

Abstract
Background
Adult-type diffuse gliomas, CNS WHO grade 4 are the most aggressive primary brain tumors and represent a particular challenge for therapeutic intervention.
Methods
In a single-center retrospective study of matched pairs of initial and post-therapeutic glioma cases with a recurrence period greater than 1 year, we performed whole exome sequencing combined with mRNA and microRNA expression profiling to identify processes that are altered in recurrent gliomas.
Results
Mutational analysis of recurrent gliomas revealed early branching evolution in 75% of the patients. High plasticity was confirmed at the mRNA and miRNA levels. SBS1 signature was reduced and SBS11 was elevated, demonstrating the effect of alkylating agent therapy on the mutational landscape. There was no evidence for secondary genomic alterations driving therapy resistance. ALK7/ACVR1C and LTBP1 were upregulated, whereas LEFTY2 was downregulated, pointing towards enhanced Tumor Growth Factor β (TGF-β) signaling in recurrent gliomas. Consistently, altered microRNA expression profiles pointed towards enhanced Nuclear Factor Kappa B and Wnt signaling that, cooperatively with TGF-β, induces epithelial to mesenchymal transition (EMT), migration, and stemness. TGF-β-induced expression of pro-apoptotic proteins and repression of antiapoptotic proteins were uncoupled in the recurrent tumor.
Conclusions
Our results suggest an important role of TGF-β signaling in recurrent gliomas. This may have clinical implications since TGF-β inhibitors have entered clinical phase studies and may potentially be used in combination therapy to interfere with chemoradiation resistance. Recurrent gliomas show high incidence of early branching evolution. High tumor plasticity is confirmed at the level of microRNA and mRNA expression profiles.
Keywords: Glioblastoma, longitudinal analysis, miRNA, tumor evolution, TGF-β signaling
Key points.
Longitudinal analysis revealed elevated TGF-β signaling in recurrent tumors.
High plasticity exists at genomic, mRNA, and microRNA expression levels.
Recurrence follows an early divergence evolution pattern distinct from the initial tumor.
Importance of Study.
Adult-type diffuse gliomas, CNS WHO grade 4 are highly heterogeneous primary brain tumors, which are refractory to standard therapy. In a single-centered retrospective longitudinal study of patients with long-term recurrence, we identified microRNAs and mRNAs that are differentially expressed in the recurrent tumor. These RNAs act in a concerted manner to induce TGF-β signaling, a pathway known to induce epithelial to mesenchymal transition (EMT), migration, and stemness in gliomas. This may open new approaches for therapeutic intervention using TGF-β inhibitors to interfere with resistance mechanisms. In addition, we provide evidence for early branching evolution of recurrent gliomas. High plasticity was confirmed at the level of miRNA and mRNA expression.
Glioblastoma IDH wild-type (GBM, CNS WHO grade 4) is the most frequent and aggressive primary brain tumor in adults.1 It is encountered as malignant neoplasm at first presentation, while its histologically indistinguishable counterpart, astrocytoma IDH-mutant, CNS WHO grade 4 (astro-IDH-mut-G4), is the converging endpoint of progression from a lower-grade astrocytoma. These diffuse glioma subtypes are characterized by extensive plasticity and considerable heterogeneity.1 Comprehensive analysis of somatic alterations and expression profiling allowed these glioma subtypes to be classified into classical, mesenchymal, neural, and proneural subtypes, which often coexist in the same tumor.2 Nevertheless, the prognosis of glioma patients is very poor across all subtypes.
Treatment options for grade 4 diffuse glioma patients have not changed over the last 15 years, and the success of targeted therapy has been limited. Thus, the standard of care for GBM patients remains surgical resection followed by concomitant DNA alkylating agent therapy with temozolomide (TMZ) and radiation therapy (RT).3 However, the time to relapse is generally short (6.9 months on average4) owing to the tumor’s ability to infiltrate normal parenchyma and its intrinsic resistance to radio-chemotherapy. Due to the lack of alternative treatment options, elucidating underlying resistance mechanisms has been a major focus of research, with the hope of new therapeutic interventions overcoming resistance.
Longitudinal analysis of recurrent gliomas revealed driver mutations in the therapy-naïve (hereafter “initial”) tumor were also retained in the post-therapeutic (hereafter “recurrent”) tumor, and there was no evidence for specific secondary alterations driving tumor recurrence.5 Instead, tumor progression was mainly a result of a highly branched evolutionary process that involved clonal expansion, genetic diversification, and clonal selection.5–8 Drivers of the proneural subtype were identified as early events since they are shared by most subclones, whereas drivers of other subtypes often occur at late recurrence.7 The extensive tumor plasticity as the basis for TMZ resistance is further corroborated by high epigenetic plasticity in recurrent tumors.9 MicroRNAs (miRNAs), short noncoding RNAs that control gene expression at the posttranscriptional level, add additional complexity to the gene regulatory networks.10 Despite miRNA deregulation being a potential TMZ resistance mechanism,11 there are no reports assessing miRNA profiles of recurrent tumors in a systematic manner.
To explore therapy resistance driving events, we performed a study using a single-centric collective of matched pairs of therapy-naïve and post-therapeutic tissue from 43 astro-IDH-mut-G4/GBM patients, selected based on a maximally long interval between resections of the initial and the recurrent tumors. This rare collective was analyzed by whole exome sequencing (WES), mRNA, and miRNA expression profiling. Here we provide evidence for a high degree of early branching (ancestral) evolution and enhanced tumor necrosis factor-α (TNF-α), Wnt, and transforming growth factor-β (TGF-β) signaling. To the best of our knowledge, this is the largest single-center retrospective study of matched cases with an exceptionally long recurrence period.
Materials and Methods
Patient Cohort
We included GBM IDH wild-type and astro-IDH-mut-G4 patients with a first surgery at diagnosis and at least one additional surgery upon recurrence following standard treatment, diagnosed at the Institute of Pathology, University of Bern 1999–2016. All tumors were resected at the Department of Neurosurgery, Inselspital University Hospital Bern. The cohort was retrospectively assembled according to the pathology and clinical files. We performed a central review of all cases according to current guidelines (WHO 20211). IDH mutation status,12 loss of heterozygosity 1p/19q,13MGMT methylation status,14 and immunohistochemical (IHC) staining for MIB1 were assessed during routine diagnostic evaluation. H3 mutations, EGFR gene amplification, and +7/−10 chromosome copy number changes were assessed by WES (see below). The initial cohort comprised 97 patients with matched formalin-fixed paraffin-embedded (FFPE) samples from the initial and recurrent tumors. A sub-cohort comprising 43 patients, with an interval of at least one year between pre-therapeutic and post-therapeutic resections was selected for molecular analysis.
Patient and clinical data of this sub-cohort are shown in Supplementary Table1. All tumors had sufficient tumor purity (>50%) by histological analysis.
