

Abstract
Lung cancer is the leading cause of cancer-related deaths. Lung cancer cells develop resistance to apoptosis by suppressing the secretion of the tumor suppressor Par-4 protein (also known as PAWR) and/or down-modulating the Par-4 receptor GRP78 on the cell surface (csGRP78). We sought to identify FDA-approved drugs that elevate csGRP78 on the surface of lung cancer cells and induce Par-4 secretion from the cancer cells and/or normal cells in order to inhibit cancer growth in an autocrine or paracrine manner. In an unbiased screen, we identified crizotinib (CZT), an inhibitor of activated ALK/MET/ROS1 receptor tyrosine kinase, as an inducer of csGRP78 expression in ALK-negative, KRAS or EGFR mutant lung cancer cells. Elevation of csGRP78 in the lung cancer cells was dependent on activation of the non-receptor tyrosine kinase SRC by CZT. Inhibition of SRC activation in the cancer cells prevented csGRP78 translocation but promoted Par-4 secretion by CZT, implying that activated SRC prevented Par-4 secretion. In normal cells, CZT did not activate SRC and csGRP78 elevation but induced Par-4 secretion. Consequently, CZT induced Par-4 secretion from normal cells and elevated csGRP78 in the ALK-negative tumor cells to cause paracrine apoptosis in cancer cell cultures and growth inhibition of tumor xenografts in mice. Thus, CZT induces differential activation of SRC in normal and cancer cells to trigger the pro-apoptotic Par-4-GRP78 axis. As csGRP78 is a targetable receptor, CZT can be repurposed to elevate csGRP78 for inhibition of ALK-negative lung tumors.
Keywords: Par-4, csGRP78, crizotinib, lung cancer, SRC, apoptosis
Introduction
Lung cancer is a leading cause of cancer-related deaths in the world [1]. A majority (80-85%) of the lung cancers are classified as non-small cell lung cancers (NSCLCs) that include adenocarcinoma, squamous cell carcinoma, and large cell carcinoma; about 10-15% are small cell lung cancers; and fewer than 5% are lung carcinoid tumors. Oncogenic mutations in KRAS, EGFR, ALK, MET, ROS1, PI3KCA, or BRAF are the key drivers of NSCLC [2]. Lung tumors initially respond to chemotherapy, but resistance rapidly develops in most patients [3,4].
Recent studies have indicated that cell-surface GRP78 (csGRP78) is a targetable receptor in cancer cells [5-11]. GRP78 resides primarily in the endoplasmic reticulum (ER) in normal cells where it plays an essential role in protein folding and cell survival [6-8]. In cancer cells, however, GRP78 is aberrantly localized to the cell surface [5-11]. This differential aberrant location on the cancer cell surface allows csGRP78 to bind ligands that will trigger downstream cell signaling pathways, including proliferation, survival, and apoptosis [10,11]. It is particularly interesting that csGRP78 operates as a receptor for α2-macroglobulin that induces tumor growth and survival [6]. The elevated expression of csGRP78 is associated with cancer stemness and metastasis [5,10]. Death-inducing natural ligands such as the tumor suppressor Prostate apoptosis response-4 (Par-4 aka PAWR) or Kringle 5, a proteolytic fragment of human plasminogen, can bind to csGRP78 and trigger cancer cell death [6,7,9,11]. This latter process led us to study mechanisms for suppressing NSCLCs using ligands that take advantage of the appearance of csGRP78 on the surfaces of these cancer cells.
Par-4 is a ubiquitously expressed leucine zipper containing protein [12]. Par-4 expression is compromised in cancer cells by diverse mechanisms, including inactivation by AKT cell survival kinase-mediated phosphorylation, methylation-dependent suppression of the Par-4 promoter, or spontaneous mutation [13-16]. Loss of Par-4 in breast cancer or glioblastoma results in therapy resistance, cancer recurrence and a decline in patient survival [17-19]. Genetic knockout of Par-4 in mice results in obesity within 3-4 months [20]. Consistent with its role as a tumor-suppressor, spontaneous tumors develop in diverse organs as Par-4 knockout mice grow older [21]. However, overexpression of Par-4 induces apoptosis in cancer cell lines, but not normal cells, and Par-4 transgenic mice exhibit a normal life span and cancer-free survival [22].
Par-4 protein functions intracellularly in the cytoplasm or nucleus, or is secreted and functions extracellularly to induce cancer cell apoptosis [12]. Our previous studies have indicated that Par-4 is secreted by normal cells via the classical ER-Golgi secretory pathway, and extracellular Par-4 binds to GRP78 on the cancer cell surface to trigger apoptosis by activation of the FADD-caspase-8-caspase-3 pathway [7]. Except for a few normal cell types such as endothelial cells and monocytes, the levels of csGRP78 in most normal cells, including normal/immortalized lung epithelial and fibroblast cells, are low and such cells are therefore resistant to the action of csGRP78 targeting killer ligands including secreted Par-4 [5-9,11].
Some lung tumors that initially respond to therapy subsequently develop treatment resistance [3,4]. Because resistance to lung cancer treatment represents an important, unsolved problem [2-4], and because csGRP78 in cancer cells is a promising and actively pursued targetable receptor [5-9,11], we are interested in identifying drugs that induce csGRP78 and render both therapy-sensitive and therapy-resistant cells vulnerable to the action of extracellular Par-4. We present evidence that crizotinib (CZT), a first-generation inhibitor of activated ALK/MET/ROS1 [23-26], paradoxically activates the non-receptor tyrosine kinase SRC in lung cancer cells that lack active ALK/MET/ROS1 (herein referred to as ALK-negative), to induce csGRP78 translocation and susceptibility to Par-4. Interestingly, SRC activated by CZT inhibits Par-4 secretion. CZT activates SRC in cancer cells but not normal cells, and therefore induces Par-4 secretion but does not elevate csGRP78 in normal cells. This differential regulation of SRC by CZT results in induction of Par-4 secretion from normal cells and csGRP78 elevation in cancer cells leading to paracrine growth inhibition of ALK-negative cancer cell cultures and xenografts in mice.
Methods
Cells
Human lung cancer cells A549, and prostate cancer cells LNCaP, mouse lung cancer cells LLC-1, and human lung fibroblast cells HEL and epithelial cells BEAS-2B were from ATCC, Gaithersburg, MD (Table S1). Human lung cancer cells H460, NCI-H1975 (H1975), H1299, were from Carla F. Kim (Harvard Medical School); NCI-H1650 (H1650), H2030, and H2009 were from Christine F. Brainson (University of Kentucky) authenticated by IDEXX CellCheck9 (North Grafton, MA). Parent A549 and their paclitaxel-resistant derivative A549TR cells were previously described [27]. Mouse KP7B lung adenocarcinoma tumor cells, derived from KRAS mutant and p53-null lung tumors that were induced by the Cre-adenovirus in C57BL/6 KRASG12D/+; p53-/- mice, were from Professor Tyler Jacks’ laboratory [28]. KP7B tumors maintain the disease relevance of the lung cancer model. Par-4+/+ and Par-4-/- mouse embryonic fibroblasts (MEFs) were from wild type and Par-4-null C57BL/6 mouse embryos [20]. Mouse adult lung fibroblasts (ALF) were isolated from C57BL/6 wild type adult mouse lungs, and p53-/- MEFs were from p53-null C57BL/6 mouse embryos [20]. The cell lines were authenticated as previously described [27].
Chemicals, recombinant proteins, and siRNA
The FDA-approved library of 1400+ compounds (L1300) was purchased from Selleck Chemicals LLC (Houston, TX). Brefeldin A (BFA) and other chemicals were purchased from Sigma Aldrich (St. Louis, MO) or Fisher Scientific (Hampton, NH) or were synthesized according to literature procedures. Purified His-Par-4 was prepared as described previously using the baculoviral protein expression system (GenScript, NJ) [27].
Chemicals were purchased from either Millipore Sigma (St. Louis, MO) or Fisher Scientific (Hampton, NH) unless otherwise specified. Solvents were purchased from commercial vendors and used without further purification unless otherwise noted. Proton nuclear magnetic resonance spectra were acquired on a Varian instrument at 400 MHz. Compounds were chromatographed on preparative layer Merck silica gel F254 (Fisher Scientific) plates unless otherwise indicated.
