

Abstract
Background
Patients with non-severe ANCA-associated vasculitis (AAV) are often prescribed immunosuppressive medications that are associated with severe side effects and a reduced quality of life. There is an unmet need for safer effective treatments for these patients. Hydroxychloroquine is being explored due to its effect in similar autoimmune conditions such as systemic lupus erythematosus.
Methods
Double-blind, placebo-controlled multicentre trial recruiting 76 patients across 20 sites. Participants will be randomised 1:1 to hydroxychloroquine or placebo in addition to standard of care immunosuppressive therapies over the course of 52 weeks. A phase II selection design will be used to determine hdroxychloroquine’s efficacy, using prednisolone dosage and Birmingham Vasculitis Activity Score as a measure of disease activity. Secondary outcomes will explore other elements of AAV progression, including disease flares and time to remission.
Discussion
This trial aims to explore Hydroxychloroquine as a treatment for patients with AAV. If effective, the need for immunosuppressive treatments such as prednisolone could be reduced. Hydroxychloroquine is safer, cheaper and has fewer adverse effects than conventional immunosuppressive treatments. This could improve patient outcomes while saving money for the NHS.
Trial registration
ISRCTN: ISRCTN79334891. Registered 07 June 2021.
EudraCT: 2018-001268-40. Registered 13 September 2019.
Clinicaltrials.gov: NCT04316494. Registered 20 March 2020.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13063-023-07108-3.
Keywords: Hydroxychloroquine, AAV, Auto-immune conditions, ANCA, Vasculitis, Prednisolone
Administrative information
| Title |
Hydroxychloroquine in ANCA Vasculitis Evaluation - A Multicentre, Randomised, Double-blind, Placebo-controlled Trial (HAVEN) |
| Trial registration |
ISRCTN: ISRCTN79334891 EudraCT: 2018-001268-40 |
| Protocol version | 6.0 dated 10/05/2022 |
| Funding | The trial is funded by the Medical Research Council (Grant Ref: MR/R006253/1). |
| Author details |
1School of Life Course and Population Sciences, Faculty of Life Sciences & Medicine. King’s College London, UK 2 Clinical Trial Manager Platform, Guy’s and St Thomas’ NHS Foundation Trust, UK 3 Louise Coote Lupus Unit, Rheumatology Department, Guy’s and St Thomas’ NHS Foundation Trust, UK 4 King’s Clinical Trial Unit, Research Management and Innovation Directorate, King’s College London, UK 5 Dept. of Immunology, School of Immunology & Microbial Sciences, King's College London, UK 6 Nuffield Department of Orthopaedics, Rheumatology and Musculoskeletal Science, University of Oxford, UK 7 Department of Medicine, University of Cambridge, UK 8 Centre for Rheumatic Disease, Kings College London, UK |
| Name and contact information for the trial sponsor |
Guy’s and St. Thomas’ NHS Foundation Trust (GSTT) Sponsor Contact: Amy Holton Address: King’s Health Partners Clinical Trials Office 16th Floor Tower Wing Guy’s Hospital London SE1 9RT Telephone: 02071885732 Fax: 02071888330 Email: amy.holton@kcl.ac.uk |
| Role of sponsor | GSTT is responsible for ensuring that the trial respects the dignity, rights, safety and well-being of participants. Neither the trial sponsor nor funder had direct involvement in the trial design, writing of the report, nor will they be involved in the analysis, management, interpretation and publication of the data. |
Background
The systemic vasculitides encompass a group of autoimmune inflammatory diseases affecting blood vessels of all sizes and in any organ or system. They are life-threatening diseases where the body’s defence system becomes overactive, causing inflammation of small blood vessels. The term ANCA-associated vasculitis (AAV) describes a subset of primary small vessel vasculitides characterized by the presence of anti-neutrophil cytoplasmic antibodies (ANCA): granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA) and eosinophilic granulomatosis with polyangiitis (EGPA).
AAV are serious multisystem autoimmune disorders that can affect any organ in the body and commonly involve the ear-nose-throat, lungs, kidneys, eyes and joints. Uncontrolled disease activity can lead to organ failure and death. There are approximately 20,000 AAV patients in the UK, with 1300 diagnosed annually [1].
Although immunosuppressive treatments have improved outcomes, mortality is double that of the general population [2], 20% have persistent uncontrolled disease and 50% relapse by 5 years [3], costing > £23 million/year for hospitalisations and reducing patients’ incomes by a quarter [4].
AAV patients have a 20 times higher prothrombotic tendency [5], 3 times higher cardiovascular risk [6] and are at higher risk of immunosuppression related infections and cancer [2]. However, early therapy withdrawal, especially of glucocorticoids, is associated with disease flares, lower quality of life and increased costs [7].
There is an unmet need for safe, effective therapies for non-severe AAV to reduce disease activity, prevent disease progression and damage accumulation and minimise drug toxicity. Safer medications are needed for non-life threatening ANCA vasculitis, where aggressive immune suppressive medications are not appropriate.
Hydroxychloroquine is an effective, safe and inexpensive therapy with an established track record of disease modifying effects in autoimmune rheumatic diseases such as systemic lupus erythematosus (SLE) and Sjögren’s syndrome [8–11]. Hydroxychloroquine is safer than other immunosuppressive treatments (including in pregnancy), and it is steroid sparing, allowing reductions in cumulative prednisolone doses and their adverse effects [9]. Hydroxychloroquine has been used to treat cutaneous vasculitis [12] and rheumatoid vasculitis [10] but has never been assessed in AAV [13]. The pleiotropic effects of hydroxychloroquine on cytokines, neutrophils and autoreactive T and B lymphocytes involved in the pathogenesis of AAV provide a mechanistic rationale for its potential effectiveness [13]. Hydroxychloroquine has beneficial effects on lipids, glucose levels and arterial stiffness [14]. Hydroxychloroquine has antimicrobial [15, 16], antithrombotic [17] and antineoplastic [18, 19] effects that could be useful in immunosuppressed AAV patients. Early treatment with hydroxychloroquine could potentially reduce progression to severe disease and the need for biologic therapy, such as rituximab, which is administered for refractory or relapsing AAV [20–22]. The HAVEN trial aims to investigate this by assessing whether the addition of hydroxychloroquine to background therapy improves clinical response and quality of life in patients with AAV.
Methods
Trial design
The HAVEN trial is a double-blind, placebo-controlled multi-centre trial following a phase II selection design. Participants will be randomised to receive hydroxychloroquine or placebo in a 1:1 ratio, in addition to standard of care (SoC) maintenance therapy with prednisolone and/or stable doses of azathioprine, methotrexate, mycophenolate, co-trimoxazole or maintenance therapy with B cell depleting therapy (rituximab). Each patient will be treated with adjunctive hydroxychloroquine 400 mg daily or matching placebo for 52 weeks with dose reduction according to actual body weight and renal function. Participants will attend 10 visits across 60 weeks (Fig. 1). At each visit, disease activity will be determined using the Birmingham Vasculitis Activity Score (BVAS), and data will be collected on other measures including further disease activity indicators, treatment adherence, clinical biomarkers and quality of life (Table 1).