Tissue Microarray and Immunohistochemistry (IHC)
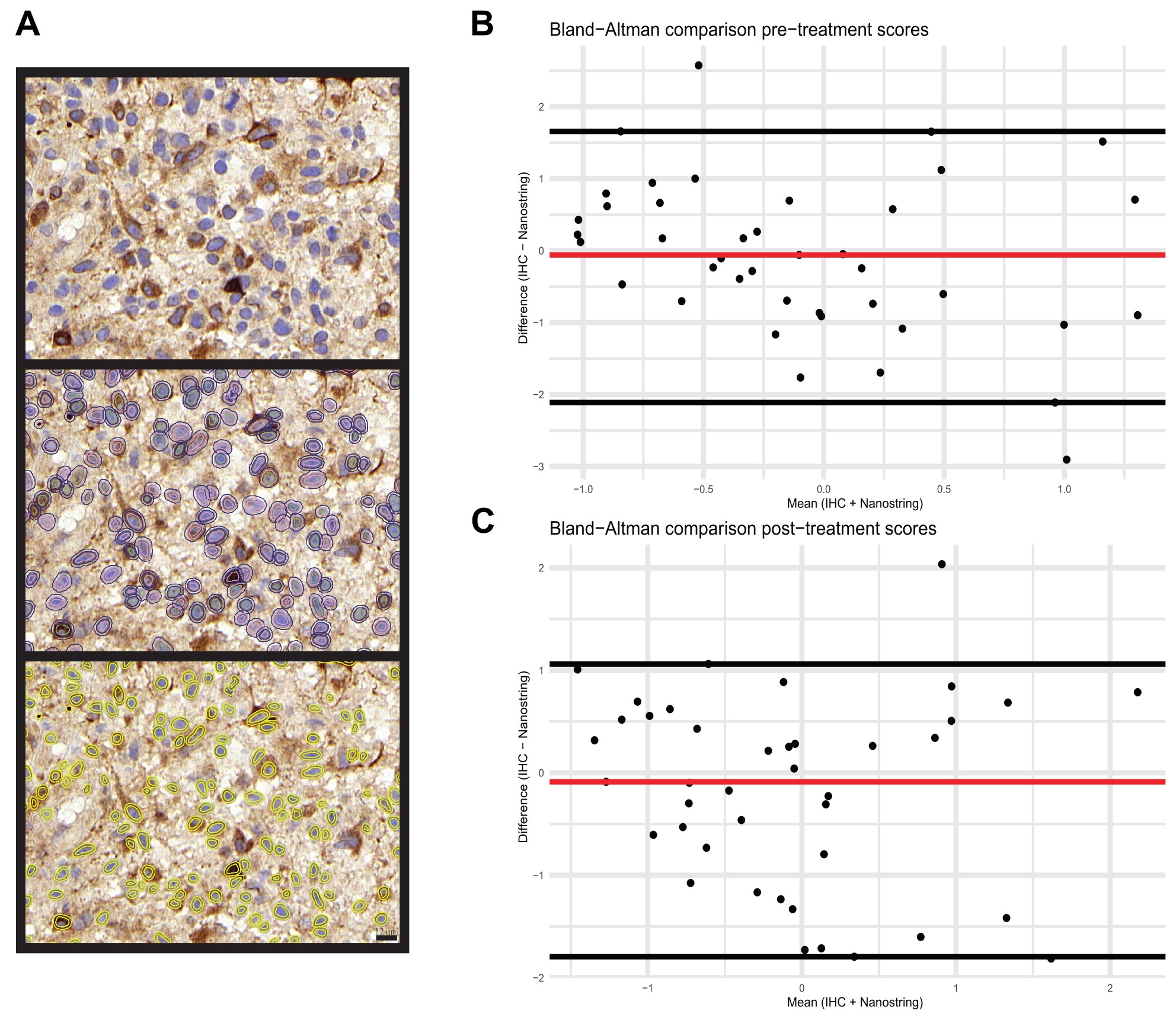
A tissue microarray (ngTMA) was constructed from at least three punches from the tumor center and, if available, from adjacent normal tissue (see Supplementary Methods). ACVR1C/ALK7, β-catenin, NF-κB, SMAD2, and SMAD4 were semiquantitatively evaluated by an experienced neuropathologist (TM) estimating the proportion of positive cells in at least three TMA punches and classifying the cases in: Negative (0%), 0%–5%, 5%–25%, 25%–50%, >50%, and positive (100%). BCL2 expression was quantitatively evaluated using QuPath.15 Initially, nuclei were detected using the StarDist2D model for nucleus segmentation.16 The detections were further split up in a central, nuclear membrane and cytoplasm compartment, and positive cells were identified by applying a threshold of mean DAB (3,3’-diaminobenzidine) intensity in the nuclear membrane compartment (Supplementary Figure1).
Nucleic Acid Extraction
Genomic DNA was extracted from FFPE punches using the QIAamp DNA Mini kit (QIAGEN) and analyzed using the Agilent NGS FFPE QC kit. 93% of samples showed sufficient DNA quality as indicated by a ΔCt value < 2. Total RNA was extracted using the RecoverAll Total Nucleic Acid Isolation Kit for FFPE (ThermoFisher). For miRNA profiling, total RNA was purified using the Zymo Research RNA Clean and Concentrator-5 kit (Zymo Research, Irvine, CA, USA).
Library Preparation and Whole Exome Sequencing
Libraries were prepared using the SureSelectXT Human All Exon V7 Low Input Reagent Kit and SureSelect XT Low Input P5 indexed adaptors (Agilent). Sequencing was performed on a NovaSeq 6000 using the S2 Reagent Kit (200 cycles). The sequencing depth was in a range of 25–130 (median 77).
Processing of WES Data
Processing and somatic variant calling of the WES data was performed using the T/N workflow from Bcbio-nextgen (v1.1.7, https://github.com/bcbio/bcbio-nextgen). Gene-level copy number alterations were determined with Sequenza v3.0.017 and GISTIC v2.0.23.18 See also Supplementary Methods.
Mutational Signatures and TMB
Mutational signatures were inferred with MutationalPatterns v3.0.019 and deconstructSigs v1.8.0,20 for the COSMIC signatures v3 (May 2019) previously described in the GLASS5 or PCWAG CNS-GBM cohort21 [SBS1, SBS3, SBS5, SBS8, SBS11, SBS15, SBS16, SBS30, and SBS40]. Paired Wilcoxon tests were used to compare signature contributions and TMB values in tumor pairs. See Supplementary Methods.
Subclonal and Phylogenetic Reconstruction and Evolutionary League Model
Tumor sample purity, average ploidy, and cancer cell fractions of mutations were estimated with ABSOLUTE v1.222 in allelic mode. Phylogenetic reconstruction and evolutionary league model inference were performed with PhylogicNDT23 (https://github.com/broadinstitute/PhylogicNDT). See Supplementary Methods.
Nanostring Expression Profiling of mRNAs and miRNAs
100ng total RNA was analyzed using the nCounter_Human_miRNA_Expression_Panel_Assay_Kit H_miRNA_V3 or the nCounter_Human_PanCancer_pathway_panel (NanoString, Seattle, USA) spiked with 30 additional genes implicated in autophagy, EMT and DNA repair processes (Supplementary Table2), as previously described.24 See Supplementary Methods for bioinformatics analysis.
Calculation of Hazard Ratio in Publicly Available Datasets
Clinical data and normalized z-score RNA-Seq data (based on all samples) from 160 patients of the TCGA glioblastoma cohort25 were downloaded on January 27, 2022. Univariable cox proportional hazard models were used to estimate the effect of gene expression using TCGAbiolinks R package.26 Genes achieving a significance level of P < .1 were included in a multivariable model. Maximally selected rank statistics were utilized to find the optimal cutoff for gene expression dichotomization. Hazard ratio was calculated using multivariable cox-regression statistical model.
