

Summary
Background
Idiopathic pulmonary fibrosis (IPF) is an incurable lung disease characterised by progressive scarring leading to alveolar stiffness, reduced lung capacity, and impeded gas transfer. We aimed to identify genetic variants associated with declining lung capacity or declining gas transfer after diagnosis of IPF.
Methods
We did a genome-wide meta-analysis of longitudinal measures of forced vital capacity (FVC) and diffusing capacity of the lung for carbon monoxide (DLCO) in individuals diagnosed with IPF. Individuals were recruited to three studies between June, 1996, and August, 2017, from across centres in the US, UK, and Spain. Suggestively significant variants were investigated further in an additional independent study (CleanUP-IPF). All four studies diagnosed cases following American Thoracic Society/European Respiratory Society guidelines. Variants were defined as significantly associated if they had a meta-analysis p<5 × 10−⁸ when meta-analysing across all discovery and follow-up studies, had consistent direction of effects across all four studies, and were nominally significant (p<0∙05) in each study.
Findings
1329 individuals with a total of 5216 measures were included in the FVC analysis. 975 individuals with a total of 3361 measures were included in the DLCO analysis. For the discovery genome-wide analyses, 7 611 174 genetic variants were included in the FVC analysis and 7 536 843 in the DLCO analysis. One variant (rs115982800) located in an antisense RNA gene for protein kinase N2 (PKN2) showed a genome-wide significant association with FVC decline (−140 mL/year per risk allele [95% CI –180 to –100]; p=9∙14 × 10−¹²).
Interpretation
Our analysis identifies a genetic variant associated with disease progression, which might highlight a new biological mechanism for IPF. We found that PKN2, a Rho and Rac effector protein, is the most likely gene of interest from this analysis. PKN2 inhibitors are currently in development and signify a potential novel therapeutic approach for IPF.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a devastating lung disease characterised by an aberrant response to lung injury leading to the deposition of scar tissue in the lung interstitium. IPF has a prevalence of between three and 60 cases per 100 000 and is more common in men, individuals older than 65 years, and people of European ancestry.1
IPF is a progressive disease in which fibrosis spreads throughout the lung leading to reduced lung capacity, poorer quality of life, and eventually death, with half of individuals dying within 3 to 5 years of diagnosis.1–3 Two measures of lung health are commonly used to monitor disease progression of IPF, forced vital capacity (FVC; the maximum volume of air that can be forcibly exhaled) and the diffusing capacity of the lung for carbon monoxide (DLCO; a measure of gas transfer between theair sacs and bloodstream).2–4
Rates of decline as measured using FVC and DLCO are highly variable between individuals, with some having a rapid decline and shorter survival times while others have relatively stable lung function and live for many years after diagnosis.2–4
There are many known genetic and environmental risk factors for IPF. Genetic associations can provide new insight into the genes and pathways relevant to disease pathology, and drug targets with supporting genetic evidence have been shown to be twice as likely to be successful during drug development.5 Previous genetic studies have identified genetic variants that implicate host defence (such as mucus and pulmonary surfactant regulation), signalling (particularly regulation of transforming growth factor β [TGFβ] signalling), cell to cell adhesion (such as desmoplakin, which plays a role in structural integrity of the epithelium), telomere maintenance (with people with IPF having shorter telomeres than age-matched healthy individuals), and spindle assembly as important processes in disease risk.6–11 Shorter telomeres have been associated with more progressive IPF.12
To date, there have been no genome-wide association studies of lung function decline in IPF. Candidate gene studies have shown that variants associated with IPF risk generally show little association with disease progression. The rs35705950 variant in the MUC5B promoter region, the strongest genetic risk factor for IPF with an odds ratio of more than 4 for each copy of the T allele, has been reported as associated with improved survival times.13 However, this variant has not been associated with lung function decline.14
Identification of genetic variants associated with disease progression, rather than disease risk, might highlight new ways to modify ongoing disease processes and yield therapeutic benefit.5
Therefore, we aimed to identify genetic variants that might highlight new biological processes involved in disease progression by performing the first genome-wide association study (GWAS) of FVC and DLCO decline in individuals with IPF.
Methods
Study design
For this study we used a two-stage design. Firstly, genome-wide variants were tested for their association with longitudinal FVC and DLCO separately in three IPF case-control studies and the results were then meta-analysed (discovery GWAS). Variants showing suggestive evidence of being associated with rate of change of FVC or DLCO in the discovery GWAS were then analysed in a fourth independent study and variants reaching genome-wide significance (with support from all studies) were reported.
Study populations
For the discovery GWAS, we used longitudinal measures of FVC and DLCO from IPF cases from three previously described studies (named as US, UK, and UUS [US, UK, and Spain]).
The US study8 (referred to as the “Chicago study” in some previous IPF GWAS) comprised three study centres that collected longitudinal data (University of Chicago, University of Pittsburgh Medical Centre, and Correlating Outcomes with Biochemical Markers to Estimate Time-progression [COMET] centres led by the University of Michigan). The COMET study did not record DLCO. Participants were enrolled between January, 2003, and January, 2012, and genotyped using the Affymetrix 6.0 SNP array.
The UK study15 comprised four centres that collected longitudinal data (the Royal Brompton and Harefield NHS Foundation Trust, London; the Prospective Study of Fibrosis in the Lung Endpoints [PROFILE] study centre at the University of Nottingham; the University of Edinburgh; and the Trent Lung Function centre, Nottingham). Participants were enrolled between June, 1996, and July, 2013, and genotyped using the Affymetrix UK BiLEVE array.
Six centres in the UUS study9 collected longitudinal data, the Anticoagulant Effectiveness in Idiopathic Pulmonary Fibrosis (ACE) study centre led by Duke University, the Royal Brompton and Harefield NHS Foundation Trust, London, the University of Chicago, the University of Nottingham, the Prednisone, Azathioprine, and N-acetylcysteine for Pulmonary Fibrosis (PANTHER) study centre led by Duke University, and the University of California, Davis centre. The ACE and PANTHER trials did not record DLCO. Participants were enrolled between June, 1996, and August, 2017, and genotyped using the Affymetrix UK Biobank array.
For the follow-up stage, the Study of Clinical Efficacy of Antimicrobial Therapy Strategy Using Pragmatic Design in Idiopathic Pulmonary Fibrosis (CleanUP-IPF) study16 was used. As CleanUP-IPF participants were followed up for a shorter time than in the other three studies (up to three measures of FVC and DLCO over 2 years), the CleanUP-IPF study was selected for the follow-up analyses and was not included in the discovery GWAS. Participants were enrolled between August, 2017, and June, 2019, and genotyped using the Affymetrix UK Biobank array (appendix pp 4–5).