Preparation of N-(2-(2-(3-(4-(4-(6-amino-5-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)pyridin-3-yl)-1H-pyrazol-1-yl)piperidin-1-yl)-3-oxopropoxy)ethoxy)ethyl)-5-(2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanamide (Biotinylated-CZT). To a solution of 60 mg (0.15 mmol) of 3-(2-(2-(5-(2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanamido)ethoxy)ethoxy)propanoic acid in 1 mL of N,N-dimethylformamide (DMF) was added successively 30 mg (0.223 mmol, 1.5 eq) of 1-hydroxybenzotriazole (HOBt), 43 mg (0.223 mmol, 1.5 eq) of N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride, 67 mg (0.15 mmol, 1 eq) of Crizotinib {3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-[1-(piperidin-4-yl)-1H-pyrazol-4-yl]-pyridin-2-amine} and 30 mg (0.3 mmol, 2 eq) of triethylamine. The mixture was stirred for 15 h at 25°C, poured into water, extracted with dichloromethane, dried over magnesium sulfate and concentrated. The crude product was purified by chromatography using 1:10 methanol-dichloromethane (Rf 0.3) to provide 74 mg (59%) of Biotinylated-CZT. 1H NMR (400 MHz, CDCl3) δ 7.74 (d, J = 1.8 Hz, 1H), 7.54 (d, J = 0.8 Hz, 1H), 7.52 (d, J = 4.7 Hz, 1H), 7.30 (dd, J = 8.9, 4.8 Hz, 1H), 7.05 (dd, J = 8.9, 7.9 Hz, 1H), 6.86 (d, J = 1.9 Hz, 1H), 6.83-7.76 (m, 1H), 6.40 (br.s, 1H), 6.06 (q, J = 6.7 Hz, 1H), 5.44-5.33 (m, 1H), 4.89 (d, J = 4.9 Hz, 2H), 4.71 (d, J = 13.6 Hz, 1H), 4.53-4.41 (m, 1H), 4.39-4.19 (m, 2H), 4.04 (d, J = 14.1 Hz, 1H), 3.82 (t, J = 6.4 Hz, 1H), 3.67-3.56 (m, 4H), 3.54 (t, J = 4.9 Hz, 2H), 3.45-3.36 (m, 2H), 3.21 (t, J = 12.9 Hz, 1H), 3.16-3.05 (m, 1H), 2.93-2.83 (m, 1H), 2.80 (t, J = 12.7 Hz, 1H), 2.74-2.52 (m, 3H), 2.28-2.10 (m, 4H), 2.08-1.87 (m, 4H), 1.85 (d, J = 6.7 Hz, 3H), 1.76-1.57 (m, 3H), 1.48-1.30 (m, 2H).
Antibodies and siRNA duplexes
The Par-4 rabbit polyclonal antibody was previously described by us [20]. GRP78 (E-4; sc-166490) antibody, GAPDH antibody (G-9; sc365062), pan-cytokeratin antibody (AE1/AE3; sc-81714) and c-SRC siRNA (human) (sc-29228) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). We purchased 4, 6-diamidino-2-phenylindole (DAPI) from Vector Laboratories, Inc., Newark, CA. Active caspase 3 antibody (D175; 9661S), Csk (C74C1; 4980S), Src (2108S), p-Src (Y416; 2101S) antibodies were from Cell Signaling Technology, Danvers, MA. The Alexa Fluor 488 conjugated goat anti-mouse secondary antibody (A55058) was purchased from Invitrogen Corporation, Waltham, MA. The β-actin antibody (A5315) was purchased from Sigma Chemical Corp. On-Targetplus human HSPA5 (GRP78) siRNA SMARTPool (L-0081198-00-0005) was purchased from Dharmacon Reagents (Horizon, Lafayette, CO).
Cell surface GRP78 detection
Cells were examined for csGRP78 as previously described [7]. Briefly, the cells were exposed to the indicated treatments and subjected without fixing to the GRP78 primary antibody (E-4) or IgG control antibody (mouse IgG isotype control 10400C; Invitrogen), followed by Alexa Fluor 488-conjugated secondary antibody. The cells were then scored for cell surface immunofluorescence under a confocal microscope or by fluorescence activated cell sorting (FACS) analysis at the Markey Cancer Center Flow Cytometry and Immune Monitoring Shared Resource Facility.
Apoptosis and cell viability assays
Cancer cell viability was measured in blinded experiments using resazurin (R7017 Sigma-Aldrich). The cells were seeded in 96-well plates and after 24 h or the indicated duration of treatment, the resazurin assay was performed as per the manufacturer’s instructions. The absorbance of the dye at 490 nm was measured using a microplate reader.
Apoptotic cells were identified by immunocytochemical (ICC) analysis for active caspase-3, and apoptotic nuclei were detected by DAPI staining. A total of three independent experiments were performed; and 100-500 cells were scored in each experiment for apoptosis using a fluorescence microscope.
Screening for drugs that activate critical components of the Par-4-csGRP78 pathway
Using the Selleck 1400+ drug library we performed a two-step screen for compounds that induced cell death on their own and/or elevated csGRP78 expression thereby rendering the target cells susceptible to extracellular Par-4. In the first step, the compounds from the library were tested in 10 μM amounts to identify drugs that induce growth inhibition of ALK-negative lung cancer cells A549, A549TR, and KP7B on their own. The top-ranking candidate drug in this screen was CZT. We confirmed with resazurin assays that CZT induced growth inhibition in these ALK-negative lung cancer cells at 10 μM concentration (Figure S1A). At this concentration, CZT was expected to cause growth inhibition either by Par-4- and csGRP78-dependent or independent mechanisms. To identify a subset of the drugs that elevated csGRP78 and promoted growth inhibition by Par-4- and csGRP78- dependent mechanisms, we included a second step of screening for Par-4 secretion, csGRP78 elevation, and apoptosis response in the presence or absence of recombinant Par-4 (100 nM). This second step in the screen used CZT at 10 μM as well as 1 μM, which reflects the amounts available in the plasma of patients treated with the drug. CZT at 10 μM but not at 1 μM induced Par-4 secretion from the lung cancer cells (Figure S1B). CZT (10 μM as well as 1 μM or 0.5 μM), but not rapamycin, which inhibits mTOR and induces autophagy [29], elevated csGRP78 expression in the ALK-negative lung cancer cells (Figure S1C, S1D). However, CZT at 1 μM sensitized the lung cancer cells to apoptosis by recombinant Par-4 (His-Par-4) (Figure S1E).
Pull down experiments
To identify the target protein for compound CZT, pull-down experiments were performed as described by us previously and subjected to western blot [30].
Animal experiments
NOD-Scid gamma IL2R-deleted (NSG) mice (from Jackson Laboratory, Bar Harbor, ME, at 8 weeks of age) were injected through the subcutaneous (s.c.) route with 2 × 106 human cancer cells. KP7B cells (2 × 106 cells) were injected s.c. into Par-4+/+ mice and Par-4-/- mice. When the tumors reached a volume of 30 mm3, the animals were administered CZT (25 mg/kg) or vehicle (as a control) by oral gavage. The gavage solution contained PEG400, Vitamin E TPGS (D-alpha-Tocopherol polyethylene glycol 1000 succinate), Poloxamer 407, PBS-1x in a ratio of 25:10:1:64. The tumors were measured using Vernier calipers every 2-3 days for period of up to 20 days. Mouse plasma samples were collected at the end of each experiment and subjected to western blot analyses. The animal procedures were performed in compliance with University of Kentucky IACUC approval.
Computational modeling and methods
Molecular modeling of CZT binding was performed using MOE and VinaMPI, a parallelized docking engine for Autodock Vina. The list of protein ascensions and other general procedures were essentially the same as in our previous computational studies for carrying out the Molecular Dynamics (MD) simulations [30-34]. Briefly, CZT was docked using VinaMPI into the binding cavities identified by MOE’s Site Finder tool and the resulting poses were refined by MD simulations. The predicted targets were c-SRC, TYK2, JAK2, PDGFRA, FGFR2, CSK, and ALK. Binding predictions for the human tyrosine kinase c-SRC (PDB code 2SRC) were further evaluated for an improved prediction of binding pose and energetics. This was done by performing an energy minimization of the predicted bound conformation from Vina docking, followed by 300 ps of dynamics and another stage of energy minimization where the protein was held fixed. The simulations were performed in MOE using the amber forcefield. The average interaction potential energy, in kcal/mol, was collected over the trajectories and used to rank binding poses. Also, the final minimized structure was used to collect a binding energy from the MOE docking tool without using a placement algorithm (i.e., the minimized ligand pose was used as the docked structure and just a score was collected).
Statistical analysis
We performed the experiments in triplicate to verify the reproducibility of these observations and summarized the data as the mean of at least 3 experiments + Standard Deviation (SD). The Statistical Analysis System software (SAS Institute, Cary, NC) was used for statistical analyses. The P values were calculated as indicated either using the Student’s t test or the two-way ANOVA model with data normality and equality of variance assumptions was employed to examine the effect of interaction between two different treatments.