In vitro Validation of ALK7 Modulation in a GBM Cell Line
150’000 authenticated U87MG cells27 were transfected using “Single piggyBac vector” system described by Michael et al.,28 except that TransIT-LT1 Transfection Reagent was used (Mirus Bio, USA). Cells were treated with TMZ (100 µM) or DMSO27 48 h post-doxycycline (DOX) induction (2 µg/ml). Viability, cytotoxicity, and apoptosis were assessed using the ApoTox-Glo™ Triplex Assay (Promega).27
Results
Sample Characteristics
Our initial cohort comprised 97 patients for which tissues were available from resections at diagnosis (initial) and at recurrence following standard treatment. The time interval between the resections was less than one year for 54 patients and 370–1890 (median 689) days for the remaining 43 patients (Supplementary Figure 2A and Supplementary Table1). There was no correlation between the extent of tumor resection and relapse interval (P = .4572). Among those patients with long-term recurrence (who were included in downstream molecular analysis), 39 had IDH wild-type and four had IDH mutation. The patient age of the sub-cohort with long-term recurrence ranged from 32 to 79 (median 57.5) years at initial resection (Supplementary Figure 2B). Brain tumor locations are indicated in Supplementary Figure 2C. All patients showed recurrence at the original tumor location. 42 patients received concomitant alkylating agent therapy with TMZ and RT and one patient received radiotherapy only. The MIB1 proliferation index was generally lower in recurrent tumors compared to the initial tumor (Supplementary Figure 2D). Of note, the proportion of tumors with hypermethylated MGMT promoter at diagnosis was significantly higher in patients with a recurrence interval greater than one year compared to patients with a shorter recurrence interval (Supplementary Figure 2E) (P = .03) consistent with its role as a predicative marker for TMZ response.29
Mutational Burden and Mutational Signatures in the Recurrent Tumor of Patients With Long-Term Relapse Interval
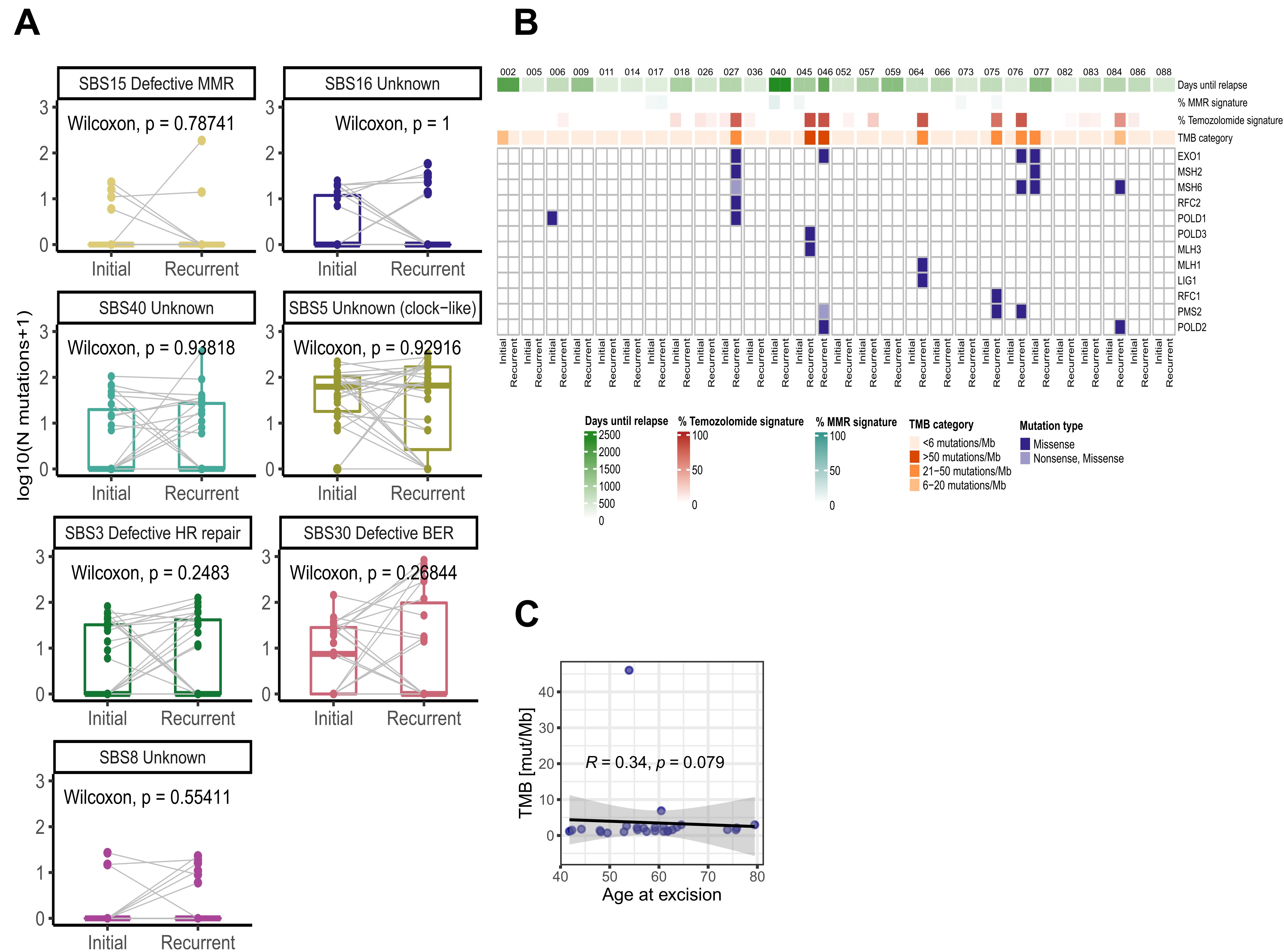
WES was performed using trios of matched initial, recurrent astro-IDH-mut-G4/GBM and normal tissue or peripheral blood from 28 patients with long-term relapse intervals (see Supplementary Figure 2F). All patients had received TMZ and concomitant RT. IDH mutational status was wild-type in 25 patients and mutant in 3 patients. We observed a range of 0.7–46.0 (median 1.6) mutations per megabase (mut/Mb) for the initial and a significantly higher range of 1.1–66.8 (median 2.4) mut/Mb for the recurrent tumors (Figure 1A). Most samples with high TMB revealed a high rate of SBS11 mutation signature (TMZ treatment) (Figure 1B and C). Although post-therapeutic mutations in MMR genes were detected in 7/28 patients, they were not associated with SBS15 (defective MMR), the latter being present only at a very low rate (Supplementary Figure 3A and B, Supplementary Table3). Instead, mutations in MMR genes were mainly present in tumors with high SBS11 signature (Supplementary Figure 3B), suggesting that the high mutational burden was most likely caused by the hypermutator activity of TMZ rather than defects in MMR genes. With the exception of patient 045, which was negative for both MLH1 and PMS2 by IHC, recurrent tumors with hypermutator phenotype were proficient for MMR proteins (data not shown). TMB was not associated with the age of the patient at the initial excision (Supplementary Figure 3C).
Figure 1.

Tumor mutational burden (TMB), mutational signature and landscape of tumors with late recurrence. (A) Log-transformed tumor mutational burden (TMB) calculated as the number of non-synonymous SNVs and INDELs per megabase. Lines indicate paired samples. Paired Wilcoxon test P-value is depicted on the box plot. (B) Contribution of selected COSMIC mutational signatures in each sample. Sample ID is indicated at the bottom of the barplot and total number of mutations (synonymous + non-synonymous) is indicated on top of the barplot (N mutations). Color indicate 9 different consensus COSMIC signatures. (C) Boxplots indicating the number of mutations contributing to the SBS1 (top) and SBS11 (bottom) signatures in the matched samples. Lines join the paired tumor samples from a given patient. Signatures with no significant change are shown in Supplementary Figure 3A. (D) Heatmap depicting mutations (SNVs&INDELs; top), amplifications (middle) and arm-level copy number gains/losses (bottom) in driver genes.
We also found a significant decrease in the contribution of the SBS1 signature (deamination of 5MeC) in recurrent tumors (Figure 1C). This observation is concordant with GLASS.5 The frequency of signatures associated with defects in different DNA repair systems was not altered between initial and recurrent tumors (Supplementary Figure 3A).
