

 This work is licensed under a
This work is licensed under a Abstract
Background
Hyperinsulinism/hyperammonemia (HI/HA) syndrome is the second most common type of congenital hyperinsulinism caused by an activating GLUD1 mutation.
Objective
The aim of this study was to determine the clinical profile and long-term neurological outcomes in children with HI/HA syndrome.
Method
This study is a retrospective review of patients with GLUD1 mutation, treated at two centers in the UK and Russia, over a 15-year period. Different risk factors for neuro-developmental disorders were analysed by Mann–Whitney U test and Fisher’s exact P test.
Results
We identified 25 cases with GLUD1 mutations (12 males). Median age of presentation was 7 months (12 h–18 months). Hypoglycaemic seizures were the presenting feature in 24 (96%) cases. Twenty four cases responded to diazoxide and protein restriction whilst one patient underwent partial pancreatectomy. In total, 13 cases (52%) developed neurodevelopmental manifestations. Epilepsy (n = 9/25, 36%), learning difficulties (n = 8/25, 32%) and speech delay (n = 8/25, 32%) were the most common neurological manifestation. Median age of presentation for epilepsy was 12 months with generalised tonic-clonic seizures being the most common (n = 4/9, 44.4%) followed by absence seizures (n = 3/9, 33.3%). Early age of presentation (P = 0.02), diazoxide dose (P = 0.04) and a mutation in exon 11 or 12 (P = 0.01) were associated with neurological disorder.
Conclusion
HI/HA syndrome is associated with wide spectrum of neurological disorders. These neurological manifestations were more frequent in cases with mutations affecting the GTP-binding site of GLUD1 in our cohort.
Keywords: hyperinsulinism/hyperammonemia, HI/HA syndrome, GLUD1, neurodevelopmental disorders, epilepsy
Introduction
Hyperinsulinism/hyperammonemia (HI/HA) syndrome is the second most common cause of congenital hyperinsulinism (CHI) after ABCC8/KCNJ11 mutations. It is caused due to an activating mutation in the GLUD1 gene, which is located on chromosome 10q23.3 (contains 13 exons) and encodes the intra-mitochondrial matrix enzyme glutamate dehydrogenase (GDH). Approximately 70% of these children have de novo mutation and 30% present with autosomal dominant inheritance (1, 2).
GDH is highly expressed in pancreatic β-cells, liver, kidney and brain, where it plays an important role in the metabolism of amino acids and ammonia (3). In pancreatic β-cells, it catalyses the oxidative deamination of glutamate to alpha-ketoglutarate and ammonia. Alpha-ketoglutarate enters the tricarboxylic acid cycle (Krebs cycle), leading to increase in ATP production, which finally results in insulin exocytosis (4). GDH is allosterically activated by leucine and inhibited by guanosine-5’-triphosphate (GTP) (5, 6). Activated mutations in the GLUD1 gene lead to loss of this allosteric inhibition by GTP, which in turn increases leucine-induced glutamate oxidation to alpha-ketoglutarate, resulting in hyperinsulinemic hypoglycaemia (7). This leucine-sensitive hypoglycaemia clinically presents as postprandial hypoglycaemia (following protein-rich food) and HA, which are two classical and persistent features of this condition.
Clinically, children with HI/HA syndrome present in late infancy, with fasting and/or postprandial (after protein-rich meal) hypoglycaemia. An elevated ammonia level, which is a striking feature, seems to be consistent even without hypoglycaemia. However, unlike the HA of urea cycle defect, these children do not have symptoms of raised ammonia (8).
Patients with HI/HA syndrome are prone to develop different neurodevelopmental disorders including epilepsy (9). Delayed presentation, recurrent hypoglycaemia, raised ammonia level and/or increased GDH activity in the brain are some of the proposed explanations for this brain damage; however, the exact pathogenesis is unclear (10).
In this study, we aimed to assess the frequency of different neurodevelopmental disorders including epilepsy in children with HI/HA syndrome due to a GLUD1 activating mutation. We further explored the significance of different risk factors that can lead to the development of neurological disorders in these children.