Results
CZT induces dose-dependent elevation of csGRP78 in ALK-negative cancer cells but not in normal/immortalized lung cells
Mutations in V-Ki-ras2 Kirsten rat sarcoma viral oncogene homologue (KRAS) are found in ~30% of patients with newly diagnosed NSCLC [2]. We therefore used KRAS-mutant A549 NSCLC cells that are resistant to apoptosis by extracellular Par-4 [35], A549TR cells that are taxane-resistant derivatives of A549 cells [27], and KP7B cells [27] as target cells to identify drugs that would elevate csGRP78 and thereby sensitize the cells to the action of secreted Par-4. An unbiased screen of an FDA-approved drug library (see Methods) for such compounds led to the identification of CZT as the top-ranked candidate. CZT is best known as an inhibitor of activated ALK/MET/ROS1 receptor tyrosine kinases and is a standard-of-care for patients with ALK+ lung tumors [23,24]. When tested for elevation of csGRP78 expression at 1 μM concentration that matches the level found in the plasma of CZT-treated patients [36], CZT induced elevation of csGRP78 in ALK-negative cancer cells that expressed mutant-KRAS (A549, A549TR, KP7B) or NRAS (H1299) but not in normal/immortalized lung cells (BEAS-2B, HEL) (Figures S1B and 1A). As CZT can also inhibit activated MET, we tested whether savolitinib (SAV), an authentic inhibitor of MET, upregulated csGRP78 expression. These studies included KRAS mutant cells (A549TR, H1299 and H460), as well as EGFR-mutant NSCLC cells (H1650 and H1975), because EGFR-mutations are found in about 30% of NSCLCs [2]. As seen in Figure 1B, CZT but not SAV induced csGRP78 expression in all the KRAS-mutant cells, as well as EGFR-mutant/wild type KRAS lung cancer cell lines that were tested. Co-treatment with CZT sensitized lung cancer cells to apoptosis by recombinant Par-4 protein, implying that CZT induces csGRP78 that is functionally active in inducing downstream signaling (Figure S1E). Moreover, tunicamycin (TU), which is known to prevent N-linked glycosylation of proteins and cause ER-stress, elevated csGRP78 (Figure S1F), implying a companion role for ER stress in this process.
Figure 1.
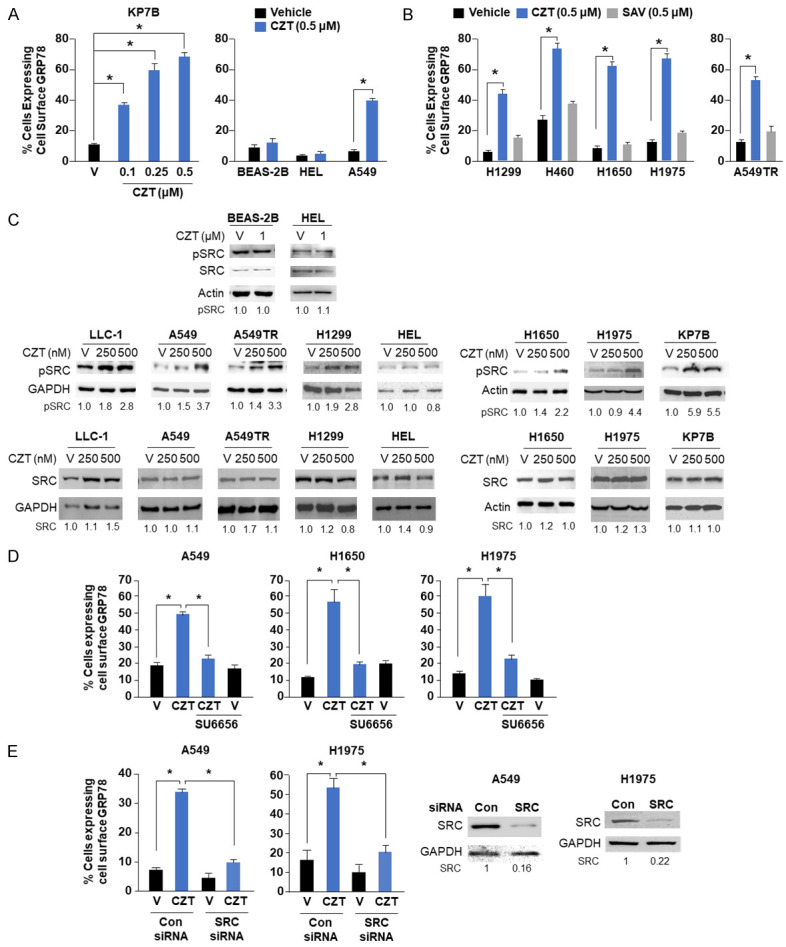
CZT induces csGRP78 in cancer cells by SRC kinase-dependent mechanism. A. CZT induces csGRP78 in cancer cells but not in normal cells. Lung cancer KP7B and A549 cells expressing mutant-Ras, and normal/immortalized lung epithelial BEAS-2B or fibroblasts HEL cells were treated with the indicated concentrations of CZT or vehicle. Cells were analyzed for csGRP78 as indicated in Methods. B. CZT but not the Met-inhibitor savolitinib (SAV) induces csGRP78 in cancer cells. Lung cancer cells with mutant-Ras (H1299 and H460) or mutant-EGFR (H1650 and H1975) were treated with 500 nM CZT, SAV, or vehicle (V). C. CZT induces phosphorylation of SRC in cancer cells, but not in normal/immortalized cells. Lung cancer cells (LLC-1, H1650, H1975, A549, A549TR, H1299) or normal/immortalized lung cells (BEAS-2B, HEL) were treated with the indicated concentrations of CZT or vehicle (v) for 24 h. Whole-cell lysates were subjected to western blot analysis for pY419-SRC (pSRC), total SRC, and GAPDH or actin as loading control. Fold change in pSRC and total SRC levels in response to CZT relative to vehicle is shown. D. SU6656 inhibits GRP78 transport to the cell surface. A549, H1650 or H1975 cells were treated with 1 µM CZT or vehicle in the presence or absence of SRC-inhibitor SU6656 (5 µM) for 24 h. E. Knockdown of SRC inhibits cell surface transport of GRP78. A549 or H1975 cells were treated with SRC-siRNA or control (CON)-siRNA for 24 h and then treated with 500 nM CZT or vehicle for 24 h (Left Panels). Knockdown of SRC was confirmed by western blot analysis (Right Panels). A, B, D and E. Percentage of csGRP78-positive cells was calculated and Mean + SD values are shown. *P < 0.001 by Student’s t test.
CZT activates SRC by an ER-stress independent mechanism
A recent study has suggested that GRP78 translocation to the cell surface is associated with the activation of SRC in some but not all cancer cell lines [37]. Phosphorylation of SRC at tyrosine 530 (Y530) by C-terminal SRC kinase (Csk) and Csk homologous kinase results in inactivation of SRC [38,39]. Dephosphorylation of Y530 by phosphatases results in autophosphorylation of SRC at Y419, leading to its activation [40,41]. To determine whether SRC activation is essential for CZT to translocate GRP78 to the cell surface, we tested the effect of CZT on SRC activation. Cancer cells, as well as normal/immortalized cells were treated with CZT or vehicle, and the levels of Y419 phosphorylation, as an indicator of SRC activation, were determined. As seen in Figure 1C, CZT elevated Y419 phosphorylation levels of SRC in cancer cells that are ALK-negative but not in normal/immortalized cells. Several other ALK-inhibitors, namely brigatinib, alectinib (second generation) and lorlatinib (third generation) [42], but not the ER-stress inducing agent TU, induced phosphorylation at Y419 in SRC (Figure S1G). As expected, the other ALK-inhibitors brigatinib and alectinib induced csGRP78 expression (Figure S1H). Moreover, the SRC-inhibitor SU6656 or siRNA mediated inhibition of SRC blocked CZT-induced translocation of GRP78 to the cell surface (Figure 1D, 1E). This outcome indicated that SRC activation is essential for csGRP78 elevation by CZT. These findings suggested that CZT activated SRC, and that SRC activation is essential for translocation of csGRP78 by CZT.
Role of ER-stress in GRP78 transport to the cell surface
As the ER-stress inducer TU failed to elevate Y419 phosphorylation levels of SRC, we further tested whether ER stress played a role in SRC activation and csGRP78 elevation by CZT. These experiments used 4-phenylbutyrate (4PB) to block the unfolded protein response and ER-stress [43]. We found that CZT-inducible csGRP78 was blocked by 4PB (Figure S2A). 4PB did not block pY419-SRC induction by CZT (Figure S2B). These findings imply that CZT-induced ER-stress is essential for csGRP78 elevation and that SRC activation by CZT may occur upstream of ER-stress in the pathway leading to cell surface translocation of GRP78.
CZT targets SRC
To determine the direct target of CZT in cancer cells, we used a molecular modeling approach followed by pull-down of the potential target proteins with a biologically active biotinylated analog of CZT. Molecular docking using the AutoDock Vina and MOE programs led to the prediction of SRC as a potential target of CZT (Figure 2A). Each of the possible binding modes identified by the docking scores and proximity to the ATP binding site, where competitive binding occurs, or binding near Y530 of SRC was used for rigorous binding calculations as described in Methods. The computationally modeled SRC-CZT binding structure revealed intermolecular interactions between CZT and SRC (Figure 2A). As SRC was found to be activated by CZT, we focused on SRC in pull-down studies.
Figure 2.
