

Abstract
Background
Alterations in transcriptional regulators of glycolytic metabolism have been implicated in brain tumor growth, but the underlying molecular mechanisms remain poorly understood.
Methods
Knockdown and overexpression cells were used to explore the functional roles of HOXA3 in cell proliferation, tumor formation, and aerobic glycolysis. Chromatin immunoprecipitation, luciferase assays, and western blotting were performed to verify the regulation of HK2 and PKM2 by HOXA3. PLA, Immunoprecipitation, and GST-pull-down assays were used to examine the interaction of HOXA3 and KDM6A.
Results
We report that transcription factor homeobox A3 (HOXA3), which is aberrantly highly expressed in glioblastoma (GBM) patients and predicts poor prognosis, transcriptionally activates aerobic glycolysis, leading to a significant acceleration in cell proliferation and tumor growth. Mechanically, we identified KDM6A, a lysine-specific demethylase, as an important cooperator of HOXA3 in regulating aerobic glycolysis. HOXA3 activates KDM6A transcription and recruits KDM6A to genomic binding sites of glycolytic genes, targeting glycolytic genes for transcriptional activation by removing the suppressive histone modification H3K27 trimethylation. Further evidence demonstrates that HOXA3 requires KDM6A for transcriptional activation of aerobic glycolysis and brain tumor growth.
Conclusions
Our findings provide a novel molecular mechanism linking HOXA3-mediated transactivation and KDM6A-coupled H3K27 demethylation in regulating glucose metabolism and GBM progression.
Keywords: aerobic glycolysis, GBM, HOXA3, KDM6A, transcriptional activation
Key Points.
HOXA3 promotes aerobic glycolysis of GBM cells and brain tumor growth.
HOXA3 interacts with KDM6A to enhance the transcription of glycolytic genes.
HOXA3 requires KDM6A for transcriptional activation of aerobic glycolysis and brain tumor growth.
Importance of the Study.
The study demonstrates that HOXA3 has a central role in transcriptional activation of aerobic glycolysis that links with KDM6A-coupled H3K27 demethylation, providing evidence for the cooperation of HOXA3 and KDM6A in regulating glucose metabolism that shifted oxidative phosphorylation toward aerobic glycolysis, and identifying that the HOXA3-KDM6A axis is a potential therapeutic target for GBM.
HOX proteins are a family of highly conserved transcription factors, including HOXA, HOXB, HOXC, and HOXD, which play important roles in embryonic development and tissue homeostasis.1 HOX genes are also associated with genetic susceptibility in many types of cancer, but there are few studies on the role of HOXA3 in cancer development.2 HOX genes promote cancer development mainly through their transcriptional regulation of downstream target genes and are involved in the regulation of tumor angiogenesis, autophagy, differentiation, apoptosis, proliferation, and metastasis.3–5 In Recent years, there has been increasing evidence that HOX proteins are also key regulators of aberrant glycolysis in the metabolic reprogramming of tumor cells. HOXA9 can act as an antitumor factor by downregulating the HIF-1α gene, which activates a series of glycolytic genes (e.g., Gluts, LHDA, HKs, and PKF) and suppresses mitochondrial oxygen consumption in cancer cells.6 In contrast, ectopic expression of HOXC8 can upregulate glycolysis-related genes to promote the progression of nasopharyngeal carcinoma.7 Different HOX proteins may play different roles in glycolytic metabolism in different types of cancer cells, and the functional role of HOXA3 in metabolic reprogramming and GBM progression is unknown.
Aerobic glycolysis is 1 of the distinctive features of GBM, and is regulated by glucose transporter proteins and some key rate-limiting enzymes.8 Recent studies have shown that epigenetic modifications, especially histone methylation modifications, are key regulators of aerobic glycolysis in GBM.9 Methylation modification of histones is dynamically regulated by histone methyltransferase (HMTs) and histone demethylase (HDMs).10 Lysine methyltransferases, such as G9a, MLL1, and EZH2, suppress the transcription of HIF-1α and its target genes to reprogram glycolytic metabolism in GBM.11–13 In addition, HDMs also play important roles in the regulation of glycolytic metabolism in GBM. For example, LSD1 and JMJD3 repress glycolysis, whereas KDM1B promotes glucose metabolism.14,15 Meanwhile, the promoter regions of HOX protein target genes are often accompanied by altered histone methylation modifications.16 Therefore, it is of scientific interest to investigate the synergistic effect of HOX protein and histone methylation modifications in the glycolytic metabolism of GBM cells.
KDM6A, a lysine-specific demethylase, specifically catalyzes the demethylation of histone H3K27 trimethylation (H3K27me3), which is the modification associated with transcriptional repression.17 KDM6A mutations frequently occurred in many solid tumors, including bladder cancer, esophageal squamous cell carcinoma, colon cancer, and multiple myeloma.18–21 According to TCGA data, GBM patients rarely have KDM6A mutations (approximately 0.4%). Earlier studies identified KDM6A as a tumor suppressor, and the deletion of KDM6A in bladder cells promoted cell proliferation and metastasis.22 Deficiency of KDM6A accelerates cancer progression and shortens mouse life span in KrasG12D mutated lung cancer mouse model.23 However, other studies have provided evidence demonstrating that KDM6A also plays an oncogenic role. Loss of KDM6A in TAL1-positive cells induces apoptosis and inhibits cell proliferation.24 Moreover, another study reveals that KDM6A promotes cell proliferation and tumor growth in ER-positive breast cancer.25 The role and mechanism of action of KDM6A in biological processes and GBM progression remain unknown.
In this study, we report that HOXA3 is upregulated in GBM specimens and cells compared with the normal groups. High expression of HOXA3 predicts poor prognosis of GBM patients. Depletion of HOXA3 in GBM cells reduces proliferation, and tumor growth and induces cell cycle arrest, which is rescued by reconstituted expression of HOXA3. Biological process studies show that HOXA3 activates aerobic glycolysis by increasing the transcription of HK2 and PKM2. Mechanistically, HOXA3 activates KDM6A transcription and recruits KDM6A to the genomic binding sites of HK2 and PKM2 for transcriptional activation by removing the suppressive histone modification H3K27 trimethylation. Our study reveals that HOXA3 has a central role in the transcriptional activation of aerobic glycolysis that links with KDM6A-coupled H3K27 demethylation. Our findings provide evidence for the cooperation of HOXA3 and KDM6A in reprogramming glycolytic metabolism and GBM progression.
Materials and Methods
Patient Samples
The glioma tissue microarrays (TMA) including 28 glioma patients were purchased from Avila Biotechnology (Xi’an, China). Patient specimens were collected in Liaocheng People’s Hospital (Liaocheng, China), with written informed consent. This project was approved by the Institute Research Ethics Committee of Jining Medical University.
