

Abstract
G4C2 repeat expansion in C9orf72 causes the most common familial frontotemporal dementia and amyotrophic lateral sclerosis (C9FTD/ALS). The pathogenesis includes haploinsufficiency of C9orf72, which forms a protein complex with Smcr8, as well as G4C2 repeat-induced gain of function including toxic dipeptide repeats (DPRs). The key in vivo disease-driving mechanisms and how loss- and gain-of-function interplay remain poorly understood. Here, we identified dysregulation of a lysosome-ribosome biogenesis circuit as an early and key disease mechanism using a physiologically relevant mouse model with combined loss- and gain-of-function across the aging process. C9orf72 deficiency exacerbates FTD/ALS-like pathologies and behaviors in C9ORF72 bacterial artificial chromosome (C9-BAC) mice with G4C2 repeats under endogenous regulatory elements from patients. Single nucleus RNA sequencing (snRNA-seq) and bulk RNA-seq revealed that C9orf72 depletion disrupts lysosomes in neurons and leads to transcriptional dysregulation of ribosomal protein genes, which are likely due to the proteotoxic stress response and resemble ribosomopathy defects. Importantly, ectopic expression of C9orf72 or its partner Smcr8 in C9FTD/ALS mutant mice promotes lysosomal functions and restores ribosome biogenesis gene transcription, resulting in the mitigation of DPR accumulation, neurodegeneration as well as FTD/ALS-like motor and cognitive behaviors. Therefore, we conclude that loss- and gain-of-function crosstalk in C9FTD/ALS converges on neuronal dysregulation of a lysosome-ribosome biogenesis circuit leading to proteotoxicity, neurodegeneration and behavioral defects.
Introduction
G4C2 hexanucleotide repeat expansion in the first intron of chromosome 9 open reading frame 72 (C9ORF72) causes the most common familial frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) (collectively, C9FTD/ALS) (1,2). FTD represents the second most common dementia in patients under age 60 years with degeneration of cortical neurons in frontal and temporal lobes. ALS is a common motor neuron (MN) disease that affects the upper and lower MNs and the corticospinal tract. Dysfunction of cognitive behaviors and language in FTD can often be detected in up to 50% of ALS patients (3,4). In addition to clinical overlap, FTD and ALS also share pathological hallmarks (5,6). It has been estimated that C9ORF72-associated ALS accounts for about 40% of familial ALS and about 5–10% of sporadic ALS cases (1,2). Therefore, mechanistic and therapeutic studies of C9FTD/ALS could have broad implications for neurodegeneration and neurodegenerative disorders.
C9FTD/ALS pathogenesis is considered to be attributed to C9ORF72 haploinsufficiency (loss of function) and gain of toxicity. The latter acts through toxic RNAs from repeat transcription as well as dipeptide repeat proteins (DPRs) due to repeat-associated non-AUG (RAN) translation (7–10). RNA foci containing sense G4C2 and antisense G2C4 are detected in patient tissues and may sequester RNA-binding proteins (RBPs) leading to a loss of normal RBP function (2,11–13). RAN translation produces various DPRs, including poly-glycine-arginine (GA) and poly-glycine-arginine (GR) from sense G4C2 transcripts, poly-proline-alanine (PA) and poly-proline-arginine (PR) from antisense G2C4, and poly-glycine-proline (GP) from both sense and antisense repeat transcripts. DPR accumulation has various pathological consequences, including deficits in mitochondrial function, ER/oxidative stress, nucleocytoplasmic transport, proteasome regulation and stress granule dynamics (7,8,10). Among these gain-of-toxicity mechanisms, ribosome biogenesis and translational dysregulation are well described. Poly(GR) and poly(PR) are enriched in the nucleolus, the site of ribosome biogenesis. They directly bind to ribosome proteins (RPs) and inhibit ribosome, disrupt stress granule dynamics and translation, leading to neurotoxicity and neurodegeneration (14–16). In spite of these studies, the early and key in vivo disease mechanisms driving gain of toxicity in C9FTD/ALS remain unclear.
C9orf72 forms a protein complex with Smcr8, both of which are downregulated in C9FTD/ALS patient tissues and mouse models (10). C9orf72 or Smcr8 depletion exacerbates the gain of toxicity (17–19), leading to more severe neurodegeneration and behavioral deficits compared to that in loss- or gain-of-function mouse models alone (18,20). These studies suggest loss- and gain-of-function crosstalk in C9FTD/ALS pathogenesis, but the underlying mechanisms remain largely unclear. C9orf72/Smcr8-containing complex regulates autophagy-lysosome functions, disruption of which leads to proteotoxic stress. We previously reported that C9orf72 or Smcr8 depletion leads to mTORC1 overactivation due to impaired lysosomal degradation and exocytosis (21), while mTORC1 activation inhibits autophagy-lysosomal functions and promotes protein synthesis (22), which further strengthens proteotoxic stress. These studies have been performed in macrophages from C9orf72 and Smcr8 knockout (KO) mice (21). Interestingly, another contributing factor to proteotoxic stress may be ribosome biogenesis dysregulation (23,24) which can occur in C9FTD/ALS due to DPR binding and sequestering of RPs, leading to abnormal orphan RP accumulation (14,15). These studies raise the possibility that DPR accumulation-mediated ribosome biogenesis defects overload a C9orf72-deficient autophagy-lysosome system and leads to exacerbated proteotoxic stress. This may underlie the gain- and loss-of-function crosstalk in the neurodegeneration characteristic of the disease.
Here, we report that C9orf72 deficiency promotes DPR accumulation and behavioral deficits in C9-BAC mice, revealing loss- and gain-of-function crosstalk. Conversely, ectopic expression of C9orf72 or Smcr8 in C9FTD/ALS mutant mice mitigates DPR accumulation, neurodegeneration and behavioral deficits. Mechanistic studies reveal that the combinatorial dysregulations in autophagy-lysosome system and ribosome biogenesis lead to exacerbated proteotoxic stress and C9FTD/ALS pathologies, which results in neurodegeneration and abnormal cognitive and motor behaviors.
Results
C9orf72 deficiency induces motor and cognitive behavioral abnormalities in C9-BAC mice
We previously reported that C9-BAC mice with complete C9orf72 loss exhibited motor behavioral deficits (20), but the effects of C9orf72 haploinsufficiency remained uncharacterized. We generated C9-BAC mice with different dose reductions of C9orf72. Western blots (WBs) from central nervous system (CNS) tissues confirmed C9orf72 and Smcr8 protein dose reduction in the background of C9-BAC mice (Fig. 1A, Supplementary Material, Fig. S1A and B). Atg101 is associated with the C9orf72/Smcr8 protein complex (25), and its protein levels were not changed, suggesting specificity of C9orf72 dose reduction (Fig. 1A).
Figure 1.

C9orf72 deficiency induces motor and cognitive behavioral abnormalities in C9-BAC mice. (A) Western blot analysis of C9orf72 and Atg101 protein in spinal cord tissues from C9-BAC mice with different C9orf72 dose reductions. β-Actin serves as the loading control. (B) Rotarod tests showed significant motor coordination defects at day 3 in C9orf72+/−;C9-BAC mice at 10–12 months and at day 4 in both C9-BAC and C9orf72+/−;C9-BAC mice at 18–20 months. (C, D) Learning curves of rotarod test reveal impaired motor learning in C9-BAC mice with C9orf72 half depletion at 10–12 months and in C9-BAC mice with or without C9orf72 half depletion at 18–20 months. Day 1 (1d) measurement is compared to that of 2d, 3d or 4d. (E) Grip strength tests reveal motor strength defects in C9-BAC mice with C9orf72 half depletion. (F) Three chamber tests show social memory defects in C9-BAC and C9orf72+/−;C9-BAC mice. (G) Preference indices in the social memory session of the three-chamber test confirmed the social memory defects in C9-BAC and C9orf72+/−;C9-BAC mice. (H) Novel object recognition defects in C9orf72+/−;C9-BAC mice but not C9-BAC mice at age of 18–20 months. All data are presented as mean ± SEM using numbers (n = 8–14) of mice as indicated. Statistical analyses were performed with one-way or two-way ANOVA with Bonferroni’s post hoc test (*P < 0.05, **P < 0.01, ***P < 0.001, n.s. represents no significant difference detected).
To examine how C9orf72 haploinsufficiency affects motor behaviors of C9-BAC mice, we raised a cohort of WT, C9-BAC and C9orf72+/−;C9-BAC mice. Previous studies reported that these C9-BAC female mice displayed more robust and consistent phenotypes than male mice (26). Therefore, we focused on female mice for behavioral studies. C9orf72 heterozygous or homozygous knockout alone in mice did not produce neurodegeneration or motor deficits (27). C9orf72 complete loss, which does not occur in C9FTD/ALS patients, led to autoimmune disorders in mice (28,29). For these reasons, and for reducing total animal usage, we excluded C9orf72+/− and C9orf72−/− mice from our behavior studies. There were no significant differences among WT, C9-BAC and C9orf72+/−;C9-BAC mice in their survival and body weights around 10–12 months old (data not shown), when motor behavior tests were started. Aged mice with different genotypes also exhibited similar body weights (Supplementary Material, Fig. S1C), although we could not track their survival due to COVID-19. We measured their motor coordination and balance using an accelerating (5–40 rpm in 5 min) rotarod test. Mice were given three to five trials per day for four consecutive days with an intertrial interval of 20 min. At 10–12 months, C9orf72+/−;C9-BAC mice exhibited decreased latency to fall at day 3, while C9-BAC mice did not (Fig. 1B and C). For the 18- to 20-month-old group, both types of mice had motor coordination defects at day 4 (Fig. 1D). WT and C9-BAC mice at 10–12 months exhibited an increased time on the rotarod during the course of these four consecutive days, suggesting a capacity to actively learn (Fig. 1C). In contrast, C9orf72+/−;C9-BAC mice did not show obvious improvement (Fig. 1C), indicating motor learnings deficits, which occurred in both C9-BAC and C9orf72+/−;C9-BAC mice at 18–20 months (Fig. 1D). Next, we performed grip strength tests and found that C9orf72 haploinsufficiency induced motor strength defects in C9-BAC mice (Fig. 1E). Together, these results suggest that C9orf72 haploinsufficiency induced motor behavioral deficits in C9-BAC mice.
To determine how C9orf72 haploinsufficiency impacts social cognitive behaviors of C9-BAC mice, we performed a three-chamber assay. C9-BAC mice exhibited comparable sociability as WT mice. Both groups showed stronger preference for the stranger mouse than the object (data not shown). While in the social novelty session, WT mice spent significantly more time interacting with stranger 2 (S2) compared to stranger 1 (S1) mice at both adult and aged stages. In contrast, there was no significant difference in social interaction time with S1 versus S2 mice for C9-BAC as well as C9orf72+/−;C9-BAC mice (Fig. 1F and G). These results suggest that C9-BAC mice with and without C9orf72 haploinsufficiency developed social memory defects. Next, we performed novel object recognition tests to measure hippocampus-dependent recognition memory. There was no difference between WT and C9-BAC mice in novel object preference (Fig. 1H). In contrast, C9orf72+/−;C9-BAC mice displayed novel object recognition defects at 18–20 months (Fig. 1H). Together, these results suggest that C9orf72 haploinsufficiency induced cognitive behavioral deficits in C9-BAC mice.