Fig. 1.
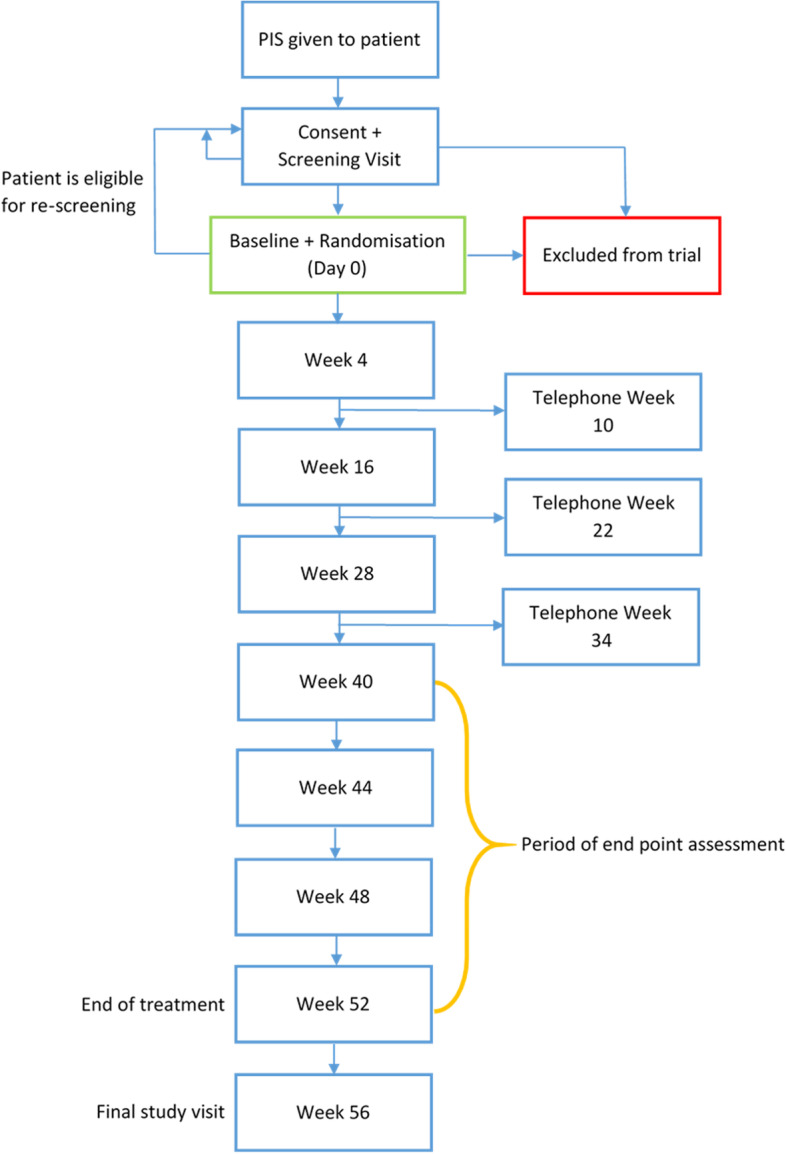
Flow chart of trial design and participant follow-up
Table 1.
Schedule of enrolment, interventions and assessments from SPIRIT checklist
| Study Visitsi | Screen | Baseline | WK4 | WK10 Telephone | WK16 | WK 22 Telephone | WK28 | WK 34 Telephone | WK40 | WK44 | WK48 | WK52 End of study treatment | Follow up WK56 | Withdrawalii |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Visit Number | 1 | 2 | 3 | - | 4 | - | 5 | - | 6 | 7 | 8 | 9 | 10 | - |
| Day (Visit window) | Up to 28 days | 0 | 28 (± 7) | 70 (± 7) | 112 (± 7) | 154 (± 7) | 196 (± 7) | 238 (± 7) | 280 (± 7) | 308 (± 7) | 336 (± 7) | 364 (± 7) | 392 (± 7) | |
| Patient information and informed consent | X | |||||||||||||
| Interventions: | ||||||||||||||
| IMP dispensing | X | X | X | X | X | |||||||||
| IMP dose recording / review of patient diary | X | X | X | X | X | X | X | X | ||||||
| Prednisolone dose recording | X | X | X | X | X | X | X | X | X | X | X | |||
| Assessments: | ||||||||||||||
| COVID symptom assessment | X | X | X | X | X | X | X | X | X | X | X | |||
| Eye screen | Xiii | |||||||||||||
| Eligibility | X | Xiv | ||||||||||||
| Randomisation | X | |||||||||||||
| Medical history, Demographics | X | |||||||||||||
| Physical exam, Weight, Vital signsv, Urine dipstick | X | X | X | X | X | X | X | X | X | X | X | |||
| ECG | X | X | ||||||||||||
| Medications | X | X | X | X | X | X | X | X | X | X | X | X | X | X |
| BVASvi | X | X | X | X | X | X | X | X | X | X | X | |||
| VDI | X | X | X | X | X | |||||||||
| GTI | X | X | X | |||||||||||
| Physician’s Global Assessment | X | X | X | X | X | X | X | X | X | X | X | |||
| Urine drug screen | X | |||||||||||||
| SF-36, EQ5D, HAQ, AAV Pro | X | Xvii | X | Xvii | X | Xvii | X | X | X | X | ||||
| FACIT score | X | X | X | X | X | X | X | |||||||
| Adverse event reporting | X | X | X | X | X | X | X | X | X | X | X | X | X | |
| Clinical Labs: | ||||||||||||||
| ANCA | X | X | X | X | X | X | X | |||||||
| ESRviii, CRP | X | X | X | X | X | X | X | |||||||
| Glucose | X | X | X | X | X | X | X | |||||||
| HbA1C | X | X | X | |||||||||||
| Lipids | X | X | X | |||||||||||
| Renal profile including creatinine and eGFRix | X | X | X | X | X | X | X | X | X | |||||
| Full blood count | X | Xx | X | X | X | X | X | X | X | |||||
| Liver function tests | X | Xx | X | X | X | X | X | X | X | |||||
| Protein: creatinine ratioxi | X | X | X | X | X | X | X | X | X | X | X | |||
| Viral screenxii | X | |||||||||||||
| Pregnancy testxiii | X | X | ||||||||||||
| Research Blood Specimens: | ||||||||||||||
| Hydroxychloroquine levels | X | X | X | X | ||||||||||
| Plasma, Serum and Cellsxiv | X | X | X | X | X | Xxv | ||||||||
i Patients will be followed up by telephone at weeks 10, 22 and 34 rather than visiting the study centre. The phone call will be structured using the AAV pro questionnaire. See section 6.13 for more details
ii To be arranged if a patient withdraws from the trial but is willing to have a final study visit. This is only necessary in instances where the patient’s last visit was more than 4 weeks ago
iii The eye screen is only required for patients whose eGFR drops below 60ml/min at any point during the trial. The eye screen should take place between week 52 and week 56 visits by a local optometrist
iv The eligibility criteria require BVAS to be scored and for female patients to have a pregnancy test at baseline as well as screening. Other screening procedures do not need to be repeated
v Vital signs to include BP, pulse, respiration rate and temperature
vi BVAS and VDI to be scored locally following training. BVAS scores will be quality checked by a central adjudication panel to ensure consistency
vii Telephone follow up interviews will only use the AAV Pro questionnaire to structure the conversation. The SF-36, EQ5D and HAQ questionnaires will not be included
viii Where ESR is not available, plasma viscosity may be used instead
ix If the patient’s eGFR drops below 60ml/min at any point of the trial they should be invited for an eye screen between week 52 and 56
x If the baseline visit is less than two weeks after screening, these tests do not need to be performed again
xi Protein:creatinine ratio only needs to be performed if urine dipstick for protein shows 1+ or more, but results can be collected if routinely performed when the urine dipstick is negative
xii Patients who test positive for hepatitis B surface antigen, hepatitis B core antibody, hepatitis C antibody or HIV-1 will be excluded
xiii Urine pregnancy test
xiv Plasma and serum collected at all sites. Cells at Guy’s Hospital only
xv Samples only collected if the patient is identified as having a flare
Setting
Patients will be recruited from approximately 20 NHS Trusts across the UK, including teaching and non-teaching hospital settings across England, Scotland and Wales. Patients will be identified and approached by members of their direct care team and will be given as much time as they need to make a decision about participation. Informed consent will be confirmed to the site’s principal investigator (PI) or a delegated sub-investigator. Patients who meet the eligibility criteria will be randomised.