Cells and Cell Culture
Primary human GBM cells (GBM01, GBM02, and GBM03) were obtained as previously reported.26 Primary GBM cells were cultured in NeurobasalTM Medium (Gibco) and supplemented with 2% B27 Neuro Mix (Thermo Fisher Scientific). SVG p12, LN229, and U118 cell lines were obtained from American Type Culture Collection. U251, U87, and HEK 293 cells were obtained from the National Infrastructure of Cell Line Resource (Beijing, China). All the cell lines were authenticated by short tandem repeat analysis. All cell lines were cultured as previously described.27
Vectors, Transfection, and Infection
The shRNA-HOXA3 and shRNA-KDM6A target site were designed and synthesized at Beijing Genomics Institute (BGI, Beijing, China) and cloned into the pLKO.1. Flag-HOXA3, Flag-HOXA3△DUF (Domain of unknown function), Flag-HOXA3△Hbox (Homeobox), HA-KDM6A, KDM6A△TPR (tetratricopeptide repeats), and KDM6A△JmjC were obtained from YouBo Biotechnology (Changsha, China) and constructed into pCDH-CMV-MCS-EF1. The plasmids transfection and lentiviruses infection were carried out as previously described.27 Primer sequences are listed in Supplementary Table S1.
Cell Viability Assays
Cell proliferation assay and BrdU immunofluorescent staining were employed to detect cell proliferation and performed as in our previous study.27
Seahorse Assay
The XF glycolysis stress test kit was used to measure glycolytic function. The glycolysis, glycolytic capacity, glycolytic reserve and nonglycolytic acidification, and oxygen consumption rate were detected by the XF-96p flux analyzer (Seahorse Bioscience, CA) as previously described.28
Chromatin Immunoprecipitation Assay
For Chromatin Immunoprecipitation (ChIP) assay, 20 × 106 GBM cells were collected and crosslinked with formaldehyde (1%). The chromatin DNA was prepared by using SimpleChIP Enzymatic Chromatin IP Kit as per the manufacturer’s instructions (CST, #9002). The chromatin was immunoprecipitated with the specific antibodies shown in Supplementary Table S2. The enrichment of each antibody was quantified by quantitative real-time PCR. The ChIP PCR sequences are shown in Supplementary Table S1.
Luciferase Assay
The HK2, PKM2, and KDM6A wild or mutate promoters were synthetic and constructed into pGL3-Bacic. The control or HOXA3 knockdown cells were transfected with the different promoter-reporter or pGL3-basic plasmids for 24 h. The cells were collected and lysed by the Renilla-LumiTM buffer. The dual luciferase activities were tested by the sea kidney luciferase reporter gene detection kit according to manufacturers’ instructions (Beyotime RG062S, Shanghai, China).
Gene Set Enrichment Assay
The GBM patient data were downloaded from the cancer genome atlas (TCGA). The data were normalized by ANOVA and divided into 2 groups (low HOXA3 and high HOXA3). Gene set enrichment assay (GSEA) was performed as described previously.29
Proximity Ligation Assay
The duolink- proximity ligation assay (PLA) assay was performed for the detection of interaction between HOXA3 and KDM6A by using Duolink In Situ Kit according to the manufacturer’s instructions. Briefly, the HOXA3 and KDM6A were added into cell lysis solutions overnight. Removed the antibodies, the cells were incubated with PLUS and MINUS PLA probes for 1 h at 37°C. The PLA signals were detected by using a confocal microscope (Nikon, Japan).
Immunoblotting and Immunoprecipitation Analysis
For western blot assay, cells were lysed by RIPA buffer (Beyotime, Shanghai, China) for 10 min on ice. The SDS-PAGE, membrane transfer, and chemiluminescence were performed as described previously.27 The interaction between HOXA3 and KDM6A in GBM cells was detected by using a Co-IP assay. Briefly, the cell lysis solutions were incubated with the different antibodies overnight and with protein A/G PLUS Agarose (Merck, New Jersey). To eliminate the interference of heavy chains, different species antibodies or Light-Chain specific secondary antibodies were used in immunoprecipitation and Western blot assays. The primary antibodies were incubated at 4°C overnight. The antibodies were listed in Supplementary Table S2.
Purification of Recombinant Proteins and GST Pulldown Assay
GST-tagged HOXA3 was constructed into pCR3.1 and expressed in BL21 E. coli. The in vitro expression of HOXA3 protein was extracted by using the T7 high-yield protein system (Promega, WI). The His-Tagged KDM6A was constructed into pcDNA3.1 and expressed in HEK 293 cells by using PEI buffer. His protein was purified by using His•Bind Purification Kit according to manufacturers’ instructions (Millipore 70239, Massachusetts). For GST-pull down, the GST or GST-HOXA3 fusion proteins were incubated with glutathione agarose (Roche, Indianapolis) for 1 h. Then, the agarose was washed and incubated with His-KDM6A protein at 4°C. The bound proteins were eluted and visualized by Immunoblotting.
Molecular Docking
The structure of the HOXA3 DUF domain and KDM6A TPR domain were predicted by alphafold (https://alphafold.ebi.ac.uk/). The molecular docking model was performed by using the Z-dock tool (https://zdock.umassmed.edu/).
Animal Studies
The BALB/c nude mice were purchased to establish subcuticular and orthotopic xenograft tumor models as previously described.29 Animal experiments were performed in compliance with the guidelines of the Institute for Laboratory Animal Research, Jining Medical University.
Immunohistochemistry and H&E Staining
The tumor tissues and mice brains were harvested and embedded for IHC staining. The IHC and H&E staining were performed as described previously.29
Statistical Analysis
Two-tailed unpaired student’s t-tests were performed by GraphPad Prism version 8.0. Clinical patient data were downloaded from oncomine, TCGA dataset, and R2 Genomic Analysis Visualization Platform (https://hgserver1.amc.nl/cgi-bin/r2/main.cgi). All the experiments were repeated at least 3 independent times. P < .05 was considered statistically significant. More detailed methods were shown in supplementary materials and methods.
Results
HOXA3 is Upregulated in GBM and Predicts Poor Prognosis of Patients
To identify the genic expression status of HOXA3, we first explored the Oncomine dataset. HOXA3 was aberrant and highly expressed in CNS cancer in 3 different databases, especially in GBM (Supplementary Figure S1A–E). To confirm this result, we detected HOXA3 expression in a glioma tissue microarray by performing IHC staining. The expression of HOXA3 was significantly increased in GBM compared with the low-grade glioma (such as astrocytoma and oligodendroglioma) (Figure 1A and B), which was consistent with French glioma data (Figure 1C). Furthermore, we examined the mRNA level of HOXA3 in our local patient specimens, including 48 GBM tissues and 7 normal tissues. HOXA3 was also upregulated in our local GBM cohort, in line with the result of TCGA GBM data (Figure 1D and E).
Figure 1.

HOXA3 is upregulated in GBM and predicts poor prognosis. (A-B) Representative immunofluorescence images and quantifications of HOXA3 staining in TMA (scale bar: 20 μm). (C) Analyses of French database about HOXA3 expression in normal tissues, low-grade glioma, and GBM. (D) qRT-PCR analysis of HOXA3 mRNA levels in our local GBM and normal tissues. (E) Analyses of HOXA3 expression in AgilenG4502A TCGA GBM database. (F) Percent overall survival from our local GBM patients with high HOXA3 expression or low HOXA3. Shown is a Kaplan–Meier curve. (G) qRT-PCR analysis of HOXA3 mRNA levels in normal glia cells, GBM cell lines, and primary GBM cells. All data were analyzed with Student’s t-test and shown as means and SD, **P < .01, ***P < .001.