C9orf72 haploinsufficiency exacerbates DPR accumulation in C9-BAC mice
It has been reported that C9ORF72 activity promotes dipeptide poly(PR) clearance in motor neurons (MNs) derived from patient induced pluripotent stem cells (30). We hypothesized that C9orf72 deficiency promotes the accumulation of DPRs, which contributes to motor and cognitive behavioral abnormalities in C9FTD/ALS mouse models. To test this hypothesis, we focused on poly(GA) and poly(GP) due to the commercial availability of their antibodies. In addition, the number of inclusions of poly(PR) and poly(PA) is relatively low in postmortem C9FTD/ALS patient brains (31,32). It has been reported that poly(GA) is toxic in various cell types and animal models (33–35); ectopic expression of poly(GA) triggered motor deficits in mice (36). Immunohistochemical (IHC) staining showed that poly(GA) and poly(GP) are mainly expressed in neurons in brains (Supplementary Material, Fig. S2). In contrast to the absence of poly(GA) in stains of WT mice, poly(GA) was clearly detected in the motor cortex and spinal cord tissues of 10- to12-month-old C9-BAC mice (Fig. 2A and C). Importantly, C9orf72 haploinsufficiency significantly increased the percentage of poly(GA)-positive cells in C9-BAC mice (Fig. 2A–D). Similar IHC stains were performed and found that poly(GP) is also expressed in the motor cortex and spinal cord tissues of C9-BAC mice at a reduced frequency compared to poly(GA) (Fig. 2E–H). We barely detected poly(GP) in WT mice, while C9orf72 haploinsufficiency significantly increased the percentage of poly(GP)-positive cells in C9-BAC mice (Fig. 2E–H). Interestingly, complete loss of C9orf72 reduced DPRs in C9-BAC mice (Fig. 2B–H), which is likely due to decreased RAN translation from the loss of ribosome machinery and will be further discussed below. Together, these results suggest that C9orf72 haploinsufficiency exacerbates DPR accumulation in C9-BAC mice.
Figure 2.

C9orf72 haploinsufficiency exacerbates DPR accumulation in C9-BAC mice. (A, C, E, G) Representative confocal images of coronal sections stained with antibodies against poly(GA) (red) and poly(GP) (green). Age-matched 10- to 12-month-old mice were used to prepare sections in the areas of the motor cortex (A, E) and spinal cord (C, G). Lower panels are enlargements of white boxed areas in upper panels. Scale bars: 8 μm (upper panels); 2 μm (lower panels). (B, D, F, H) Quantification of the relative numbers of GA- and GP-positive cells out of total cells among age-matched mice with different genotypes as indicated. Error bars represent SEM of four independent experiments; 12–16 CNS sections from each genotype were counted in each experiment. Statistical analyses were performed with one-way ANOVA with Bonferroni’s post hoc test (n = 4–6 mice, *P < 0.05, **P < 0.01, ***P < 0.001), n.s. represents no significant difference detected.
C9orf72depletion induces transcriptional downregulation of ribosomal protein genes in C9-BAC mice
To investigate molecular mechanisms underlying C9orf72 deficiency-induced pathologies and behavioral abnormalities in C9-BAC mice, we performed single nucleus RNA sequencing (snRNA-seq) on frontal cortex tissues from 10-month-old mice. It is technically challenging to effectively isolate viable single cells in aged adult mouse brains, so we chose the single nucleus as opposed to the single cell approach. Given relatively mild phenotypes induced by C9orf72 haploinsufficiency, we included C9orf72−/−;C9-BAC in our snRNA-seq studies. We collected frontal cortex tissues from WT and C9-BAC mice with different dose reductions of C9orf72 (n = 4 per group) to control for variability in the dissection. We captured about 17 975 single nuclei and sequenced a median of 1885 genes per nucleus. Considering that nuclear RNAs were profiled, 54.2% of unique molecular identifiers were mapped to exons and 31.5% to introns. Therefore, the gene expression profiles of nuclei likely reflected nascent transcripts and the cellular transcriptome. After unbiased clustering of nuclear profiles, cell-type identity was defined based on the top differentially expressed genes (DEGs) and expression of known cell-type marker genes. We identified 15 primary cell types (Fig. 3A), including four types of excitatory neurons (Ex1–4), three types of inhibitory neurons (In1–3), medium spiny neurons, astrocytes (Ast), oligodendrocytes (Olig1–2), oligodendrocyte precursor cells, microglia, enteric glia, and vascular endothelial cells (VECs). These cell cluster markers and their constituent cell types were well separated (Fig. 3A), suggesting that our snRNA-seq data were of high integrity.
Figure 3.

Single nucleus RNA-sequencing (snRNA-seq) analysis reveals transcriptional downregulation of ribosomal protein (RP) genes in mutant mice. (A, B) UMAP visualization reveals 15 clusters in the frontal cortex from 10-month-old WT (5093), C9-BAC (3210), C9orf72+/−;C9-BAC (5090) and C9orf72−/−;C9-BAC (4582) mice. (C) Differentiated expressed genes (DEGs) in individual cell clusters between WT and C9orf72+/−;C9-BAC mice. The right axis indicates the numbers of DEGs that overlap with neurodegenerative disease-related genes, which are obtained from the Neurodegenerative Disease Variation Database (NDDVD). (D, E) Heatmap illustrating RP gene expression changes in WT, C9-BAC, C9orf72+/−;C9-BAC and C9orf72−/−;C9-BAC mice within individual excitatory neurons (D) and microglia (E). (F) RT-PCR analysis of Rps6 and Rps7a mRNA expression in frontal cortex. Z-score value is used for visualization in snRNA-seq data.
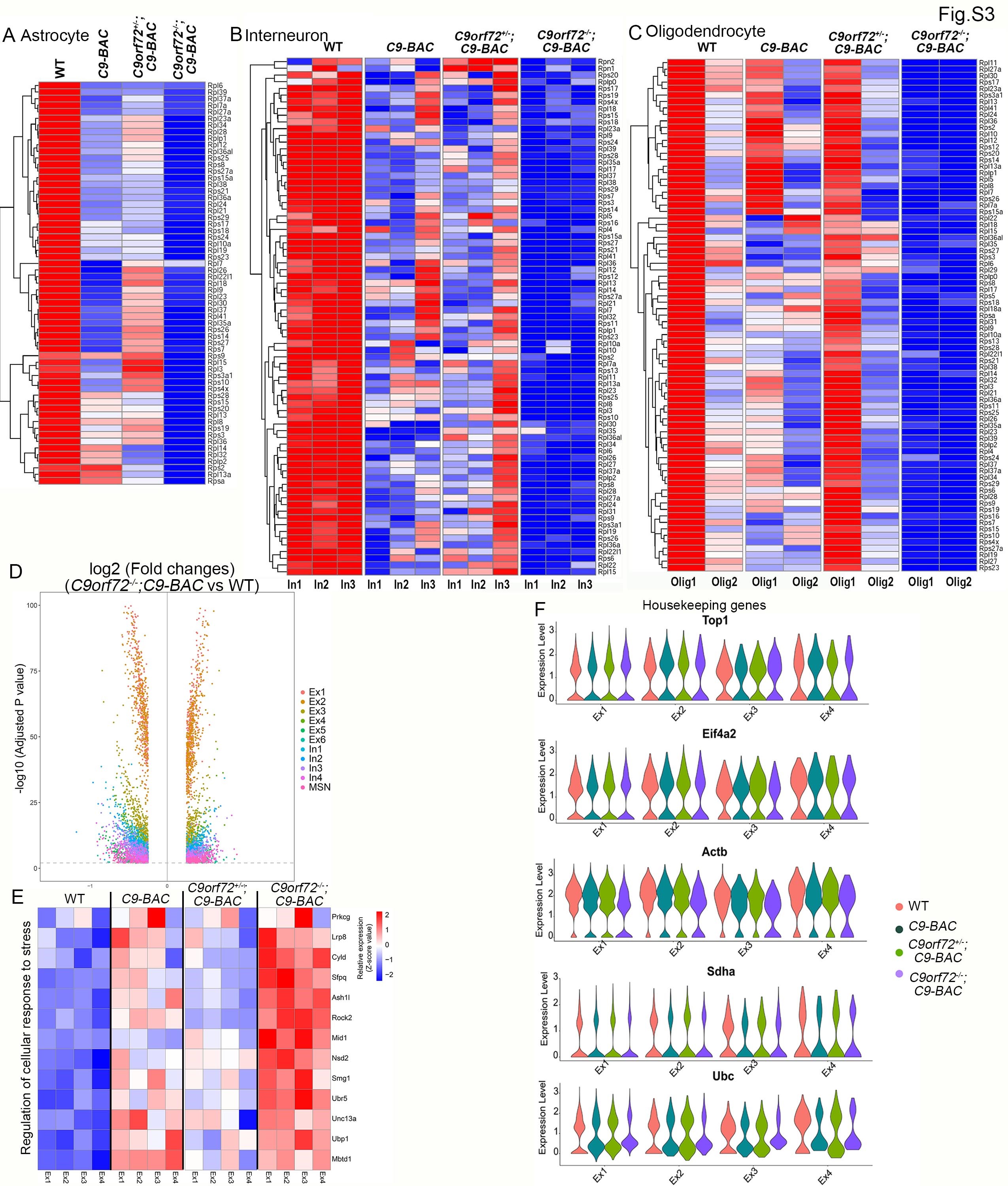
There were no significant changes in the numbers of individual cell types among WT and C9-BAC mice with different dose reduction of C9orf72 (Fig. 3B). In all groups, excitatory neurons accounted for the most abundant cell types. These results suggest that there is no significant reduction of specific cell types, which is consistent with the absence of neurodegeneration in mutant brains at this stage (Supplementary Material, Fig. S7). Next, we examined DEGs in each cell type between WT and C9orf72+/−;C9-BAC mice (Fig. 3C), which is the most representative genotype for modeling C9FTD/ALS. DEGs in C9orf72+/−;C9-BAC mice overlap with neurodegenerative disease-related genes, which are obtained from Neurodegeneration Disease Variation Database (NDDVD) (Fig. 3C). Hierarchical clustering based on log-transformed relative (fold) changes of DEGs in each cell type revealed that the top downregulated genes in C9-BAC with different C9orf72 dose reduction belong to the ribosomal protein (RP) genes, which are most severely downregulated in C9orf72−/−;C9-BAC brains at different cell types, including excitatory neurons (Fig. 3D), microglia (Fig. 3E), astrocytes (Supplementary Material, Fig. S3A), interneurons (Supplementary Material, Fig. S3B) and oligodendrocytes (Supplementary Material, Fig. S3C). The number of upregulated and downregulated DEGs was similar between WT and C9orf72−/−;C9-BAC mice (Supplementary Material, Fig. S3D). We also identified upregulated genes enriched in the cellular stress response in C9orf72−/−;C9-BAC mice, providing a DEG example for an alternative pathway to proteotoxic stress (Supplementary Material, Fig. S3E). In addition, the expression of the housekeeping genes is identical among different genotypes (Supplementary Material, Fig. S3F). These results support the idea that there is no systematic bias of our snRNA-seq analysis. Furthermore, RT-PCR analysis confirmed the upregulation of ribosome biogenesis genes Rps6 and Rps7a in C9orf72−/−;C9-BAC mice (Fig. 3F). The loss of ribosome biogenesis machinery due to RP gene downregulation is expected to decrease RAN translation. Indeed, we found that poly(GA) and poly(GP) were significantly reduced in C9orf72−/−;C9-BAC compared to C9orf72+/−;C9-BAC mice (Fig. 2D, F, G and H). Together, these results suggest that C9orf72 depletion induced a robust transcriptional downregulation of RP genes in C9-BAC brains, connecting lysosomal dysfunctions with ribosome biogenesis defects in C9FTD/ALS.