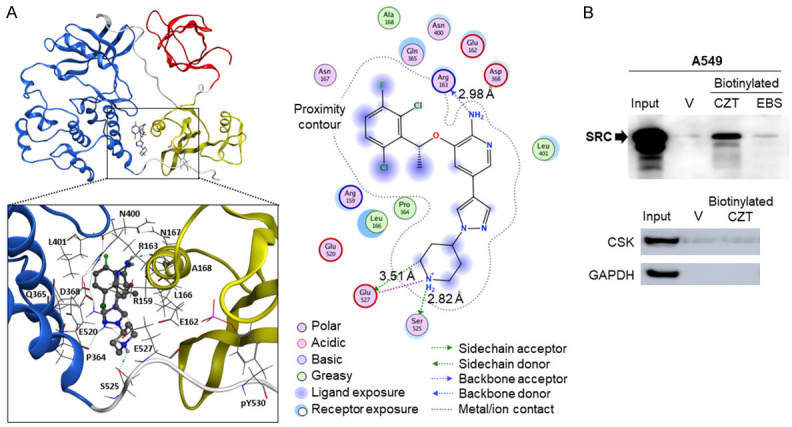
CZT binds to SRC. A. CZT binding (dashed circle) to SRC. CZT binds near the C-terminal domain of SRC and may disrupt the inactive state by preventing phosphorylation on the nearby tyrosine. Key interactions among CZT and SRC residues are shown. B. Pull-down experiments indicate that CZT binds to SRC but not to CSK. Whole-cell lysates of A549 cells were incubated with vehicle, biotinylated-CZT or biotinylated-ebastine (EBS), as a negative control, and streptavidin beads. Bound proteins were eluted from the beads with D-biotin. Eluates were subjected to western bot analysis with SRC antibody (Top Panel), or with CSK and GAPDH control antibody (Bottom Panel).
We generated a biotinylated-CZT analog and confirmed that it induces csGRP78 (Figure S3). In pull-down studies (described in Methods), we identified SRC as a target of CZT (Figure 2B). As indicated above, CZT binds to SRC and induces SRC activation, as evidenced by pY419 phosphorylation. By contract, CZT did not bind to CSK (Figure 2B), implying that SRC activation was associated with binding of CZT to SRC and not binding and sequestration of CSK by CZT.
CZT induces Par-4 secretion in normal cells but not in cancer cells
The original strategy was to combine CZT, which upregulated csGRP78 in cancer cells, with chloroquine (CQ), which causes secretion of Par-4 from normal cells [44], as a combination therapy for inhibiting tumor growth in mice. Prior to conducting the mouse experiments, we sought to rule out the possibility of Par-4 secretion triggered by CZT alone as it could potentially confound the findings with the CZT and CQ combination. To determine whether CZT induces Par-4 secretion, we treated various normal cells and cancer cells with CZT and examined the cell culture conditioned medium (CM) for secreted Par-4. As seen in Figure 3A, CZT alone induced Par-4 secretion in normal cells but not in cancer cells. Moreover, treatment of normal C57BL/6 mice that did not carry any tumors with CZT resulted in increased secretion of Par-4 in the plasma (Figure 3A). As CZT activated SRC in cancer cells but not in normal cells, we tested whether activated SRC prevented Par-4 secretion in cancer cells. Various ALK-negative cancer cell lines were treated with CZT in the presence or absence of the SRC inhibitor SU6656 and the CM was tested for secreted Par-4. These experiments indicated that SRC inhibition restored Par-4 secretion by CZT in the cancer cells (Figure S4A), implying that activated SRC prevents Par-4 secretion in cancer cells.
Figure 3.
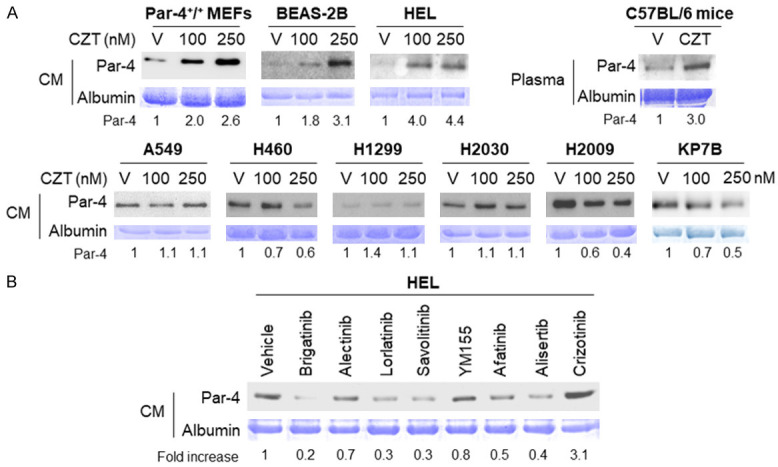
CZT induces Par-4 secretion in normal/immortalized cells. A. CZT induces Par-4 secretion in normal/immortalized cells (MEFs, HEL, BEAS-2B) and mice but not in lung cancer cells. Cells were treated with vehicle (V) or CZT (at indicated nM concentrations) for 24 h. C57BL/6 mice were treated with V or CZT for 5 days. Aliquots of the conditioned medium (CM) from the cells or mouse plasma samples were then subjected to western blot analysis for Par-4. The corresponding gels were stained with Coomassie blue, and the intensity of Par-4 bands were normalized relative to albumin. Fold change in Par-4 in response to CZT relative to vehicle is shown. B. CZT induces Par-4 secretion in normal/immortalized cells, but several other drugs do not. HEL cells were treated with vehicle or the indicated drugs (at 500 nM) for 24 h. Aliquots of the CM from the cells were then subjected to western blot analysis for Par-4. The corresponding gels were stained with Coomassie blue, and the intensity of Par-4 bands were normalized relative to albumin. Fold change in Par-4 in response to each drug relative to vehicle is shown.
To interrogate if other ALK-inhibitors induce Par-4 secretion in normal cells, we tested the effect brigatinib, alectinib, lorlatinib, and CZT as control, as well as savolitinib (MET-inhibitor), YM-155 (survivin-inhibitor), afatinib (EGFR-inhibitor), and alicertib (Arora kinase inhibitor) in HEL cells. These experiments indicated that CZT but none of the other drugs induced Par-4 secretion from normal cells (Figure 3B). As Par-4 secretion has been previously shown to be induced via the classical ER-Golgi secretory pathway [7,30,45], we tested whether CZT also induces Par-4 secretion in normal cells via the classical pathway. Accordingly, HEL or BEAS-2B cells were tested for Par-4 secretion by CZT in the presence or absence of brefeldin A (BFA), which inhibits the classical ER-Golgi pathway. As seen in Figure S4B, BFA inhibited Par-4 secretion into the CM of normal cells treated with CZT. These findings imply that CZT induces Par-4 secretion in normal cells via the classical pathway.
CZT-induced secreted Par-4 from normal cells inhibits viability of CZT-treated lung cancer cells
As CZT translocates GRP78 to the cancer cell surface and induces Par-4 secretion from normal cells, we tested whether the CM from CZT-treated normal cells inhibited viability of CZT-treated cancer cells. CZT induced the secretion of Par-4 into the CM of Par-4+/+ MEFs but not Par-4-/- MEFs (Figure 4A). As our previous studies (44) indicated that p53 function is essential for Par-4 secretion, we used p53-/- MEFs to test whether CZT induces Par-4 secretion by a p53-dependent mechanism. As seen in the Figure 4A, CZT failed to induce Par-4 secretion in these p53-null MEFs, implying that similar to previous findings, Par-4 secretion induced by Crizotinib is p53-dependent. CZT alone failed to inhibit the viability of ALK-negative lung cancer cells (Figure 4B). On the other hand, when the cancer cells were pretreated with CZT to elevate csGRP78 and then treated with CM (+IgG antibody) from Par-4+/+ MEFs treated with CZT, we noted the inhibition of cancer cell viability (Figure 4C). The effect of the CM was neutralized by the Par-4 antibody or GRP78 antibody relative to the control IgG antibody (Figure 4C). The CM from Par-4-/- MEFs treated with CZT failed to inhibit cancer cell viability (Figure 4C). Similarly, the CM from CZT-treated mouse adult lung fibroblast (ALF) cells but not vehicle-treated ALF cells inhibited the viability of lung cancer cells (Figure S5A). To further confirm the role of csGRP78 on cancer cells in the action of secreted Par-4, we knocked-down GRP78 expression in lung cancer cells using two different siRNAs and then treated the cells with CZT and/or recombinant Par-4. These experiments indicated that knockdown of GRP78 blocked the growth inhibitory effect of recombinant Par-4 in cells treated with CZT (Figure S5B).
Figure 4.