Materials and methods
We retrospectively analysed all cases of HI/HA syndrome due to an activating mutation in the GLUD1 gene, presenting in paediatric endocrine centres in London and Moscow over a period of 15 years (2003–2018). Local research and development ethical approval for retrospective data collection was obtained as per institutional requirements of contributing centres (Great Ormond Street Hospital, London, UK, and Endocrinology Research Centre, Moscow, Russia (protocol No 18 from 11.10.17)).
The clinical spectrum of these children including gestational age, birth weight, presence of neonatal newborn complications, age of presentation, presenting complaints, family history, neurological manifestation and treatment response was reviewed. HI was confirmed in all cases biochemically on provocation tests suggestive of detectable insulin (>2.0 mIU/L) and c-peptide (>100 pmol/L) at the time of hypoglycaemia (<3.0 mmol/L) and suppressed fatty acids and ketone bodies.
Gene testing for Russian patients included GCG, GLUD1, WFS1, HNF1A, GCK, INS, HNF1B, ABCC8, HNF4A, RFX6, PTF1A, NEUROD1, AKT2, ZFP57, INSR, EIF2AK3, PPARG, PAX4, PDX1, GLIS3, KCNJ11, SLC16A1, FOXP3, BLK, CEL, KLF11, GCGR and HADH. For UK patients, Sanger sequencing of GLUD1 gene was performed as a first-line test as all the patients presented with strong clinical suspicion of HI/HA syndrome.
Neurological assessment of mental status, motor and sensory function, cranial nerves and reflexes by an experienced neurologist and detailed neurodevelopmental assessment by a community paediatrician as per local protocol were performed in every case. Of the 25 genetically confirmed cases of HI/HA, electroencephalography (EEG) was performed in 14/25 cases and MRI brain was performed in 12/25 cases. A spectrum of neurological disorders was assessed using percentages. Suggested risk factors for neurodevelopmental disorder and epilepsy included gender, birth weight, gestational age, prematurity, age of presentation, ammonia level at presentation, glucose infusion requirement at presentation, dose of diazoxide, history of asphyxia and mutation site. Children with and without neurodevelopmental disorders and those with and without epilepsy were divided into separate groups. Non-binary data were analysed with the help of Mann–Whitney U test and binary data by Fisher’s exact P test with P value < 0.05 considered as significant.
Results
We included 25 cases of HI/HA cases due to an activating GLUD1 mutation (13 females and 12 males). The median current age of the children enrolled is 8 years. Most of the children were born at term with normal birth weight (Table 1). The vast majority presented with hypoglycaemic seizures during the first year of life (median age of presentation is 7 months); presentation after the first year of life occurred only in three cases.
Table 1.
Neurodevelopmental disorders in children with HI/HA syndrome.
| Case | Exon | Protein change | Nucleotide change | Age of presentation | Gestational age in weeks | Birth weight (grams) | Epilepsy | Learning difficulties | Speech delay | Motor delay | Abnormal movement | Vision problem |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 01a | 11 | p.(Ser498Leu) | c.1506C>T | 4 months | 39 | 3628 | Yes | Yes | Yes | Yes | No | No |