We further explored the clinical prognostic character of HOXA3 expression in GBM. In our local patient cohort, GBM patients with low HOXA3 expression had a longer survival time than those with high HOXA3 expression (Figure 1G). The analysis of TCGA and R2 French database further confirmed that HOXA3 predicted a poor prognosis in GBM (Supplementary Figure S1F and G). In line with the results of GBM specimens, HOXA3 expression was significantly increased in GBM cell lines and primary GBM cells compared with normal glial cells (Figure 1H).
HOXA3 is Essential for Cell Proliferation and Tumor Growth in GBM
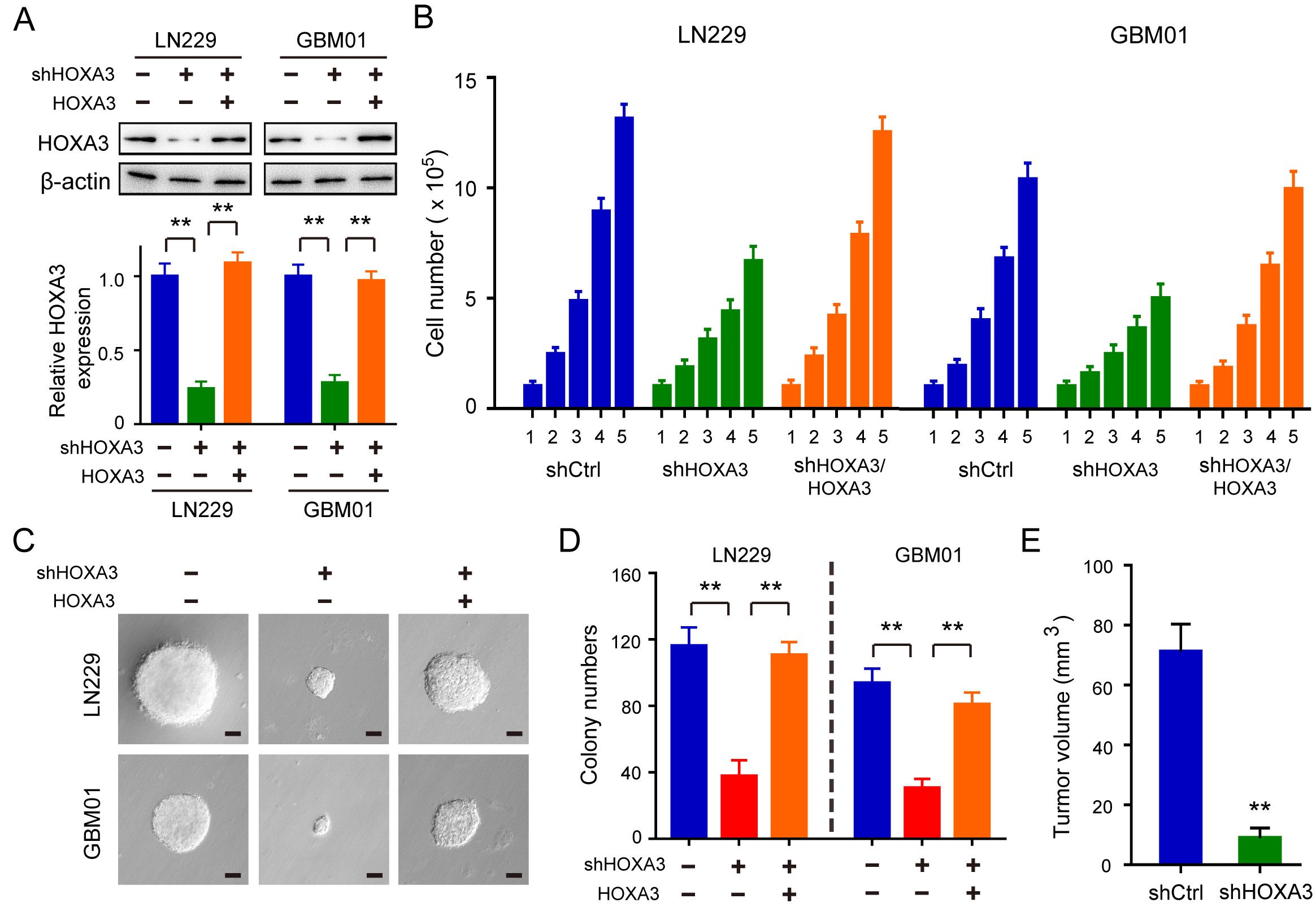
To explore the functional role of HOXA3 in GBM cell proliferation and tumor growth, we constructed stable HOXA3 knockdown cells with shRNA (Figure 2A). In GBM cell lines and primary cells, we observed a significant inhibitory effect of HOXA3 knockdown on cell proliferation (Figure 2B). The BrdU assay further demonstrated that HOXA3 was essential for cell proliferation, as HOXA3 silencing cells showed a pronounced reduction in DNA synthesis (Figure 2C). Since DNA synthesis is closely correlated with the cell cycle, we examined the changes in cell cycle procession by performing flow cytometry. In HOXA3 silencing cells, the cell cycle was arrested in the G1 phase with the downregulation of cyclin E and CDK2 (Figure 2D).
Figure 2.

HOXA3 promotes GBM cell proliferation, tumor growth, and aerobic glycolysis. (A) Immunoblotting of HOXA3 in the indicated cells transduced with shHOXA3 or shRNA scramble. (B) Growth assay of the indicated GBM cells with HOXA3 knockdown or control. (C) Representative immunofluorescence images and quantifications of BrdU staining in GBM cells with HOXA3 knockdown or control. (D) Immunoblotting of cyclin E and CDK2 in the indicated GBM cells (up). Cell cycle analyses in GBM cells transduced with shHOXA3 or shRNA scramble (below). (E) Orthotopic tumor growth abilities of LN229 cells expressing Ctrl or shHOXA3 and survival rates of mice (n = 5). (F) Glycolytic rate of the indicated GBM cells transduced with shCtrl, shHOXA3, or shHOXA3/HOXA3. ECAR extracellular acidification rate. (G) Cellular oxygen consumption rate of the indicated GBM cells transduced with shCtrl, shHOXA3, or shHOXA3/HOXA3. All data were shown as the mean ± SD (n = 3–5), *P <.05, **P < .01.
To further identify the important role of HOXA3 in the regulation of cell proliferation, we reconstituted HOXA3 in the HOXA3 silencing cells (Supplementary Figure S2A). Cell growth assays showed that the inhibitory effect of cell proliferation induced by HOXA3 knockdown was recovered by reconstituting the expression of HOXA3 in culture medium and soft agar (Supplementary Figure S2B–D). In addition, we established orthotopic xenograft tumor models to determine the effect of HOXA3 on the regulation of tumor growth. Knockdown of HOXA3 significantly suppressed the growth of brain tumors and increased the survival time of mice (Figure 2E, Supplementary Figure S2E). All these results suggest that HOXA3 is required for GBM cell proliferation and tumor growth.
