, Simoes ICM, Rosa HS, Khan S, Karkucinska-Wieckowska A, Wieckowski MR. A diet induced maladaptive increase in hepatic mitochondrial DNA precedes OXPHOS defects and may contribute to non-alcoholic fatty liver disease. Cells 8: 1222, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Mandishona E, MacPhail AP, Gordeuk VR, Kedda MA, Paterson AC, Rouault TA, Kew MC. Dietary iron overload as a risk factor for hepatocellular carcinoma in Black Africans. Hepatology 27: 1563–1566, 1998. [DOI] [PubMed] [Google Scholar]
- 253.Mano J Reactive carbonyl species: Their production from lipid peroxides, action in environmental stress, and the detoxification mechanism. Plant Physiol Biochem 59: 90–97, 2012. [DOI] [PubMed] [Google Scholar]
- 254.Mano J, Biswas MS, Sugimoto K. Reactive carbonyl species: A missing link in ROS signaling. Plants (Basel) 8: 391, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Ma-On C, Sanpavat A, Whongsiri P, Suwannasin S, Hirankarn N, Tangkijvanich P, Boonla C. Oxidative stress indicated by elevated expression of Nrf2 and 8-OHdG promotes hepatocellular carcinoma progression. Med Oncol 34: 57, 2017. [DOI] [PubMed] [Google Scholar]
- 256.Mari M, Colell A, Morales A, von Montfort C, Garcia-Ruiz C, Fernandez-Checa JC. Redox control of liver function in health and disease. Antioxid Redox Signal 12: 1295–1331, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Maslak E, Gregorius A, Chlopicki S. Liver sinusoidal endothelial cells (LSECs) function and NAFLD; NO-based therapy targeted to the liver. Pharmacol Rep 67: 689–694, 2015. [DOI] [PubMed] [Google Scholar]
- 258.Massoud O,CharltonM.Nonalcoholicfattyliverdisease/nonalcoholic steatohepatitis and hepatocellular carcinoma. Clin Liver Dis 22: 201–211, 2018. [DOI] [PubMed] [Google Scholar]
- 259.Matsumoto M, Zhang J, Zhang X, Liu J, Jiang JX, Yamaguchi K, Taruno A, Katsuyama M, Iwata K, Ibi M, Cui W, Matsuno K, Marunaka Y, Itoh Y, Torok NJ, Yabe-Nishimura C. The NOX1 isoform of NADPH oxidase is involved in dysfunction of liver sinusoids in nonalcoholic fatty liver disease. Free Radic Biol Med 115: 412–420, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.McCarroll SA, Murphy CT, Zou S, Pletcher SD, Chin CS, Jan YN, Kenyon C, Bargmann CI, Li H. Comparing genomic expression patterns across species identifies shared transcriptional profile in aging. Nat Genet 36: 197–204, 2004. [DOI] [PubMed] [Google Scholar]
- 261.McKeown-Longo PJ, Higgins PJ. Integration of canonical and noncanonical pathways in TLR4 signaling: Complex regulation of the wound repair program. Adv Wound Care (New Rochelle) 6: 320–329, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.McLoughlin MR, Orlicky DJ, Prigge JR, Krishna P, Talago EA, Cavigli IR, Eriksson S, Miller CG, Kundert JA, Sayin VI, Sabol RA, Heinemann J, Brandenberger LO, Iverson SV, Bothner B, Papagian-nakopoulos T, Shearn CT, Arner ESJ, Schmidt EE. TrxR1, Gsr, and oxidative stress determine hepatocellular carcinoma malignancy. Proc Natl Acad Sci U S A 116: 11408–11417, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.McMahan RH, Porsche CE, Edwards MG, Rosen HR. Free fatty acids differentially downregulate chemokines in liver sinusoidal endothelial cells: Insights into non-alcoholic fatty liver disease. PLoS One 11: e0159217, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Meakin PJ, Chowdhry S, Sharma RS, Ashford FB, Walsh SV, McCrimmon RJ, Dinkova-Kostova AT, Dillon JF, Hayes JD, Ashford ML. Susceptibility of Nrf2-null mice to steatohepatitis and cirrhosis upon consumption of a high-fat diet is associated with oxidative stress, perturbation of the unfolded protein response, and disturbance in the expression of metabolic enzymes but not with insulin resistance. Mol Cell Biol 34: 3305–3320, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Mello T, Zanieri F, Ceni E, Galli A. Oxidative stress in the healthy and wounded hepatocyte: A cellular organelles perspective. Oxid Med Cell Longev 2016: 8327410, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Mesquita A, Weinberger M, Silva A, Sampaio-Marques B, Almeida B, Leao C, Costa V, Rodrigues F, Burhans WC, Ludovico P. Caloric restriction or catalase inactivation extends yeast chronological lifespan by inducing H2O2 and superoxide dismutase activity. Proc Natl Acad Sci U S A 107: 15123–15128, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Meyer J, Balaphas A, Fontana P, Morel P, Robson SC, Sadoul K, Gonelle-Gispert C, Buhler L. Platelet interactions with liver sinusoidal endothelial cells and hepatic stellate cells lead to hepatocyte proliferation. Cells 9, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Mittler R ROS are good. Trends Plant Sci 22: 11–19, 2017. [DOI] [PubMed] [Google Scholar]
- 269.Miura K, Yang L, van Rooijen N, Ohnishi H, Seki E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am J Physiol Gastrointest Liver Physiol 302: G1310–G1321, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Mohanty P, Ghanim H, Hamouda W, Aljada A, Garg R, Dandona P. Both lipid and protein intakes stimulate increased generation of reactive oxygen species by polymorphonuclear leukocytes and mononuclear cells. Am J Clin Nutr 75: 767–772, 2002. [DOI] [PubMed] [Google Scholar]
- 271.Molina DK, DiMaio VJ. Normal organ weights in men: Part II-the brain, lungs, liver, spleen, and kidneys. Am J Forensic Med Pathol 33: 368–372, 2012. [DOI] [PubMed] [Google Scholar]
- 272.Montosi G, Garuti C, Iannone A, Pietrangelo A. Spatial and temporal dynamics of hepatic stellate cell activation during oxidant-stress-induced fibrogenesis. Am J Pathol 152: 1319–1326, 1998. [PMC free article] [PubMed] [Google Scholar]