C9orf72 or Smcr8 mitigates lysosomal abnormalities in C9FTD/ALS mutant mice
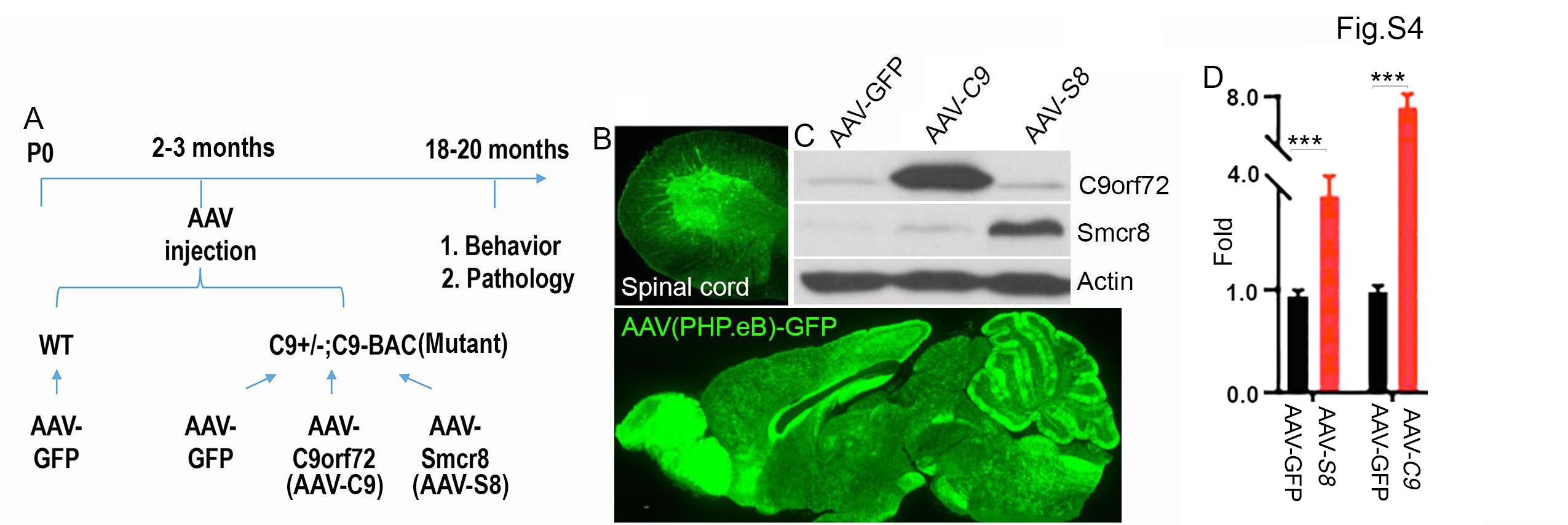
After C9orf72 loss-of-function perturbation, we investigated whether C9orf72 ectopic expression can mitigate C9FTD/ALS-like pathologies and behaviors. We previously showed that C9orf72 forms a protein complex with Smcr8 (25), and Smcr8 deficiency exacerbates DPR accumulation in C9-BAC mice (19). Therefore, our gain-of-function perturbation includes both C9orf72 and Smcr8. We raised a cohort of WT and C9FTD/ALS mice (C9orf72+/−;C9-BAC) and performed retro-orbital injection of AAV-PHP.eB viruses expressing C9orf72 or Smcr8 at 2–3 months followed by analysis at 18–20 months (Supplementary Material, Fig. S4A). AAV-PHP.eB viruses can cross the blood–brain barrier and effectively express target genes in the CNS (37), especially in the brain and spinal cord (Supplementary Material, Fig. S4C). WBs confirmed the ectopic expression of C9orf72 and Smcr8 (Supplementary Material, Fig. S4C and D). From here on, we refer C9orf72+/−;C9-BAC as mutant, C9orf72+/−;C9-BAC with AAV-PHP.eB-C9orf72 as AAV-C9 and C9orf72+/−;C9-BAC with AAV-PHP.eB-Smcr8 as AAV-S8.
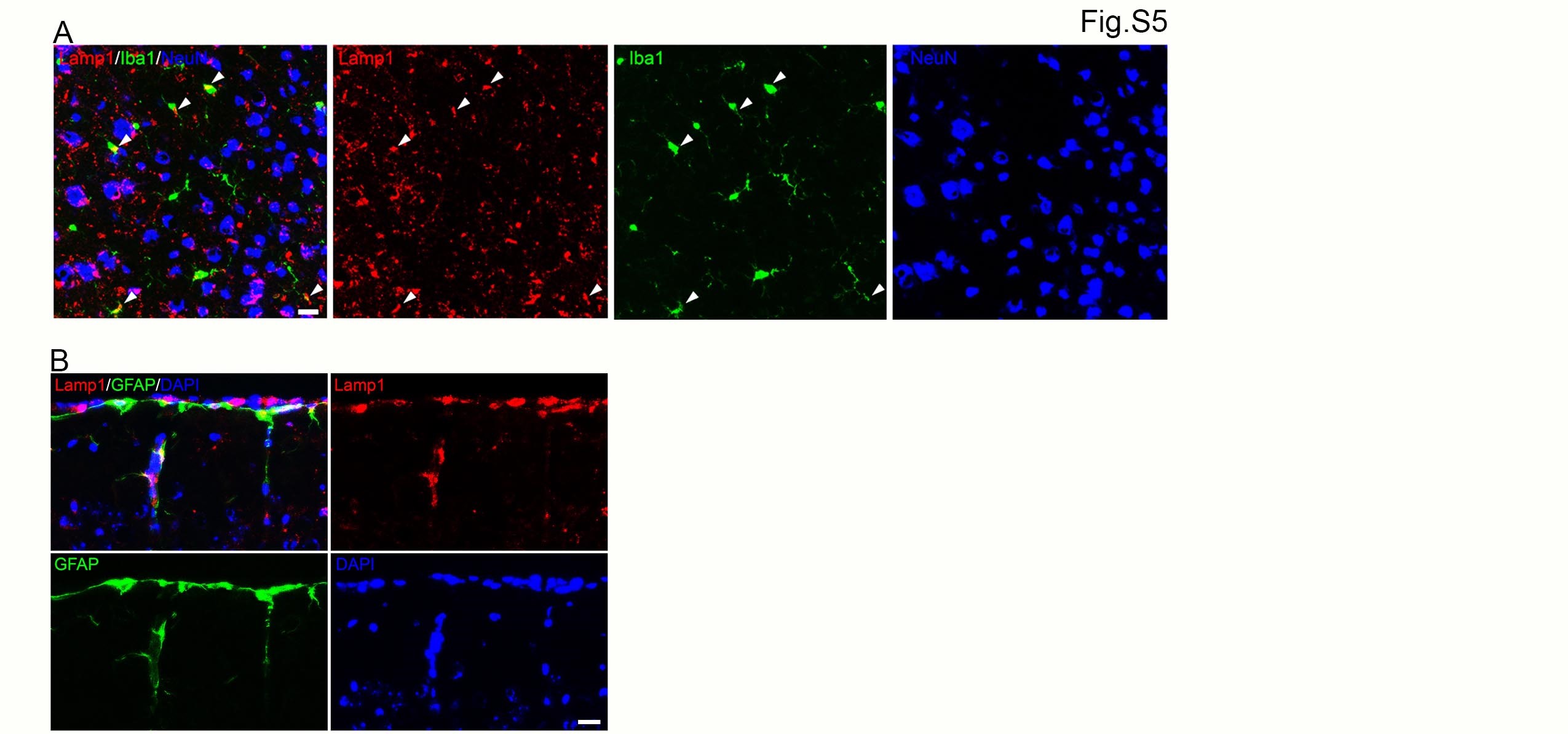
We previously reported that the C9orf72/Smcr8-containing complex regulates autophagy-lysosomal functions (25), and their individual or combined KO led to enlarged lysosomes and impaired lysosomal functions in macrophages (21). Therefore, we examined lysosomal morphology by IHC staining on motor cortex tissues with antibodies against Lamp1. Despite its localization in microglia and astrocytes, Lamp1 is mainly found in NeuN-positive neurons (Supplementary Material, Fig. S5), which are the most abundant cells in brains. Compared to WT controls, mutant brains exhibited a significant increase in the intensity of Lamp1, suggesting increased proteotoxic stress (Fig. 4A and B). AAV-C9 and AAV-S8 expression reduced the aberrant Lamp1 upregulation (Fig. 4A and B). Lipofuscin is an autofluorescent lipopigment and its accumulation is an indication of aging-related lysosome dysfunction (38,39). Altered lysosomal morphology prompted us to examine its function by focusing on lipofuscin intensity and its distribution pattern. Compared to WT controls, mutant motor cortices displayed more intense lipofuscin that is mitigated in AAV-C9 or AAV-S8 brains (Fig. 4C). Statistical analysis showed that mutant brains displayed a significant increase in the percentage of lipofuscin-positive cells, which is reduced by AAV-C9 or AAV-S8 (Fig. 4D). Lipofuscin aggregation is considered an effector of aging and represents a risk factor for neurodegeneration (39). Therefore, we monitored relative ratios between sparse and aggregated lipofuscin. There is a decrease in the percentage of sparse lipofuscin-positive cells in mutants, which were restored by AAV-C9 or AAV-S8 (Fig. 4E and F). Compared to WT, mutant motor cortices exhibited an increase in the percentage of cells with lipofuscin aggregations, which is significantly mitigated in AAV-C9 or AAV-S8 brains (Fig. 4E and F). Together, these results suggest that ectopic expression of C9orf72 or Smcr8 mitigated the aberrant lysosomal morphologies and functions in mutant brains.
