

Abstract
Troponin I (TnI) is a key regulator of cardiac contraction and relaxation with TnI Ser-23/24 phosphorylation serving as a myofilament mechanism to modulate cardiac function. Basal cardiac TnI Ser-23/24 phosphorylation is high such that both increased and decreased TnI phosphorylation may modulate cardiac function. While the effects of increasing TnI Ser-23/24 phosphorylation on heart function are well established, the effects of decreasing TnI Ser-23/24 phosphorylation are not clear. To understand the in vivo role of decreased TnI Ser-23/24 phosphorylation, mice expressing TnI with Ser-23/24 mutated to alanine (TnI S23/24A) that lack the ability to be phosphorylated at these residues were subjected to echocardiography and pressure-volume hemodynamic measurements in the absence or presence of physiological (pacing increasing heart rate or adrenergic stimulation) or pathological (transverse aortic constriction (TAC)) stress. In the absence of pathological stress, the lack of TnI Ser-23/24 phosphorylation impaired systolic and diastolic function. TnI S23/24A mice also had an impaired systolic and diastolic response upon stimulation increased heart rate and an impaired adrenergic response upon dobutamine infusion. Following pathological cardiac stress induced by TAC, TnI S23/24A mice had a greater increase in ventricular mass, worse diastolic function, and impaired systolic and diastolic function upon increasing heart rate. These findings demonstrate that mice lacking the ability to phosphorylate TnI at Ser-23/24 have impaired in vivo systolic and diastolic cardiac function, a blunted cardiac reserve and a worse response to pathological stress supporting decreased TnI Ser23/24 phosphorylation is a modulator of these processes in vivo.
Keywords: Troponin I, phosphorylation, cardiac, in vivo, heart rate, sympathetic
Graphical Abstract
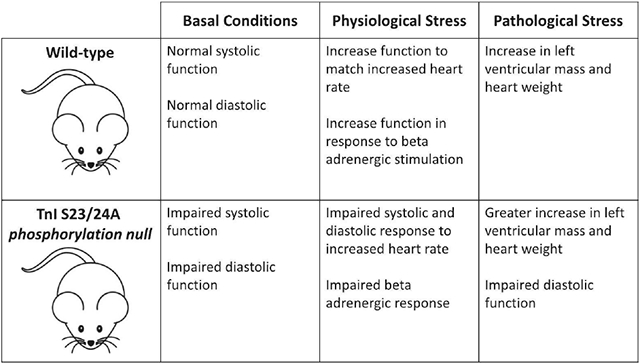
1. Introduction
The ability of the heart to increase cardiac output is essential to match the increased demand in both short-term physiological stress and to long-term cardiac health [1, 2]. Cardiac output increases when stroke volume and/or heart rate is increased. This ability to increase cardiac output is the heart’s cardiac reserve and is highly influenced by sympathetic tone, the Frank-Starling relationship between sarcomere length and force, and the Bowditch effect of increased force upon increased heart rate [3]. All three of these governing parameters strongly influence stroke volume and/or heart rate such that these parameters are critical to consider as part of the heart’s reserve to increase in vivo cardiac function.
At the level of the myocardium, cardiac stroke volume is dependent upon myocyte contraction and relaxation. Contraction and relaxation of the myocyte are regulated through calcium and myofilament dependent mechanisms that control myosin binding to actin, subsequent cross-bridge cycling, sarcomeric shortening, and force production. Contraction is initiated upon increased intracellular calcium binding to troponin C that induces alterations of the myofilament regulatory protein interactions to result in an active myofilament conformation promoting the binding of myosin to actin and force production. Critical to this active myofilament conformation are the movement of tropomyosin, the release of the troponin inhibitory subunit (TnI) from actin to expose the myosin binding sites on actin and the subsequent binding of TnI to troponin C. Similarly, myocyte relaxation results from active myofilament processes. Upon the release of calcium from troponin C, altered interactions within the myofilament induce an inhibitory myofilament conformation that blocks myosin binding to actin [2, 4]. This inhibitory myofilament conformation includes the return of tropomyosin to its inhibitory conformation, the release of TnI from troponin C and subsequent inhibitory binding of TnI to actin. Myocyte contraction and relaxation can therefore be modulated either by altering intracellular calcium or by altering any of the regulatory myofilament proteins that modulate myosin binding to actin and force production resulting from calcium binding to troponin C (myofilament calcium sensitivity) [5]. Generally, increasing myofilament calcium sensitivity increases activation of these myofilament processes at the same calcium concentration to result in increased force generation but impairs myocyte relaxation. In contrast, decreasing calcium sensitivity decreases force generation and enhances myocyte relaxation.
The phosphorylation of TnI is a key mechanism to modulate calcium sensitivity and regulate in vivo heart function in both health and disease [6–8]. Biochemical, molecular, and in vivo studies have demonstrated increasing TnI Ser-23 and Ser-24 (Ser-23/24) phosphorylation is a critical myofilament regulatory mechanism that decreases myofilament calcium sensitive force production and accelerates myofilament deactivation to result in accelerated in vivo myocardial relaxation and enhanced diastolic function [9–20]. In the resting heart, approximately 40% of cardiac TnI is basally phosphorylated at Ser-23/24 [21–24]. As a result of this high basal phosphorylation, TnI can also undergo decreased phosphorylation which occurs in end stage heart failure [22, 23, 25]. While the effects of increasing TnI Ser-23/24 phosphorylation are well established, the effects of decreasing TnI Ser-23/24 phosphorylation are based upon a limited number of contradictory reports and its effects are less clear. Studies directly inhibiting cardiac TnI Ser-23/24 phosphorylation in the heart by transgenic expression of the phosphorylation null TnI S23/24A demonstrated that the loss of Ser-23/24 phosphorylation increased [34] or did not alter [35] myofilament calcium sensitivity and did not alter in vivo cardiac function [35, 36]. Similarly, decreasing TnI Ser-23/24 phosphorylation from the basal state by gene transfer of TnI S23/24A did not alter myocyte calcium sensitivity or relaxation parameters [17]. With the clear effect of increased TnI Ser-23/24 phosphorylation to decrease calcium sensitivity and accelerate active relaxation, why the loss of TnI Ser-23/24 phosphorylation did not alter cardiac function is unclear.
Altered muscle calcium sensitivity has been supported to affect in vivo systolic and diastolic heart function, however the lack of altered in vivo function upon expression of the phosphorylation null TnI S23/24A contradicts this dogma. The myofilament mechanisms that regulate in vivo myocardial function involve multiple regulatory protein conformational changes such that calcium sensitivity may not always predict in vivo function [37]. To further investigate the loss of TnI Ser-23/24 phosphorylation on in vivo cardiac function we designed specific experiments to evaluate the basal in vivo cardiac function as well as major factors that modulate in vivo heart function, including physiological (rate and sympathetic) and pathological (trans aortic constriction) stress, in mice lacking the ability of cardiac TnI Ser-23/24 to undergo phosphorylation.
2. Methods
2.1. Animals.
All procedures performed in this study were approved by The Ohio State University’s Institutional Animal Care and Use Committee in accordance with NIH guidelines. Mice were housed at The Ohio State University in a temperature-controlled animal facility with a 12-hr light/dark cycle and free access to food and water. All studies were conducted on adult male mice (3 – 6 months old) of FVBN background. Mice transgenic for the over-expression of TnI serine 23 and serine 24 mutated to alanine on a TnI null background (TnI S23/24A) were previously generated [34]. The TnI S23/24A mouse model is transgenic for overexpression of TnI S23/24A and homozygous for knock-out of the wild type cardiac TnI gene as was previously demonstrated to result in cardiac specific expression of only the phosphorylation null TnI S23/24A in the heart [34]. Non-transgenic littermate mice negative for TnI S23/24A but containing knock-out of one wild type cardiac TnI gene (WT / TnI KO) were not different in body weight, heart weight, echocardiography cardiac structure (left ventricular anterior wall thickness at systole, left ventricular anterior wall thickness at diastole or left ventricular mass) or echocardiography cardiac function (diameter at systole, diameter at diastole, or ejection fraction) from that of wild type (FVBN WT) mice purchased from Jackson Laboratories (Suppl. Table 1). Since we observed no difference between non-transgenic littermates and purchased wild type FVBN mice, we used purchased FVBN wild type mice as WT control mice for all experiments.
2.2. Protein electrophoresis and western blot.
Excised mouse ventricular tissue was solubilized in denaturing buffer (8 M urea, 2 M thiourea, 3% SDS, 75 mM DTT, 0.03% bromophenol blue and 50 mM Tris-HCl, ph 6.8), heated for five minutes at 80°C and clarified by centrifugation at 21,000 g for five minutes. TnI isoform expression was determined by Western blot similar to that previously described [38]. Briefly, proteins were separated on 12% (29:1) Laemmli gel and transferred to PVDF. Following blocking with 1% BSA in TBS, membranes were incubated with a mouse anti-TnI antibody (Fitzgerald; clone C5) that recognizes cardiac, slow skeletal and fast skeletal TnI isoforms. Following washes, membranes were incubated with a Dylight labeled fluorescent secondary antibody (Jackson ImmunoResearch Laboratories) and visualized on an Azure 600 imaging system (Azure Biosystems). Cardiac and slow skeletal TnI (ssTnI) bands in resulting images were quantified using Image Quant TL 7.0 (GE Healthcare) and the percent ssTnI determined as the (ssTnI / (cTnI + ssTnI)) x 100. Myosin isoform expression was determined by SDS-PAGE similar to that previously described [39, 40]. Briefly, proteins were separated on 6% (37.5:1) gel cross-linked with DATD cooled to 4°C and run until the dye front ran off the bottom. The resultant gel was stained with Coomassie Blue. The alpha and beta myosin heavy chain (MHC) bands were quantified using Image Quant TL 7.0 (GE Healthcare) and the percent beta MHC determined as the (beta MHC / (alpha MHC + beta MHC)) x 100.
2.3. Resting Heart Rate.
Resting heart rate was determined in conscious, unanesthetized mice using the ECGenie system (Mouse Specifics, Inc). Briefly, mice were placed on the lead plate electrode and a continuous ECG reading was collected for 10-12 minutes. Excluding the first 5 minutes, a 5-10 second selection of the recordings demonstrating clear peaks and minimal noise was analyzed offline with the LabChart8 (ADInstruments) ECG analysis module.
2.4. Echocardiography.
Echocardiography was performed using a Vevo 3100 Visual Sonics (VisualSonics) system. Mice were lightly anesthetized with 1.5% isoflurane and the ventricular chamber dimensions were determined through M-mode using the parasternal short axis view as previously described [41].
2.5. Hemodynamic measurements.
Cardiac pressure / volume measurements were assessed via a closed chest approach using a 1.4 French Millar pressure catheter (ADInstruments) advanced into the left ventricle through the right carotid artery as previously described [42]. Briefly, mice were anaesthetized with ketamine (100 mg/kg) and xylazine (20 mg/kg) and placed in the supine position on a heating pad. Following a midline neck incision, the underlying muscles were pulled away to expose the carotid artery. Using a 4-0 suture the artery was tied and the pressure volume catheter was advanced through the artery into the left ventricle of the heart. After 5-10 minutes of stabilization, values were determined upon stimulating the heart at increasing frequency (6-9Hz) by electrodes placed into the intercostal space across the heart. Stimulation was then stopped and mice were then injected with 10mg/kg dobutamine intraperitoneal. Measurements of cardiac function in response to beta-adrenergic stimulation were taken at the maximum heart rate. Measurements and analysis were performed on LabChart7 (ADInstruments).
