

Selective vulnerability of excitatory neurons in Alzheimer’s disease (AD): AD is the most common form of dementia; however, the pathogenesis of AD is largely unknown. One of the characteristic features of AD is the formation of intracellular neurofibrillary tangles (NFTs). NFTs are abnormal accumulates of misfolded tau protein, which may eventually cause neuronal death and neurodegeneration (Jack et al., 2018). In the early stages of AD progression, not all neurons are equally vulnerable to tau aggregates. Previous studies have shown that large pyramidal neurons in the entorhinal cortex (EC) are specifically vulnerable to pathological tau accumulation (Fu et al., 2017). This selective vulnerability of excitatory neurons to tau pathology is one of the fundamental questions needed to be answered in AD research.
Techniques such as single-nucleus RNA sequencing enable us to profile the transcriptomics of cells on a large-scale and allow us to uncover subtypes of excitatory neuronal populations which may play important roles in the onset and progression of AD. For example, one recent study has identified a particular subtype of excitatory neurons expressing the RAR-related Orphan Receptor B gene as more vulnerable in the EC by comparing transcriptomic profiles of postmortem human brains from different AD stages (Leng et al., 2021). Although single-nucleus RNA sequencing allows us to compare differentially expressed genes among different subtypes of neurons, thereby allowing us to distinguish vulnerable neurons in AD, we still cannot directly attribute these differences to the presence of pathological components such as NFTs since we are unable to target and profile neurons bearing NFTs at the whole transcriptome level. However, we can validate results from transcriptomic studies by using additional models of AD including tau mouse models, to further inquire about the molecular mechanisms underlying the selective vulnerability of certain excitatory neurons to tau pathology in AD.
Grid cells, which are located in layer II/III of the EC, have been recently found to be preferentially vulnerable to tau pathology (Fu et al., 2017). This subtype of excitatory neurons has been identified to regulate animals’ spatial navigation and memory. In EC-tau mice (a tau transgenic mouse model), tau accumulation has been demonstrated to cause grid cell dysfunction and neuronal death (Fu et al., 2017). Interestingly, grid cells also highly express wolframin (WFS1) protein. Delpech et al. (2021) have recently shown WFS1-positive (WFS1+) neurons make up 6.5% of total cells in human EC layer II. Moreover, the proportion of WFS1+ neurons dramatically decreased in the postmortem human brain from individuals with clinical dementia rating (CDR) scores over 2 (2.5% for CDR2 and 2.7% for CDR3), and PHF1+ (pS396/404) p-tau started to accumulate in ECII WFS1+ neurons at early stages with mild cognitive decline (Delpech et al., 2021). In summary, the protein level of WFS1 and the number of WFS1+ neurons are significantly reduced in both AD-like mouse model brains and human post-mortem AD, suggesting these WFS1+ neurons become vulnerable to tau pathology when their WFS1 level decreases. Why WFS1+ neurons are particularly vulnerable to tau pathology and the role of WFS1 in tau pathology associated neurodegeneration, however, remains unknown.
Lessons from Wolfram Syndrome: Similarities between Wolfram Syndrome and AD point to a new role for WFS1 in neurodegeneration: WFS1 is an endoplasmic reticulum (ER) membrane protein encoded by the WFS1 gene which is highly expressed in the human brain and pancreas. Mutations in the human WFS1 gene have been identified as a causative factor for Wolfram Syndrome, a rare and severe neurodegenerative disease. Though no loss-of-function mutations of WFS1 have been found in AD, both Wolfram Syndrome and AD display similarities in terms of ER Ca2+ dyshomeostasis, ER stress, and autophagy dysfunction (Li et al., 2020). It is worth investigating whether WFS1 deficiency in AD also affects Ca2+ homeostasis, ER stress, and autophagy in a similar way to that in Wolfram Syndrome.
Since WFS1+ cells have been found to colocalize with pathological tau (Delpech et al., 2021), we first investigated whether WFS1 deficiency promotes tau aggregation or inhibits tau degradation and clearance. By knocking out WFS1 in PS19 mice (P301S transgenic tau mice (Yoshiyama et al., 2007)), tau aggregates were found significantly increased in layers II/III of the EC, and the learning and memory of those mice were also impaired. However, tau pathology decreased dramatically when WFS1 was overexpressed in the EC region of PS19 mice three months after the injection of human WFS1 virus, compared with age matched PS19 mice injected with a control GFP virus. Importantly, astrogliosis, synaptic dysfunction and neurodegeneration associated with tau pathology were also reversed in PS19 mice by overexpressing WFS1 (Chen et al., 2022). To further confirm whether the WFS1 protein directly interacts with tau protein in neurons, the Duolink proximity ligation assay was performed. In both the EC-tau mice and human postmortem brain samples, WFS1 was found to interact with total tau (recognized by the Tau46 antibody) and pathological tau (recognized by the AT8, pS202/T205 tau antibody), indicating WFS1 may be involved in tau aggregation, propagation, and degradation by directly interacting with tau protein (Chen et al., 2022).
