

Abstract
Objective.
Systemic sclerosis (SSc; scleroderma) is a chronic disease that affects the skin and various internal organs. Dermal fibrosis is a major component of this disease. The mechanisms that promote dermal fibrosis remain elusive. Elevations in tissue adenosine levels and the subsequent engagement of the profibrotic A2B adenosine receptor (ADORA2B) have been shown to regulate fibrosis in multiple organs including the lung, kidney, and penis; however, the role of ADORA2B in dermal fibrosis has not been investigated. We undertook this study to test our hypothesis that elevated expression of ADORA2B in the skin drives the development of dermal fibrosis.
Methods.
We assessed the involvement of ADORA2B in the regulation of dermal fibrosis using a well-established mouse model of dermal fibrosis. Using an orally active ADORA2B antagonist, we demonstrated how inhibition of ADORA2B results in reduced dermal fibrosis in 2 distinct experimental models. Finally, using human dermal fibroblasts, we characterized the expression of adenosine receptors.
Results.
We demonstrated that levels of ADORA2B were significantly elevated in dermal fibrosis and that the therapeutic blockade of this receptor in vivo using an ADORA2B antagonist could reduce the production of profibrotic mediators in the skin and attenuate dermal fibrosis. Antagonism of ADORA2B resulted in reduced numbers of arginase-expressing macrophages and myofibroblasts and in reduced levels of the extracellular matrix proteins fibronectin, collagen, and hyaluronan.
Conclusion.
These findings identify ADORA2B as a potential profibrotic regulator in dermal fibrosis and suggest that ADORA2B antagonism may be a useful approach for the treatment of SSc.
Dermal fibrosis is a prominent feature of scleroderma (systemic sclerosis [SSc]), which is a chronic and complex autoimmune disease that affects the skin and various internal organs (1). It has been proposed that the dermal fibrosis seen in SSc is driven in part by pathways that are similar to those found in normal wound healing, including the regulation of inflammation (2), the recruitment and proliferation of fibroblasts (3), and extracellular matrix production (3). Despite efforts to understand the immunologic and pathologic pathways involved in this disease, the mechanisms that promote dermal fibrosis in SSc remain elusive, and there are no effective therapies to halt the progression of dermal fibrosis in SSc patients.
Adenosine is a signaling molecule that is produced in response to tissue injury and inflammation (4). Extracellular adenosine can regulate numerous cellular responses through engagement of 4 cell surface adenosine receptors (A1 adenosine receptor [ADORA1], ADORA2A, ADORA2B, and ADORA3) (5). Consistent with the notion that dermal fibrosis is associated with the activation of wound healing pathways, chronic elevations in adenosine have been implicated in the regulation of dermal fibrosis through the engagement of ADORA2A on dermal fibroblasts (6). Indeed, ADORA2A has been implicated in several processes pertinent to the regulation of wound healing and fibrosis (6). Similarly, ADORA2B has also been implicated in processes associated with wound healing and fibrosis (7,8). These include the ability of this receptor to regulate profibrotic activities on fibroblasts (9) and macrophages (10). Moreover, work using genetically modified mice and selective ADORA2B antagonists has demonstrated that this receptor is effective in regulating the progression of pulmonary fibrosis (11,12), kidney fibrosis (13), and fibrosis in the penis (14). However, the role of ADORA2B in dermal fibrosis has not been systematically investigated.
The goal of this study was to assess the involvement of ADORA2B in the regulation of dermal fibrosis using a well-established mouse model of dermal fibrosis that is elicited by the subcutaneous (SC) injection of bleomycin (15) as well as by using the TSK1 mouse model (16). Using the bleomycin model, we characterized the expression of the adenosine receptors and demonstrated that levels of ADORA2B were significantly elevated in dermal fibrosis. Moreover, the therapeutic blockade of this receptor in vivo using an ADORA2B antagonist was shown to reduce the production of profibrotic mediators in the skin and to attenuate dermal fibrosis both in the bleomycin model and in TSK1 mice. Experiments using human fibroblasts from normal or SSc patients revealed high expression of ADORA2B. We also found increased hyaluronan synthase 2 (HAS-2) expression and increased HA levels in SSc skin. Collectively, this study identifies ADORA2B as a potential profibrotic regulator in dermal fibrosis and suggests that ADORA2B antagonism may be a useful approach for the treatment of SSc.
MATERIALS AND METHODS
Mouse model of bleomycin-induced skin fibrosis and TSK1 mice.
Male wild-type C57BL/6J mice were acquired from Envigo. Bleomycin (0.02 units/day/mouse; Teva Parenteral Medicines) dissolved in saline or saline alone was administered to 8-week-old mice by daily SC injections in 2 distinct sites on shaved backs of mice. On day 28, mice were killed, and lesional skin was obtained for protein lysates, total RNA, and histology (15). Each group consisted of 10 mice. In experiments using the ADORA2B antagonist GS-6201, the drug was provided mixed in chow starting on day 15 until the end of the study. Control mice received chow with vehicle. TSK1 mice were obtained from The Jackson Laboratory. Eight-week-old TSK1 mice were fed chow containing vehicle or GS-6201 for 30 days until the end of the study. Animal experiments were approved by the Animal Welfare Committee (AWC) of UTHealth (protocol no. AWC-13–043).
Histochemical studies and immunohistochemistry.
Five-micrometer thick sections of paraffin-embedded skin tissues were stained with hematoxylin and eosin, Masson’s trichrome (both from Sigma-Aldrich), or picrosirius red (Abcam). Skin fibrosis was quantified by measuring the thickness of the dermis, defined as the distance from the epidermal–dermal junction to the dermal–adipose layer junction, at 6 randomly selected sites/microscopic fields in each skin sample (15). To analyze the accumulated collagen content in the lesional skin, deparaffinized sections were stained with Masson’s trichrome. Fibrosis in picrosirius red–stained skin was quantified using ImageJ and Macro Language software (National Institutes of Health; https://imagej.nih.gov/ij/docs/examples/stained-sections/index.html). Data were calculated as percentage of total area.
Immunohistochemistry was performed using antibodies against fibronectin (FN; Abcam), α-smooth muscle actin (α-SMA; Sigma-Aldrich), and arginase 1 (Bioss) or a biotinylated HA-binding protein (EMD Millipore). Bound antibodies were detected with secondary antibodies from a Vectastain kit (Vector). Fibroblasts were identified by their spindle morphology, and inflammatory cells were identified by round morphology. For immunofluorescence-stained slides, Vector Red (Vector) or Alexa Fluor 488 (Life Technologies) was used to visualize the primary antibody. Sections were mounted with medium containing DAPI or propidium iodide (Abcam) as counterstain.