2.6. Transverse aortic Constriction.
Transverse aortic constriction (TAC) was performed as previously described [43]. Animals were anesthetized with 1% isoflurane, placed in a right lateral position and the manubrium excised from the upper thorax aperture to the sternal angle. The lobes of the thymus gland were separated between the interlobar incisures to allow direct visualization of the transverse aorta (without entering the pleural space). A silk thread was then inserted underneath the transverse aorta between the right innominate artery and the left carotid artery, a 27.5 gauge needle placed over the artery and a double knot made with a 2-3 mm diameter loop. The needle was immediately removed after ligation, leaving the aortic constriction in place and the chest closed. Pain management was provided with ibuprofen treatment (30 mg/kg in the drinking water) started the day prior to surgery and buprenorphine treatment (0.1 mg/kg) started at the time of surgery.
2.7. Histology.
Following hemodynamic measurements and while still under anesthesia, hearts were extracted, weighed and divided with half of the ventricle embedded in Optimal Cutting Temperature (OCT) medium (Tissue-Tek) and half processed for SDS-PAGE as described above. Following exsanguination, the tibia was exposed and measured [41]. Ventricle cryosections of 8 μm were cut on a cryostat (Bright) and stained with hematoxylin and eosin [44], rabbit anti-mouse fibronectin (1:40, Abcam, catalogue number ab23750) and detected by subsequent incubation with Alexa 555 goat anti-rabbit IgG (1:200, Life Technologies, catalogue number A21429) [45] or Alexa 488 goat anti-mouse IgG (1:200, Life Technologies, catalogue number A11029) as a marker of degenerating myocytes that contain intracellular serum proteins [45]. Stained sections were imaged on a Nikon Eclipse 800 microscope under a 10x objective using a Nikon DS-Ri2 Digital camera driven by Nikon Br Elements software. Fibronectin quantification was performed using Adobe Photoshop CS6, at a tolerance of 70 and is reported as the percentage of cross-sectional area [45].
2.8. Statistics.
All data is presented as mean +/-standard error of the mean. The comparison of 2 groups was determined by unpaired t test. The comparison of two groups over multiple heart rates was determined by two-way ANOVA with Sidak’s multiple comparisons test. The comparison of one group over multiple heart rates was determined by one-way ANOVA with Tukey’s multiple comparisons test. Statistical significance was defined as P<0.05. Statistics were performed in Prism 9.
3. Results
To investigate the role of decreased TnI Ser-23/24 phosphorylation on basal in vivo heart function, we employed TnI phosphorylation null transgenic mice expressing 100% of the cardiac TnI containing residues Ser-23 and Ser-24 mutated to alanine (TnI S23/24A) that lacks the ability to be phosphorylated at these residues [34]. We performed Western blot to quantify cardiac and slow skeletal TnI expression in the heart. Similar to the original report of this mouse line [34], Western blot demonstrated knock-out of the cardiac TnI gene and transgenic expression of the phosphorylation-null cardiac TnI S23/24A and did not alter expression levels of slow skeletal TnI in the heart (Suppl. Fig. 1) [34]. Excised whole heart weight was not different between TnI S23/24A and WT mice (Heart weight normalized to tibia length: WT = 0.00675 ± 0.000174 g mm−1, TnI S23/24A = 0.00690 ± 0.000103 g mm−1; p>0.05) (Fig. 1A). Likewise, there was no difference in conscious resting heart rate between TnI S23/24A and WT mice (Resting heart rate: WT = 729 + 24.6 BPM, TnI S23/24A = 746 ± 14.3 BPM; p>0.05) (Fig. 1B). Sequential ProQ Diamond phospho protein specific and Sypro Ruby stained gels of total left ventricular proteins demonstrated no difference in cardiac TnI expression or total protein, myosin binding protein C, troponin T, tropomyosin, or myosin regulatory light chain phosphorylation of TnI S23/24A compared to WT mouse hearts (Suppl. Fig. 2 and Suppl. Table 2) indicating the expression of TnI S23/24A did not cause compensatory changes in cardiac muscle proteins. M-mode echocardiography measurements demonstrated no difference in cardiac structure or calculated cardiac performance in TnI S23/24A compared to WT mice (Fig. 1C–F; Table 1). Hemodynamic measurements from mice paced to 480 beats per minute demonstrated TnI S23/24A mice had impaired systolic (decreased dP/dtmax) and impaired diastolic (less negative dP/dtmin, longer Tau and decreased maximal diastolic volume) function compared to WT mice (Fig. 2, Table 2). Interestingly, the impaired systolic function in the TnI S23/24A mice occurred without altered end systolic pressure, diastolic pressure, stroke volume or cardiac output (Table 2). Together these data demonstrate the loss of cardiac TnI Ser-23/24 phosphorylation in the heart results in impaired systolic and diastolic function at basal conditions in vivo.
Figure 1. Cardiac structure and calculated performance of WT and TnI S23/24A mice.
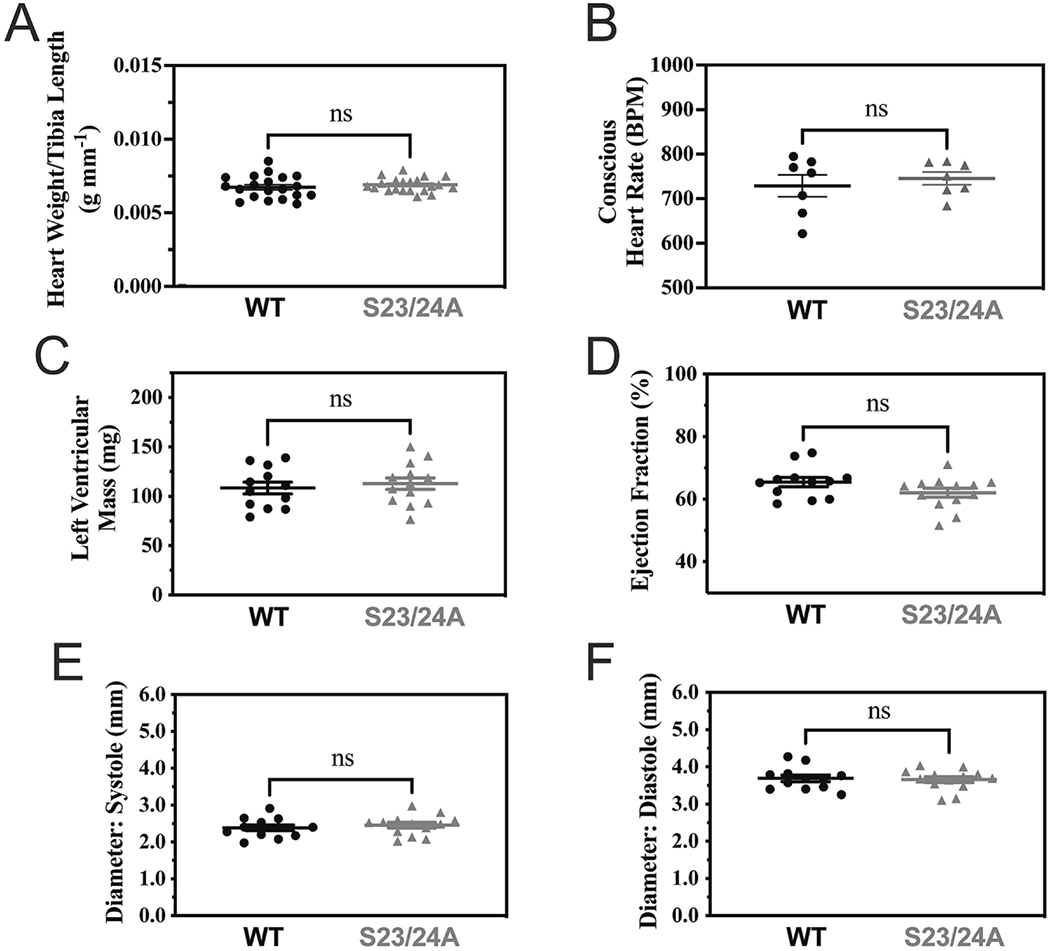
(A) Excised heart weight normalized to tibia length of WT (black circle) and TnI S23/24A (grey triangle) mice. n = 20 mice for WT and n = 21 mice for TnI S23/24A. (B) Conscious Heart Rate of WT and TnI S23/24A mice. Echocardiography measurements of WT and TnI S23/24A mice for (C) left ventricular mass, (D) ejection fraction, (E) left ventricular diameter at systole and (F) left ventricular diameter at diastole. n = 12 mice for WT and n = 13 mice for TnI S23/24A. ns, P>0.05; *P<0.05, WT versus TnI S23/24A mice by unpaired t test. Error bars are standard error of the mean.
Table 1.
Cardiac Echocardiography.
| ECHOCARDIOGRAPHY | |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Parameter | Units | WT | S23/24A | t test | WT-TAC | S23/24A-TAC | t test |
| Number | 12 | 13 | 6 | 9 | |||
| HR | BPM | 361 ± 18.0 | 345 ± 12.7 | 0.464 | 398 ± 25.4 | 380 ± 15.6 | 0.520 |
| EF | % | 65.5 ± 1.46 | 62.1 ± 1.43 | 0.112 | 52.5 ± 2.17 | 46.9 ± 4.92 | 0.394 |
| LVESD | mm | 2.39 ± 0.0767 | 2.46 ± 0.0766 | 0.523 | 2.72 ± 0.0866 | 2.83 ± 0.194 | 0.681 |
| LVEDD | mm | 3.69 ± 0.0885 | 3.66 ± 0.0783 | 0.762 | 3.71 ± 0.149 | 3.68 ± 0.153 | 0.882 |
| LV Mass | mg | 108 ± 5.90 | 113 ± 5.88 | 0.607 | 144 ± 20.7 | 187 ± 12.2 | 0.078 |
| LVAW;s | mm | 1.47 ± 0.0441 | 1.48 ± 0.0478 | 0.901 | 1.45 ± 0.0888 | 1.74 ± 0.146 | 0.159 |
| LVAW;d | mm | 0.946 ± 0.0324 | 1.01 ± 0.0411 | 0.262 | 1.10 ± 0.0772 | 1.41 ± 0.122 | 0.076 |
WT, wild-type mice; S23/24A, TnI S23/24A mice; WT-TAC, WT mice plus TAC; S23/24A-TAC, TnI S23/24A mice plus TAC, HR, heart rate; EF, ejection fraction; LVESD, left ventricular end systolic diameter; LVEDD, left ventricular end diastolic diameter; LV Mass, left ventricular mass; LVAW;s, left ventricular anterior wall thickness at end systole; LVAW;d, left ventricular anterior wall thickness at end diastole; BPM, beats per minute; %, percent; mm, millimeter; mg, milligram. Values are mean +/−standard error of the mean.
P<0.05, WT versus TnI S23/24A mice by unpaired t test.
Figure 2. Cardiac hemodynamic function of WT and TnI S23/24A mice.