Figure 4.

C9orf72 and Smcr8 rescued lysosomal abnormalities in C9FTD/ALS mutant mice. (A) Representative confocal image of motor cortex from 20-month-old mice stained with antibodies against Lamp1 (red) and Hoechst stains for nuclei (blue). The lower panels are enlargements of white boxed areas in the upper panels. Scale bars: 200 μm (upper panels), 20 μm (lower panels). (B) Quantification of Lamp1 staining signal intensity normalized to WT. Note that AAV-C9orf72 or AAV-Smcr8 expression rescued the aberrantly increased Lamp1 intensity in C9TFD/ALS mutant mice. (C, E) Representative confocal images of lipofuscin accumulation in a 20-month-old mouse motor cortex. Hoechst stains nuclei (blue). Right panels in E are enlargements of white boxed areas in the left panels. Scale bars: 20 μm in C and 10 μm in E right panels. (D) Quantification of the percentage of lipofuscin-positive cells. (F) Quantification of the percentage of sparse and aggregated lipofuscin-positive cells out of total lipofuscin-positive cells. Mutant: C9orf72+/−;C9-BAC; AAV-C9: C9orf72+/−;C9-BAC with AAV-PHP.eB-C9orf72; AAV-S8: C9orf72+/−;C9-BAC with AAV-PHP.eB-Smcr8. Data information: For all analyses, data are presented as mean ± SEM. N = 6 mice with one-way ANOVA with Bonferroni’s post hoc test (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
C9orf72 or Smcr8 mitigates gene dysregulations in C9FTD/ALS mutant mice
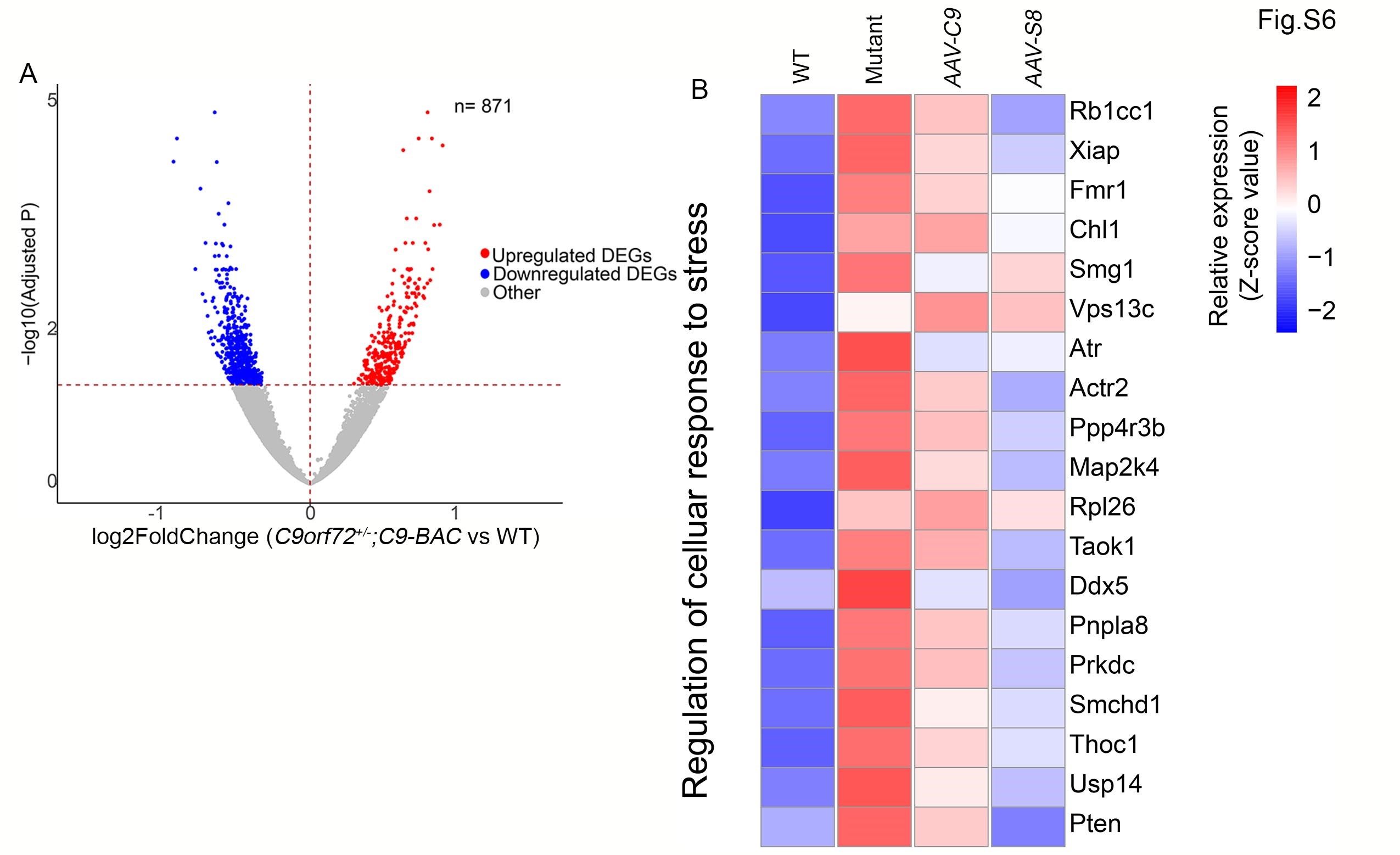
It has been reported that aberrant ribosome biogenesis leads to proteotoxicity and induces stress responses including the transcriptional downregulation of RP genes (40,41). DPRs bind and sequester RPs in the nucleolus (14,15), and inhibit ribosome machine functions (16). Therefore, we interpret the transcriptional downregulation of RP genes by C9orf72 depletion as a stress response to the DPR-mediated ribosome biogenesis disruption in C9-BAC mice. To examine how ectopic expression of C9orf72 or Smcr8 impacts gene expression in mutant brains, we performed bulk RNA-seq using a 20-month-old mouse motor cortex. Hierarchical clustering based on log-transformed relative (fold) changes of DEGs revealed that AAV-C9 and AAV-S8 partially reversed both downregulated and upregulated genes in mutant brains (Fig. 5A). Among those 564 downregulated genes in the mutant motor cortex, ribosome biogenesis and lysosomal functions are the two pathways mainly enriched (Fig. 5B and C). Importantly, a significant portion of these downregulated genes in both pathways were normalized close to WT conditions (Fig. 5B and C). Again the number of the upregulated and downregulated DEGs in C9orf72+/−;C9-BAC compared to WT mice was similar (Supplementary Material, Fig. S6A), and mutant brains exhibited an upregulation of cellular stress response genes (Supplementary Material, Fig. S6B), suggesting no systematic bias of our bulk RNA-seq analysis. Together, these results suggest that ectopic expression of C9orf72 or Smcr8 in mutant mice mitigates transcriptional downregulation of genes involved in ribosome biogenesis and lysosomal functions.
Figure 5.

C9orf72 and Smcr8 reduced ribosome biogenesis and lysosome gene dysregulations in C9FTD/ALS mutant mice. Heatmaps illustrate that AAV-C9orf72 or AAV-Smcr8 reduced the aberrant downregulation of ribosome biogenesis and lysosome genes in the mutant motor cortex at 20 months. Genes with FDR P < 0.05 are plotted based on hierarchical clustering analysis. Z-score value is used for visualization in bulk RNA-seq data. For all analyses, data are presented as mean ± SEM. N = 3–5 mice with one-way ANOVA with Bonferroni’s post hoc test (**P < 0.01 and ***P < 0.001).
C9orf72 or Smcr8 mitigates DPR accumulation and neurodegeneration in mutant mice
Our C9orf72 loss-of-function perturbation showed that C9orf72 haploinsufficiency exacerbated DPR accumulation in 10- to 12-month-old C9-BAC mice. Following this, we investigated how ectopic expression of C9orf72 or Smcr8 impacts DPR accumulation. There are generally more DPR foci in mutant mice at 20 versus 12-months, suggesting aging-dependent DPR accumulation. Importantly, the percentage of poly(GA)- and poly(GP)-positive cells is significantly reduced in AAV-C9 and AAV-S8 mice cortices compared to mutant motor cortices (Fig. 6A–D). These results suggest that ectopic expression of C9orf72 or Smcr8 mitigates DPR accumulation in C9FTD/ALS mutant brains.
Figure 6.

C9orf72 and Smcr8 reduced DPR accumulation in C9FTD/ALS mutant mice. (A, C) Representative confocal images of motor cortex coronal sections stained with antibodies against poly(GA) (red in A) and poly(GP) (red in C) in 20-month-old WT and mutant mice with or without the expression of AAV-C9orf72 or AAV-Smcr8. White arrows represent poly(GA) and poly(GP)-positive foci. White arrows represent poly(GA) and poly(GP)-positive foci. Scale bars: 20 μm. (B, D) Quantification of the percentage of GA- and GP-positive cells out of total cells. Note that AAV-C9orf72 or AAV-Smcr8 significantly mitigated DPR accumulation in C9TFD/ALS mutant mice. For all analyses, data are presented as mean ± SEM. N = 3–6 mice with one-way ANOVA with Bonferroni’s post hoc test (**P < 0.01, ***P < 0.001 and ****P < 0.0001).