- 273.Mooli RGR, Mukhi D, Chen Z, Buckner N, Ramakrishnan SK. An indispensable role for dynamin-related protein 1 in beige and brown adipogenesis. J Cell Sci 133: jcs247593, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Mooli RGR, Mukhi D, Watt M, Edmunds L, Xie B, Capooci J, Reslink M, Eze C, Mills A, Stolz DB, Jurczak M, Ramakrishnan SK. Sustained mitochondrial biogenesis is essential to maintain caloric restriction-induced beige adipocytes. Metabolism 107: 154225, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NFkappaB signaling. Cell Res 21: 103–115, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Morimoto M, Zern MA, Hagbjork AL, Ingelman-Sundberg M, French SW. Fish oil, alcohol, and liver pathology: Role of cytochrome P450 2E1. Proc Soc Exp Biol Med 207: 197–205, 1994. [DOI] [PubMed] [Google Scholar]
- 277.Moshfegh CM, Collins CW, Gunda V, Vasanthakumar A, Cao JZ, Singh PK, Godley LA, Case AJ. Mitochondrial superoxide disrupts the metabolic and epigenetic landscape of CD4(+) and CD8(+) T-lymphocytes. Redox Biol 27: 101141, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, Haczeyni F, Teoh NC, Savard C, Ioannou GN, Masters SL, Schroder K, Cooper MA, Feldstein AE, Farrell GC. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol 66: 1037–1046, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Muller M, Ahumada-Castro U, Sanhueza M, Gonzalez-Billault C, Court FA, Cardenas C. Mitochondria and calcium regulation as basis of neurodegeneration associated with aging. Front Neurosci 12: 470, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 417: 1–13, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Murphy MP, Hartley RC. Mitochondria as a therapeutic target for common pathologies. Nat Rev Drug Discov 17: 865–886, 2018. [DOI] [PubMed] [Google Scholar]
- 282.Murphy MP, Holmgren A,Larsson NG,Halliwell B,Chang CJ,Kalyanaraman B, Rhee SG, Thornalley PJ, Partridge L, Gems D, Nystrom T, Belousov V, Schumacker PT, Winterbourn CC. Unraveling the biological roles of reactive oxygen species. Cell Metab 13: 361–366, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Nassir F, Ibdah JA. Role of mitochondria in nonalcoholic fatty liver disease. Int J Mol Sci 15: 8713–8742, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Nemeth E, Baird AW, O’Farrelly C. Microanatomy of the liver immune system. Semin Immunopathol 31: 333–343, 2009. [DOI] [PubMed] [Google Scholar]
- 285.Niedzwiecki MM, Walker DI, Vermeulen R, Chadeau-Hyam M, Jones DP, Miller GW. The exposome: Molecules to populations. Annu Rev Pharmacol Toxicol 59: 107–127, 2019. [DOI] [PubMed] [Google Scholar]
- 286.Niki E, Yoshida Y. Biomarkers for oxidative stress: Measurement, validation, and application. J Med Invest 52 (Suppl): 228–230, 2005. [DOI] [PubMed] [Google Scholar]
- 287.Nohl H, Gille L, Staniek K. Intracellular generation of reactive oxygen species by mitochondria. Biochem Pharmacol 69: 719–723, 2005. [DOI] [PubMed] [Google Scholar]
- 288.Nordgren M, Fransen M. Peroxisomal metabolism and oxidative stress. Biochimie 98: 56–62, 2014. [DOI] [PubMed] [Google Scholar]
- 289.Oberkampf M, Guillerey C, Mouries J, Rosenbaum P, Fayolle C, Bobard A, Savina A, Ogier-Denis E, Enninga J, Amigorena S, Leclerc C, Dadaglio G. Mitochondrial reactive oxygen species regulate the induction of CD8(+) T cells by plasmacytoid dendritic cells. Nat Commun 9: 2241, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH, Subramanian V, Mukundan L, Ferrante AW, Chawla A. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab 7: 496–507, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Oliveira CP, Gayotto LC, Tatai C, Della Nina BI, Lima ES, Abdalla DS, Lopasso FP, Laurindo FR, Carrilho FJ. Vitamin C and vitamin E in prevention of Nonalcoholic Fatty Liver Disease (NAFLD) in choline deficient diet fed rats. Nutr J 2: 9, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Orford KW, Scadden DT. Deconstructing stem cell self-renewal: Genetic insights into cell-cycle regulation. Nat Rev Genet 9: 115–128, 2008. [DOI] [PubMed] [Google Scholar]
- 293.Orr AL, Quinlan CL, Perevoshchikova IV, Brand MD. A refined analysis of superoxide production by mitochondrial sn-glycerol 3-phosphate dehydrogenase. J Biol Chem 287: 42921–42935, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Orr WC, Sohal RS. Extension of life-span by overexpression of superoxide dismutase and catalase in Drosophila melanogaster. Science 263: 1128–1130, 1994. [DOI] [PubMed] [Google Scholar]
- 295.Owusu-Ansah E, Banerjee U. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature 461: 537–541, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Paardekooper LM, Dingjan I, Linders PTA, Staal AHJ, Cristescu SM, Verberk W, van den Bogaart G. Human monocyte-derived dendritic cells produce millimolar concentrations of ROS in phagosomes per second. Front Immunol 10: 1216, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev 87: 315–424, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Paiva CN, Bozza MT. Are reactive oxygen species always detrimental to pathogens? Antioxid Redox Signal 20: 1000–1037, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Paniker NV, Srivastava SK, Beutler E. Glutathione metabolism of the red cells. Effect of glutathione reductase deficiency on the stimulation of hexose monophosphate shunt under oxidative stress. Biochim Biophys Acta 215: 456–460, 1970. [DOI] [PubMed] [Google Scholar]
- 300.Park HS, Jung HY, Park EY, Kim J, Lee WJ, Bae YS. Cutting edge: Direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-kappa B. J Immunol 173: 3589–3593, 2004. [DOI] [PubMed] [Google Scholar]
