

Abstract
Myocardial infarction (MI) is defined as evidence of myocardial necrosis consistent with prolonged ischemia. In response to MI, the myocardium undergoes a series of wound healing events that initiate inflammation and shift to anti‐inflammation before transitioning to tissue repair that culminates in scar formation to replace the region of the necrotic myocardium. The overall response to MI is determined by two major steps, the first of which is the secretion of proteases by infiltrating leukocytes to breakdown extracellular matrix (ECM) components, a necessary step to remove necrotic cardiomyocytes. The second step is the generation of new ECM that comprises the scar; and this step is governed by the cardiac fibroblasts as the major source of new ECM synthesis. The leukocyte component resides in the middle of the two‐step process, contributing to both sides as the leukocytes transition from pro‐inflammatory to anti‐inflammatory and reparative cell phenotypes. The balance between the two steps determines the final quantity and quality of scar formed, which in turn contributes to chronic outcomes following MI, including the progression to heart failure. This review will summarize our current knowledge regarding the cardiac wound healing response to MI, primarily focused on experimental models of MI in mice.
This article is categorized under:
Cardiovascular Diseases > Molecular and Cellular Physiology
Immune System Diseases > Molecular and Cellular Physiology
Keywords: cardiac remodeling, heart attack, human, inflammation, mice
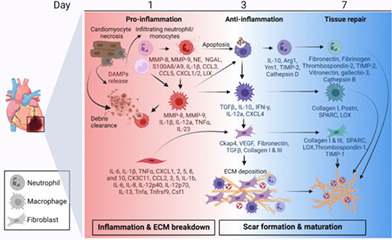
Cardiac wound healing after myocardial infarction is a balance between the breakdown of existing and the construction of new extracellular matrix, with inflammation feeding into both sides.
1. INTRODUCTION
Myocardial infarction (MI) is defined as evidence of cardiomyocyte necrosis consistent with the presence of prolonged ischemia due to a blocked coronary artery (Thygesen, Alpert, White, & Joint ESC/ACCF/AHA/WHF Task Force for the Redefinition of Myocardial Infarction, 2007). In the clinic, MI diagnosis includes the detection of cardiac biomarkers (e.g., troponin) above the 99th percentile of the upper reference limit, along with other evidence of ischemia. Ischemia is demonstrated by the presence of symptoms, electrocardiogram changes, or imaging showing loss of viable myocardium or regional wall motion abnormality (Thygesen et al., 2007). MI is also diagnosed in humans following sudden unexpected cardiac death with symptoms of ischemia or evidence of fresh thrombus at autopsy. The current optimum therapy for MI includes timely reperfusion, along with medical treatment including angiotensin‐converting enzyme inhibitors, beta‐blockers, and statins (Ibanez et al., 2018; Members et al., 1996; Roe et al., 2005). Even when reperfusion occurs, there is an increased risk of developing heart failure, such that ischemic heart disease is a major contributor to heart failure with reduced ejection fraction (Vedin et al., 2017). The total measurable extent of cardiomyocyte death is an important factor in the overall risk of heart failure development (Teringova & Tousek, 2017).
Clinically, not everyone receives timely therapy. About 25% of patients with MI will not be reperfused due to a variety of explanations, including delayed presentation or diagnosis, and lack of success in reperfusion (Lindsey, de Castro Brás, et al., 2021). In addition, up to 50% of those provided reperfusion therapy will experience the phenomenon of no‐reflow, which is a state of myocardial tissue hypoperfusion due to impaired microvascular flow that occurs in the presence of a patent epicardial coronary artery (Niccoli et al., 2010; Rezkalla & Kloner, 2002). There is currently a lack of effective treatments for no‐reflow, in part due to its multifactorial nature and in part due to the hemodynamic instability of therapies that require very high doses not easily tolerated by patients (Niccoli et al., 2010). Combined, this results in a significant number of patients with MI who will undergo adverse cardiac remodeling and in some cases death due to rupture of the left ventricle (LV; Reed et al., 2017). For these patients, the risk of progression to heart failure is high, and the majority of heart failure with reduced ejection fraction have MI as the underlying etiology (Cahill & Kharbanda, 2017; Ho et al., 2013; Roger, 2013).
In animal models of MI, the permanent occlusion of nonreperfused MI recapitulates the phenotype of the patient with no‐reflow after MI who ultimately progresses to heart failure and will be the focus of this review (Mouton, Rivera, & Lindsey, 2018b). As the mouse MI model of permanent occlusion has been extensively evaluated, we will primarily use this model in our discussion. All permanent MI animal models undergo a similar series of events; the major differences among them are the tolerance to ischemia and the temporal space in which each phase of wound healing occurs (Dewald et al., 2004; Frangogiannis, 2014; Guo et al., 2012; Yeap et al., 2013; Box 1).
BOX 1.
For the majority of heart failure with reduced ejection fraction cases, myocardial infarction is the underlying etiology. Patients with no reflow can progress to heart failure and have higher chances of adverse cardiac remodeling andmortality.
After MI, the myocardium undergoes a series of events that initiates inflammation and shift to anti‐inflammation before transitioning to tissue repair that culminates in scar formation to replace the necrotic myocardium. The overall response to MI is determined by two major steps, the first of which is the inflammatory response that includes the secretion of proteases to break down the extracellular matrix (ECM) and is a necessary step to clear necrotic cardiomyocytes. The second step is the generation of new ECM that comprises the scar; and this step is governed by the cardiac fibroblasts as the major source of new ECM synthesis. Leukocytes are central to both steps, contributing to the proteases in the first step and stimulating the fibroblasts in the second step. Leukocytes transition from pro‐inflammatory to anti‐inflammatory and reparative cell phenotypes over the first week of MI. The balance between their phenotypes over the two steps of MI response determines the final quantity and quality of scar formed, which in turn contributes to chronic outcomes following MI, including the progression to heart failure (Becirovic‐Agic et al., 2022; Chalise, Daseke, Kalusche, et al., 2022).
Necrotic myocytes release complement and other mediators such as damage‐associated molecular patterns (DAMPs) that initiate an early robust inflammatory response (Dobaczewski et al., 2006). Macrophages and neutrophils in the circulation and spleen respond by infiltrating the infarct site (B Gowda et al., 2021; Dutta et al., 2015; Halade et al., 2018; Kain & Halade, 2020). Neutrophils quickly predominate, such that by MI day 1 the neutrophil is the primary cell type in the infarct (Daseke et al., 2021; Ma et al., 2016). The macrophage numbers peaks at MI day 3–5 (Mouton, DeLeon‐Pennell, Rivera Gonzalez, et al., 2018). Lymphocytes also contribute to the MI response and are present in lower numbers than neutrophils or macrophages (Zaidi, Aguilar, Troncoso, Ilatovskaya, & DeLeon‐Pennell, 2021a; Zaidi, Corker, et al., 2021). T and B cells role in infarct repair has been recently reviewed in detail (Hofmann & Frantz, 2015; Ilatovskaya et al., 2019; Porsch et al., 2021). While neutrophils and monocytes increase early in response to myocardial injury, lymphocytes actually decrease to induce lymphopenia, and evaluations that add more to our understanding of how leukocytes communicate to coordinate MI repair are certainly warranted (Ma et al., 2022). Cardiac fibroblasts are present throughout the time course, peaking in proliferative capacity at MI day 3 (Mouton et al., 2019). Inflammation peaks between MI day 1 and 3, followed by anti‐inflammation leading to tissue repair and scar formation over the course of MI (Becirovic‐Agic et al., 2022; Tenkorang et al., 2019). The early phase of response is crucial for MI wound healing, and we will discuss here the roles of the neutrophil, macrophage, and fibroblast to coordinate ECM turnover to regulate wound healing.
2. TEMPORAL EVOLUTION OF NEUTROPHIL PHENOTYPES
Neutrophils are among the first leukocytes to the ischemic injury after MI and serve as a crucial cell type to initiate and coordinate the early removal of necrotic debris. Neutrophil numbers peak at MI day 1 and return toward baseline values by day 7 (Ma et al., 2016). Neutrophil activation involves an increase in cell surface adhesion molecule expression that allows circulating neutrophils to enter the infarct region and mobilization of newly formed neutrophils from the bone marrow. Exactly how immune cells home to the infarct is not entirely clear. The most discussed and accepted possibility is that extravasation occurs at the border zone, and leukocytes then migrate to the necrotic site (Daseke et al., 2021; Prabhu & Frangogiannis, 2016). Another possible route is through transportation occurring through lymphatic vessels, which is not impeded by arterial occlusion (Klaourakis et al., 2021; Lindsey et al., 2001; Swirski & Nahrendorf, 2013). The lymphatic route has been shown as a mechanism to bring CD4+ T cells to the infarct zone (Castell et al., 2019; Hampton & Chtanova, 2016; Hofmann et al., 2012).
Cardiomyocyte necrosis releases complement, DAMPs (e.g., high mobility group box 1 [HMGB‐1], S100A8/A9, mitochondrial DNA, and adenosine triphosphate) and other signaling molecules that activate cell adhesion molecule expression on circulating neutrophils to trigger extravasation into the infarct region and stimulate maturation and recruitment of further neutrophils from the bone marrow (Chalise, Becirovic‐Agic, Daseke, et al., 2022; Chalise, Becirovic‐Agic, & Lindsey, 2022b). Reduced secretion of CXCL12 in the bone marrow allows mobilization of myeloid cells to the circulation through CXCL12/CXCR4 (fusion, CD184) axis (Christopher et al., 2009). CXCL12/CXCR4 axis is very important in early neutrophil and monocyte mobilization from the bone marrow to the circulation and hence for overall MI wound healing. CXCR4 blockade by use of antagonist improves tissue repair after MI (Hess et al., 2020; Wang et al., 2019). Neutrophil aging is an intrinsic circadian process, regulating both entry into and clearance out of tissue, that can be modified by inflammation, with Bmal1 and CXCR2 extending while CXCR4 shortens the half‐life (Adrover et al., 2019).
Once extravasated, neutrophils shed L‐selectin (CD62L) to allow diapedesis to the infarct and activation by binding toll‐like receptor (TLR)‐4 (Hafezi‐Moghadam et al., 2001). Activated neutrophils are characterized as CD62Llow and CXCR4high (Deniset & Kubes, 2016; Vafadarnejad et al., 2020). Inhibition of neutrophil recruitment by using CD18 antibody reduces infarct size and improves cardiac remodeling in the reperfused MI mouse model (Faxon et al., 2002). Neutrophils, once activated, traverse to the infarct region where they degranulate and release a number of proteases (e.g., matrix metalloproteinase [MMP]‐8 and MMP‐9 and neutrophil elastase) to breakdown ECM into fragments to remove necrotic cardiomyocytes (Becirovic‐Agic et al., 2021). Neutrophils also release a number of pro‐inflammatory cytokines and chemokines that serve to amplify the inflammatory signal and herald in the macrophages (Chalise et al., 2022b).
While the net effect of neutrophils is destructive, overall, neutrophil presence is required and essential for MI resolution (Horckmans et al., 2017). A number of groups have shown that complete blockade of neutrophil entry using anti‐inflammatory strategies increases the incidence of rupture in mouse models of permanent occlusion MI (Huang & Frangogiannis, 2018; Sholter & Armstrong, 2000). Clinical trials targeting immune responses such as methylprednisolone, cyclosporine, or canakinumab (IL‐1β antibody) failed to show any significant difference between placebo and treatment (Cung et al., 2015; Giugliano et al., 2003; Madias & Hood Jr, 1982; Ridker et al., 2017). Angiotensin‐converting enzymes and beta blockers are therapeutically indicated and work in part by modulating inflammation (Pfeffer et al., 1995; Pfeffer & Pfeffer, 1988). Neutrophil depletion started before MI worsens cardiac physiology after MI by altering the role of the macrophage (Horckmans et al., 2017). While macrophage numbers are elevated in the neutrophil‐depleted mice due to increased infiltration, at the same time, there is a reduction in their conversion to a reparative phenotype and reduced phagocytic capacity. Neutrophil gelatinase‐associated lipocalin (NGAL) was identified in the MI neutrophil secretome as being the driver of the effect on macrophages, and NGAL alone could rescue the neutrophil‐depleted phenotype and induce macrophage phagocytosis. Neutrophil depletion, therefore, was detrimental to cardiac repair (Horckmans et al., 2017).