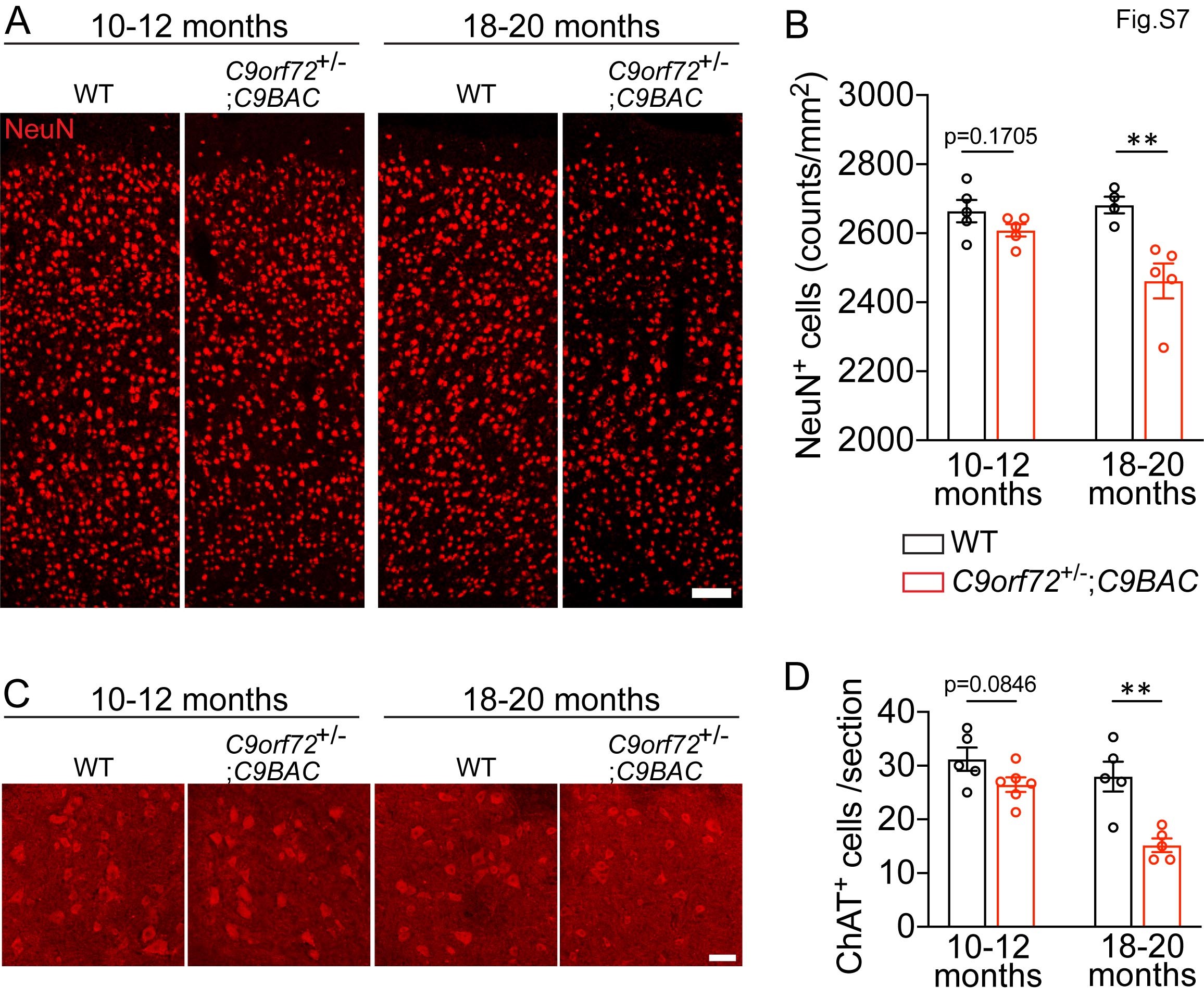
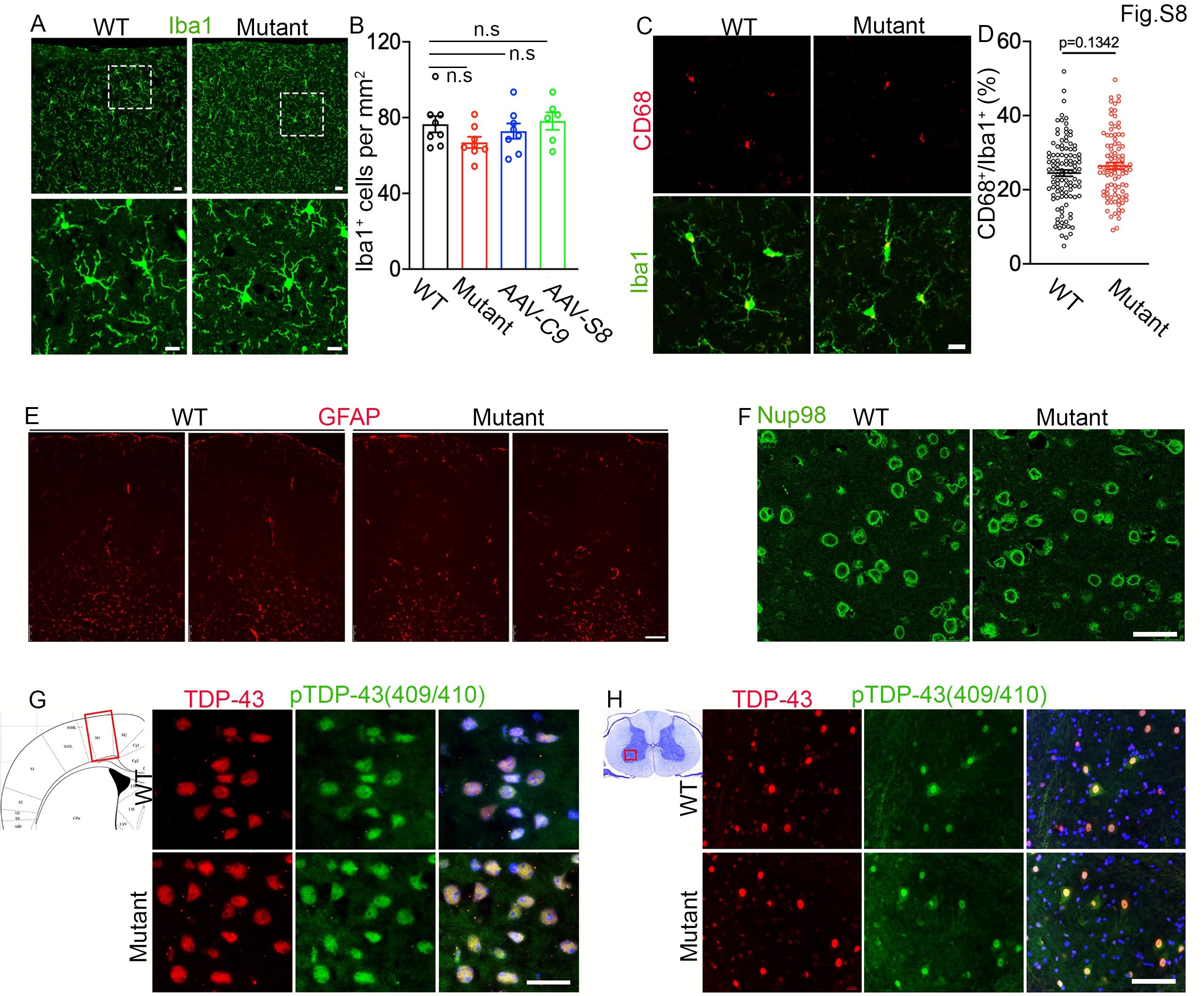
DPR foci reduction in AAV-C9 and AAV-S8 mice prompted us to examine their impacts on neurodegeneration. We firstly focused on the motor cortex. IHC staining of NeuN showed that there was a significant decrease in NeuN-positive cells in mutant mice compared to WT controls in 18- to 20- but not 10- to 12-month-old mice (Supplementary Material, Fig. S7A and B). Similarly, ChAT (choline acetyltransferse)-positive MNs were reduced in mutant brains in 18- to 20- but not 10- to 12-month-old mice (Supplementary Material, Fig. S7C and D). These results suggest that C9FTD/ALS-like cortical and motor neurodegeneration can be modeled in C9orf72+/−;C9-BAC mice. There were no significant changes in the number of Iba1+ microglia and their activation (Supplementary Material, Fig. S8A–D), nor were there in GFAP+ astrocyte distribution pattern (Supplementary Material, Fig. S8E) in mutant brains compared to WT controls. This suggests that cell degeneration is specific to neurons in mutant mice. We also did not find abnormal expression patterns of nuclear pore protein Nup98 or TDP-43 in mutant brains (Supplementary Material, Fig. S8F–H). Importantly, ectopic expression of C9orf72 leads to an increase in the number of NeuN-positive cells per area, while AAV-S8 also displayed an increased trend for NeuN-positive cells (Fig. 7B). Next, we examined ventral horn regions (leftmost panel in Fig. 7C) of 20-month-old mice. IHC staining of ChAT revealed a significant decrease in the ChAT-positive MNs in mutants compared to controls (Fig. 7C and D), suggesting MN degeneration in mutant mice. Both C9orf72 and Smcr8 expression significantly restored the ChAT-positive MNs (Fig. 7D). These results suggest that ectopic expression of C9orf72 or Smcr8 is sufficient to reduce DPR accumulation and neurodegeneration in C9FTD/ALS mice.
Figure 7.

C9orf72 and Smcr8 reduced neurodegeneration in C9FTD/ALS mutant mice. (A) Representative confocal images of motor cortex coronal sections (diagram in leftmost panel) stained with antibodies against NeuN (red) in 20-month-old WT and mutant mice with or without the expression of AAV-C9orf72 or AAV-Smcr8. Scale bars: 200 μm. (B) Quantification of NeuN-positive cells per mm2. (C) Representative confocal images of spinal cord ventral horn sections (diagram in the leftmost panel) stained with antibodies against ChAT (red) in 20-month-old mice. Scale bars: 50 μm. (D) Quantification of ChAT-positive cells per section. For all analyses, data are presented as mean ± SEM. N = 4–7 mice with one-way ANOVA with Bonferroni’s post hoc test (*P < 0.05, **P < 0.01 and **P < 0.001, n.s., not significant).
C9orf72 or Smcr8 mitigates motor and cognitive behavioral deficits in C9FTD/ALS mutant mice
We previously reported that complete C9orf72 KO induced motor behavioral deficits in C9-BAC mice (20). Current studies (Fig. 1B–H) further showed that C9orf72 haploinsufficiency can induce motor learning and motor strength deficits in C9-BAC mice. Conversely, we examined how ectopic expression of C9orf72 or Smcr8 impact motor behaviors in C9orf72+/−;C9-BAC mutant mice. Open field tests showed no significant differences in total distance traveled and in time spent in the center zone among WT and mutant mice with or without AAV-C9 or AAV-S8 (Fig. 8A and B), suggesting normal locomotion and anxiety. To confirm the anxiety data, we performed an elevated plus maze test. There were no significant differences in entries into open arms and in time spent in open arms among WT and mutant mice with and without C9orf72 or Smcr8 expression (Fig. 8C and D). These results suggest that there are no increased anxiety defects in mutant mice. We next performed grip strength assays and found that mutant mice exhibited reduced motor strength compared to controls (Fig. 8E). Rotarod assays showed that WT mice displayed a continuous increase in falling latency, suggesting a normal active learning process. In contrast, mutant mice had a significant delay in their motor learning (Fig. 8F). These results suggest that C9orf72+/−;C9-BAC mutant mice have ALS-like motor behavioral deficits. Importantly, both motor strength and motor learning deficits were significantly mitigated by ectopic expression of C9orf72 or Smcr8 (Fig. 8E and F).
Figure 8.

C9orf72 and Smcr8 mitigated motor behavioral deficits in C9FTD/ALS mutant mice. (A, B) Open field assays revealed no differences in total distance traveled (A) and in time spent in the center (B) among WT, mutant and rescue group mice. (C, D) Elevated plus maze assays revealed no difference in entries into open arm (C) and in time spent in open arms (D) among WT, mutant and rescue group mice. (E) Grip strength tests showed that AAV-C9orf72 or AAV-Smcr8 significantly mitigated motor strength defects in C9TFD/ALS mutant mice. (F) Rotarod tests showed that AAV-C9orf72 or AAV-Smcr8 significantly mitigated motor strength defects in C9TFD/ALS mutant mice. Behavior tests were performed at 18–20 months. For all analyses, data are presented as mean ± SEM. N = 11–17 mice with one-way ANOVA with Bonferroni’s post hoc test (**P < 0.01, ***P < 0.001 and ****P < 0.0001).