- 301.Peng KY, Watt MJ, Rensen S, Greve JW, Huynh K, Jayawardana KS, Meikle PJ, Meex RCR. Mitochondrial dysfunction-related lipid changes occur in nonalcoholic fatty liver disease progression. J Lipid Res 59: 1977–1986, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Perillo B, Di Donato M, Pezone A, Di Zazzo E, Giovannelli P, Galasso G, Castoria G, Migliaccio A. ROS in cancer therapy: The bright side of the moon. Exp Mol Med 52: 192–203, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Perkins A, Nelson KJ, Parsonage D, Poole LB, Karplus PA. Peroxiredoxins: Guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem Sci 40: 435–445, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Perrone A, Giovino A, Benny J, Martinelli F. Advanced glycation end products (AGEs): Biochemistry, signaling, analytical methods, and epigenetic effects. Oxid Med Cell Longev 2020: 3818196, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Perumpail BJ, Li AA, John N, Sallam S, Shah ND, Kwong W, Cholankeril G, Kim D, Ahmed A. The role of vitamin E in the treatment of NAFLD. Diseases 6: 86, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm 2010, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Piccoli C, Scrima R, Quarato G, D’Aprile A, Ripoli M, Lecce L, Boffoli D, Moradpour D, CapitanioN.HepatitisCvirusproteinexpression causes calcium-mediated mitochondrial bioenergetic dysfunction and nitro-oxidative stress. Hepatology 46: 58–65, 2007. [DOI] [PubMed] [Google Scholar]
- 308.Pinto M, Moraes CT. Mechanisms linking mtDNA damage and aging. Free Radic Biol Med 85: 250–258, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Poisson J, Lemoinne S, Boulanger C, Durand F, Moreau R, Valla D, Rautou PE. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J Hepatol 66: 212–227, 2017. [DOI] [PubMed] [Google Scholar]
- 310.Poli G, Albano E, Dianzani MU. The role of lipid peroxidation in liver damage. Chem Phys Lipids 45: 117–142, 1987. [DOI] [PubMed] [Google Scholar]
- 311.Pollock JD, Williams DA, Gifford MA, Li LL, Du X, Fisherman J, Orkin SH, Doerschuk CM, Dinauer MC. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet 9: 202–209, 1995. [DOI] [PubMed] [Google Scholar]
- 312.Ponsero AJ, Igbaria A, Darch MA, Miled S, Outten CE, Winther JR, Palais G, D’Autreaux B, Delaunay-Moisan A, Toledano MB. Endoplasmic reticulum transport of glutathione by Sec61 is regulated by Ero1 and Bip. Mol Cell 67: 962–973 e965, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Poprac P, Jomova K, Simunkova M, Kollar V, Rhodes CJ, Valko M. Targeting free radicals in oxidative stress-related human diseases. Trends Pharmacol Sci 38: 592–607, 2017. [DOI] [PubMed] [Google Scholar]
- 314.Postic C, Dentin R, Girard J. Role of the liver in the control of carbohydrate and lipid homeostasis. Diabetes Metab 30: 398–408, 2004. [DOI] [PubMed] [Google Scholar]
- 315.Pratt DA, Tallman KA, Porter NA. Free radical oxidation of polyunsaturated lipids: New mechanistic insights and the development of peroxyl radical clocks. Acc Chem Res 44: 458–467, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Presa N, Clugston RD, Lingrell S, Kelly SE, Merrill AH Jr, Jana S, Kassiri Z, Gomez-Munoz A, Vance DE, Jacobs RL, van der Veen JN. Vitamin E alleviates non-alcoholic fatty liver disease in phosphatidylethanolamine N-methyltransferase deficient mice. Biochim Biophys Acta Mol Basis Dis 1865: 14–25, 2019. [DOI] [PubMed] [Google Scholar]
- 317.Pryor WA, Houk KN, Foote CS, Fukuto JM, Ignarro LJ, Squadrito GL, Davies KJ. Free radical biology and medicine: It’s a gas, man! Am J Physiol Regul Integr Comp Physiol 291: R491–R511, 2006. [DOI] [PubMed] [Google Scholar]
- 318.Qureshi AA, Zuvanich EG, Khan DA, Mushtaq S, Silswal N, Qureshi N. Proteasome inhibitors modulate anticancer and anti-proliferative properties via NF-kB signaling, and ubiquitin-proteasome pathways in cancer cell lines of different organs. Lipids Health Dis 17: 62, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Peroxynitrite Radi R., a stealthy biological oxidant. J Biol Chem 288: 26464–26472, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Radi R Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc Natl Acad Sci U S A 115: 5839–5848, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Rafiei H, Omidian K, Bandy B. Dietary polyphenols protect against oleic acid-induced steatosis in an in vitro model of NAFLD by modulating lipid metabolism and improving mitochondrial function. Nutrients 11: 541, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Rang FJ, Boonstra J. Causes and consequences of age-related changes in DNA methylation: A role for ROS? Biology (Basel) 3: 403–425, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Reyes-Gordillo K, Shah R, Muriel P. Oxidative stress and inflammation in hepatic diseases: current and future therapy. Oxid Med Cell Longev 2017: 3140673, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Rhee SG. Redox signaling: Hydrogen peroxide as intracellular messenger. Exp Mol Med 31: 53–59, 1999. [DOI] [PubMed] [Google Scholar]
- 325.Ricci JE, Munoz-Pinedo C, Fitzgerald P, Bailly-Maitre B, Perkins GA, Yadava N, Scheffler IE, Ellisman MH, Green DR. Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell 117: 773–786, 2004. [DOI] [PubMed] [Google Scholar]
- 326.Rifkind JM, Mohanty JG, Nagababu E. The pathophysiology of extracellular hemoglobin associated with enhanced oxidative reactions. Front Physiol 5: 500, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Rivera CA, Bradford BU, Hunt KJ, Adachi Y, Schrum LW, Koop DR, Burchardt ER, Rippe RA, Thurman RG. Attenuation of CCl(4)-induced hepatic fibrosis by GdCl(3) treatment or dietary glycine. Am J Physiol Gastrointest Liver Physiol 281: G200–G207, 2001. [DOI] [PubMed] [Google Scholar]