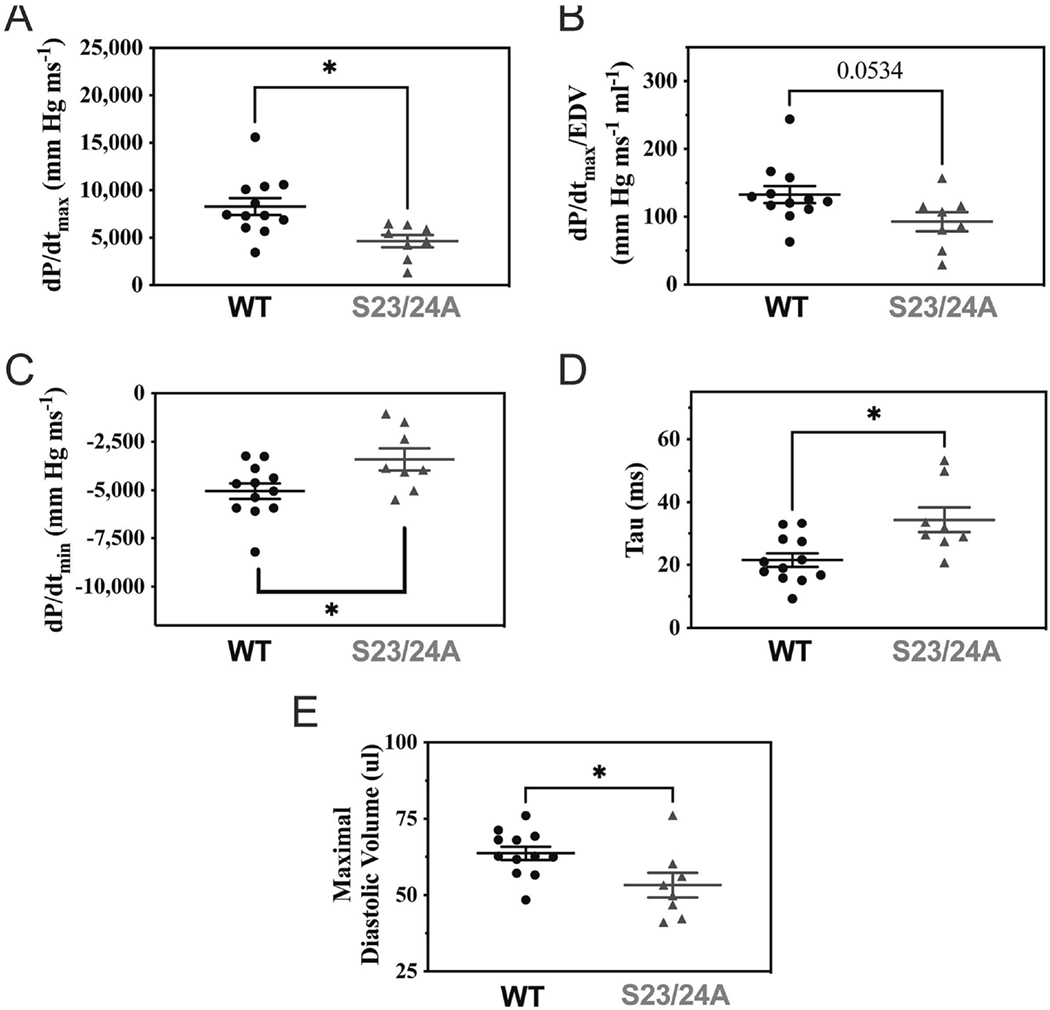
In vivo left ventricular pressure-volume hemodynamic measurements of WT (black circle) and TnI S23/24A (grey triangle) mice for (A) dP/dtmax, (B) dP/dtmax/EDV, (C) dP/dtmin, (D) Tau and (E) maximal diastolic volume. n = 12 mice for WT and n = 8 mice for TnI S23/24A. *P<0.05, WT versus TnI S23/24A mice by unpaired t test. Error bars are standard error of the mean.
Table 2.
Cardiac Hemodynamics.
| HEMODYNAMICS | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Parameter | Units | WT | S23/24A | t test | WT+Dob | S23/24A+Dob | t test | WT-TAC | S23/24A-TAC | t test | |||||
| Number | 12 | 8 | 6 | 9 | 6 | 6 | |||||||||
| HR | BPM | 483 ± 2.00 | 482 ± 1.16 | 0.687 | 459 ± 22.9 | 326 ± 12.7 | <0.000 | * | 478 ± 2.62 | 478 ± 2.80 | 0.956 | ||||
| dP/dTmax | mm Hg ms-1 | 8,286 ± 899 | 4,641 ± 645 | 0.008 | * | 11,336 ± 1,064 | 9,035 ± 581 | 0.060 | 6,011 ± 1,041 | 3,763 ± 738 | 0.109 | ||||
| dP/dTmax/EDV | mm Hg ms-1 ml-1 | 133 ± 12.6 | 92.8 ± 14.3 | 0.053 | 402 ± 73.4 | 336 ± 28.6 | 0.356 | 93.6 ± 11.9 | 70.5 ± 13.0 | 0.219 | |||||
| dP/dTmin | mm Hg ms-1 | −5,045 ± 402 | −3,408 ± 570 | 0.026 | * | −5,604 ± 476 | −4,243 ± 504 | 0.086 | −4,655 ± 1,119 | −2,808 ± 659 | 0.185 | ||||
| tau | ms | 21.5 ± 2.16 | 34.4 ± 4.00 | 0.006 | * | 16.1 ± 3.38 | 32.3 ± 4.97 | 0.028 | * | 24.3 ± 5.36 | 36.8 ± 11.2 | 0.338 | |||
| ESP | mmHg | 108 ± 6.23 | 95.9 ± 9.80 | 0.278 | 117 ± 15.9 | 125 ± 12.6 | 0.689 | 118 ± 23.3 | 110 ± 15.0 | 0.792 | |||||
| EDP | mmHg | 24.3 ± 3.73 | 31.2 ± 3.18 | 0.209 | 18.6 ± 3.72 | 28.8 ± 4.52 | 0.125 | 22.7 ± 5.60 | 30.3 ± 7.99 | 0.452 | |||||
| Vmin | μl | 36.1 ± 3.69 | 26.6 ± 1.61 | 0.074 | 21.3 ± 2.55 | 16.2 ± 0.757 | 0.043 | * | 35.5 ± 7.27 | 38.7 ± 3.58 | 0.703 | ||||
| Vmax | μl | 63.7 ± 2.15 | 53.3 ± 4.01 | 0.022 | * | 32.1 ± 3.38 | 28.8 ± 2.22 | 0.406 | 65.1 ± 2.77 | 56.1 ± 1.51 | 0.018 | * | |||
| SV | μl | 27.6 ± 3.63 | 23.3 ± 1.64 | 0.377 | 10.9 ± 1.71 | 12.6 ± 1.76 | 0.514 | 29.6 ± 5.22 | 17.4 ± 2.62 | 0.065 | |||||
| CO | μl/min | 13,311 ± 1,764 | 11,220 ± 789 | 0.369 | 4,835 ± 611 | 4,172 ± 659 | 0.499 | 14,096 ± 2,487 | 8,356 ± 1,286 | 0.068 | |||||
WT, wild-type mice; S23/24A, TnI S23/24A mice; WT+Dob, WT mice plus dobutamine; S23/24A+Dob, TnI S23/24A mice plus dobutamine; WT-TAC, WT mice plus TAC; S23/24A-TAC, TnI S23/24A mice plus TAC, HR, heart rate; dP/dTmax, maximal rate of left ventricular pressure rise; dP/dTmax/EDV, maximal rate of left ventricular pressure rise / end diastolic volume; dP/dTmin, maximal rate of left ventricular pressure decrease; tau, isovolumic relaxation constant; ESP, end systolic pressure; EDP, end diastolic pressure; Vmin, minimal systolic volume; Vmax, maximal diastolic volume; SV, stroke volume; CO, cardiac output; BPM, beats per minute; mmHg, millimeters of mercury; ms, millisecond; μl, microliter. Values are mean +/- standard error of the mean.
P<0.05, WT versus TnI S23/24A mice by unpaired t test.
The ability of the heart to increase its cardiac output upon increased demand is cardiac reserve. A robust cardiac reserve is critical to normal heart function and is impaired in disease [46–48]. To further investigate the effect of decreased TnI Ser-23/24 phosphorylation on key parameters that influence cardiac function, we measured cardiac function in TnI S23/24A mice upon increasing heart rate or beta-adrenergic stimulation. Upon pacing induced increase in heart rate from 360 to 540 beats per minute, TnI S23/24A mice demonstrated the inability to increase systolic function (decreased dP/dtmax and dP/dtmax/EDV), diastolic function (less negative dP/dtmin, longer tau, and increased end diastolic pressure) and cardiac output to the same degree as WT mice (Fig. 3A–F). Pacing was stopped and the beta adrenergic agonist dobutamine was injected once the mice stabilized at their anesthetized heart rate. Following beta-adrenergic stimulation, TnI S23/24A mice demonstrated the inability to increase heart rate to the same degree as WT mice (Table 2). Furthermore, TnI S23/24A mice demonstrated the inability to increase diastolic function (longer Tau) compared to WT mice (Fig. 3G). This stimulation response impairment occurred in the presence of a decrease in minimal systolic volume (Table 2). Following dobutamine, dP/dtmax trended to decrease and dP/dtmin trended to be less negative in TnI S23/24A compared to WT mice, however neither reached the level of statistical significance (Fig. 3H–I). Together these data demonstrate the lack of cardiac TnI Ser-23/24 phosphorylation results in an impaired ability to increase heart function upon increased demand (increasing heart rate and dobutamine stimulation) indicative of an impaired cardiac reserve.
Figure 3. Cardiac reserve of WT and TnI S23/24A mice.
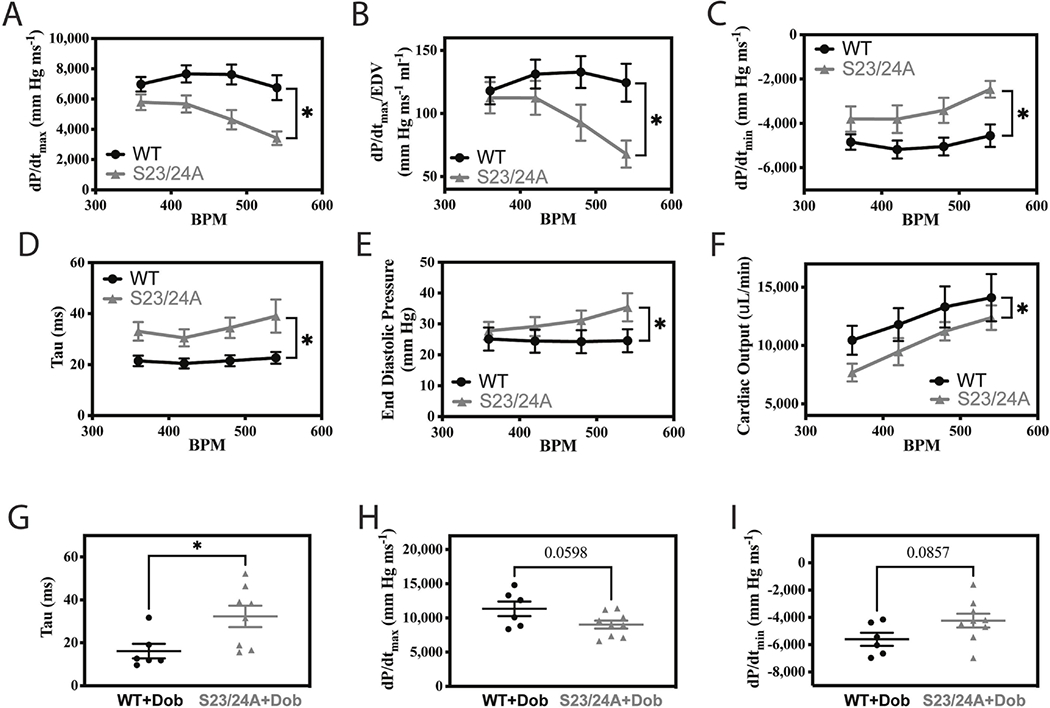
In vivo left ventricular pressure-volume hemodynamic measurements of WT (black circle) and TnI S23/24A (grey triangle) mice: Upon increasing heart rate from 360 to 540 beats per minute for (A) dP/dtmax, (B) dP/dtmax/EDV, (C) dP/dtmin, (D) Tau, (E) end diastolic pressure and (F) cardiac output. n = 12 mice at each heart rate for WT and n = 6–10 mice at each heart rate for TnI S23/24A. *P<0.05, WT versus TnI S23/24A mice by two-way analysis of variance. Following dobutamine stimulation for (G) Tau, (H) dP/dtmax and (I) dP/dtmin. n = 6 mice for WT and n = 9 mice for TnI S23/24A. *P<0.05, WT versus TnI S23/24A mice by unpaired t test. Error bars are standard error of the mean.