All studies were imputed using the Haplotype Reference Consortium17 and diagnosed cases followed the American Thoracic Society/European Respiratory Society guidelines.3,18
This research was conducted using previously published work with appropriate ethics approval. The PROFILE study (which provided samples for the UK and UUS studies) had institutional ethics approval at the University of Nottingham (NCT01134822; ethics reference 10/H0402/2) and Royal Brompton and Harefield NHS Foundation Trust (NCT01110694; ethics reference 10/H0720/12). UK samples were recruited across multiple sites with individual ethics approval (University of Edinburgh Research Ethics Committee [The Edinburgh Lung Fibrosis Molecular Endotyping Study, NCT04016181] 17/ES/0075, and Nottingham Research Ethics Committee 09/H0403/59). For individuals recruited at the University of Chicago, consenting patients with IPF who were prospectively enrolled in the institutional review board (IRB) approved interstitial lung disease (ILD) registry (IRB 14163A) were included. Individuals recruited at the University of Pittsburgh Medical Centre had ethics approval from the University of Pittsburgh Human Research Protection Office (reference STUDY20030223: Genetic Polymorphisms in IPF). This study also included individuals from clinical trials with ethics approval (ACE [NCT00957242], PANTHER [NCT00650091], COMET [NCT01071707], and CleanUP-IPF [NCT02759120] studies). Individuals for the ACE and PANTHER trials were recruited through the Idiopathic Pulmonary Fibrosis Clinical Research Network (NCT00517933, NCT00650091, and NCT00957242), which is a multicentre network that recruits individuals to IPF studies. Each centre has appropriate ethics approval (IRBs 09–220-B and 09–214-B for the University of Chicago, where the genotyping of all individuals was conducted), which was overseen by the Duke Clinical Research Institute, which acted as the Data Coordinating Centre. CleanUP-IPF samples were genotyped under University of Virginia ethics approval (IRB 20845).
Quality control
For quality control, we excluded individuals who did not meet the Affymetrix genotyping quality measures,8,19,15 who did not have IPF, whose genetic sex did not match their recorded sex at birth, who were heterozygosity outliers, or who had non-European ancestry based on genetic principal components, duplicates, and up to second-degree relatives of other people in the study. We only included individuals with at least two longitudinal measures. No exclusions were made based on the time span between measurements. For duplicates or relatives, the individual with the more complete phenotype data was kept (when this was the same, the individual from the smaller study was kept).
In our analysis we took enrolment to the study as a proxy for time of diagnosis. Historical measures of FVC and DLCO were available for a small subset of individuals; however, we excluded measures that were more than a year before enrolment. Because most centres only recorded longitudinal measures for 3 to 5 years, to reduce biases in fitting longitudinal models with sparse data at later timepoints, we only included FVC and DLCO measures taken within 3 years of enrolment. We only included variants with a minor allele frequency greater than 1%, an imputation quality greater than 0∙5, and in Hardy-Weinberq equilibrium (p≥10−⁶).
GWAS
We performed GWASs using a longitudinal linear mixed model (appendix p 5) with random slope and intercept with a (Time × SNP) interaction term, where SNP (single nucleotide plymorphism) is the genetic variant being tested, and adjusting for agesex, the first ten genetic principal components (to account for population stratification) and study centre, as follows:
where is
and is
where i is each individual and j is each timepoint, e is a normally distributed random variable for level 1, and u is a normally distributed random variable for level 2. Absolute values of FVC and DLCO were used. SNP was the genetic dosage for that variant (ie, equal to 0 for those with two copies of the reference allele, 1 for heterozygotes, and 2 for those with two copies of the effect allele), Time was the time in years after enrolment that the measure was taken, Sex was coded as 1 for males and 0 for females, PC1 to PC10 were the first 10 genetic principal components (included to adjust for population stratification), and Centre was a categorical variable for each study centre. Age was treated as a time-varying covariate and centred on 72 years (the median age at baseline). Because we are interested in whether the genetic variant affects the rate of change of FVC or DLCO, our effect estimate of interest is γ11, which is the effect size estimate of the (SNP × Time) variable.
The genome-wide discovery analysis was performed in each of the three studies (US, UK, and UUS) separately and results were meta-analysed across studies using a fixed effect inverse variance weighted meta-analysis using the METAL software (the version released on March 25, 2011).19 Genomic control was applied to the meta-analysis results where λ was greater than 1. Only variants that were included in at least two of the studies were included in the meta-analysis.
Independent variants were followed up in the CleanUP-IPF study if they had p<10−⁵ in the discovery meta-analysis, and had consistent direction of effects and reached nominal significance (p<0∙05) in each of the contributing GWAS studies. Conditional analyses were performed using GCTA-COJO (v1.90.2) to identify whether there were multiple independent association signals at each association locus (appendix p 5).
Variants were defined as significantly associated with FVC or DLCO if they were genome-wide significant when meta-analysing across all studies (p<5 × 10−⁸), had consistent direction of effects, and reached nominal significance (p<0∙05) in each of the contributing studies.
Gene prioritisation and characterisation of association signals
Credible sets were calculated for each associated risk signal to generate a set of variants that were 95% certain to contain the true causal variant (under the assumption that there is only one causal variant and that we have measured it; appendix p 6).
To identify putative causal genes from association signals, we performed seven analyses to prioritise genes of interest. (1) Nearest gene: the nearest gene is often found to be the gene the genetic signal acts through;20 we therefore included nearest gene as part of our gene prioritisation analyses. (2) Annotation: to determine the functional annotation of the variants in the credible sets, we used the Ensebl Variant Effect Predictor (v105).21 (3) Gene expression: to determine whether the association signals were associated with gene expression, we investigated whether the variants in the credible sets were associated with gene expression using publicly available eQTL resources: eQTLgen22 (whole blood samples from up to 31 684 individuals) and GTEx23 version 7 (49 tissues including lung from between 73 to 706 individuals). Colocalisation analyses were performed to determine whether the same variant was likely to be driving the association with FVC or DLCO decline and gene expression (appendix p 6). (4) Physical DNA interactions: to identify genes that lie in regions of the DNA that physically interact with the region of DNA showing an association with either FVC or DLCO decline, we used the HUGIN Hi-C database24 (appendix p 7). (5) Identification of relevant mendelian diseases: because we hypothesise that genes associated with relevant phenotypes are more likely to be the gene of interest, we used online resources (Orphanet25 and Online Mendelian Inheritance in Man26) to identify nearby genes associated with relevant mendelian diseases (appendix p 7). (6) Rare variant associated diseases: to identify genes associated with relevant respiratory or fibrotic phenotypes through rare variant changes or accumulation of rare variants, we investigated nearby genes using the AstraZeneca PheWAS Portal27 (appendix p 7). (7) Mouse knockout models: we investigated genes using the International Mouse Phenotyping Consortium Web Portal28 to identify nearby genes that exhibit relevant phenotypes when knocked out in mice (appendix p 7).
Variants in the credible set were investigated for whether they had been reported as associated with any other trait in previous GWAS (appendix p 7).29 We investigated the effect these variants had on IPF susceptibility in a GWAS of 4125 patients with IPF versus 20 464 individuals without IPF.11 Variants previously reported as associated with IPF susceptibility were investigated for their effect on FVC and DLCO decline.
Finally, the combined effect of multiple variants in a gene and enrichment in biological pathways was tested using Vegas230 (v2.01.17; appendix p 8).