HOXA3 Promotes Aerobic Glycolysis by Activating the Transcription of HK2 and PKM2
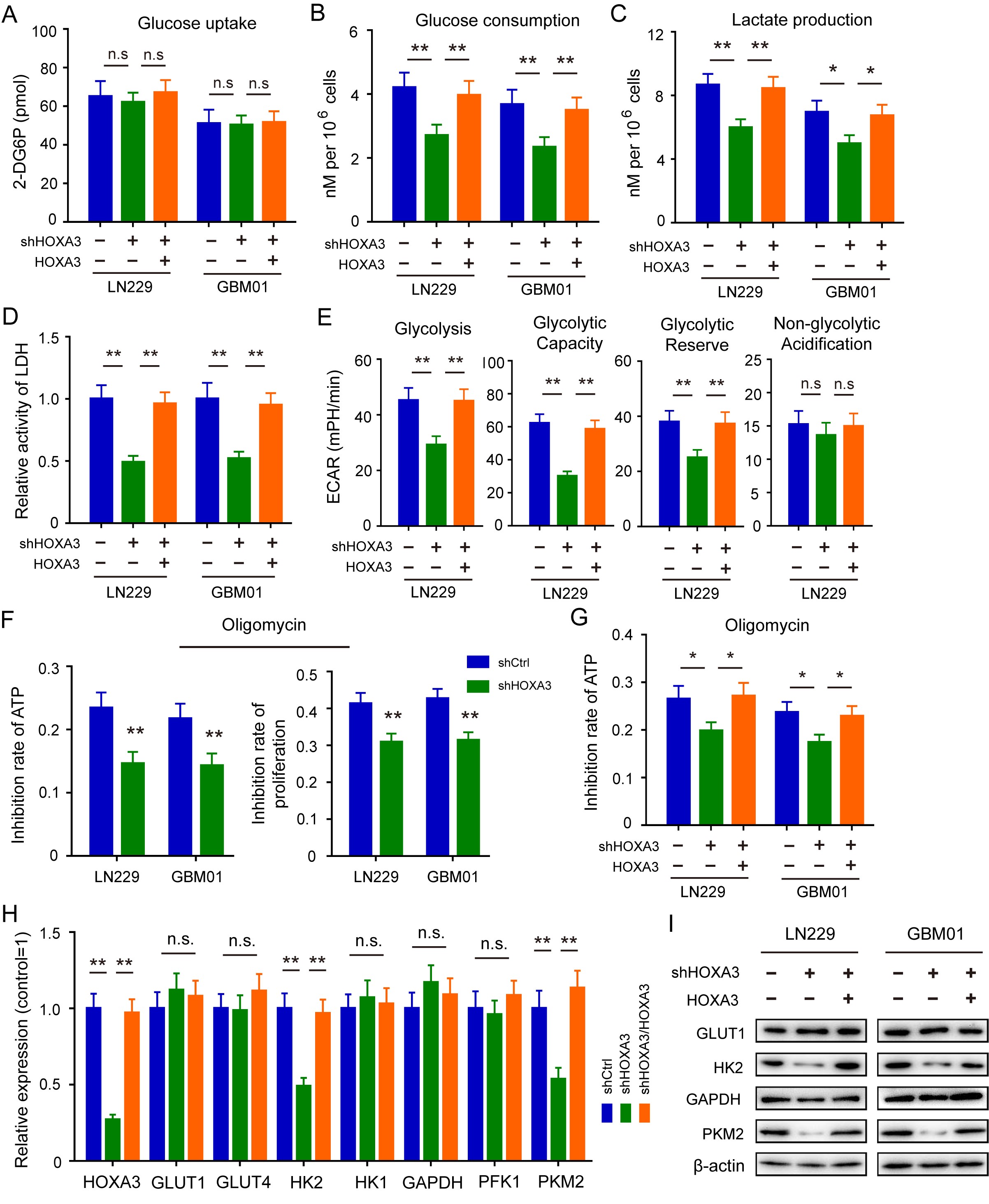
There is increasing evidence that HOX proteins are key regulators of aerobic glycolysis of tumor cells. Here, we determined whether HOXA3 had a similar functional role in GBM cells. As shown by the results of metabolite measurement, glucose consumption and lactate production but not glucose uptake was obviously inhibited in HOXA3 depletion cells, and these effects were rescued by HOXA3 restoration (Supplementary Figure S3A–D). Since GBM cells preferentially employ aerobic glycolysis to generate energy, we examined the effects of HOXA3 in regulating glycolysis by using a seahorse assay. We found a pronounced inhibition of glycolysis in HOXA3 silencing cells, which was rescued by HOXA3 restoration (Figure 2F, Supplementary Figure S3E). Consistent with this finding, HOXA3 depletion significantly attenuated the inhibitory rate of ATP and cell proliferation induced by ATP synthase inhibitor oligomycin (Supplementary Figure S3F and G). In addition, the knockdown of HOXA3 enhanced mitochondrial respiration in GBM cells, as evidenced by oxygen consumption rate assay (Figure 2G). All these results suggested that HOXA3 drives glucose metabolism from oxidative phosphorylation toward aerobic glycolysis in GBM cells. We next detected the expression of key glycolysis-associated genes after HOXA3 silencing or restoration, the results showed that HOXA3 depletion remarkably reduced the expression of HK2 and PKM2, but not GLUT1, HK1, and PFK1 (Supplementary Figure S3H and I, Supplementary Figure S4A and B).
As HOXA3 is a transcription factor, we examined whether HOXA3 activated the transcription of HK2 and PKM2. We firstly identified the potential binding sites of HOXA3 on HK2 and PKM2 (Figure 3A and D). ChIP and quantitative PCR demonstrated pronounced levels of HOXA3 at the HK2 promoter containing the −42 to −33 region in GBM cells (Figure 3B, Supplementary Figure S4C), and HOXA3 knockdown obviously decreased HOXA3 enrichment at this promoter (Figure 3C). Similarly, there was significant enrichment of HOXA3 at PKM2 promoter containing 1 to 11 regions in GBM cells (Figure 3E, Supplementary Figure S4D). Depletion of HOXA3 markedly reduced HOXA3 levels at the PKM2 promoter (Figure 3F). We further performed dual luciferase experiments to verify whether HOXA3 regulates the transcription of HK2 and PKM2. We found that HOXA3 silencing significantly inhibited the activities of HK2 and PKM2 promoters but failed to affect the activities of these promoters with HOXA motif mutants (Figure 3G and H).
Figure 3.

HOXA3 promotes aerobic glycolysis by activating the transcription of HK2 and PKM2. (A) Predicted sequences of HOXA motif in HK2 promoter by JASPAR. (B) ChIP-qPCR analysis of HOXA3 at different regions of HK2 promoter in LN229 cells. (C) ChIP-qPCR analysis of HOXA3 at the indicated HK2 promoter in GBM cells expressing shCtrl or shHOXA3. (D) Predicted sequences of HOXA motif in PKM2 promoter by JASPAR. (E) ChIP-qPCR analysis of HOXA3 at different regions of PKM2 promoter in LN229 cells. (F) ChIP-qPCR analysis of HOXA3 at the indicated PKM2 promoter in GBM cells expressing shCtrl or shHOXA3. (G–H) The indicated promoter-reporter constructs were co-transfected with shCtrl or shHOXA3 vector into HEK293T cells. The promoter activities of HK2 (G) and PKM2 (H) were presented. (I) Immunoblotting of Flag, HK2, and PKM2 in the indicated GBM cells expressing HOXA3-Flag or HOXA3△Hbox-Flag. (J) Glycolytic rate of the indicated GBM cells expressing HOXA3-Flag or HOXA3△Hbox-Flag. All data were shown as means and SD (n = 3), **P < .01, ***P < .001.