Diastolic dysfunction contributes to the disease state in both heart failure with preserved and reduced ejection fraction [47, 49, 50]. Furthermore, the level of TnI Ser-23/24 phosphorylation is decreased in end stage heart failure compared to the non-diseased heart [22, 23, 25]. To investigate the effect of ablating TnI Ser-23/24 phosphorylation during pathological stress, we subjected TnI S23/24A mice to transaortic constriction (TAC). Following TAC, serial echocardiography was conducted at 0, 2, 6, 8, 10 and 12 weeks. At 12 weeks post-TAC mice were subjected to terminal hemodynamic measurements, the hearts excised, weighed, and ventricles separated for SDS-PAGE and histological analysis. Necropsy at 12 weeks post-TAC demonstrated TnI S23/24A TAC mice had increased heart weight compared to WT TAC mice (heart weight normalized to tibia length: WT TAC = 0.00757 ± 0.000883 g mm-1, TnI S23/24A TAC = 0.0114 ± 0.00117 g mm-1; p<0.05) while both mice had similar a body weight (WT TAC = 25.5 ± 1.52 mg, TnI S23/24A TAC = 27.5 ± 1.21 mg; p>0.05) (Fig. 4A–B). Hematoxylin and eosin staining of ventricular sections demonstrated no difference in gross histology between TnI S23/24A TAC and WT TAC mouse hearts (Fig. 4C). To further investigate myocyte death and fibrosis, ventricular sections were stained for IgG and fibronectin. IgG is excluded from healthy cardiac myocytes and its entry into the myocyte identifies ongoing myocyte degeneration. Fibronectin is an extracellular matrix protein normally present between myocytes that is increased as a major component of pathological interstitial or replacement fibrosis. Staining for IgG demonstrated no intracellular IgG present in any of the stained sections from either TnI S23/24A TAC or WT TAC mouse ventricles (data not shown). Likewise, staining for fibronectin demonstrated no difference in the percent of ventricular fibronectin between TnI S23/24A TAC and WT TAC mouse hearts (% Fibronectin: WT TAC = 32.1 ± 2.75%, TnI S23/24A TAC = 28.8 ± 2.87%: p>0.05) (Fig. 4D). Disease related hypertrophic stimulus can increase beta myosin heavy chain expression that can alter cardiac function as a result of its different force generating characteristics [51, 52]. SDS-PAGE analysis of ventricular homogenates demonstrated no difference in beta myosin heavy chain expression between TnI S23/24A TAC and WT TAC mice (% beta myosin heavy chain: WT TAC = 13.5 ± 3.68%, TnI S23/24A TAC = 9.10 ± 2.00%: p>0.05) (Fig. 4E). Serial echocardiography conducted at 0, 2, 6, 8, 10 and 12 weeks post-TAC demonstrated both TnI S23/24A TAC and WT TAC mice exhibited increased left ventricular mass, increased anterior wall thickness during diastole, and decreased ejection fraction over the 12 weeks following TAC indicating the development of cardiac disease (Fig. 5). While there was no statistically significant difference in echocardiography measurements between TnI S23/24A TAC and WT TAC mice at the 12 week time point (Table 1), over the 12 week period TnI S23/24A TAC mice demonstrated worse TAC mediated hypertrophic remodeling and cardiac impairment (further increased left ventricular mass, anterior wall thickness during systole and diastole, and ejection fraction) compared to WT TAC mice (Fig. 5). Hemodynamic measurements demonstrated that TnI S23/24A TAC mice had a more severe impairment in the maximal diastolic volume compared to WT TAC mice (Fig. 6A, Table 2). Following TAC, TnI S23/24A TAC mice also trended to have worse dP/dtmax, dP/dtmin, end diastolic pressure, stroke volume, and cardiac output, however this did not reach the level of significance (Fig. 6B, Table 2). Upon pacing induced increase in the heart rate from 360 to 540 beats per minute, TnI S23/24A TAC mice demonstrated worse systolic function (decreased dP/dtmax), diastolic function (less negative dP/dtmin, decreased maximal diastolic volume) and decreased cardiac output compared to WT TAC mice (Fig. 6C–F). Together these data demonstrate that while both TnI S23/24A and WT mice developed cardiac dysfunction in response to TAC, the inability to phosphorylate cardiac TnI Ser-23/24 resulted in further increased ventricular mass and worsened cardiac function in response to TAC pathological stress, indicative of worse cardiac disease development.
Figure 4. Morphology of WT and TnI S23/24A mice following TAC.
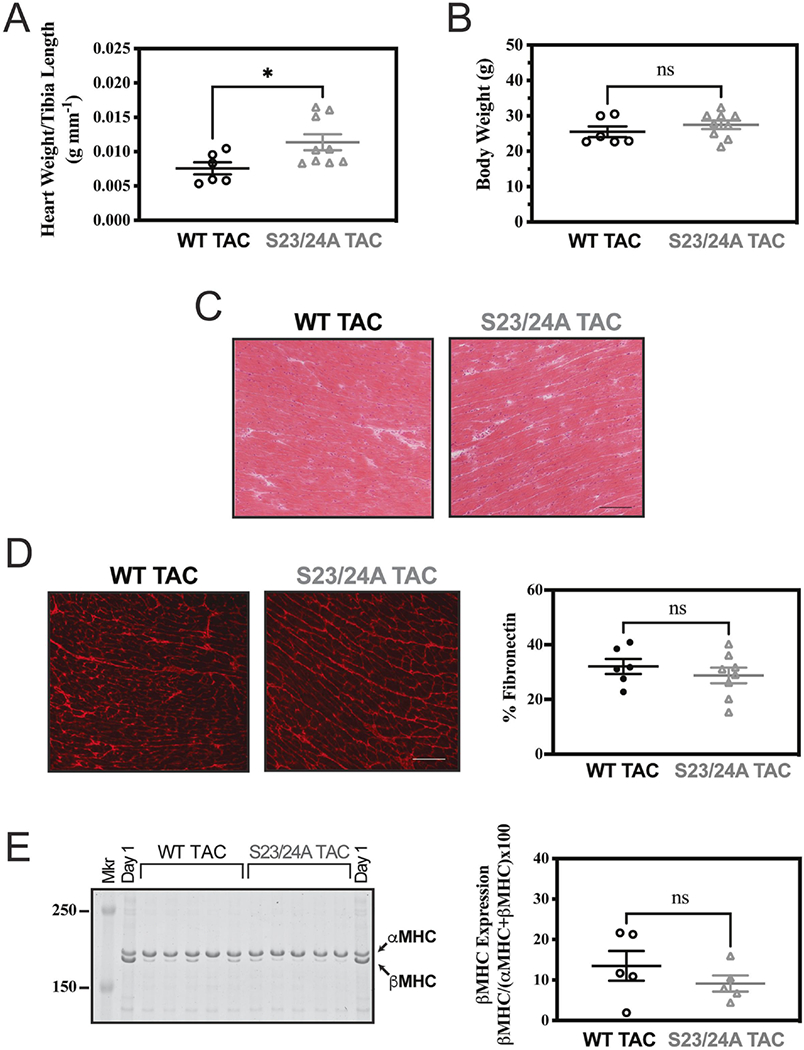
Excised heart cardiac morphology of WT TAC (black circle) and S23/24A TAC (grey triangle) mice at 12 weeks after TAC for (A) heart weight and (B) body weight. n = 6 mice for WT TAC and n = 9 mice for S23/24A TAC. (C) Representative hematoxylin and eosin stained ventricle sections (Bar = 100 um). (D) Representative fibronectin stained ventricle sections (Bar = 100 um) and fibronectin quantification. n = 6 mice for WT TAC and n = 8 mice for S23/24A TAC. (E) Coomassie stained gel of mouse ventricle homogenate and beta myosin heavy chain quantification. αMHC, alpha myosin heavy chain; βMHC, beta myosin heavy chain; Mkr, Molecular Weight Marker; Day 1, 1 day old mouse heart expressing both αMHC and βMHC. n = 5 hearts for WT TAC and n = 5 hearts for S23/24A TAC. ns, P>0.05; *P<0.05, WT TAC versus S23/24A TAC by unpaired t test. Error bars are standard error of the mean.
Figure 5. Cardiac structure and derived function of WT and TnI S23/24A mice following TAC.
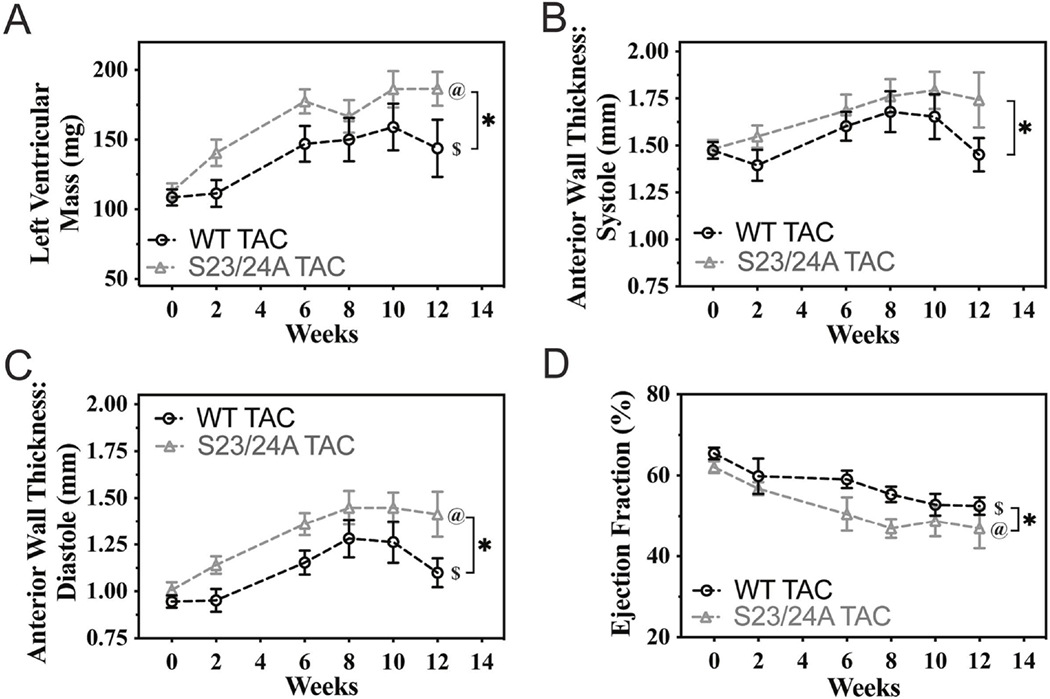
Echocardiography measurement of WT TAC (black circle) and S23/24A TAC (grey triangle) mice before and at 2, 6, 8, 10 and 12 weeks after TAC for (A) left ventricular mass, (B) left ventricular anterior wall thickness at systole, (C) left ventricular anterior wall thickness at diastole and (D) ejection fraction. n = 6-12 mice per week for WT TAC and n = 9-13 mice per week S23/24A TAC. $P<0.05, WT TAC versus time; @P<0.05, S23/24A TAC versus time by one-way analysis of variance. *P<0.05, WT TAC versus S23/24A TAC by two-way analysis of variance. Error bars are standard error of the mean.