| 02a | 7 | p.(His315Tyr) | c.956C>T | 7 months | Not known | Not known | No | No | No | No | No | No |
| 03b | 7 | p.(Arg322His) | c.965G>A | 8 months | 38 | 1170 | No | No | No | No | No | No |
| 04a | 6 | p.(Arg274Cys) | c.820C>T | 9 months | Not known | 2000 | No | No | No | No | No | No |
| 05b | 11 | p.(Ser498Leu) | c.1493C>T | 3 days | 38 | Not known | No | Yes | Yes | Yes | Yes (ataxia) | Yes (central vision) |
| 06b | 7 | p.(Arg318Ile) | c.953G>T | 1 year | 42 | Not known | No | No | No | No | No | No |
| 07a | 6 | p.(Arg274Cys) | c.820C>T | 10 months | Not known | 3628 | No | No | No | No | No | No |
| 08b | 11 | p.(Pro489Leu) | c.1466C>T | 4 days | Not known | Not known | No | Yes | No | No | No | No |
| 09b | 6 | p.(Arg274Cys) | c.820C>T | 18 months | 42 | Not known | Yes | No | No | No | Yes (tics) | No |
| 10b |
11 | p.(Ser498Leu) | c.1493C>T | 12 hours | 40 | Not known | Yes | Yes | No | No | No | Yes (bilateral cataract) |
| 11b | 7 | p.(Arg318Lys) | c.953G>A | 4 months | 40 | 4960 | Yes | Yes | No | No | No | No |
| 12b | 11 | p.(Ser498Leu) | c.1493C>T | 12 hours | 39 | 3939 | No | No | No | No | No | No |
| 13c | 6 | p.(Arg274Cys) | c.820C>T | 13 months | 40 | 3790 | No | No | No | No | No | No |
| 14c | 7 | p.(Arg322His) | c.965G>A | 4 months | 41 | 4360 | No | No | No | No | No | No |
| 15c | 11 | p.(Ser498Leu) | c.1493C>T | 9 months | 41 | 3850 | Yes | Yes | Yes | Yes | No | No |
| 16c | 12 | p.(Gly499Arg) | c.1495G>A | 7 days | 36 | 4200 | Yes | No | Yes | Yes | No | No |
| 17c | 6 | p.(Arg274Cys) | c.820C>T | 10 months | 40 | 2290 | No | No | Yes | No | Yes (ataxia) | No |
| 18c | 7 | p.(Arg322His) | c.965G>A | 13 months | 40 | 3700 | No | No | No | No | No | No |
| 19c | 7 | p.(Arg322His) | c.965G>A | 8 months | 40 | 3840 | No | No | No | No | No | No |
| 20c | 11 | p.(Ser498Leu) | c.1493C>T | 2 months | 38 | 3680 | Yes | Yes | Yes | Yes | Yes (ataxia, dystonia) | No |
| 21c | 11 | p.(Ile497Met) | c.1491A>G | 10 months | 39 | 2870 | No | No | No | No | No | No |
| 22c | 12 | p.(Gly499Val) | c.1496G>T | 15 days | Not known | 3000 | No | No | No | Yes | No | Yes (optic nerve atrophy) |
| 23c | 11 | p.(Ser498Leu) | c.1493C>T | 4 days | 38 | 3020 | Yes | Yes | Yes | No | Yes (spasticity) | Yes (myopia, amblyopia) |
| 24c | 11 | p.(Asn463Asp) | c.1387A>G | 1 months | 40 | 3580 | Yes | No | Yes | Yes | Yes (dystonia) | No |
| 25c | 11 | P.(Ser498Leu) | c.1493C>T | 9 months | 39 | 3100 | No | No | No | No | No | No |
aGenetic testing done by others; bGenetic testing done by Exeter university; cGenetic testing done by Russian team.
More than half of the patients experience neurodevelopmental disorders (56%) with epilepsy being the most common (36%), followed by learning difficulties, speech delay, motor delay, abnormal movement and vision problem (Fig. 1). A total of 14 cases had an EEG, with 6/14 (43%) showing abnormality. MRI brain was performed in 12 cases with 6/12 showing abnormal findings (50%).
Figure 1.
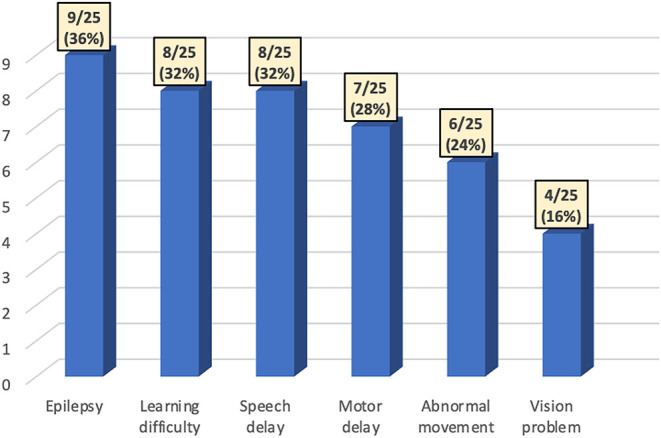
Neurodevelopmental disorders in children with HI/HA syndrome.