Sensitivity analyses
To investigate variants associated with either FVC or DLCO decline further, we performed eight sensitivity analyses. (1) For short-term progression, we repeated the longitudinal mixed model analysis only including data within 1 year of diagnosis. (2) To assess the clinical use of associated variants, we calculated the 1-year trend of FVC for each person (in terms of percentage change) and classified individuals as progressive if they had a 1-year decline in FVC of 10% or more or died within the first year. We then fitted a logistic regression model to test the association between the genetic variant and this binary trait (appendix p 8). (3) We investigated non-linear effects for time by allowing for polynomial time and interaction effects (appendix p 9). (4) For baseline lung function, we tested whether the variant was associated with the first measure of FVC or DLCO using a linear regression model (appendix p 9). (5) For effect in the general population, we used 32 013 unrelated European individuals with longitudinal spirometry measures (no inclusions or exclusions were made based on disease status) in the UK Biobank, and we tested whether the variant was associated with a decline of FVC, forced expiratory volume in 1 second (FEV1), or FEV1 divided by FVC (appendix p 9). (6) For the effect of drop-out, we tested the association of the variant with survival times using a Cox proportional hazards model (appendix pp 9–10) and then performed a joint model combining the longitudinal linear mixed model and the Cox proportional hazards model (appendix p 10). (7) To investigate whether the variant was associated with age at baseline, we used a linear regression model (appendix p 10). (8) For treatment response, longitudinal analyses were performed allowing for an interaction between treatment, genetic variant, and time (appendix pp 10–11). Analyses were performed using the CleanUP-IPF study and the effect of associated genetic variants on nintedanib, pirfenidone, and anti-microbial therapy (co-trimoxazole or doxycycline) response were investigated.
In all circumstances, the sensitivity analyses were performed in the UK, UUS, US, and CleanUP-IPF studies separately and then meta-analysed across studies.
Role of the funding source
The funders of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the manuscript.
Results
1329 individuals passed the quality control measures for the FVC longitudinal analysis and 975 individuals passed quality control measures for inclusion in the DLCO longitudinal analysis (appendix pp 25–26). 711 individuals were included in both analyses, 337 included only in the FVC analysis and 18 included only in the DLCO analysis. In total, there were 5216 measures of FVC and 3361 measures of DLCO (table; appendix pp 27–32).
Table:
Characteristics of individuals included in FVC and DLCO analyses
| Discovery GWAS |
Follow-up |
Total | |||
|---|---|---|---|---|---|
| US | UK | UUS | CleanUP-IPF | ||
| FVC | |||||
| Sample size | 142 | 314 | 592 | 281 | 1329 |
| Study centres | 3 | 4 | 6 | 1 | 14 |
| Sex | |||||
| Female | 34 (23∙9%) | 90 (28∙7%) | 143 (24∙2%) | 57 (20∙3%) | 324 (24∙4%) |
| Male | 108 (76∙1%) | 224 (71∙3%) | 449 (75∙8%) | 224 (79∙7%) | 1005 (75∙6%) |
| Mean age at baseline, years | 65∙7 (8∙3) | 71∙7 (8∙2) | 68∙8 (8∙0) | 71∙3 (7∙4) | 69∙7 (8∙2) |
| Total number of visits | 604 | 1440 | 2516 | 656 | 5216 |
| Mean number of visits per person | 4∙3 (1∙8) | 4∙6 (2∙1) | 4∙3 (1∙8) | 2∙3 (0∙5) | 3∙9 (1∙9) |
| Maximum number of visits | 13 | 12 | 14 | 3 | 14 |
| Number of deaths | 27 (19∙0%) | 110 (35∙0%) | 136 (23∙0%) | 11 (3∙9%) | 284 (21∙4%) |
| DLCO | |||||
| Sample size | 75 | 293 | 361 | 246 | 975 |
| Study centres | 2 | 4 | 4 | 1 | 11 |
| Sex | |||||
| Female | 15 (20∙0%) | 79 (27∙0%) | 88 (24∙4%) | 54 (22∙0%) | 236 (24∙2%) |
| Male | 60 (80∙0%) | 214 (73∙0%) | 273 (75∙6%) | 192 (78∙1%) | 739 (75∙8%) |
| Mean age at baseline, years | 67∙5 (8∙5) | 71∙5 (8∙2) | 69∙2 (8∙1) | 71∙1 (7∙4) | 70∙2 (8∙1) |
| Total number of visits | 251 | 1146 | 1398 | 566 | 3361 |
| Mean number of visits per person | 3∙3 (1∙4) | 3∙9 (1∙7) | 3∙9 (1∙7) | 2∙3 (0∙5) | 3∙4 (1∙7) |
| Maximum number of visits | 7 | 11 | 13 | 3 | 13 |
| Number of deaths | 17 (22∙7%) | 94 (32∙1%) | 112 (31∙0%) | 9 (3∙7%) | 232 (23∙8%) |
Data are n, n (%), or mean (SD). CleanUP-IPF=Clinical Efficacy of Antimicrobial Therapy Strategy Using Pragmatic Design in Idiopathic Pulmonary Fibrosis study. DLCO=diffusing capacity of the lung for carbon monoxide. FVC=forced vital capacity. GWAS=genome-wide association study. USS=US, UK, and Spain.
7 611 174 variants were included in the FVC longitudinal analysis and 7 536 843 variants in the DLCO longitudinal analysis (figure 1; appendix p 33). There were 24 independent variants in the FVC analysis and 30 independent variants in the DLCO analysis, with p<10−⁵ in the discovery meta-analysis plus p<0∙05 and consistent direction of effect in each of the contributing studies that were investigated further in the CleanUP-IPF dataset (appendix pp 12–15). No variants had p<10⁻⁵ in both the FVC and DLCO analyses (appendix p 34).
Figure 1: Manhattan plots for longitudinal FVC (A) and DLCO (B) genome-wide meta-analyses.
