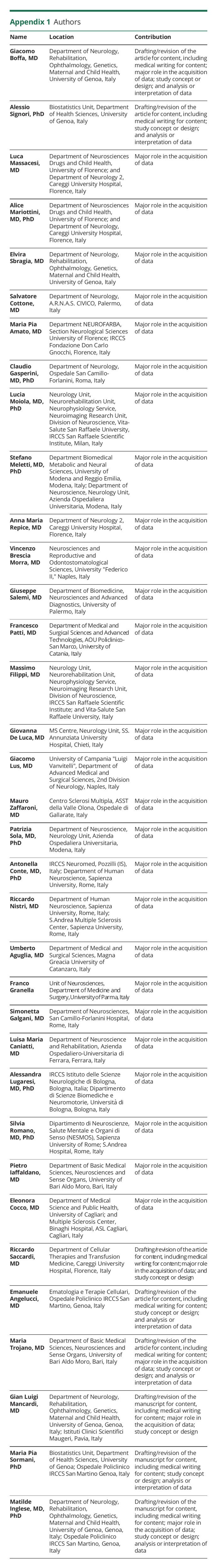
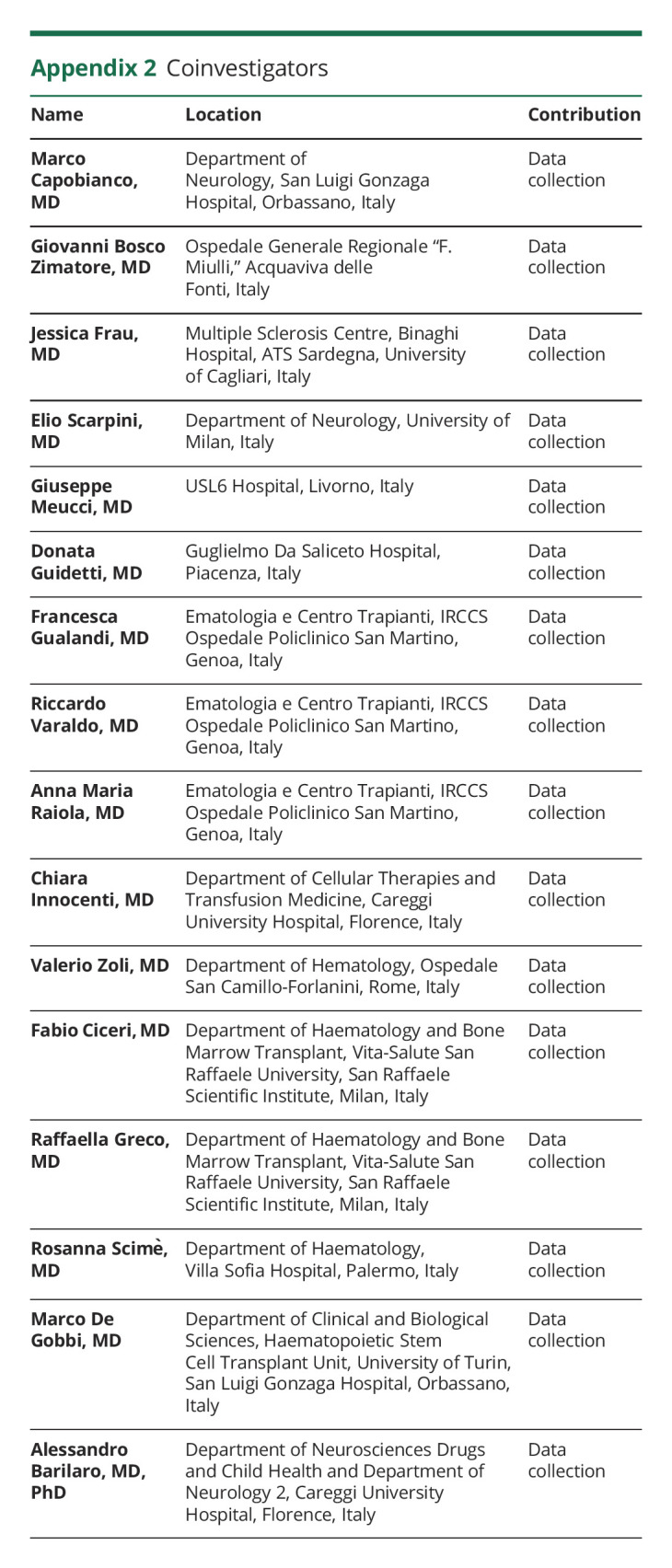
Giacomo Boffa
Giacomo Boffa, MD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,*,
Alessio Signori
Alessio Signori, PhD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,*,
Luca Massacesi
Luca Massacesi, MD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Alice Mariottini
Alice Mariottini, MD, PhD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Elvira Sbragia
Elvira Sbragia, MD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Salvatore Cottone
Salvatore Cottone, MD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Maria Pia Amato
Maria Pia Amato, MD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Claudio Gasperini
Claudio Gasperini, MD, PhD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Lucia Moiola
Lucia Moiola, MD, PhD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Stefano Meletti
Stefano Meletti, MD, PhD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Anna Maria Repice
Anna Maria Repice, MD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Vincenzo Brescia Morra
Vincenzo Brescia Morra, MD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Giuseppe Salemi
Giuseppe Salemi, MD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Francesco Patti
Francesco Patti, MD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Massimo Filippi
Massimo Filippi, MD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Giovanna De Luca
Giovanna De Luca, MD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Giacomo Lus
Giacomo Lus, MD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Mauro Zaffaroni
Mauro Zaffaroni, MD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Patrizia Sola
Patrizia Sola, MD, PhD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Antonella Conte
Antonella Conte, MD, PhD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Riccardo Nistri
Riccardo Nistri, MD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Umberto Aguglia
Umberto Aguglia, MD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Franco Granella
Franco Granella
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Simonetta Galgani
Simonetta Galgani, MD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Luisa Maria Caniatti
Luisa Maria Caniatti, MD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Alessandra Lugaresi
Alessandra Lugaresi, MD, PhD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Silvia Romano
Silvia Romano, MD, PhD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Pietro Iaffaldano
Pietro Iaffaldano, MD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Eleonora Cocco
Eleonora Cocco, MD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Riccardo Saccardi
Riccardo Saccardi, MD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Emanuele Angelucci
Emanuele Angelucci, MD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Maria Trojano
Maria Trojano, MD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Giovanni Luigi Mancardi
Giovanni Luigi Mancardi, MD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Maria Pia Sormani
Maria Pia Sormani, PhD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,
Matilde Inglese
Matilde Inglese, MD, PhD