Median age of epilepsy onset was 12 months (from 4 weeks to 8 years). Among 13 children with epilepsy, 3 cases presented with generalised tonic-clonic (GTC) seizures, 2 cases with absence seizures, 1 child with a combination of GTC and absence seizures and in 3 cases the type of seizures were not specified. Monotherapy (sodium valproate, lamotrigine) was effective in 6/13 cases (Table 2).
Table 2.
Clinical spectrum in HI/HA children with epilepsy.
| Case no | Gender | Age of presentation | Seizure type | EEG | MRI | Treatment responded | F/H of epilepsy | Asphyxia, sepsis |
|---|---|---|---|---|---|---|---|---|
| 1 | F | 6 months | GTC and absence | Not known | Normal | Lamotrigine, levetiracetam | No | No |
| 9 | F | 12 months | Absence | Normal | Normal | Lamotrigine | No | No |
| 10 | F | 2 years | Absence | Not known | Normal | Lamotrigine | No | No |
| 11 | M | 8 years | GTC | Normal | Normal | Lamotrigine, valproate | Yes | No |
| 15 | M | 15 months | Not known | Abnormal | Not done | Valproate | No | No |
| 16 | F | 11 months | GTC | Abnormal | abnormal | Valproate | No | No |
| 20 | F | 10 months | GTC | Abnormal | Abnormal | Valproate, carbamazepine | No | No |
| 23 | M | 4 years | Not known | Abnormal | Abnormal | Valproate | No | No |
| 24 | M | 1 month | Not known | Abnormal | Abnormal | Valproate | No | No |
EEG, electroencephalogram; GTC, generalised tonic-clonic;, F/H, family history; F, female; M, male.
Neurodevelopmental disorders were associated with early age of presentation (P = 0.02), higher diazoxide requirement (P = 0.04) and a mutation in exon 11 or 12 (P = 0.01). No relevant risk factors for epilepsy were identified (Table 3).
Table 3.
Risk factors for neurodevelopmental disorders.
| Risk factors | Neurodevelopmental disorders, P value | Epilepsy, P value |
|---|---|---|
| Age of presentation (weeks) | 0.02 | 0.16 |
| Gestational age (weeks) | 0.77 | 0.32 |
| Birth weight (gram) | 0.96 | 0.30 |
| Ammonia level | 0.62 | 0.92 |
| Glucose requirement at presentation (mg/kg/min) | 0.07 | 0.54 |
| Diazoxide dosage (mg/kg/day) | 0.04 | 0.22 |
| Gender | 0.58 | 0.56 |
| Prematurity | 0.68 | 0.68 |
| Asphyxia | 0.09 | 0.09 |
| Mutation in exon 11 or 12 | 0.01 | 0.06 |
Almost all patients (24/25) responded very well to diazoxide (median dose 7 mg/kg/day) along with dietary advice (to have mixed meals with carbs and protein and avoiding pure protein-rich meal). One patient underwent partial pancreatectomy due to uncontrolled hypoglycaemia while on octreotide (diazoxide was not used in that index case as it was not available at that time).
Ammonia level was found to be raised in almost all the cases where data were available (17/25) both at normoglycaemia and hypoglycaemia (data were unavailable in 8/25 children but was reported to be raised on referral).