For immunofluorescence experiments with human tissues, fluorescein isothiocyanate–conjugated anti–α-SMA (Sigma-Aldrich) was used. Antibodies against ADORA2B (Thermo Scientific) and HAS-2 (Santa Cruz Biotechnology) were conjugated to Texas Red (Santa Cruz Biotechnology). Slides were counter-stained with DAPI. All imaging was performed with a Leica DM4000 (Leica Microsystems) equipped with a monochrome camera (DFC 3000G; Leica). For immunohistochemistry experiments with human tissues, antibodies against α-SMA (clone 1A4 mouse monoclonal; Sigma-Aldrich), ADORA2B, and HAS-2 were used. α-SMA was visualized with Vector Blue (Vector) using an ImmPRESS-AP Anti-Mouse IgG (alkaline phosphatase) Polymer Detection Kit (Vector). ADORA2B and HAS-2 were visualized with ImmPACT Diaminobenzidine Peroxidase Substrate (Vector) after incubation using an ImmPRESS HRP Anti-Rabbit IgG (peroxidase) Polymer Detection Kit (Vector). Morphometric evaluation of cells positive for α-SMA and ADORA2B or of cells positive for α-SMA and HAS-2 was performed by a scientist (KP) who was blinded with regard to group status. Evaluations were performed using a total of 10 control or 10 SSc double-stained skin samples. Demographic characteristics of the SSc patients and controls who donated tissue used for immunohistochemistry are presented in Supplementary Table 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40554/abstract.
Determination of messenger RNA (mRNA) levels by quantitative reverse transcription–polymerase chain reaction (RT-PCR).
Total RNA was isolated from lesional skin tissue using TRIzol reagent (Life Technologies) and purified with an RNeasy Mini kit (Qiagen). Quantitative RT-PCR was performed using validated gene expression assays for ADORA1, ADORA2A, ADORA2B, ADORA3, COL1A1, interleukin-6 (IL-6), monocyte chemoattractant protein 1 (MCP-1), HAS-2, and FN (Sigma-Aldrich). Cyclophilin A (PPIA) and β-actin were used as endogenous controls to normalize transcript levels of total RNA of each sample.
The following primers were used for this study: ADORA1 forward 5′-TGTGCCCGGAAATGTACTGG-3′, reverse 5′-TCTGTGGCCCAATGTTGATAAG-3′; ADORA2A forward 5′-TTCCACTCCGGTACAATGGC-3′, reverse 5′-CGATGGCGAATGACAGCAC-3′; ADORA2B forward 5′-GCGTCCCGCTCAGGTATAAAG-3′, reverse 5′-CGGAGTCAATCCAATGCCAAAG-3′; ADORA3 forward 5′-ACGGACTGGCTGAACATCAC-3′, reverse 5′-AGACAATGAAATAGACGGTGGTG-3′; COL1A1 forward 5′-GCTCCTCTTAGGGGCCACT-3′, reverse 5′-CCACG TCTCACCATTGGGG-3′; IL-6 forward 5′-ACCGCTATGAAGTTCCTCTC-3′, reverse 5′-CTCCGACTTGTGAAGTGGTA-3′; MCP-1 forward 5′-AGCATCCACGTGTTGGCTC-3′, reverse 5′-TGGGATCATCTTGCTGGTG-3′; HAS-2 forward 5′-TGTGAGAGGTTTCTATGTGTCCT-3′, reverse 5′-ACCGTACAGTCCAAATGAGAAGT-3′; PPIA forward 5′-GAGCTGTTTGCAGACAAAGTTC-3′, reverse 5′-CCCTGGCACATGAATCCTGG-3′; β-actin forward 5′-GGCTGTATTCCCCTCCATCG-3′, reverse 5′-CCAGTTGGTAACAATGCCATGT-3′. Data were analyzed with GraphPad Prism software using the 2–ΔΔCt method.
Quantification of tissue collagen.
The collagen content of skin was determined using a Sircol Collagen Assay (Biocolor). Collagen content was normalized to total protein content (Bradford assay; Bio-Rad).
Plasma MCP-1 assay.
MCP-1 enzyme-linked immunosorbent assays (ELISAs) were performed on plasma using a Quantikine ELISA kit (R&D Systems). Results are given as the mean ± SEM pg/ml.
Primary normal human dermal fibroblasts (HDFs).
Primary normal HDFs were obtained from Lonza (batch no. 0000214247) and cultured according to instructions provided by Lonza. These cells originated from a 41-year-old woman undergoing either reduction mammaplasty or abdominoplasty. RNA isolation and RT-PCR for human adenosine receptors were performed as previously described (17). Cells were washed twice in Hanks’ balanced salt solution and incubated in serum-free basal medium (Lonza) with or without the agonist 5′-N-ethylcarboxamidoadenosine (NECA) or the selective ADORA2B antagonist CVT-6694 (18) for 18 hours. The concentration of IL-6 in cell medium was then determined using ELISA kits (Thermo Fisher). In experiments using fibroblasts isolated from patients with SSc or healthy controls, 5 SSc and 5 control fibroblast cell lines from age- and sex-matched individuals were used. All experiments were performed in passage 6. Demographic characteristics of the SSc patients and controls who donated primary cells are summarized in Supplementary Table 2, http://onlinelibrary.wiley.com/doi/10.1002/art.40554/abstract.
Availability of data and material.
The data sets used and/or analyzed during the current study are available on reasonable request from the corresponding author.
RESULTS
Elevated fibroproliferative lesions in the skin following bleomycin exposure.
To demonstrate our ability to consistently induce fibrosis in the skin of mice, we injected bleomycin SC into a cohort of animals and assessed various fibrosis end points. Masson’s trichrome staining of sections of skin from mice exposed to phosphate buffered saline or bleomycin demonstrated a consistent increase in dermal fibrosis in mice exposed to bleomycin (representative findings are shown in Supplementary Figure 1A, http://onlinelibrary.wiley.com/doi/10.1002/art.40554/abstract). These observations were consistent with increased quantifiable metrics of fibrosis including increased transcripts for collagen and FN in the skin of mice exposed to bleomycin (see Supplementary Figures 1B and C). These findings demonstrate our ability to consistently induce dermal fibrosis in mice using bleomycin.