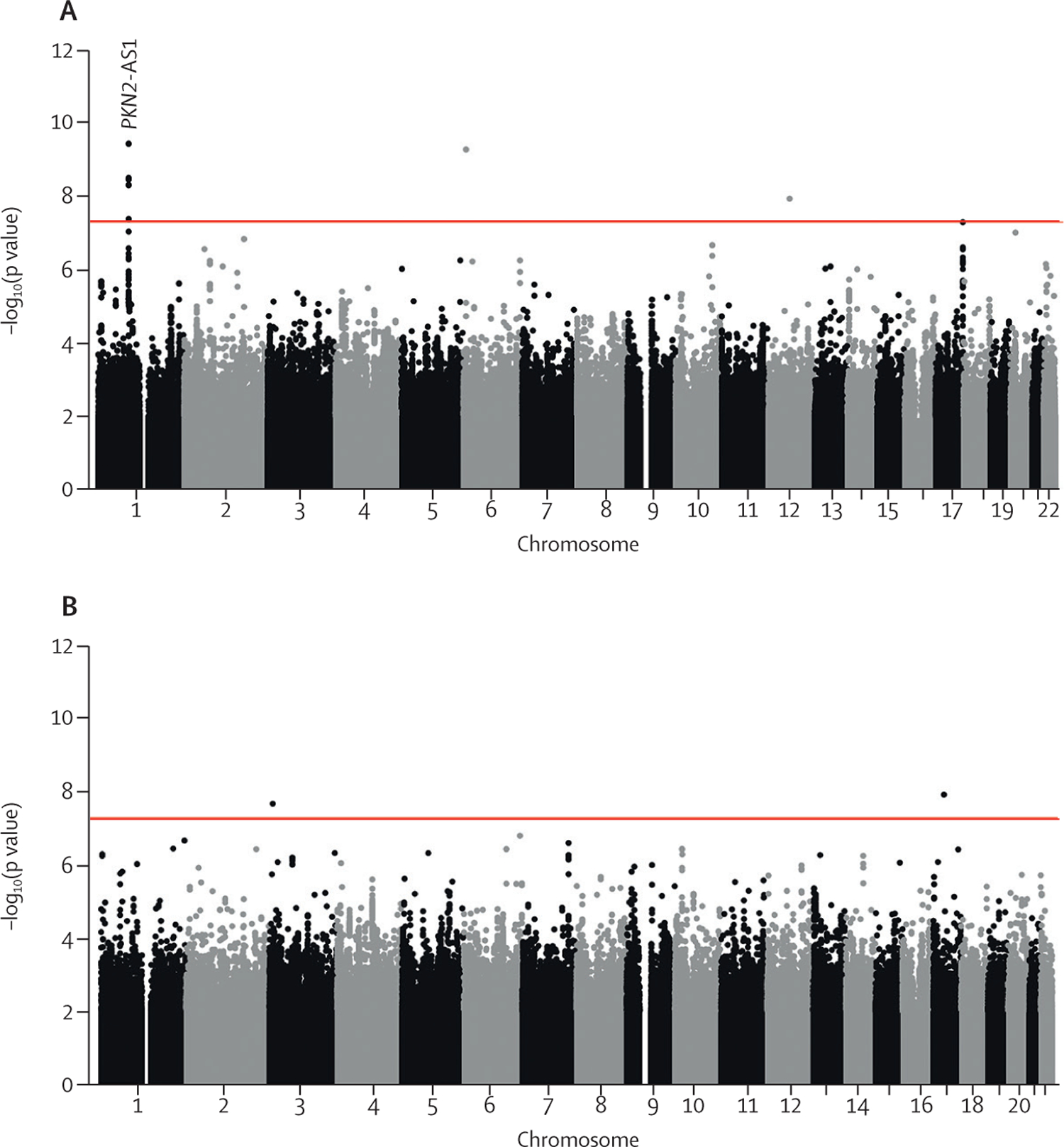
Each point represents a variant, with chromosomal position on the x axis and –log10(p value) on the y axis. The red horizontal line shows the genome-wide significance threshold of p=5 × 10−8. DLCO=diffusing capacity of the lung for carbon monoxide. FVC=forced vital capacity. PKN2-AS1=protein kinase N2 gene in introns of the antisense RNA.
One variant, rs115982800, met genome-wide significance (p=3∙68 × 10−10) in the discovery GWAS meta-analysis, with consistent direction of effects and nominal significance (p<0∙05) in all discovery cohorts (figure 2). This variant was also significant in CleanUP-IPF (p=0∙007) with a consistent effect. Following meta-analysis of the discovery GWAS and CleanUP-IPF, this variant was associated with an annual FVC decline of 140 mL/year per copy of the risk allele A (95% CI −180 to −100; p=9∙14 × 10−12; appendix pp 35–37). The credible set for this association signal contained seven genetic variants with three highly correlated variants accounting for 25∙7% of the posterior probability each (appendix p 16). These variants were located on chromosome 1 upstream of the Protein Kinase N2 gene (PKN2) in introns of the antisense RNA PKN2-AS1 (figure 3). One of the three variants in the credible set with high posterior probability (rs115590681) was located in an open chromatin region.
Figure 2: Forest plot for rs115982800.
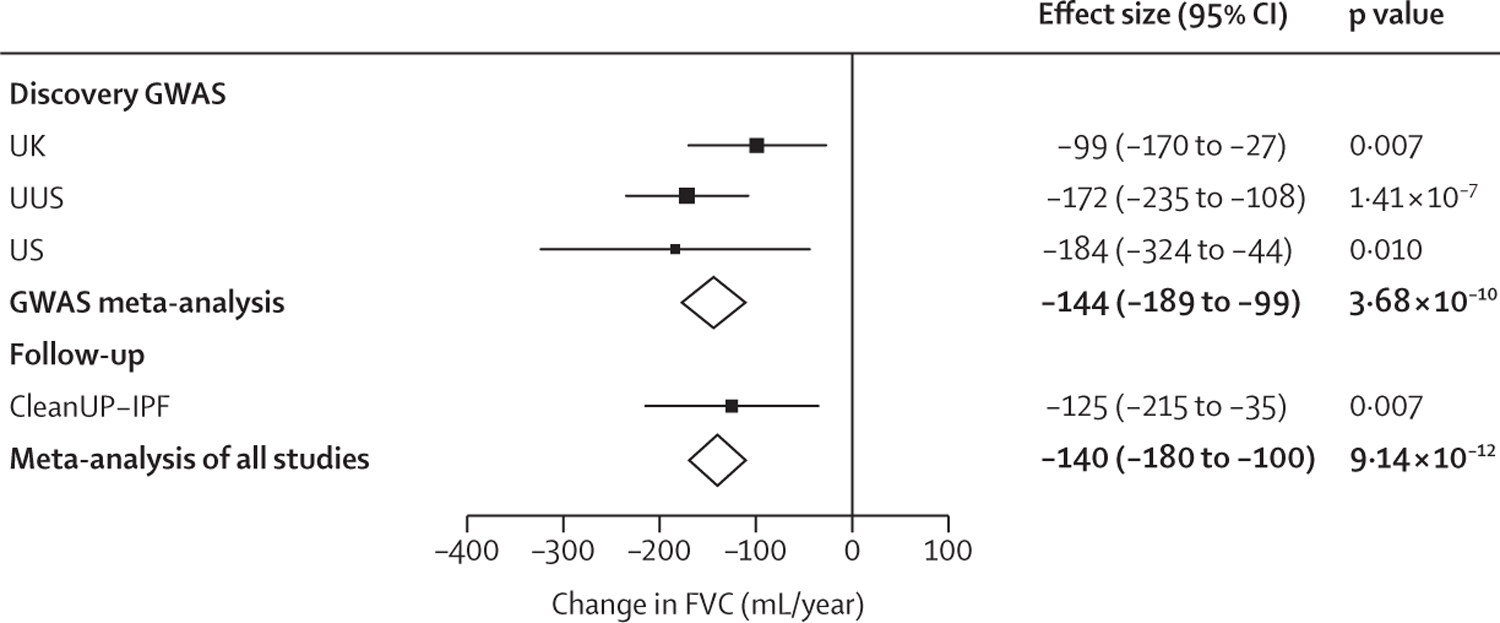
Effect size estimates for each study are shown with squares and the bars show the 95% CI. The size of the square is relative to the weight given to each study in the meta-analysis (which is based on the inverse of the standard error). Meta-analysis results are shown by diamonds. Effect estimates to the left of the vertical line suggest that allele A is associated with a more rapid decline in lung function. CleanUP-IPF=Clinical Efficacy of Antimicrobial Therapy Strategy Using Pragmatic Design in Idiopathic Pulmonary Fibrosis study. FVC=forced vital capacity. GWAS=genome-wide association study. USS=US, UK, and Spain.
Figure 3: Region plot for the locus significantly associated with FVC decline.