- 328.Romero-Puertas MC, Sandalio LM. Nitric oxide level is self-regulating and also regulates its ROS partners. Front Plant Sci 7: 316, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Rose P, Moore PK, Zhu YZ. H2S biosynthesis and catabolism: New insights from molecular studies. Cell Mol Life Sci 74: 1391–1412, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Ruart M, Chavarria L, Camprecios G, Suarez-Herrera N, Montironi C, Guixe-Muntet S, Bosch J, Friedman SL, Garcia-Pagan JC, Hernandez-Gea V. Impaired endothelial autophagy promotes liver fibrosis by aggravating the oxidative stress response during acute liver injury. J Hepatol 70: 458–469, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Saadati S, Sadeghi A, Mansour A, Yari Z, Poustchi H, Hedayati M, Hatami B, Hekmatdoost A. Curcumin and inflammation in non-alcoholic fatty liver disease: A randomized, placebo controlled clinical trial. BMC Gastroenterol 19: 133, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Sakellariou GK, Pearson T, Lightfoot AP, Nye GA, Wells N, Giakoumaki II, Vasilaki A, Griffiths RD, Jackson MJ, McArdle A. Mitochondrial ROS regulate oxidative damage and mitophagy but not age-related muscle fiber atrophy. Sci Rep 6: 33944, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Sakon S, Xue X, Takekawa M, Sasazuki T, Okazaki T, Kojima Y, Piao JH, Yagita H, Okumura K, Doi T, Nakano H. NF-kappaB inhibits TNF-induced accumulation of ROS that mediate prolonged MAPK activation and necrotic cell death. EMBO J 22: 3898–3909, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Salomone F, Godos J, Zelber-Sagi S. Natural antioxidants for nonalcoholic fatty liver disease: Molecular targets and clinical perspectives. Liver Int 36: 5–20, 2016. [DOI] [PubMed] [Google Scholar]
- 335.Salzano S, Checconi P, Hanschmann EM, Lillig CH, Bowler LD, Chan P, Vaudry D, Mengozzi M, Coppo L, Sacre S, Atkuri KR, Sahaf B, Herzenberg LA, Herzenberg LA, Mullen L, Ghezzi P. Linkage of inflammation and oxidative stress via release of glutathionylated peroxiredoxin-2, which acts as a danger signal. Proc Natl Acad Sci U S A 111: 12157–12162, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Sans J, Aguilera AM, Faundez P, Troncoso P, Fernandez V, Videla LA. Influence of copper-(II) on colloidal carbon-induced Kupffer cell-dependent oxygen uptake in rat liver: Relation to hepatotoxicity. Free Radic Res 30: 489–498, 1999. [DOI] [PubMed] [Google Scholar]
- 337.Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, Luketic VA, Shiffman ML, Clore JN. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 120: 1183–1192, 2001. [DOI] [PubMed] [Google Scholar]
- 338.Sanz A Mitochondrial reactive oxygen species: Do they extend or shorten animal lifespan? Biochim Biophys Acta 1857: 1116–1126, 2016. [DOI] [PubMed] [Google Scholar]
- 339.Sato K, Gosho M, Yamamoto T, Kobayashi Y, Ishii N, Ohashi T, Nakade Y, Ito K, Fukuzawa Y, Yoneda M. Vitamin E has a beneficial effect on nonalcoholic fatty liver disease: A meta-analysis of randomized controlled trials. Nutrition 31: 923–930, 2015. [DOI] [PubMed] [Google Scholar]
- 340.Sato M, Suzuki S, Senoo H. Hepatic stellate cells: Unique characteristics in cell biology and phenotype. Cell Struct Funct 28: 105–112, 2003. [DOI] [PubMed] [Google Scholar]
- 341.Sayiner M, Koenig A, Henry L, Younossi ZM. Epidemiology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis in the united states and the rest of the world. Clin Liver Dis 20: 205–214, 2016. [DOI] [PubMed] [Google Scholar]
- 342.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg 4. EMBO J 26: 1749–1760, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol 24: R453–R462, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Schreck R, Rieber P, Baeuerle PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J 10: 2247–2258, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Schuster S, Cabrera D, Arrese M, Feldstein AE. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol 15: 349–364, 2018. [DOI] [PubMed] [Google Scholar]
- 346.Schwarz DS, Blower MD. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell Mol Life Sci 73: 79–94, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Schweikl H, Gallorini M, Forstner M, Petzel C, Bolay C, Hiller KA, Cataldi A, Krifka S, Buchalla W. Flavin-containing enzymes as a source of reactive oxygen species in HEMA-induced apoptosis. Dent Mater 33: e255–e271, 2017. [DOI] [PubMed] [Google Scholar]
- 348.Selye H Forty years of stress research: Principal remaining problems and misconceptions. Can Med Assoc J 115: 53–56, 1976. [PMC free article] [PubMed] [Google Scholar]
- 349.Selye H A syndrome produced by diverse nocuous agents. J Neuropsychiatry Clin Neurosci 10: 230–231, 1998. [DOI] [PubMed] [Google Scholar]
- 350.Semchyshyn HM. Reactive carbonyl species in vivo: Generation and dual biological effects. ScientificWorldJournal 2014: 417842, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell 48: 158–167, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, Wang CR, Schumacker PT, Licht JD, Perlman H, Bryce PJ, Chandel NS. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 38: 225–236, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Structure Senoo H. and function of hepatic stellate cells. Med Electron Microsc 37: 3–15, 2004. [DOI] [PubMed] [Google Scholar]
- 354.Senoo H, Mezaki Y, Fujiwara M. The stellate cell system (vitamin A-storing cell system). Anat Sci Int 92: 387–455, 2017. [DOI] [PubMed] [Google Scholar]
- 355.Seo K, Shin SM. Induction of Lipin1 by ROS-Dependent SREBP-2 Activation. Toxicol Res 33: 219–224, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Serviddio G, Bellanti F, Stanca E, Lunetti P, Blonda M, Tamborra R, Siculella L, Vendemiale G, Capobianco L, Giudetti AM. Silybin exerts antioxidant effects and induces mitochondrial biogenesis in liver of rat with secondary biliary cirrhosis. Free Radic Biol Med 73: 117–126, 2014. [DOI] [PubMed] [Google Scholar]