Elevated ADORA2B expression in dermal fibrosis.
Adenosine has been shown to play a detrimental role in several organs where aberrant tissue remodeling and the development of fibrosis are consequences of chronic injury (7,8). Our laboratory and those of others have attributed the profibrotic effects of adenosine to the engagement of ADORA2B (11); however, the involvement of this receptor in bleomycin-induced skin fibrosis has not been examined. To gain insight into the potential role of ADORA2B in dermal fibrosis, we examined transcript levels of ADORA2B and other adenosine receptors in RNA lysates generated from mice exposed to bleomycin. Results demonstrated that there was a significant increase in ADORA2B transcript levels in the fibrotic skin of mice exposed to bleomycin (see Supplementary Figure 1F, http://onlinelibrary.wiley.com/doi/10.1002/art.40554/abstract). Interestingly, transcript levels for ADORA1, ADORA2A, or ADORA3 (see Supplementary Figures 1D, E, and G) were not increased in this model. These findings suggest that ADORA2B is increased in association with dermal fibrosis.
Inhibition of dermal fibrosis by an ADORA2B antagonist.
Previous studies have shown that treatment with ADORA2B antagonists during stages of active fibrosis can attenuate fibrosis in the lung (11,12), kidney (13), and penis (14). These findings led us to hypothesize that ADORA2B antagonism would attenuate dermal fibrosis following bleomycin exposure. To test this hypothesis, we examined the therapeutic efficacy of an ADORA2B antagonist in attenuating bleomycin-induced skin fibrosis. Mice were exposed to SC bleomycin, and a cohort was provided with chow containing the ADORA2B antagonist GS-6201 beginning on day 15 following bleomycin administration, a stage when skin fibrosis is already evident (15). On day 28, the skin of mice with exposure to bleomycin and GS-6201 treatment was examined for metrics of skin fibrosis. Mice exposed to bleomycin and treated with the ADORA2B antagonist GS-6201 exhibited reduced dermal fibrosis compared to bleomycin-exposed control chow–treated mice, as observed histologically in Masson’s trichrome–stained skin sections (Figure 1A). These qualitative observations were validated with quantifiable analysis of skin fibrosis, including reduced dermal thickness assessed morphometrically (Figure 1B) and reduced collagen transcript levels (Figure 1C). We next stained skin sections with α-SMA to identify myofibroblasts. There were increased numbers of myofibroblasts within fibrotic lesions in mice exposed to bleomycin, and these increased numbers were attenuated in bleomycin-exposed mice treated with GS-6201 (Figure 1D).
Figure 1.

Attenuation of skin fibrosis by GS-6201. A, Masson’s trichrome–stained skin sections from representative mice treated with phosphate buffered saline (PBS) plus control chow, PBS plus GS-6201, bleomycin (bleo) plus control chow, or bleomycin plus GS-6201. Bar = 500 μm. B, Dermal thickness assessed morphometrically from Masson’s trichrome–stained skin sections from mice treated as shown in A. C, COL1A1 transcript levels in skin sections from mice treated as shown in A. PPIA = cyclophilin A. D, Immunofluorescence for α-smooth muscle actin (α-SMA; red) counterstained with DAPI (blue) in skin sections from mice treated as shown in A. Arrowheads indicate myofibroblasts. Bar = 25 μm. In B and C, values are the mean ± SEM. * = P < 0.05; *** = P < 0.001 versus PBS plus control chow. # = P < 0.05; ## = P < 0.01 versus bleomycin plus control chow, by analysis of variance.
Taken together, these observations demonstrate that treatment with GS-6201 starting on day 15 after the initiation of bleomycin exposure is able to halt the progression of dermal fibrosis. These experiments suggest a potential therapeutic role for GS-6201 in the treatment of skin fibrosis.
ADORA2B antagonism attenuates FN expression and reduces the number of alternatively activated macrophages found in dermal fibrosis.
Further analysis of fibrotic lesions showed increased expression of the extracellular matrix molecule FN in mice exposed to SC bleomycin. Levels of FN were attenuated in bleomycin-exposed mice treated with GS-6201 (Figures 2A and C), further indicating the ability of this ADORA2B antagonist to regulate dermal fibrosis in this model.
Figure 2.
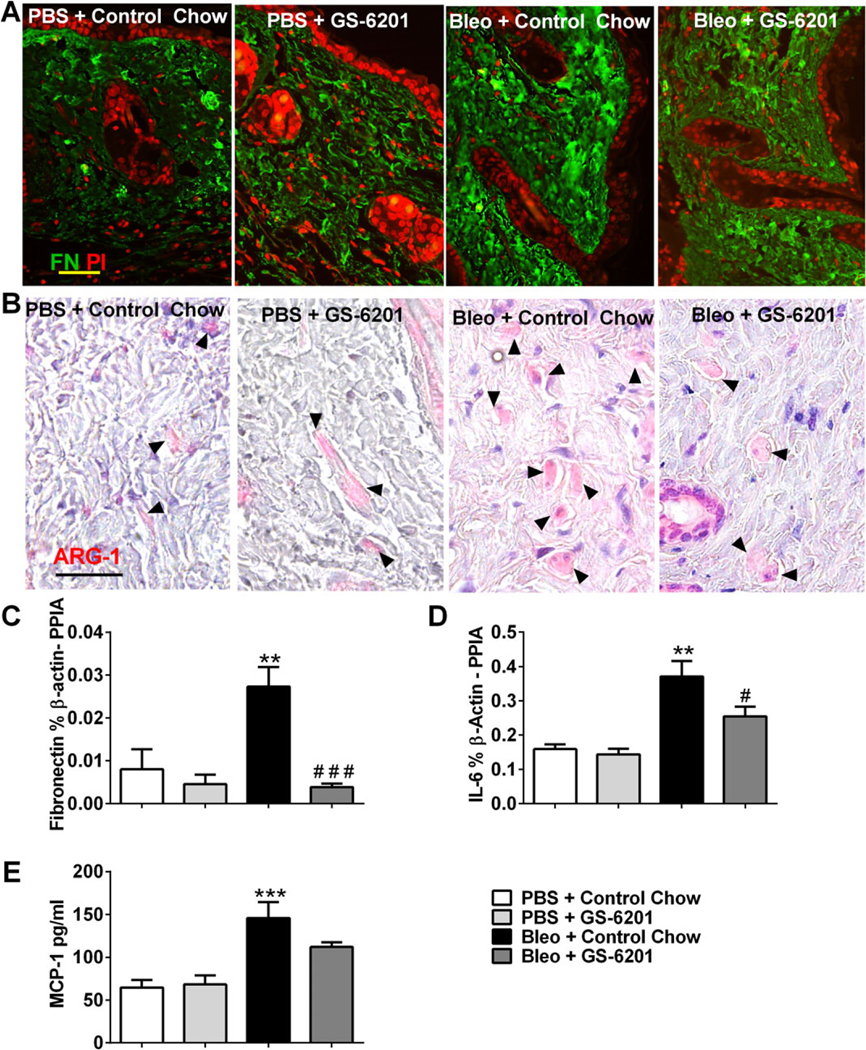
GS-6201 reduces fibronectin (FN) levels and numbers of alternatively activated macrophages. A and B, Immunofluorescence for FN (green) and counterstaining with propidium iodide (PI; red) (A) and immunohistochemical staining for arginase 1 (ARG-1) and counterstaining with hematoxylin (B) in skin sections from mice treated with phosphate buffered saline (PBS) plus control chow, PBS plus GS-6201, bleomycin (bleo) plus control chow, or bleomycin plus GS-6201. Arrowheads in B indicate ARG-1–positive cells. Bars = 50 μm. C–E, Transcript levels of FN (C) and interleukin-6 (IL-6) (D) and protein levels of monocyte chemoattractant protein 1 (MCP-1) (E) in skin sections from mice in the 4 treatment groups. Values are the mean ± SEM. ** = P < 0.01; *** = P < 0.001 versus PBS plus control chow. # = P < 0.05; ### = P < 0.001 versus bleomycin plus control chow, by analysis of variance. PPIA = cyclophilin A.