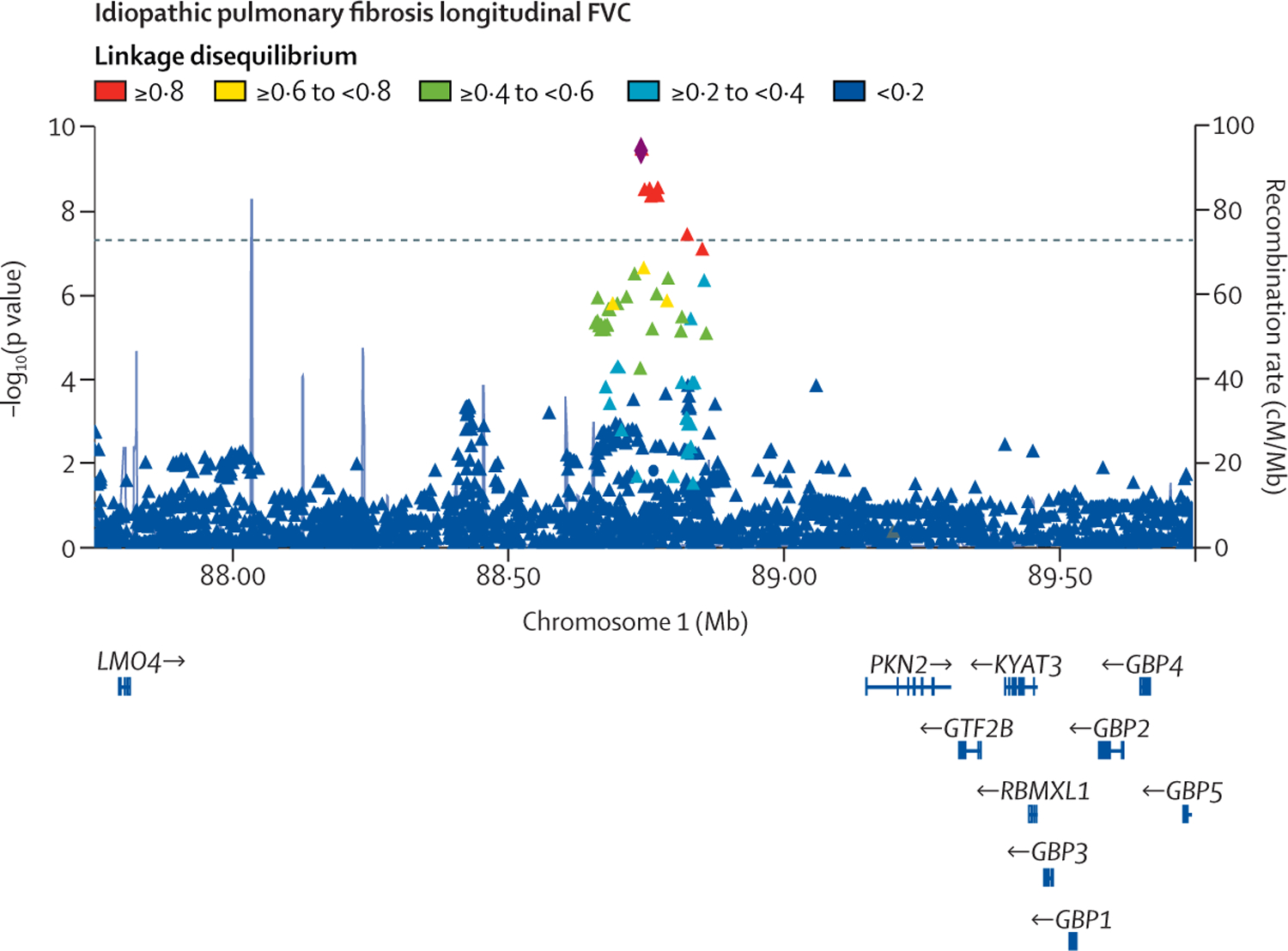
Each point represents a variant, with chromosomal position on the x axis and −log10(p value) on the y axis.
The dashed horizontal line shows the genome-wide significance threshold of p=5 × 10−8. Variants are coloured by linkage disequilibrium with the sentinel variant (rs115982800) depicted as a diamond. Light blue lines show the recombination rate and gene positions are shown below the x axis. The plot was created using LocusZoom.31 FVC=forced vital capacity.
Sensitivity analyses showed that the association with rs115982800 remained when restricting measurements to 1 year after diagnosis (appendix p 38). Variant rs115982800 showed an association with clinically defined progressive IPF when the four studies were meta-analysed together; however, there were inconsistent effects across the four studies (appendix p 39). There was evidence of non-linear effects in the UUS study only (appendix p 39). The variant rs115982800 was not significantly associated with survival time; however, this finding might be due to statistical power and incorporating survival as part of a joint model did reduce the estimated effect of rs115982800 on longitudinal FVC (appendix pp 38, 41–42). The variant showed no association with baseline FVC, baseline DLCO, or with age at baseline (appendix pp 40–41). There was no evidence of a treatment interaction between rs115982800 and response to nintedanib, pirfenidone, or anti-microbial therapy (appendix p 17); however, the rs115982800 variant does show an association with increased odds of reporting dosulepin medication in UK Biobank individuals. The variant shows no association with IPF risk or with lung function decline in the UK Biobank (appendix p 18).
Given that the genome-wide significant association signal might be involved in long-distance gene regulation through an open chromatin, we performed gene-prioritisation on all genes within 3 Mb of the sentinel variant. PKN2 was the nearest gene and the location of the association signal in an antisense RNA gene for PKN2 implicated PKN2 as a gene of interest. The fragment of DNA that contains the longitudinal FVC association signal appears to physically interact with many other parts of the DNA across multiple tissues and cell types (appendix p 43).24 Both PKN2 and GBP5 are included on a diagnostic panel of 44 autoinflammatory diseases (which includes ILD autoinflammatory disorders) but are not directly linked to a specific ILD (appendix pp 19–21).25 Rare variants in both PKN2 (percentage predicted FEV1) and LMO4 (FEV1 and FVC) had p<5 × 10−6 with lung function and rare variants in HFM1 show an association with the level of IGF-1 (which is known to promote pulmonary fibrosis; appendix pp 19–21).27 The FVC association signal did not colocalise with expression of any genes and no genes showed a relevant phenotype when knocked out in mouse models28 (appendix pp 19–21, 44). Collectively, PKN2 had the strongest evidence of being the gene that this association signal acts through (appendix p 45).
Variants previously reported as associated with IPF risk did not show an association with FVC or DLCO decline (appendix p 22).
When using the genome-wide summary statistics, no genes or pathways were found to be significantly enriched in genetic associations with FVC or DLCO decline after Bonferroni corrections for multiple testing (appendix pp 23, 46). The gene TMEM105 showed a borderline significant association with FVC decline in the gene-based analysis; however, this gene includes variants in the FVC decline association signal on chromosome 17 that was not supported in the analysis in CleanUP-IPF (appendix pp 12–13). “GO:0051302_regulation_of_cell_ division” was the only pathway term with empirical p<10−4 in the FVC or DLCO analysis (genes in this pathway were enriched with DLCO decline, p=8∙80 × 10−5). This pathway contained 271 genes, many known to be associated with IPF risk including growth factor-regulating genes (especially for TGFβ signalling), spindle assembly genes, and WNT signalling (appendix p 24).
Discussion
By performing the first GWAS of decline in lung health in individuals diagnosed with IPF, we have identified a genetic variant associated with declining lung capacity after IPF diagnosis, which lies within an antisense RNA for PKN2.