Discussion
HI/HA syndrome is a distinctive form of diazoxide responsive CHI, which is characterised by fasting/postprandial recurrent hypoglycaemia, asymptomatic HA and association with neurological manifestations including epilepsy (11, 12, 13, 14, 15). Children with HI/HA syndrome often present after 4–6 months of life and usually have normal birth weight (7, 16, 17). This was also observed in our study where the median age of presentation was 7 months and the median birth weight was 3654 g. One of the striking findings in our cohort was that 8 of the 25 cases (32%) presented within the neonatal period. This is a higher frequency of neonatal presentation of HI/HA syndrome compared to previously reported studies where it ranges from 15 to 18% (9, 18, 19). Interestingly, all these children with neonatal presentation in our cohort had a mutation within exon 11 or 12. This association was also observed by Su and coworkers (9) who reported three out of four cases of HI/HA syndrome with neonatal presentation having a mutation in exon 11 or 12 of GLUD1. This suggests that an activating GLUD1 mutation within these two exons may result in a more severe phenotype leading to an earlier presentation.
Children with HI/HA syndrome are prone to develop various neurological manifestations including epilepsy (7, 10, 11, 20, 21). Epilepsy has been reported in children with HI/HA syndrome with frequency ranging from 46 to 64% (18, 19, 21, 22). In our study, epilepsy was the most reported neurological disorder with an overall frequency of 32%. Similar to our findings, many other studies have suggested GTC seizures and absence seizures, as the most common type of epilepsy associated with HI/HA syndrome (18, 19). Bahi-Buisson and coworkers (18) reported that 79% of HI/HA with epilepsy respond to monotherapy while the remainder of patients required combination therapy. This is comparable to our study where 46% of epilepsy responded well to monotherapy. We did not find an association between epilepsy and suggested risk factors (age of presentation, gender, gestational age, prematurity, birthweight, asphyxia, ammonia level, glucose requirement at presentation, diazoxide dosage and mutation in exon 11 or 12). The limited number of patients could be a limiting factor for revealing statistical significance. Previous studies have suggested an association between GLUD1 mutations affecting exon 6 or 7 with epilepsy (9, 18, 19).
Learning disabilities, speech delay, motor delay, abnormal movements (tics, dystonia, ataxia, spasticity) and vision problems were other neurological disorders observed in our cohort. Among these, learning disabilities are well reported in previous studies (18, 20, 21, 23). In our series, 32% of patients had learning disabilities, which were frequently associated with epilepsy. An interesting finding in our study was the development of abnormal movements disorder in six cases (24%). Bahi-Buisson and coworkers also reported this finding in 2 out of 22 cases (9%) (18).
The pathophysiology of these neurodevelopmental disorders including epilepsy resulting from an activating GLUD1 mutation is not well understood and is likely to be complex. Many theories have been proposed including chronic HA, late presentation, recurrent hypoglycaemia and increased GDH activity in the brain. However, none of these can fully explain the underlying mechanism. We tried to improve understanding by evaluating the association of different risk factors with neurodevelopmental disorders and epilepsy in our study. High ammonia level, which should be detrimental to the developing brain, was not associated with neurological manifestation including epilepsy (P = 0.62 and P = 0.92, respectively). GLUD1 patients hardly mimic any signs and symptoms of HA (11). Many authors like in our study failed to establish the association between HA and neurodevelopmental disorders in HI/HA syndrome (9, 18). Moreover, lowering blood ammonia with sodium benzoate and N-carbamoyl glutamate led to no improvement (24). Early age of presentation and higher dose of diazoxide were associated with neurological manifestation in our cohort (P = 0.02 and P = 0.04, respectively). We also observed a statistically significant association between mutations in exon 11 or 12 and neurological disorder, whereas previous studies reported the association of exon 6 or 7 with epilepsy (9, 18, 19) or no association with the location of a mutation at all (11, 18, 21, 25).
This may indicate that children with activating GLUD1 mutations located within exon 11 or 12 may have severe disease, with increased GDH activity in different body tissues including the brain and pancreas. Overactivity of GDH in the pancreas might explain the reason for severe phenotype of CHI leading to early presentation and higher dose of diazoxide. Similarly, increased activity of GDH in the brain leading to disequilibrium between glutamate and γ-aminobutyric acid could account for higher frequency of neurological disorders (26, 27, 28).