- 357.Severi T, van Malenstein H, Verslype C, van Pelt JF. Tumor initiation and progression in hepatocellular carcinoma: Risk factors, classification, and therapeutic targets. Acta PharmacolSin 31:1409–1420,2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Shah V, Haddad FG, Garcia-Cardena G, Frangos JA, Mennone A, Groszmann RJ, Sessa WC. Liver sinusoidal endothelial cells are responsible for nitric oxide modulation of resistance in the hepatic sinusoids. J Clin Invest 100: 2923–2930, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Shang L, Chen S, Du F, Li S, Zhao L, Wang X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci U S A 108: 4788–4793, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Shetty S, Lalor PF, Adams DH. Liver sinusoidal endothelial cells - gatekeepers of hepatic immunity. Nat Rev Gastroenterol Hepatol 15: 555–567, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Shi C, Wang Q, Rao Z, Shi Y, Wei S, Wang H, Lu X, Wang P, Lu L, Zhou H, Cheng F. Diabetes induces hepatocyte pyroptosis by promoting oxidative stress-mediated NLRP3 inflammasome activation during liver ischaemia and reperfusion injury. Ann Transl Med 8: 739, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci U S A 91: 10771–10778, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Shih VF, TsuiR, Caldwell A, Hoffmann A.Asingle NFkappaB system for both canonical and non-canonical signaling. Cell Res 21: 86–102, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Sies H Oxidative stress: A concept in redox biology and medicine. Redox Biol 4: 180–183, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Sies H, Berndt C, Jones DP. Oxidative stress. Annu Rev Biochem 86: 715–748, 2017. [DOI] [PubMed] [Google Scholar]
- 366.Sies H, Cadenas E. Oxidative stress: Damage to intact cells and organs. Philos Trans R Soc Lond B Biol Sci 311: 617–631, 1985. [DOI] [PubMed] [Google Scholar]
- 367.Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol 21: 363–383, 2020. [DOI] [PubMed] [Google Scholar]
- 368.Singh CK, Chhabra G, Ndiaye MA, Garcia-Peterson LM, Mack NJ, Ahmad N. The role of sirtuins in antioxidant and redox signaling. Antioxid Redox Signal 28: 643–661, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Singh M, Kapoor A, Bhatnagar A. Oxidative and reductive metabolism of lipid-peroxidation derived carbonyls. Chem Biol Interact 234: 261–273, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Singh R, Czaja MJ. Regulation of hepatocyte apoptosis by oxidative stress. J Gastroenterol Hepatol 22 (Suppl 1): S45–S48, 2007. [DOI] [PubMed] [Google Scholar]
- 371.Singh R, Wang Y, Xiang Y, Tanaka KE, Gaarde WA, Czaja MJ. Differential effects of JNK1 and JNK2 inhibition on murine steatohepatitis and insulin resistance. Hepatology 49: 87–96, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Sivell C Nonalcoholic fatty liver disease: a silent epidemic. Gastroenterol Nurs 42: 428–434, 2019. [DOI] [PubMed] [Google Scholar]
- 373.Siwecka N, Rozpedek W, Pytel D, Wawrzynkiewicz A, Dziki A, Dziki L, Diehl JA, Majsterek I. Dual role of endoplasmic reticulum stress-mediated unfolded protein response signaling pathway in carcinogenesis. Int J Mol Sci 20: 4354, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Song MJ, Malhi H. The unfolded protein response and hepatic lipid metabolism in non alcoholic fatty liver disease. Pharmacol Ther 203: 107401, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Sorato E, Menazza S, Zulian A, Sabatelli P, Gualandi F, Merlini L, Bonaldo P, Canton M, Bernardi P, Di Lisa F. Monoamine oxidase inhibition prevents mitochondrial dysfunction and apoptosis in myoblasts from patients with collagen VI myopathies. Free Radic Biol Med 75: 40–47, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Srinivas US, Tan BWQ, Vellayappan BA, Jeyasekharan AD. ROS and the DNA damage response in cancer. Redox Biol 25: 101084, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Staniek K, Nohl H. Are mitochondria a permanent source of reactive oxygen species? Biochim Biophys Acta 1460: 268–275, 2000. [DOI] [PubMed] [Google Scholar]
- 378.Starkov AA, Fiskum G, Chinopoulos C, Lorenzo BJ, Browne SE, Patel MS, Beal MF. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J Neurosci 24: 7779–7788, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Stone JR, Yang S. Hydrogen peroxide: A signaling messenger. Antioxid Redox Signal 8: 243–270, 2006. [DOI] [PubMed] [Google Scholar]
- 380.Sun B, Karin M. NF-kappaB signaling, liver disease and hepatoprotective agents. Oncogene 27: 6228–6244, 2008. [DOI] [PubMed] [Google Scholar]
- 381.Sun C, Zhang F, Ge X, Yan T, Chen X, Shi X, Zhai Q. SIRT1 improves insulin sensitivity under insulin-resistant conditions by repressing PTP1B. Cell Metab 6: 307–319, 2007. [DOI] [PubMed] [Google Scholar]
- 382.Sun L, Wu Q, Nie Y, Cheng N, Wang R, Wang G, Zhang D, He H, Ye RD, Qian F. A role for MK2 in enhancing neutrophil-derived ROS production and aggravating liver ischemia/reperfusion injury. Front Immunol 9: 2610, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Sunny NE, Bril F, Cusi K. Mitochondrial adaptation in nonalcoholic fatty liver disease: novel mechanisms and treatment strategies. Trends Endocrinol Metab 28: 250–260, 2017. [DOI] [PubMed] [Google Scholar]
- 384.Sunshine H, Iruela-Arispe ML. Membrane lipids and cell signaling. Curr Opin Lipidol 28: 408–413, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Sutton A, Nahon P, Pessayre D, Rufat P, Poire A, Ziol M, Vidaud D, Barget N, Ganne-Carrie N, Charnaux N, Trinchet JC, Gattegno L, Beaugrand M. Genetic polymorphisms in antioxidant enzymes modulate hepatic iron accumulation and hepatocellular carcinoma development in patients with alcohol-induced cirrhosis. Cancer Res 66: 2844–2852, 2006. [DOI] [PubMed] [Google Scholar]
- 386.Suzuki T, Yamamoto M. Stress-sensing mechanisms and the physiological roles of the Keap1-Nrf2 system during cellular stress. J Biol Chem 292: 16817–16824, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Svineng G, Ravuri C, Rikardsen O, Huseby NE, Winberg JO. The role of reactive oxygen species in integrin and matrix metalloproteinase expression and function. Connect Tissue Res 49: 197–202, 2008. [DOI] [PubMed] [Google Scholar]
- 388.Szabo C, Ischiropoulos H, Radi R. Peroxynitrite: Biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov 6: 662–680, 2007. [DOI] [PubMed] [Google Scholar]