PKN2, which had the strongest evidence of being the causal gene for the association signal, is a Rho and Rac effector protein known to regulate cell cycle progression, actin cytoskeleton assembly, cell migration, cell adhesion, tumour cell invasion, and transcription activation signalling processes.32 RhoA is associated with IPF susceptibility through regulating TGFβ signalling, and other RhoGEFs for RhoA have been implicated in GWAS of IPF risk (eg, AKAP13).15 Protein analyses showed that PKN2 is linked to fibrotic processes in chronic atrial fibrillation.33 IPF is a chronic lung disease characterised by fibroblast proliferation, activation, and differentiation into myofibroblasts. Studies have shown that mouse embryonic fibroblasts depend on PKN2 for proliferation, growth, and motility,34 and cancer-associated fibroblasts depend on PKN2 for activation, differentiation, and motility.35 Additionally, PKN2 has been suggested to play critical roles in actin stress fibre formation in NIH-3T3 cells.36 PKN2 inhibitors are currently in development for cancer therapy37 and the PKN2 inhibitor fostamatinib has been suggested as a drug repurposing candidate for the treatment of acute respiratory distress syndrome in patients with severe COVID-19.38
The rs115982800 variant was not associated with lung function decline in the UK Biobank general population and has not been previously reported for association with other respiratory traits. It is likely that genetic variants associated with decline in IPF reflect specific underlying disease pathology and so it is perhaps not surprising that the same effect is not seen in the general population or in studies of other diseases. However, there are few published studies of lung function decline and we cannot rule out that overlap of associations might be observed when larger datasets are available for analysis.
The variant we identified as associated with FVC decline also shows an association with increased dosulepin use in the UK Biobank. Dosulepin is an antidepressant that has previously been reported as a potential risk factor for IPF.39
There was no evidence of an interaction between rs115982800 and any of the treatments investigated. The treatment an individual is taking is likely to affect the rate of FVC decline. The studies used in this analysis recruited participants over a long period of time with changing treatment recommendations. The fact that the rs115982800 association with FVC decline was consistent across all four studies when different treatments were in use suggests that this association is unlikely to be driven by the effects of specific treatments. The results from the CleanUP-IPF study, which recruited during a period when immunosuppression was not widely used for IPF, supports the notion that the association of rs115982800 is not significantly influenced by immunosuppressive effects in the discovery studies. However, treatment effects in the discovery studies might have accounted for other signals that met p<10−5 and which were not supported in the CleanUP-IPF study. Furthermore, it is possible that treatment effects unaccounted for in the US, UK, and UUS studies might have obscured other signals.
There are several strengths to this study. First, the identified association signal shows a consistent association and effect size estimate across four independent studies, suggesting that this variant is robustly associated with FVC decline after diagnosis of IPF. Second, sensitivity analyses showed that the signal remained when restricting FVC measurements to the first year after diagnosis, and the variant also showed an association with clinically defined progressive IPF. Third, by applying a mixed model, we were able to maximise the power of the analysis by incorporating over 5000 measures of FVC and 3000 measures of DLCO. We used a linear mixed effects model because we were not aiming to model lung function trajectories; rather, we were interested in genetic variants associated with declining FVC and DLCO. Although it is unlikely that FVC or DLCO trajectories follow a linear trend, assuming linearity simplifies the model (increasing power and aiding model convergence) while still being able to distinguish between individuals with a generally declining trend against those with relatively stable disease. Sensitivity analyses did show a nominal association with non-linear effects; however, the effect size was small and a significant non-linear effect was only observed in one study.
There are also limitations to the analysis. First, our model assumes non-informative dropout, which is unlikely to be true, especially for DLCO. Sensitivity analyses showed that although rs115982800 is not associated with survival times, incorporating censoring due to death through a joint model does remove the association with longitudinal FVC, meaning censoring due to death might have some effect on the analysis. The joint model coefficients are estimated together, and it is possible the effect of the variant is being distributed across the longitudinal and time-to-event parts of the model. This distribution of effect might be especially true given the complexity of the joint model and if rs115982800 explains little of the variance. Furthermore, rs115982800 showed consistent effect size estimates across all four studies with varying levels of censoring, including the CleanUP-IPF study in which there was very little censoring, suggesting that this association is not purely caused by informative dropout. Second, the credible set included a variant lying in an open chromatin region and multiple regions were significant in the Hi-C analyses. This finding suggests that the signal could be involved in regulating many genes in a tissue-specific or cell-specific manner. Third, to reduce confounding we only included Europeans, so the findings presented here might not be generalisable to other ancestries. The rs115982800_A allele is low frequency in most populations and rare in African or Asian populations.40 Fourth, we have only investigated the effects of common genetic variants, and there might be rare variants associated with progressive forms of pulmonary fibrosis that have not been included in this study. Finally, although we have maximised the available power of the analysis by incorporating multiple measurements, larger studies are needed to detect variants with smaller effect sizes or for rarer variants associated with lung function decline in individuals with IPF. The smaller sample size analysis means that there was lower statistical power in the DLCO analysis, which might explain why no association signals were identified.
In summary, by using thousands of lung health measures we have shown a genetic association with worsening lung capacity in IPF. These results highlight the role of PKN2 in disease progression and might aid the development of new and desperately needed treatments.
Supplementary Material
Research in context.
Evidence before this study
Idiopathic pulmonary fibrosis (IPF) is a devastating disease whereby the lungs become scarred; this scarring leads to a reduced lung capacity, poorer rates of gas transfer, and is eventually fatal. However, disease progression is highly variable, and it is not clear why this is. We searched Web of Science using the terms “idiopathic pulmonary fibrosis” AND “genome-wide association study”, for English language articles published from database inception to March 23, 2022. To date, genome-wide association studies (GWAS) have identified 20 genetic loci associated with susceptibility to IPF. These genetic loci implicate genes involved with host defence, regulation of TGFβ signalling, telomere maintenance, cell to cell adhesion, and spindle assembly as important biological processes involved in the pathogenesis of IPF. The GWAS variant with the strongest effect on disease risk is found in the promoter region of the MUC5B gene (rs35705950). Generally, variants associated with IPF susceptibility show little or no association with disease progression, apart from the risk allele at rs35705950, which has been reported as having an association with improved survival times but not with lung function decline. Shorter telomere length has also been reported to be associated with more progressive IPF. No study has conducted a genome-wide analysis of genetic associations with rates of lung function decline in IPF.
Added value of this study
Although genetic variants associated with disease risk have been widely studied, little has been reported about the effect of genetics on progression of IPF. Here we present a GWAS of progressive IPF in which we identify genetic variants associated with longitudinal measures of lung health after diagnosis of IPF. We identify a genetic locus associated with a more rapid decline in lung capacity that lies in the RNA antisense gene of protein kinase N2 (PKN2).
Implications of all the available evidence
The novel genetic locus associated with a more rapid decline in lung capacity in individuals with IPF implicates a Rho and Rac effector protein. Effective treatments for IPF are desperately needed. There are currently PKN2 inhibitors under development, so our analysis highlights a potential therapeutic target for IPF. We also show that genetic determinants of IPF progression appear to be distinct from those that drive IPF susceptibility.