Prolonged pro‐inflammation is also detrimental and has negative effects on cardiac physiology and survival. While reduced neutrophil recruitment improves MI repair, complete blockade, or depletion of neutrophils prior to MI is detrimental to repair and resolution (Jolly et al., 1986; Wu et al., 2006). Neutrophils are required for creating the functional space for infarct scar formation by degrading damaged tissue and aiding in its removal (Chalise et al., 2022b). Excessive neutrophils also have negative effects on infarct repair due to overactive tissue degradation (Arruda‐Olson et al., 2009; Chalise et al., 2022c). Administration of S100A8/A9, a neutrophil cytosolic protein that acts as a DAMP, exacerbates infarct wall thinning and impairs LV physiology (Chalise et al., 2022a; Marinković et al., 2019; Marinković et al., 2020). Similarly, Inhibition of MMP‐12 prevents neutrophil apoptosis from occurring and results in excessive infarct wall thinning and exacerbated LV remodeling due to the prolonged presence of neutrophils (Iyer et al., 2015; A. J. Mouton, Rivera Gonzalez, Kaminski, Moore, & Lindsey, 2018d). MMP‐12 induces polarization of thneutrophil signalome towards an apoptotic signature with upregulation in IL‐4 secretion and FOXO1 signaling and downregulation in WNT signaling. (Chalise et al., 2022d) The neutrophil, therefore, has a complex cellular role in MI remodeling.
One reason for this complexity is that neutrophils across the MI time continuum undergo a full shift in cell polarization from early pro‐inflammation to anti‐inflammation to repair (M. J. Daseke et al., 2019; Ma et al., 2016). The change in MI microenvironment across the first week of MI dictates neutrophil phenotype, as neutrophils polarize in response to the environment into which they enter. Single‐cell analysis revealed distinct neutrophil profiles across the response to MI. At MI day 1, neutrophils show higher expression of CD62L, CXCL3, CCL6, and CD177 that shifts to the expression of ICAM1, tumor necrosis factor (TNF) α, and interleukin (IL)‐23a at day 3 indicating activated or aged neutrophils (Prince et al., 2017; Vafadarnejad et al., 2020). At day 5, neutrophils express higher nuclear receptor 4A2 (NR4A2) indicating resolution of inflammation with increased apoptosis (Vafadarnejad et al., 2020). The MI neutrophil signaling map is shown in Figure 1.
FIGURE 1.
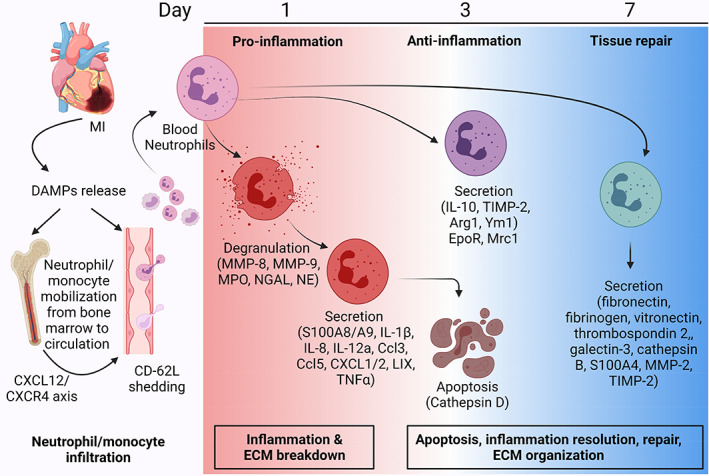
Myocardial infarction (MI) neutrophil signaling network. After MI, necrotic cardiomyocytes release damage‐associated molecular patterns (DAMPs) that stimulate neutrophil infiltration from the circulation and mobilization of new neutrophils from the bone marrow, regulated by CXCL12/CXCR4 signaling. Blood neutrophils infiltrate the infarct, shedding CD‐62L along the way. MI neutrophils are polarized to a pro‐inflammatory phenotype at MI day 1. Pro‐inflammatory neutrophils undergo degranulation to release various proteases such as matrix metalloproteinase (MMP)‐8, MMP‐9, myeloperoxidase (MPO), neutrophil gelatinase‐associated lipocalin (NGAL), and neutrophil elastase. Pro‐inflammatory neutrophils also secrete pro‐inflammatory proteins (e.g., S100A8/A9, interleukin (IL)‐1β, IL‐8, IL‐12a, CCL3, CCL5, CXCL1/2, LIX, and tumor necrosis factor (TNF)α). With the change in tissue microenvironment to anti‐inflammatory status, pro‐inflammatory neutrophils undergo apoptosis and newly infiltrating neutrophils polarize to an anti‐inflammatory phenotype. Anti‐inflammatory neutrophils are characterized by increased expression of IL‐10, arginase (Arg)1, Ym1, tissue inhibitor of matrix metalloproteinase (TIMP)‐2, and mannose receptor C‐type 1 (Mrc1). By MI day 7, neutrophils show a reparative phenotype with the predominant secretion of extracellular matrix (ECM) proteins such as fibronectin, fibrinogen, vitronectin, thrombospondin 2, galectin‐3, cathepsin B, S100A4, MMP‐2, and TIMP‐2. Created with Biorender.com
2.1. Pro‐inflammatory neutrophils
Activated neutrophils in the infarct show a pro‐inflammatory phenotype. Upon entry to the ischemic site, neutrophils degranulate to release tissue degrading proteases, reactive oxygen species, chemokines, and cytokines (Kain & Halade, 2020). The overall phenotype of neutrophils at MI day 1 is pro‐inflammatory, with invasion and degranulation being primary features. Proteomic analysis showed high upregulation of S100A9, fibrinogen, histones, and activin A indicating positive regulation of secretion (M. J. Daseke et al., 2019). The predominant MMPs are MMP‐8 and MMP‐9, although recently MMP‐12 was shown to be expressed by neutrophils (Horckmans et al., 2017; Iyer et al., 2015; Kain & Halade, 2020). Neutrophils also secrete myeloperoxidase, NGAL, and neutrophil elastase in response to MI. MMP‐9 secreted by neutrophils in turn stimulates the production of CXCR4 to further increase cell mobilization through a positive feedback loop (Gopalkrishna et al., 2021; Jin et al., 2008). In the presence of the pro‐inflammatory stimulus, fibronectin induces neutrophil degranulation to release MMP‐9, which then breaks down fibronectin to its 120 kDa fragment to further stimulate MMP‐9 release in a positive feedback loop that involves fibronectin signaling (M. J. Daseke et al., 2019).
Neutrophils on MI days 1 and 3 are characterized by the expression of pro‐inflammatory cytokines and chemokines, including IL‐1b, IL‐8, IL‐12a, CC chemokines (CCL)3 and CCL5, CXCL1/2, lipopolysaccharide‐induced CXC chemokine (LIX), TNFα (Kain & Halade, 2020). Pro‐inflammatory neutrophils also show low expression of anti‐inflammatory proteins, including macrophage mannose receptor 1 (MRC1/CD206) and IL‐10 (Ma et al., 2016). The release of pro‐inflammatory mediators creates a gradient for chemotaxis to stimulate the entry of more neutrophils from circulation to the site of MI as part of the amplification process. This gradient also stimulates the entry of macrophages (Kumar et al., 2018). Pro‐inflammatory neutrophil numbers correlate with the extent of infarct wall thinning (Ma et al., 2016). Pro‐inflammatory neutrophils can sustain inflammation by forming neutrophil extracellular traps (NETs), which allows proinflammatory neutrophils homing to a specific region by the release of chromatin fibers and histones (Chalise et al., 2022a; Hofbauer et al., 2019; Ma, 2021).
At the same time, the presence of pro‐inflammatory neutrophils is also essential in cardiac wound healing, as inflammation is required before resolution and repair can be initiated. For the timely resolution of inflammation, it is important that pro‐inflammatory neutrophils undergo apoptosis to remove them after cardiac wound healing has been initiated. Neutrophil apoptosis occurs through the activation of caspase 3, and MMP‐12 can stimulate neutrophil apoptosis directly (Iyer et al., 2015). MMP‐9 reduces macrophage phagocytosis of apoptotic neutrophils, and MMP‐9 also reduces neutrophil apoptosis indicated by reduced caspase 9 (DeLeon‐Pennell et al., 2016). MMP‐9, therefore, prevents neutrophils from becoming apoptotic and prevents macrophages from phagocytosing apoptotic neutrophils. Induction of neutrophil apoptosis also initiates the polarization of the anti‐inflammatory neutrophil phenotype, indicating there is a built‐in shut‐off mechanism to temporally limit inflammation.
2.2. Anti‐inflammatory and reparative neutrophils
Within the infarct zone, by MI day 3 the progression to anti‐inflammation and repair commences. The infarct environment shows increased expression of anti‐inflammatory molecules and growth factors such as IL‐10, transforming growth factor β (TGF‐β), and vascular endothelial growth factor (VEGF), and this is the environment into which new neutrophils infiltrate. The overall phenotype of the neutrophil at MI day 3 is apoptotic, with cathepsin activity and ECM reorganization being primary features. Because neutrophils are transcriptionally active but terminally differentiated cells, apoptosis of the pro‐inflammatory neutrophils reduces active inflammation and alters the tissue microenvironment, which in turn programs new incoming neutrophils toward an anti‐inflammatory phenotype (Lakschevitz et al., 2015; Rosales, 2018). The anti‐inflammatory neutrophil is characterized by high expression of Ly6G and CD206, which continue to increase until MI day 7 (Ma et al., 2016). CD206 in neutrophils is locally produced, as neutrophils in circulation on MI days 1, 3, or 5 are all CD206 negative (Ma et al., 2016). Proteomic analysis shows high expression of cathepsin D, Epo‐R, α‐synuclein, fibronectin, and fibrinogen indicating positive regulation of vasoconstriction (M. J. Daseke et al., 2019). By MI day 5, neutrophil recruitment is reduced, and the neutrophils present have reduced pro‐inflammatory and elevated anti‐inflammatory gene expression. By MI day 7, the overall phenotype of the neutrophil is reparative, with neutrophils contributing to ECM synthesis and reorganization as the top enriched pathway (M. J. Daseke et al., 2019).
Cathepsins degrade ECM and stimulate apoptosis and autophagy, providing shut‐off valves to prevent extended neutrophil activity through upregulation of caspase 8 and downregulation of pro‐survival BCL‐2 protein (Chwieralski et al., 2006; Conus et al., 2012). Upregulation of fibronectin amplifies TNFα stimulated neutrophil apoptosis and augments macrophage uptake of apoptotic neutrophils, thus serving a dual role in promoting neutrophil removal from the MI (M. J. Daseke et al., 2019; Kettritz et al., 1999; McCutcheon et al., 1998).
The overall phenotype of the neutrophil at MI day 7 is reparative, with ECM reorganization being the primary feature. The direct role of neutrophils in tissue repair has been understudied, and neutrophils on MI days 5 and 7 have increased ECM protein expression, including fibrinogen, fibronectin, thrombospondin‐2, galectin‐3, MMP‐2, tissue inhibitor of metalloproteinase (TIMP)‐2, and vitronectin (M. J. Daseke et al., 2019). Neutrophils at MI day 7 also express cathepsin B and S100A4.
Fibrinogen stimulates fibroblast proliferation, indicating upregulation of neutrophils to fibroblast cross‐talk (Gray et al., 1993). The increase in TIMP‐2 indicates that neutrophils at later times of MI prevent further ECM breakdown by reducing their expression of MMP‐8 and MMP‐9 while upregulating TIMPs. At the same time, the increased expression of MMP‐2 in the day 7 neutrophils indicates that it may also serve a neo‐homeostatic role to help maintain the infarct scar, as MMP‐2 is associated with cardiomyocyte homeostasis (DeCoux et al., 2014). In vivo, exogenous IL‐4 infusion started 24 h after MI reduced neutrophil expression of CCL3, IL‐12a, TGF‐β1, and TNFα on MI day 3 (Daseke, Tenkorang, et al., 2020). Neutrophils stimulated in vitro with IL‐4 show a strong anti‐inflammatory polarization with increased expression of anti‐inflammatory markers [i.e., arginase (ARG)1, CD206, TGF‐β1, and chitinase‐3‐like protein 3 (YM1); Liu et al., 2021].