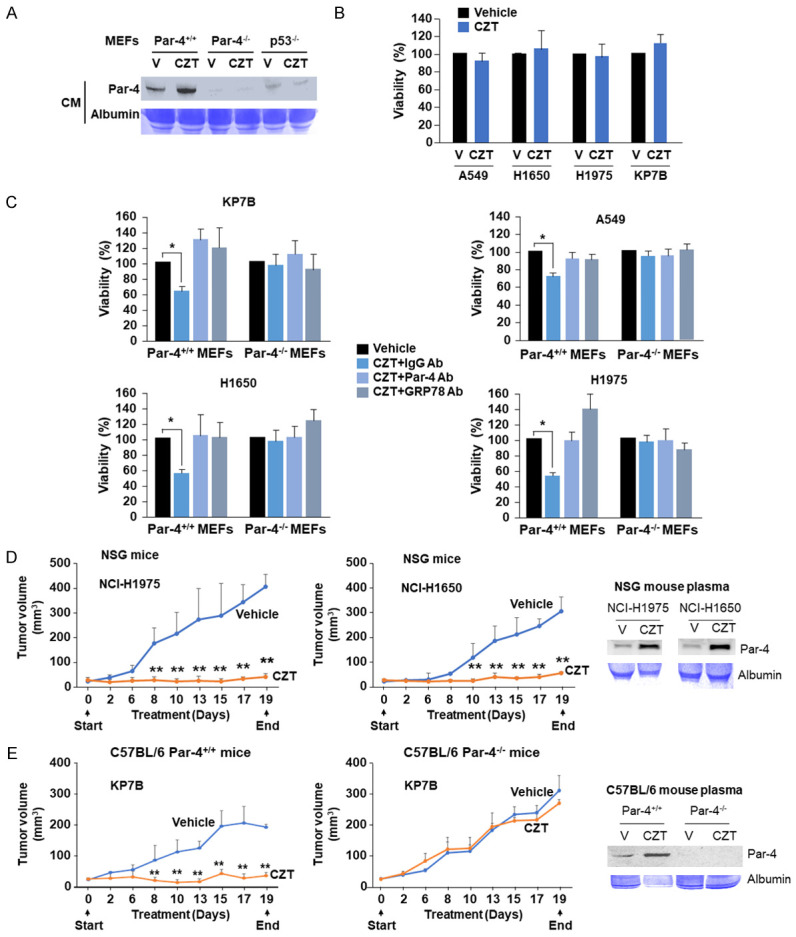
Par-4 secreted by normal cells treated with CZT inhibits cancer cell viability and tumor growth. (A) CZT induces Par-4 secretion in mouse embryonic fibroblasts (MEFs) from Par-4+/+ mice but not from Par-4-/- or p53-/- mice. The indicated MEFs were treated with CZT (1 µM) or vehicle (V) for 24 h, and the CM was examined for Par-4 secretion by western blot analysis. (B) CZT by itself does not inhibit cell viability of lung cancer cells. Lung cancer cells were treated with CZT (1 µM) or vehicle (V) for 24 h, and cell viability was examined by resazurin assays. (C) CM from CZT treated Par-4+/+ MEFs, but not Par-4-/- MEFs, inhibits cancer cell viability. Lung cancer cells A549, H1650, H1975, and KP7B were pretreated with CZT or vehicle (V) for 24 h and washed three times. The cells were then treated with the CM from normal cells Par-4+/+ or Par-4-/- MEFs treated with CZT. Prior to treating the cancer cells with the CM, we pre-incubated the CM for 30 min with Par-4 antibody, GRP78 antibody or control IgG. After 24 h of treatment, the cells were subjected to resazurin assays. Mean + SD values are shown. *P < 0.001 by Student’s t test. (D) CZT treatment inhibits the growth of xenografts derived from lung tumor cells. NSG mice were injected s.c. with H1650 or H1975 cells. When the tumors grew to 30 mm3, the mice were administered CZT (25 mg/kg body weight) or vehicle (n=5 per group) once every day for 19 consecutive days by oral gavage. (E) Inhibition of lung cancer growth by CZT in mice is Par-4-dependent. C57BL/6 mice that were either Par-4+/+ (wild type) or Par-4-/- were injected s.c. with KP7B cells, and the tumors were examined for response to CZT or vehicle. (D, E) Tumor volumes were determined at the indicated time intervals. Mean + SD is shown. **P < 0.01 by ANOVA test for tumor volumes at corresponding days in the vehicle control and CZT treatment groups of mice. Plasma samples from the mice treated with vehicle (V) or CZT collected at the end of the experiments were subjected to western blot analysis for Par-4 or Coomassie blue staining for albumin.
CZT-induced secreted Par-4 from normal cells causes apoptosis in CZT-treated lung cancer cells
As secreted Par-4 is known to inhibit viability of cancer cells by induction of apoptosis [7,44,46], we tested whether the CM from CZT-treated normal cells causes apoptosis in cancer cells. The CM from CZT-treated Par-4+/+ MEFs but not Par-4-/- MEFs induced apoptosis of the cancer cells that were treated with CZT (Figure S6A). On the other hand, transfer of CM from the CZT-treated Par-4+/+ cells to vehicle-treated cancer cells did not induce apoptosis in the cancer cells (Figure S6A). The effect of the CM from CZT-treated MEFs was neutralized by the Par-4 antibody and GRP78 antibody relative to IgG control antibody (Figure S6A). Lung cancer A549 cells that were co-cultured with normal lung HEL cells, but not those that were grown separately, underwent apoptosis when treated with CZT (Figure S6B). Importantly, the CM from CZT-treated HEL cells but not vehicle-treated HEL cells (Figure S6C) induced apoptosis of CZT-treated lung cancer cells in a Par-4 and GRP78-dependent manner. Moreover, organoid cultures of mouse lung cancer cells from genetically engineered bronchiolar mutant-EGFR (L858R; T790M) lung bronchiolar tumors grown with mouse primary endothelial cells underwent growth inhibition with CZT (described in Figure S6D). Collectively, these findings indicate that CZT-induced secreted Par-4 from normal cells causes apoptosis and growth inhibition in CZT-treated lung cancer cells.
CZT inhibits the growth of ALK-negative tumor cell implants in mice
As CZT induces Par-4 secretion from normal cell culture and mice and also induces plasma membrane translocation of GRP78 in cancer cells, we tested whether tumors grown from ALK-negative cells in mice are responsive to CZT. Tumors grown from the human lung cancer cells in the flanks of immunodeficient NSG mice were tested for their response to CZT or vehicle. As seen in Figure 4D, CZT induced Par-4 secretion in the plasma of the mice and CZT remarkably inhibited the growth of the tumors. To ascertain that the tumor growth inhibitory effect of CZT is Par-4-dependent, immunocompetent C57BL/6 Par-4+/+ or Par-4-/- mice were implanted with syngeneic mouse tumor KP7B cells. When the tumors grew to 30 mm3, the mice were administered CZT or vehicle. As seen in Figure 4E, CZT inhibited the growth of tumors in Par-4+/+ mice but not in Par-4-/- mice. As expected, Par-4 was secreted in the plasma of Par-4+/+ mice but not Par-4-/- mice (Figure 4E). Collectively, these findings indicate that CZT inhibits the growth of tumors in a Par-4-dependent manner.
Discussion
Previous studies identified several small-molecule secretagogues of Par-4 that induce Par-4 secretion mainly from normal cells [30,35,44-46]. Although secreted Par-4 induces paracrine apoptosis of cancer cells and inhibits lung metastasis, none of the previously identified secretagogues inhibited the growth of tumor xenografts in the flanks of mice [44]. Resistance to secreted Par-4 was attributed to the lack of adequate expression of the Par-4 receptor csGRP78 in tumors [35]. The present study led to the discovery that the FDA-approved drug, CZT is a new secretagogue of Par-4 that induces Par-4 secretion in normal cells but not in cancer cells. Importantly, CZT induced the cell surface expression of GRP78 in cancer cells (and not in normal cells) overcoming the limitations seen with earlier Par-4 secretagogues. These opposing effects in normal and cancer cells result in activation of the paracrine Par-4-csGRP78 axis for the desired selective inhibition of tumor cell growth without affecting the growth of normal cells (Figure 5). These findings were validated in cell culture using the CM from CZT-treated normal cells to induce apoptosis in CZT-treated cancer cells. Validation was corroborated as tumors grown in the flanks of mice produced CZT-induced systemic expression of Par-4 and inhibited the growth of lung tumor cell xenografts. CZT inhibited tumor growth in Par-4+/+ mice but not in Par-4-null mice, implying a Par-4-dependent mechanism of CZT action. Moreover, in cancer cell cultures, CZT itself did not inhibit the growth of the cancer cells, but CM from CZT-treated Par-4+/+ MEFs (but not Par-4-/- MEFs) induced growth inhibition in the cancer cells. The growth inhibitory action of the CM was neutralized by the Par-4 and GRP78 antibodies, as well as by the siRNA knockdown of GRP78 in the cancer cells. These findings implied that Par-4 was the effector moiety in the CM that induced apoptosis via its receptor csGRP78 on cancer cells. CZT is a known inhibitor of activated ALK/MET/ROS1, and our studies indicate that it can activate the secreted Par-4-csGRP78 paracrine pathway to inhibit the growth of ALK-negative lung cancer cells that express oncogenic KRAS or EGFR or lack p53 function. As KRAS and EGFR mutations together account for about 60% of the ALK-negative NSCLCs in patients, our findings extend the potential target range of CZT to a majority of lung tumors.
Figure 5.
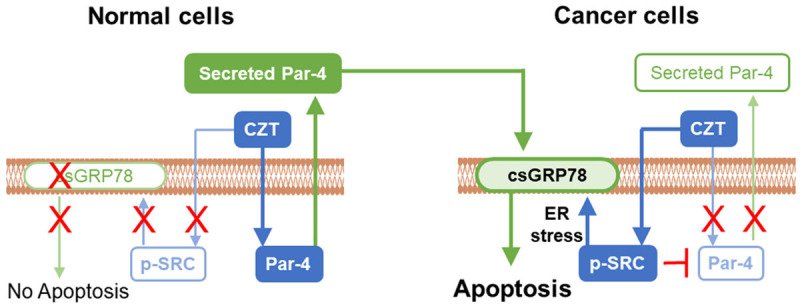
Model for differential effects of CZT in normal and cancer cells. CZT activates SRC in cancer cells but not normal cells by directly interacting with it, and activated SRC is required for translocation of GRP78 from the ER to the cancer cell surface. On the other hand, induction of Par-4 secretion in normal cells but not in cancer cells by CZT results in paracrine apoptosis and growth inhibition of cancer cells in co-culture studies and mouse xenograft models. As CZT does not activate SRC in normal cells, csGRP78 is not elevated and normal cells are resistant to apoptosis and growth inhibition by secreted Par-4.