Mutational Landscape of Tumors With Late Recurrence
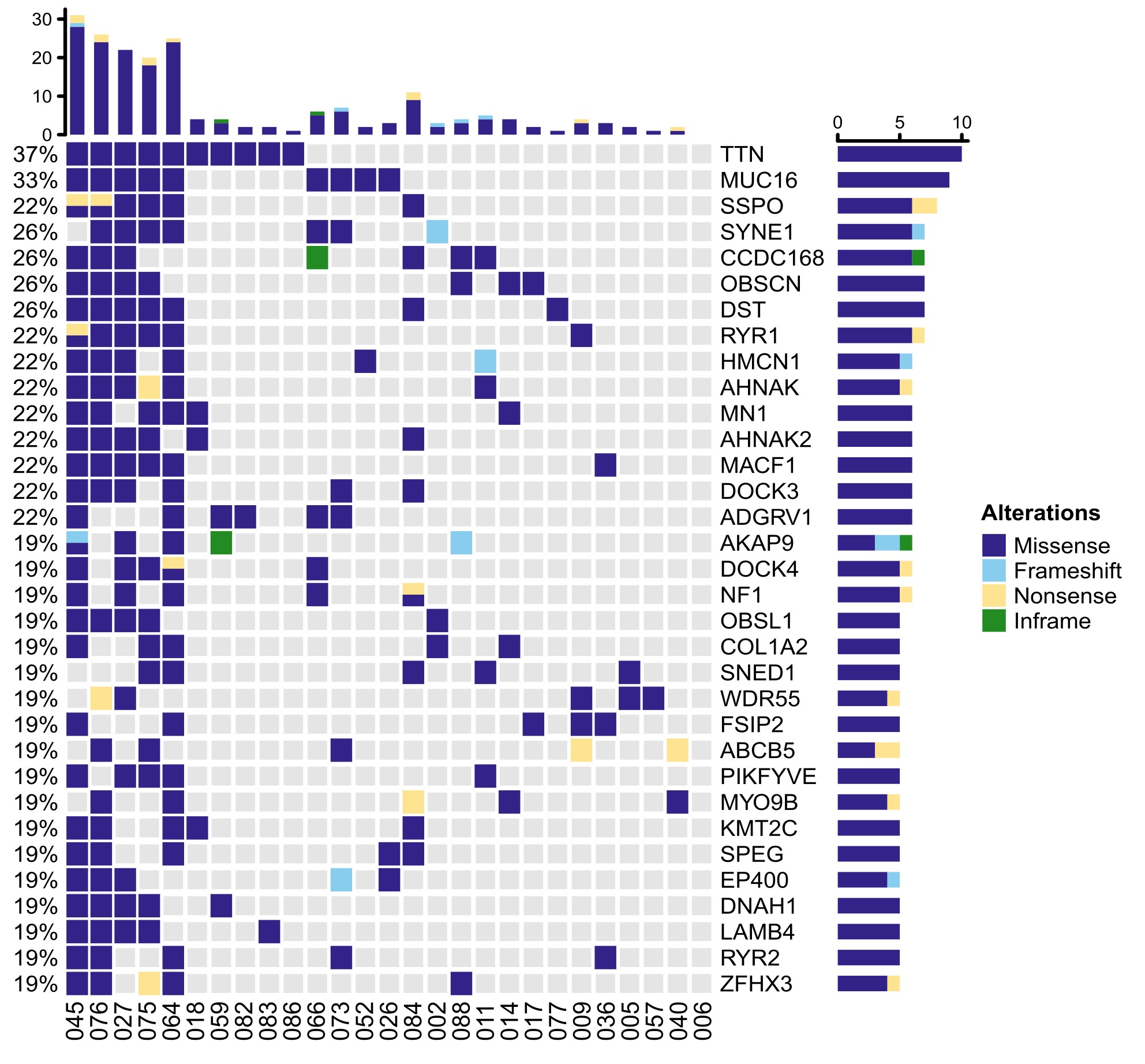
Most tumors showed a copy number gain at chromosome 7 and a copy number loss at chromosome 10, which is characteristic of GBM IDH wild-type (Figure 1D and Supplementary Figure4 and Supplementary Figure 5). Interestingly, an almost identical copy number profile was retained in the recurrent tumor in most cases (Supplementary Figure 4A and B). Likewise, known drivers of astro-IDH-mut-G4/GBM occurring in the initial tumors including IDH1 (2/2), PTEN (5/7), PIK3CA (4/6), PIK3R1 (3/3), and TP53 (5/5) mutations were often retained in the recurrent tumors. In contrast, mutations in EGFR (9/11), NF1 (7/10), RB1 (3/4) and amplifications of EGFR (4/8), PDGFRA (3/3), and MDM2 (2/3) were either private to the initial or recurrent tumors (Figure 1D and Supplementary Table3). LTBP4 was exclusively private to the recurrent tumor and present in 14% of astro-IDH-mut-G4/GBM (Supplementary Table3). The prevalence of these alterations in initial tumors in our dataset was similar to those of the TCGA dataset, which comprises mainly therapy-naïve, initial tumors with shorter recurrence. The only exception was NF1 mutations, which were more abundant in our dataset (5/27 initial tumors:19%) compared to the TCGA dataset (9.3%). Mutations that were most frequently private to recurrent tumors were also enriched in cases with high TMB, and there was no evidence for recurrent driver alterations (Supplementary Figure 6 and Supplementary Table3).
Tumor Evolution of Recurrent Astro-IDH-Mut-G4/GBM With Long-Term Therapy Response
To identify early and late genetic events in grade 4 gliomas with long recurrence periods, we determined the tumor developmental trajectories across the cohort. We found that PTEN mutations were early events whereas potentially targetable alterations in EGFR, mTOR, and NF1 appeared later during tumor development (Figure 2A). Longitudinal analysis of the paired tumors revealed a persistent mutational cluster in only 5/20 (25%) of patients (Figure 2C) Instead, in 15/20 (75%) cases, the dominant subclone in the recurrent tumor was a direct linear descendant of the founding clone, suggesting early branching as the predominant pattern of evolution (Figure 2B). Mutational shifts of early branching tumors from first presentation to recurrence were confirmed by immunohistochemistry or Sanger sequencing (Supplementary Figure7). A similar finding was obtained by Wang et al. who reported that the dominant subclone of the initial tumor was absent in the recurrence in 59% of cases.30 Consequently, patient 052, who carries an actionable EGFR p.A289V mutation in the major subclone of the initial tumor (Supplementary Figure 8A), may not profit from the EGFR inhibitor lapatinib, since this mutation is absent in the recurrent tumor. In contrast, patient 086, who carries a truncal BRAF p.V600E mutation, would be more likely to respond to a secondary therapy with BRAF inhibitors (Supplementary Figure 8B).
Figure 2.

Acquisition of driver alterations over time in tumors with late recurrence. (A) Estimated cohort-level timing of mutational events in 20 tumors with late recurrence. (B) Fish plots showing tumors with early branching evolution. (C) Fish plots showing tumors with ongoing evolution of the initial subclone.
TNF-ɑ, Wnt, and TGF-β Signaling Pathways are Enhanced in the Recurrent Tumor
To identify pathways altered in recurrent tumors, longitudinal mRNA and miRNA expression analyses were performed on the matched initial and recurrent tumor tissues of 43 patients and normal tissues adjacent to the initial tumors from 10 patients using the NanoString Pancancer panel (800 genes) spiked with 30 additional genes implicated in TMZ resistance (Supplementary Table2). As expected, mRNA and miRNA expression patterns of samples derived from normal adjacent brain tissue were clustered separately from those derived from tumor tissues (Figure 3A). However, neither initial-recurrent tumor groups nor individual patient pairs were clustered together, confirming high plasticity of recurrent tumors at the transcript level (Supplementary Figure 9).
Figure 3.

Differential pattern of mRNA and miRNA expression upon late recurrence. (A) Principal Component Analysis of mRNA (upper) and microRNA (lower) expression profiles. Note that the PCA plot from initial (I) and recurrent (R) samples almost completely overlap, but cluster separately from normal tissues (N). (B) Volcano plot indicating significantly up- (right) and down- (left) regulated mRNAs (upper panel) and miRNAs (lower panel). Only q-value significant DE genes are labeled with gene names. (C) Expression levels of significantly dysregulated mRNAs (upper) and miRNAs (lower) in primary, recurrent and adjacent normal tissues. ns (P > .05), * (P ≤ .05), ** (P ≤ .01), *** (P ≤ .001), **** (P ≤ .0001). (D) Scatter plots indicating expression (normalized by Transcript per Million, TPM) of ALK7 (up) and RASAL1 (bottom) in initial and recurrent of matched GLASS cases. P-value of the Wilcoxon matched-pairs signed rank test is indicated on top of each graph. Main protein coding transcript of each gene is assessed (transcript ID: ENST00000243349.13 and ENST00000548055.2, for ALK7 and RASAL1, respectively).