Alternatively activated macrophages have been implicated in the regulation of fibrosis in various organs in part by the ability of ADORA2B on these cells to elicit the production of profibrotic molecules (19). To begin to assess whether a similar pathway may be active in dermal fibrosis, we stained tissue sections with arginase 1, a marker for alternatively activated macrophages (Figure 2B). Staining for arginase 1 demonstrated increased numbers of alternatively activated macrophages in bleomycin-exposed skin (Figure 2B). Interestingly, arginase 1–positive alternatively activated macrophages appeared to be less prevalent in the dermis of bleomycin-exposed mice treated with GS-6201 (Figure 2B). We have previously demonstrated that engagement of ADORA2B on alternatively activated macrophages regulates the production of the profibrotic molecule IL-6 (10,20) and of the immunoregulatory molecule MCP-1 (21). To determine if these molecules were regulated by ADORA2B signaling during dermal fibrosis, we measured IL-6 and MCP-1 transcripts in the skin of mice exposed to bleomycin with and without GS-6201 treatment. Results demonstrated significant elevations in IL-6 (Figure 2D) and MCP-1 (Figure 2E) in the skin of mice exposed to bleomycin. Furthermore, there was a significant reduction in IL-6 transcript levels in bleomycin-exposed mice treated with GS-6201 (Figure 2D) and a trend toward reduction in MCP-1 transcripts (Figure 2E). These findings demonstrate that GS-6201 treatment is able to reduce numbers of alternatively activated macrophages and attenuate fibrosis mediators in the skin of mice exposed to bleomycin.
Attenuation of HA pathways following treatment with an ADORA2B antagonist.
Recent studies have shown that engagement of ADORA2B leads to enhanced HA production through increased expression of HAS-2 (22). HA has been implicated in mediating fibrosis through its interaction with fibroblasts (22); however, this profibrotic molecule has not been examined in dermal fibrosis. To address this gap in knowledge, we examined levels of HA and HAS-2 in the skin of mice exposed to bleomycin and determined the impact of GS-6201 treatment on these levels (Figure 3). Staining for HA revealed increased signals in bleomycin-exposed mice that were attenuated in bleomycin-exposed mice treated with GS-6201 (Figure 3A). These observations were consistent with increased HAS-2 expression in bleomycin-exposed mice that was attenuated in bleomycin-exposed mice treated with GS-6201 (Figure 3B). These findings suggest that ADORA2B-dependent up-regulation of HAS-2, and subsequently of HA, may impact dermal fibrosis (Figures 3B and C).
Figure 3.

Increased levels of dermal hyaluronan (HA) in bleomycin (bleo)–exposed mice. A, Immunofluorescence for HA (red) and counterstaining with DAPI (blue) in skin sections from mice treated with phosphate buffered saline (PBS) plus control chow, PBS plus GS-6201, bleomycin plus control chow, or bleomycin plus GS-6201. Bar = 75 μm. B and C, Transcript levels of HA synthase 2 (HAS-2) (B) and A2B adenosine receptor (ADORA2B) (C) in skin sections from mice in the 4 treatment groups. Values are the mean ± SEM. * = P < 0.05 versus PBS plus control chow. ## = P < 0.01 versus bleomycin plus control chow, by analysis of variance. PPIA = cyclophilin A.
Attenuation of skin fibrosis in the TSK1 mouse following treatment with an ADORA2B antagonist.
To investigate whether a similar antifibrotic effect could be achieved in a secondary model of dermal fibrosis, GS-6201 was administered to TSK1 mice, mice with genetically induced dermal fibrosis caused by fibrillin 1 mutation in which fibrotic changes are observed in the hyperdermal layer (16). Female TSK1 mice age 6–8 weeks were fed chow containing vehicle or GS-6201 for 30 days. Skin was collected on day 30 for fibrosis examination. As observed in the Masson’s trichrome–stained sections, TSK1 mice treated with GS-6201 presented with reduced dermal fibrosis at the hyperdermal layer (Figure 4A). Analysis blinded with regard to dermal thickness confirmed a significant reduction in hyperdermal layer thickness (Figure 4B). These changes were consistent with reduced COL1A1 and FN expression levels (Figures 4D and E). In addition, we observed that both IL-6 and MCP-1 transcript expression levels were reduced in the skin of mice treated with GS-6201 (Figures 4F and G), which suggests that the ADORA2B antagonist is able to attenuate fibrosis mediators and inflammation markers associated with alternatively activated macrophages in TSK1 mice. However, HAS-2 transcript levels were not significantly altered in mice treated with GS-6201 compared to control mice (Figure 4H). Taken together, our findings demonstrate that treatment with GS-6201 can halt progression of dermal fibrosis in TSK1 mice.
Figure 4.