CZT is a first generation ALK-inhibitor for ALK-positive NSCLCs [23]. Although the ALK-positive tumors initially respond well to the treatment, the tumors eventually develop resistance to this drug [24]. Acquired resistance to CZT may result from amplification of the ELM4-ALK gene fusion, secondary mutations in the kinase domain of ALK or activation of alternate bypass pathways via the growth factor receptor signaling [24]. As the findings of this study would predict induction of Par-4 secretion from the normal tissue cells in the patients with ALK-positive tumors treated with CZT, the above mechanisms of acquired resistance may render the tumors refractory to apoptosis by the secreted-Par-4 csGRP78 axis. The precise identity of those resistance mechanisms in ALK-resistant tumors (that were previously ALK-responsive) to secreted Par-4 has not been addressed. It is apparent, however, from the present study that the majority of the NSCLCs that are ALK-negative (i.e., never ALK-responsive) and exhibit oncogenic KRAS or EGFR, do not express those underlying resistance mechanisms over the duration of treatment in cell culture or mouse tumor models. It is important to note that unlike a few other studies that used high pharmacologic concentrations (>10 μM) of CZT to report off-target growth inhibitory effects of CZT [47,48], our experiments intentionally used 1 μM CZT that represents the physiological concentration noted in patients undergoing CZT treatment [36]. Although CZT may exert direct growth inhibitory effects in lung cancer cells with amplified MET [49,50], our studies indicated that the MET inhibitor savolitinib failed to induce csGRP78 (Figure 1B) and that CZT did not on its own inhibit the viability of the lung cancer cells in culture or tumors in Par-4-/- mice (Figure 4B and 4E). Together, these observations indicate that the effect of CZT is Par-4-dependent and not associated with direct inhibition of MET. A long-term follow-up of the mice in remission after their tumors are inhibited by CZT will be undertaken in future studies to determine whether the tumors regrow after withdrawal of CZT and whether combination with other treatments would overcome resistance to CZT monotherapy. Moreover, sotorasib-resistant KRAS G12C NSCLC cells expressing amplified MET are resistant to CZT [51], and future studies will also determine whether certain driver mutations in KRAS or EGFR would render lung cancer cells refractory to the growth inhibitory action of the Par-4-csGRP78 axis activated by CZT.
We noted that CZT activates SRC in cancer cells but not in normal cells. Such differential activation of SRC results in csGRP78 elevation in cancer cells but not normal cells, and prevents Par-4 secretion by cancer cells but not normal cells. Importantly, CZT can elevate csGRP78 in other diverse cancer cells including prostate cancer (Burikhanov, unpublished results), implying that CZT may find application in inducing apoptosis and growth inhibition in diverse tumors. The requirement of SRC activation for CZT to induce csGRP78 elevation is consistent with the role of SRC activation in triggering a signaling cascade that prevents retrograde trafficking of proteins from the Golgi to the ER and promotes GRP78 translocation from the Golgi to the cell surface [37,52]. It is particularly striking that GRP78 translocation to the cell surface but not SRC activation by CZT was inhibited by reducing unfolded protein response (UPR) and ER-stress with 4PB, an outcome that implied that activation of ER-stress is either co-parallel or occurs downstream of SRC activation by CZT and is essential for csGRP78 elevation. Interestingly, we noted that the N-linked glycosylation inhibitor TU, which causes accumulation of unfolded proteins and activates ER-stress, can induce csGRP78 but not SRC activation, implying that alternate pathways are available in cancer cells for GRP78 transport to the cell surface.
Several reports have suggested that chemotherapy and radiation may directly or indirectly cause activation of SRC that confers malignant phenotypes and resistance to treatment in cancer cells [53-56]. Early-phase clinical trials with SRC inhibitors, dasatinib and bosutinib, as monotherapies suggested only modest activity in breast and prostate cancers, indicating that combination therapies may be more effective [57] than monotherapies for certain cancers. Interestingly, this study suggested that SRC activation results in induction of csGRP78, and that SRC activation and csGRP78 that are paradoxically induced by cancer therapeutics can be harnessed for inhibition of tumor growth. Although we found that CZT binds to SRC in cancer cells, it is possible that CZT may have other unidentified targets in cancer and normal cells. As csGRP78 is targetable by various approaches using killer proteins, antibodies, and CAR-T cells [5-11,27,58-60], selective upregulation of csGRP78 in cancer cells but not normal cells by repurposing CZT could be exploited to overcome resistance in tumors.
Like other Par-4 secretagogues, CZT induced Par-4 secretion via the classical ER-Golgi pathway (Figure S4B) that was dependent on wild type p53 function (Figure 4A). It was, however, interesting to find that CZT was unique in promoting Par-4 secretion relative to the second or third generation ALK/MET/ROS1 inhibitors, the MET-inhibitor savolitinib, the survivin inhibitor YM-155, the EGFR-inhibitor afatinib, and the Arora kinase inhibitor alicertib, all of which failed to induce Par-4 secretion (Figure 3B). Future studies will define the precise differences in the mechanism of action of these other inhibitors that, similar to CZT, promoted csGRP78 elevation in cancer cells but, unlike CZT, failed to induce Par-4 secretion in normal cells.
In summary, we identified CZT as a selective small-molecule that induced Par-4 secretion from normal cells and csGRP78 translocation in cancer cells, which renders the cancer cells vulnerable to paracrine apoptosis and growth inhibition by Par-4. This effect was validated in mice in which normal cells secreted Par-4 and caused inhibition of tumor growth in a Par-4-dependent manner. Normal cells did not show induction of csGRP78 under the influence of CZT and were resistant to the action of the drug.
Conclusion
Our study extended the target range of CZT to NSCLC that are ALK-negative and express oncogenic KRAS or EGFR, as seen in a majority of lung cancer patients. Mechanistically, CZT activated SRC selectively in cancer cells but not normal cells and this activation facilitated csGRP78 translocation in cancer cells and Par-4 secretion from normal cells, a combination of effects that proved lethal to NSCLC cancer cells. This previously unrecognized, bifunctional attribute of CZT could provide a means to elevate csGRP78 and target it in a broad range of lung tumors that afflict a large number of patients.
Acknowledgements
We acknowledge the generous gifts of KP7B cells from Professor Tyler Jacks and Carla Kim for the H1659, H2030 and H2009 cells. The University of Kentucky Markey Cancer Center’s Research Communications Office assisted with preparation of the manuscript. This work was supported by NIH/NCI grants R01 CA165469, R01 CA187273, and R21 CA179283 (to V.M.R.). N.A. was supported by a scholarship (ID 13137-13-1) from Coordenação de Aperfeiçoamento Superior (CAPES), Brazil. C.F.B. was supported by R01 CA237643, American Cancer Society Research Scholar Grant 133123-RSG-19-081-01-TBG. The research was also supported by NCI Administrative Supplement 3P30CA177558-07S2 (to B. Mark Evers, Markey Cancer Center), and by the following University of Kentucky Markey Cancer Center Shared Resource Facilities (P30 CA177558), including Biostatistics and Bioinformatics Shared Resource Facility and Flow Cytometry and Immune Monitoring Shared Resource Facility.