Whilst it is too early to speculate on why mutations in exons 11 and 12 have a more dramatic effect on the neurological system, understanding the role of the exons is important. Exons 6 and 7 encode the NAD-binding domain that forms catalytic cleft. Exons 11 and 12 encode the antenna, a unique allosteric domain. Mutations confined to exons 11 and 12 interfere with GTP and ATP inhibition of GDH, which leads to increased activity of the enzyme (23).
We can also make an assumption that mutations in exon 11 and 12 lead to hypoglycaemia-induced brain damage due to severe pancreatic disease. However, we doubt hypoglycaemic brain damage to be the main reason for neurological manifestation in these children. All patients are well-controlled with no recurrent hypoglycaemia, most of them show normal brain MRI, and those with abnormal MRI findings are not suggestive of hypoglycaemic brain damage. Our cohort presented with GTC and absence seizures in contrast to hypoglycaemia-induced focal epilepsy.
In this study, the small number of patients, due to the rarity of the condition, and the absence of some data (ammonia level, MRI results) in several cases have lessened statistical significance. Even in the cases where the association was found, we cannot claim robust results. Another limitation is that this clinical study cannot reveal the underlying cause of the link between mutation site and neurological disorders, and further functional tests are needed. We are also unable to exclude the neurological impact of subclinical hypoglycaemic episodes that could have happened before the clinical presentation of the HI.
Conclusion
Children with HI/HA syndrome due to an activating mutation in the GLUD1 gene are prone to neurodevelopmental disorders. Epilepsy is the most common, followed by learning difficulties and speech delay. Except well-known risk factors, we revealed that mutations in exon 11 and 12 are more likely to lead to neurodevelopmental disorders.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This work did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Author contribution statement
Sommayya Aftab: collected data, literature review and wrote manuscript. Diliara Gubaeva: collected data, analysed data; Antonia Dastamani: literature review; Ellada Sotiridou: collected data; Clare Gilbert: collected data; Jayne Houghton: collection of genetic results and analysis; Sarah E Flanagan: collection of genetic results and analysis; Anatoliy Tyulpakov: collection of genetic results and analysis; Maria Melikyan: collected data, analysed data and wrote manuscript; Pratik Shah: conceived the idea, collected data and wrote manuscript; M Melikyan and P Shah: joint last authors.
References
- 1.Aka S, Alanay Y, Boodhansingh KE, Stanley CA, Semiz S. Seizures and diagnostic difficulties in hyperinsulinism–hyperammonemia syndrome. Turkish Journal of Pediatrics 201658541–544. ( 10.24953/turkjped.2016.05.014) [DOI] [PubMed] [Google Scholar]
- 2.Demirbilek H, Hussain K. Congenital hyperinsulinism: diagnosis and treatment update. Journal of Clinical Research in Pediatric Endocrinology 2017969–87. ( 10.4274/jcrpe.2017.S007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hudson RC, Daniel RM. l-glutamate dehydrogenases: distribution, properties and mechanism. Comparative Biochemistry and Physiology: B, Comparative Biochemistry 1993106767–792. ( 10.1016/0305-0491(9390031-y) [DOI] [PubMed] [Google Scholar]