Ten sites were initially approached to join the trial but this was expanded to 20 in September 2021 in order to address delays faced as a result of the COVID-19 pandemic. This expansion has allowed the trial to reach a larger pool of AAV patients. Sites with larger vasculitis clinics will be expected to recruit between 4 and 10 patients, with smaller community sites recruiting between 2 and 5 patients.
Participants
Patients with ANCA-associated vasculitis (AAV) attending NHS vasculitis clinics will be approached by their direct care team to take part in the trial. Once the patient has provided consent, they will be screened against the following criteria:
Inclusion criteria
Are at least 18 years of age at screening
Have a clinical diagnosis of granulomatosis polyangiitis (GPA) or a diagnosis of microscopic polyangiitis (MPA) or a diagnosis of eosinophilic granulomatosis with polyangiitis (EGPA) according to the Chapel Hill Consensus Conference Definitions
Have a Birmingham Vasculitis Activity Score (version 3) [23] > 3 with minor BVAS items only (no major BVAS items). BVAS should be > 3 at screening and at randomisation
Patients should be receiving maintenance therapy at a stable dose for 4 weeks prior to randomisation. Maintenance therapy is defined as prednisolone and/or azathioprine, methotrexate, mycophenolate, co-trimoxazole or maintenance rituximab therapy
Patients receiving corticosteroids for reasons other than vasculitis must be on a stable regimen for 4 weeks prior to randomisation
- A female patient is eligible to enter the study if she is:
- Not pregnant or nursing
- Of non-childbearing potential (i.e. women who have had a hysterectomy, are postmenopausal, defined as ≥ 1 year without menses, have both ovaries surgically removed or have documented tubal ligation or other permanent sterilisation procedure); or
- Of childbearing potential. These women must have a negative urine pregnancy test at screening and at baseline and be using at least one effective method of contraception
Periodic abstinence (e.g. calendar, ovulation, symptothermal, post-ovulation methods) and withdrawal are not acceptable methods of contraception. Consistent and correct use of one of the following acceptable methods of birth control for 1 month prior to the start of the study agent, during the study and 16 weeks after the last dose of study agent:
Oral contraceptive, either combined or progestogen alone
Injectable progestogen
Implants of levonorgestrel or etonogestrel
Estrogenic vaginal ring
Percutaneous contraceptive patches
Intrauterine device (IUD) or intrauterine system (IUS) with < 1% failure rate as stated in the product label
-
7.
No contraindications to hydroxychloroquine therapy
-
8.
Willing and able to give written informed consent to participate in the trial
-
9.
Patients should have sufficient English in order to provide informed consent and complete the patient questionnaires
Exclusion criteria
Patients currently taking hydroxychloroquine or related antimalarial such as mepacrine or chloroquine
Patients with estimated glomerular filtration rate (eGFR) < 30 ml/min
Patients weighing < 40 kg
Sensitivity, anaphylaxis or allergy to hydroxychloroquine or any other 4-aminoquinoline compound
Known glucose 6 phosphate dehydrogenase deficiency
Known lactose intolerance
Evidence of plaque psoriasis
- Concomitant use of the following medications within the last 6 months:
- Tumour necrosis factor inhibitor treatment (e.g. etanercept)
- Cyclophosphamide
- Abatacept
- Alemtuzumab
- Any experimental or biological therapies
- Intravenous, intramuscular or sub-cutaneous immunoglobulin
- Plasma exchange
- Antithymocyte globulin
- Tamoxifen
- Live vaccines
B cell depleting therapy (rituximab) for remission induction within the last 6 months. Rituximab maintenance therapy is permitted
Severe or rapidly progressive ANCA vasculitis with at least one major BVAS item
Have clinical evidence of significant unstable or uncontrolled acute or chronic diseases not due to vasculitis (i.e. cardiovascular, pulmonary, hematologic, gastrointestinal, hepatic, renal, neurological, malignancy or infectious disease) which, in the opinion of the principal investigator, could confound the results of the study or put the patient at undue risk
Patients taking long-term macrolide antibiotics for a chronic condition. This does not include topical preparations
Have a history of malignant neoplasm within the last 5 years, except for adequately treated cancers of the skin (basal or squamous cell) or carcinoma in situ of the uterine cervix
Have current drug or alcohol abuse or dependence or a history of drug or alcohol abuse or dependence within 364 days prior to randomisation. A urine drug screen should be performed and confirmed negative prior to study entry
Have active hepatitis B, hepatitis C or HIV-1. Patients with positive hepatitis B core antibodies may be eligible providing the viral load is undetectable and/or the patient is receiving prophylactic antiviral agents
Have a grade 3 or greater laboratory abnormality based on the Common Terminology Criteria for Adverse Events (CTCAE) toxicity scale (version 5), unless considered by the investigator to be related to the underlying disease or induction therapy
Screening 12-lead electrocardiogram (ECG) that demonstrates clinically relevant abnormalities that may affect patient safety or interpretation of study results, including: QT interval corrected using the same consistent formula at each visit (QTc) > 470 msec for female > 450 msec for male patients demonstrated by at least two ECGs
Participation in any other interventional trial within the last 6 months
Have a current symptomatic COVID-19 infection
Have been admitted to the ICU in the past 6 months due to a COVID-19 infection
At trial entry, eligible participants will have uncontrolled disease, which is defined as BVAS > 3. Over the course of the study, a participant may have a limited or severe flare, persistent disease, or they may enter remission. A limited flare is defined as a new or worsening minor item on the BVAS with no new major items, whereas a severe flare includes a new or worsening major item. Persistent disease indicates the presence of one or more ongoing items on the BVAS with no new or worsening items, while remission is defined as a BVAS of 0 on two consecutive visits planned to be at least 4 weeks apart.