As neutrophils directly regulate inflammatory response and tissue microenvironment in MI, targeting neutrophils or neutrophil components could be therapeutically viable. In cancer, training neutrophils toward granulopoiesis has a positive effect (Kalafati et al., 2020). Inhibition of neutrophil dimer protein S100A8/A9 improves infarct size after MI (Gebhardt et al., 2006; Volz et al., 2012; B. Zhao et al., 2021). Resolution promoting factors (resolvin D, annexin A1, and MMP‐12) impact neutrophil infiltration and pro‐inflammation (A. J. Mouton et al., 2018b; Pullen et al., 2020; Tourki et al., 2020). Selective transdifferentiation of neutrophils from an inflammatory to reparative phenotype may provide an improved response to MI. A significant challenge lies in identifying the appropriate time point to start the therapy, as pro‐inflammatory neutrophils and subsequent signaling are essential for MI resolution. Sex as a biological variable may also need to be considered. While neutrophil numbers were lower in female mice at MI day 1, the neutrophils from the female mice showed higher per cell release of MMP‐9 to maintain tissue clearance rates (K. Y. DeLeon‐Pennell & Lindsey, 2019; DeLeon‐Pennell et al., 2018). The net effect of sex differences on remodeling remains to be fully elucidated.
3. TEMPORAL EVOLUTION OF MACROPHAGE PHENOTYPES
Along with neutrophils, macrophages are the other leukocytes that predominate the infarct region and include bone marrow‐derived monocytes and macrophages as well as resident macrophages. Blood monocytes infiltrate the LV infarct in response to chemotactic signals and initially differentiate to a pro‐inflammatory profile. Macrophages display one of two types: (1) the CCR2+ subtype that regulates monocyte recruitment through myeloid differentiation primary response 88 and (2) the CCR2− subtype that inhibits further monocyte recruitment and offers cardioprotection after MI through homeostatic functions (Bajpai et al., 2019; Frantz & Nahrendorf, 2014; Hulsmans et al., 2017; Pinto et al., 2012). The early pro‐inflammatory macrophage phenotypically evolves over the first week of MI (Figure 2; A. J. Mouton, DeLeon‐Pennell, Gonzalez, et al., 2018). Pro‐inflammatory macrophages amplify the inflammatory signaling cascade, while anti‐inflammatory and reparative macrophages phagocytose apoptotic neutrophils to shut off inflammation, and secrete growth factors and ECM components to initiate scar formation, and stimulate neovascularization in the infarct zone.
FIGURE 2.
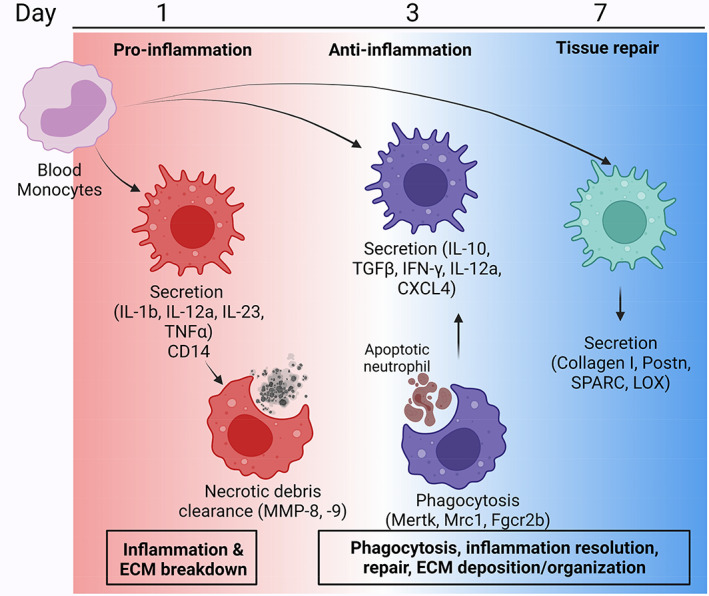
MI macrophage signaling network. Blood monocytes infiltrate the infarct area and are initially polarized to pro‐inflammatory macrophages by processes similar to neutrophils. Pro‐inflammatory macrophages highly express CD14 and a variety of cytokines (e.g., IL‐1β, IL‐12a, IL‐23, and TNFα). On MI day 1, macrophages aid the neutrophils in necrotic debris clearance by secreting tissue degrading proteases such as MMP‐8 and MMP‐9. By MI day 3, macrophages transition to an anti‐inflammatory phenotype and phagocytose apoptotic neutrophils through cell surface expression of Mertk, Mrc1, and Fgcr2b. Anti‐inflammatory macrophages secrete IL‐10 and TGF‐β, along with IFN‐γ, IL‐12a, and CXCL4, indicating a mixed profile. By MI day 7, macrophages continue to transition toward a reparative phenotype with the secretion of ECM proteins such as collagen I, periostin (Postn), secreted protein acidic and rich in cysteine (SPARC) and lysyl oxidase (LOX). Created with BioRender.com
3.1. Pro‐inflammatory macrophages
The overall phenotype of the macrophage at MI day 1 is pro‐inflammatory, with cytokine upregulation and ECM degradation being primary features. Similar to neutrophils, pro‐inflammatory macrophages express cytokines and chemokines, including IL‐1b, IL‐6, and TNFα (Liu et al., 2021). CD14 is also upregulated on MI day 1. Th1‐related cytokines activate M1 macrophages and induce pro‐inflammatory activity. In addition to cytokine release, another major role of pro‐inflammatory macrophages is to secrete MMP‐8 and MMP‐9 to coordinate necrotic tissue removal in conjunction with the neutrophils. Enrichment analysis of the day 1 MI macrophage transcriptome showed upregulation of degranulation or release of tissue degrading proteases, and downregulation of ECM organization. Day 1 macrophages balance neutrophil degranulation by secreting murinoglobulin, an attenuator, and galectin‐3, a stimulator, of MMP‐9 release (Chalise et al., 2022c).
In vivo, macrophages at MI day 1 are activated by binding TLR ligands to TLR in a MYD88‐dependent manner to induce transcription of interferon (IFN)γ and TNFα. Macrophages stimulated by pro‐inflammatory cytokines secrete pro‐inflammatory cytokines to maintain the inflammatory condition that extends neutrophil survival (Kumar et al., 2018). At MI day 1, pro‐inflammatory macrophages (F4/80lowLy6Chigh) predominantly exhibit NF‐κB and MAPK signal activation to increase IL‐1β, IL‐12a, CXCL4, ILR‐2, MMP‐8, and IL‐24 expression (A. J. Mouton, DeLeon‐Pennell, Gonzalez, et al., 2018). Hypoxic response is indicated in M1 macrophages at day 1 with differential expression of HIF‐1α. Glycolysis, a metabolic hallmark of the pro‐inflammatory macrophages, is upregulated at MI day 1 (Zhang et al., 2019; M. Zhao et al., 2020).
Pro‐inflammatory macrophages associate with adverse cardiac remodeling after MI. In humans with acute MI, peak in pro‐inflammatory monocytes (CD14+CD16−) in the blood is negatively associated with recovery of ejection fraction 6 months after MI (Tsujioka et al., 2009). Peak circulating monocyte counts were higher in patients with acute MI who had pump failure or LV aneurysm, with monocyte counts correlating positively with LV end‐diastolic volumes and negatively with ejection fraction (Maekawa et al., 2002). Similar results have been observed in mouse models of MI with increased macrophages correlated to higher incidences of LV rupture (Hanna et al., 2020). ApoEnull mice after MI have exacerbated infiltration and prolonged presence of pro‐inflammatory macrophages resulting in reduced ejection fraction measured at MI day 21 (Panizzi et al., 2010). When the ApoEnull mice were treated with siRNA targeting CCR2, they were protected from heart failure and had improved MI wound healing (Panizzi et al., 2010). An excessive inflammatory response caused by pro‐inflammatory macrophages therefore can be deleterious and appropriate targeting of this macrophage subtype may improve MI outcomes.
3.2. Anti‐inflammatory and reparative macrophages
At about MI day 3, macrophages begin to polarize with the overall phenotype of the macrophage being anti‐inflammatory, with proliferation, phagocytosis, and metabolic reprogramming being primary features (A. J. Mouton, DeLeon‐Pennell, Gonzalez, et al., 2018). These cells are characterized by high expression of IFNγ, IL‐12a, and CXCL4, indicating a combination of pro‐inflammation and anti‐inflammation. Anti‐inflammatory macrophages can be stimulated in vitro by IL‐4, IL‐10, or IL‐13 (Liu et al., 2021). While IL‐4 is not endogenously expressed at high levels after MI, cells in the heart express IL‐4 receptors and can respond to IL‐4 stimulation (Daseke, Tenkorang, et al., 2020). Anti‐inflammatory genes expressed by macrophages include ARG1, IL‐10, CD206, TGFβ, and YM1 (Alvarez‐Argote & O'Meara, 2021). By MI day 7, the macrophages convert to a reparative phenotype, producing collagen, periostin (Postn), secreted protein acidic and rich in cysteine(SPARC), and lysyl oxidase (A. J. Mouton et al., 2019).
Engulfing of apoptotic neutrophils by macrophages involves GAS6 binding and MerTK activation through upregulation of AKT signaling (de Couto et al., 2019; DeBerge et al., 2017). Acute inhibition of CD47 during reperfused MI promotes macrophage phagocytosis and improves cardiac repair (Zhang et al., 2017). Neutrophil phagocytosis by macrophages reduces IL‐23 release and triggers an anti‐inflammatory response (Rosales, 2018). The anti‐inflammatory macrophages stimulate fibroblast proliferation and ECM synthesis to initiate scar formation. TGF‐β secreted by macrophages initiates the proliferative phase and induces repair and regeneration of ECM in the infarct region (A. Hanna & Frangogiannis, 2019). Anti‐inflammatory macrophages also communicate with endothelial cells through the release of growth factors (e.g., platelet‐derived growth factor) to stimulate the formation of new blood vessels necessary to vascularize the infarct scar (Humeres & Frangogiannis, 2019).
As macrophages play various crucial physiological roles which are essential for infarct wound healing, complete depletion or early inhibition of macrophages holds less therapeutic potential. Depletion of macrophages by clodronate liposomes or genetic ablation both impair wound repair (Alvarez‐Argote & O'Meara, 2021). In humans, pro‐inflammatory M1 macrophages (CD14+CD16−) were lower in patients with thrombus after MI compared to patients without thrombus (Ben‐Mordechai et al., 2013; Frantz et al., 2013). In patients with ST‐segment elevation MI, blood CD14+CD16− monocyte counts negatively correlate with MI resolution (Peet et al., 2020). Increasing macrophages proliferation by administration of macrophage colony‐stimulating factor (M‐CSF, CSF‐1) reduces MI cardiac remodeling (Morimoto et al., 2007), while administering an inducer of granulocyte–macrophage colony‐stimulating factor (GM‐CSF) has the opposite effect (Maekawa et al., 2004).
Optimization or refinement of macrophage subtype kinetics may be beneficial in the MI response. A blunted or accelerated pro‐inflammatory phase or an enhanced or early anti‐inflammatory phase may provide an improved balance of phenotypes that allow the essential inflammation while promoting repair. CCR2 inhibition targets pro‐inflammatory monocytes (Ly‐6Chigh) and is beneficial for MI repair (Peet et al., 2020). Similarly, RNA inhibition of the IRF5 transcription factor reduces pro‐inflammatory macrophage numbers without altering anti‐inflammatory or reparative macrophages (Wynn & Vannella, 2016). IL‐10, an anti‐inflammatory agent, administered 24 h after MI, improved ejection fraction and reduced cardiac dilation by reducing macrophage numbers, and shifting the phenotype to anti‐inflammation and repair (Jung et al., 2017). IL‐10 induces phagocytosis by acting through an IL‐10‐STAT3‐galectin 3 axis to upregulate production of osteopontin (Shirakawa et al., 2018). Macrophage‐induced phagocytosis is essential for tissue repair by clearing apoptotic cells and coordinating ECM reorganization during scar formation (Shirakawa et al., 2018). Effective strategies targeting the macrophages, therefore, will need to consider the necessity of inflammation.
4. TEMPORAL EVOLUTION OF FIBROBLAST PHENOTYPES
The cardiac fibroblast is a resident cell type that actively participates in homeostasis of the myocardium by providing continuous low‐grade turnover of ECM, contributing to vascular maintenance, and aiding in the transduction of electrical signals (Baudino et al., 2006; Camelliti et al., 2005; Daseke, Tenkorang‐Impraim, et al., 2020; Gaudesius et al., 2003; Humeres & Frangogiannis, 2019; Miragoli et al., 2007; A. J. Mouton et al., 2019; Vasquez et al., 2011) Following MI, the fibroblast transitions from supporting homeostatic functions to actively participating in all phases of infarct wound healing (Humeres & Frangogiannis, 2019; A. J. Mouton et al., 2019). While ECM synthesis and deposition is the major and most known role of the fibroblast during infarct healing, the fibroblast polarizes and shows a spectrum of phenotypes in response to changes in the microenvironment over the MI time continuum (Figure 3; A. J. Mouton et al., 2019). During the early inflammatory phase, ECM degradation is favored over ECM deposition as the removal of debris is necessary before scar formation (Nielsen et al., 2019; Tenkorang et al., 2019). Consequently, fibroblasts polarize to support ECM degradation and necrotic debris removal (A. J. Mouton et al., 2019; Nielsen et al., 2019). During the anti‐inflammatory and reparative phase, changes in the microenvironment stimulate fibroblast proliferation and ECM synthesis resulting in scar formation (A. J. Mouton et al., 2019; Nielsen et al., 2019).