- 389.Takami T, Sakaida I. Iron regulation by hepatocytes and free radicals. J Clin Biochem Nutr 48: 103–106, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Tan BL, Norhaizan ME, Liew WP. Nutrients and oxidative stress: Friend or foe? Oxid Med Cell Longev 2018: 9719584, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Tang T, Sui Y, Lian M, Li Z, Hua J. Pro-inflammatory activated Kupffer cells by lipids induce hepatic NKT cells deficiency through activation-induced cell death. PLoS One 8: e81949, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Tang W, Jiang YF, Ponnusamy M, Diallo M. Role of Nrf2 in chronic liver disease. World J Gastroenterol 20: 13079–13087, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Tao R, Wei D, Gao H, Liu Y, DePinho RA, Dong XC. Hepatic FoxOs regulate lipid metabolism via modulation of expression of the nicotinamide phosphoribosyltransferase gene. J Biol Chem 286: 14681–14690, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Tarantino G, Citro V, Finelli C. What non-alcoholic fatty liver disease has got to do with obstructive sleep apnoea syndrome and viceversa? J Gastrointestin Liver Dis 23: 291–299, 2014. [DOI] [PubMed] [Google Scholar]
- 395.Tarao K, Nozaki A, Ikeda T, Sato A, Komatsu H, Komatsu T, Taguri M, Tanaka K. Real impact of liver cirrhosis on the development of hepatocellular carcinoma in various liver diseases-meta-analytic assessment. Cancer Med 8: 1054–1065, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Taylor RS, Taylor RJ, Bayliss S, Hagstrom H, Nasr P, Schattenberg JM, Ishigami M, Toyoda H, Wai-Sun Wong V, Peleg N, Shlomai A, Sebastiani G, Seko Y, Bhala N, Younossi ZM, Anstee QM, McPherson S, Newsome PN. Association between fibrosis stage and outcomes of patients with nonalcoholic fatty liver disease: A systematic review and meta-analysis. Gastroenterology 158: 1611–1625 e1612, 2020. [DOI] [PubMed] [Google Scholar]
- 397.Teodoro JS, Duarte FV, Gomes AP, Varela AT, Peixoto FM, Rolo AP, Palmeira CM. Berberine reverts hepatic mitochondrial dysfunction in high-fat fed rats: A possible role for SirT3 activation. Mitochondrion 13: 637–646, 2013. [DOI] [PubMed] [Google Scholar]
- 398.Terlecky SR, Terlecky LJ, Giordano CR. Peroxisomes, oxidative stress, and inflammation. World J Biol Chem 3: 93–97, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Terman A, Kurz T. Lysosomal iron, iron chelation, and cell death. Antioxid Redox Signal 18: 888–898, 2013. [DOI] [PubMed] [Google Scholar]
- 400.Tessier FJ. The Maillard reaction in the human body. The main discoveries and factors that affect glycation. Pathol Biol (Paris) 58: 214–219, 2010. [DOI] [PubMed] [Google Scholar]
- 401.Theodotou M, Fokianos K, Moniatis D, Kadlenic R, Chrysikou A, Aristotelous A, Mouzouridou A, Diakides J, Stavrou E. Effect of resveratrol on non-alcoholic fatty liver disease. Exp Ther Med 18: 559–565, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Tinggi U Selenium: Its role as antioxidant in human health. Environ Health Prev Med 13: 102–108, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Traber MG, Atkinson J. Vitamin E, antioxidant and nothing more. Free Radic Biol Med 43: 4–15, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Trefts E, Gannon M, Wasserman DH. The liver. Curr Biol 27: R1147–R1151, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Tsukamoto H, Horne W, Kamimura S, Niemela O, Parkkila S, Yla-Herttuala S, Brittenham GM. Experimental liver cirrhosis induced by alcohol and iron. J Clin Invest 96: 620–630, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Tsutsui H, Nishiguchi S. Importance of Kupffer cells in the development of acute liver injuries in mice. Int J Mol Sci 15: 7711–7730, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Ueno T, Komatsu M. Autophagy in the liver: Functions in health and disease. Nat Rev Gastroenterol Hepatol 14: 170–184, 2017. [DOI] [PubMed] [Google Scholar]
- 408.Ursini F, Maiorino M, Forman HJ. Redox homeostasis: The Golden Mean of healthy living. Redox Biol 8: 205–215, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Utzschneider KM, Kahn SE. Review: The role of insulin resistance in nonalcoholic fatty liver disease. J Clin Endocrinol Metab 91: 4753–4761, 2006. [DOI] [PubMed] [Google Scholar]
- 410.Vaka VR, McMaster KM, Cunningham MW Jr, Ibrahim T, Hazlewood R, Usry N, Cornelius DC, Amaral LM, LaMarca B. Role of mitochondrial dysfunction and reactive oxygen species in mediating hypertension in the reduced uterine perfusion pressure rat model of preeclampsia. Hypertension 72: 703–711, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Valerio LG Jr, Parks T, Petersen DR. Alcohol mediates increases in hepatic and serum nonheme iron stores in a rat model for alcohol-induced liver injury. Alcohol Clin Exp Res 20: 1352–1361, 1996. [DOI] [PubMed] [Google Scholar]
- 412.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 39: 44–84, 2007. [DOI] [PubMed] [Google Scholar]
- 413.Velarde MC, Flynn JM, Day NU, Melov S, Campisi J. Mitochondrial oxidative stress caused by Sod2 deficiency promotes cellular senescence and aging phenotypes in the skin. Aging (Albany NY) 4: 3–12, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Volani C, Doerrier C, Demetz E, Haschka D, Paglia G, Lavdas AA, Gnaiger E, Weiss G. Dietary iron loading negatively affects liver mitochondrial function. Metallomics 9: 1634–1644, 2017. [DOI] [PubMed] [Google Scholar]
- 415.Wan D, Jiang W, Hao J. Research advances in how the cGAS-STING pathway controls the cellular inflammatory response. Front Immunol 11: 615, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Wang K Molecular mechanisms of hepatic apoptosis. Cell Death Dis 5: e996, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Wang L, Wang X, Xie G, Wang L, Hill CK, DeLeve LD. Liver sinusoidal endothelial cell progenitor cells promote liver regeneration in rats. J Clin Invest 122: 1567–1573, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Wang ML, Yu XJ, Li XG, Pang DZ, Su Q, Saahene RO, Li HB, Mao XY, Liu KL, Fu LY, Li Y, Zhu GQ, Kang YM. Blockade of TLR4 within the paraventricular nucleus attenuates blood pressure by regulating ROS and inflammatory cytokines in prehypertensive rats. Am J Hypertens 31: 1013–1023, 2018. [DOI] [PubMed] [Google Scholar]