Figure 6. Cardiac hemodynamic function and reserve of WT and TnI S23/24A mice following TAC.
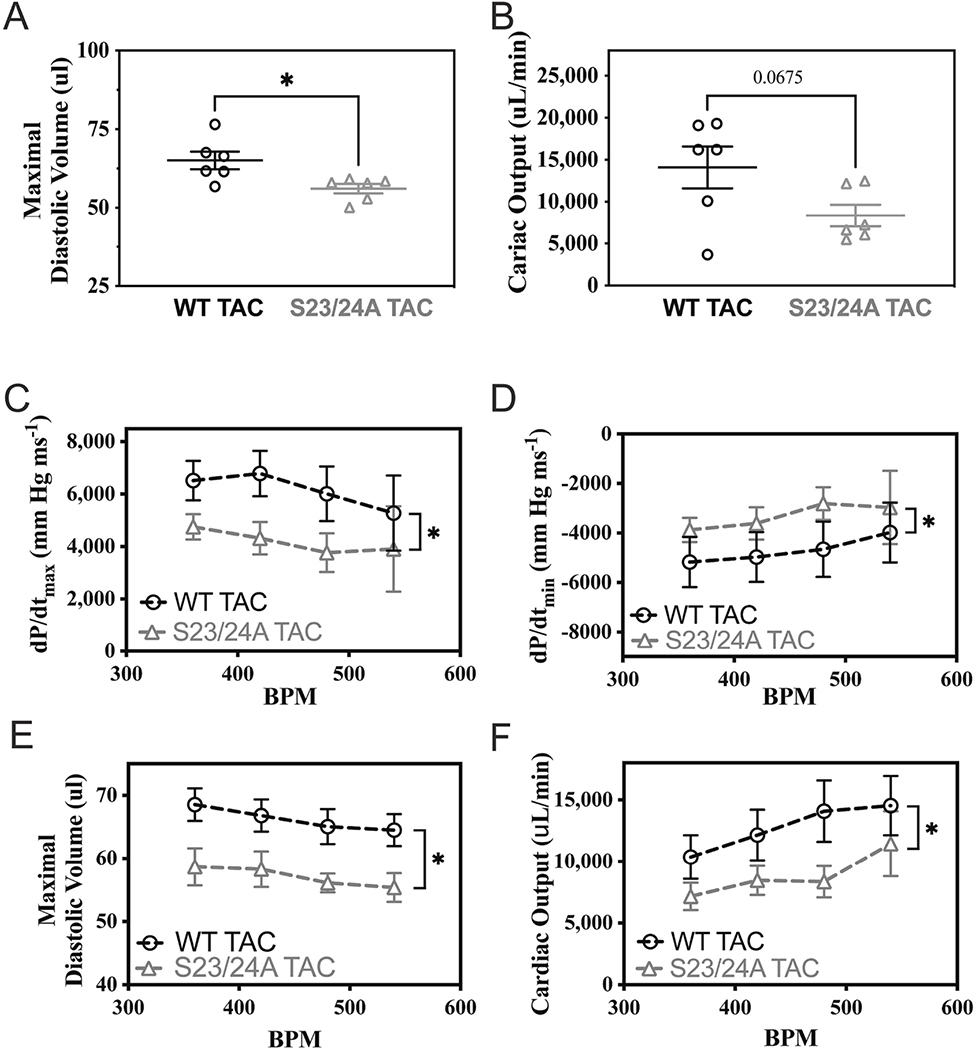
In vivo left ventricular pressure-volume hemodynamic measurements of WT TAC (black circle) and S23/24A TAC (grey triangle) mice at 12 weeks after TAC for (A) maximal diastolic volume and (B) cardiac output. n = 6 mice for WT TAC and n = 6 mice for S23/24A TAC. *P<0.05, WT TAC versus S23/24A TAC by unpaired t test. Hemodynamic measurements upon increasing heart rate from 360 to 540 beats per minute for (C) dP/dtmax, (D) dP/dtmin, (E) maximal diastolic volume and (F) cardiac output. n = 5-6 mice at each heart rate for WT TAC and n = 3-7 mice at each heart rate for TnI S23/24A TAC. *P<0.05, WT TAC versus S23/24A by two-way analysis of variance. Error bars are standard error of the mean.
4. Discussion
4.1. Key Findings.
Increasing the phosphorylation of TnI at Ser-23/24 results in decreased calcium sensitivity of the cardiac muscle, accelerated cardiac muscle relaxation, and accelerated in vivo diastolic function [9–20], however the loss of TnI Ser-23/24 phosphorylation was reported to not affect in vivo heart function [36]. To thoroughly investigate the role of decreased TnI Ser-23/24 phosphorylation on in vivo cardiac function we employed phosphorylation null mice transgenic for the cardiac specific expression of TnI containing residues Ser-23/24 mutated to alanine (TnI S23/24A) that lack the ability to be phosphorylated at these sites and therefore results in no cardiac TnI Ser-23/24 phosphorylation in vivo [34]. Measuring in vivo function in TnI S23/24A transgenic mice subjected to physiological (increasing heart rate or adrenergic stimulation) or pathological (TAC) stress we demonstrate that the loss of cardiac TnI Ser-23/24 phosphorylation: 1) impairs in vivo systolic and diastolic function; 2) impairs the ability to increase systolic and diastolic function upon increasing heart rate and adrenergic stimulation; 3) further increases ventricular weight, impairs diastolic function, and blunts the ability to increase function upon increased demand following TAC mediated cardiac stress. Together these data support decreased cardiac TnI Ser-23/24 phosphorylation as a mechanism that regulates in vivo cardiac function, key modulators of cardiac reserve, and the pathological development of cardiac disease.
4.2. The lack of cardiac TnI Ser-23/24 phosphorylation impairs in vivo systolic and diastolic function.
Troponin I is a myofilament regulatory protein essential to translating the calcium activation signal into an activated myofilament structure that allows myosin’s interaction with actin. Troponin I is therefore a critical protein in the regulation of cardiac contraction and relaxation [53].
Increasing the phosphorylation of TnI at Ser-23/24 is a key physiological and pathological modulator of this myofilament regulation and in vivo heart function [2, 7, 53]. TnI Ser-23/24 phosphorylation accelerates myocardial relaxation by decreasing calcium binding to troponin C and weakening the TnI-troponin C interaction [11] that results in decreased calcium sensitivity [7, 9, 11, 15]. This allows for a faster deactivation of the thin filament and an accelerated cross-bridge detachment rate [11] that speeds active myocardial relaxation without affecting the elasticity of the ventricle or passive filling. In the non-diseased heart, approximately 40% of TnI is basally phosphorylated at Ser-23/24 [21–23] providing the potential for both increased and decreased TnI Ser-23/24 phosphorylation as mechanisms to modulate in vivo heart function. The phosphorylation of cardiac TnI at Ser-23/24 is increased as part of the heart’s mechanism to modulate function in response to physiological stress [7, 20]. Multiple studies have defined the biochemical, muscle, cellular, and in vivo effects of increased TnI Ser23/24 phosphorylation. Increasing the phosphorylation of TnI at Ser-23/24 decreases calcium dependent myofilament ATPase activity, decreases myocyte calcium sensitive force development (calcium sensitivity), accelerates myofibril relaxation, accelerates myocyte re-lengthening, and accelerates in vivo myocardial relaxation [9–20].
The phosphorylation of TnI at Ser-23/24 is decreased in heart failure supporting a role for the decrease of TnI Ser-23/24 phosphorylation to regulate cardiac function [22, 23, 25]. While studies have investigated the effect of expressing slow skeletal TnI (that lacks the 32 amino terminal amino acids containing Ser-23/24) in the heart [26–28], slow skeletal TnI also contains a number of other amino acids that are different from cardic TnI and that have been demonstrated to modify contractile function [29] [30–33] such that these studies were not able to distinguish between the effects of Ser-23/24 phosphorylation loss and other amino acid modifications. While there have been studies that investigated the effects of inhibiting TnI Ser-23/24 phosphorylation in combination with inhibited phosphorylation at other TnI phosphorylation sites, many of these studies focused on understanding the functional effects that result from kinase signaling mediated phosphorylation when TnI Ser-23/24 cannot be phosphorylated and did not directly compare the loss of TnI Ser-23/24 to the basally phosphorylated state. Only a few studies have directly investigated the effect of decreasing only TnI Ser-23/24 phosphorylation without modifying other phosphorylated residues. Pi et al. generated TnI S23/24A phosphorylation null mice and investigated the inability of TnI Ser-23/24 to be phosphorylated on cardiac function following various kinase stimulation [34]. While Pi et al. did not directly compare the basal state of TnI S23/24A and WT mice, their data shows increased calcium sensitivity of skinned myofibrils from TnI S23/24A compared to myofibrils from WT mice [34]. Contrary to this finding, Stelzer et al. reported no difference in calcium sensitivity between skinned ventricular myocardium from TnI S23/24A and WT mice [35]. Yasuda et al. employed gene transfer into cardiac myocytes to investigate the role of TnI Ser-23/24 in adrenergic signaling. Following TnI S23/24A gene transfer into rat myocytes, Yasuda et al. observed no difference in calcium sensitivity, but a trend to slower myocyte re-lengthening (relaxation) in intact myocytes that was not significantly different from WT TnI gene transfer [17]. It is important to consider that gene transfer in this study only accounted for approximately 50% TnI S23/24A expression, which needs to be considered in these findings. Two studies have reported in vivo effects following the loss of TnI Ser-23/24 phosphorylation compared to the basal TnI phosphorylation state. Stelzer et al. reported in vivo echocardiography parameters were not different between TnI S23/24A and WT mice [35]. Subsequently, Gresham and Stelzer investigated the role of TnI and myosin binding protein C in the adrenergic response and reported there was no in vivo functional difference between TnI S23/24A and WT mice at basal conditions or in response to dobutamine as measured by echocardiography and left ventricular hemodynamic function [36]. Based upon these studies, the role of decreased TnI Ser-23/24 phosphorylation on cardiac muscle calcium sensitivity is unclear and the loss of TnI Ser-23/24 phosphorylation does not affect in vivo cardiac function.
To directly investigate the role of decreasing TnI Ser-23/24 phosphorylation as a mechanism to modulate heart function, we designed specific experiments to directly compare cardiac function in TnI S23/24A mice that lack that ability to be phosphorylated at TnI Ser-23/24 and WT mice. Similar to the report by Stelzer et al. [35], we observed no functional difference between TnI S23/24A and WT mouse function by echocardiography (Fig.1C–F, Table 1). We do however demonstrate TnI S23/24A mice have impaired in vivo hemodynamic systolic and diastolic function (Fig. 2, Table 2). This functional impairment occurred in the absence of morphological changes (Figs. 1, Table 1). These results demonstrate that the loss of basal TnI Ser-23/24 phosphorylation is detrimental to in vivo systolic and diastolic cardiac function.