FIGURE 3.
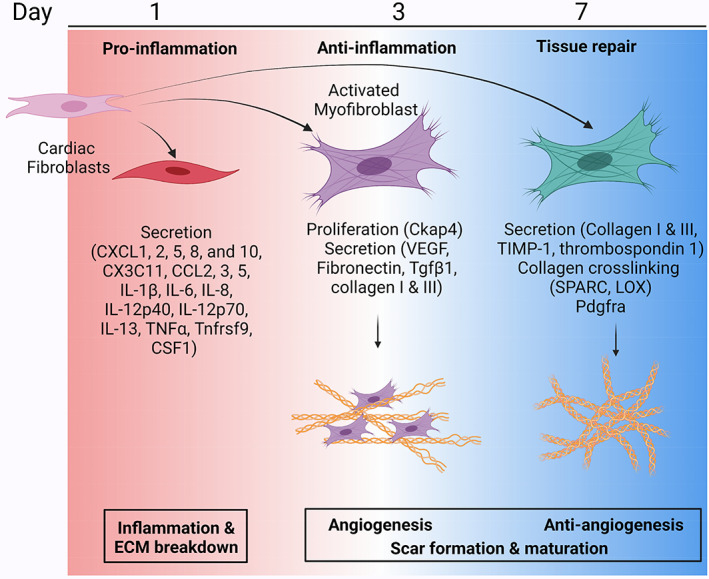
MI fibroblast signaling network. In response to MI, cardiac fibroblasts initially polarize to a proinflammatory phenotype at MI day 1 and secrete cytokines and chemokines (e.g., CXCL1, 2, 5, 8, and 10, CX3C11, CCL2, 3, 5, IL‐1β, IL‐6, IL‐8, IL‐12p40, IL‐12p70, IL‐13, TNFα, Tnfrsf9, and CSF1) to facilitate leukocyte infiltration. By MI day 3, fibroblasts polarize to a more proliferative phenotype (Ckap4 expression) with characteristics of an activated myofibroblast (α‐smooth muscle actin expression and Tgfβ1). Activated myofibroblasts secrete ECM proteins, including collagen I and III, as well as fibronectin that initiates ECM deposition for scar formation. Vascular endothelial growth factor (VEGF) secretion increases at MI day 3, indicating an angiogenic role for fibroblasts. By MI day 7, fibroblasts continue to secrete more ECM proteins (collagen I and III, TIMP‐1, and collagen crosslinking proteins such as SPARC and LOX for scar maturation. Reparative fibroblasts at MI day 7 demonstrate an anti‐angiogenic profile, indicated by expression of thrombospondin 1. Created with Biorender.com
4.1. Pro‐inflammatory fibroblasts
The overall phenotype of the fibroblast at MI day 1 is pro‐inflammatory, with cytokine upregulation and anti‐migration being primary features. Cardiac fibroblasts both contribute and respond to pro‐inflammatory signaling during MI (Daseke, Tenkorang, et al., 2020). DAMPs released by necrotic myocytes and leukocytes stimulate fibroblasts through pattern recognition receptors (TLRs, NLRs, IL‐1R1, RAGE) to produce and release a range of chemokines (CXCL1, CXCL2, CXCL5, CXCL8, CXCL10, CX3C11, CCL2, CCL3, and CCL5) and cytokines (IL‐1β, IL‐6, IL‐8, IL‐12p40, IL‐12p70, IL‐13, TNFRSF9, and TNFα; Li et al., 2021; Turner, 2016; Wu et al., 2014). The MI day 1 fibroblast also produces colony‐stimulating factor (CSF)‐1, which is needed for macrophage differentiation (Mouton, DeLeon‐Pennell, Gonzalez, et al., 2018). The production of pro‐inflammatory cytokines by fibroblasts stimulates the influx of leukocytes into the infarct zone (Altara et al., 2016; Dewald et al., 2005; Frangogiannis, 2015; Montecucco et al., 2012; Mylonas et al., 2017; C. L. Wu et al., 2021). Cultured fibroblasts also produce pro‐inflammatory chemokines and cytokines in response to IFNγ and TNFα, indicating a positive feedback loop is initiated in the presence of a pro‐inflammatory environment (Daseke, Tenkorang, et al., 2020; Pappritz et al., 2018; Turner et al., 2007). By transcriptomics, the most upregulated pathway in MI day 1 fibroblast is chemokine‐ and cytokine‐mediated inflammation indicating that cardiac fibroblast actively regulates the inflammatory response (A. J. Mouton et al., 2019). Pro‐inflammatory fibroblasts also display a pro‐survival, anti‐proliferative, and anti‐migratory phenotype. The expression of anti‐apoptotic BCL2 is increased in MI day 1 fibroblast indicating a pro‐survival phenotype, which would also prevent neutrophils from undergoing apoptosis (A. J. Mouton et al., 2019). This in conjunction with a reduced migration rate and absence of proliferation favors ECM degradation (A. J. Mouton et al., 2019; Nielsen et al., 2019). Thus, pro‐inflammatory fibroblasts promote leukocyte infiltration, ECM degradation, and necrotic debris removal.
4.2. Anti‐inflammatory and reparative fibroblasts
The inflammation starts to subside around MI day 3 (Daseke, Tenkorang, et al., 2020). The overall phenotype of the fibroblast at MI day 3 is proliferative, with promotion of angiogenesis and ECM production being primary features. The shift from an inflammatory to an anti‐inflammatory environment induces transdifferentiation of the fibroblasts into myofibroblasts, which start to proliferate and migrate into the infarcted area, resulting in increased wound contraction, and repair capacity (de Boer et al., 2019; Ma et al., 2017; A. J. Mouton et al., 2019). The main function of the fibroblast is to synthesize and secrete ECM, a function fundamental for the formation of the collagen‐rich scar that replaces the necrotic myocytes (Daseke, Tenkorang, et al., 2020; Humeres & Frangogiannis, 2019; Nielsen et al., 2019). Even though replacing myocytes with scar tissue results in loss of contractility and is suboptimal compared to the pre‐MI state, the formation of an infarct scar is crucial for structural integrity to prevent the formation of LV aneurysm and cardiac rupture and is an optimal response to MI (de Boer et al., 2019).
TGF‐β1, secreted by most myocardial cell types, including cardiomyocytes and macrophages, is the central mediator of myofibroblast activation (M. Becirovic‐Agic et al., 2021; A. Hanna & Frangogiannis, 2019; Ma et al., 2017). IL‐1, corticotrophin‐1, and fibroblast growth factor are additional factors that can stimulate fibroblast migration into the infarct, but their effect on infarct scar formation is still uncertain (Dobaczewski et al., 2010; Freed et al., 2011; Ma et al., 2017; Mitchell et al., 2007). The myofibroblast is commonly defined as a postinjury fibroblast, a definition loosely based on the binary expression of alpha‐smooth muscle actin (Ma et al., 2017). This terminology, however, is limiting since it does not include all the different phenotypes that the fibroblasts display during the MI time continuum (Daseke, Tenkorang, et al., 2020). MI day 3 fibroblasts have a proliferative and pro‐angiogenic phenotype (A. J. Mouton et al., 2019). Cholesterol biosynthesis, necessary for membrane synthesis and proliferation, is the major upregulated pathway in the MI day 3 fibroblast. Increased proliferation in combination with downregulation of pro‐apoptotic genes (Caspase 3) explains the increased fibroblast numbers detected around MI day 3.
Angiogenesis, cell migration involved in sprouting angiogenesis, and blood vessel endothelial cell migration are additional processes upregulated in the MI day 3 fibroblasts. In vitro, the secretome from day 3 fibroblasts stimulates endothelial cell tube formation, supporting the pro‐angiogenic phenotype (A. J. Mouton et al., 2019). Collagen content in the infarcted area does not increase measurably until 3–7 days after MI and peaks after 3–6 weeks (de Boer et al., 2019; Virag & Murry, 2003). The fibroblasts that we typically think of, a prominent producer of ECM, are seen on MI day 7 (Nielsen et al., 2019). The overall phenotype of the fibroblast at MI day 7 is scar‐forming, with ECM production and anti‐angiogenesis being primary features. The secretome of day 7 fibroblasts is rich in collagen I alpha 1 and 2 chains, SPARC, and lysyl oxidase. At the same time, day 7 fibroblasts also secrete TIMP‐1, TIMP‐3, and THBS1, which inhibits ECM degradation and angiogenesis (A. J. Mouton et al., 2019). Thus, the anti‐inflammatory and reparative fibroblasts support scar formation thorough ECM deposition and revascularization.
Fibroblasts are very changeable in character and therefore amenable to therapy (Gourdie et al., 2016). Promoting pro‐angiogenic fibroblasts early on may be a way to accelerate revascularization and infarct healing. To do this, a better understanding of the factors that promote different fibroblast phenotypes is warranted. TGF‐β1, in addition to stimulating ECM synthesis and deposition, is a very potent suppressor of inflammation and may therefore be used to promote infarct healing (A. Hanna & Frangogiannis, 2019). On the other hand, excessive ECM deposition may induce arrhythmias and diastolic dysfunction, showing the importance of having the right amount at the right time (Prabhu & Frangogiannis, 2016; Zamilpa & Lindsey, 2010).
5. CONCLUSIONS
The cardiac response to MI involves a series of wound healing events categorized as temporal shifts from pro‐inflammation to anti‐inflammation to repair and scar formation (Figure 4). The neutrophil, macrophage, and fibroblast all evolve in phenotype along this time continuum. Understanding the balance in interactions among these three cell types will provide insight into both the mechanisms involved as well as provide targets for therapeutic modulation. With increased awareness of the importance of rigor and reproducibility, a number of resources have been provided by the research community to aid in MI research (Lindsey, Brunt, et al., 2021; Lindsey, de Castro Brás, et al., 2021).
FIGURE 4.
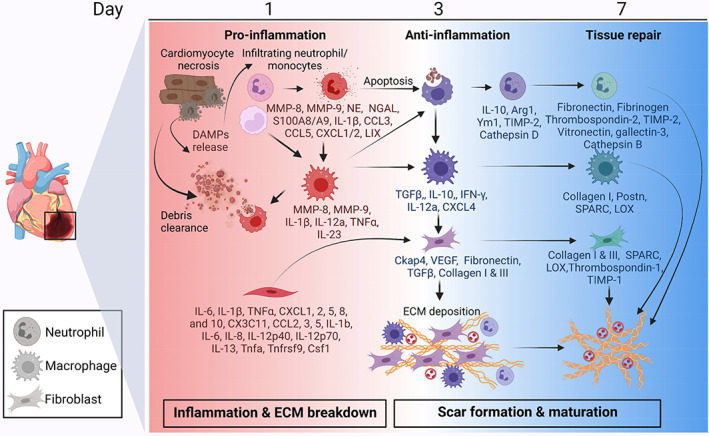
Cardiac wound healing after MI is a balance between the breakdown of existing ECM and the construction of new ECM to form an infarct scar. Cell phenotypes transition from early pro‐inflammatory to anti‐inflammatory and then reparative polarization phenotypes, with overlap across the time continuum. Pro‐inflammatory cells clear necrotic tissue, while anti‐inflammatory cells turn off the inflammatory response and reparative cells promote scar formation and maturation through secretion of ECM and ECM‐modifying proteins. Created with Biorender.com
Fine‐tuning the system, rather than total ablation or overexpression of components, will likely yield a better outcome. Strategies that temper inflammation or accelerate the transition to turn off inflammation may provide a means to modify the response. Using resolution‐promoting factors is one such arena of promising research (Kain et al., 2014; A. J. Mouton et al., 2018b; Tourki et al., 2020, Chalise et al., 2022d). While clinical treatment of acute MI has improved dramatically over the last 40 years, preventing the progression of heart failure remains a significant clinical issue (Box 2).
BOX 2.
Fine‐tuning the system, rather than total ablation or overexpression of components, will likely yield a better outcome in myocardial infarction resolution.