- 419.Wang T, Weinman SA. Interactions between hepatitis C virus and mitochondria: impact on pathogenesis and innate immunity. Curr Pathobiol Rep 1: 179–187, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Wang X, Rao H, Zhao J, Wee A, Li X, Fei R, Huang R, Wu C, Liu F, Wei L. STING expression in monocyte-derived macrophages is associated with the progression of liver inflammation and fibrosis in patients with nonalcoholic fatty liver disease. Lab Invest 100: 542–552, 2020. [DOI] [PubMed] [Google Scholar]
- 421.Wang Y, Branicky R, Noe A, Hekimi S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J Cell Biol 217: 1915–1928, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Wang Z, Dou X, Li S, Zhang X, Sun X, Zhou Z, Song Z. Nuclear factor (erythroid-derived 2)-like 2 activation-induced hepatic very-low-density lipoprotein receptor overexpression in response to oxidative stress contributes to alcoholic liver disease in mice. Hepatology 59: 1381–1392, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Wang Z, Li Z, Ye Y, Xie L, Li W. Oxidative stress and liver cancer: etiology and therapeutic targets. Oxid Med Cell Longev 2016: 7891574, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Wang Z, Liu Y, Zhao X, Liu S, Liu Y, Wang D. Aronia melanocarpa prevents alcohol-induced chronic liver injury via regulation of Nrf2 signaling in C57BL/6 mice. Oxid Med Cell Longev 2020: 4054520, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Wasik U, Milkiewicz M, Kempinska-Podhorodecka A, Milkiewicz P. Protection against oxidative stress mediated by the Nrf2/Keap1 axis is impaired in Primary Biliary Cholangitis. Sci Rep 7: 44769, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Watanabe K, Shibuya S, Ozawa Y, Nojiri H, Izuo N, Yokote K, Shimizu T. Superoxide dismutase 1 loss disturbs intracellular redox signaling, resulting in global age-related pathological changes. Biomed Res Int 2014: 140165, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Wei PL, Huang CY, Chang YJ. Propyl gallate inhibits hepatocellular carcinoma cell growth through the induction of ROS and the activation of autophagy. PLoS One 14: e0210513, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Wei Y, Wang D, Gentile CL, Pagliassotti MJ. Reduced endoplasmic reticulum luminal calcium links saturated fatty acid-mediated endoplasmic reticulum stress and cell death in liver cells. Mol Cell Biochem 331: 31–40, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Wesselink E, Koekkoek WAC, Grefte S, Witkamp RF, van Zanten ARH. Feeding mitochondria: Potential role of nutritional components to improve critical illness convalescence. Clin Nutr 38: 982–995, 2019. [DOI] [PubMed] [Google Scholar]
- 430.Wheeler ML, Defranco AL. Prolonged production of reactive oxygen species in response to B cell receptor stimulation promotes B cell activation and proliferation. J Immunol 189: 4405–4416, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Williams CD, Bajt ML, Sharpe MR, McGill MR, Farhood A, Jaeschke H. Neutrophil activation during acetaminophen hepatotoxicity and repair in mice and humans. Toxicol Appl Pharmacol 275: 122–133, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Williams JA, Ni HM, Ding Y, Ding WX. Parkin regulates mitophagy and mitochondrial function to protect against alcohol-induced liver injury and steatosis in mice. Am J Physiol Gastrointest Liver Physiol 309: G324–G340, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Win S, Than TA, Zhang J, Oo C, Min RWM, Kaplowitz N. New insights into the role and mechanism of c-Jun-N-terminal kinase signaling in the pathobiology of liver diseases. Hepatology 67: 2013–2024, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, Hoffman HM, Feldstein AE. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 59: 898–910, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Wu D, Cederbaum AI. Ethanol-induced apoptosis to stable HepG2 cell lines expressing human cytochrome P-4502E1. Alcohol Clin Exp Res 23: 67–76, 1999. [PubMed] [Google Scholar]
- 436.Wu JR, You RI, Hu CT, Cheng CC, Rudy R, Wu WS. Hydrogen peroxide inducible clone-5 sustains NADPH oxidase-dependent reactive oxygen species-c-jun N-terminal kinase signaling in hepatocellular carcinoma. Oncogenesis 8: 40, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Wyck S, Herrera C, Requena CE, Bittner L, Hajkova P, Bollwein H, Santoro R. Oxidative stress in sperm affects the epigenetic reprogramming in early embryonic development. Epigenetics Chromatin 11: 60, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Wymann MP, Schneiter R. Lipid signalling in disease. Nat Rev Mol Cell Biol 9: 162–176, 2008. [DOI] [PubMed] [Google Scholar]
- 439.Xie WH, Ding J, Xie XX, Yang XH, Wu XF, Chen ZX, Guo QL, Gao WY, Wang XZ, Li D. Hepatitis B virus X protein promotes liver cell pyroptosis under oxidative stress through NLRP3 inflammasome activation. Inflamm Res 69: 683–696, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Xu D, Xu M, Jeong S, Qian Y, Wu H, Xia Q, Kong X. The role of Nrf2 in liver disease: Novel molecular mechanisms and therapeutic approaches. Front Pharmacol 9: 1428, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Xu R, Chen MY, Liang W, ChenY, Guo MY. Zinc deficiency aggravation of ROS and inflammatory injury leading to renal fibrosis in mice. Biol Trace Elem Res 199 (2): 622–632, 2021. [DOI] [PubMed] [Google Scholar]