Current understanding predicts altered calcium sensitivity to result in opposite effects on systolic and diastolic function. Based upon the lusitropic effect of decreased calcium sensitivity resulting from increased TnI Ser-23/24 phosphorylation, we predicted decreasing TnI Ser23/24 phosphorylation from the basal state would increase calcium sensitivity to impair active relaxation without affecting passive filling but would enhance contraction. In agreement with this understanding, we observe that TnI S23/24A mice have impaired diastolic function, however, contrary to this current understanding, TnI S23/24A mice have impaired systolic in vivo cardiac function (Fig. 2, Table 2). The in vivo effects of TnI S23/24A on systolic function can result from a direct effect of calcium sensitivity on muscle force or an indirect effect of impaired diastolic function. The direct ability of calcium to activate the myofilament involves changes in multiple myofilament regulatory protein interactions that are not completely understood. While muscle calcium sensitivity is measured at a steady-state calcium equilibrium, intracellular calcium in vivo is constantly changing such that activating calcium is never at equilibrium. As such, muscle calcium sensitivity measurements do not necessarily account for the kinetic changes involved in the myofilament regulation of cardiac function in vivo. Post-translational modification of any myofilament regulatory protein can alter protein-protein interactions to differentially affect steady-state and kinetic thin filament activation [54, 55]. The in vivo effects of decreased TnI Ser-23/24 phosphorylation therefore may not be simply described by its effect on steady-state calcium sensitivity. Further complicating the mechanistic understanding of TnI Ser-23/24 phosphorylation on in vivo systolic function, systolic function is indirectly dependent on preload that is determined by ventricular filling [1, 46, 56]. In vivo, depressed diastolic function can impair ventricular filling such that cardiac contraction is depressed by the Frank-Starling mechanism resulting in impaired systolic function. Thus, TnI S23/24A mediated impairment of diastolic function and decreased maximal diastolic volume (Table 2) may also indirectly impair systolic function. Whether the expression of TnI S23/24A directly or indirectly alters systolic function, our data supports the loss of TnI Ser-23/24 phosphorylation is detrimental to in vivo systolic function. The details of how decreased TnI Ser-23/24 phosphorylation impairs both systolic and diastolic cardiac function necessitate further detailed investigation.
4.3. The loss of cardiac TnI Ser-23/24 phosphorylation impairs in vivo cardiac systolic and diastolic reserve.
The ability of the heart to increase cardiac output in response to elevated demand is the heart’s cardiac reserve. At the organ level, the ability to increase cardiac output is highly influenced by sympathetic tone, the Frank-Starling relationship between sarcomere length and force, and the Bowditch effect of increased force upon increased heart rate [3]. Although these factors are interdependent, each of these factors has its own distinct mechanism to modify systolic and diastolic function in response to physiological stress and elevated demand. The phosphorylation of TnI Ser23-24 is altered with all 3 of these factors suggesting its role in cardiac reserve [7, 12, 16, 18, 20, 57–60].
To further explore the effects of decreased TnI Ser-23/24 phosphorylation on in vivo heart function, we investigated the effects of TnI S23/24A on the mechanisms that modulate cardiac reserve by increasing heart rate and adrenergic stimulation. We found TnI S23/24A mice demonstrated worse systolic and diastolic function upon increasing heart rate (Fig. 3A–E). This worse systolic and diastolic function in TnI S23/24A mice upon increasing heart rate is consistent with the TnI S23/24A mediated impairment of relaxation (Fig. 2C–D) resulting in slowed ventricular filling, and decreased maximal diastolic volume (Fig. 2E). The effect is exacerbated by the increasingly shorter ventricular filling time when heart rate is increased. This finding is the first to demonstrate TnI Ser-23/24 phosphorylation is necessary for a normal in vivo response to increasing heart rate in vivo.
While TnI S23/24A mice were able to increase in vivo function in response to physiological stress by dobutamine (adrenergic signaling), this response was blunted in TnI S23/24A mice compared to WT mice (Fig. 3G–I, Table 2). While TnI S23/24A mice did increase their heart rate upon dobutamine, TnI S23/24A mice were not able to increase their heart rate to the same degree as that of WT mice (Table 2). Since key determinants of in vivo function are dependent upon heart rate, the blunted TnI S23/24A diastolic function following dobutamine (Fig. 3G) may have resulted from this impaired ability to increase heart rate upon dobutamine. Whether TnI S23/24A mice are not able to fully increase function following dobutamine because of impaired function or because of an inability to increase heart rate, both explanations demonstrate an impaired response to adrenergic stimulation when TnI Ser-23/24 are not able to be phosphorylated. As TnI S23/24A mice were still able to increase function in response to adrenergic stimulation, other additional mechanisms must also be involved in the adrenergic response such as those involving myosin binding protein C, titin, and / or myosin regulatory light chain [61–63]. This blunted adrenergic in vivo function for TnI S23/24A mice is similar to that reported in mice transgenic for the loss of myosin binding protein C phosphorylation [36, 61, 64, 65] and supports that TnI and myosin binding protein C function jointly upon adrenergic stimulation as previously suggested by muscle mechanic experiments [66]. Together these findings support an essential role of TnI Ser-23/24 phosphorylation for in vivo heart rate and adrenergic mediated cardiac function responses.
4.4. The loss of cardiac TnI Ser-23/24 phosphorylation further increases ventricular weight, impairs diastolic function, and blunts the ability to increase function following TAC mediated cardiac stress.
Myocardial relaxation is impaired in both systolic and diastolic heart failure with detrimental functional effects [47, 49, 50]. The phosphorylation of TnI at Ser-23/24 is increased following myocardial infarction [67] and is decreased in end state heart failure [22, 23, 25] suggesting a role for TnI Ser-23/24 phosphorylation in cardiac pathology. While increased TnI Ser-23/24 phosphorylation has been demonstrated to have beneficial effects in a hypertrophic cardiomyopathy model [19], the functional effects of decreased TnI Ser-23/24 phosphorylation during pathological cardiac stress and its role in disease development are unknown.
To investigate decreased TnI Ser-23/24 phosphorylation during chronic pathological stress, we subjected TnI S23/24A mice to TAC as a model of pressure overload-induced cardiomyopathy. TnI Ser-23/24A mice subjected to 12 weeks of TAC exhibited a larger left ventricular mass (Fig. 5A), decreased maximal diastolic volume (Fig. 6A), inability to increase systolic and diastolic function to a similar magnitude as WT mice upon increasing heart rate (Fig. 6C–F) and a larger increase in heart weight compared to WT mice subjected to TAC (Fig. 4A). This decreased maximal diastolic volume and greater increase in left ventricular mass and heart weight likely results from the inability of TnI S23/24A mice to decrease calcium sensitivity by TnI Ser-23/24 phosphorylation in response to TAC mediated stress. The TnI Ser-23/24 mediated increase in calcium sensitivity during TAC further slows myocardial relaxation to further decrease maximal diastolic volume, which is consistent with hypertrophic remodeling associated with increased calcium sensitivity [49, 68]. Heart weight was not increased in TnI S23/24A mice in the absence of TAC (Fig. 1A) suggesting the increased calcium sensitivity resulting from the loss of TnI S23/24A alone is not sufficient to induce increased heart weight, possibly due to compensation by post-translational modifications not detected in our global assessment of myofilament phosphorylation (Suppl. Fig. 2 and Suppl. Table 2). Upon the added stress of TAC, TnI Ser-23/24 mediated increase in calcium sensitivity may reach a threshold level that is no longer able to be compensated for and is sufficient to induce cardiac hypertrophic remodeling of TnI S23/24A TAC compared to WT TAC mice. Together these findings demonstrate that losing the ability to phosphorylate TnI Ser-23/24 is further detrimental during chronic pathological cardiac stress and suggests a role for altered TnI Ser-23/24 phosphorylation in cardiac pathology.
4.5. Study considerations.
Our finding of depressed in vivo systolic and diastolic function in TnI S23/24A mice at the basal state (Fig. 2) contradicts the findings from Gresham and Stelzer [36]. One possible explanation for this discrepancy is the difference in experimental design addressing different questions between our two studies, Gresham and Stelzer compared 4 groups of mice while we directly compared 2 groups of mice. Furthermore, our studies measured different hemodynamic parameters. Finally, Gresham and Stelzer [36] conducted their hemodynamic measurements in mice anesthetized with isoflurane under ventilation, while our hemodynamic measurements were conducted under ketamine and xylazine anesthesia, at paced heart rates and in spontaneously breathing mice. It is possible that we observed impaired hemodynamics in the TnI S23/24A mouse while Gresham and Stelzer did not, due to a combination of these differing factors.
Although we demonstrate impaired in vivo basal cardiac function and worse diastolic function following TAC upon the loss of TnI Ser-23/24 phosphorylation in the TnI S23/24A mouse, additional points must be considered when applying these findings to understand the effect of decreased TnI Ser-23/24 phosphorylation. The TnI S23/24A mouse utilizes the phosphorylation null mutation of serine to alanine to ablate Ser-23/24 phosphorylation. Pseudo-phosphorylation aspartic acid mutations have been demonstrated to structurally and functionally mimic phosphate [18, 69–71] and the phosphorylation null TnI S23/24A alanine mutations do not differ from TnI containing serine at residues 23/24 in myofilament calcium sensitivity or troponin calcium dissociation [14, 69, 72]. We cannot, however, rule out possible effects of the alanine amino acid substitution itself and must consider this in the interpretation of our functional outcomes. Additionally, the TnI S23/24A mouse results in the constitutive loss of TnI Ser-23/24 phosphorylation while decreased TnI Ser-23/24 phosphorylation is transient. Expression of the phosphorylation null cardiac TnI S23/24A is driven by the alpha myosin heavy chain promoter and therefore becomes the sole TnI expressed by 2 weeks after birth when cardiac TnI is also knocked out [34, 73, 74]. This mouse model therefore generates a constitutive loss of TnI Ser-23/24 phosphorylation in the heart from this time. The mouse heart is developed by this time, however the constitutive loss of Ser-23/24 phosphorylation may induce compensatory changes. While we did not observe compensatory alteration of total cardiac protein, myosin binding protein C phosphorylation, troponin T, tropomyosin, or regulatory light chain phosphorylation (Suppl. Fig. 2 and Suppl. Table 2), we cannot rule out all possible compensatory muscle alterations that may affect cardiac function. Future experiments testing the transient effects of decreasing TnI Ser-23/24 phosphorylation by employing an inducible model of TnI S23/24A expression at different time points will be significant to investigate the effects of transiently decreasing TnI Ser-23/24 phosphorylation.
Our evaluation of cardiac function did not include Doppler or speckle tracking echocardiography to measure blood flow or cardiac strain, such measurements may detect further functional differences between these mice. While we used dP/dt max/EDV to normalize for preload, we did not perform occlusion to directly evaluate pre-load dependence in the hemodynamic measurements. The conductance signal for our hemodynamics measurements were calibrated using known volumes of blood. This method can produce inaccurate values because the cylinders of blood do not exactly replicate the electric field distribution of the mouse heart and there can be variability between disease states [75]. This could explain why the left ventricular volumes from hemodynamics are a bit larger than those calculated by echocardiography. We concluded that TAC induced hypertrophic remodeling based on excised heart weight and left ventricular wall measurements from echocardiography, but we did not assess cardiomyocyte size. Additionally, performing a more severe aortic constriction or evaluating the mice for longer than 12 weeks post-TAC could result in a greater difference in cardiac structure or function between WT TAC and S23/24A TAC groups.