To examine cognitive behaviors, we first started with a novel object recognition test to measure hippocampus-dependent recognition memory (Fig. 9A). Animal tracks showed that WT mice spent more time with the novel object versus the familiar one, a pattern which is disrupted in mutant mice (Fig. 9B). Statistical analysis showed that C9orf72+/−;C9-BAC mutant mice exhibited reduced preference for the novel object, which is significantly brought back by ectopic expression of C9orf72 or Smcr8 (Fig. 9C). Next, we performed a three-chamber test to examine sociability and social novelty. Compared to the WT control, C9orf72+/−;C9-BAC mutant mice exhibited a significant decrease in the discrimination index of sociability and social memory. Both of these were restored in AAV-C9 or AAV-S8 mice (Fig. 9D–G). Together, these results suggest that ectopic expression of C9orf72 or Smcr8 can mitigate cognitive behavioral deficits in C9FTD/ALS mutant mice.
Figure 9.

C9orf72 and Smcr8 mitigated cognitive behavioral deficits in C9FTD/ALS mutant mice. (A) Diagram of the novel object recognition test. (B) Representative animal tracks in the novel object recognition test. (C) The preference index in the novel object recognition test. (D, F) Representative animal tracks in the sociability and social novelty tests. (E, G) The discrimination index in sociability (E) and social novelty (G) tests. Three chamber assays showed that AAV-C9orf72 or AAV-Smcr8 significantly mitigated sociability and social novelty defects in C9FTD/ALS mutant mice. Behavior tests were performed at 18–20 months. For all analyses, data are presented as mean ± SEM. N = 14–17 mice with one-way ANOVA with Bonferroni’s post hoc test (*P < 0.05 and **P < 0.01). (H) A working model suggests that DPRs localize to nucleolus, bind to ribosomal proteins (RPs), disrupt normal ribosome biogenesis and likely yield excess ‘orphan’ RPs that are not incorporated into ribosome machine leading to the overloading of autophagy-lysosome function. C9orf72 haploinsufficiency disrupts autophagy-lysosomal degradation and further exacerbates the DPR-mediated gain of toxicity. Therefore, loss of function and gain of function in C9FTD/ALS crosstalk and converge on proteotoxicity leading to neurodegeneration and behavioral defects.
Discussion
Here we reported dysregulation of a lysosome-ribosome biogenesis circuit, which mediates loss- and gain-of-function crosstalk and drives C9FTD/ALS-like neurodegeneration and behaviors. Ectopic expression of C9orf72 or Smcr8 promotes lysosomal functions and restores transcriptional dysregulation of ribosome biogenesis genes. This in turn leads to the mitigation of DPR accumulation, neurodegeneration, and FTD/ALS-like motor and cognitive behaviors.
Our genome-wide unbiased studies revealed that transcriptional downregulation of RP genes is a major outcome of C9orf72 depletion in C9-BAC mice, which shows lysosome and ribosome biogenesis defects converge to cause proteotoxic stress. Recent research on ribosomopathies (23,24), a group of human disorders caused by mutations in ribosome biogenesis factors (42–45), corroborates this idea, showing that ribosome biogenesis impairment leads to proteotoxic stress, which can be relieved by promoting autophagy-lysosomal functions (23,24). Similarly, accumulated DPRs localize to the nucleolus, the site of ribosome biogenesis, and bind to RPs, causing proteotoxic stress (14,15). This can be further exacerbated by C9orf72 deficiency-mediated lysosome impairment. Therefore, it appears that proteotoxicity is a converging point for both DPR mediated ribosome biogenesis defects and C9orf72 haploinsufficiency-mediated lysosomal impairment (Fig. 9H), which underlies the crosstalk between gain of function and loss of function. Aberrant ribosome biogenesis leads to proteotoxicity and induces stress responses, including the transcriptional downregulation of RP genes (40,41). We found a transcriptional downregulation of RP genes upon C9orf72 depletion in C9-BAC mice. Furthermore, complete C9orf72 loss led to reduced, rather increased, DPR accumulation, which is likely caused by impaired RAN translation due to the loss of ribosome biogenesis machines. These results suggest that transcriptional downregulation of RP genes is likely a stress response to the aberrant ribosome biogenesis in C9FTD/ALS mice. Importantly, C9orf72- or Smcr8-mediated lysosomal promotion not only restores RP transcription, but also mitigates DPR accumulation, neurodegeneration and behavioral abnormalities in C9FTD/ALS mice, suggesting the functional importance of this lysosome-ribosome biogenesis circuit. These studies suggest that C9FTD/ALS is a ribosomopathy-like disorder, which echoes the nucleolar stress in C9orf72 and sporadic ALS MNs preceding TDP-43 mislocalization (46). Future studies should investigate if abnormal accumulation of orphan RPs due to nucleolar dysfunctions by DPRs (23,24) overload the autophagy-lysosomal degradation system, leading to proteotoxic stress and neurodegeneration in C9FTD/ALS.
The C9-BAC mice (26) used in this study were reported to have inconsistent survival and motor behavioral phenotypes from different laboratories (47,48). Our C9FTD/ALS mouse (C9orf72+/−;C9-BAC) studies are different from previous research for the following reasons. First, we changed the background of the original C9-BAC mice from FVB to C57BL/6, as FVB mice are more sensitive to excitotoxicity and exhibit more extensive neuronal death compared to C57BL/6 mice (49,50). Second, our mice combined C9orf72+/− heterozygous with C9-BAC, while previous studies used C9-BAC mice alone. Third, we performed studies at 18–20 months, while previous studies used about 1-year-old C9-BAC mice. In these C9orf72+/−;C9-BAC mice, we identified C9FTD/ALS-like pathologies, including DPR accumulation and neurodegeneration, as well as cognitive and motor behavioral abnormalities. However, we did not detect microglia and TDP-43 abnormalities in these mice. We did not detect robust poly(PR) and poly(PA) signals in CNS tissues of C9orf72+/−;C9-BAC mice, which is consistent with the reports that the number of inclusions for poly(PR) and poly(PA) is relatively low in postmortem patient brains (31,32). Due to COVID-19, we were not able to monitor the survival of C9orf72+/−;C9-BAC mice.
Collectively, our studies identified dysregulation of a lysosome-ribosome biogenesis circuit in C9FTD/ALS mouse models in vivo. Functional studies showed that breaking this vicious circuit is capable of arresting DPR pathologies and neurodegeneration, leading to the mitigation of cognitive and motor behavioral abnormalities. Interventions that promote lysosomal functions, normalize ribosome defects, or have their combinations could form the basis of therapeutic strategies for treating C9FTD/ALS.
Materials and Methods
Animals
All experimental procedures used in this study were approved by the Institutional Animal Care and Use Committee at the University of Southern California. C9orf72 knockout (Cat#: 027068) and C9-BAC mice (Cat#: 029099) were ordered from Jackson laboratory.
Virus injection
Mice were given retro-orbital injection of AAV virus as previously described (51). Mice were briefly anesthetized with 3% isoflurane. Then, 200 μL of AAVPhP.eB-CMV-C9orf72-EGFP or AAVPhP.eB-CMV-Smcr8-EGFP with a titer of 1.1E + 14 gc/ml (Penn Vector Core, Philadelphia) was injected into the retro-orbital sinus with a 30 G needle and a 0.5 ml syringe. The mice were placed on the heating pad for recovery.
Behavioral testing
The experimenter was blind to the animal’s genotype during all tests. A cohort of mice was analyzed using the following behavioral assays. Open field: The subject mouse was placed in the empty arena (40 cm × 40 cm) and allowed to freely explore for 15 min. The total distance traveled and time spent in the center zone were recorded and automatically measured by using Smart v3.0 (Panlab Havard Apparatus). Elevated plus maze: The subject mouse was placed in the center platform of the elevated plus maze apparatus (open arms: 25 × 5 × 0.5 cm; closed arms: 25 × 5 × 16 cm) facing the open arm and allowed to freely explore for 10 min. The number of entries and time spent in center zone, open arms and closed arms were recorded and measured by using SMART v3.0 (Panlab Havard Apparatus). Rotarod test: An accelerating rotarod (Panlab) was used to analyze motor coordination and balance. Mice were trained three times on the rotarod with 4 rpm speed 1 day before testing. During test conditions, we measured the latency (time) to the mouse falling from the rotating beam while ramping up the speed from 4 to 40 rpm over a 5-min trial period. Mice were given three or five trials per day, with an intertrial interval of 20 min. The average of the three trials was used to evaluate latency to fall. Grip strength: Grip strength of front paws was measured using a grip strength meter (Bioseb). Each mouse was held by the tail and lowered toward the apparatus. Front paws were allowed to grasp the assembly. The mouse was then pulled backwards in the horizontal plane until the pull bar was released. The trial was repeated four times, and the highest force generated by pulling the animal away from the wire mesh was recorded. Three-chamber social interaction test: This was performed in a Plexiglas box containing three compartments connected by small openings that allowed mice free access to each compartment. The subject mouse was first placed in the middle chamber with side doors open to allow it to freely explore the three empty chambers. After 10 min of habituation, the mouse was gently guided to the middle chamber and side doors were closed. A stranger mouse was placed in the inverted wire cage in one side chamber and an empty wire cage was placed in the other side chamber. Then, the side doors were opened and the subject mouse was allowed to freely explore the chambers for 10 min. After this period, the subject mouse was again guided into the middle chamber and the side doors were closed. A second stranger mouse was placed in the previous empty wire cage. The side doors were opened, and the subject mouse was allowed to freely explore for another 10 min. The amount of time that the subject mouse spent sniffing each wire cage was quantified and the preference index was calculated as (Ts1 − Te)/Ts1 + Te) * 100% and (Ts2—Ts1)/Ts2 + Ts1) * 100%. Here, Te, Ts1 and Ts2 represent the time spent exploring empty, stranger 1 and stranger 2 wire cage, respectively. Novel object test: This test consists of habituation, familiarization and test phases. In the habituation phase, subject mice were placed in the center of a clean mouse cage and allowed to explore freely for 5 min. After 24 h, the familiarization phase was performed. Two identical objects were taped to the floor along the long axis, 10 cm away from the south and north walls. The mouse was placed in the center of the cage facing the east or west wall and allowed to explore for 10 min. The test phase was performed 24 h after the familiarization phase. One of the identical objects was replaced with a novel object with a different shape but similar size. The mouse was placed in the center of the cage facing the east or west wall and allowed to explore for 10 min. The apparatus and objects were thoroughly cleaned with 75% ethanol to remove the olfactory cues between each trial. The entire test phase was videotaped, and the travel of the subject mouse was manually documented. The preference index was calculated as (Tn − Tf)/(Tn + Tf) × 100%, where Tn and Tf represent the time spent exploring novel and familiar objects, respectively.