AUTHOR CONTRIBUTIONS
Upendra Chalise: Conceptualization (lead); project administration (lead); resources (supporting); supervision (lead); writing – original draft (lead); writing – review and editing (lead). Mediha Becirovic‐Agic: Conceptualization (supporting); funding acquisition (supporting); project administration (supporting); resources (supporting); supervision (supporting); writing – original draft (supporting); writing – review and editing (supporting). Merry L. Lindsey: Conceptualization (lead); funding acquisition (lead); project administration (lead); writing – original draft (supporting); Writing – review and editing (lead).
FUNDING INFORMATION
We acknowledge funding from the National Institutes of Health under award numbers GM115458 and HL137319; the Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development under award number 5I01BX000505; and the Swedish Society for Medical Research under award number P19‐0144.
CONFLICT OF INTEREST
All authors have read the journal authorship agreement and policy on disclosure of potential conflicts of interest and have nothing to disclose.
RELATED WIREs ARTICLES
ACKNOWLEDGMENTS
The content is solely the responsibility of the authors and does not necessarily represent the official views of any of the funding agencies. All authors have reviewed and approved the article.
Chalise, U. , Becirovic‐Agic, M. , & Lindsey, M. L. (2023). The cardiac wound healing response to myocardial infarction. WIREs Mechanisms of Disease, 15(1), e1584. 10.1002/wsbm.1584
Edited by: Mark Mercola, Editor
Funding information National Institutes of Health, Grant/Award Numbers: GM115458, HL137319; Svenska Sällskapet för Medicinsk Forskning, Grant/Award Number: P19‐0144; US Department of Veterans Affairs, Grant/Award Number: 5I01BX000505
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- Adrover, J. M. , del Fresno, C. , Crainiciuc, G. , Cuartero, M. I. , Casanova‐Acebes, M. , Weiss, L. A. , Huerga‐Encabo, H. , Silvestre‐Roig, C. , Rossaint, J. , Cossío, I. , Lechuga‐Vieco, A. V. , García‐Prieto, J. , Gómez‐Parrizas, M. , Quintana, J. A. , Ballesteros, I. , Martin‐Salamanca, S. , Aroca‐Crevillen, A. , Chong, S. Z. , Evrard, M. , … Hidalgo, A. (2019). A neutrophil timer coordinates immune defense and vascular protection. Immunity, 50(2), 390–402.e10. 10.1016/J.immuni.2019.01.002 [DOI] [PubMed] [Google Scholar]
- Altara, R. , Mallat, Z. , Booz, G. W. , & Zouein, F. A. (2016). The CXCL10/CXCR3 Axis and cardiac inflammation: Implications for immunotherapy to treat infectious and noninfectious diseases of the heart. Journal of Immunology Research, 2016, 4396368. 10.1155/2016/4396368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez‐Argote, S. , & O'Meara, C. C. (2021). The evolving roles of cardiac macrophages in homeostasis, regeneration, and repair. International Journal of Molecular Sciences, 22(15), 7923. 10.3390/ijms22157923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arruda‐Olson, A. M. , Reeder, G. S. , Bell, M. R. , Weston, S. A. , & Roger, V. L. (2009). Neutrophilia predicts death and heart failure after myocardial infarction: A community‐based study. Circulation. Cardiovascular Quality and Outcomes, 2(6), 656–662. 10.1161/CIRCOUTCOMES.108.831024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajpai, G. , Bredemeyer, A. , Li, W. , Zaitsev, K. , Koenig, A. L. , Lokshina, I. , Mohan, J. , Ivey, B. , Hsiao, H. M. , Weinheimer, C. , Kovacs, A. , Epelman, S. , Artyomov, M. , Kreisel, D. , & Lavine, K. J. (2019). Tissue resident CCR2− and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circulation Research, 124(2), 263–278. 10.1161/CIRCRESAHA.118.314028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudino, T. A. , Carver, W. , Giles, W. , & Borg, T. K. (2006). Cardiac fibroblasts: Friend or foe? American Journal of Physiology. Heart and Circulatory Physiology, 291(3), H1015–H1026. 10.1152/ajpheart.00023.2006 [DOI] [PubMed] [Google Scholar]
- Becirovic‐Agic, M. , Chalise, U. , Daseke, M. J., 2nd , Konfrst, S. , Salomon, J. D. , Mishra, P. K. , & Lindsey, M. L. (2021). Infarct in the heart: What's MMP‐9 got to do with it? Biomolecules, 11(4), 491. 10.3390/biom11040491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becirovic‐Agic, M. , Chalise, U. , Jung, M. , Rodriguez‐Paar, J. R. , Konfrst, S. R. , Flynn, E. R. , Salomon, J. D. , Hall, M. E. , & Lindsey, M. L. (2022). Faster skin wound healing predicts survival after myocardial infarction. American Journal of Physiology. Heart and Circulatory Physiology, 322(4), H537–H548. 10.1152/ajpheart.00612.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben‐Mordechai, T. , Holbova, R. , Landa‐Rouben, N. , Harel‐Adar, T. , Feinberg, M. S. , Abd Elrahman, I. , Blum, G. , Epstein, F. H. , Silman, Z. , Cohen, S. , & Leor, J. (2013). Macrophage subpopulations are essential for infarct repair with and without stem cell therapy. Journal of the American College of Cardiology, 62(20), 1890–1901. 10.1016/j.jacc.2013.07.057 [DOI] [PubMed] [Google Scholar]
- Cahill, T. J. , & Kharbanda, R. K. (2017). Heart failure after myocardial infarction in the era of primary percutaneous coronary intervention: Mechanisms, incidence and identification of patients at risk. World Journal of Cardiology, 9(5), 407. 10.4330/wjc.v9.i5.407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camelliti, P. , Borg, T. K. , & Kohl, P. (2005). Structural and functional characterisation of cardiac fibroblasts. Cardiovascular Research, 65(1), 40–51. 10.1016/j.cardiores.2004.08.020 [DOI] [PubMed] [Google Scholar]
- Castell, S. D. , Harman, M. F. , Morón, G. , Maletto, B. A. , & Pistoresi‐Palencia, M. C. (2019). Neutrophils which migrate to lymph nodes modulate CD4+ T cell response by a PD‐L1 dependent mechanism. Frontiers in Immunology, 10, 105. 10.3389/fimmu.2019.00105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalise, U. , Becirovic‐Agic, M. , Daseke, M. J., 2nd , Konfrst, S. R. , Rodriguez‐Paar, J. R. , Feng, D. , Salomon, J. D. , Anderson, D. R. , Cook, L. M. , & Lindsey, M. L. (2022). S100A9 is a functional effector of infarct wall thinning after myocardial infarction. American Journal of Physiology. Heart and Circulatory Physiology, 322(2), H145–H155. 10.1152/ajpheart.00475.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalise, U. , Becirovic‐Agic, M. , & Lindsey, M. L. (2022). Neutrophil crosstalk during cardiac wound healing after myocardial infarction. Current Opinion in Physiology, 24, 100485. 10.1016/j.cophys.2022.100485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalise, U. , Daseke, M. J. , Kalusche, W. J. , Konfrst, S. R. , Rodriguez‐Paar, J. R. , Flynn, E. R. , Cook, L. M. , Becirovic‐Agic, M. , & Lindsey, M. L. (2022). Macrophages secrete murinoglobulin‐1 and galectin‐3 to regulate neutrophil degranulation after myocardial infarction. Molecular Omics, 18(3), 185–196. 10.1039/d1mo00519g [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalise, U., Becirovic‐Agic, M., Konfrst, S. R., Rodriguez‐Paar, J. R., Cook, L. M., & Lindsey, M. L. (2022). MMP‐12 polarizes neutrophil signalome towards an apoptotic signature. Journal of Proteomics, 264, 104636. 10.1016/j.jprot.2022.104636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopher, M. J. , Liu, F. , Hilton, M. J. , Long, F. , & Link, D. C. (2009). Suppression of CXCL12 production by bone marrow osteoblasts is a common and critical pathway for cytokine‐induced mobilization. Blood, The Journal of the American Society of Hematology, 114(7), 1331–1339. 10.1182/blood-2008-10-184754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chwieralski, C. E. , Welte, T. , & Bühling, F. (2006). Cathepsin‐regulated apoptosis. Apoptosis, 11(2), 143–149. 10.1007/s10495-006-3486-y [DOI] [PubMed] [Google Scholar]
- Conus, S. , Pop, C. , Snipas, S. J. , Salvesen, G. S. , & Simon, H. U. (2012). Cathepsin D primes caspase‐8 activation by multiple intra‐chain proteolysis. The Journal of Biological Chemistry, 287(25), 21142–21151. 10.1074/jbc.M111.306399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cung, T.‐T. , Morel, O. , Cayla, G. , Rioufol, G. , Garcia‐Dorado, D. , Angoulvant, D. , Bonnefoy‐Cudraz, E. , Guérin, P. , Elbaz, M. , Delarche, N. , Coste, P. , Vanzetto, G. , Metge, M. , Aupetit, J. F. , Jouve, B. , Motreff, P. , Tron, C. , Labeque, J. N. , Steg, P. G. , … Ovize, M. (2015). Cyclosporine before PCI in patients with acute myocardial infarction. New England Journal of Medicine, 373(11), 1021–1031. 10.1056/JEJMoa1505489 [DOI] [PubMed] [Google Scholar]
- Daseke, M. J., 2nd , Chalise, U. , Becirovic‐Agic, M. , Salomon, J. D. , Cook, L. M. , Case, A. J. , & Lindsey, M. L. (2021). Neutrophil signaling during myocardial infarction wound repair. Cellular Signalling, 77, 109816. 10.1016/j.cellsig.2020.109816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daseke, M. J., 2nd , Tenkorang, M. A. A. , Chalise, U. , Konfrst, S. R. , & Lindsey, M. L. (2020). Cardiac fibroblast activation during myocardial infarction wound healing: Fibroblast polarization after MI. Matrix Biology, 91–92, 109–116. 10.1016/j.matbio.2020.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daseke, M. J., 2nd , Tenkorang‐Impraim, M. A. A. , Ma, Y. , Chalise, U. , Konfrst, S. R. , Garrett, M. R. , DeLeon‐Pennell, K. , & Lindsey, M. L. (2020). Exogenous IL‐4 shuts off pro‐inflammation in neutrophils while stimulating anti‐inflammation in macrophages to induce neutrophil phagocytosis following myocardial infarction. Journal of Molecular and Cellular Cardiology, 145, 112–121. 10.1016/j.yjmcc.2020.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daseke, M. J. , Valerio, F. M. , Kalusche, W. J. , Ma, Y. , DeLeon‐Pennell, K. Y. , & Lindsey, M. L. (2019). Neutrophil proteome shifts over the myocardial infarction time continuum. Basic Research in Cardiology, 114(5), 1–13. 10.1007/s00395-019-0746-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer, R. A. , de Keulenaer, G. , Bauersachs, J. , Brutsaert, D. , Cleland, J. G. , Diez, J. , Du, X. J. , Ford, P. , Heinzel, F. R. , Lipson, K. E. , McDonagh, T. , Lopez‐Andres, N. , Lunde, I. G. , Lyon, A. R. , Pollesello, P. , Prasad, S. K. , Tocchetti, C. G. , Mayr, M. , Sluijter, J. P. G. , … Heymans, S. (2019). Towards better definition, quantification and treatment of fibrosis in heart failure. A scientific roadmap by the Committee of Translational Research of the heart failure association (HFA) of the European Society of Cardiology. European Journal of Heart Failure, 21(3), 272–285. 10.1002/ejhf.1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Couto, G. , Jaghatspanyan, E. , DeBerge, M. , Liu, W. , Luther, K. , Wang, Y. , Tang, J. , Thorp, E. B. , & Marbán, E. (2019). Mechanism of enhanced MerTK‐dependent macrophage Efferocytosis by extracellular vesicles. Arteriosclerosis, Thrombosis, and Vascular Biology, 39(10), 2082–2096. 10.1161/atvbaha.119.313115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerge, M. , Zhang, S. , Glinton, K. , Grigoryeva, L. , Hussein, I. , Vorovich, E. , Ho, K. , Luo, X. , & Thorp, E. B. (2017). Efferocytosis and outside‐in signaling by cardiac phagocytes. Links to repair, cellular programming, and intercellular crosstalk in heart. Frontiers in Immunology, 8, 1428. 