Intervention
Participants will be blinded and receive either 400mg daily hydroxychloroquine or placebo tablets taken orally in addition to standard of care therapies. The dose will be reduced to 200 mg daily for the duration of the trial for patients who weigh < 50 kg or who have an eGFR of 30–50 ml/min.
After study treatment, participants whose eGFR dropped below 60 ml/min at any point of the trial will be asked to attend an optometrist appointment to assess near visual acuity and visual fields.
Participants will be withdrawn from the investigational medicinal product (IMP) if they develop end stage renal failure, require a critical care admission, or if they are admitted to hospital with a COVID-19 infection. The patient may resume treatment once recovered if admitted due to COVID-19. The PI may also discontinue treatment in the event of an adverse event (AE) in accordance with best medical judgement and patients may ask to withdraw from the trial at any time.
Participants will be asked to return any unused IMP and/or empty packaging at each trial visit. The study drug returns will be returned to pharmacy for accountability. Participants will also be prompted to complete a weekly diary to record compliance. If compliance is ≤ 80%, the investigator should counsel the patient and ensure steps are taken to improve compliance. Any instances of a patient with > 120% compliance (considered an overdose) will be escalated to the trial management group (TMG) and the trial steering committee (TSC) in addition to the patient receiving counselling. Secondary levels of monitoring are provided by the DMC as part of interim data reviews and blood hydroxychloroquine levels.
Blood samples will be collected at four visits across the trial in order to analyse the levels of hydroxychloroquine in the blood. This analysis will be completed after the trial has ended to maintain blinding to treatment allocation. Samples will also be collected and stored for future research into AAV.
Randomisation and blinding
Participants will be randomised 1:1 to the intervention or placebo with minimisation for ANCA (positive vs negative), age (</≥ 60 years), previous rituximab therapy (yes/no) and smoking (current smoker/non-smoker) to ensure balanced groups. Authorised staff members at each trial site will recruit participants and enter their details into a web-based randomisation system hosted by King’s Clinical Trial Unit (KCTU) to obtain treatment allocation. Blinded allocation details will be sent by email automatically to key trial site staff.
Both participants and research staff will be blinded to treatment allocation. Only the data monitoring committee (DMC) will have access to unblinded data during the trial period to safeguard patient interests. Blinded treatment supplies with unique treatment pack identifiers will be allocated through the King’s Clinical Trials Unit Intervention Management System. Evidence of unblinding of research staff completing data collection will be investigated.
Emergency code breaking services will be provided by ESMS Global with services available 24/7. Direct care teams and clinical trial personnel are permitted to receive unblinding information in the instance that the medical care of the patient is dependent on knowing the treatment allocation. Where possible, the PI will be contacted to confirm whether unblinding is necessary.
Outcome measures
The primary outcome is the proportion of patients at any point during the final 12 weeks of the study (± 7 days) with uncontrolled AAV disease activity (defined as BVAS > 3) OR with controlled AAV disease activity (BVAS ≤ 3) but prednisolone dose for AAV > 7.5 mg daily OR with controlled AAV disease activity (BVAS ≤ 3) but any corticosteroid use > 7.5mg daily for any reason*.
The following secondary outcomes will be examined:
Median cumulative number of visits where BVAS = 0 experienced by each patient (excluding screening, baseline and week 56)
Proportion of patients with deemed treatment failure at week 52
Mean cumulative dosage of prednisolone*
Total number of adverse events per treatment arm*
Median cumulative number of infections experienced by each patient
Median number of vasculitis flares (major and minor) experienced by each patient (excluding those as screening, baseline and week 56)
Median time from randomisation to remission (BVAS = 0 on two consecutive visits at least 4 weeks apart)
Median time to the first severe flare with starting time points of randomisation and remission (if achieved)
Median time to the first limited flare with starting time points of randomisation and remission (if achieved)
Proportion of patients categorised as having a severe flare at each time point (excluding screening, baseline and week 56)
Proportion of patients categorised as having a limited flare at each time point (excluding screening, baseline and week 56)
Median absolute Vasculitis Damage Index (VDI) [24] at each time point and the median relative change in VDI from baseline to each time point
The following exploratory outcomes will be examined:
Proportion of patient with incidence of new diabetes mellitus*
Proportion of patients with dyslipidaemia
Median Functional Assessment of Chronic Illness Therapy (FACIT) [25] score at each time point and the median relative change in FACIT score from baseline to week 52/56*
Median scores in quality-of-life measures: SF-36 [26], EQ5D [27], Health Assessment Questionnaire (HAQ) [28] and AAV Patient Reported Outcome (PRO) [29] at each time point and the median relative change in the same quality of life measures from baseline to week 52/56*
Median Glucocorticoid Toxicity Index (GTI) [30] at each time point and the median relative change in GTI from baseline to week 52/56
Median Physician’s Global Assessment (PGA) [31] at each time point and the median relative change in PGA from baseline to week 52/56*
Mean ANCA titres at each time point and the median relative change in ANCA titres from baseline to each time point*
Proportion of patients with medicine compliance of ≤ 80%
Mean absolute values and relative change from baseline to each time point in renal variables: serum creatinine, serum albumin, urine protein to creatinine ratio*
Participants wishing to stop trial medication will be asked to continue trial visits and data collection if willing. Otherwise, they will be withdrawn from the trial.
In the case that a participant withdraws more than 4 weeks since their last hospital visit, they will be invited to attend a withdrawal visit or a telephone interview. Data contributing to the asterisked (*) outcome measures above will be collected if feasible during this visit. Data collected prior to participant withdrawal will be included in the statistical analysis where possible.
It is anticipated that the withdrawal rate for this cohort of patients will be < 5% [21, 32, 33]. No specific plans have been made to ensure participant retention as this low withdrawal rate is based on similar studies in AAV patients. However, participants will be contacted via the telephone between hospital visits which are longer than 4 weeks apart to provide regular contact and encourage participants to report AEs.
Sample size calculation
As AAV is uncommon, the required sample size was optimised using a phase II selection theory approach (also known as ‘pick the winner’) as proposed by Simon et al. [34]. The treatment which is ranked as having the lower proportion of patients with uncontrolled AAV (BVAS > 3) (or controlled but with > 7.5 mg prednisolone or other corticosteroid daily) in the final 12 weeks of the study will be selected as the ‘winning treatment’.
A sample size of 72 patients (36 in each arm) will be required to provide 90% probability of correctly ranking hydroxychloroquine as superior to placebo, assuming 50% of patients in the placebo group have uncontrolled AAV (or controlled but with > 7.5 mg prednisolone or other corticosteroid daily) in the final 12 weeks of the study and assuming that the true reduction in this rate due to hydroxychloroquine is 15%. This trial is not designed to demonstrate a statistically significant difference between the two arms, so the type I error rate does not apply.
Seventy-six participants will be recruited to allow for a 5% drop-out rate. This drop-out rate is based on similar trials which had a < 5% drop-out rate [21, 32, 33].