1From the Department of Neurology, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (G.B., E.S., G.L.M., M.I.), University of Genoa; Biostatistics Unit (A.S., M.P.S.), Department of Health Sciences, University of Genoa; Department of Neurosciences Drugs and Child Health (L. Massacesi, A.M.), University of Florence; and Department of Neurology 2 (L. Massacesi, A.M., A.M.R.), Careggi University Hospital, Florence; Department of Neurology (S.C.), A.R.N.A.S. CIVICO, Palermo; Department NEUROFARBA (M.P.A.), Section Neurological Sciences University of Florence; IRCCS Fondazione Don Carlo Gnocchi, (M.P.A) Florence; Department of Neurology (C.G.), Ospedale San Camillo-Forlanini, Rome; Neurology Unit (L. Moiola, M.F.), Neurorehabilitation Unit (F.M.), Neurophysiology Service (F.M.), Neuroimaging Research Unit, Division of Neuroscience (F.M.), IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University (F.M.), Milan; Department Biomedical Metabolic and Neural Sciences (S.M.), University of Modena and Reggio Emilia, Modena, Italy; Department of Neuroscience, Neurology Unit (S.M., P.S.), Azienda Ospedaliera Universitaria, Modena; Neurosciences and Reproductive and Odontostomatological Sciences (V.B.M.), University “Federico II,” Naples; Department of Biomedicine, Neurosciences and Advanced Diagnostics (G.S.), University of Palermo; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), AOU Policlinico-San Marco, University of Catania; MS Centre, Neurology Unit (G.D.L.), SS. Annunziata University Hospital, Chieti; Department of Advanced Medical and Surgical Sciences (G.L.), 2nd Division of Neurology, University of Campania “Luigi Vanvitelli,” Naples; Centro Sclerosi Multipla (M.Z.), ASST della Valle Olona, Ospedale di Gallarate, Italy; IRCCS Neuromed (A.C.), Pozzilli (IS); Department of Human Neuroscience (A.C., R.N.) and Dipartimento di Neuroscienze, Salute Mentale e Organi di Senso (NESMOS) (S.R.) Sapienza University, Rome; S.Andrea Multiple Sclerosis Center (R.N.), Sapienza University, Rome; S.Andrea Hospital (S.R.), Rome; Department of Medical and Surgical Sciences (U.A.), Magna Greacia University of Catanzaro; Unit of Neurosciences, Department of Medicine and Surgery (F.G.), University of Parma; Department of Neurosciences (S.G.), San Camillo-Forlanini Hospital, Rome; Department of Neuroscience and Rehabilitation (L.M.C.), Azienda Ospedaliero-Universitaria di Ferrara; IRCCS Istituto delle Scienze Neurologiche di Bologna (A.L.); Dipartimento di Scienze Biomediche e Neuromotorie (A.L.), Università di Bologna; Department of Basic Medical Sciences, Neurosciences and Sense Organs (P.I., M.T.), University of Bari Aldo Moro; Department of Medical Science and Public Health (E.C), University of Cagliari, Cagliari; Multiple Sclerosis Center, Binaghi Hospital, ASL Cagliari; Department of Cellular Therapies and Transfusion Medicine (R.S.), Careggi University Hospital, Florence; Ematologia e Terapie Cellulari (E.A.), Ospedale Policlinico IRCCS San Martino (M.P.S.), Genoa; Istituti Clinici Scientifici Maugeri (G.L.M.), Pavia; Ospedale Policlinico IRCCS San Martino (M.I.), Genoa, Italy.
1,✉;
on behalf of the Italian BMT-MS Study Group and the Italian MS Register1,
Marco Capobianco,
Giovanni Bosco Zimatore
Giovanni Bosco Zimatore, MD
,
Jessica Frau,
Elio Scarpini,
Giuseppe Meucci,
Donata Guidetti,
Francesca Gualandi,
Riccardo Varaldo,
Anna Maria Raiola,
Chiara Innocenti,
Valerio Zoli,
Fabio Ciceri,
Raffaella Greco,
Rosanna Scim`e,
Marco De Gobbi,
Alessandro Barilaro
Alessandro Barilaro, MD, PhD