HOXA3 functions as a transcription factor depending on its Hbox domain that binds to DNA. We constructed HOXA3 with Hbox domain deletion overexpressing GBM cells (HOXA3△Hbox) to determine whether HOXA3 promoted aerobic glycolysis by its transcriptional activity. In HOXA3△Hbox GBM cells, we observed no change in the expression of HK2 and PKM2 compared with the control cells (Figure 3I). In line with these results, HOXA3 with Hbox deletion did not promote glycolysis or increase lactate production (Figure 3J, Supplementary Figure S4E). All the results demonstrated that HOXA3 promotes aerobic glycolysis by activating the transcription of HK2 and PKM2.
HOXA3 Activates the Transcription of KDM6A and Modifies H3K27me3 of HK2 and PKM2 Promoters
To further identify the underlying mechanism by which HOXA3 transcriptionally activates aerobic glycolysis, we downloaded TCGA GBM patient data and performed GSEA analyses. We found that high HOXA3 expression led to higher expression of a large number of methylation genes including those critical for the regulation of histone methylation. (Figure 4A, Supplementary Figure S5A). ChIP-qPCR revealed that HOXA3 silencing significantly increased the H3K27me3 but not H3K4me3 or H3K9me3 levels at the promoters of HK2 and PKM2 in LN229 cells (Figure 4B). The histone modification H3K27me3 is a transcriptional repressive factor and is regulated by histone methyltransferases and demethylases, including EZH2, KDM6A, and KDM6B. In HOXA3 silencing cells, we observed a pronounced decrease of KDM6A but not EZH2 or KDM6B compared with control cells (Figure 4C, Supplementary Figure S5B). Further ChIP-qPCR demonstrated pronounced levels of HOXA3 at the KDM6A promoter containing the −965 to −955 region in GBM cells (Figure 4D, Supplementary Figure S5C), and HOXA3 knockdown clearly decreased HOXA3 enrichment at this promoter (Figure 4E). Dual luciferase experiments showed that HOXA3 silencing significantly inhibited the activities of the KDM6A promoter but failed to affect the activities of the promoter with the HOX motif mutant (Figure 4F).
Figure 4.

HOXA3 activates the transcription of KDM6A and modifies H3K27me3 of HK2 and PKM2 promoter. (A) Gene set enrichment assay of IlluminaHiSeq (n = 172) the cancer genome atlas (TCGA) showed significant enrichment of gene sets involved in the methylation with the abundant genes being upregulated (cutoff: median). (B) ChIP-qPCR analysis of H3K4me3, H3K9me3, and H3K27me3 at HK2 and PKM2 promoter in GBM cells expressing shCtrl or shHOXA3. (C) Immunoblotting of HOXA3, EZH2, KDM6A, and H3K27me3 in the indicated GBM cells expressing shCtrl, shHOXA3, or shHOXA3/HOXA3. (D) Predicted sequences of HOXA motif in KDM6A promoter by JASPAR. (E) ChIP-qPCR analysis of HOXA3 at the indicated KDM6A promoter in GBM cells expressing shCtrl or shHOXA3. (F) The KDM6A promoter-reporter constructs were co-transfected with shCtrl or shHOXA3 vector into HEK293T cells. The promoter activities were presented. (G) Immunoblotting of HOXA3, KDM6A, HK2, and PKM2 in the indicated GBM cells expressing shCtrl, shHOXA3, or shHOXA3/KDM6A. (H) ChIP-qPCR analysis of H3K27me3 at HK2 and PKM2 promoter in GBM cells expressing shCtrl, shHOXA3, or shHOXA3/KDM6A. All data were shown as means and SD (n = 3), **P < .01.
The results demonstrated that HOXA3 activated the transcription of KDM6A and reduced H3K27me3 at HK2 and PKM2 promoters. Next, we reconstituted the expression of KDM6A in HOXA3 silencing cells and detected the expression of HK2 and PKM2. Unexpectedly, the expression levels of HK2 and PKM2 were not significantly recovered in KDM6A-restored cells (Figure 4G, Supplementary Figure S5D). Consistent with this result, the histone modification H3K27me3 at HK2 and PKM2 promoters was not decreased in KDM6A restoration cells (Figure 4H), suggesting that HOXA3 was essential for KDM6A to modify H3K27me3 at HK2 and PKM2 promoters.
HOXA3 Interacts With KDM6A to Enhance the Transcription of Glycolytic Genes
ChIP-qPCR showed pronounced levels of KDM6A at the promoters of HK2 and PKM2 (Supplementary Figure S6A). STRING dataset identified a potential interaction between HOXA3 and KDM6A, suggesting that HOXA3 cooperates with KDM6A to activate the transcription of glycolytic genes (Supplementary Figure S6B). The duolink-PLA assay was performed to determine whether HOXA3 could interact with KDM6A in GBM cells. The results showed significantly red dots caused by HOXA3 and KDM6A interaction (Figure 5A). The Co-IP results further confirmed that HOXA3 could interact with KDM6A, but not EZH2 and KDM6B in GBM cells (Figure 5B, Supplementary Figure S6C and D). In addition, exogenous HOXA3 coprecipitated with KDM6A from the lysate of HEK293T cells that expressed Flag-tagged HOXA3 and HA-tagged KDM6A (Figure 5C). Incubation of purified GST-tagged bacterially purified HOXA3 with the intracellular domain of KDM6A (His-KDM6A) proteins revealed that the 2 proteins directly interacted with each other (Figure 5D).
Figure 5.

HOXA3 directly interacts with KDM6A. (A) proximity ligation assay (PLA) assay of HOXA3-KDM6A expression in GBM cells using anti-HOXA3 and anti-KDM6A antibodies. (B) Immunoblot analysis of Co-IP of HOXA3 and KDM6A in extracts from LN229 and GBM01 cells. (C) Immunoblot analysis of Flag and HA in Flag-immunoprecipitated HOXA3 fragments from 293T cells expressing HOXA3-Flag and KDM6A-HA. (D) GST-pull-down analysis of the interaction between HOXA3 and KDM6A. (E) Immunoblot analysis of Co-IP of the interaction between different domains of HOXA3 and KDM6A. (F) Immunoblot analysis of Co-IP of the interaction between HOXA3 and different domains of KDM6A. (G) Molecular docking analysis of DUF domain of HOXA3 and TPR domain of KDM6A.