Data management and monitoring
A web-based electronic data capture system has been designed using the InferMed Macro 4 system hosted by the King’s College London Clinical Trials Unit (KCTU). No identifiable data beyond participant initials, age at consent and year of birth will be entered on the electronic case report form (eCRF), and no data will be collected unless a participant has signed a consent form. Participants will be assigned a unique trial number upon entry to the trial, ensuring that all trial data is pseudonymised.
Source data worksheets will be used to collect data, and these will be transcribed to the eCRF. The FACIT, SF-36, EQ5D, HAQ and AAV Pro questionnaires will be used to collect quality of life data. Lab sample results will be transcribed to the eCRF from medical records.
Trial monitoring will be provided by the King’s Health Partners Clinical Trials Office (KHP-CTO). The clinical research associate (CRA) will perform source data verification and raise queries with the trial site staff. Missing data and discrepancies will be reviewed and queried by the trial’s data manager to ensure data quality prior to each extract.
Quality assurance
All clinicians scoring BVAS and VDI will be trained and certified prior to recruitment of participants. An adjudication committee blinded to treatment group will scrutinise all the BVAS and VDI scores and major and minor relapses. The adjudication committee will provide advice and further training where appropriate to ensure consistency across sites. The committee is chaired by Prof Raashid Luqmani, one of the sites’ principal investigators. He is the leading expert on BVAS and VDI.
Safety
All serious adverse events (SAEs), serious adverse reactions (SARs) and suspected unexpected serious adverse reactions (SUSARs) related to trial procedures will be reported immediately (and no later than 24 h) by the investigator to the KHP-CTO and chief investigator (CI) for review in accordance with their Pharmacovigilance Policy. Adverse events related to disease activity will be recorded as part of BVAS and so will not be reported separately. All data regarding the occurrence of adverse events will be made available to the DMC for review.
Trial oversight
The trial has a DMC, TSC and TMG.
The DMC meet every 6 months. The committee has access to unblinded accumulating comparative data and is comprised of independent members. The DMC is responsible for monitoring the accumulating data and making recommendations to the TSC on whether the trial should continue as planned. Only the DMC and a partially unblinded statistician has access to the unblinded accumulating data.
The TSC meet following each DMC meeting and is responsible for oversight of the trial in order to safeguard the interests of trial participants. The TSC provides advice to the CI, KHP-CTO, the funder and sponsor on all aspects of the trial through its independent chair. The BVAS adjudication committee feed into the TSC as needed.
The TMG is responsible for the day-to-day running and management of the trial and meet on a monthly basis. Sites will be invited to attend the TMG to inform them of trial amendments and provide a forum for feedback and recruitment information.
Statistical analysis
Due to the use of a selection theory (pick the winner) approach, the main analysis will not include inferential analysis. Instead, the main analysis will involve presenting the descriptive statistics for each treatment arm to demonstrate which treatment is selected (i.e. which treatment group has a lower proportion of participants meeting the primary endpoint criteria). Due to this trial design, no interim analyses have been planned.
The descriptive statistics follow those specified in the outcome measures. The primary outcome and secondary/exploratory outcomes specifying a proportion will be described as the number and proportion of participants meeting that outcome. Secondary/exploratory outcomes specifying median will be described as median and range. Secondary/exploratory outcomes specifying mean will be described as mean and standard deviation.
Subgroup/exploratory analyses
The reporting of descriptive statistics for the primary endpoint and ranking of the two treatment arms will be repeated for the following subgroups:
Those deemed compliant with treatment (consumption of > 80% of provided tablets)
Those deemed non-compliant with treatment (consumption of ≤ 80% of provided tablets)
These analyses will only be repeated if enough participants meet these criteria for each treatment arm (≥ 5 participants per group).
Additionally, exploratory inferential analyses have been planned. This will involve tests of two proportions for those outcomes specifying a proportion. For the other outcomes, unpaired t-tests and Mann Whitney U tests will be used dependent on the resultant outcome distribution. For all tests, a one-sided significance level of 10% will be used to determine results of potential interest.
Considerations
Loss to follow-up is not expected as trials in a similar cohort only had a < 5% drop-out rate [21, 32, 33]. However, individual time points may experience missing data due to logistical issues. The number and proportion of participants with missing outcome data at each time point will be reported. The main analysis for this trial only considers time points in the final 12 weeks of the trial. Changes in this endpoint at each of the four visits during this period will be investigated but there is likely to be a low variability across each visit. As a result, participants with missing outcome data in one or two visits are unlikely to substantially bias the results. A sensitivity analysis will be utilised to confirm the lack of bias due to missingness in these last 12 weeks.
Patient and public involvement
Prior to recruitment, two patients known to the CI were asked to review the trial protocol and patient information sheet. Adjustments were made to the trial documents where this was feasible. A patient representative is asked to attend each meeting of the HAVEN TSC to ensure that feedback is obtained on potential protocol amendments and issues that may arise with recruitment.
During the COVID-19 pandemic, the Vasculitis UK charity was approached to ask their members for feedback about their views on attending clinical trial visits during the pandemic. The feedback received was largely positive, but concerns about travelling on public transport were a major theme. The trial was amended to increase reimbursement for travel fees so participants could travel privately.
Adjustments made because of COVID-19
The coronavirus pandemic delayed the trial opening and as a result the trial has been extended to cover this lost time. An amendment was made to the protocol in light of the impact of the pandemic. Firstly, the eligibility criteria was updated to exclude patients with a current symptomatic infection and those who had been admitted to critical care in the past 6 months as a result of a COVID-19 infection. Greater flexibility was added to trial visit windows to allow for participant isolation.
Discussion
This trial aims to investigate whether the administration of hydroxychloroquine in addition to standard of care maintenance therapy can improve the clinical response of AAV by moderating disease activity, reducing disease flares and preventing disease progression, culminating in an improved quality of life for patients with this disease. The importance of developing additional treatment options for AAV cannot be understated. Although a recent meta-analysis has demonstrated a decline in mortality risk (likely due to improvements in therapeutics and patient care) [2, 35, 36], the mortality within this population is still approximately double that of the general population. Alongside this, both physical and mental quality of life for this cohort is poor compared to the general population [37], and there is little evidence that this has improved under the more recent therapeutic developments [20].
Systemic vasculitis, including AAV, is associated with increased healthcare costs due to increases in hospital care and expensive medications [4, 38, 39]. Biological therapies used for relapsing AAV cost in excess of £6000 annually per patient. On an individual level, chronic autoimmune rheumatic disorders have been associated with higher unemployment, particularly as disease duration increases [40]. AAV is unlikely to be an exception with patient reports of reduced income and hindered careers [4, 40] as a result of their disease. The increasing cost of living in the UK means that new treatments that improve patient quality of life and ability to work are greatly needed for these patients. Alongside this, the NHS has seen a decade of budget cuts, which means that inexpensive treatments would reduce the healthcare burden of these patients. Hydroxychloroquine has the benefit of being a relatively cheap medication with an estimated cost of < £70 annually per patient and it has been shown to modify disease trajectory in other related-autoimmune conditions [8–11], including SLE. Shared immunopathology between AAV and these more common autoimmune conditions suggests that Hydroxychloroquine could be just as beneficial in this patient cohort.