Reduction of dermal fibrosis by GS-6201 in TSK1 mice. Eight-week-old female TSK1 mice were fed control chow or chow containing GS-6201 for 30 days. A, After 30 days, skin was collected for Masson’s trichrome staining or picrosirius red staining to evaluate skin thickness and collagen deposition. Arrows indicate extent of dermal fibrosis at the hyperdermal layer. Bar = 500 μm. B, Skin thickness at the hyperdermal layer was measured and quantified. C, Picrosirius red–stained skin was quantified, and data were expressed as percentage of the total area. D–H, Quantitative reverse transcription–polymerase chain reaction was performed to determine transcript levels of COL1A1 (D), fibronectin (E), interleukin-6 (IL-6) (F), monocyte chemoattractant protein 1 (MCP-1) (G), and hyaluronan synthase 2 (HAS-2) (H) in skin collected from TSK1 mice fed control chow or chow containing GS-6201. In B–H, values are the mean ± SEM. * = P < 0.05 versus control. PPIA = cyclophilin A.
High expression of ADORA1 and ADORA2B in normal HDFs.
Our experiments to determine the expression levels of adenosine receptors in normal HDFs demonstrated that ADORA1 and ADORA2B, but not ADORA2A or ADORA3, were highly expressed in normal HDFs (Figure 5A). We found that normal HDFs treated with NECA, an adenosine receptor analog (18), led to increased levels of IL-6 in media, and that this was blocked by CVT-6694, a selective ADORA2B antagonist (Figure 5B). Interestingly, primary dermal fibroblasts cultured from normal donors or patients diagnosed as having SSc revealed that ADORA2B was the primary adenosine receptor expressed in dermal fibroblasts (Figure 5C). However, no significant differences in adenosine receptor expression were observed between normal and SSc dermal fibroblasts. Furthermore, assessment of mRNA expression levels for adenosine deaminase (ADA) and adenosine kinase, 2 enzymes involved in the degradation of adenosine, revealed a significant increase in adenosine kinase expression but not in ADA expression in SSc samples compared to normal samples (Figure 5D). ADA is normally responsible for extracellular adenosine degradation, while adenosine kinase usually degrades intracellular adenosine. In addition, there were no significant differences in CD73 expression levels, but HAS-2 expression levels were elevated in SSc fibroblasts compared to normal fibroblasts (Figure 5D).
Figure 5.

Adenosine receptors and A2B adenosine receptor (ADORA2B) response in normal human dermal fibroblasts (HDFs). A, Adenosine receptor expression levels in normal HDFs (n = 3 experiments). B, Interleukin-6 (IL-6) release from normal HDFs (n = 4 experiments). Cells were incubated with 5′-N-ethylcarboxamidoadenosine (NECA) (10 μM) with or without CVT-6694 (100 nM) for 18 hours. C and D, Adenosine receptor expression (C) and transcript levels for adenosine deaminase (ADA), adenosine kinase (AK), CD73, and hyaluronan synthase 2 (HAS-2) (D) in primary HDFs from 5 normal donors and 5 patients diagnosed as having systemic sclerosis (SSc). Values are the mean ± SEM. In B, *** = P < 0.001 versus control; ### = P < 0.001 versus NECA alone, by analysis of variance. In D, * = P < 0.05 versus HDFs from normal donors, by t-test.
Similarly, immunofluorescence staining and subsequent morphometric evaluation revealed coexpression of ADORA2B and α-SMA in fibroblasts in normal and SSc skin (Figure 6A), with an increased number of ADORA2B-positive myofibroblasts in SSc skin compared to control skin (Figure 6A). In sections immunohistochemically stained for HAS-2 and α-SMA, we observed increased HAS-2–positive myofibroblasts in SSc skin compared to control skin (Figure 6B), which was consistent with increased HA deposition in SSc skin sections (Figure 6C).
Figure 6.
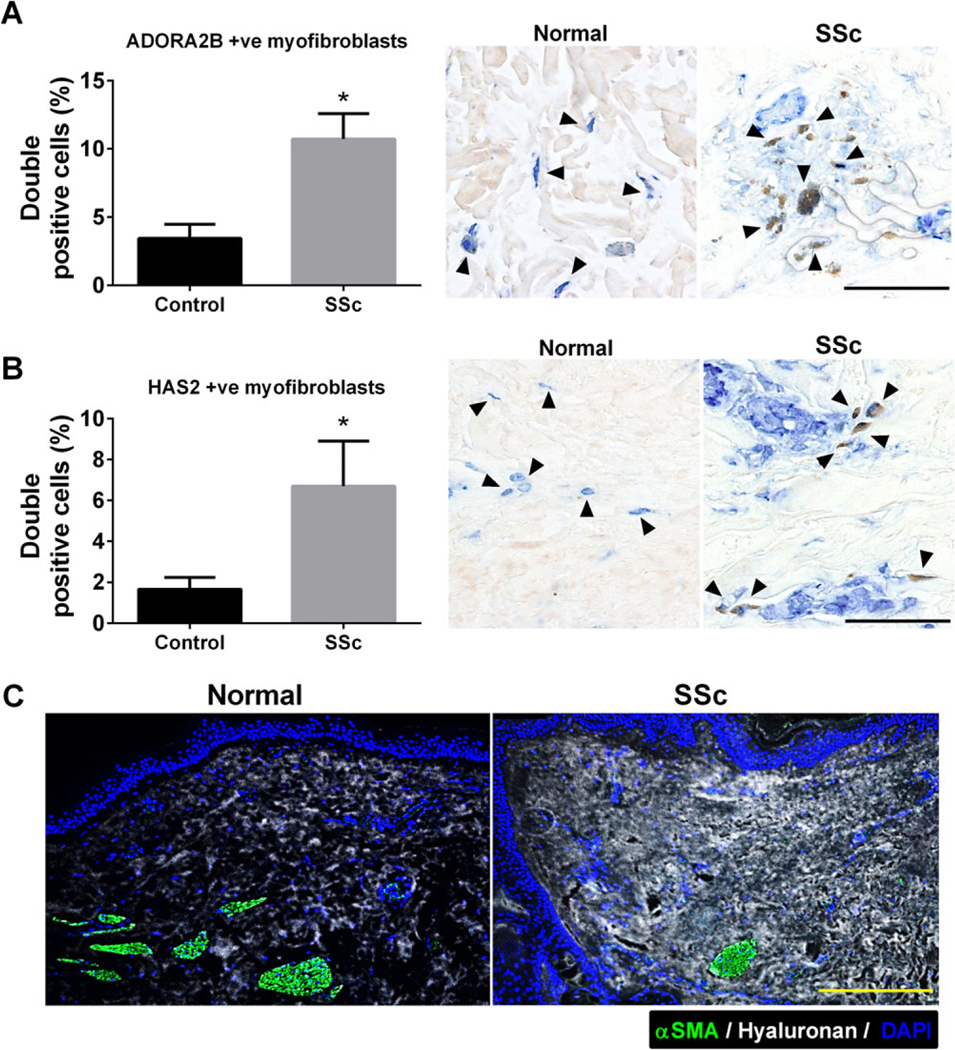
Expression of A2B adenosine receptor (ADORA2B), hyaluronan, and hyaluronan synthase 2 (HAS-2) in human normal and systemic sclerosis (SSc) skin tissue. A and B, Immunofluorescence and morphometric evaluation of cells double positive for α-smooth muscle actin (α-SMA; blue) and ADORA2B (brown) (A) and of cells double positive for α-SMA and HAS-2 (brown) (B). Arrowheads in A indicate α-SMA and ADORA2B double-positive cells; arrowheads in B indicate α-SMA and HAS-2 double-positive cells. Bars = 100 μm. Values are the mean ± SEM. * = P < 0.05 versus control, by Student’s 2-tailed t-test. C, Staining for α-SMA (green) and hyaluronan (white) with DAPI counterstaining (blue). Bar = 50 μm.