10.3389/fimmu.2017.01428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCoux, A. , Lindsey, M. L. , Villarreal, F. , Garcia, R. A. , & Schulz, R. (2014). Myocardial matrix metalloproteinase‐2: Inside out and upside down. Journal of Molecular and Cellular Cardiology, 77, 64–72. 10.1016/j.yjmcc.2014.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLeon‐Pennell, K. Y. , & Lindsey, M. L. (2019). Somewhere over the sex differences rainbow of myocardial infarction remodeling: Hormones, chromosomes, inflammasome, oh my. Expert Review of Proteomics, 16(11–12), 933–940. 10.1080/14789450.2019.1664293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLeon‐Pennell, K. Y. , Mouton, A. J. , Ero, O. K. , Ma, Y. , Padmanabhan Iyer, R. , Flynn, E. R. , Espinoza, I. , Musani, S. K. , Vasan, R. S. , Hall, M. E. , Fox, E. R. , & Lindsey, M. L. (2018). LXR/RXR signaling and neutrophil phenotype following myocardial infarction classify sex differences in remodeling. Basic Research in Cardiology, 113(5), 40. 10.1007/s00395-018-0699-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLeon‐Pennell, K. Y. , Tian, Y. , Zhang, B. , Cates, C. A. , Iyer, R. P. , Cannon, P. , Shah, P. , Aiyetan, P. , Halade, G. V. , Ma, Y. , Flynn, E. , Zhang, Z. , Jin, Y. F. , Zhang, H. , & Lindsey, M. L. (2016). CD36 is a matrix Metalloproteinase‐9 substrate that stimulates neutrophil apoptosis and removal during cardiac remodeling. Circulation. Cardiovascular Genetics, 9(1), 14–25. 10.1161/CIRCGENETICS.115.001249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deniset, J. F. , & Kubes, P. (2016). Recent advances in understanding neutrophils. F1000Research, 5, 2912. 10.12688/f1000research.9691.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewald, O. , Ren, G. , Duerr, G. D. , Zoerlein, M. , Klemm, C. , Gersch, C. , Tincey, S. , Michael, L. H. , Entman, M. L. , & Frangogiannis, N. G. (2004). Of mice and dogs: Species‐specific differences in the inflammatory response following myocardial infarction. The American Journal of Pathology, 164(2), 665–677. 10.1016/s0002-9440(10)63154-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewald, O. , Zymek, P. , Winkelmann, K. , Koerting, A. , Ren, G. , Abou‐Khamis, T. , Michael, L. H. , Rollins, B. J. , Entman, M. L. , & Frangogiannis, N. G. (2005). CCL2/monocyte chemoattractant protein‐1 regulates inflammatory responses critical to healing myocardial infarcts. Circulation Research, 96(8), 881–889. 10.1161/01.RES.0000163017.13772.3a [DOI] [PubMed] [Google Scholar]
- Dobaczewski, M. , Bujak, M. , Li, N. , Gonzalez‐Quesada, C. , Mendoza, L. H. , Wang, X. F. , & Frangogiannis, N. G. (2010). Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circulation Research, 107(3), 418–428. 10.1161/circresaha.109.216101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobaczewski, M. , Bujak, M. , Zymek, P. , Ren, G. , Entman, M. L. , & Frangogiannis, N. G. (2006). Extracellular matrix remodeling in canine and mouse myocardial infarcts. Cell and Tissue Research, 324(3), 475–488. 10.1007/s00441-005-0144-6 [DOI] [PubMed] [Google Scholar]
- Dutta, P. , Hoyer, F. F. , Grigoryeva, L. S. , Sager, H. B. , Leuschner, F. , Courties, G. , Borodovsky, A. , Novobrantseva, T. , Ruda, V. M. , Fitzgerald, K. , Iwamoto, Y. , Wojtkiewicz, G. , Sun, Y. , da Silva, N. , Libby, P. , Anderson, D. G. , Swirski, F. K. , Weissleder, R. , & Nahrendorf, M. (2015). Macrophages retain hematopoietic stem cells in the spleen via VCAM‐1. The Journal of Experimental Medicine, 212(4), 497–512. 10.1084/jem.20141642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faxon, D. P. , Gibbons, R. J. , Chronos, N. A. , Gurbel, P. A. , Sheehan, F. , & Investigators, H.‐M. (2002). The effect of blockade of the CD11/CD18 integrin receptor on infarct size in patients with acute myocardial infarction treated with direct angioplasty: The results of the HALT‐MI study. Journal of the American College of Cardiology, 40(7), 1199–1204. https://doi.org/10.1016/s0735‐1097(02)02136‐8 [DOI] [PubMed] [Google Scholar]
- Frangogiannis, N. G. (2014). The inflammatory response in myocardial injury, repair, and remodelling. Nature Reviews. Cardiology, 11(5), 255–265. 10.1038/nrcardio.2014.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangogiannis, N. G. (2015). Interleukin‐1 in cardiac injury, repair, and remodeling: Pathophysiologic and translational concepts. Discoveries (Craiova), 3(1), e41. 10.15190/d.2015.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantz, S. , Hofmann, U. , Fraccarollo, D. , Schäfer, A. , Kranepuhl, S. , Hagedorn, I. , Nieswandt, B. , Nahrendorf, M. , Wagner, H. , Bayer, B. , Pachel, C. , Schön, M. P. , Kneitz, S. , Bobinger, T. , Weidemann, F. , Ertl, G. , & Bauersachs, J. (2013). Monocytes/macrophages prevent healing defects and left ventricular thrombus formation after myocardial infarction. The FASEB Journal, 27(3), 871–881. 10.1096/fj.12-214049 [DOI] [PubMed] [Google Scholar]
- Frantz, S. , & Nahrendorf, M. (2014). Cardiac macrophages and their role in ischaemic heart disease. Cardiovascular Research, 102(2), 240–248. 10.1093/cvr/cvu025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed, D. H. , Chilton, L. , Li, Y. , Dangerfield, A. L. , Raizman, J. E. , Rattan, S. G. , Visen, N. , Hryshko, L. V. , & Dixon, I. M. (2011). Role of myosin light chain kinase in cardiotrophin‐1‐induced cardiac myofibroblast cell migration. American Journal of Physiology. Heart and Circulatory Physiology, 301(2), H514–H522. 10.1152/ajpheart.01041.2010 [DOI] [PubMed] [Google Scholar]
- Gaudesius, G. , Miragoli, M. , Thomas, S. P. , & Rohr, S. (2003). Coupling of cardiac electrical activity over extended distances by fibroblasts of cardiac origin. Circulation Research, 93(5), 421–428. 10.1161/01.Res.0000089258.40661.0c [DOI] [PubMed] [Google Scholar]
- Gebhardt, C. , Németh, J. , Angel, P. , & Hess, J. (2006). S100A8 and S100A9 in inflammation and cancer. Biochemical Pharmacology, 72(11), 1622–1631. 10.1016/j.bcp.2006.05.017 [DOI] [PubMed] [Google Scholar]
- Giugliano, G. R. , Giugliano, R. P. , Gibson, C. M. , & Kuntz, R. E. (2003). Meta‐analysis of corticosteroid treatment in acute myocardial infarction. The American Journal of Cardiology, 91(9), 1055–1059. 10.1016/s0002-9149(03)00148-6 [DOI] [PubMed] [Google Scholar]
- Gopalkrishna, S. , Johnson, J. , Jaggers, R. M. , Dahdah, A. , Murphy, A. J. , Hanssen, N. M. , & Nagareddy, P. R. (2021). Neutrophils in cardiovascular disease: Warmongers, peacemakers or both? Cardiovascular Research, 118(12), 2596–2609. 10.1093/cvr/cvab302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourdie, R. G. , Dimmeler, S. , & Kohl, P. (2016). Novel therapeutic strategies targeting fibroblasts and fibrosis in heart disease. Nature Reviews. Drug Discovery, 15(9), 620–638. 10.1038/nrd.2016.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowda, S. B , Gowda, D. , Kain, V. , Chiba, H. , Hui, S. P. , Chalfant, C. E. , Parcha, V. , Arora, P. , & Halade, G. V. (2021). Sphingosine‐1‐phosphate interactions in the spleen and heart reflect extent of cardiac repair in mice and failing human hearts. American Journal of Physiology. Heart and Circulatory Physiology, 321(3), H599–H611. 10.1152/ajpheart.00314.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray, A. J. , Bishop, J. E. , Reeves, J. T. , & Laurent, G. J. (1993). A alpha and B beta chains of fibrinogen stimulate proliferation of human fibroblasts. Journal of Cell Science, 104(Pt 2), 409–413. 10.1242/jcs.104.2.409 [DOI] [PubMed] [Google Scholar]
- Guo, Y. , Flaherty, M. P. , Wu, W. J. , Tan, W. , Zhu, X. , Li, Q. , & Bolli, R. (2012). Genetic background, gender, age, body temperature, and arterial blood pH have a major impact on myocardial infarct size in the mouse and need to be carefully measured and/or taken into account: Results of a comprehensive analysis of determinants of infarct size in 1,074 mice. Basic Research in Cardiology, 107(5), 288. 10.1007/s00395-012-0288-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafezi‐Moghadam, A. , Thomas, K. L. , Prorock, A. J. , Huo, Y. , & Ley, K. (2001). L‐selectin shedding regulates leukocyte recruitment. The Journal of Experimental Medicine, 193(7), 863–872. 10.1084/jem.193.7.863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halade, G. V. , Kain, V. , & Ingle, K. A. (2018). Heart functional and structural compendium of cardiosplenic and cardiorenal networks in acute and chronic heart failure pathology. American Journal of Physiology. Heart and Circulatory Physiology, 314(2), H255–h267. 10.1152/ajpheart.00528.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton, H. R. , & Chtanova, T. (2016). The lymph node neutrophil. Proceedings of the Seminars in Immunology . 10.1016/j.smim.2016.03.008 [DOI] [PubMed]
- Hanna, A. , & Frangogiannis, N. G. (2019). The role of the TGF‐β superfamily in myocardial infarction. Frontiers in Cardiovascular Medicine, 6, 140. 10.3389/fcvm.2019.00140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna, A. , Shinde, A. V. , & Frangogiannis, N. G. (2020). Validation of diagnostic criteria and histopathological characterization of cardiac rupture in the mouse model of nonreperfused myocardial infarction. American Journal of Physiology. Heart and Circulatory Physiology, 319(5), H948–H964. 10.1152/ajpheart.00318.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess, A. , Derlin, T. , Koenig, T. , Diekmann, J. , Wittneben, A. , Wang, Y. , Wester, H. J. , Ross, T. L. , Wollert, K. C. , Bauersachs, J. , Bengel, F. M. , & Thackeray, J. T. (2020). Molecular imaging‐guided repair after acute myocardial infarction by targeting the chemokine receptor CXCR4. European Heart Journal, 41(37), 3564–3575. 10.1093/eurheartj/ehaa598 [DOI] [PubMed] [Google Scholar]
- Ho, J. E. , Lyass, A. , Lee, D. S. , Vasan, R. S. , Kannel, W. B. , Larson, M. G. , & Levy, D. (2013). Predictors of new‐onset heart failure: Differences in preserved versus reduced ejection fraction. Circulation: Heart Failure, 6(2), 279–286. 10.1161/CIRCHEARTFAILURE.112.972828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofbauer, T. M. , Mangold, A. , Scherz, T. , Seidl, V. , Panzenböck, A. , Ondracek, A. S. , Müller, J. , Schneider, M. , Binder, T. , Hell, L. , & Hell, L. (2019). Neutrophil extracellular traps and fibrocytes in ST‐segment elevation myocardial infarction. Basic Research in Cardiology, 114(5), 1–15. 10.1007/s00395-019-0740-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann, U. , Beyersdorf, N. , Weirather, J. , Podolskaya, A. , Bauersachs, J. , Ertl, G. , Kerkau, T. , & Frantz, S. (2012). Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation, 125(13), 1652–1663. 10.1161/CIRCULATIONAHA.111.044164 [DOI] [PubMed] [Google Scholar]
- Hofmann, U. , & Frantz, S. (2015). Role of lymphocytes in myocardial injury, healing, and remodeling after myocardial infarction. Circulation Research, 116(2), 354–367. 10.1161/CIRCRESAHA.116.304072 [DOI] [PubMed] [Google Scholar]