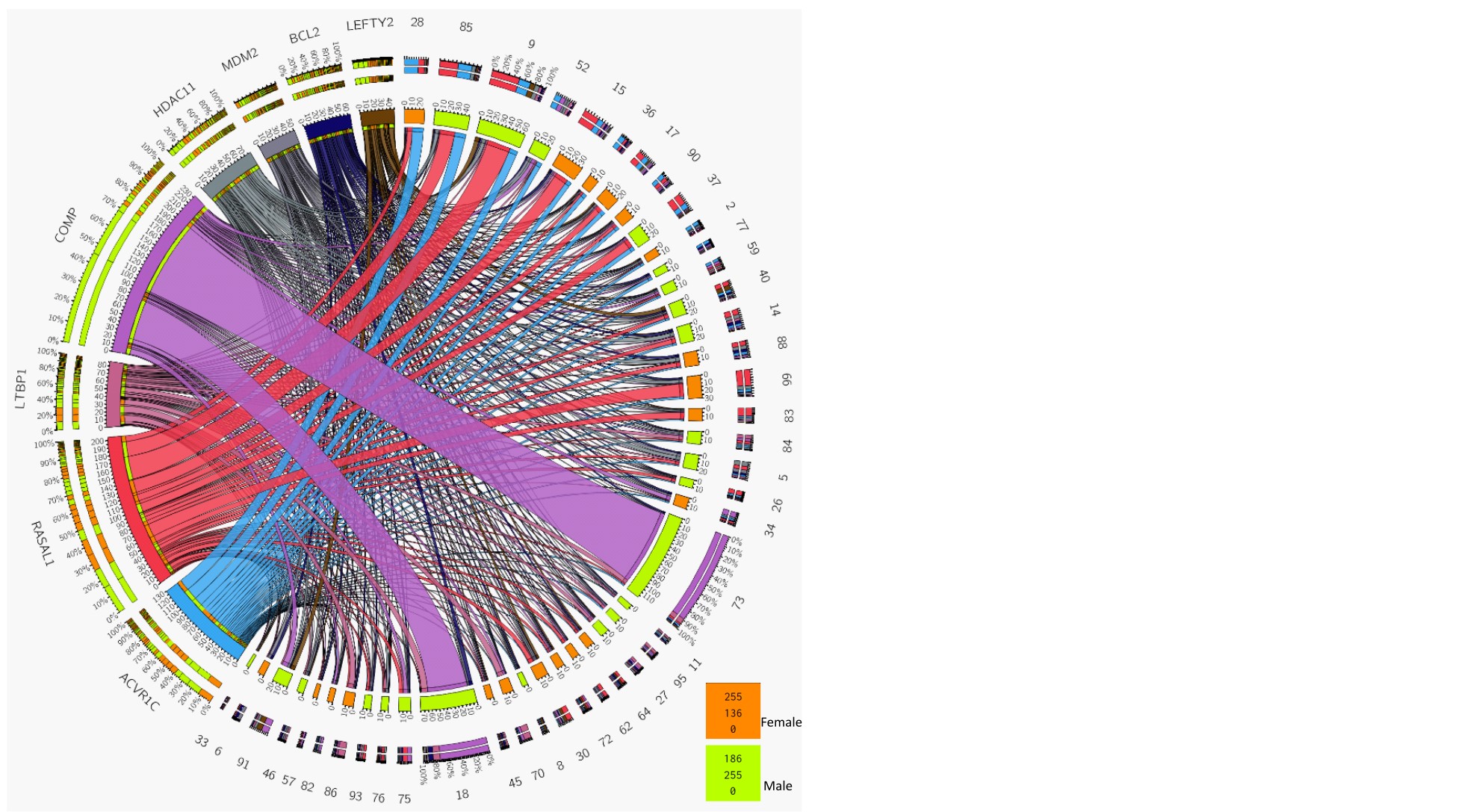
Differential gene expression analysis of initial and recurrent tumor pairs indicated upregulation of ALK7 (ACVR1C), RASAL1, COMP, LTBP1, HDAC11, and BCL2 and downregulation of MDM2 and LEFTY2 in recurrent tumors (Figure 3B–C, upper; Supplementary Table 4). Notably, with the exception of COMP, all genes were dysregulated in multiple patients as indicated by circos plot (Supplementary Figure10). Upregulation of ALK7 and RASAL1 in the recurrent tumors were confirmed in the RNA-seq dataset (Synapse) from the GLASS consortium containing recurrent tumors with a short- and long-term relapse period (Figure 3D). In analyses of TCGA data, only LTBP1 (P = .027) was significantly associated with worse survival (Supplementary Figure11). Interestingly, among miRNAs that were significantly dysregulated in the recurrent tumor (Figure 3B and C, lower), miR-129-5p, miR-1246, miR-494-3p, and miR-19b-3p have been linked to TMZ resistance by targeting Wnt signaling,31 CCNG2,32 p-AKT33 and PTEN,34 respectively.
Gene set enrichment analysis for GO, KEGG, and Reactome of differentially expressed mRNAs revealed a significant enrichment of transmembrane protein kinase activity (NES = +1.5), and TGF-β signaling (NES = +1.3) among others (Figure 4A, upper). Biological function analysis based on regulated pathways for differentially expressed mRNAs revealed a significant regulation of disease-specific pathways, cellular immune response, cellular growth, proliferation, development, and apoptosis, consistent with increased TMZ resistance (Figure 4B, upper). Gene network analysis allowed the identification of potential master regulators of differentially regulated mRNAs leading to enhanced protein phosphorylation (Supplementary Figure 12). Interestingly, differentially regulated mRNAs and predicted targets of differentially expressed miRNA revealed an almost complete overlap of biological functions (Figure 4B, lower), although different pathways were induced (Figure 4A, lower), suggesting that mRNAs and miRNAs act in a concerted manner. Integrated analysis of dysregulated miRNAs and their inversely correlating miRNA targets allowed the identification of miRNA/mRNA negative regulation pairs, with miR-19b-3p, predicted to be most frequently engaged in binding to dysregulated mRNAs (Figure 4C and D). TNF was the highest induced transcript (log ratio = +1.051) in recurrent tumors by the miRNA/mRNA negative regulation pairs (mainly due to the miR-19b-3p downregulation) (Figure 4E). Geneset enrichment analysis of the deregulated miRNAs pointed towards an enriched Wnt pathway (Figure 4A, lower). Accordingly, biological function based on differentially regulated mRNAs, and oppositely regulated miRNAs, pointed towards enhanced disease-specific pathways, proliferation, and developmental pathways (Supplementary Figure 13). Enhanced expression of SMAD2/3 (effector of TGF-β signaling), β-catenin (effector of Wnt signaling), and NF-κB (effector of TNF) in the recurrent tumor were confirmed by IHC (Supplementary Figure 14).
Figure 4.

Deregulated pathways upon late recurrence (A) Gene set enrichment analysis of the mRNA (upper panel) and miRNA (lower panel) datasets for GO, KEGG and Reactome pathways. X axis indicates the Normalized Enrichment Score (NES). Positive NES indicates upregulation and negative NES, downregulated on a given pathway in recurrence. Grayscale indicates level of adjusted significance (q value) and dot size is proportional to the number of contributed leading edge genes. (B) Radar graph scored based on pathway analysis using IPA for mRNA data (upper panel) and miRNA data (lower panel). (C) Integration of mRNA and miRNA dataset and its translation into united deregulated pathways. Differentially regulated miRNAs with their inversely regulated putative mRNA targets. (D) Word cloud of differentially regulated miRNAs engaged in binding to their theoretical mRNA targets. (E) Word cloud of affected putative mRNA targets. Size implies number of targets, and level of impact by miR deregulation on putative targets.
ALK7 is Enhanced in the Recurrent Tumor
ALK7, a member of the type-I TGF-β receptor subfamily implicated in EMT,35 was most significantly upregulated in recurrent tumors (Figure 3B and C, upper). Pairwise comparison of matched initial and recurrent tumors confirmed ALK7 upregulation (Figure 5A). ALK7 expression at the protein level showed a significant positive correlation (R2 = 0.2614, P < .001) with Nanostring normalized counts (Figure 5B). Overexpression of a dominant negative form of ALK7, containing a p.K222R kinase dead mutation28 (ALK7-DN) in U87 GBM cells conferred enhanced TMZ-induced cytotoxicity and apoptosis relative to cells stably transfected with ALK7 wild-type construct (ALK7-WT), suggesting that ALK7 overexpression is a resistance mechanism in recurrent tumors (Figure 5C). In non-neoplastic cells, ALK7 induces apoptosis through upregulation of pro-apoptotic Bax and downregulation of antiapoptotic factors BCL2, BCL-XL, and XIAP.35 Consistent with these findings, overexpression of a constitutively active form of ALK7 containing a p.T194D mutation28 (ALK7-CA), but not ALK7-DN or ALK7-WT, strongly induced apoptosis and reduced viability in our GBM cell line model (Figure 5D). However, TMZ-induced viability, but not TMZ-induced cytotoxicity or TMZ-induced apoptosis was significantly enhanced in ALK7-CA cells (Figure 5C).