DISCUSSION
Extracellular adenosine has been associated with the progression of several chronic diseases, including diseases in which fibrosis is a major component (7,8). Mice deficient in ADA exhibit progressive and chronic elevations in adenosine levels due to the absence of the enzyme that breaks down adenosine (23). These mice spontaneously develop fibrosis in many organs including the lung (24,25), liver (24), kidney (13), penis (14), and skin (26). These findings led to the hypothesis that chronic elevations in adenosine serve as a profibrotic signal (7) and initiated a series of studies to investigate the mechanisms involved. With regard to skin fibrosis, substantial amounts of experimental work have demonstrated that adenosine generation (27) and its signaling through ADORA2A constitute a major profibrotic regulator in dermal fibrosis (6). Fernandez and colleagues have demonstrated that antagonism of ADORA2A in the ADA-deficient mouse model can attenuate adenosine-driven skin fibrosis (26). In addition, this group has shown that direct engagement of ADORA2A on dermal fibroblasts can access numerous profibrotic pathways (28–30). Thus, adenosine clearly plays a role in regulating dermal fibrosis through ADORA2A expressed on dermal fibroblasts.
Work from our laboratories has shown that ADORA2B plays an important role in the regulation of fibrosis in several organs (8). Moreover, the highly selective ADORA2B antagonist GS-6201 has been used in these studies to show that ADORA2B plays an important role in the regulation of pulmonary fibrosis (11). Treatment with GS-6201 was able to attenuate pulmonary fibrosis in ADA-deficient mice (11) and mice in which pulmonary fibrosis was induced by bleomycin (11,12). Importantly, in these studies, GS-6201 was given to mice after pulmonary fibrosis was well-established (12), demonstrating that blockade of this pathway was therapeutic. A major finding of the current study was that treatment with this same antagonist was also effective in attenuating dermal fibrosis in the SC bleomycin model and in TSK1 mice. Similar to studies on pulmonary fibrosis, GS-6201 was effective in attenuating several metrics of dermal fibrosis even though treatment was not initiated until late in the model. These findings provide the first evidence that ADORA2B plays a role in regulating dermal fibrosis, and they suggest that targeting this receptor may be an attractive approach for the treatment of fibrotic dermal disorders such as SSc.
Additional evidence to support a role for ADORA2B in dermal fibrosis was our finding that levels of this receptor were elevated in skin samples from mice exposed to SC bleomycin. Elevations in ADORA2B levels are commonly seen in fibrotic tissues in both mice and humans (12,20), where various profibrotic activities have been identified on different cell types. For example, ADORA2B levels are found to be elevated on myofibroblasts (9), where ADORA2B signaling can promote both the differentiation of myofibroblasts and the production of fibrosis mediators. In addition to its importance for myofibroblasts, ADORA2B has recently been shown to be important in regulating the function of a subtype of macrophages known as alternatively activated macrophages (10). Alternatively activated macrophages have been suggested to play an important role in the regulation of fibrosis in many organs (19), and a recent study has demonstrated that the expression of ADORA2B on this cell type contributes to the progression of pulmonary fibrosis in mice (10). ADORA2B is known to induce the production of the profibrotic molecule IL-6 from alternatively activated macrophages both in mice (10) and in humans (20) with pulmonary fibrosis, and genetically removing ADORA2B from macrophages is associated with reduced IL-6 production and reduced pulmonary fibrosis (10).
Similarly, in the current study, we demonstrate that IL-6 levels are elevated in the SC bleomycin model in conjunction with increased numbers of alternatively activated macrophages. Moreover, we show that treatment with an ADORA2B antagonist is associated with fewer alternatively activated macrophages and reduced production of IL-6 both in the SC bleomycin model and in TSK1 mice. Taken together, these results suggest that ADORA2B signaling on alternatively activated macrophages in fibrotic skin may regulate IL-6 production, which could regulate dermal fibrosis. In support of these studies, blockade of IL-6 was shown to lead to significant reduction in skin thickening in a randomized controlled trial in SSc patients (31).
We recently found that ADORA2B-driven fibrotic responses in the lungs of mice are also associated with increased HA signaling (22). Similarly, we demonstrated in the current study that there are substantial increases both in the enzyme that produces hyaluronan, HAS-2, and in HA itself in fibrotic lesions of mice exposed to bleomycin and in skin samples from patients with SSc. Moreover, treatment with GS-6201 was associated with decreased HAS-2 expression and with a reduction in HA in fibrotic skin. HA signaling contributes to the progression of fibrosis by stimulating profibrotic activities on fibroblasts (22), and our findings suggest that the ADORA2B-dependent regulation of this pathway may contribute to dermal fibrosis. These findings echo our observations in experimental models of lung injury in which treatment with GS-6201 or depletion of ADORA2B in myeloid cells reduced the extent of fibrotic deposition in the lungs and attenuated vascular remodeling and pulmonary hypertension (10,12). Further studies by our group identified increased HA as a modulator of enhanced fibrotic deposition and vascular remodeling (10,22) that can be targeted therapeutically (32). These observations have important implications for SSc, in which the development of interstitial lung disease or pulmonary hypertension is a serious complication (33,34).