We next explored the mechanism of how WFS1 deficiency affected tau aggregation and clearance. A previous study of Wolfram Syndrome has shown that WFS1 defects may induce chronic ER stress and further cause the impairment of the ER-associated degradation in pancreatic β-cells (Li et al., 2020). In neurons, we also found evidence linking WFS1 deficiency to chronic ER stress and autophagy-lysosome pathway (ALP) dependent ER-associated degradation. After knocking out WFS1 in PS19 mice, ATF4 and CHOP, two chronic ER stress markers, were significantly increased in the EC. Meanwhile, p62, a critical autophagosome cargo protein in ALP, started to accumulate in the tau aggregated cells. Moreover, autophagy related marker cathepsin D and lysosome biogenesis regulator transcription factor EB were also significantly reduced. On the contrary, overexpressing WFS1 could reverse those phenotypes in PS19 mice. These results suggest that WFS1 deficiency may induce chronic ER stress, and further affect the degradation and clearance of tau aggregates via the ALP pathway in vivo (Chen et al., 2022).
WFS1: a new regulator of ER stress, and autophagy in AD: ER stress is regulated by three unfolded protein response (UPR) signaling pathways, which are initiated from three ER membrane sensors: protein kinase RNA-like endoplasmic reticulum kinase (PERK), inositol-requiring enzyme 1, and the activating transcription factor 6. In Wolfram Syndrome, WFS1 deficiency has been reported to be involved in the upregulation of all three UPR pathways (Li et al., 2020). Though WFS1 deficiency has been identified to affect the PERK/ATF4/CHOP pathway in AD (Chen et al., 2022), it is unknown whether it is also involved in other two UPR signaling pathways (Figure 1). Moreover, although evidence of ER stress response in AD is apparent, whether it is a causative factor of AD remains in question. Importantly, opposing studies on ER stress response in AD from different groups contribute to this debate. For example, Halliday et al. (2017) observed the neuroprotective effects of PERK pathway inhibitors in both tauopathy and prion-diseased mice, while Bruch et al. (2017) found that PERK activation may mitigate tau pathology in vitro and in vivo. However, considering the potential relationships between WFS1, ER stress, and AD pathogenesis, it will still be of great interest to further investigate the role of WFS1 in ER stress pathways in AD.
Figure 1.
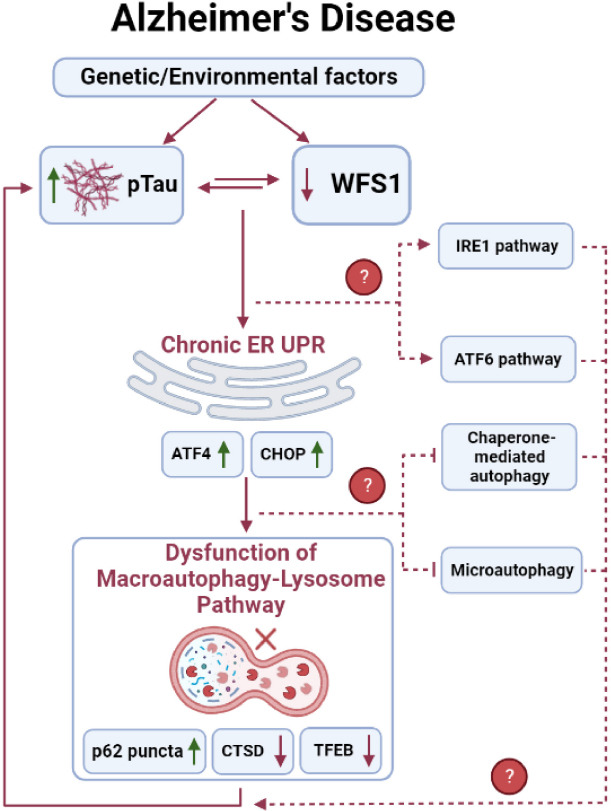
The role of WFS1 in Tau clearance in Alzheimer’s disease.