HOXA3 contains the Hbox domain and DUF domain, and DUF seems to function as a protein-binding domain by bioinformatics analysis. We generated different vectors coding the full HOXA3 sequence or the deletion sequences lacking either the Hbox domain or the DUF domain. The three different Flag-tagged constructs were transfected with His-tagged KDM6A into 293T cells. Co-IP assays revealed that KDM6A coimmunoprecipitated with deleted HOXA3 sequence, which contains the DUF domain but not the Hbox domain (Figure 5E). Besides this, we also generated different vectors coding the full KDM6A sequence or the deletion sequences lacking either TPR domain or JmjC domain, and transfected them with Flag-tagged HOXA3 into 293T cells. We found that HOXA3 coimmunoprecipitated with deleted KDM6A sequence, which contains TPR domain but not the JmjC domain (Figure 5F). Further molecular docking results demonstrated that the DUF domain of HOXA3 could directly interact with the TPR domain of KDM6A (Figure 5G). Promoter reporter assay showed that HOXA3 could cooperate with KDM6A to enhance the activities of HK2 and PKM2 promoters (Supplementary Figure S6E). All these results demonstrated that HOXA3 cooperates with KDM6A to enhance the expression of HK2 and PKM2.
HOXA3 Requires KDM6A for Transcriptional Activation of Aerobic Glycolysis and Brain Tumor Growth
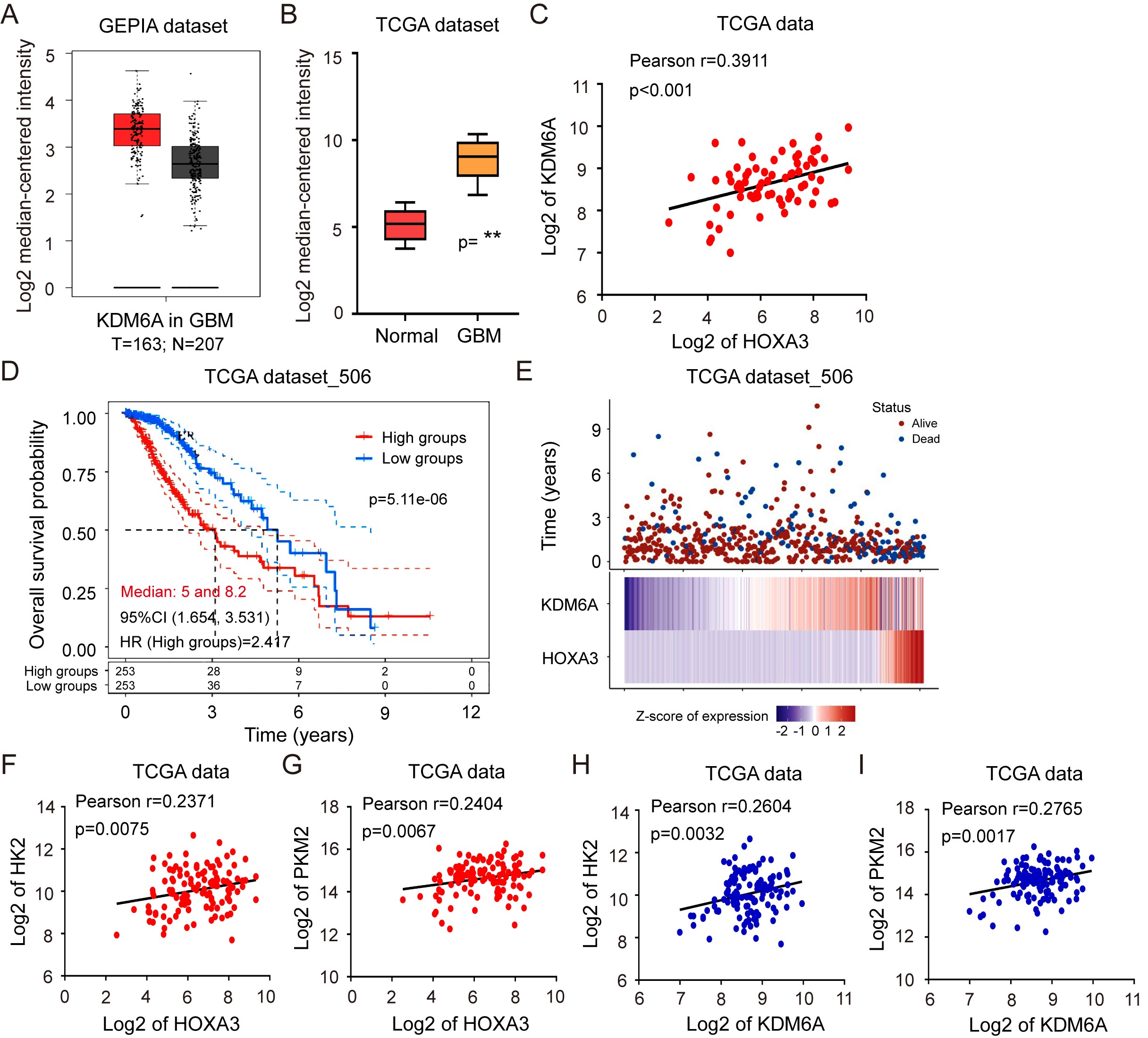
KDM6A was upregulated in GBM and positively correlated with HOXA3 expression according to TCGA data analysis (Supplementary Figure S7A–C). Furthermore, Glioma patients with high coexpression of HOXA3 and KDM6A revealed a poorer prognosis (Supplementary Figure S7D and E). Besides this, HOXA3 was positively correlated with HK2 and PKM2 in clinical GBM specimens (Supplementary Figure S7F and G). Meanwhile, KDM6A was positively correlated with HK2 and PKM2 in clinical GBM specimens (Supplementary Figure S7H and I). These results suggested that HOXA3 might require KDM6A to transcriptionally regulate aerobic glycolysis. In support of the above hypothesis, the knockdown of KDM6A expression significantly repressed the ability of HOXA3 to promote the expression of HK2 and PKM2 (Figure 6A). In line with these results, KDM6A silencing obviously reduced the ability of HOXA3 to promote aerobic glycolysis in GBM cells (Figure 6B).
Figure 6.

HOXA3 requires KDM6A for transcriptional activation of aerobic glycolysis and brain tumor growth. (A) Immunoblotting of HOXA3, KDM6A, HK2, and PKM2 in the indicated GBM cells expressing Ctrl, HOXA3, or HOXA3/shKDM6A. (B) Glycolytic rate of the indicated GBM cells expressing HOXA3 or HOXA3/shKDM6A. (C-D) ChIP-qPCR analysis of HOXA3, KDM6A, and H3K27me3 at HK2 (C) and PKM2 (D) promoter in GBM cells expressing HOXA3 or HOXA3/shKDM6A. (E) Orthotopic tumorigenesis abilities of LN229 cells expressing Ctrl, HOXA3, or HOXA3/shKDM6A and survival rates of mice (n = 6). (F) Model for HOXA3-KDM6A cooperation in transactivation of glycolysis and GBM progression. All data were shown as means and SD, **P < .01.
To gain the molecular mechanism of the cooperation, we investigated the possibility that HOXA3 might recruit KDM6A to the target gene promoters and remove the suppressive histone modification H3K27me3. We detected HK2 and PKM2 promoters, which were enriched with high levels of HOXA3 and KDM6A. As expected, in the HOXA3 overexpressed GBM cells, KDM6A silencing markedly reduced the levels of KDM6A but not HOXA3 at the promoters of HK2 and PKM2, with a corresponding increase in H3K27me3 levels (Figure 6C and D). Moreover, animal studies showed that KDM6A knockdown significantly inhibited the effect of HOXA3 to promote brain tumor growth and prolonged the mice’s survival (Figure 6E). Taken together, our results demonstrated that HOXA3 activated the transcription of KDM6A and recruited KDM6A to glycolytic gene promoters and remove H3K27me3 to promote aerobic glycolysis and GBM progression (Figure 6F).