One key finding from studies administering hydroxychloroquine to treat rheumatoid arthritis/SLE is the potential glucocorticoid sparing effect. Glucocorticoids to induce disease remission remain the standard of care treatment for AAV, although treatment regimens have been modified over the years to minimise the cumulative dosage administered [41]. However, glucocorticoids are associated with a wide range of physical, mental and social adverse effects including the induction of long-term complications such as hypertension, diabetes, osteoporosis, cataracts and cardiovascular events [42, 43]. Similarly, the risk of infections is high, with evidence suggesting that the continued high mortality in patients may be due to treatment-related adverse events rather than AAV itself [44].
A review of off-label use of hydroxychloroquine in patients with AAV found that patients had fewer relapses, improved symptoms and a reduced need for glucocorticoids [13], and it is anticipated that these benefits will be seen in the HAVEN trial. However, a recent study by Lane et al. has shown that hydroxychloroquine increases the cardiovascular risk to rheumatoid arthritis patients after 30 days of continuous use, and this is worsened with concomitant use of azithromycin [45]. The HAVEN protocol was amended to exclude patients taking macrolide antibiotics for long-term conditions and to reintroduce an ECG at week 16 to monitor cardiovascular safety. Previous research conducted on COVID-19 patients [46] had led to this ECG being removed.
COVID-19 had a large impact on the HAVEN trial. Hydroxychloroquine received widespread media attention in the early months of the COVID-19 pandemic. Tweets sent by then-US President Donald J Trump advocating the use of hydroxychloroquine were estimated to have been seen by approximately 78 million people [47]. Awareness of hydroxychloroquine was amplified by the media’s reporting on the results of the RECOVERY trial [48] and news of the World Health Organization’s halt of the SOLIDARITY trial [49]. Concerns were raised that this news would impact recruitment to the HAVEN trial, especially given that the patient cohort was shielding, deemed to be at high risk for COVID, and reportedly avoiding medical appointments or undertaking laboratory tests [50]. The trial patient information sheet was amended to allay patient concerns about the safety of hydroxychloroquine, particularly in the light that the doses in the RECOVERY trial were 10 times greater than the doses used in HAVEN [48].
With concerns of slow recruitment, a consideration when it came to determining recruitment targets was the rarity of AAV. Approximately 1 in 3000 people within the UK population have AAV [1] meaning that this condition fits the definition for a rare disease as defined by the UK Rare Disease Framework. As a result, specific considerations were made when determining the sample size for this trial. A typical study design, with a stated type 1 error/significance level, would require at least 200 patients to compare the primary outcome in patients administered hydroxychloroquine or placebo using statistical tests with a sufficient power. For a novel therapeutic trial in a rare disease, this sample size would have been difficult to achieve, so an alternative pick the winner design was used [34]. This design describes the outcome measures in each treatment group and ranks the treatment groups but does not statistically compare them. If hydroxychloroquine ranks better than placebo (i.e. fewer patients have uncontrolled AAV or controlled AAV with high medication usage in the final 12 weeks), an extended study would be needed to accurately estimate the efficacy of hydroxychloroquine unless this effect size is substantially larger than the expected 15% used to design this trial.
Despite concerns about the impact of COVID-19 on recruitment, the HAVEN trial hopes to show that the addition of hydroxychloroquine as an adjunctive therapy to standard of care therapies does reduce disease activity in patients with AAV.
Trial status
The trial is currently working to protocol version 6.0, dated 10 May 2022. The protocol was written in line with SPIRIT guidance.
Recruitment began on 17 December 2020 and is due to close 31 December 2024.
Supplementary Information
Acknowledgements
This research was supported by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care.
AEL and AD also acknowledge funding support from the NIHR Applied Research Collaboration (ARC) South London at King’s College Hospital NHS Foundation Trust and the Royal College of Physicians.
We would like to acknowledge all of the members of the research teams at participating sites, as well as the members of the DMC (Professor Ariane Herrick, Professor Caroline Dore and Professor Ed Vital) and TSC (Professor Michael Ehrenstein, Mr Nigel Davies, James Hancocks, Professor Justin Mason, Dr Sanjeev Patel and Dr Gayathri Rajakaruna) for their involvement in the HAVEN trial. We would also like to acknowledge John Mills, chair of the Vasculitis UK charity, and the charity members who helped guide our COVID-19 response.
We would also like to thank every patient who has participated in the trial for giving up their time for us.
Dissemination
It is intended that the results of the trial will be reported and disseminated at international conferences and in peer-reviewed scientific journals. The chief investigator will ensure that the results are analysed, written up, reported and disseminated upon completion of the trial. Patients recruited to the trial will receive a summarised version of the results at their request.
Abbreviations
- AAV
ANCA-associated vasculitis
- AAV PRO
ANCA-associated vasculitis patient reported outcome
- AE
Adverse event
- ANCA
Anti-neutrophil cytoplasmic antibodies
- BVAS
Birmingham Vasculitis Activity Score
- CI
Chief investigator
- CRA
Clinical research associate
- CTCAE
Common Terminology Criteria for Adverse Events
- DMC
Data monitoring committee
- ECG
Electrocardiogram
- eCRF
Electronic case report form
- eGFR
Estimated glomerular filtration rate
- EGPA
Eosinophilic granulomatosis with polyangiitis
- FACIT
Functional assessment of chronic illness therapy
- GPA
Granulomatosis with polyangiitis
- GSTT
Guy’s and St Thomas’s NHS Foundation Trust
- GTI
Glucocorticoid Toxicity Index
- HAQ
Health Assessment Questionnaire
- IMP
Investigational medicinal product
- IUD
Intrauterine device
- IUS
Intrauterine system
- KCL
King’s College London
- KCTU
King’s Clinical Trials Unit
- KHP-CTO
King’s Health Partners Clinical Trials Office
- MPA
Microscopic polyangiitis
- PGA
Physician’s Global Assessment
- PI
Principal investigator
- SAE
Serious adverse event
- SAR
Serious adverse reaction
- SLE
Systemic lupus erythematosus
- SoC
Standard of care
- SUSAR
Suspected unexpected serious adverse reaction
- TMG
Trial management group
- TSC
Trial steering committee
- VDI
Vasculitis Damage Index
Authors’ contributions
DDC is the CI for this trial and led the proposal and protocol development. LA, FR, NBH, ACas, SS, NM, LN, ACap, SJ, DS, RL, DJ and JG all contributed significantly to the protocol. AD, AEL, FR and NBH planned the statistical analysis. AEL and LA wrote the first draft of the manuscript, and this was edited by DDC and AD. All authors were offered the opportunity to read and approve the final manuscript.
Funding
This trial is funded by the Medical Research Council (Grant Ref: MR/R006253/1). The funder has had no direct involvement in the trial design or writing of the report nor will they be involved in the analysis, management, interpretation and publication of the data.
Availability of data and materials
The full trial protocol is available on request to the corresponding author.