Schematic diagram outlining the potential mechanisms underlying the role of WFS1 in regulating tau clearance. CTSD: Cathepsin D; ER: endoplasmic reticulum; IRE1: inositol-requiring enzyme 1; pTau: phosphorylated tau; TFEB: transcription factor EB; UPR: unfolded protein response. Created with BioRender.com.
As discussed above, ER stress may further trigger the ALP dependent ER-associated degradation and downstream autophagy pathway through UPR. Autophagy mainly consists of three categories: macroautophagy, chaperone-mediated autophagy (CMA), and microautophagy. WFS1 deficiency has been found to impair the macroautophagy via chronic ER stress (Chen et al., 2022), however, it is not clear whether WFS1 deficiency is also involved in the dysfunction of CMA and microautophagy in AD and other neurodegenerative diseases. Unlike macroautophagy, CMA selectively degrades cytosolic proteins that contain a KFERQ-like motif which can be recognized by the molecular chaperone heat-shock cognate protein 70. The target protein will then form a complex with heat-shock cognate protein 70 and binds with lysosome-associated membrane protein type 2A on the lysosomal surface for further degradation. Both amyloid precursor protein and tau can be a target of the CMA pathway: amyloid precursor protein contains a KFERQ-like motif (Park et al., 2016), while tau contains QVEVK and KDRVQ motif which can be targeted by CMA pathway (Wang et al., 2009). In contrast, microautophagy has more diverse morphologies and types which directly engulf the cargo by lysosomal action (Oku and Sakai, 2018). Mutations in tau could prevent their degradation by microautophagy and in turn become dependent on macroautophagy for degradation of such proteins. Post-translationally modified tau, such as acetylated tau can also undergo microautophagy and if blocked, re-route to macroautophagy (Caballero et al., 2021). However, the role of CMA and microautophagy in amyloid precursor protein/amyloid β or tau clearance is still under investigation. Future studies investigating the role of WFS1 in tau clearance via the CMA or microautophagy pathway are also warranted to reveal their potential relationship (Figure 1).
Conclusions and perspectives: Although we demonstrate WFS1 plays an important role in inhibiting tau pathology, one important question remains. It is still unknown why WFS1+ excitatory neurons in EC layer-II are more vulnerable than other types of neurons if WFS1 has a neuroprotective function. In general, selective vulnerability might result from low levels of protective factors or high levels of toxic factors, and both intrinsic and extrinsic factors may contribute to this selective neuronal vulnerability. We hypothesize that there are specific gene signatures and signaling pathways, which may contribute to the selective vulnerability of WFS1+ EX neurons to tau pathology in the EC. We will further investigate if WFS1+ EX neurons intrinsically express less tau aggregation protector genes and more genes encoding tau co-aggregators and aggregation-prone proteins than WFS1-negative neurons.
Considering the short average life expectancy (30–40 years of age) of Wolfram Syndrome, no Wolfram Syndrome patients have been reported to develop AD, in which aging is the most important known risk factor. However, cognitive disability has been found in 32% of Wolfram Syndrome patients (Li et al., 2020). Both Wolfram Syndrome and AD share common physiopathological signaling pathways (Li et al., 2020). Thus, reduced WFS1 caused by unknown factors may increase the neuronal vulnerability to tau pathology and neurodegeneration in AD pathogenesis, even if Wolfram Syndrome patients do not show AD pathology.
Interestingly, we also found the protein level and the number of WFS1+ cells were significantly reduced not only in AD, but also in other neurodegenerative diseases such as frontotemporal lobar degeneration with tau pathology, frontotemporal lobar degeneration with transactive response DNA binding protein, and diffuse Lewy body disease (Chen et al., 2022). This indicates that WFS1+ neurons in the EC may be vulnerable in other types of neurodegenerative diseases as well. Although the downstream pathways of WFS1 deficiency remain to be determined for other proteinopathies, WFS1 may be a common therapeutic target in proteinopathies besides AD.
Taken altogether, as an important regulator of both ER stress and autophagy pathways, WFS1 could be a potential target for developing novel drugs to promote the degradation and clearance of misfolded proteins in AD and other neurodegenerative diseases.
The present work was supported by awards K01-AG056673, R56-AG066782-01 and R01-AG075092-01 (to HF) from the National Institute on Aging of the National Institutes of Health. The work was also supported by the award of the W81XWH1910309 (to HF) from the Department of Defense.