Discussion
GBM, the most common aggressive and primary brain cancer, yields damaging clinical outcomes despite extensive searches for a therapeutic scheme. The poor prognosis, survival of about 14 months following initial diagnosis, lends an urgent need for a better understanding of the central players that contribute to the growth and malignancy of GBM.30 Here, we showed that HOXA3 was very highly expressed in human GBM and predicted poor prognosis of patients. Moreover, we demonstrated that HOXA3 promoted cell proliferation, cell cycle process, and tumor growth in GBM, consistent with the previous results that HOXA3 promoted cell proliferation in lymphoma and non-small-cell lung carcinoma.31,32 This finding provides HOXA3 as a potential therapeutic target for the effective antiproliferative treatment of GBM.
GBM undergoes metabolic reprogramming to fulfill the high energy and nutrient demands of tumor growth. A well-known metabolic reprogramming of tumor cells is the Warburg effect, in which tumor cells primarily initiate glycolysis to produce energy even in the case of sufficient oxygen.33There is increasing evidence that HOX proteins are also key regulators of aberrant glycolysis in the metabolic reprogramming of tumor cells. In our study, we found that HOXA3 silencing remarkably decreased glucose consumption, lactate production, and attenuated inhibitory rate of ATP and cell proliferation induced by ATP synthase inhibitor oligomycin, suggesting that HOXA3 leaded glucose metabolism towards aerobic glycolysis in GBM cells. Mechanically, HOXA3 activated the transcription of HK2 and PKM2 by binding to the HOXA motif of their promoter regions, which were essential for glycolysis and cell proliferation in GBM. Our findings suggested that HOXA3 promoted GBM growth by remodeling glucose metabolism.
GSEA of TCGA GBM patient data demonstrated that HOXA3 was positively associated with the methylation process. Our results revealed that HOXA3 knockdown significantly increased the H3K27me3 but not H3K4me3 and H3K9me3 levels at the promoters of HK2 and PKM2. The histone modification H3K27me3 is a transcriptional repressive factor and is regulated by histone methyltransferases and demethylases, including EZH2, KDM6A, and KDM6B.34 The histone H3K27M mutation frequently occurs in diffuse intrinsic pontine gliomas but rarely in GBM, leading to a global reduction of H3K27me3.35 In GBM, erasure of H3K27me3 might depend on the activity of KDM6A or KDM6B, but not H3 K27 mutant. The depletion of H3K27me3 promotes cell proliferation and epithelial-mesenchymal transition (EMT), but the role of H3K27me3 in metabolic reprogramming remains poorly understood. Here, we reported that HOXA3 activated the transcription of KDM6A and reduced H3K27me3 at HK2 and PKM2 promoters. Besides this, reconstituted the expression of KDM6A in HOXA3 silencing cells could not decrease H3K27me3 at HK2 and PKM2 promoter, suggesting that HOXA3 is essential for KDM6A to modify H3K27me3 at HK2 and PKM2 promoters.
KDM6A consists of a TPR protein–protein interaction domain (6-tetratricopeptide repeat) and a JmjC catalytic domain. KDM6A seems to require some cofactors to bind to genomic binding sites and modify the H3K27me3 as it is without a DNA binding domain. Previous study has demonstrated that HNF1A could recruit KDM6A to EMT genes to activate differentiated acinar cell programs.36 In our study, we showed that HOXA3 could directly interact with KDM6A and recruit KDM6A to genomic binding sites of glycolytic genes, targeting glycolytic genes for transcriptional activation by removing the suppressive histone modification H3K27 trimethylation. Of note, our data revealed that the DUF domain of HOXA3 could directly interact with the TPR domain of KDM6A. This interesting finding suggests that the DUF domain of HOXA3 functions as a protein–protein interaction domain. In addition, KDM6A silencing obviously reduced the ability of HOXA3 to promote glycolysis and tumor growth, suggesting that HOXA3 requires KDM6A for transcriptional activation of aerobic glycolysis and brain tumor growth.
In conclusion, our data present evidence for cooperation between HOXA3 and KDM6A in transcriptional regulation of glucose metabolism, providing a novel molecular mechanism linking HOXA3-mediated transactivation and KDM6A-coupled H3K27 demethylation in glucose metabolism and GBM development.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Contributor Information
Rui Yang, Institute of Precision Medicine, Jining Medical University, Jining 272067, China.
Guanghui Zhang, Cancer Center, Medical Research Institute, Southwest University, Chongqing 400716, China; State Key Laboratory of Silkworm Genome Biology, Southwest University, Chongqing 400716, China; Engineering Research Center for Cancer Biomedical and Translational Medicine, Southwest University, Chongqing 400716, China.
Zhen Dong, Cancer Center, Medical Research Institute, Southwest University, Chongqing 400716, China; State Key Laboratory of Silkworm Genome Biology, Southwest University, Chongqing 400716, China; Engineering Research Center for Cancer Biomedical and Translational Medicine, Southwest University, Chongqing 400716, China.
Shanshan Wang, Institute of Precision Medicine, Jining Medical University, Jining 272067, China.
Yanping Li, Institute of Precision Medicine, Jining Medical University, Jining 272067, China.
Fuming Lian, Institute of Precision Medicine, Jining Medical University, Jining 272067, China.
Xiaoran Liu, Institute of Precision Medicine, Jining Medical University, Jining 272067, China.
Haibin Li, Institute of Precision Medicine, Jining Medical University, Jining 272067, China.
Xiaonan Wei, Institute of Precision Medicine, Jining Medical University, Jining 272067, China.
Hongjuan Cui, Cancer Center, Medical Research Institute, Southwest University, Chongqing 400716, China; State Key Laboratory of Silkworm Genome Biology, Southwest University, Chongqing 400716, China; Engineering Research Center for Cancer Biomedical and Translational Medicine, Southwest University, Chongqing 400716, China.
Funding
This work was supported by National Nature Science Foundation of China (No. 82002639 to R.Y., 82101867 to S.W., 82002961 to Y.L.), Shandong Provincial Natural Science Foundation, China (No. ZR2020QH235 to R.Y.), and Faculty Start-up Funds from Jining Medical University (to R.Y.).
Conflict of Interest
The authors declare no competing interests.
Authorship Statement
R.Y. contributed to the design of the work, the performance of experiments, interpretation of data, and writing the paper. H.C. contributed to the design of the work and interpretation of data. G.Z., S.W., Y.L., X.L., and X.W. contributed to performance of experiments. Z.D. and F.L. contributed to interpretation of data.