- Horckmans, M. , Ring, L. , Duchene, J. , Santovito, D. , Schloss, M. J. , Drechsler, M. , Weber, C. , Soehnlein, O. , & Steffens, S. (2017). Neutrophils orchestrate post‐myocardial infarction healing by polarizing macrophages towards a reparative phenotype. European Heart Journal, 38(3), 187–197. 10.1093/eurheartj/ehw002 [DOI] [PubMed] [Google Scholar]
- Huang, S. , & Frangogiannis, N. G. (2018). Anti‐inflammatory therapies in myocardial infarction: Failures, hopes and challenges. British Journal of Pharmacology, 175(9), 1377–1400. 10.1111/bph.14155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulsmans, M. , Clauss, S. , Xiao, L. , Aguirre, A. D. , King, K. R. , Hanley, A. , Hucker, W. J. , Wülfers, E. M. , Seemann, G. , Courties, G. , Iwamoto, Y. , Sun, Y. , Savol, A. J. , Sager, H. B. , Lavine, K. J. , Fishbein, G. A. , Capen, D. E. , Da Silva, N. , Miquerol, L. , … Courties, G. (2017). Macrophages facilitate electrical conduction in the heart. Cell, 169(3), 510–522.e520. 10.1016/j.cell.2017.03.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humeres, C. , & Frangogiannis, N. G. (2019). Fibroblasts in the infarcted, remodeling, and failing heart. JACC: Basic to Translational Science, 4(3), 449–467. 10.1016/j.jacbts.2019.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibanez, B. , James, S. , Agewall, S. , Antunes, M. J. , Bucciarelli‐Ducci, C. , Bueno, H. , Caforio, A. L. P. , Crea, F. , Goudevenos, J. A. , Halvorsen, S. , Hindricks, G. , Kastrati, A. , Lenzen, M. J. , Prescott, E. , Roffi, M. , Valgimigli, M. , Varenhorst, C. , Vranckx, P. , Widimský, P. , & ESC Scientific Document Group . (2018). 2017 ESC guidelines for the management of acute myocardial infarction in patients presenting with ST‐segment elevation: The task force for the management of acute myocardial infarction in patients presenting with ST‐segment elevation of the European Society of Cardiology (ESC). European Heart Journal, 39(2), 119–177. 10.1093/eurheartj/ehx393 [DOI] [PubMed] [Google Scholar]
- Ilatovskaya, D. V. , Pitts, C. , Clayton, J. , Domondon, M. , Troncoso, M. , Pippin, S. , & DeLeon‐Pennell, K. Y. (2019). CD8+ T‐cells negatively regulate inflammation post‐myocardial infarction. American Journal of Physiology. Heart and Circulatory Physiology, 317(3), H581–H596. 10.1152/ajpheart.00112.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer, R. P. , Patterson, N. L. , Zouein, F. A. , Ma, Y. , Dive, V. , de Castro Brás, L. E. , & Lindsey, M. L. (2015). Early matrix metalloproteinase‐12 inhibition worsens post‐myocardial infarction cardiac dysfunction by delaying inflammation resolution. International Journal of Cardiology, 185, 198–208. 10.1016/j.ijcard.2015.03.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, F. , Zhai, Q. , Qiu, L. , Meng, H. , Zou, D. , Wang, Y. , Li, Q. , Yu, Z. , Han, J. , Li, Q. , & Zhou, B. (2008). Degradation of BM SDF‐1 by MMP‐9: The role in G‐CSF‐induced hematopoietic stem/progenitor cell mobilization. Bone Marrow Transplantation, 42(9), 581–588. 10.1038/bmt.2008.222 [DOI] [PubMed] [Google Scholar]
- Jolly, S. R. , Kane, W. J. , Hook, B. G. , Abrams, G. D. , Kunkel, S. L. , & Lucchesi, B. R. (1986). Reduction of myocardial infarct size by neutrophil depletion: Effect of duration of occlusion. American Heart Journal, 112(4), 682–690. 10.1016/j.ijcard.2015.03.054 [DOI] [PubMed] [Google Scholar]
- Jung, M. , Ma, Y. , Iyer, R. P. , DeLeon‐Pennell, K. Y. , Yabluchanskiy, A. , Garrett, M. R. , & Lindsey, M. L. (2017). IL‐10 improves cardiac remodeling after myocardial infarction by stimulating M2 macrophage polarization and fibroblast activation. Basic Research in Cardiology, 112(3), 33. 10.1007/s00395-017-0622-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kain, V. , & Halade, G. V. (2020). Role of neutrophils in ischemic heart failure. Pharmacology & Therapeutics, 205, 107424. 10.1016/j.pharmthera.2019.107424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kain, V. , Prabhu, S. D. , & Halade, G. V. (2014). Inflammation revisited: Inflammation versus resolution of inflammation following myocardial infarction. Basic Research in Cardiology, 109(6), 444. 10.1007/s00395-014-0444-7 [DOI] [PubMed] [Google Scholar]
- Kalafati, L. , Kourtzelis, I. , Schulte‐Schrepping, J. , Li, X. , Hatzioannou, A. , Grinenko, T. , Hagag, E. , Sinha, A. , Has, C. , Dietz, S. , de Jesus Domingues, A. M. , Nati, M. , Sormendi, S. , Neuwirth, A. , Chatzigeorgiou, A. , Ziogas, A. , Lesche, M. , Dahl, A. , Henry, I. , … Chavakis, T. (2020). Innate immune training of granulopoiesis promotes anti‐tumor activity. Cell, 183(3), 771–785.e12. 10.1016/j.cell.2020.09.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettritz, R. , Xu, Y.‐X. , Kerren, T. , Quass, P. , Klein, J. , Luft, F. C. , & Haller, H. (1999). Extracellular matrix regulates apoptosis in human neutrophils. Kidney International, 55(2), 562–571. 10.1046/j.1523-1755.1999.00280.x [DOI] [PubMed] [Google Scholar]
- Klaourakis, K. , Vieira, J. M. , & Riley, P. R. (2021). The evolving cardiac lymphatic vasculature in development, repair and regeneration. Nature Reviews Cardiology, 18(5), 368–379. 10.1038/s41569-020-00489-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, K. P. , Nicholls, A. J. , & Wong, C. H. (2018). Partners in crime: Neutrophils and monocytes/macrophages in inflammation and disease. Cell and Tissue Research, 371(3), 551–565. 10.1007/s00441-017-2753-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakschevitz, F. S. , Visser, M. B. , Sun, C. , & Glogauer, M. (2015). Neutrophil transcriptional profile changes during transit from bone marrow to sites of inflammation. Cellular & Molecular Immunology, 12(1), 53–65. 10.1038/cmi.2014.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z. , Nguyen, T. T. , & Valaperti, A. (2021). Human cardiac fibroblasts produce pro‐inflammatory cytokines upon TLRs and RLRs stimulation. Molecular and Cellular Biochemistry, 476(9), 3241–3252. 10.1007/s11010-021-04157-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey, M. , Wedin, K. , Brown, M. D. , Keller, C. , Evans, A. J. , Smolen, J. , Burns, A. R. , Rossen, R. D. , Michael, L. , & Entman, M. (2001). Matrix‐dependent mechanism of neutrophil‐mediated release and activation of matrix metalloproteinase 9 in myocardial ischemia/reperfusion. Circulation, 103(17), 2181–2187. 10.1161/01.cir103.17.2181 [DOI] [PubMed] [Google Scholar]
- Lindsey, M. L. , de Castro Brás, L. E. , DeLeon‐Pennell, K. Y. , Frangogiannis, N. G. , Halade, G. V. , O'Meara, C. C. , Spinale, F. G. , Kassiri, Z. , Kirk, J. A. , Kleinbongard, P. , Ripplinger, C. M. , & Brunt, K. R. (2021). Reperfused vs. nonreperfused myocardial infarction: When to use which model. American Journal of Physiology. Heart and Circulatory Physiology, 321(1), H208–H213. 10.1152/ajpheart.00234.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey, M. L. , Brunt, K. R. , Kirk, J. A. , Kleinbongard, P. , Calvert, J. W. , de Castro Brás, L. E. , DeLeon‐Pennell, K. , del Re, D. , Frangogiannis, N. G. , Frantz, S. , Gumina, R. J. , Halade, G. V. , Jones, S. P. , Ritchie, R. H. , Spinale, F. G. , Thorp, E. B. , Ripplinger, C. M. , & Kassiri, Z. (2021). Guidelines for in vivo mouse models of myocardial infarction. American Journal of Physiology. Heart and Circulatory Physiology, 321(6), H1056–H1073. 10.1152/ajpheart.00459.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X. , Zhang, J. , Zeigler, A. C. , Nelson, A. R. , Lindsey, M. L. , & Saucerman, J. J. (2021). Network analysis reveals a distinct Axis of macrophage activation in response to conflicting inflammatory cues. Journal of Immunology, 206(4), 883–891. 10.4049/jimmunol.1901444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, Y. (2021). Role of neutrophils in cardiac injury and repair following myocardial infarction. Cell, 10(7), 1676. 10.3390/cells10071676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, Y. , Iyer, R. P. , Jung, M. , Czubryt, M. P. , & Lindsey, M. L. (2017). Cardiac fibroblast activation post‐myocardial infarction: Current knowledge gaps. Trends in Pharmacological Sciences, 38(5), 448–458. 10.1016/j.tips.2017.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, Y. , Yabluchanskiy, A. , Iyer, R. P. , Cannon, P. L. , Flynn, E. R. , Jung, M. , Henry, J. , Cates, C. A. , Deleon‐Pennell, K. Y. , & Lindsey, M. L. (2016). Temporal neutrophil polarization following myocardial infarction. Cardiovascular Research, 110(1), 51–61. 10.1093/cvr/cvw024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, Y. , Yang, X. , Villalba, N. , Chatterjee, V. , Reynolds, A. , Spence, S. , Wu, M. H. , & Yuan, S. Y. (2022). Circulating lymphocyte trafficking to the bone marrow contributes to lymphopenia in myocardial infarction. American Journal of Physiology. Heart and Circulatory Physiology, 322(4), H622–H635. 10.1152/ajpheart.00003.2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madias, J. E. , & Hood, W. B., Jr. (1982). Effects of methylprednisolone on the ischemic damage in patients with acute myocardial infarction. Circulation, 65(6), 1106–1113. 10.1161/01.cir.65.6.1106 [DOI] [PubMed] [Google Scholar]
- Maekawa, Y. , Anzai, T. , Yoshikawa, T. , Asakura, Y. , Takahashi, T. , Ishikawa, S. , Mitamura, H. , & Ogawa, S. (2002). Prognostic significance of peripheral monocytosis after reperfused acute myocardial infarction: A possible role for left ventricular remodeling. Journal of the American College of Cardiology, 39(2), 241–246. 10.1016/s0735-1097(01)01721-1 [DOI] [PubMed] [Google Scholar]
- Maekawa, Y. , Anzai, T. , Yoshikawa, T. , Sugano, Y. , Mahara, K. , Kohno, T. , Takahashi, T. , & Ogawa, S. (2004). Effect of granulocyte‐macrophage colony‐stimulating factor inducer on left ventricular remodeling after acute myocardial infarction. Journal of the American College of Cardiology, 44(7), 1510–1520. 10.1016/j.jacc.2004.05.083 [DOI] [PubMed] [Google Scholar]
- Marinković, G. , Grauen Larsen, H. , Yndigegn, T. , Szabo, I. A. , Mares, R. G. , de Camp, L. , Weiland, M. , Tomas, L. , Goncalves, I. , Nilsson, J. , Jovinge, S. , & Schiopu, A. (2019). Inhibition of pro‐inflammatory myeloid cell responses by short‐term S100A9 blockade improves cardiac function after myocardial infarction. European Heart Journal, 40(32), 2713–2723. 10.1093/eurheartj/ehz461 [DOI] [PubMed] [Google Scholar]
- Marinković, G. , Koenis, D. S. , de Camp, L. , Jablonowski, R. , Graber, N. , de Waard, V. , de Vries, C. J. , Goncalves, I. , Nilsson, J. , Jovinge, S. , & Schiopu, A. (2020). S100A9 links inflammation and repair in myocardial infarction. Circulation Research, 127(5), 664–676. 10.1161/CIRCRESAHA.120.315865 [DOI] [PubMed] [Google Scholar]
- McCutcheon, J. C. , Hart, S. P. , Canning, M. , Ross, K. , Humphries, M. J. , & Dransfield, I. (1998). Regulation of macrophage phagocytosis of apoptotic neutrophils by adhesion to fibronectin. Journal of Leukocyte Biology, 64(5), 600–607. 10.1002/jlb.64.5.600 [DOI] [PubMed] [Google Scholar]