4.6. Summary
Our current findings demonstrate the loss of cardiac TnI Ser-23/24 phosphorylation results in impaired basal cardiac function, impaired response to increasing heart rate and adrenergic stimulation, and worse hypertrophy and diastolic dysfunction following pathological stress. These findings support that cardiac TnI Ser-23/24 phosphorylation is necessary for the adult heart to maintain basal in vivo cardiac function and to increase cardiac function upon physiological stress. Furthermore, the development of greater hypertrophic remodeling and decreased maximal diastolic volume in the absence of TnI Ser-23/24 phosphorylation following TAC supports a role for TnI Ser-23/24 phosphorylation in the progression of pathology, such as heart failure, when TnI Ser23/24 phosphorylation is decreased. Together these findings support the role of decreased TnI Ser-23/24 phosphorylation as a mechanism to regulate in vivo cardiac function.
Supplementary Material
Highlights.
The loss of cardiac TnI Ser-23/24 phosphorylation impairs in vivo systolic and diastolic function.
The loss of cardiac TnI Ser-23/24 phosphorylation impairs the ability to increase systolic and diastolic function upon increasing heart rate and adrenergic stimulation.
The loss of cardiac TnI Ser-23/24 further increases ventricular mass, impairs diastolic function and blunts the ability to increase function upon increased demand following TAC mediated cardiac stress.
Funding
This work was supported by the National Institutes of Health R01 HL114940 (B.J.B), R01 AG060542 (M.T.Z), and T32 HL134616 (L.S and S.S).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: none declared.
References
- [1].Solaro RJ, Integration of myofilament response to Ca2+ with cardiac pump regulation and pump dynamics, Am J Physiol 277(6 Pt 2) (1999) S155–63. [DOI] [PubMed] [Google Scholar]
- [2].Solaro RJ, Westfall M, Physiology of the myocardium, in: Sellke FW (Ed.), Surgery of the Chest, Elsevier Sanders, Philadelphia, 2005, pp. 767–779. [Google Scholar]
- [3].Janssen PM, Myocardial contraction-relaxation coupling, Am J Physiol Heart Circ Physiol 299(6) (2010) H1741–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kobayashi T, Solaro RJ, Calcium, thin filaments, and the integrative biology of cardiac contractility, Annu Rev Physiol 67 (2005) 39–67. [DOI] [PubMed] [Google Scholar]
- [5].Janssen PM, Biesiadecki BJ, Ziolo MT, Davis JP, The Need for Speed: Mice, Men, and Myocardial Kinetic Reserve, Circ Res 119(3) (2016) 418–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Solaro RJ, Modulation of cardiac Myofilament activity by protein phosphorylation., in: Page E, Fozzard H, Solaro RJ (Eds.), Handbook of Physiology: Section 2. The Cardiovascular System, Oxford University Press, New York, 2001, pp. 264–300. [Google Scholar]
- [7].Solaro RJ, Moir AJ, Perry SV, Phosphorylation of troponin I and the inotropic effect of adrenaline in the perfused rabbit heart, Nature 262(5569) (1976) 615–7. [DOI] [PubMed] [Google Scholar]
- [8].Solaro RJ, Henze M, Kobayashi T, Integration of troponin I phosphorylation with cardiac regulatory networks, Circ Res 112(2) (2013) 355–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kentish JC, McCloskey DT, Layland J, Palmer S, Leiden JM, Martin AF, Solaro RJ, Phosphorylation of troponin I by protein kinase A accelerates relaxation and crossbridge cycle kinetics in mouse ventricular muscle, Circ Res 88(10) (2001) 1059–65. [DOI] [PubMed] [Google Scholar]
- [10].Nixon BR, Thawornkaiwong A, Jin J, Brundage EA, Little SC, Davis JP, Solaro RJ, Biesiadecki BJ, AMP-activated protein kinase phosphorylates cardiac troponin I at Ser-150 to increase myofilament calcium sensitivity and blunt PKA-dependent function, J Biol Chem 287(23) (2012) 19136–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rao V, Cheng Y, Lindert S, Wang D, Oxenford L, McCulloch AD, McCammon JA, Regnier M, PKA phosphorylation of cardiac troponin I modulates activation and relaxation kinetics of ventricular myofibrils, Biophys J 107(5) (2014) 1196–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ramirez-Correa GA, Cortassa S, Stanley B, Gao WD, Murphy AM, Calcium sensitivity, force frequency relationship and cardiac troponin I: critical role of PKA and PKC phosphorylation sites, J Mol Cell Cardiol 48(5) (2010) 943–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hanft LM, Biesiadecki BJ, McDonald KS, Length dependence of striated muscle force generation is controlled by phosphorylation of cTnI at serines 23/24, J Physiol 591(18) (2013) 4535–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rao VS, Korte FS, Razumova MV, Feest ER, Hsu H, Irving TC, Regnier M, Martyn DA, N-terminal phosphorylation of cardiac troponin-I reduces length-dependent calcium sensitivity of contraction in cardiac muscle, J Physiol 591(2) (2013) 475–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhang R, Zhao J, Mandveno A, Potter JD, Cardiac troponin I phosphorylation increases the rate of cardiac muscle relaxation, Circ Res 76(6) (1995) 1028–35. [DOI] [PubMed] [Google Scholar]
- [16].Takimoto E, Soergel DG, Janssen PM, Stull LB, Kass DA, Murphy AM, Frequency- and afterload-dependent cardiac modulation in vivo by troponin I with constitutively active protein kinase A phosphorylation sites, Circ Res 94(4) (2004) 496–504. [DOI] [PubMed] [Google Scholar]
- [17].Yasuda S, Coutu P, Sadayappan S, Robbins J, Metzger JM, Cardiac transgenic and gene transfer strategies converge to support an important role for troponin I in regulating relaxation in cardiac myocytes, Circ Res 101(4) (2007) 377–86. [DOI] [PubMed] [Google Scholar]
- [18].Sakthivel S, Finley NL, Rosevear PR, Lorenz JN, Gulick J, Kim S, VanBuren P, Martin LA, Robbins J, In vivo and in vitro analysis of cardiac troponin I phosphorylation, J Biol Chem 280(1) (2005) 703–14. [DOI] [PubMed] [Google Scholar]
- [19].Alves ML, Dias FAL, Gaffin RD, Simon JN, Montminy EM, Biesiadecki BJ, Hinken AC, Warren CM, Utter MS, Davis RTR, Sakthivel S, Robbins J, Wieczorek DF, Solaro RJ, Wolska BM, Desensitization of myofilaments to Ca2+ as a therapeutic target for hypertrophic cardiomyopathy with mutations in thin filament proteins, Circ Cardiovasc Genet 7(2) (2014) 132–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kranias EG, Solaro RJ, Phosphorylation of troponin I and phospholamban during catecholamine stimulation of rabbit heart, Nature 298(5870) (1982) 182–4. [DOI] [PubMed] [Google Scholar]
- [21].Ayaz-Guner S, Zhang J, Li L, Walker JW, Ge Y, In vivo phosphorylation site mapping in mouse cardiac troponin I by high resolution top-down electron capture dissociation mass spectrometry: Ser22/23 are the only sites basally phosphorylated, Biochemistry 48(34) (2009) 8161–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zakhary DR, Moravec CS, Stewart RW, Bond M, Protein kinase A (PKA)-dependent troponin-I phosphorylation and PKA regulatory subunits are decreased in human dilated cardiomyopathy, Circulation 99(4) (1999) 505–10. [DOI] [PubMed] [Google Scholar]
- [23].Zhang P, Kirk JA, Ji W, dos Remedios CG, Kass DA, Van Eyk JE, Murphy AM, Multiple reaction monitoring to identify site-specific troponin I phosphorylated residues in the failing human heart, Circulation 126(15) (2012) 1828–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhang J, Guy MJ, Norman HS, Chen YC, Xu Q, Dong X, Guner H, Wang S, Kohmoto T, Young KH, Moss RL, Ge Y, Top-down quantitative proteomics identified phosphorylation of cardiac troponin I as a candidate biomarker for chronic heart failure, J Proteome Res 10(9) (2011) 4054–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bodor GS, Oakeley AE, Allen PD, Crimmins DL, Ladenson JH, Anderson PA, Troponin I phosphorylation in the normal and failing adult human heart, Circulation 96(5) (1997) 1495–500. [DOI] [PubMed] [Google Scholar]
- [26].Wolska BM, Vijayan K, Arteaga GM, Konhilas JP, Phillips RM, Kim R, Naya T, Leiden JM, Martin AF, de Tombe PP, Solaro RJ, Expression of slow skeletal troponin I in adult transgenic mouse heart muscle reduces the force decline observed during acidic conditions, J Physiol 536(Pt 3) (2001) 863–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Fentzke RC, Buck SH, Patel JR, Lin H, Wolska BM, Stojanovic MO, Martin AF, Solaro RJ, Moss RL, Leiden JM, Impaired cardiomyocyte relaxation and diastolic function in transgenic mice expressing slow skeletal troponin I in the heart, J Physiol 517 (Pt 1)(Pt 1) (1999) 143–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Carley AN, Taglieri DM, Bi J, Solaro RJ, Lewandowski ED, Metabolic efficiency promotes protection from pressure overload in hearts expressing slow skeletal troponin I, Circ Heart Fail 8(1) (2015) 119–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Westfall MV, Albayya FP, Metzger JM, Functional analysis of troponin I regulatory domains in the intact myofilament of adult single cardiac myocytes, J Biol Chem 274(32) (1999) 22508–16. [DOI] [PubMed] [Google Scholar]
- [30].Day SM, Westfall MV, Fomicheva EV, Hoyer K, Yasuda S, La Cross NC, D'Alecy LG, Ingwall JS, Metzger JM, Histidine button engineered into cardiac troponin I protects the ischemic and failing heart, Nat Med 12(2) (2006) 181–9. [DOI] [PubMed] [Google Scholar]
- [31].Westfall MV, Albayya FP, Turner II, Metzger JM, Chimera analysis of troponin I domains that influence Ca(2+)-activated myofilament tension in adult cardiac myocytes, Circ Res 86(4) (2000) 470–7. [DOI] [PubMed] [Google Scholar]
- [32].Westfall MV, Turner I, Albayya FP, Metzger JM, Troponin I chimera analysis of the cardiac myofilament tension response to protein kinase A, Am J Physiol Cell Physiol 280(2) (2001) C324–32. [DOI] [PubMed] [Google Scholar]
- [33].Dargis R, Pearlstone JR, Barrette-Ng I, Edwards H, Smillie LB, Single mutation (A162H) in human cardiac troponin I corrects acid pH sensitivity of Ca2+-regulated actomyosin S1 ATPase, J Biol Chem 277(38) (2002) 34662–5. [DOI] [PubMed] [Google Scholar]
- [34].Pi Y, Zhang D, Kemnitz KR, Wang H, Walker JW, Protein kinase C and A sites on troponin I regulate myofilament Ca2+ sensitivity and ATPase activity in the mouse myocardium, J Physiol 552(Pt 3) (2003) 845–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Stelzer JE, Patel JR, Walker JW, Moss RL, Differential roles of cardiac myosin-binding protein C and cardiac troponin I in the myofibrillar force responses to protein kinase A phosphorylation, Circ Res 101 (5) (2007) 503–11. [DOI] [PubMed] [Google Scholar]