Western blot
Mouse tissues or cultured cells were eluted in SDS-PAGE sampling buffer by boiling for 10 min followed by WB analysis. For individual studies, the densitometry of individual blot signals from three independent WB experiments was quantified using Image J software. The individual values for each blot signals were normalized to respective controls followed by the statistical analysis among different samples (Student’s t-test). The following antibodies were used, including antibodies against C9orf72 (22637–1-AP, Proteintech), rabbit anti-SMCR8 (ab121682, Abcam) and rabbit anti-ATG101 (SAB4200175, Sigma).
Immunohistochemistry
Mouse brains and spinal cords were fixed in 4% paraformaldehyde, pH 7.4 for 24 h, and incubated in a 30% sucrose/PBS solution for 2 days, then embedded in Tissue-Tek OCT compound (Sakura). Coronal sections were sliced at 40 μm using a cryostat (Leica CM1950). Sections were washed in PBS three times (5 min each time) and then incubated with blocking solution (5% normal goat serum, 1% BSA, 0.3% Triton X-100 in PBS) for 2 h at room temperature. Sections were then incubated with antibodies against poly(GA) (MABN889, Millipore; 24 492–1-AP, Proteintech), poly(GP) (23978–1-AP, Proteintech), Lamp1 (1D4B, DSHB), pS6 (5364, Cell Signaling Technology), NeuN (MAB377, Millipore), ChAT (ab178850, Abcam), NUP98 (ab50610, Abcam), Phospho-TDP43 (Ser409/410) (22309–1-AP, Proteintech), TDP-43 (12892–1-AP, Proteintech), Iba1 (019–19 741, FUJIFILM Wako Chemicals), Olig2 (NBP1–28667SS, Novus Bio), GFAP (MCA1957T, Bio-rad) and CD68 (MAB3402, Millipore) in blocking solution overnight at 4°C. After washing in PBS three times (5 min each time), sections were incubated with species-specific fluorescently conjugated secondary antibodies (1:200, Invitrogen) and Hoechst 33342 (1:1000) in blocking solution for 2 h at room temperature. After washing in PBS three times (5 min each time), sections were mounted on glass slides with mounting medium and coverslipped. Images of stained sections were acquired using a Leica DMI6000 CS confocal microscope with a 10×, 20× or 60× objective lens. All images were taken using identical laser power, gain and offset values. Number of fluorescent positive signals Figs 2A–H,4C–F,5A–C,6A–D were calculated manually, and the immunofluorescent intensity was analyzed with ImageJ Fig. 4A and B.
Nuclear isolation and snRNA-seq
Mice were euthanized by CO2 inhalation and then decapitated. Frontal cortices were cut using a vibratome (10 111 N, Ted Pella) in ice-cold dissection buffer (60 mm NaCl, 3 mm KCl, 1.25 mM NaH2PO4, 25 mm NaHCO3, 115 mm sucrose, 10 mm glucose, 7 mm MgCl2, 0.5 mm CaCl2; saturated with 5% CO2 balanced O2; pH = 7.4). Nuclear isolation was performed as previously described with minor modifications. Briefly, samples from three mice of each group were combined and Dounce-homogenized with four strokes of a loose pestle and four strokes of a tight pestle in ice-cold detergent lysis buffer (0.1% Triton-X, 0.32 M sucrose, 10 mm HEPES, 5 mm CaCl2, 3 mM MgAc, 0.1 mm EDTA and 1 mm DTT in nuclease-free water, pH 8.0). The lysate was centrifuged at 3200 × g for 10 min at 4°C and the pellet was resuspended with 3 ml low sucrose buffer (0.32 M sucrose, 10 mm HEPES, 5 mm CaCl2, 3 mm MgAc, 0.1 mm EDTA and 1 mm DTT in nuclease-free water, pH 8.0). The nuclei were isolated and purified by centrifugation in a sucrose density gradient at 3200 × g for 20 min at 4°C, and then resuspended with resuspension solution (0.4 mg/ml BSA, 0.2 U/μL RNAse inhibitor in DPBS). Isolated nuclei were loaded into the 10X Chromium system with a targeted recovery of 10 000 nuclei to be barcoded for snRNA-seq using a Single Cell 3’ Library Kit v2 (PN-120267, 10× Genomics). Sequencing was performed on the Illumina Novaseq System. Raw read counts were analyzed using the Seurat R package.
snRNA-Seq analysis
Demultiplexing and alignment of sequencing reads to the mouse transcriptome were performed using Cell Ranger software (version 3.0.2, 10X Genomics). We used the option ‘—forcecells 9000’ in ‘cellranger count’ to extract a reasonable number of cell barcodes in samples, as we found that the automatic estimate of Cell Ranger was inaccurate. The top 2000 genes were identified by variable feature selection based on a variance stabilizing transformation (‘vst’). Then, 50 principal components (PCs) were utilized to calculate a k-nearest neighbors (KNN) graph based on the Euclidean distance in PCA space, and the first 30 PCs were accordingly selected for the subsequent analysis according to the Jackstraw function. Clusters were then visualized using a Uniform Manifold Approximation and Projection (UMAP) plot. To annotate the cell types by gene markers, MAST was used to perform differential gene expression analysis by comparing nuclei in each cluster to the rest of the nuclear profiles. Genes with FDR < 0.05 and log fold change ≥1 were selected as cell-type markers. To identify genes differentially expressed in between two groups in each cell type, FindMarkers of Seurat were used with the MAST method. To estimate the relative contribution of differentially expressed genes by each cell types, we downsampled the cell numbers to 500 from each cluster for 10 times before performing differential expression analysis. Genes with log2 (fold change of expression) of at least 0.25 and FDR < 0.01 were selected as differentially expressed.
RNA-Seq
Two-four biological replicates were subjected to RNA-seq. One microgram of RNA sample quality was assessed by Bioanalyzer 2100 Eukaryote Total RNA Pico (Agilent Technologies, CA, USA) and quantified by Qubit RNA HS assay (ThermoFisher). Libraries were constructed with TruSeq Stranded mRNA library kits (Illumina Inc., San Diego, CA, USA) based on the manufacturer’s recommendations. Library concentration was measured by qPCR and library quality evaluated by Tapestation High Sensitivity D1000 screentapes (Agilent Technologies, CA, USA). Equimolar pooling of libraries was performed based on qPCR values. Libraries were sequenced on a HiSeq with a read length configuration of 150 PE targeting 40 M total reads per sample (20 M each direction) by Admera Health, LLC.
Data availability
RNAseq and snRNA-seq data have been deposited in GEO under accession number GSE206666.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank Chen laboratory colleagues for stimulating discussions. We are grateful for Bridget Samuels’ critical reading of the manuscript.
Conflict of Interest statement. The authors declare that there are no competing financial interests that might be perceived as affecting the objectivity of these studies.
Declaration of Interests: The authors declare no competing interests.
Contributor Information
Li Ma, Center for Craniofacial Molecular Biology, University of Southern California (USC), Los Angeles, CA 90033, USA.
Chen Liang, Center for Craniofacial Molecular Biology, University of Southern California (USC), Los Angeles, CA 90033, USA.
Jing Wang, Department of Human Genetics, David Geffen School of Medicine, University of California Los Angeles, Los Angeles, CA 90095, USA.
Qing Chang, Center for Craniofacial Molecular Biology, University of Southern California (USC), Los Angeles, CA 90033, USA.
Yuan Wang, Center for Craniofacial Molecular Biology, University of Southern California (USC), Los Angeles, CA 90033, USA.
Wei Zhang, Center for Craniofacial Molecular Biology, University of Southern California (USC), Los Angeles, CA 90033, USA.
Yuanning Du, Center for Craniofacial Molecular Biology, University of Southern California (USC), Los Angeles, CA 90033, USA.
Jotham Sadan, Center for Craniofacial Molecular Biology, University of Southern California (USC), Los Angeles, CA 90033, USA.
Jian-Fu Chen, Center for Craniofacial Molecular Biology, University of Southern California (USC), Los Angeles, CA 90033, USA.
Funding
Chen laboratory is supported by funds from the Associate Dean of Research Fund from the Center for Craniofacial Molecular Biology, Herman Ostrow School of Dentistry at the University of Southern California, and grants R01DE030901 (J.C.), R21AG075665 (J.C.), R21AG070681 from the National Institute of Health.
Authors’ Contributions
L.M., C.L., Q.C., Y.W. and Y.D. performed all experiments. J.W. analyzed all bioinformatic data. W.Z. and J.S. helped with the manuscript writing. J.-F.C. designed and interpreted the experiments and wrote the manuscript.