Figure 5.

ALK7 is upregulated in the TMZ-resistant tumor of patients with late recurrence (A) Pairwise comparison of ALK7 mRNA expression. (B) Top: Correlation of tumor-specific ALK7 IHC scores and Nanostring normalized counts from tissue punches. Bottom: Example of TMA punches from the tumor center of initial (left) and recurrent (right) tumor from patient 063 by ALK7 IHC. (C) TMZ response of U87 cells overexpressing ALK7 wild-type (ALK7-WT), dominant negative mutant (ALK7-DN) or constitutive active mutant (ALK7-CA) induced by DOX. Fold change in viability, cytotoxicity and apoptosis following treatment with 100uM TMZ or DMSO is shown after 48 hrs. (D) Viability (relative fluorescent units, RFU) and apoptosis (relative luminescent units, RLU) of cells in the absence of TMZ induced by DOX for 48 hrs. ns (P > .05), ** (P ≤ .01), *** (P ≤ .001). (E) Inverse Pearson correlation of ALK7 and BAX in our cohort of primary and recurrent gliomas. (F) Pairwise comparison of BCL2 mRNA expression. (G) Correlation of BCL2 IHC scores and Nanostring normalized counts from tissue punches (top) and an example of TMA punches from the tumor center of initial (left) and recurrent (right) tumor from patient 027 by BCL2 IHC. (H) Model of concerted TGFβ signaling pathway. ALK7, LTBP1, RASAL1 upregulation and LEFTY2 downregulation translate into increased TGFβ induced EMT and stemness. To overcome TGFβ-induced apoptosis, recurrent tumor cells downregulate pro-apoptotic proteins BAX and BAD and upregulate BCL2 (see E and Supplementary Figure 15). Dashed arrows indicate mechanisms that are uncoupled in recurrent tumors.
GBM Cells Overexpressing ALK7 are Protected From Apoptosis
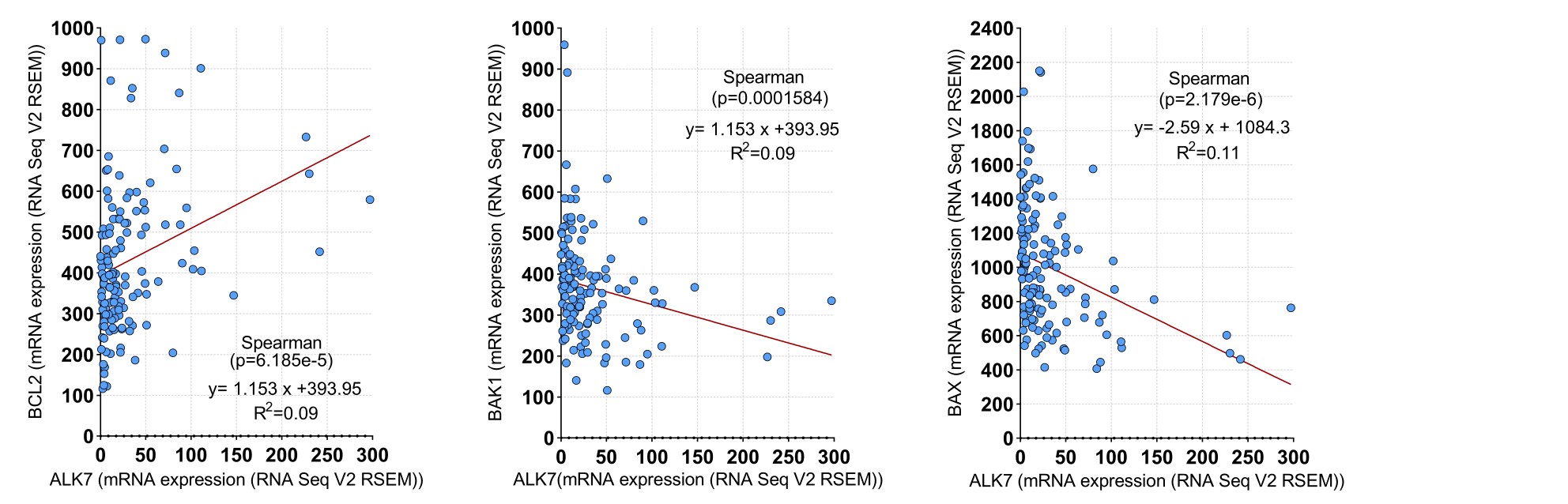
ALK7 mRNA expression was directly correlated with BCL2 expression and inversely correlated with BAK1 and BAX expression in the TCGA dataset of initial grade 4 gliomas (Supplementary Figure 15), suggesting that the apoptosis supporting role of ALK7 is reversed in recurrent tumors. In line with TCGA data, ALK7 and BAX mRNA expression was inversely correlated in our dataset of long-term responders (Figure 5E). BCL2 was also significantly upregulated in recurrent tumors (Figure 3B and C, upper), which was confirmed by pairwise comparison of matched initial and recurrent tissues (Figure 5F and G).
Discussion
We performed a comprehensive analysis of a single-center cohort of patients with matched initial and recurrent tumors resected with a relapse time interval greater than one year. We provide evidence for a highly branched evolution of recurrent astro-IDH-mut-G4/GBM with a high proportion of patients showing early divergent (ancestral) evolution defined by the outgrowth of distinct subclones in the initial and recurrent tumors, which have been branched from a common ancestor long before the diagnosis.7 This type of evolution results in the disappearance of the initial clone(s) following therapy and the outgrowth of a new clone from an ancestor cell, which shares only truncal mutations with the initial clone. In contrast, patient collectives from other studies, which also include short-term survivors, tend to show dominance of clones in the initial tumor that progress during recurrence.5 While assigning the long relapse interval as inclusion criteria which may be a better reflection of tumor evolution, it limited the number of eligible cases to include. Highly branched clonal evolution of astro-IDH-mut-G4/GBM at the mutational level is further corroborated by the finding that high plasticity also exists for mRNA and miRNA profiles (our data), as well as epigenetic profiles.9 This may have clinical implications, since potentially actionable alterations, such as those in EGFR, NF1, or MTOR, may dominate the initial tumor, but may disappear during recurrence rendering targeted therapy ineffective.
In agreement with previous studies,5 there was no evidence for recurring secondary alterations at the genomic level driving TMZ/RT resistance. Instead, known drivers of gliomas were often private to either the initial or the recurrent tumor. Some of these alterations are drivers of proneural, classical, or mesenchymal subtypes,6 but we noted no significant enrichment for a certain subtype, which is in line with previous findings.7,36TP53 mutations appeared early during tumor evolution in line with previous findings,7 but a discrepancy exists for PTEN mutations, which was an early event in our study and a late event in the study by Kim et al.7 Whether this is due to a bias for late recurrence in our patient cohort, remains to be shown. In contrast to the high mutational plasticity, the copy number profile remained largely unchanged during progression. The new WHO classification,1 recommends consulting copy number profiles for the classification of adult-type diffuse gliomas, grade 4. Thus, our finding, that copy number profiles are retained in recurrent tumors even in cases with ancestral evolution, is important in the context of the new classification guidelines, since they also apply to the recurrent tumor.
It was previously shown that TMZ treatment is associated with a significant increase in the mutational burden,5 which was also confirmed by our study. We found in our cohort of long-term responders that SBS11 (TMZ treatment) and SBS1 (deamination of 5-methylcytosine) were the only mutational signatures that were altered in recurrent tumors. Decreased 5-methylcytosine can be secondary to temporal enhanced BER activity to repair N7-MeG and N3-MeA cytotoxic DNA adducts, introduced into the DNA by alkylating therapy.37 However, no evidence for altered SBS15 signature (defective MMR) was observed.