Footnotes
C-Editors: Zhao M, Liu WJ, Qiu Y; T-Editor: Jia Y
References
- 1.Bruch J, Xu H, Rosler TW, De Andrade A, Kuhn PH, Lichtenthaler SF, Arzberger T, Winklhofer KF, Muller U, Hoglinger GU. PERK activation mitigates tau pathology in vitro and in vivo. EMBO Mol Med. 2017;9:371–384. doi: 10.15252/emmm.201606664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Caballero B, Bourdenx M, Luengo E, Diaz A, Sohn PD, Chen XU, Wang C, Juste YR, Wegmann S, Patel B, Young ZT, Kuo SY, Rodriguez-Navarro JA, Shao H, Lopez MG, Karch CM, Goate AM, Gestwicki JE, Hyman BT, Gan L, et al. Acetylated tau inhibits chaperone-mediated autophagy and promotes tau pathology propagation in mice. Nat Commun. 2021;12:2238. doi: 10.1038/s41467-021-22501-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen S, Acosta D, Li L, Liang J, Chang Y, Wang C, Fitzgerald J, Morrison C, Goulbourne CN, Nakano Y, Villegas NC, Venkataraman L, Brown C, Serrano GE, Bell E, Wemlinger T, Wu M, Kokiko-Cochran ON, Popovich P, Flowers XE, et al. Wolframin is a novel regulator of tau pathology and neurodegeneration. Acta Neuropathol. 2022;143:547–569. doi: 10.1007/s00401-022-02417-4. [DOI] [PubMed] [Google Scholar]
- 4.Delpech JC, Pathak D, Varghese M, Kalavai SV, Hays EC, Hof PR, Johnson WE, Ikezu S, Medalla M, Luebke JI, Ikezu T. Wolframin-1-expressing neurons in the entorhinal cortex propagate tau to CA1 neurons and impair hippocampal memory in mice. Sci Transl Med. 2021;13:eab.e8455. doi: 10.1126/scitranslmed.abe8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fu H, Rodriguez GA, Herman M, Emrani S, Nahmani E, Barrett G, Figueroa HY, Goldberg E, Hussaini SA, Duff KE. Tau pathology induces excitatory neuron loss, grid cell dysfunction, and spatial memory deficits reminiscent of early Alzheimer’s disease. Neuron. 2017;93:533–541. doi: 10.1016/j.neuron.2016.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Halliday M, Radford H, Zents KAM, Molloy C, Moreno JA, Verity NC, Smith E, Ortori CA, Barrett DA, Bushell M, Mallucci GR. Repurposed drugs targeting eIF2α-P-mediated translational repression prevent neurodegeneration in mice. Brain. 2017;140:1768–1783. doi: 10.1093/brain/awx074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jack CR, Jr, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, Holtzman DM, Jagust W, Jessen F, Karlawish J, Liu E, Molinuevo JL, Montine T, Phelps C, Rankin KP, Rowe CC, Scheltens P, Siemers E, Snyder HM, Sperling R. NIA-AA research framework:toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14:535–562. doi: 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leng K, Li E, Eser R, Piergies A, Sit R, Tan M, Neff N, Li SH, Rodriguez RD, Suemoto CK, Leite REP, Ehrenberg AJ, Pasqualucci CA, Seeley WW, Spina S, Heinsen H, Grinberg LT, Kampmann M. Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease. Nat Neurosci. 2021;24:276–287. doi: 10.1038/s41593-020-00764-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li L, Venkataraman L, Chen S, Fu H. Function of WFS1 and WFS2 in the central nervous system:implications for Wolfram Syndrome and Alzheimer’s disease. Neurosci Biobehav Rev. 2020;118:775–783. doi: 10.1016/j.neubiorev.2020.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oku M, Sakai Y. Three distinct types of microautophagy based on membrane dynamics and molecular machineries. Bioessays. 2018;40:e1800008. doi: 10.1002/bies.201800008. [DOI] [PubMed] [Google Scholar]
- 11.Park JS, Kim DH, Yoon SY. Regulation of amyloid precursor protein processing by its KFERQ motif. BMB Rep. 2016;49:337–342. doi: 10.5483/BMBRep.2016.49.6.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, Martinez-Vicente M, Kruger U, Kaushik S, Wong E, Mandelkow EM, Cuervo AM, Mandelkow E. Tau fragmentation, aggregation and clearance:the dual role of lysosomal processing. Hum Mol Genet. 2009;18:4153–4170. doi: 10.1093/hmg/ddp367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]