Disclosure of conflict of interest
VMR is owner of a start-up company Parcure, LLC, in Lexington, KY, USA. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
- 4BP
4-phenylbutyrate-block protein unfolding
- Afatinib
EGFR inhibitor
- Alectinib
ALK inhibitor
- Alicertib
Arora kinase inhibitor
- Brigatinib
ALK inhibitor
- CM
conditioned medium
- CQ
chloroquine
- CsGRP78
cell-surface GRP78
- Csk
C-terminal SRC kinase
- CZT
Crizotinib-an inhibitor of activated ALK/MET/ROS1 pathways
- DAPI
4, 6-diamidino-2-phenylindole-a nuclear stain
- ER
endoplasmic reticulum
- FACS
fluorescence-activated cell sorting
- GRP78
protein involved in ER-mediated protein folding
- ICC
immunocytochemical
- KRAS
Kirsten rat sarcoma virus
- Lorlatinib
ALK inhibitor Lorlatinib, inhibits ALK and ROS1
- MEF
mouse embryonic fibroblasts
- NSCLC
non-small cell lung cancer
- NSG
NOD-Scid/gamma IL2R-deleted mice
- Par-4
prostate apoptosis response-4/PAWR
- R7017
resazurin-cell viability assay
- SAV
savolitinib-MET inhibitor
- SRC
non-receptor tyrosine kinase/c-Src
- SU6656
SRC inhibitor
- YM-155
survivin inhibitor
- BFA
classical secretory pathway inhibitor brefeldin A
Supporting Information
References
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022;72:7–33. doi: 10.3322/caac.21708. [DOI] [PubMed] [Google Scholar]
- 2.Skoulidis F, Heymach JV. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat Rev Cancer. 2019;19:495–509. doi: 10.1038/s41568-019-0179-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin JJ, Shaw AT. Resisting resistance: targeted therapies in lung cancer. Trends Cancer. 2016;2:350–364. doi: 10.1016/j.trecan.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Devarakonda S, Govindan R. Targeting resistance to targeted therapies: combating a resilient foe. Clin Cancer Res. 2018;24:6112–6114. doi: 10.1158/1078-0432.CCR-18-3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Araujo N, Hebbar N, Rangnekar VM. GRP78 is a targetable receptor on cancer and stromal cells. EBioMedicine. 2018;33:2–3. doi: 10.1016/j.ebiom.2018.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gonzalez-Gronow M, Selim MA, Papalas J, Pizzo SV. GRP78: a multifunctional receptor on the cell surface. Antioxid Redox Signal. 2009;11:2299–2306. doi: 10.1089/ARS.2009.2568. [DOI] [PubMed] [Google Scholar]
- 7.Burikhanov R, Zhao Y, Goswami A, Qiu S, Schwarze SR, Rangnekar VM. The tumor suppressor Par-4 activates an extrinsic pathway for apoptosis. Cell. 2009;138:377–388. doi: 10.1016/j.cell.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ni M, Zhang Y, Lee AS. Beyond the endoplasmic reticulum: atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem J. 2011;434:181–188. doi: 10.1042/BJ20101569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee AS. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer. 2014;14:263–276. doi: 10.1038/nrc3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hernandez I, Cohen M. Linking cell-surface GRP78 to cancer: from basic research to clinical value of GRP78 antibodies. Cancer Lett. 2022;524:1–14. doi: 10.1016/j.canlet.2021.10.004. [DOI] [PubMed] [Google Scholar]
- 11.Ge R, Kao C. Cell surface GRP78 as a death receptor and an anticancer drug target. Cancers (Basel) 2019;11:1787. doi: 10.3390/cancers11111787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shrestha-Bhattarai T, Rangnekar VM. Cancer-selective apoptotic effects of extracellular and intracellular Par-4. Oncogene. 2010;29:3873–3880. doi: 10.1038/onc.2010.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cook J, Krishnan S, Ananth S, Sells SF, Shi Y, Walther MM, Linehan WM, Sukhatme VP, Weinstein MH, Rangnekar VM. Decreased expression of the pro-apoptotic protein Par-4 in renal cell carcinoma. Oncogene. 1999;18:1205–1208. doi: 10.1038/sj.onc.1202416. [DOI] [PubMed] [Google Scholar]
- 14.Goswami A, Burikhanov R, de Thonel A, Fujita N, Goswami M, Zhao Y, Eriksson JE, Tsuruo T, Rangnekar VM. Binding and phosphorylation of par-4 by akt is essential for cancer cell survival. Mol Cell. 2005;20:33–44. doi: 10.1016/j.molcel.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 15.Moreno-Bueno G, Fernandez-Marcos PJ, Collado M, Tendero MJ, Rodriguez-Pinilla SM, Garcia-Cao I, Hardisson D, Diaz-Meco MT, Moscat J, Serrano M, Palacios J. Inactivation of the candidate tumor suppressor Par-4 in endometrial cancer. Cancer Res. 2007;67:1927–1934. doi: 10.1158/0008-5472.CAN-06-2687. [DOI] [PubMed] [Google Scholar]
- 16.Hebbar N, Wang C, Rangnekar VM. Mechanisms of apoptosis by the tumor suppressor Par-4. J Cell Physiol. 2012;227:3715–3721. doi: 10.1002/jcp.24098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alvarez JV, Pan TC, Ruth J, Feng Y, Zhou A, Pant D, Grimley JS, Wandless TJ, Demichele A I-SPY 1 TRIAL Investigators. Chodosh LA. Par-4 downregulation promotes breast cancer recurrence by preventing multinucleation following targeted therapy. Cancer Cell. 2013;24:30–44. doi: 10.1016/j.ccr.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jagtap JC, Parveen D, Shah RD, Desai A, Bhosale D, Chugh A, Ranade D, Karnik S, Khedkar B, Mathur A, Natesh K, Chandrika G, Shastry P. Secretory prostate apoptosis response (Par)-4 sensitizes multicellular spheroids (MCS) of glioblastoma multiforme cells to tamoxifen-induced cell death. FEBS Open Bio. 2014;5:8–19. doi: 10.1016/j.fob.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y, Gilbert MR, Kyprianou N, Rangnekar VM, Horbinski C. The tumor suppressor prostate apoptosis response-4 (Par-4) is regulated by mutant IDH1 and kills glioma stem cells. Acta Neuropathol. 2014;128:723–732. doi: 10.1007/s00401-014-1334-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Araujo N, Sledziona J, Noothi SK, Burikhanov R, Hebbar N, Ganguly S, Shrestha-Bhattarai T, Zhu B, Katz WS, Zhang Y, Taylor BS, Liu J, Chen L, Weiss HL, He D, Wang C, Morris AJ, Cassis LA, Nikolova-Karakashian M, Nagareddy PR, Melander O, Evers BM, Kern PA, Rangnekar VM. Tumor suppressor Par-4 regulates complement factor C3 and obesity. Front Oncol. 2022;12:860446. doi: 10.3389/fonc.2022.860446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.García-Cao I, Duran A, Collado M, Carrascosa MJ, Martín-Caballero J, Flores JM, Diaz-Meco MT, Moscat J, Serrano M. Tumour-suppression activity of the proapoptotic regulator Par4. EMBO Rep. 2005;6:577–583. doi: 10.1038/sj.embor.7400421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao Y, Burikhanov R, Qiu S, Lele SM, Jennings CD, Bondada S, Spear B, Rangnekar VM. Cancer resistance in transgenic mice expressing the SAC module of Par-4. Cancer Res. 2007;67:9276–9285. doi: 10.1158/0008-5472.CAN-07-2124. [DOI] [PubMed] [Google Scholar]
- 23.Devarakonda S, Ganesh B, Mann J, Govindan R. Crizotinib: an orphan drug for treating non-small-cell lung cancer. Expert Opin Orphan Drugs. 2015;3:1209–1218. [Google Scholar]
- 24.Isozaki H, Takigawa N, Kiura K. Mechanisms of acquired resistance to ALK inhibitors and the rationale for treating ALK-positive lung cancer. Cancers (Basel) 2015;7:763–783. doi: 10.3390/cancers7020763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farshbaf M, Khosroushahi AY, Mojarad-Jabali S, Zarebkohan A, Valizadeh H, Walker PR. Cell surface GRP78: an emerging imaging marker and therapeutic target for cancer. J Control Release. 2020;328:932–941. doi: 10.1016/j.jconrel.2020.10.055. [DOI] [PubMed] [Google Scholar]
- 26.Dagogo-Jack I, Shaw AT. Crizotinib resistance: implications for therapeutic strategies. Ann Oncol. 2016;27(Suppl 3):iii42–iii50. doi: 10.1093/annonc/mdw305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hebbar N, Burikhanov R, Shukla N, Qiu S, Zhao Y, Elenitoba-Johnson KSJ, Rangnekar VM. A naturally generated decoy of the prostate apoptosis response-4 protein overcomes therapy resistance in tumors. Cancer Res. 2017;77:4039–4050. doi: 10.1158/0008-5472.CAN-16-1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DuPage M, Dooley AL, Jacks T. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc. 2009;4:1064–1072. doi: 10.1038/nprot.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim EJ, Jeong JH, Bae S, Kang S, Kim CH, Lim YB. mTOR inhibitors radiosensitize PTEN-deficient non-small-cell lung cancer cells harboring an EGFR activating mutation by inducing autophagy. J Cell Biochem. 2013;114:1248–1256. doi: 10.1002/jcb.24465. [DOI] [PubMed] [Google Scholar]
- 30.Burikhanov R, Sviripa VM, Hebbar N, Zhang W, Layton WJ, Hamza A, Zhan CG, Watt DS, Liu C, Rangnekar VM. Arylquins target vimentin to trigger Par-4 secretion for tumor cell apoptosis. Nat Chem Biol. 2014;10:924–926. doi: 10.1038/nchembio.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robinson DR, Wu YM, Lin SF. The protein tyrosine kinase family of the human genome. Oncogene. 2000;19:5548–5557. doi: 10.1038/sj.onc.1203957. [DOI] [PubMed] [Google Scholar]