Acknowledgments
RJA and PLM are Action for Pulmonary Fibrosis Mike Bray Research Fellows. JMO reports National Institutes of Health (NIH) National Heart, Lung, and Blood Institute grants R56HL158935 and K23HL138190. BG-G is supported by Wellcome Trust grant 221680/Z/20/Z. CF is supported by the Instituto de Salud Carlos III (PI20/00876) and the Spanish Ministry of Science and Innovation (grant RTC-2017-6471-1), co-financed by the European Regional Development Funds (A way of making Europe) from the EU. RGJ and LVW report funding from the Medical Research Council (MR/V00235X/1). LVW holds a GlaxoSmithKline Asthma + Lung UK Chair in Respiratory Research (C17-1). The research was partially supported by the National Institute for Health Research (NIHR) Leicester Biomedical Research Centre; the views expressed are those of the author(s) and not necessarily those of the National Health Service, the NIHR, or the Department of Health. AA reports NIH National Heart, Lung, and Blood Institute grant K23HL146942. NK reports grants from the NIH (R21HL161723, R01HL127349, R01HL141852, U01HL145567, and UH2HL123886). This research includes use of the UK Biobank through application 648 and used the SPECTRE High Performance Computing Facility at the University of Leicester.
Funding
Action for Pulmonary Fibrosis, Medical Research Council, Wellcome Trust, and National Institutes of Health National Heart, Lung, and Blood Institute.
Footnotes
Declaration of interests
LVW reports research funding from GlaxoSmithKline and Orion Pharma, and consultancy for Galapagos, outside of the submitted work. JMO reports personal fees from Boehringer Ingelheim, Genentech, United Therapeutics, AmMax Bio, and Lupin Pharmaceuticals, outside of the submitted work. RGJ is a trustee of Action for Pulmonary Fibrosis and reports personal fees from AstraZeneca, Biogen, Boehringer Ingelheim, Bristol Myers Squibb, Chiesi, Daewoong, Galapagos, Galecto, GlaxoSmithKline, Heptares, NuMedii, PatientMPower, Pliant, Promedior, Redx, Resolution Therapeutics, Roche, Veracyte, and Vicore, outside of the submitted work. AA reports personal fees from Boehringer Ingelheim and Genentech, outside of the submitted work. NK served as a consultant to Biogen, Boehringer Ingelheim, Third Rock, Pliant, Samumed, NuMedii, TheraVance, Indalo, LifeMax, Three Lake Partners, Optikira, AstraZeneca, Rohbar, Veracyte, Augmanity, Gilead, Chiesi, Arrowhead, CSL-Behring, Galapagos, and Thyron over the past 3 years; reports Equity in Pliant and Thyron; reports being a scientific founder of Thyron; grants from Veracyte, Boehringer Ingelheim, Bristol Myers Squibb, and the Three Lakes Foundation; non-financial support from MiRagen and AstraZeneca; and has intellectual property on novel biomarkers and therapeutics in idiopathic pulmonary fibrosis licensed to Biotech. MDT reports grants or contracts from research collaborations with GlaxoSmithKline and Orion Pharma. RBH reports grants or contracts from Galapagos for serving on a trial adjudication outcome committee for an IPF trial, from AstraZeneca for serving on a COVID-19 vaccine safety committee, and from Boehringer Ingelheim for an IPF service provision consultation. WAF and EO are employees of GlaxoSmithKline. All other authors declare no competing interests.
Data sharing
Full summary statistics for the FVC and DLCO genome-wide meta-analyses can be accessed from https://github.com/genomicsITER/ PFgenetics.
Contributor Information
Richard J Allen, Department of Health Sciences, University of Leicester, Leicester, UK.
Justin M Oldham, Division of Pulmonary and Critical Care Medicine, University of Michigan, Ann Arbor, MI, USA.
David A Jenkins, Division of Informatics, Imaging & Data Sciences, University of Manchester, Manchester, UK.
Olivia C Leavy, Department of Health Sciences, University of Leicester, Leicester, UK.
Beatriz Guillen-Guio, Department of Health Sciences, University of Leicester, Leicester, UK.
Carl A Melbourne, Department of Health Sciences, University of Leicester, Leicester, UK.
Shwu-Fan Ma, Division of Pulmonary & Critical Care Medicine, University of Virginia, Charlottesville, VA, USA.
Jonathan Jou, Department of Surgery, University of Illinois College of Medicine at Peoria, Peoria, IL, USA.
John S Kim, Division of Pulmonary & Critical Care Medicine, University of Virginia, Charlottesville, VA, USA.
William A Fahy, Discovery Medicine, GlaxoSmithKline, Stevenage, UK.
Eunice Oballa, Discovery Medicine, GlaxoSmithKline, Stevenage, UK.
Richard B Hubbard, Division of Epidemiology and Public Health, University of Nottingham, Nottingham, UK; National Institute for Health Research, Nottingham Biomedical Research Centre, Nottingham University Hospitals NHS Trust, Nottingham, UK.
Vidya Navaratnam, Division of Epidemiology and Public Health, University of Nottingham, Nottingham, UK; National Institute for Health Research, Nottingham Biomedical Research Centre, Nottingham University Hospitals NHS Trust, Nottingham, UK; Queensland Lung Transplant Service, The Prince Charles Hospital, Brisbane, QLD, Australia.
Rebecca Braybrooke, Division of Epidemiology and Public Health, University of Nottingham, Nottingham, UK; National Institute for Health Research, Nottingham Biomedical Research Centre, Nottingham University Hospitals NHS Trust, Nottingham, UK.
Gauri Saini, Respiratory Medicine, Nottingham University Hospitals NHS Trust, Nottingham, UK.
Katy M Roach, Department of Respiratory Sciences, University of Leicester, Glenfield Hospital, Leicester, UK.
Martin D Tobin, Department of Health Sciences, University of Leicester, Leicester, UK.
Nik Hirani, Centre for Inflammation Research, Queen’s Medical Research Institute, University of Edinburgh, Edinburgh, UK.
Moira K B Whyte, Centre for Inflammation Research, Queen’s Medical Research Institute, University of Edinburgh, Edinburgh, UK.
Naftali Kaminski, Section of Pulmonary, Critical Care and Sleep Medicine, Yale School of Medicine, New Haven, CT, USA.
Yingze Zhang, Division of Pulmonary, Allergy and Critical Care Medicine, The University of Pittsburgh, Pittsburgh, PA, USA.
Fernando J Martinez, Division of Pulmonary and Critical Care Medicine, Weill Cornell Medicine, New York, NY, USA.
Angela L Linderholm, Department of Internal Medicine, University of California Davis, Davis, CA, USA.
Ayodeji Adegunsoye, Section of Pulmonary and Critical Care Medicine, University of Chicago, Chicago, IL, USA.
Mary E Strek, Section of Pulmonary and Critical Care Medicine, University of Chicago, Chicago, IL, USA.
Toby M Maher, National Heart and Lung Institute, Imperial College London, London, UK; Royal Brompton and Harefield Hospitals, London, UK; Division of Pulmonary and Critical Care Medicine, University of Southern California, Los Angeles, CA, USA.
Philip L Molyneaux, National Heart and Lung Institute, Imperial College London, London, UK; Royal Brompton and Harefield Hospitals, London, UK.
Carlos Flores, Research Unit, Hospital Universitario Nuestra Señora de Candelaria, Santa Cruz de Tenerife, Spain; CIBER de Enfermedades Respiratorias, Instituto de Salud Carlos III, Madrid, Spain; Genomics Division, Instituto Tecnológico y de Energías Renovables, Santa Cruz de Tenerife, Spain.