- 4.Kelly A, Ng D, Ferry RJ, Grimberg A, Koo-McCoy S, Thornton PS, Stanley CA. Acute insulin responses to leucine in children with the hyperinsulinism/hyperammonemia syndrome. Journal of Clinical Endocrinology and Metabolism 2001863724–3728. ( 10.1210/jcem.86.8.7755) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li C, Najafi H, Daikhin Y, Nissim IB, Collins HW, Yudkoff M, Matschinsky FM, Stanley CA. Regulation of leucine-stimulated insulin secretion and glutamine metabolism in isolated rat islets. Journal of Biological Chemistry 20032782853–2858. ( 10.1074/jbc.M210577200) [DOI] [PubMed] [Google Scholar]
- 6.Fahien LA, MacDonald MJ, Kmiotek EH, Mertz RJ, Fahien CM. Regulation of insulin release by factors that also modify glutamate dehydrogenase. Journal of Biological Chemistry 198826313610–13614. ( 10.1016/S0021-9258(1868285-7) [DOI] [PubMed] [Google Scholar]
- 7.Hsu BY, Kelly A, Thornton PS, Greenberg CR, Dilling LA, Stanley CA. Protein-sensitive and fasting hypoglycemia in children with the hyperinsulinism/hyperammonemia syndrome. Journal of Pediatrics 2001138383–389. ( 10.1067/mpd.2001.111818) [DOI] [PubMed] [Google Scholar]
- 8.Palladino AA &Stanley CA. The hyperinsulinism/hyperammonemia syndrome. Reviews in Endocrine and Metabolic Disorders 201011171–178. ( 10.1007/s11154-010-9146-0) [DOI] [PubMed] [Google Scholar]
- 9.Su C, Liang XJ, Li WJ, Wu D, Liu M, Cao BY, Chen JJ, Qin M, Meng X, Gong CX. Clinical and molecular spectrum of glutamate dehydrogenase gene defects in 26 Chinese congenital hyperinsulinemia patients. Journal of Diabetes Research 20182018 2802540. ( 10.1155/2018/2802540) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bahi-Buisson N, El Sabbagh S, Soufflet C, Escande F, Boddaert N, Valayannopoulos V, Bellane´-Chantelot C, Lascelles K, Dulac O, Plouin Pet al. Myoclonic absence epilepsy with photosensitivity and a gain of function mutation in glutamate dehydrogenase. Seizure 200817658–664. ( 10.1016/j.seizure.2008.01.005) [DOI] [PubMed] [Google Scholar]
- 11.MacMullen C, Fang J, Hsu BY, Kelly A, de Lonlay-Debeney P, Saudubray JM, Ganguly A, Smith TJ, Stanley CA. & Hyperinsulinism/Hyperammonemia Contributing Investigators. Hyperinsulinism/hyperammonemia syndrome in children with regulatory mutation in the inhibitory guanosine triphosphate binding domain of glutamate dehydrogenase. Journal of Clinical Endocrinology and Metabolism 2001861782–1787. ( 10.1210/jcem.86.4.7414) [DOI] [PubMed] [Google Scholar]
- 12.Weinzimer SA, Stanley CA, Berry GT, Yudkoff M, Tuchman M, Thornton PS. A syndrome of congenital hyperinsulinism and hyperammonemia. Journal of Pediatrics 1997130661–664. ( 10.1016/s0022-3476(9770256-7) [DOI] [PubMed] [Google Scholar]
- 13.Zammarchi E, Filippi L, Novembre E, Donati MA. Biochemical evaluation of a patient with a familial form of leucine-sensitive hypoglycemia and concomitant hyperammonemia. Metabolism: Clinical and Experimental 199645957–960. ( 10.1016/s0026-0495(9690262-0) [DOI] [PubMed] [Google Scholar]
- 14.Parini R, Colombo F, Lombardi AM, Menni F, Beccaria L. Hyperinsulinism plus hyperammonemia. Journal of Pediatrics 1998133800–801. ( 10.1016/s0022-3476(9870158-1) [DOI] [PubMed] [Google Scholar]
- 15.Huijmans JG, Duran M, de Klerk JB, Rovers MJ, Scholte HR. Functional hyperactivity of hepatic glutamate dehydrogenase as a cause of the hyperinsulinism/hyperammonemia syndrome: effect of treatment. Pediatrics 2000106596–600. ( 10.1542/peds.106.3.596) [DOI] [PubMed] [Google Scholar]