The trial team are responsible for the final trial dataset. This dataset and the statistical code can be made available on reasonable request after consideration by the trial management group.
Declarations
Ethics approval and consent to participate
This trial will adhere to the UK Framework for Health and Social Care research. Prior to participation, all subjects provide informed consent and are informed in advance that they can withdraw from the trial at any time without penalty.
The trial was approved by the London – Riverside Research Ethics committee.
Consent for publication
Not applicable
Competing interests
DDC reports participating in a speaker bureau for Vifor Pharma.
The other authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Annastazia E Learoyd and Lauren Arnold are joint first authors.
References
- 1.Watts RA, Scott DGI. Epidemiology of the vasculitides. Semin Respir Crit Care Med. 2004;25:455–464. doi: 10.1055/s-2004-836139. [DOI] [PubMed] [Google Scholar]
- 2.Flossmann O, Berden A, De Groot K, et al. Long-term patient survival in ANCA-associated vasculitis. Ann Rheum Dis. 2011;70:488–494. doi: 10.1136/ard.2010.137778. [DOI] [PubMed] [Google Scholar]
- 3.Pagnoux C, Mahr A, Hamidou MA, et al. Azathioprine or methotrexate maintenance for ANCA-associated vasculitis. N Engl J Med. 2008;359:2790–2803. doi: 10.1056/NEJMoa0802311. [DOI] [PubMed] [Google Scholar]
- 4.Hoffman GS, Drucker Y, Cotch MF, et al. Wegener’s granulomatoisis: patient-reported effects of disease on health, function, and income. Arthritis Rheum. 1998;41:2257–2262. doi: 10.1002/1529-0131(199812)41:12<2257::AID-ART22>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 5.Merkel PA, Lo GH, Holbrook JT, et al. Brief Communication: High incidence of venous thrombotic events among patients with Wegener granulomatosis: The Wegener’s Clinical Occurrence of Thrombosis (WeCLOT) Study. Ann Intern Med. 2005;142:620. doi: 10.7326/0003-4819-142-8-200505030-00011. [DOI] [PubMed] [Google Scholar]
- 6.Suppiah R, Judge A, Batra R, et al. A model to predict cardiovascular events in patients with newly diagnosed Wegener’s granulomatosis and microscopic polyangiitis. Arthritis Care Res (Hoboken) 2011;63:588–596. doi: 10.1002/acr.20433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walsh M, Merkel PA, Mahr A, et al. Effects of duration of glucocorticoid therapy on relapse rate in antineutrophil cytoplasmic antibody–associated vasculitis: a meta-analysis. Arthritis Care Res. 2010;62:1166–1173. doi: 10.1002/acr.20176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.The Canadian Hydroxychloroquine Study Group A randomized study of the effect of withdrawing hydroxychloroquine sulfate in systemic lupus erythematosus. N Engl J Med. 1991;324:150–154. doi: 10.1056/NEJM199101173240303. [DOI] [PubMed] [Google Scholar]
- 9.Petri M. Hydroxychloroquine use in the baltimore lupus cohort: effects on lipids, glucose and thrombosis. Lupus. 1996;5:S16–S22. [PubMed] [Google Scholar]
- 10.Makol A, Matteson EL, Warrington KJ. Rheumatoid vasculitis: an update. Curr Opin Rheumatol. 2015;27:63–70. doi: 10.1097/BOR.0000000000000126. [DOI] [PubMed] [Google Scholar]
- 11.Mumcu G, Biçakçigil M, Yilmaz N, et al. Salivary and serum B-cell activating factor (BAFF) levels after hydroxychloroquine treatment in primary Sjögren’s syndrome. Oral Heal Prev Dent. 2013;11:229–234. doi: 10.3290/j.ohpd.a30172. [DOI] [PubMed] [Google Scholar]
- 12.Jachiet M, Flageul B, Deroux A, et al. The clinical spectrum and therapeutic management of hypocomplementemic urticarial vasculitis: data from a French nationwide study of fifty-seven patients. Arthritis Rheum. 2015;67:527–534. doi: 10.1002/art.38956. [DOI] [PubMed] [Google Scholar]
- 13.Casian A, Sangle S, D’Cruz D. The role of hydroxychloroquine in anti-neutrophil cytoplasmic antibody–positive and negative vasculitis. Ann Rheum Dis. 2016;75:1086–1087. [Google Scholar]
- 14.Tanay A, Leibovitz E, Frayman A, et al. Vascular elasticity of systemic lupus erythematosus patients is associated with steroids and hydroxychloroquine treatment. Ann N Y Acad Sci. 2007;1108:24–34. doi: 10.1196/annals.1422.003. [DOI] [PubMed] [Google Scholar]
- 15.Rolain JM, Colson P, Raoult D. Recycling of chloroquine and its hydroxyl analogue to face bacterial, fungal and viral infections in the 21st century. Int J Antimicrob Agents. 2007;30:297–308. doi: 10.1016/j.ijantimicag.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feldman CH, Hiraki LT, Winkelmayer WC, et al. Serious infections among adult medicaid beneficiaries with systemic lupus erythematosus and lupus nephritis. Arthritis Rheum. 2015;67:1577–1585. doi: 10.1002/art.39070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruiz-Irastorza G, Egurbide MV, Pijoan JI, et al. Effect of antimalarials on thrombosis and survival in patients with systemic lupus erythematosus. Lupus. 2006;15:577–583. doi: 10.1177/0961203306071872. [DOI] [PubMed] [Google Scholar]
- 18.Lagneaux L, Delforge A, Carlier S, et al. Early induction of apoptosis in B-chronic lymphocytic leukaemia cells by hydroxychloroquine: activation of caspase-3 and no protection by survival factors. Br J Haematol. 2001;112:344–352. doi: 10.1046/j.1365-2141.2001.02553.x. [DOI] [PubMed] [Google Scholar]
- 19.Rahim R, Strobl JS. Hydroxychloroquine, chloroquine, and all-trans retinoic acid regulate growth, survival, and histone acetylation in breast cancer cells. Anti-Cancer Drugs. 2009;20:736–745. doi: 10.1097/CAD.0b013e32832f4e50. [DOI] [PubMed] [Google Scholar]
- 20.Stone JH. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010;363:221–232. doi: 10.1056/NEJMoa0909905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones RB, Cohen Tervaert JW, Hauser T, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med. 2010;363:211–220. doi: 10.1056/NEJMoa0909169. [DOI] [PubMed] [Google Scholar]
- 22.Guillevin L, Pagnoux C, Karras A, et al. Rituximab versus azathioprine for maintenance in ANCA-associated vasculitis. N Engl J Med. 2014;371:1771–1780. doi: 10.1056/NEJMoa1404231. [DOI] [PubMed] [Google Scholar]
- 23.Mukhtyar C, Lee R, Brown D, et al. Modification and validation of the Birmingham Vasculitis Activity Score (version 3) Ann Rheum Dis. 2009;68:1827–1832. doi: 10.1136/ard.2008.101279. [DOI] [PubMed] [Google Scholar]
- 24.Exley A, Bacon P, Luqmani R, et al. Development and initial validation of the Vasculitis Damage Index for the standardized clinical assessment of damage in the systemic vasculitides. Arthritis Rheum. 1997;40:371–380. doi: 10.1002/art.1780400222. [DOI] [PubMed] [Google Scholar]