Messenger RNA expression profiling revealed that ALK7 and LTBP1 were significantly upregulated while LEFTY2 was significantly downregulated in recurrent tumors. Most strikingly, all these genes contribute to enhanced TGF-β signaling. ALK7 is a brain-enriched type-I-receptor family member of the TGF-β superfamily, which is induced by Activin B, Activin AB, and Nodal28 and exerts its functions by interacting with type II receptors leading to enhanced SMAD2/3 phosphorylation and nuclear translocation, promoting proliferation38 and EMT28 and our in vitro results indicate that it was also involved in TMZ resistance (Figure 5C and H). In addition, ALK7 signaling has a protumorigenic role by controlling glycolysis and ROS production.39LTBP1 encodes a protein binding to TGF-β leading to its activation. Interestingly, LTBP1 expression in the initial tumor is associated with an unfavorable prognosis. Since LTBP1 is also part of the transcriptional signature of relapsed tumors, it may be implicated in both gliogenesis and tumor relapse. Another member of the LTBP family, LTBP4, was previously shown to be mutated in 11% of patients and was always private to the recurrent tumors.7 We identified LTBP4 mutations in 14% of glioma cases, and again, all were private to the recurrent tumors. Left-right determination factor LEFTY2 is a member of the transforming growth factor β (TGF-β) superfamily. It is activated by the TGF-β signaling and confers its repression by a negative feedback loop.40 Thus, reduced expression of LEFTY2 may result in constitutive TGF-β activity, and tumorigenesis (Figure 5H). The TGF-β signal is often reduced during tumor initiation to overcome its antiproliferative and pro-apoptotic effects but is reactivated during progression to enhance EMT and stemness.35,40–42 This dichotomous function results in a non-monotonic expression pattern of normal brain tissue, initial and recurrent tumor, a finding also confirmed in our study. How can recurrent tumors overcome the pro-apoptotic signal? We show that antiapoptotic protein BCL2 is induced whereas pro-apoptotic BAX is reduced in recurrent gliomas, suggesting that TGF-β-induced apoptosis is uncoupled during recurrence. It remains to be shown if early branching evolution may support the dichotomous pattern of TGF-β signaling since tumor clones with reduced TGF-β activity in the initial tumor may be replaced by new clones with enhanced TGF-β activity.
Interestingly, dysregulated mRNAs and miRNAs may be implicated in similar cellular processes although they seem to induce different pathways. Thus, enhanced TGF-β signaling elicited by dysregulated mRNAs and enhanced TNF-α and Wnt signaling elicited by dysregulated miRNAs, may cooperatively induce SMAD2 phosphorylation to enhance EMT.41 RASAL1 and HDAC overexpression have been associated with an unfavorable prognosis of GBM patients. RASAL1 overexpression induces proliferation and migration, and confers enhanced EMT by inducing MEK/ERK signaling.42 Overexpression of histone deacetylase (HDAC) proteins is associated with radiotherapy resistance,43 but it is unknown if this is due to EMT.
Conclusions
Combined expression profiling and mutational profiling of recurrent adult-type diffuse gliomas, CNS WHO grade 4 with long-term recurrence provided evidence for early branching evolution in the majority of cases, which may have clinical implications. In addition, our results suggest a major role of the TGF-β signaling in recurrent tumors. Thus, administering TGF-β inhibitors in conjunction with standard therapy may interfere with TMZ/RT resistance mechanisms.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors gratefully acknowledge the molecular pathology team of the clinical genomics lab, University Hospital Inselspital, and the Translational Research Unit of the Institute of Pathology, University of Bern, for excellent technical support. D.Hanahan, Laboratory of translational oncology, EPFL, generously provided ALK7 plasmids.
Contributor Information
Elham Kashani, Institute of Pathology, University of Bern, Bern, Switzerland; Graduate School for Cellular and Biomedical Sciences, University of Bern, Bern, Switzerland.
Désirée Schnidrig, Department for BioMedical Research, University of Bern, Bern, Switzerland; SIB Swiss Institute of Bioinformatics, Lausanne, Switzerland.
Ali Hashemi Gheinani, Urological Diseases Research Center, Boston Children’s Hospital, Boston, MA, USA; Department of Surgery, Harvard Medical School, Boston, MA, USA; Broad Institute of MIT and Harvard, Cambridge, MA, USA.
Martina Selina Ninck, Institute of Pathology, University of Bern, Bern, Switzerland.
Philipp Zens, Institute of Pathology, University of Bern, Bern, Switzerland; Graduate School for Health Sciences, University of Bern, Bern, Switzerland.
Theoni Maragkou, Institute of Pathology, University of Bern, Bern, Switzerland.
Ulrich Baumgartner, Cell and Molecular Biology Department, QIMR Berghofer Medical Research Institute, Sid Faithfull Brain Cancer Laboratory, Brisbane, Australia; School of Biomedical Sciences, University of Queensland, Brisbane, Australia.
Philippe Schucht, Department of Neurosurgery, University Hospital Bern, Bern, Switzerland.
Gunnar Rätsch, SIB Swiss Institute of Bioinformatics, Lausanne, Switzerland; ETH Zurich, Zurich, Switzerland.
Mark A Rubin, Department for BioMedical Research, University of Bern, Bern, Switzerland; Bern Center for Precision Medicine, Bern, Switzerland.
Sabina Berezowska, Institute of Pathology, University of Bern, Bern, Switzerland; Institute of Pathology, Lausanne University Hospital and University of Lausanne, Lausanne, Switzerland.
Charlotte K Y Ng, Department for BioMedical Research, University of Bern, Bern, Switzerland; SIB Swiss Institute of Bioinformatics, Lausanne, Switzerland; Bern Center for Precision Medicine, Bern, Switzerland.
Erik Vassella, Institute of Pathology, University of Bern, Bern, Switzerland.
SOCIBP consortium:
Andrej Benjak, Rémy Bruggmann, Federico Comoglio, André Kahles, Irene Keller, Charlotte K Y Ng, Salvatore Piscuoglio, Laurie Prélot, Gunnar Rätsch, Mark A Rubin, Désirée Schnidrig, Senija Selimovic-Hamza, and Tinu M Thomas
Funding
This work was supported by a grant from the Swiss National Science Foundation (grant number 31003A_175656; to E.V.). The SOCIBP (SwissMolecularPathologyBreakthroughPlatform) is a driver project funded by the Swiss Personalized Health Network (SPHN).
Ethics Approval and Consent to Participate
The study was approved by the Cantonal Ethics Commission of the Canton of Bern (KEK-BE:2018-00663), which waived the requirement for written informed consent.
Data Availability
The datasets supporting the conclusions of this article is available in the European Genome-Phenotype Archive (EGA) under the accession EGAS00001006022 (for WES Bam files) and is provided at the time of submission as additional files (for post-processed WES data and gene expression datasets).
Conflict of Interest:
The authors declare no conflicts of interest associated with this manuscript.
Authorship Statemet:
E.K., D.S., A.H.G., C.K.Y.N., and E.V. designed the study and the experiments. E.K. performed experiments. E.K., D.S., A.H.G., P.Z., C.K.Y.N., and E.V. performed the analysis of the results. E.V. developed the concept and acquired funding. D.S. performed bioinformatic analysis of WES dataset, with contributions from G.R., M.A.R., and the SOCIBP consortium. E.K., A.H.G., and U.B. performed bioinformatics analysis of Nanostring data. M.S.N. and S.B. constructed the TMA and performed the pathology review. E.K. and T.M. performed the immunohistochemical evaluation. P.Z. performed immunohistochemical confirmation using Digital Pathology. P.S. provided clinical data. E.V. and E.K. wrote the initial draft of the manuscript.
SOCIBP consortium (SwissMolecularPathologyBreakthroughPlatform).