Consistent with these results, in experiments using normal HDFs we demonstrate that both ADORA1 and ADORA2B are highly expressed in these cells and that activation of ADORA2B leads to increased levels of the profibrotic cytokine IL-6. Interestingly, our data from primary cells isolated from SSc patients or controls did not show increased ADORA1 signals. This may be explained by the origin of the normal HDFs obtained from Lonza. These cells originated from 1 donor, a 41-year-old female who underwent reduction mammaplasty or abdominoplasty, while our results from primary cells represent average signals from cells obtained from the forearms of 5 controls and 5 SSc patients. Thus, we believe that our data from isolated primary fibroblasts may be more representative than the data from normal HDFs.
Our experiments using immunofluorescence showed expression of ADORA2B in fibroblasts in both normal and SSc tissue. These findings are significant as this response is likely augmented in fibrotic normal HDFs in which increased ADORA2B expression is expected (8). This is consistent with previously published data showing increased IL-6 levels from SSc skin lesions following exposure to ATP (35).
It is also important to point out that although our experiments in mice did not show increased ADORA2A levels, increased expression of ADORA2A has been shown in fibroblasts from SSc patients (36). Furthermore, activation of ADORA2A by CGS-21680 has been shown to stimulate fibroblast-to-myofibroblast differentiation in human SSc fibroblasts (36). In contrast, ADORA2A-deficient mice or mice treated with the ADORA2A antagonist ZM241385 have exaggerated dermal fibrosis due to abrogated production of transforming growth factor β and connective tissue growth factor (26,29). These studies clearly show an important role of adenosine signaling through ADORA2A in dermal fibrosis.
ADORA2A and ADORA2B are both coupled to adenylyl cyclase by the stimulatory G protein subunit (Gas) and can induce intracellular cAMP levels. ADORA2B has the lowest affinity to extracellular adenosine; therefore, it is normally activated in pathologic conditions due to elevated extracellular adenosine (37). Previous studies in lymphocytes indicate that surface expression of ADORA2A is partially responsible for surface expression of ADORA2B in lymphocytes (38), indicating that activation of ADORA2A promotes ADORA2B signaling. The different turnover rate may contribute to the low ADORA2A expression in dermal fibroblasts. However, because ADORA2A has much higher affinity than ADORA2B, we believe that ADORA2A and ADORA2B both play potential roles during the pathogenesis of dermal fibrosis. Despite this, our results reveal that ADORA2B is the adenosine receptor most abundantly expressed both in normal and in SSc fibroblasts, yet no differences were identified in adenosine receptor expression between normal and SSc samples.
Taken together, these results point to the potential benefit of ADORA2A antagonists to treat human dermal fibrosis, in addition to ADORA2B antagonism. Interestingly, our data also show increased expression of adenosine kinase but not ADA in SSc skin samples. These results are significant since adenosine kinase metabolizes adenosine back to AMP intracellularly, while ADA degrades adenosine to inosine extracellularly. These results may suggest that in SSc, increased intracellular adenosine levels are present that lead to increased expression of adenosine kinase.
In conclusion, we have shown that the ADORA2B antagonist GS-6201 is effective in reducing dermal fibrosis in a mouse model of SC bleomycin exposure. These findings suggest that this receptor could play a role in regulating dermal fibrosis and that targeting this receptor may prove beneficial in the treatment of disorders such as SSc, in which dermal fibrosis is prominent. These studies were not able to resolve the relative difference between the ADORA2A and ADORA2B signaling pathways in dermal fibrosis; therefore, additional studies are needed to address this issue and to identify when and how best to promote the use of adenosine-based therapeutics for the treatment of dermal fibrosis.
Supplementary Material
ACKNOWLEDGMENT
We thank Kelly A. Volcik (UTHealth) for her help in revising the manuscript.
Supported by Gilead Sciences, Inc.
ROLE OF THE STUDY SPONSOR
Gilead Sciences, Inc. had no role in the study design or in the collection, analysis, or interpretation of the data, the writing of the manuscript, or the decision to submit the manuscript for publication. Publication of this article was not contingent upon approval by Gilead Sciences, Inc.
Footnotes
Dr. Zhong owns stock or stock options in Gilead Sciences, Inc.
REFERENCES
- 1.Charles C, Clements P, Furst DE. Systemic sclerosis: hypothesis-driven treatment strategies. Lancet 2006;367:1683–91. [DOI] [PubMed] [Google Scholar]
- 2.Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest 2007;117:557–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Varga J, Whitfield ML. Transforming growth factor-β in systemic sclerosis (scleroderma). Front Biosci (Schol Ed) 2009;1:226–35. [DOI] [PubMed] [Google Scholar]
- 4.Fredholm BB. Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death Differ 2007;14: 1315–23. [DOI] [PubMed] [Google Scholar]
- 5.Blackburn MR, Vance CO, Morschl E, Wilson CN. Adenosine receptors and inflammation. Handb Exp Pharmacol 2009:215–69. [DOI] [PubMed] [Google Scholar]
- 6.Chan ES, Cronstein BN. Adenosine in fibrosis. Mod Rheumatol 2010;20:114–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou Y, Schneider DJ, Blackburn MR. Adenosine signaling and the regulation of chronic lung disease. Pharmacol Ther 2009;123:105–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karmouty-Quintana H, Xia Y, Blackburn MR. Adenosine signaling during acute and chronic disease states. J Mol Med (Berl) 2013;91:173–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhong H, Belardinelli L, Maa T, Zeng D. Synergy between A2B adenosine receptors and hypoxia in activating human lung fibroblasts. Am J Respir Cell Mol Biol 2005;32:2–8. [DOI] [PubMed] [Google Scholar]