- 32.Hamza A, Abdulhameed MD, Zhan CG. Understanding microscopic binding of human microsomal prostaglandin E synthase-1 with substrates and inhibitors by molecular modeling and dynamics simulation. J Phys Chem B. 2008;112:7320–7329. doi: 10.1021/jp8007688. [DOI] [PubMed] [Google Scholar]
- 33.Hamza A, Zhao X, Tong M, Tai HH, Zhan CG. Novel human mPGES-1 inhibitors identified through structure-based virtual screening. Bioorg Med Chem. 2011;19:6077–6086. doi: 10.1016/j.bmc.2011.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou Z, Yuan Y, Zhou S, Ding K, Zheng F, Zhan CG. Selective inhibitors of human mPGES-1 from structure-based computational screening. Bioorg Med Chem Lett. 2017;27:3739–3743. doi: 10.1016/j.bmcl.2017.06.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burikhanov R, Shrestha-Bhattarai T, Qiu S, Shukla N, Hebbar N, Lele SM, Horbinski C, Rangnekar VM. Novel mechanism of apoptosis resistance in cancer mediated by extracellular PAR-4. Cancer Res. 2013;73:1011–1019. doi: 10.1158/0008-5472.CAN-12-3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Balis FM, Thompson PA, Mosse YP, Blaney SM, Minard CG, Weigel BJ, Fox E. First-dose and steady-state pharmacokinetics of orally administered crizotinib in children with solid tumors: a report on ADVL0912 from the Children’s Oncology Group Phase 1/Pilot Consortium. Cancer Chemother Pharmacol. 2017;79:181–187. doi: 10.1007/s00280-016-3220-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsai YL, Ha DP, Zhao H, Carlos AJ, Wei S, Pun TK, Wu K, Zandi E, Kelly K, Lee AS. Endoplasmic reticulum stress activates SRC, relocating chaperones to the cell surface where GRP78/CD109 blocks TGF-beta signaling. Proc Natl Acad Sci U S A. 2018;115:E4245–E4254. doi: 10.1073/pnas.1714866115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bolen JB, Veillette A, Schwartz AM, DeSeau V, Rosen N. Activation of pp60c-src protein kinase activity in human colon carcinoma. Proc Natl Acad Sci U S A. 1987;84:2251–2255. doi: 10.1073/pnas.84.8.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Okada M. Regulation of the SRC family kinases by Csk. Int J Biol Sci. 2012;8:1385–1397. doi: 10.7150/ijbs.5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wheeler DL, Iida M, Dunn EF. The role of Src in solid tumors. Oncologist. 2009;14:667–678. doi: 10.1634/theoncologist.2009-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bjorge JD, Jakymiw A, Fujita DJ. Selected glimpses into the activation and function of Src kinase. Oncogene. 2000;19:5620–5635. doi: 10.1038/sj.onc.1203923. [DOI] [PubMed] [Google Scholar]
- 42.Spagnuolo A, Maione P, Gridelli C. Evolution in the treatment landscape of non-small cell lung cancer with ALK gene alterations: from the first- to third-generation of ALK inhibitors. Expert Opin Emerg Drugs. 2018;23:231–241. doi: 10.1080/14728214.2018.1527902. [DOI] [PubMed] [Google Scholar]
- 43.Eigner K, Filik Y, Mark F, Schütz B, Klambauer G, Moriggl R, Hengstschläger M, Stangl H, Mikula M, Röhrl C. The unfolded protein response impacts melanoma progression by enhancing FGF expression and can be antagonized by a chemical chaperone. Sci Rep. 2017;7:17498. doi: 10.1038/s41598-017-17888-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burikhanov R, Hebbar N, Noothi SK, Shukla N, Sledziona J, Araujo N, Kudrimoti M, Wang QJ, Watt DS, Welch DR, Maranchie J, Harada A, Rangnekar VM. Chloroquine-inducible Par-4 secretion is essential for tumor cell apoptosis and inhibition of metastasis. Cell Rep. 2017;18:508–519. doi: 10.1016/j.celrep.2016.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burikhanov R, Shrestha-Bhattarai T, Hebbar N, Qiu S, Zhao Y, Zambetti GP, Rangnekar VM. Paracrine apoptotic effect of p53 mediated by tumor suppressor Par-4. Cell Rep. 2014;6:271–277. doi: 10.1016/j.celrep.2013.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ghosalkar J, Sonawane V, Pisal T, Achrekar S, Pujari R, Chugh A, Shastry P, Joshi K. Prostate apoptosis response-4 (Par-4): a novel target in pyronaridine-induced apoptosis in glioblastoma (GBM) cells. Cancers (Basel) 2022;14:3198. doi: 10.3390/cancers14133198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu P, Zhao L, Kepp O, Kroemer G. Crizotinib - a tyrosine kinase inhibitor that stimulates immunogenic cell death. Oncoimmunology. 2019;8:1596652. doi: 10.1080/2162402X.2019.1596652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ji C, Zhang L, Cheng Y, Patel R, Wu H, Zhang Y, Wang M, Ji S, Belani CP, Yang JM, Ren X. Induction of autophagy contributes to crizotinib resistance in ALK-positive lung cancer. Cancer Biol Ther. 2014;15:570–577. doi: 10.4161/cbt.28162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kubo T, Yamamoto H, Lockwood WW, Valencia I, Soh J, Peyton M, Jida M, Otani H, Fujii T, Ouchida M, Takigawa N, Kiura K, Shimizu K, Date H, Minna JD, Varella-Garcia M, Lam WL, Gazdar AF, Toyooka S. MET gene amplification or EGFR mutation activate MET in lung cancers untreated with EGFR tyrosine kinase inhibitors. Int J Cancer. 2009;124:1778–1784. doi: 10.1002/ijc.24150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tanizaki J, Okamoto I, Okamoto K, Takezawa K, Kuwata K, Yamaguchi H, Nakagawa K. MET tyrosine kinase inhibitor crizotinib (PF-02341066) shows differential antitumor effects in non-small cell lung cancer according to MET alterations. J Thorac Oncol. 2011;6:1624–1631. doi: 10.1097/JTO.0b013e31822591e9. [DOI] [PubMed] [Google Scholar]
- 51.Suzuki S, Yonesaka K, Teramura T, Takehara T, Kato R, Sakai H, Haratani K, Tanizaki J, Kawakami H, Hayashi H, Sakai K, Nishio K, Nakagawa K. KRAS inhibitor resistance in MET-amplified KRAS (G12C) non-small cell lung cancer induced by RAS- and non-RAS-mediated cell signaling mechanisms. Clin Cancer Res. 2021;27:5697–5707. doi: 10.1158/1078-0432.CCR-21-0856. [DOI] [PubMed] [Google Scholar]
- 52.Bard F, Mazelin L, Péchoux-Longin C, Malhotra V, Jurdic P. Src regulates Golgi structure and KDEL receptor-dependent retrograde transport to the endoplasmic reticulum. J Biol Chem. 2003;278:46601–46606. doi: 10.1074/jbc.M302221200. [DOI] [PubMed] [Google Scholar]
- 53.Kopetz S, Morris VK, Parikh N, Overman MJ, Jiang ZQ, Maru D, Elvin P, Gallick G. Src activity is modulated by oxaliplatin and correlates with outcomes after hepatectomy for metastatic colorectal cancer. BMC Cancer. 2014;14:660. doi: 10.1186/1471-2407-14-660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Higuchi M, Ishiyama K, Maruoka M, Kanamori R, Takaori-Kondo A, Watanabe N. Paradoxical activation of c-Src as a drug-resistant mechanism. Cell Rep. 2021;34:108876. doi: 10.1016/j.celrep.2021.108876. [DOI] [PubMed] [Google Scholar]
- 55.Fatherree JP, Guarin JR, McGinn RA, Naber SP, Oudin MJ. Chemotherapy-induced collagen iv drives cancer cell motility through activation of Src and focal adhesion kinase. Cancer Res. 2022;82:2031–2044. doi: 10.1158/0008-5472.CAN-21-1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim RK, Cui YH, Yoo KC, Kim IG, Lee M, Choi YH, Suh Y, Lee SJ. Radiation promotes malignant phenotypes through SRC in breast cancer cells. Cancer Sci. 2015;106:78–85. doi: 10.1111/cas.12574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mayer EL, Krop IE. Advances in targeting SRC in the treatment of breast cancer and other solid malignancies. Clin Cancer Res. 2010;16:3526–3532. doi: 10.1158/1078-0432.CCR-09-1834. [DOI] [PubMed] [Google Scholar]
- 58.Liu R, Li X, Gao W, Zhou Y, Wey S, Mitra SK, Krasnoperov V, Dong D, Liu S, Li D, Zhu G, Louie S, Conti PS, Li Z, Lee AS, Gill PS. Monoclonal antibody against cell surface GRP78 as a novel agent in suppressing PI3K/AKT signaling, tumor growth, and metastasis. Clin Cancer Res. 2013;19:6802–6811. doi: 10.1158/1078-0432.CCR-13-1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dadey DYA, Kapoor V, Hoye K, Khudanyan A, Collins A, Thotala D, Hallahan DE. Antibody targeting GRP78 enhances the efficacy of radiation therapy in human glioblastoma and non-small cell lung cancer cell lines and tumor models. Clin Cancer Res. 2017;23:2556–2564. doi: 10.1158/1078-0432.CCR-16-1935. [DOI] [PubMed] [Google Scholar]
- 60.Hebbar N, Epperly R, Vaidya A, Thanekar U, Moore SE, Umeda M, Ma J, Patil SL, Langfitt D, Huang S, Cheng C, Klco JM, Gottschalk S, Velasquez MP. CAR T cells redirected to cell surface GRP78 display robust anti-acute myeloid leukemia activity and do not target hematopoietic progenitor cells. Nat Commun. 2022;13:587. doi: 10.1038/s41467-022-28243-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.