Members: Andrej Benjak, Rémy Bruggmann, Federico Comoglio, André Kahles, Irene Keller, Charlotte K.Y.Ng, Salvatore Piscuoglio, Laurie Prélot, Gunnar Rätsch, Mark A.Rubin, Désirée Schnidrig, Senija Selimovic-Hamza, Tinu M.Thomas.
References
- 1. Louis DN, Perry A, Wesseling P, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 2021;23(8):1231–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Neftel C, Laffy J, Filbin MG, et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell. 2019;178(4):835–849.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. [DOI] [PubMed] [Google Scholar]
- 4. Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359(5):492–507. [DOI] [PubMed] [Google Scholar]
- 5. Barthel FP, Johnson KC, Varn FS, et al. Longitudinal molecular trajectories of diffuse glioma in adults. Nature. 2019;576(7785): 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Verhaak RGW, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim H, Zheng S, Amini SS, et al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res. 2015;25(3):316–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Park AK, Kim P, Ballester LY, Esquenazi Y, Zhao Z. Subtype-specific signaling pathways and genomic aberrations associated with prognosis of glioblastoma. Neuro Oncol. 2019;21(1):59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Klughammer J, Kiesel B, Roetzer T, et al. The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space. Nat Med. 2018;24(10): 1611–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Piwecka M, Rolle K, Belter A, et al. Comprehensive analysis of microRNA expression profile in malignant glioma tissues. Mol Oncol. 2015;9(7):1324–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Banelli B, Carra E, Barbieri F, et al. The histone demethylase KDM5A is a key factor for the resistance to temozolomide in glioblastoma. Cell Cycle. 2015;14(21):3418–3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hewer E, Vajtai I, Dettmer MS, Berezowska S, Vassella E. Combined ATRX/IDH1 immunohistochemistry predicts genotype of oligoastrocytomas. Histopathology. 2016;68(2):272–278. [DOI] [PubMed] [Google Scholar]
- 13. Mariani L, Deiana G, Vassella E, et al. Loss of heterozygosity 1p36 and 19q13 is a prognostic factor for overall survival in patients with diffuse WHO grade 2 gliomas treated without chemotherapy. J Clin Oncol. 2006;24(29):4758–4763. [DOI] [PubMed] [Google Scholar]
- 14. Vassella E, Vajtai I, Bandi N, et al. Primer extension based quantitative polymerase chain reaction reveals consistent differences in the methylation status of the MGMT promoter in diffusely infiltrating gliomas (WHO grade II-IV) of adults. J Neurooncol. 2011;104(1):293–303. [DOI] [PubMed] [Google Scholar]
- 15. Bankhead P, Loughrey MB, Fernández JA, et al. QuPath: open source software for digital pathology image analysis. Sci Rep. 2017;7(1):1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schmidt U, Weigert M, Broaddus C, Myers G. Cell detection with star-convex polygons. Lect Notes Comput Sci (including Subser Lect Notes Artif Intell Lect Notes Bioinformatics). 2018;11071 LNCS:265–273. doi: 10.1007/978-3-030-00934-2_30. [DOI] [Google Scholar]
- 17. Favero F, Joshi T, Marquard AM, et al. Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann Oncol. 2015;26(1):64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mermel CH, Schumacher SE, Hill B, et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12(4):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Blokzijl F, Janssen R, van Boxtel R, Cuppen E. MutationalPatterns: comprehensive genome-wide analysis of mutational processes. Genome Med. 2018;10(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016;17(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alexandrov LB, Kim J, Haradhvala NJ, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578(7793):94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Carter SL, Cibulskis K, Helman E, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30(5):413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Leshchiner I, Livitz D, Gainor JF, et al. Comprehensive analysis of tumour initiation, spatial and temporal progression under multiple lines of treatment. bioRxiv. Published online February 16, 2019:508127. doi: 10.1101/508127. [DOI] [Google Scholar]
- 24. Baumgartner U, Berger F, Hashemi Gheinani A, et al. miR-19b enhances proliferation and apoptosis resistance via the EGFR signaling pathway by targeting PP2A and BIM in non-small cell lung cancer. Mol Cancer. 2018;17(1):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hoadley KA, Yau C, Hinoue T, et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell. 2018;173(2):291–304.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Colaprico A, Silva TC, Olsen C, et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016;44(8):e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haemmig S, Baumgartner U, Glück A, et al. MiR-125b controls apoptosis and temozolomide resistance by targeting TNFAIP3 and NKIRAS2 in glioblastomas. Cell Death Dis. 2014;5(6):e1279–e1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Michael IP, Saghafinia S, Tichet M, et al. ALK7 signaling manifests a homeostatic tissue barrier that is abrogated during tumorigenesis and metastasis. Dev Cell. 2019;49(3):409–424.e6. [DOI] [PubMed] [Google Scholar]
- 29. Hegi ME, Diserens A-C, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. [DOI] [PubMed] [Google Scholar]
- 30. Wang J, Cazzato E, Ladewig E, et al. Clonal evolution of glioblastoma under therapy. Nat Genet. 2016;48(7):768–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zeng A, Yin J, Li Y, et al. miR-129-5p targets Wnt5a to block PKC/ERK/NF-κB and JNK pathways in glioblastoma. Cell Death Dis. 2018;9(3):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang H, Wu B, Wang J, et al. Methylation associated miR-1246 contributes to poor prognosis in gliomas treated. Clin Neurol Neurosurg. 2021;200(July 2020):106344. [DOI] [PubMed] [Google Scholar]
- 33. Qiu G, Tong W, Jiang C, et al. Long noncoding RNA WT1-AS inhibit cell malignancy via miR-494-3p in glioma. Technol Cancer Res Treat. 2020;19(2020):153303382091975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jia Z, Wang K, Zhang A, et al. MiR-19a and miR-19b overexpression in gliomas. Pathol Oncol Res. 2013;19(4):847–853. [DOI] [PubMed] [Google Scholar]
- 35. Xu G, Zhou H, Wang Q, Auersperg N, Peng C. Activin receptor-like kinase 7 induces apoptosis through up-regulation of Bax and down-regulation of Xiap in normal and malignant ovarian epithelial cell lines. Mol Cancer Res. 2006;4(4):235–246. [DOI] [PubMed] [Google Scholar]
- 36. Kwon SM, Kang SH, Park CK, et al. Recurrent glioblastomas reveal molecular subtypes associated with mechanistic implications of drug-resistance. PLoS One. 2015;10(10):e0140528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Grin I, Ishchenko AA. An interplay of the base excision repair and mismatch repair pathways in active DNA demethylation. Nucleic Acids Res. 2016;44(8):3713–3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. De Silva T, Ye G, Liang YY, et al. Nodal promotes glioblastoma cell growth. Front Endocrinol. 2012;3(2012):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li H, Chen J, Wang X, et al. Nodal induced by hypoxia exposure contributes to dacarbazine resistance and the maintenance of stemness in melanoma cancer stem-like cells. Oncol Rep. 2018;39(6):2855–2864. [DOI] [PubMed] [Google Scholar]
- 40. Matsumoto T, Chino H, Akiya M, et al. Requirements of LEFTY and Nodal overexpression for tumor cell survival under hypoxia in glioblastoma. Mol Carcinog. 2020;59(12):1409–1419. [DOI] [PubMed] [Google Scholar]
- 41. Yoshimatsu Y, Kimuro S, Pauty J, et al. TGF-beta and TNF-alpha cooperatively induce mesenchymal transition of lymphatic endothelial cells via activation of Activin signals. PLoS One. 2020;15(5):e0232356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chang RX, Cui AL, Dong L, et al. Overexpression of RASAL1 indicates poor prognosis and promotes invasion of ovarian cancer. Open Life Sci. 2019;14(1):133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Takashima Y, Kawaguchi A, Yamanaka R. Promising prognosis marker candidates on the status of epithelial-mesenchymal transition and glioma stem cells in glioblastoma. Cells. 2019;8(11):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets supporting the conclusions of this article is available in the European Genome-Phenotype Archive (EGA) under the accession EGAS00001006022 (for WES Bam files) and is provided at the time of submission as additional files (for post-processed WES data and gene expression datasets).