References
- 1. Deschamps J, Duboule D. Embryonic timing, axial stem cells, chromatin dynamics, and the Hox clock. Genes & Dev. 2017;31(14):1406–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shah N, Sukumar S. The Hox genes and their roles in oncogenesis. Nat Rev Cancer. 2010;10(5):361–371. [DOI] [PubMed] [Google Scholar]
- 3. Zhu H, Dai W, Li J, et al. HOXD9 promotes the growth, invasion and metastasis of gastric cancer cells by transcriptional activation of RUFY3. J Exp Clin Cancer Res. 2019;38(1):412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carrabotta M, Laginestra MA, Durante G, et al. Integrated molecular characterization of patient-derived models reveals therapeutic strategies for treating CIC-DUX4 sarcoma. Cancer Res. 2022;82(4):708–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen YQ, Yang TQ, Zhou B, et al. HOXA5 overexpression promotes osteosarcoma cell apoptosis through the p53 and p38α MAPK pathway. Gene. 2019;689:18–23. [DOI] [PubMed] [Google Scholar]
- 6. Zhou L, Wang Y, Zhou M, et al. HOXA9 inhibits HIF-1α-mediated glycolysis through interacting with CRIP2 to repress cutaneous squamous cell carcinoma development. Nat Commun. 2018;9(1):1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jiang Y, Yan B, Lai W, et al. Repression of Hox genes by LMP1 in nasopharyngeal carcinoma and modulation of glycolytic pathway genes by HoxC8. Oncogene. 2015;34(50):6079–6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Venneti S, Thompson CB. Metabolic reprogramming in brain tumors. Annu Rev Pathol. 2017;12:515–545. [DOI] [PubMed] [Google Scholar]
- 9. Dong Z, Cui H. Epigenetic modulation of metabolism in glioblastoma. Semin Cancer Biol. 2019;57:45–51. [DOI] [PubMed] [Google Scholar]
- 10. Wood A, Shilatifard A. Posttranslational modifications of histones by methylation. Adv Protein Chem. 2004;67:201–222. [DOI] [PubMed] [Google Scholar]
- 11. Bao L, Chen Y, Lai HT, et al. Methylation of hypoxia-inducible factor (HIF)-1α by G9a/GLP inhibits HIF-1 transcriptional activity and cell migration. Nucleic Acids Res. 2018;46(13):6576–6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Heddleston JM, Wu Q, Rivera M, et al. Hypoxia-induced mixed-lineage leukemia 1 regulates glioma stem cell tumorigenic potential. Cell Death Differ. 2012;19(3):428–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pang B, Zheng XR, Tian JX, et al. EZH2 promotes metabolic reprogramming in glioblastomas through epigenetic repression of EAF2-HIF1α signaling. Oncotarget. 2016;7(29):45134–45143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ene CI, Edwards L, Riddick G, et al. Histone demethylase Jumonji D3 (JMJD3) as a tumor suppressor by regulating p53 protein nuclear stabilization. PLoS One. 2012;7(12):e51407e51407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hu J, Sun T, Wang H, et al. MiR-215 Is induced post-transcriptionally via HIF-drosha complex and mediates glioma-initiating cell adaptation to hypoxia by targeting KDM1B. Cancer Cell. 2016;29(1):49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Paço A, Freitas R. HOX genes as transcriptional and epigenetic regulators during tumorigenesis and their value as therapeutic targets. Epigenomics. 2019;11(13):1539–1552. [DOI] [PubMed] [Google Scholar]
- 17. Lan F, Bayliss PE, Rinn JL, et al. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature. 2007;449(7163):689–694. [DOI] [PubMed] [Google Scholar]
- 18. Chen X, Lin X, Pang G, et al. Significance of KDM6A mutation in bladder cancer immune escape. BMC Cancer. 2021;21(1):635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gao YB, Chen ZL, Li JG, et al. Genetic landscape of esophageal squamous cell carcinoma. Nat Genet. 2014;46(10):1097–1102. [DOI] [PubMed] [Google Scholar]
- 20. Jung SH, Kim SY, An CH, et al. Clonal structures of regionally synchronous gastric adenomas and carcinomaS. Clin Cancer Res. 2018;24(19):4715–4725. [DOI] [PubMed] [Google Scholar]
- 21. Chen J, Gao XM, Zhao H, et al. A highly heterogeneous mutational pattern in POEMS syndrome. Leukemia. 2021;35(4):1100–1107. [DOI] [PubMed] [Google Scholar]
- 22. Liu L, Cui J, Zhao Y, et al. KDM6A-ARHGDIB axis blocks metastasis of bladder cancer by inhibiting Rac1. Mol Cancer. 2021;20(1):77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu Q, Tian Y, Zhang J, et al. In vivo CRISPR screening unveils histone demethylase UTX as an important epigenetic regulator in lung tumorigenesis. Proc Natl Acad Sci USA. 2018;115(17):E3978–E3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Benyoucef A, Palii CG, Wang C, et al. UTX inhibition as selective epigenetic therapy against TAL1-driven T-cell acute lymphoblastic leukemia. Genes Dev. 2016;30(5):508–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xie G, Liu X, Zhang Y, et al. UTX promotes hormonally responsive breast carcinogenesis through feed-forward transcription regulation with estrogen receptor. Oncogene. 2017;36(39):5497–5511. [DOI] [PubMed] [Google Scholar]
- 26. Zhang C, Zhang X, Xu R, et al. Retraction Note: TGF-β2 initiates autophagy via Smad and non-Smad pathway to promote glioma cells’ invasion. J Exp Clin Cancer Res. 2021;40(1):399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang R, Li X, Wu Y, et al. EGFR activates GDH1 transcription to promote glutamine metabolism through MEK/ERK/ELK1 pathway in glioblastoma. Oncogene. 2020;39(14):2975–2986. [DOI] [PubMed] [Google Scholar]
- 28. Li Y, Liang R, Sun M, et al. AMPK-dependent phosphorylation of HDAC8 triggers PGM1 expression to promote lung cancer cell survival under glucose starvation. Cancer Lett. 2020;478:82–92. [DOI] [PubMed] [Google Scholar]
- 29. Yang R, Wang M, Zhang G, et al. POU2F2 regulates glycolytic reprogramming and glioblastoma progression via PDPK1-dependent activation of PI3K/AKT/mTOR pathway. Cell Death Dis. 2021;12(5):433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ostrom QT, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2014-2018. Neuro Oncol. 2021;23( suppl 212):iii1–iii105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang L, Sui M, Wang X. miR‑338‑3p suppresses the malignancy of T-cell lymphoblastic lymphoma by downregulating HOXA3. Mol Med Rep. 2019;20(3):2127–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lin S, Zhang R, An X, et al. LncRNA HOXA-AS3 confers cisplatin resistance by interacting with HOXA3 in non-small-cell lung carcinoma cells. Oncogenesis. 2019;8(11):60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhou W, Wahl DR. Metabolic abnormalities in glioblastoma and metabolic strategies to overcome treatment resistance. Cancers. 2019;11(9):1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. He C, Sun J, Liu C, Jiang Y, Hao Y. Elevated H3K27me3 levels sensitize osteosarcoma to cisplatin. Clin Epigenetics. 2019;11(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Louis DN, Perry A, Wesseling P, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro oOncol. 2021;23(8):1231–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kalisz M, Bernardo E, Beucher A, et al. HNF1A recruits KDM6A to activate differentiated acinar cell programs that suppress pancreatic cancer. EMBO J. 2020;39(9):e102808. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.