Imre Noth, Division of Pulmonary & Critical Care Medicine, University of Virginia, Charlottesville, VA, USA.
R Gisli Jenkins, National Heart and Lung Institute, Imperial College London, London, UK.
Louise V Wain, Department of Health Sciences, University of Leicester, Leicester, UK; National Institute for Health Research, Leicester Respiratory Biomedical Research Centre, Glenfield Hospital, Leicester, UK.
References
- 1.Lederer DJ, Martinez FJ. Idiopathic pulmonary fibrosis. N Engl J Med 2018; 378: 1811–23. [DOI] [PubMed] [Google Scholar]
- 2.Fujimoto H, Kobayashi T, Azuma A. Idiopathic pulmonary fibrosis: treatment and prognosis. Clin Med Insights Circ Respir Pulm Med 2016; 9 (suppl 1): 179–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med 2018; 198: e44–68. [DOI] [PubMed] [Google Scholar]
- 4.Kottmann RM, Hogan CM, Phipps RP, Sime PJ. Determinants of initiation and progression of idiopathic pulmonary fibrosis. Respirology 2009; 14: 917–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nelson MR, Tipney H, Painter JL, et al. The support of human genetic evidence for approved drug indications. Nat Genet 2015; 47: 856–60. [DOI] [PubMed] [Google Scholar]
- 6.Seibold MA, Wise AL, Speer MC, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med 2011; 364: 1503–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fingerlin TE, Murphy E, Zhang W, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet 2013; 45: 613–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noth I, Zhang Y, Ma SF, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med 2013; 1: 309–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allen RJ, Guillen-Guio B, Oldham JM, et al. Genome-wide association study of susceptibility to idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2020; 201: 564–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dhindsa RS, Mattsson J, Nag A, et al. Identification of a missense variant in SPDL1 associated with idiopathic pulmonary fibrosis. Commun Biol 2021; 4: 392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allen RJ, Stockwell A, Oldham JM, et al. Genome-wide association study across five cohorts identifies five novel loci associated with idiopathic pulmonary fibrosis. medRxiv 2021; published online Dec 7. 10.1101/2021.12.06.21266509 (preprint). [DOI] [PMC free article] [PubMed]
- 12.Courtwright AM, El-Chemaly S. Telomeres in interstitial lung disease: the short and the long of it. Ann Am Thorac Soc 2019; 16: 175–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peljto AL, Zhang Y, Fingerlin TE, et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA 2013; 309: 2232–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Newton CA, Oldham JM, Ley B, et al. Telomere length and genetic variant associations with interstitial lung disease progression and survival. Eur Respir J 2019; 53: 1801641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allen RJ, Porte J, Braybrooke R, et al. Genetic variants associated with susceptibility to idiopathic pulmonary fibrosis in people of European ancestry: a genome-wide association study. Lancet Respir Med 2017; 5: 869–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinez FJ, Yow E, Flaherty KR, et al. Effect of antimicrobial therapy on respiratory hospitalization or death in adults with idiopathic pulmonary fibrosis: the CleanUP-IPF randomized clinical trial. JAMA 2021; 325: 1841–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.The Haplotype Reference Consortium. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet 2016; 48: 1279–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183: 788–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010; 26: 2190–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forgetta V, Jiang L, Vulpescu NA, et al. An effector index to predict target genes at GWAS loci. Hum Genet 2022; published online Feb 11. 10.1007/s00439-022-02434-z. [DOI] [PubMed]
- 21.McLaren W, Gil L, Hunt SE, et al. The ensembl variant effect predictor. Genome Biol 2016; 17: 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Võsa U, Claringbould A, Westra H, et al. Unraveling the polygenic architecture of complex traits using blood eQTL meta-analysis. bioRxiv 2018; published online Oct 19. 10.1038/s41588-021-00913-z (preprint). [DOI]
- 23.Battle A, Brown CD, Engelhardt BE, Montgomery SB. Genetic effects on gene expression across human tissues. Nature 2017; 550: 204–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martin JS, Xu Z, Reiner AP, et al. HUGIn: Hi-C unifying genomic interrogator. Bioinformatics 2017; 33: 3793–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weinreich SS, Mangon R, Sikkens JJ, Teeuw ME, Cornel MC. [Orphanet: a European database for rare diseases]. Ned Tijdschr Geneeskd 2008; 152: 518–19 (in Dutch). [PubMed] [Google Scholar]
- 26.Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res 2005; 33 (suppl 1): D514–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Q, Dhindsa RS, Carss K, et al. Rare variant contribution to human disease in 281,104 UK Biobank exomes. Nature 2021; 597: 527–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koscielny G, Yaikhom G, Iyer V, et al. The International Mouse Phenotyping Consortium Web Portal, a unified point of access for knockout mice and related phenotyping data. Nucleic Acids Res 2014; 42: D802–09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghoussaini M, Mountjoy E, Carmona M, et al. Open Targets Genetics: systematic identification of trait-associated genes using large-scale genetics and functional genomics. Nucleic Acids Res 2021; 49: D1311–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mishra A, Macgregor S. VEGAS2: Software for more flexible gene-based testing. Twin Res Hum Genet 2015; 18: 86–91. [DOI] [PubMed] [Google Scholar]
- 31.Pruim RJ, Welch RP, Sanna S, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 2010; 26: 2336–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stelzer G, Rosen R, Plaschkes I, et al. The GeneCards suite: from gene data mining to disease genome sequence analysis. Curr Protoc Bioinformatics 2016; 54: 1.30.1–1.30.33. [DOI] [PubMed] [Google Scholar]
- 33.Zhang P, Wang W, Wang X, et al. Protein analysis of atrial fibrosis via label-free proteomics in chronic atrial fibrillation patients with mitral valve disease. PLoS One 2013; 8: e60210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quétier I, Marshall JJT, Spencer-Dene B, et al. Knockout of the PKN family of rho effector kinases reveals a non-redundant role for PKN2 in developmental mesoderm expansion. Cell Rep 2016; 14: 440–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murray ER, Menezes S, Henry JC, et al. Disruption of pancreatic stellate cell myofibroblast phenotype promotes pancreatic tumor invasion. Cell Rep 2022; 38: 110227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vincent S, Settleman J. The PRK2 kinase is a potential effector target of both Rho and Rac GTPases and regulates actin cytoskeletal organization. Mol Cell Biol 1997; 17: 2247–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scott F, Fala AM, Pennicott LE, et al. Development of 2-(4-pyridyl)-benzimidazoles as PKN2 chemical tools to probe cancer. Bioorg Med Chem Lett 2020; 30: 127040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kost-Alimova M, Sidhom EH, Satyam A, et al. A high-content screen for mucin-1-reducing compounds identifies fostamatinib as a candidate for rapid repurposing for acute lung injury. Cell Rep Med 2020; 1: 100137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hubbard R, Venn A, Smith C, Cooper M, Johnston I, Britton J. Exposure to commonly prescribed drugs and the etiology of cryptogenic fibrosing alveolitis: a case-control study. Am J Respir Crit Care Med 1998; 157: 743–47. [DOI] [PubMed] [Google Scholar]
- 40.1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015; 526: 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.