- 442.Yan B, Chen G, Saigal K, Yang X, Jensen ST, Van Waes C, Stoeckert CJ, Chen Z. Systems biology-defined NF-kappaB regulons, interacting signal pathways and networks are implicated in the malignant phenotype of head and neck cancer cell lines differing in p53 status. Genome Biol 9: R53, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Yan C, Zhang Y, Zhang X, Aa J, Wang G, Xie Y. Curcumin regulates endogenous and exogenous metabolism via Nrf2-FXR-LXR pathway in NAFLD mice. Biomed Pharmacother 105: 274–281, 2018. [DOI] [PubMed] [Google Scholar]
- 444.Yang H, Yang T, Heng C, Zhou Y, Jiang Z, Qian X, Du L, Mao S, Yin X, Lu Q. Quercetin improves nonalcoholic fatty liver by ameliorating inflammation, oxidative stress, and lipid metabolism in db/db mice. Phytother Res 33: 3140–3152, 2019. [DOI] [PubMed] [Google Scholar]
- 445.Yang J, Fernandez-Galilea M, Martinez-Fernandez L, Gonzalez-Muniesa P, Perez-Chavez A, Martinez JA, Moreno-Aliaga MJ. Oxidative stress and non-alcoholic fatty liver disease: Effects of omega-3 fatty acid supplementation. Nutrients 11: 872, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Yang SF, Chang CW, Wei RJ, Shiue YL, Wang SN, Yeh YT. Involvement of DNA damage response pathways in hepatocellular carcinoma. Biomed Res Int 2014: 153867, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Yang W, Tao Y, Wu Y, Zhao X, Ye W, Zhao D, Fu L, Tian C, Yang J, He F, Tang L. Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nat Commun 10: 1076, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A 113: E4966–E4975, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Yang WS, Stockwell BR. Ferroptosis: Death by lipid peroxidation. Trends Cell Biol 26: 165–176, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Yang Y, Sharma R, Sharma A, Awasthi S, Awasthi YC. Lipid peroxidation and cell cycle signaling: 4-Hydroxynonenal, a key molecule in stress mediated signaling. Acta Biochim Pol 50: 319–336, 2003. [PubMed] [Google Scholar]
- 451.Ye R, Jung DY, Jun JY, Li J, Luo S, Ko HJ, Kim JK, Lee AS. Grp78 heterozygosity promotes adaptive unfolded protein response and attenuates diet-induced obesity and insulin resistance. Diabetes 59: 6–16, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Yi SW, Choi JS, Yi JJ, Lee YH, Han KJ. Risk factors for hepatocellular carcinoma by age, sex, and liver disorder status: A prospective cohort study in Korea. Cancer 124: 2748–2757, 2018. [DOI] [PubMed] [Google Scholar]
- 453.Yoboue ED, Sitia R, Simmen T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis 9: 331, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Yuan K, Lei Y, Chen HN, Chen Y, Zhang T, Li K, Xie N, Wang K, Feng X, Pu Q, Yang W, Wu M, Xiang R, Nice EC, Wei Y, Huang C. HBV-induced ROS accumulation promotes hepatocarcinogenesis through Snail-mediated epigenetic silencing of SOCS3. Cell Death Differ 23: 616–627, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Zakhari S Overview: How is alcohol metabolized by the body? Alcohol Res Health 29: 245–254, 2006. [PMC free article] [PubMed] [Google Scholar]
- 456.Zhan SS, Jiang JX, Wu J, Halsted C, Friedman SL, Zern MA, Torok NJ. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology 43: 435–443, 2006. [DOI] [PubMed] [Google Scholar]
- 457.Zhang CY, Yuan WG, He P, Lei JH, Wang CX. Liver fibrosis and hepatic stellate cells: Etiology, pathological hallmarks and therapeutic targets. World J Gastroenterol 22: 10512–10522, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu Y, Dong W. ROS and ROS-mediated cellular signaling. Oxid Med Cell Longev 2016: 4350965, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Zhang K, Li L, Qi Y, Zhu X, Gan B, DePinho RA, Averitt T, Guo S. Hepatic suppression of Foxo1 and Foxo3 causes hypoglycemia and hyperlipidemia in mice. Endocrinology 153: 631–646, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Zhang K, Wang S, Malhotra J, Hassler JR, Back SH, Wang G, Chang L, Xu W, Miao H, Leonardi R, Chen YE, Jackowski S, Kaufman RJ. The unfolded protein response transducer IRE1alpha prevents ER stress-induced hepatic steatosis. EMBO J 30: 1357–1375, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Zhang NP, Liu XJ, Xie L, Shen XZ, Wu J. Impaired mitophagy triggers NLRP3 inflammasome activation during the progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis. Lab Invest 99: 749–763, 2019. [DOI] [PubMed] [Google Scholar]
- 462.Zhang T, Ono K, Tsutsuki H, Ihara H, Islam W, Akaike T, Sawa T. Enhanced Cellular Polysulfides Negatively Regulate TLR4 Signaling and Mitigate Lethal Endotoxin Shock. Cell Chem Biol 26: 686–698 e684, 2019. [DOI] [PubMed] [Google Scholar]
- 463.Zhao L, Zhang N, Yang D, Yang M, Guo X, He J, Wu W, Ji B, Cheng Q, Zhou F. Protective effects of five structurally diverse flavonoid sub-groups against chronic alcohol-induced hepatic damage in a mouse model. Nutrients 10: 1754, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Zhao RZ, Jiang S, Zhang L, Yu ZB. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int J Mol Med 44: 3–15, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Zheltova AA, Kharitonova MV, Iezhitsa IN, Spasov AA. Magnesium deficiency and oxidative stress: An update. Biomedicine (Taipei) 6: 20, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Zhong Z, Zhai Y, Liang S, Mori Y, Han R, Sutterwala FS, Qiao L. TRPM2 links oxidativestress to NLRP3 inflammasome activation. Nat Commun 4: 1611, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Zhou Z, Xu MJ, Cai Y, Wang W, Jiang JX, Varga ZV, Feng D, Pacher P, Kunos G, Torok NJ, Gao B. Neutrophil-hepatic stellate cell interactions promote fibrosis in experimental steatohepatitis. Cell Mol Gastroenterol Hepatol 5: 399–413, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Zhuang A, Yap FY, Bruce C, Leung C, Plan MR, Sullivan MA, Herath C, McCarthy D, Sourris KC, Kantharidis P, Coughlan MT, Febbraio MA, Hodson MP, Watt MJ, Angus P, Schulz BL, Forbes JM. Increased liver AGEs induce hepatic injury mediated through an OST48 pathway. Sci Rep 7: 12292, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]