- 16.Kapoor RR, Flanagan SE, Arya VB, Shield JP, Ellard S, Hussain K. Clinical and molecular spectrum of 300 patients with congenital hyperinsulinism. European Journal of Endocrinology 2013168557–564. ( 10.1530/EJE-12-0673) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Lonlay P, Fournet JC, Touati G, Groos MS, Martin D, Sevin C, Delagne V, Mayaud C, Chigot V, Sempoux Cet al. Heterogeneity of persistent hyperinsulinaemic hypoglycaemia. A series of 175 cases. European Journal of Pediatrics 200216137–48. ( 10.1007/s004310100847) [DOI] [PubMed] [Google Scholar]
- 18.Bahi-Buisson N, Roze E, Dionisi C, Escande F, Valayannopouslos V, Feillet F, Heinrichs C, Chadefax-Vekemans B, Dan B, Lonlay PD. Neurological aspects of hyperinsulinism–hyperammonaemia syndrome. Developmental Medicine and Child Neurology 200850945–949. ( 10.1111/j.1469-8749.2008.03114.x) [DOI] [PubMed] [Google Scholar]
- 19.Kapoor RR, Flanagan SE, Fulton P, Chakrapani A, Chadefaux B, Ben-Omran T, Banerjee I, Shield JP, Ellard S, Hussain K. Hyperinsulinism–hyperammonaemia syndrome: novel mutations in the GLUD1 gene and genotype–phenotype correlations. European Journal of Endocrinology 2009161731–735. ( 10.1530/EJE-09-0615) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miki Y, Taki T, Ohura T, Kato H, Yanagisawa M, Hayashi Y. Novel missense mutations in the glutamate dehydrogenase gene in the congenital hyperinsulinism–hyperammonemia syndrome. Journal of Pediatrics 200013669–72. ( 10.1016/s0022-3476(0090052-0) [DOI] [PubMed] [Google Scholar]
- 21.Raizen DM, Brooks-Kayal A, Steinkrauss L, Tennekoon GI, Stanley CA, Kelly A. Central nervous system hyperexcitability associated with glutamate dehydrogenase gain of function mutations. Journal of Pediatrics 2005146388–394. ( 10.1016/j.jpeds.2004.10.040) [DOI] [PubMed] [Google Scholar]
- 22.Grimaldi M, Karaca M, Latini L, Brioudes E, Schalch T, Maechler P. Identification of the molecular dysfunction caused by glutamate dehydrogenase S445L mutation responsible for hyperinsulinism/hyperammonemia. Human Molecular Genetics 2017263453–3465. ( 10.1093/hmg/ddx213) [DOI] [PubMed] [Google Scholar]
- 23.Stanley CA, Fang J, Kutyna K, Hsu BY, Ming JE, Glaser B, Poncz M. Molecular basis and characterization of the hyperinsulinism/hyperammonemia syndrome: predominance of mutations in exons 11 and 12 of the glutamate dehydrogenase gene. HI/HA Contributing Investigators. Diabetes 200049667–673. ( 10.2337/diabetes.49.4.667) [DOI] [PubMed] [Google Scholar]
- 24.De Lonlay P, Benelli C, Fouque F, Ganguly A, Aral B, Dionisi-Vici C, Touati G, Heinrichs C, Rabier D, Kamoun Pet al. Hyperinsulinism and hyperammonemia syndrome: report of twelve unrelated patients. Pediatric Research 200150353–357. ( 10.1203/00006450-200109000-00010) [DOI] [PubMed] [Google Scholar]
- 25.Santer R, Kinner M, Passarge M, Superti-Furga A, Mayatepek E, Meissner T, Schneppenheim R, Schaub J. Novel missense mutations outside the allosteric domain of glutamate dehydrogenase are prevalent in European patients with the congenital hyperinsulinism–hyperammonemia syndrome. Human Genetics 200110866–71. ( 10.1007/s004390000432) [DOI] [PubMed] [Google Scholar]
- 26.Owens DF, Kriegstein AR. Maturation of channels and receptors: consequences for excitability. International Review of Neurobiology 20014543–87. ( 10.1016/s0074-7742(0145006-9) [DOI] [PubMed] [Google Scholar]
- 27.Kriegstein AR, Owens DF. GABA may act as a self-limiting trophic factor at developing synapses. Science STKE 20012001pe1. ( 10.1126/stke.2001.95.pe1) [DOI] [PubMed] [Google Scholar]
- 28.Owens DF, Kriegstein AR. Developmental neurotransmitters? Neuron 200236989–991. ( 10.1016/s0896-6273(0201136-4) [DOI] [PubMed] [Google Scholar]