- 10.Karmouty-Quintana H, Philip K, Acero LF, Chen NY, Weng T, Molina JG, et al. Deletion of ADORA2B from myeloid cells dampens lung fibrosis and pulmonary hypertension. FASEB J 2015;29:50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun CX, Zhong H, Mohsenin A, Morschl E, Chunn JL, Molina JG, et al. Role of A2B adenosine receptor signaling in adenosine-dependent pulmonary inflammation and injury. J Clin Invest 2006;116:2173–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karmouty-Quintana H, Zhong H, Acero L, Weng T, Melicoff E, West JD, et al. The A2B adenosine receptor modulates pulmonary hypertension associated with interstitial lung disease. FASEB J 2012;26:2546–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang W, Zhang Y, Wang W, Dai Y, Ning C, Luo R, et al. Elevated ecto-5’-nucleotidase-mediated increased renal adenosine signaling via A2B adenosine receptor contributes to chronic hypertension. Circ Res 2013;112:1466–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wen J, Jiang X, Dai Y, Zhang Y, Tang Y, Sun H, et al. Increased adenosine contributes to penile fibrosis, a dangerous feature of priapism, via A2B adenosine receptor signaling. FASEB J 2010; 24:740–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu M, Schneider DJ, Mayes MD, Assassi S, Arnett FC, Tan FK, et al. Osteopontin in systemic sclerosis and its role in dermal fibrosis. J Invest Dermatol 2012;132:1605–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walker MA, Harley RA, LeRoy EC. Inhibition of fibrosis in TSK mice by blocking mast cell degranulation. J Rheumatol 1987;14: 299–301. [PubMed] [Google Scholar]
- 17.Zhong H, Belardinelli L, Maa T, Feoktistov I, Biaggioni I, Zeng D. A(2B) adenosine receptors increase cytokine release by bronchial smooth muscle cells. Am J Respir Cell Mol Biol 2004; 30:118–25. [DOI] [PubMed] [Google Scholar]
- 18.Kalla RV, Elzein E, Perry T, Li X, Gimbel A, Yang M, et al. Selective, high affinity A(2B) adenosine receptor antagonists: N-1 monosubstituted 8-(pyrazol-4-yl)xanthines. Bioorg Med Chem Lett 2008;18:1397–401. [DOI] [PubMed] [Google Scholar]
- 19.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 2011;11:723–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou Y, Murthy JN, Zeng D, Belardinelli L, Blackburn MR. Alterations in adenosine metabolism and signaling in patients with chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. PLoS One 2010;5:e9224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pedroza M, Schneider DJ, Karmouty-Quintana H, Coote J, Shaw S, Corrigan R, et al. Interleukin-6 contributes to inflammation and remodeling in a model of adenosine mediated lung injury. PLoS One 2011;6:e22667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karmouty-Quintana H, Weng T, Garcia-Morales LJ, Chen NY, Pedroza M, Zhong H, et al. Adenosine A2B receptor and hyaluronan modulate pulmonary hypertension associated with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 2013;49:1038–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blackburn MR. Too much of a good thing: adenosine overload in adenosine-deaminase-deficient mice. Trends Pharmacol Sci 2003; 24:66–70. [DOI] [PubMed] [Google Scholar]
- 24.Chunn JL, Mohsenin A, Young HW, Lee CG, Elias JA, Kellems RE, et al. Partially adenosine deaminase-deficient mice develop pulmonary fibrosis in association with adenosine elevations. Am J Physiol Lung Cell Mol Physiol 2006;290:L579–87. [DOI] [PubMed] [Google Scholar]
- 25.Chunn JL, Molina JG, Mi T, Xia Y, Kellems RE, Blackburn MR. Adenosine-dependent pulmonary fibrosis in adenosine deaminase-deficient mice. J Immunol 2005;175:1937–46. [DOI] [PubMed] [Google Scholar]
- 26.Fernandez P, Trzaska S, Wilder T, Chiriboga L, Blackburn MR, Cronstein BN, et al. Pharmacological blockade of A2A receptors prevents dermal fibrosis in a model of elevated tissue adenosine. Am J Pathol 2008;172:1675–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fernandez P, Perez-Aso M, Smith G, Wilder T, Trzaska S, Chiriboga L, et al. Extracellular generation of adenosine by the ectonucleotidases CD39 and CD73 promotes dermal fibrosis. Am J Pathol 2013;183:1740–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perez-Aso M, Fernandez P, Mediero A, Chan ES, Cronstein BN. Adenosine 2A receptor promotes collagen production by human fibroblasts via pathways involving cyclic AMP and AKT but independent of Smad2/3. FASEB J 2014;28:802–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chan ES, Fernandez P, Merchant AA, Montesinos MC, Trzaska S, Desai A, et al. Adenosine A2A receptors in diffuse dermal fibrosis: pathogenic role in human dermal fibroblasts and in a murine model of scleroderma. Arthritis Rheum 2006;54:2632–42. [DOI] [PubMed] [Google Scholar]
- 30.Chan ES, Liu H, Fernandez P, Luna A, Perez-Aso M, Bujor AM, et al. Adenosine A(2A) receptors promote collagen production by a Fli1- and CTGF-mediated mechanism. Arthritis Res Ther 2013;15:R58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khanna D, Denton CP, Jahreis A, van Laar JM, Frech TM, Anderson ME, et al. Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): a phase 2, randomised, controlled trial. Lancet 2016;387:2630–40. [DOI] [PubMed] [Google Scholar]
- 32.Collum SD, Chen NY, Hernandez AM, Hanmandlu A, Sweeney H, Mertens TC, et al. Inhibition of hyaluronan synthesis attenuates pulmonary hypertension associated with lung fibrosis. Br J Pharmacol 2017;174:3284–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wells AU, Steen V, Valentini G. Pulmonary complications: one of the most challenging complications of systemic sclerosis. Rheumatology (Oxford) 2009;48 Suppl 3:iii40–4. [DOI] [PubMed] [Google Scholar]
- 34.Proudman SM, Stevens WM, Sahhar J, Celermajer D. Pulmonary arterial hypertension in systemic sclerosis: the need for early detection and treatment. Int Med J 2007;37:485–94. [DOI] [PubMed] [Google Scholar]
- 35.Lo Monaco A, Gulinelli S, Castellino G, Solini A, Ferrari D, La Corte R, et al. Increased sensitivity to extracellular ATP of fibroblasts from patients affected by systemic sclerosis. Ann Rheum Dis 2007;66:1124–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lazzerini PE, Natale M, Gianchecchi E, Capecchi PL, Montilli C, Zimbone S, et al. Adenosine A2A receptor activation stimulates collagen production in sclerodermic dermal fibroblasts either directly and through a cross-talk with the cannabinoid system. J Mol Med 2012;90:331–42. [DOI] [PubMed] [Google Scholar]
- 37.Liu H, Xia Y. Beneficial and detrimental role of adenosine signaling in diseases and therapy. J Appl Physiol (1985) 2015;119: 1173–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moriyama K, Sitkovsky MV. Adenosine A2A receptor is involved in cell surface expression of A2B receptor. J Biol Chem 2010; 285:39271–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.