- 25.Webster K, Cella D, Yost K. The Functional Assessment of Chronic Illness Therapy (FACIT) Measurement System: properties, applications, and interpretation. Health Qual Life Outcomes. 2003;16:79. doi: 10.1186/1477-7525-1-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ware JJ, Sherbourne C. The MOS 36-item short-form health survey (SF-36). I. Conceptual framework and item selection. Med Care. 1992;30:473–483. [PubMed] [Google Scholar]
- 27.EuroQol Group EuroQol--a new facility for the measurement of health-related quality of life. Health Policy (New York) 1990;16:199–208. doi: 10.1016/0168-8510(90)90421-9. [DOI] [PubMed] [Google Scholar]
- 28.Bruce B, Fries J. The Stanford Health Assessment Questionnaire: dimensions and practical applications. Health Qual Life Outcomes. 2003;1:20. doi: 10.1186/1477-7525-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robson JC, Dawson J, Doll H, et al. Validation of the ANCA-associated vasculitis patient-reported outcomes (AAV-PRO) questionnaire. Ann Rheum Dis Rhemautic Dis. 2018;77:1157–1164. doi: 10.1136/annrheumdis-2017-212713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miloslavsky EM, Naden RP, Bijlsma J, et al. Development of a Glucocorticoid Toxicity Index (GTI) using multicriteria decision analysis. Ann Rheum Dis. 2017;76:543–546. doi: 10.1136/annrheumdis-2016-210002. [DOI] [PubMed] [Google Scholar]
- 31.Stone JH, Hoffman GS, Merkel PA, et al. A disease-specific activity index for Wegener’s granulomatosis: modification of the Birmingham Vasculitis Activity Score. International Network for the Study of the Systemic Vasculitides (INSSYS) Arthritis Rheum. 2001;44:912–920. doi: 10.1002/1529-0131(200104)44:4<912::AID-ANR148>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 32.Jayne D, Rasmussen N, Andrassy K, et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med. 2009;349:36–44. doi: 10.1056/NEJMoa020286. [DOI] [PubMed] [Google Scholar]
- 33.De Groot K, Rasmussen N, Bacon PA, et al. Randomized trial of cyclophosphamide versus methotrexate for induction of remission in early systemic antineutrophil cytoplasmic antibody–associated vasculitis. Arthritis Rheum. 2005;52:2461–2469. doi: 10.1002/art.21142. [DOI] [PubMed] [Google Scholar]
- 34.Simon R, Wittes R, Ellenberg S. Randomized phase II clinical trials. Cancer Treat Rep. 1985;69:1375–1381. [PubMed] [Google Scholar]
- 35.Tan JA, Dehghan N, Chen W, et al. Mortality in ANCA-associated vasculitis: ameta-analysis of observational studies. Ann Rheum Dis. 2017;76:1566–1574. doi: 10.1136/annrheumdis-2016-210942. [DOI] [PubMed] [Google Scholar]
- 36.Wallace ZS, Fu X, Harkness T, et al. All-cause and cause-specific mortality in ANCA-associated vasculitis: overall and according to ANCA type. Rheumatology. 2020;59:2308–2315. doi: 10.1093/rheumatology/kez589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Basu N, McClean A, Harper L, et al. The characterisation and determinants of quality of life in ANCA associated vasculitis. Ann Rheum Dis. 2014;73:207–211. doi: 10.1136/annrheumdis-2012-202750. [DOI] [PubMed] [Google Scholar]
- 38.Thorpe CT, Thorpe JM, Jiang T, et al. Healthcare utilization and expenditures for United States Medicare beneficiaries with systemic vasculitis. Semin Arthritis Rheum. 2018;47:507–519. doi: 10.1016/j.semarthrit.2017.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Raimundo K, Farr AM, Kim G, et al. Clinical and economic burden of antineutrophil cytoplasmic antibody-associated vasculitis in the United States. J Rheumatol. 2015;42:2383–2391. doi: 10.3899/jrheum.150479. [DOI] [PubMed] [Google Scholar]
- 40.Mau W, Listing J, Huscher D, et al. Employment across chronic inflammatory rheumatic diseases and comparison with the general population. J Rheumatol. 2005;32:721–728. [PubMed] [Google Scholar]
- 41.Walsh M, Merkel PA, Peh C-A, et al. Plasma exchange and glucocorticoids in severe ANCA-associated vasculitis. N Engl J Med. 2020;382:622–631. doi: 10.1056/NEJMoa1803537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tesar V, Hruskova Z. Limitations of standard immunosuppressive treatment in ANCA-associated vasculitis and lupus nephritis. Nephron Clin Pr. 2014;128:205–215. doi: 10.1159/000368569. [DOI] [PubMed] [Google Scholar]
- 43.Robson JC, Dawson J, Cronholm PF, et al. Patient perceptions of glucocorticoids in anti-neutrophil cytoplasmic antibody-associated vasculitis. Rheumatol Int. 2018;38:675–682. doi: 10.1007/s00296-017-3855-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Little M, Nightingale P, Verburgh CA, et al. Early mortality in systemic vasculitis: relative contribution of adverse events and active vasculitis. Ann Rheum Dis. 2010;69:1036–1043. doi: 10.1136/ard.2009.109389. [DOI] [PubMed] [Google Scholar]
- 45.Lane JCE, Weaver J, Kostka K, et al. Risk of hydroxychloroquine alone and in combination with azithromycin in the treatment of rheumatoid arthritis: a multinational, retrospective study. Lancet Rheumatol. 2020;2:e698–e711. doi: 10.1016/S2665-9913(20)30276-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kamp TJ, Hamdan MH, January CT. Chloroquine or hydroxychloroquine for COVID-19: is cardiotoxicity a concern? J Am Heart Assoc. 2020;9:16887. doi: 10.1161/JAHA.120.016887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Niburski K, Niburski O. Impact of Trump’s promotion of unproven COVID-19 treatments and subsequent Internet trends: observational study. J Med Internet Res. 2020;22:e20044. doi: 10.2196/20044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.The RECOVERY Collaborative Group Effect of hydroxychloroquine in hospitalized patients with COVID-19. N Engl J Med. 2020;383:2030–2040. doi: 10.1056/NEJMoa2022926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mahase E. Covid-19: WHO halts hydroxychloroquine trial to review links with increased mortality risk. BMJ. 2020;369:m2126. doi: 10.1136/bmj.m2126. [DOI] [PubMed] [Google Scholar]
- 50.Kronbichler A, Geetha D, Smith RM, et al. The COVID-19 pandemic and ANCA-associated vasculitis – reports from the EUVAS meeting and EUVAS education forum. Autoimmun Rev. 2021;20:102986. doi: 10.1016/j.autrev.2021.102986. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The full trial protocol is available on request to the corresponding author.
The trial team are responsible for the final trial dataset. This dataset and the statistical code can be made available on reasonable request after consideration by the trial management group.