References
- 1. Renton, A.E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J.R., Schymick, J.C., Laaksovirta, H., van Swieten, J.C., Myllykangas, L. et al. (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron, 72, 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. DeJesus-Hernandez, M., Mackenzie, I.R., Boeve, B.F., Boxer, A.L., Baker, M., Rutherford, N.J., Nicholson, A.M., Finch, N.A., Flynn, H., Adamson, J. et al. (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron, 72, 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ferrari, R., Kapogiannis, D., Huey, E.D. and Momeni, P. (2011) FTD and ALS: a tale of two diseases. Curr. Alzheimer Res., 8, 273–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Strong, M.J. (2008) The syndromes of frontotemporal dysfunction in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler., 9, 323–338. [DOI] [PubMed] [Google Scholar]
- 5. Neumann, M., Sampathu, D.M., Kwong, L.K., Truax, A.C., Micsenyi, M.C., Chou, T.T., Bruce, J., Schuck, T., Grossman, M., Clark, C.M. et al. (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science, 314, 130–133. [DOI] [PubMed] [Google Scholar]
- 6. Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H., Mann, D., Tsuchiya, K., Yoshida, M., Hashizume, Y. and Oda, T. (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun., 351, 602–611. [DOI] [PubMed] [Google Scholar]
- 7. Gao, F.-B., Almeida, S. and Lopez-Gonzalez, R. (2017) Dysregulated molecular pathways in amyotrophic lateral sclerosis-frontotemporal dementia spectrum disorder. EMBO J., 36, 2931–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ling, S.-C., Polymenidou, M. and Cleveland, D.W. (2013) Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron, 79, 416–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Taylor, J.P., Brown, R.H. and Cleveland, D.W. (2016) Decoding ALS: from genes to mechanism. Nature, 539, 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gendron, T.F. and Petrucelli, L. (2018) Disease mechanisms of C9ORF72 repeat expansions. Cold Spring Harb Perspect Med, 8, a024224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Conlon, E.G., Lu, L., Sharma, A., Yamazaki, T., Tang, T., Shneider, N.A. and Manley, J.L. (2016) The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains. elife, 5, 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gendron, T.F., Bieniek, K.F., Zhang, Y.-J., Jansen-West, K., Ash, P.E.A., Caulfield, T., Daughrity, L., Dunmore, J.H., Castanedes-Casey, M., Chew, J. et al. (2013) Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol., 126, 829–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Haeusler, A.R., Donnelly, C.J., Periz, G., Simko, E.A.J., Shaw, P.G., Kim, M.-S., Maragakis, N.J., Troncoso, J.C., Pandey, A., Sattler, R., Rothstein, J.D. and Wang, J. (2014) C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature, 507, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lopez-Gonzalez, R., Lu, Y., Gendron, T.F., Karydas, A., Tran, H., Yang, D., Petrucelli, L., Miller, B.L., Almeida, S. and Gao, F.-B. (2016) Poly(GR) in C9ORF72-related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron, 92, 383–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang, Y.-J., Gendron, T.F., Ebbert, M.T.W., O'Raw, A.D., Yue, M., Jansen-West, K., Zhang, X., Prudencio, M., Chew, J., Cook, C.N. et al. (2018) Poly(GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat. Med., 24, 1136–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Loveland, A.B., Svidritskiy, E., Susorov, D., Lee, S., Park, A., Zvornicanin, S., Demo, G., Gao, F.-B. and Korostelev, A.A. (2022) Ribosome inhibition by C9ORF72-ALS/FTD-associated poly-PR and poly-GR proteins revealed by cryo-EM. Nat. Commun., 13, 2776–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Boivin, M., Pfister, V., Gaucherot, A., Ruffenach, F., Negroni, L., Sellier, C. and Charlet-Berguerand, N. (2020) Reduced autophagy upon C9ORF72 loss synergizes with dipeptide repeat protein toxicity in G4C2 repeat expansion disorders. EMBO J., 39, e100574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhu, Q., Jiang, J., Gendron, T.F., McAlonis-Downes, M., Jiang, L., Taylor, A., Diaz Garcia, S., Ghosh Dastidar, S., Rodriguez, M.J., King, P. et al. (2020) Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nat. Neurosci., 23, 615–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liang, C., Shao, Q., Zhang, W., Yang, M., Chang, Q., Chen, R. and Chen, J.-F. (2019) Smcr8 deficiency disrupts axonal transport-dependent lysosomal function and promotes axonal swellings and gain of toxicity in C9ALS/FTD mouse models. Hum. Mol. Genet., 28, 3940–3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shao, Q., Liang, C., Chang, Q., Zhang, W., Yang, M. and Chen, J.-F. (2019) C9orf72 deficiency promotes motor deficits of a C9ALS/FTD mouse model in a dose-dependent manner. Acta Neuropathol Commun, 7, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shao, Q., Yang, M., Liang, C., Ma, L., Zhang, W., Jiang, Z., Luo, J., Lee, J.-K., Liang, C. and Chen, J.-F. (2019) C9orf72 and smcr8 mutant mice reveal MTORC1 activation due to impaired lysosomal degradation and exocytosis. Autophagy, 72, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Laplante, M. and Sabatini, D.M. (2012) mTOR signaling in growth control and disease. Cell, 149, 274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Recasens-Alvarez, C., Alexandre, C., Kirkpatrick, J., Nojima, H., Huels, D.J., Snijders, A.P. and Vincent, J.-P. (2021) Ribosomopathy-associated mutations cause proteotoxic stress that is alleviated by TOR inhibition. Nat. Cell Biol., 23, 127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Baumgartner, M.E., Dinan, M.P., Langton, P.F., Kucinski, I. and Piddini, E. (2021) Proteotoxic stress is a driver of the loser status and cell competition. Nat. Cell Biol., 23, 136–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang, M., Liang, C., Swaminathan, K., Herrlinger, S., Lai, F., Shiekhattar, R. and Chen, J.-F. (2016) A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci. Adv., 2, e1601167–e1601167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu, Y., Pattamatta, A., Zu, T., Reid, T., Bardhi, O., Borchelt, D.R., Yachnis, A.T. and Ranum, L.P.W. (2016) C9orf72 BAC mouse model with motor deficits and neurodegenerative features of ALS/FTD. Neuron, 90, 521–534. [DOI] [PubMed] [Google Scholar]
- 27. Koppers, M., Blokhuis, A.M., Westeneng, H.-J., Terpstra, M.L., Zundel, C.A.C., Vieira de Sá, R., Schellevis, R.D., Waite, A.J., Blake, D.J., Veldink, J.H., van den Berg, L.H. and Pasterkamp, R.J. (2015) C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann. Neurol., 78, 426–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. O'Rourke, J.G., Bogdanik, L., Yáñez, A., Lall, D., Wolf, A.J., Muhammad, A.K.M.G., Ho, R., Carmona, S., Vit, J.P., Zarrow, J. et al. (2016) C9orf72 is required for proper macrophage and microglial function in mice. Science, 351, 1324–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Burberry, A., Suzuki, N., Wang, J.-Y., Moccia, R., Mordes, D.A., Stewart, M.H., Suzuki-Uematsu, S., Ghosh, S., Singh, A., Merkle, F.T. et al. (2016) Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci. Transl. Med., 8, 347ra93–347ra93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shi, Y., Lin, S., Staats, K.A., Li, Y., Chang, W.-H., Hung, S.-T., Hendricks, E., Linares, G.R., Wang, Y., Son, E.Y. et al. (2018) Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med., 24, 313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mackenzie, I.R.A., Frick, P., Grässer, F.A., Gendron, T.F., Petrucelli, L., Cashman, N.R., Edbauer, D., Kremmer, E., Prudlo, J., Troost, D. and Neumann, M. (2015) Quantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta Neuropathol., 130, 845–861. [DOI] [PubMed] [Google Scholar]
- 32. Mackenzie, I.R.A., Frick, P. and Neumann, M. (2014) The neuropathology associated with repeat expansions in the C9ORF72 gene. Acta Neuropathol., 127, 347–357. [DOI] [PubMed] [Google Scholar]
- 33. May, S., Hornburg, D., Schludi, M.H., Arzberger, T., Rentzsch, K., Schwenk, B.M., Grässer, F.A., Mori, K., Kremmer, E., Banzhaf-Strathmann, J. et al. (2014) C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol., 128, 485–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang, Y.-J., Gendron, T.F., Grima, J.C., Sasaguri, H., Jansen-West, K., Xu, Y.-F., Katzman, R.B., Gass, J., Murray, M.E., Shinohara, M. et al. (2016) C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat. Neurosci., 19, 668–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mizielinska, S., Grönke, S., Niccoli, T., Ridler, C.E., Clayton, E.L., Devoy, A., Moens, T., Norona, F.E., Woollacott, I.O.C., Pietrzyk, J. et al. (2014) C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science, 345, 1192–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schludi, M.H., Becker, L., Garrett, L., Gendron, T.F., Zhou, Q., Schreiber, F., Popper, B., Dimou, L., Strom, T.M., Winkelmann, J. et al. (2017) Spinal poly-GA inclusions in a C9orf72 mouse model trigger motor deficits and inflammation without neuron loss. Acta Neuropathol., 134, 241–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chan, K.Y., Jang, M.J., Yoo, B.B., Greenbaum, A., Ravi, N., Wu, W.-L., Sánchez-Guardado, L., Lois, C., Mazmanian, S.K., Deverman, B.E. and Gradinaru, V. (2017) Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci., 20, 1172–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ahmed, Z., Sheng, H., Xu, Y.-F., Lin, W.-L., Innes, A.E., Gass, J., Yu, X., Wuertzer, C.A., Hou, H., Chiba, S. et al. (2010) Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am. J. Pathol., 177, 311–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Moreno-García, A., Kun, A., Calero, O., Medina, M. and Calero, M. (2018) An Overview of the Role of Lipofuscin in Age-Related Neurodegeneration. Front. Neurosci., 12, 464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Albert, B., Kos-Braun, I.C., Henras, A.K., Dez, C., Rueda, M.P., Zhang, X., Gadal, O., Kos, M. and Shore, D. (2019) A ribosome assembly stress response regulates transcription to maintain proteome homeostasis. elife, 8, e45002, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tye, B.W., Commins, N., Ryazanova, L.V., Wühr, M., Springer, M., Pincus, D. and Churchman, L.S. (2019) Proteotoxicity from aberrant ribosome biogenesis compromises cell fitness. elife, 8, e43002, 1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mills, E.W. and Green, R. (2017) Ribosomopathies: there's strength in numbers. Science, 358, eaan2755. [DOI] [PubMed] [Google Scholar]
- 43. Yelick, P.C. and Trainor, P.A. (2015) Ribosomopathies: global process, tissue specific defects. Rare Dis, 3, e1025185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Khajuria, R.K., Munschauer, M., Ulirsch, J.C., Fiorini, C., Ludwig, L.S., McFarland, S.K., Abdulhay, N.J., Specht, H., Keshishian, H., Mani, D.R. et al. (2018) Ribosome levels selectively regulate translation and lineage commitment in human hematopoiesis. Cell, 173, 90–103.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Farley-Barnes, K.I., Ogawa, L.M. and Baserga, S.J. (2019) Ribosomopathies: old concepts. New Controversies. Trends Genet., 35, 754–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Aladesuyi Arogundade, O., Nguyen, S., Leung, R., Wainio, D., Rodriguez, M. and Ravits, J. (2021) Nucleolar stress in C9orf72 and sporadic ALS spinal motor neurons precedes TDP-43 mislocalization. Acta Neuropathol Commun, 9, 26–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mordes, D.A., Morrison, B.M., Ament, X.H., Cantrell, C., Mok, J., Eggan, P., Xue, C., Wang, J.-Y., Eggan, K. and Rothstein, J.D. (2020) Absence of survival and motor deficits in 500 repeat C9ORF72 BAC mice. Neuron, 108, 775–783.e4. [DOI] [PubMed] [Google Scholar]
- 48. Nguyen, L., Laboissonniere, L.A., Guo, S., Pilotto, F., Scheidegger, O., Oestmann, A., Hammond, J.W., Li, H., Hyysalo, A., Peltola, R. et al. (2020) Survival and motor phenotypes in FVB C9-500 ALS/FTD BAC transgenic mice reproduced by multiple labs. Neuron, 108, 784–796.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schauwecker, P.E. (2003) Genetic basis of kainate-induced excitotoxicity in mice: phenotypic modulation of seizure-induced cell death. Epilepsy Res., 55, 201–210. [DOI] [PubMed] [Google Scholar]
- 50. Schauwecker, P.E. (2002) Modulation of cell death by mouse genotype: differential vulnerability to excitatory amino acid-induced lesions. Exp. Neurol., 178, 219–235. [DOI] [PubMed] [Google Scholar]
- 51. Yardeni, T., Eckhaus, M., Morris, H.D., Huizing, M. and Hoogstraten-Miller, S. (2011) Retro-orbital injections in mice. Lab Anim (NY), 40, 155–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNAseq and snRNA-seq data have been deposited in GEO under accession number GSE206666.