- [36].Gresham KS, Stelzer JE, The contributions of cardiac myosin binding protein C and troponin I phosphorylation to beta-adrenergic enhancement of in vivo cardiac function, J Physiol 594(3) (2016) 669–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kooij V, Saes M, Jaquet K, Zaremba R, Foster DB, Murphy AM, Dos Remedios C, van der Velden J, Stienen GJ, Effect of troponin I Ser23/24 phosphorylation on Ca2+-sensitivity in human myocardium depends on the phosphorylation background, J Mol Cell Cardiol 48(5) (2010) 954–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Salhi HE, Walton SD, Hassel NC, Brundage EA, de Tombe PP, Janssen PM, Davis JP, Biesiadecki BJ, Cardiac troponin I tyrosine 26 phosphorylation decreases myofilament Ca2+ sensitivity and accelerates deactivation, J Mol Cell Cardiol 76 (2014) 257–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Biesiadecki BJ, Tachampa K, Yuan C, Jin JP, de Tombe PP, Solaro RJ, Removal of the cardiac troponin I N-terminal extension improves cardiac function in aged mice, J Biol Chem 285(25) (2010) 19688–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Dias FA, Urboniene D, Yuzhakova MA, Biesiadecki BJ, Pena JR, Goldspink PH, Geenen DL, Wolska BM, Ablation of iNOS delays cardiac contractile dysfunction in chronic hypertension, Front Biosci (Elite Ed) 2 (2010) 312–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Shettigar V, Zhang B, Little SC, Salhi HE, Hansen BJ, Li N, Zhang J, Roof SR, Ho HT, Brunello L, Lerch JK, Weisleder N, Fedorov VV, Accornero F, Rafael-Fortney JA, Gyorke S, Janssen PM, Biesiadecki BJ, Ziolo MT, Davis JP, Rationally engineered Troponin C modulates in vivo cardiac function and performance in health and disease, Nat Commun 7 (2016) 10794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Pinckard KM, Shettigar VK, Wright KR, Abay E, Baer LA, Vidal P, Dewal RS, Das D, Duarte-Sanmiguel S, Hernandez-Saavedra D, Arts PJ, Lehnig AC, Bussberg V, Narain NR, Kiebish MA, Yi F, Sparks LM, Goodpaster BH, Smith SR, Pratley RE, Lewandowski ED, Raman SV, Wold LE, Gallego-Perez D, Coen PM, Ziolo MT, Stanford KI, A Novel Endocrine Role for the BAT-Released Lipokine 12,13-diHOME to Mediate Cardiac Function, Circulation 143(2) (2021) 145–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kumar V, McElhanon KE, Min JK, He X, Xu Z, Beck EX, Simonetti OP, Weisleder N, Raman SV, Non-contrast estimation of diffuse myocardial fibrosis with dual energy CT: A phantom study, J Cardiovasc Comput Tomogr 12(1) (2018) 74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Howard ZM, Dorn LE, Lowe J, Gertzen MD, Ciccone P, Rastogi N, Odom GL, Accornero F, Chamberlain JS, Rafael-Fortney JA, Micro-dystrophin gene therapy prevents heart failure in an improved Duchenne muscular dystrophy cardiomyopathy mouse model, JCI Insight 6(7) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lowe J, Kadakia FK, Zins JG, Haupt M, Peczkowski KK, Rastogi N, Floyd KT, Gomez-Sanchez EP, Gomez-Sanchez CE, Elnakish MT, Rafael-Fortney JA, Janssen PML, Mineralocorticoid Receptor Antagonists in Muscular Dystrophy Mice During Aging and Exercise, J Neuromuscul Dis 5(3) (2018) 295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Chattopadhyay S, Alamgir MF, Nikitin NP, Rigby AS, Clark AL, Cleland JG, Lack of diastolic reserve in patients with heart failure and normal ejection fraction, Circ Heart Fail 3(1) (2010) 35–43. [DOI] [PubMed] [Google Scholar]
- [47].Mishra S, Kass DA, Cellular and molecular pathobiology of heart failure with preserved ejection fraction, Nat Rev Cardiol 18(6) (2021) 400–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Waddingham PH, Bhattacharyya S, Zalen JV, Lloyd G, Contractile reserve as a predictor of prognosis in patients with non-ischaemic systolic heart failure and dilated cardiomyopathy: a systematic review and meta-analysis, Echo Res Pract 5(1) (2018) 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Chung JH, Martin BL, Canan BD, Elnakish MT, Milani-Nejad N, Saad NS, Repas SJ, Schultz JEJ, Murray JD, Slabaugh JL, Gearinger RL, Conkle J, Karaze T, Rastogi N, Chen MP, Crecelius W, Peczkowski KK, Ziolo MT, Fedorov VV, Kilic A, Whitson BA, Higgins RSD, Smith SA, Mohler PJ, Binkley PF, Janssen PML, Etiology-dependent impairment of relaxation kinetics in right ventricular end-stage failing human myocardium, J Mol Cell Cardiol 121 (2018) 81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Reddy YN, Borlaug BA, Heart Failure With Preserved Ejection Fraction, Curr Probl Cardiol 41(4) (2016) 145–88. [DOI] [PubMed] [Google Scholar]
- [51].Lopez JE, Myagmar BE, Swigart PM, Montgomery MD, Haynam S, Bigos M, Rodrigo MC, Simpson PC, beta-myosin heavy chain is induced by pressure overload in a minor subpopulation of smaller mouse cardiac myocytes, Circ Res 109(6) (2011) 629–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Pandya K, Cowhig J, Brackhan J, Kim HS, Hagaman J, Rojas M, Carter CW Jr., Mao L, Rockman HA, Maeda N, Smithies O, Discordant on/off switching of gene expression in myocytes during cardiac hypertrophy in vivo, Proc Natl Acad Sci U S A 105(35) (2008) 13063–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Perry SV, Troponin I: inhibitor or facilitator, Mol Cell Biochem 190(1-2) (1999) 9–32. [PubMed] [Google Scholar]
- [54].Salhi HE, Hassel NC, Siddiqui JK, Brundage EA, Ziolo MT, Janssen PM, Davis JP, Biesiadecki BJ, Myofilament Calcium Sensitivity: Mechanistic Insight into TnI Ser-23/24 and Ser-150 Phosphorylation Integration, Front Physiol 7 (2016) 567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Siddiqui JK, Tikunova SB, Walton SD, Liu B, Meyer M, de Tombe PP, Neilson N, Kekenes-Huskey PM, Salhi HE, Janssen PM, Biesiadecki BJ, Davis JP, Myofilament Calcium Sensitivity: Consequences of the Effective Concentration of Troponin I, Front Physiol 7 (2016) 632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Little WC, Kitzman DW, Cheng CP, Diastolic dysfunction as a cause of exercise intolerance, Heart Fail Rev 5(4) (2000) 301–6. [DOI] [PubMed] [Google Scholar]
- [57].Monasky MM, Biesiadecki BJ, Janssen PM, Increased phosphorylation of tropomyosin, troponin I, and myosin light chain-2 after stretch in rabbit ventricular myocardium under physiological conditions, J Mol Cell Cardiol 48(5) (2010) 1023–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Wijnker PJ, Sequeira V, Foster DB, Li Y, Dos Remedios CG, Murphy AM, Stienen GJ, van der Velden J, Length-dependent activation is modulated by cardiac troponin I bisphosphorylation at Ser23 and Ser24 but not by Thr143 phosphorylation, Am J Physiol Heart Circ Physiol 306(8) (2014) H1171–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Haizlip KM, Milani-Nejad N, Brunello L, Varian KD, Slabaugh JL, Walton SD, Gyorke S, Davis JP, Biesiadecki BJ, Janssen PM, Dissociation of Calcium Transients and Force Development following a Change in Stimulation Frequency in Isolated Rabbit Myocardium, Biomed Res Int 2015 (2015) 468548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Haizlip KM, Bupha-Intr T, Biesiadecki BJ, Janssen PM, Effects of increased preload on the force-frequency response and contractile kinetics in early stages of cardiac muscle hypertrophy, Am J Physiol Heart Circ Physiol 302(12) (2012) H2509–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Barefield D, Sadayappan S, Phosphorylation and function of cardiac myosin binding protein-C in health and disease, J Mol Cell Cardiol 48(5) (2010) 866–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Hidalgo C, Granzier H, Tuning the molecular giant titin through phosphorylation: role in health and disease, Trends Cardiovasc Med 23(5) (2013) 165–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kamm KE, Stull JT, Signaling to myosin regulatory light chain in sarcomeres, J Biol Chem 286(12) (2011) 9941–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Tong CW, Stelzer JE, Greaser ML, Powers PA, Moss RL, Acceleration of crossbridge kinetics by protein kinase A phosphorylation of cardiac myosin binding protein C modulates cardiac function, Circ Res 103(9) (2008) 974–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Sadayappan S, Gulick J, Osinska H, Barefield D, Cuello F, Avkiran M, Lasko VM, Lorenz JN, Maillet M, Martin JL, Brown JH, Bers DM, Molkentin JD, James J, Robbins J, A critical function for Ser-282 in cardiac Myosin binding protein-C phosphorylation and cardiac function, Circ Res 109(2) (2011) 141–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Hanft LM, Cornell TD, McDonald CA, Rovetto MJ, Emter CA, McDonald KS, Molecule specific effects of PKA-mediated phosphorylation on rat isolated heart and cardiac myofibrillar function, Arch Biochem Biophys 601 (2016) 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Nixon BR, Walton SD, Zhang B, Brundage EA, Little SC, Ziolo MT, Davis JP, Biesiadecki BJ, Combined troponin I Ser-150 and Ser-23/24 phosphorylation sustains thin filament Ca(2+) sensitivity and accelerates deactivation in an acidic environment, J Mol Cell Cardiol 72 (2014) 177–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Biesiadecki BJ, Davis JP, Troponin abnormality and systolic function of the heart, in: Jin J-P (Ed.), Advances in Troponin Research, Nova Sciences 2014, pp. 233–258. [Google Scholar]
- [69].Dohet C, al-Hillawi E, Trayer IP, Ruegg JC, Reconstitution of skinned cardiac fibres with human recombinant cardiac troponin-I mutants and troponin-C, FEBS Lett 377(2) (1995) 131–4. [DOI] [PubMed] [Google Scholar]
- [70].Finley N, Abbott MB, Abusamhadneh E, Gaponenko V, Dong W, Gasmi-Seabrook G, Howarth JW, Rance M, Solaro RJ, Cheung HC, Rosevear PR, NMR analysis of cardiac troponin C-troponin I complexes: effects of phosphorylation, FEBS Lett 453(1–2) (1999) 107–12. [DOI] [PubMed] [Google Scholar]
- [71].Howarth JW, Meller J, Solaro RJ, Trewhella J, Rosevear PR, Phosphorylation-dependent conformational transition of the cardiac specific N-extension of troponin I in cardiac troponin, J Mol Biol 373(3) (2007) 706–22. [DOI] [PubMed] [Google Scholar]
- [72].Ward DG, Brewer SM, Gallon CE, Gao Y, Levine BA, Trayer IP, NMR and mutagenesis studies on the phosphorylation region of human cardiac troponin I, Biochemistry 43(19) (2004) 5772–81. [DOI] [PubMed] [Google Scholar]
- [73].Palermo J, Gulick J, Colbert M, Fewell J, Robbins J, Transgenic remodeling of the contractile apparatus in the mammalian heart, Circ Res 78(3) (1996) 504–9. [DOI] [PubMed] [Google Scholar]
- [74].Pi Y, Kemnitz KR, Zhang D, Kranias EG, Walker JW, Phosphorylation of troponin I controls cardiac twitch dynamics: evidence from phosphorylation site mutants expressed on a troponin I-null background in mice, Circ Res 90(6) (2002) 649–56. [DOI] [PubMed] [Google Scholar]
- [75].Pacher P, Nagayama T, Mukhopadhyay P, Batkai S, Kass DA, Measurement of cardiac function using pressure-volume conductance catheter technique in mice and rats, Nat Protoc 3(9) (2008) 1422–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.