- Members, C. , Ryan, T. J. , Anderson, J. L. , Antman, E. M. , Braniff, B. A. , Brooks, N. H. , Califf, R. M. , Hillis, L. D. , Hiratzka, L. F. , Rapaport, E. , Riegel, B. J. , Russell, R. O. , Smith, E. E., Jr. , & Rapaport, E. (1996). ACC/AHA guidelines for the management of patients with acute myocardial infarction: A report of the American College of Cardiology/American Heart Association task force on practice guidelines (Committee on Management of Acute Myocardial Infarction). Journal of the American College of Cardiology, 28(5), 1328–1419. 10.1016/s0735-1097(96)00392-0 [DOI] [PubMed] [Google Scholar]
- Miragoli, M. , Salvarani, N. , & Rohr, S. (2007). Myofibroblasts induce ectopic activity in cardiac tissue. Circulation Research, 101(8), 755–758. 10.1161/circresaha.107.160549 [DOI] [PubMed] [Google Scholar]
- Mitchell, M. D. , Laird, R. E. , Brown, R. D. , & Long, C. S. (2007). IL‐1beta stimulates rat cardiac fibroblast migration via MAP kinase pathways. American Journal of Physiology. Heart and Circulatory Physiology, 292(2), H1139–H1147. 10.1152/ajpheart.00881.2005 [DOI] [PubMed] [Google Scholar]
- Montecucco, F. , Braunersreuther, V. , Lenglet, S. , Delattre, B. M. , Pelli, G. , Buatois, V. , Guilhot, F. , Galan, K. , Vuilleumier, N. , Ferlin, W. , Fischer, N. , Vallée, J. P. , Kosco‐Vilbois, M. , & Mach, F. (2012). CC chemokine CCL5 plays a central role impacting infarct size and post‐infarction heart failure in mice. European Heart Journal, 33(15), 1964–1974. 10.1093/eurheartj/ehr127 [DOI] [PubMed] [Google Scholar]
- Morimoto, H. , Takahashi, M. , Shiba, Y. , Izawa, A. , Ise, H. , Hongo, M. , Hatake, K. , Motoyoshi, K. , & Ikeda, U. (2007). Bone marrow‐derived CXCR4+ cells mobilized by macrophage colony‐stimulating factor participate in the reduction of infarct area and improvement of cardiac remodeling after myocardial infarction in mice. The American Journal of Pathology, 171(3), 755–766. 10.2353/ajpath.2007.061276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouton, A. J. , DeLeon‐Pennell, K. Y. , Rivera Gonzalez, O. J. , Flynn, E. R. , Freeman, T. C. , Saucerman, J. J. , Garrett, M. R. , Ma, Y. , Harmancey, R. , & Lindsey, M. L. (2018). Mapping macrophage polarization over the myocardial infarction time continuum. Basic Research in Cardiology, 113(4), 26. 10.1007/s00395-018-0686-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouton, A. J. , Ma, Y. , Rivera Gonzalez, O. J. , Daseke, M. J., 2nd , Flynn, E. R. , Freeman, T. C. , Garrett, M. R. , DeLeon‐Pennell, K. , & Lindsey, M. L. (2019). Fibroblast polarization over the myocardial infarction time continuum shifts roles from inflammation to angiogenesis. Basic Research in Cardiology, 114(2), 6. 10.1007/s00395-019-0715-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouton, A. J. , Rivera Gonzalez, O. J. , Kaminski, A. R. , Moore, E. T. , & Lindsey, M. L. (2018). Matrix metalloproteinase‐12 as an endogenous resolution promoting factor following myocardial infarction. Pharmacological Research, 137, 252–258. 10.1016/j.phrs.2018.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouton, A. J. , Rivera, O. J. , & Lindsey, M. L. (2018). Myocardial infarction remodeling that progresses to heart failure: A signaling misunderstanding. American Journal of Physiology ‐ Heart and Circulatory Physiology, 315, H71–H79. 10.1152/ajpheart.00131.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mylonas, K. J. , Turner, N. A. , Bageghni, S. A. , Kenyon, C. J. , White, C. I. , McGregor, K. , Kimmitt, R. A. , Sulston, R. , Kelly, V. , Walker, B. R. , Porter, K. E. , Chapman, K. E. , & Gray, G. A. (2017). 11β‐HSD1 suppresses cardiac fibroblast CXCL2, CXCL5 and neutrophil recruitment to the heart post MI. The Journal of Endocrinology, 233(3), 315–327. 10.1530/joe-16-0501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niccoli, G. , Kharbanda, R. K. , Crea, F. , & Banning, A. P. (2010). No‐reflow: Again prevention is better than treatment. European Heart Journal, 31(20), 2449–2455. 10.1093/eurheartj/ehq299 [DOI] [PubMed] [Google Scholar]
- Nielsen, S. H. , Mouton, A. J. , DeLeon‐Pennell, K. Y. , Genovese, F. , Karsdal, M. , & Lindsey, M. L. (2019). Understanding cardiac extracellular matrix remodeling to develop biomarkers of myocardial infarction outcomes. Matrix Biology, 75‐76, 43–57. 10.1016/j.matbio.2017.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panizzi, P. , Swirski, F. K. , Figueiredo, J.‐L. , Waterman, P. , Sosnovik, D. E. , Aikawa, E. , Libby, P. , Pittet, M. , Weissleder, R. , & Nahrendorf, M. (2010). Impaired infarct healing in atherosclerotic mice with ly‐6ChiMonocytosis. Journal of the American College of Cardiology, 55(15), 1629–1638. 10.1016/j.jacc.2009.08.089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappritz, K. , Savvatis, K. , Koschel, A. , Miteva, K. , Tschöpe, C. , & Van Linthout, S. (2018). Cardiac (myo)fibroblasts modulate the migration of monocyte subsets. Scientific Reports, 8(1), 5575. 10.1038/s41598-018-23881-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peet, C. , Ivetic, A. , Bromage, D. I. , & Shah, A. M. (2020). Cardiac monocytes and macrophages after myocardial infarction. Cardiovascular Research, 116(6), 1101–1112. 10.1093/cvr/cvz336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer, J. M. , Fischer, T. A. , & Pfeffer, M. A. (1995). Angiotensin‐converting enzyme inhibition and ventricular remodeling after myocardial infarction. Annual Review of Physiology, 57(1), 805–826. 10.1146/annurev.ph.57.030195.004105 [DOI] [PubMed] [Google Scholar]
- Pfeffer, J. M. , & Pfeffer, M. A. (1988). Angiotensin converting enzyme inhibition and ventricular remodeling in heart failure. The American Journal of Medicine, 84(3), 37–44. 10.1016/0002-9343(88)90203-3 [DOI] [PubMed] [Google Scholar]
- Pinto, A. R. , Paolicelli, R. , Salimova, E. , Gospocic, J. , Slonimsky, E. , Bilbao‐Cortes, D. , Godwin, J. W. , & Rosenthal, N. A. (2012). An abundant tissue macrophage population in the adult murine heart with a distinct alternatively‐activated macrophage profile. PLoS One, 7(5), e36814. 10.1371/journal.pone.0036814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porsch, F. , Mallat, Z. , & Binder, C. J. (2021). Humoral immunity in atherosclerosis and myocardial infarction: From B cells to antibodies. Cardiovascular Research, 117(13), 2544–2562. 10.1093/cvr/cvab285 [DOI] [PubMed] [Google Scholar]
- Prabhu, S. D. , & Frangogiannis, N. G. (2016). The biological basis for cardiac repair after myocardial infarction: From inflammation to fibrosis. Circulation Research, 119(1), 91–112. 10.1161/circresaha.116.303577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince, L. R. , Prosseda, S. D. , Higgins, K. , Carlring, J. , Prestwich, E. C. , Ogryzko, N. V. , Rahman, A. , Basran, A. , Falciani, F. , Taylor, P. , Renshaw, S. A. , Whyte, M. , & Taylor, P. (2017). NR4A orphan nuclear receptor family members, NR4A2 and NR4A3, regulate neutrophil number and survival. Blood, The Journal of the American Society of Hematology, 130(8), 1014–1025. 10.1182/blood-2017-03-770164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullen, A. B. , Kain, V. , Serhan, C. N. , & Halade, G. V. (2020). Molecular and cellular differences in cardiac repair of male and female mice. Journal of the American Heart Association, 9(8), e015672. 10.1161/jaha.119.015672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed, G. W. , Rossi, J. E. , & Cannon, C. P. (2017). Acute myocardial infarction. Lancet, 389(10065), 197–210. 10.1016/S0140-6736(16)30677-8 [DOI] [PubMed] [Google Scholar]
- Rezkalla, S. H. , & Kloner, R. A. (2002). No‐reflow phenomenon. Circulation, 105(5), 656–662. 10.1161/hc0502.102867 [DOI] [PubMed] [Google Scholar]
- Ridker, P. M. , Everett, B. M. , Thuren, T. , MacFadyen, J. G. , Chang, W. H. , Ballantyne, C. , Fonseca, F. , Nicolau, J. , Koenig, W. , Anker, S. D. , Kastelein, J. J. P. , Cornel, J. H. , Pais, P. , Pella, D. , Genest, J. , Cifkova, R. , Lorenzatti, A. , Forster, T. , Kobalava, Z. , … CANTOS Trial Group . (2017). Antiinflammatory therapy with canakinumab for atherosclerotic disease. New England Journal of Medicine, 377(12), 1119–1131. 10.1056/NEJMoa1707914 [DOI] [PubMed] [Google Scholar]
- Roe, M. T. , Parsons, L. S. , Pollack, C. V. , Canto, J. G. , Barron, H. V. , Every, N. R. , Rogers, W. J. , Peterson, E. D. , & National Registry of Myocardial Infarction Investigators . (2005). Quality of care by classification of myocardial infarction: Treatment patterns for ST‐segment elevation vs non‐ST‐segment elevation myocardial infarction. Archives of Internal Medicine, 165(14), 1630–1636. 10.1001/archarchnite.165.14.1630 [DOI] [PubMed] [Google Scholar]
- Roger, V. L. (2013). Epidemiology of heart failure. Circulation Research, 113(6), 646–659. 10.1161/CIRCRESAHA.113.300268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosales, C. (2018). Neutrophil: A cell with many roles in inflammation or several cell types? Frontiers in Physiology, 9, 113. 10.3389/fphys.2018.00113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakawa, K. , Endo, J. , Kataoka, M. , Katsumata, Y. , Yoshida, N. , Yamamoto, T. , Isobe, S. , Moriyama, H. , Goto, S. , Kitakata, H. , Hiraide, T. , Fukuda, K. , & Sano, M. (2018). IL (interleukin)‐10‐STAT3‐galectin‐3 axis is essential for osteopontin‐producing reparative macrophage polarization after myocardial infarction. Circulation, 138(18), 2021–2035. 10.1161/CIRCULATIONAHA.118.035047 [DOI] [PubMed] [Google Scholar]
- Sholter, D. E. , & Armstrong, P. W. (2000). Adverse effects of corticosteroids on the cardiovascular system. The Canadian Journal of Cardiology, 16(4), 505–511. 10.1186/1710-1492-9-30 [DOI] [PubMed] [Google Scholar]
- Swirski, F. K. , & Nahrendorf, M. (2013). Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science, 339(6116), 161–166. 10.1126/science.1230719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenkorang, M. A. , Chalise, U. , Daseke, I. , Michael, J. , Konfrst, S. R. , & Lindsey, M. L. (2019). Understanding the mechanisms that determine extracellular matrix remodeling in the infarcted myocardium. Biochemical Society Transactions, 47(6), 1679–1687. 10.1042/BST20190113 [DOI] [PubMed] [Google Scholar]
- Teringova, E. , & Tousek, P. (2017). Apoptosis in ischemic heart disease. Journal of Translational Medicine, 15(1), 87. 10.1186/s12967-017-1191-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thygesen, K. , Alpert, J. S. , White, H. D. , & Joint ESC/ACCF/AHA/WHF Task Force for the Redefinition of Myocardial Infarction . (2007). Universal definition of myocardial infarction. Circulation, 116(22), 2634–2653. 10.1161/CIRCULATIONAHA.107.187397 [DOI] [PubMed] [Google Scholar]
- Tourki, B. , Kain, V. , Pullen, A. B. , Norris, P. C. , Patel, N. , Arora, P. , Leroy, X. , Serhan, C. N. , & Halade, G. V. (2020). Lack of resolution sensor drives age‐related cardiometabolic and cardiorenal defects and impedes inflammation‐resolution in heart failure. Molecular Metabolism, 31, 138–149. 10.1016/j.molmet.2019.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujioka, H. , Imanishi, T. , Ikejima, H. , Kuroi, A. , Takarada, S. , Tanimoto, T. , Kitabata, H. , Okochi, K. , Arita, Y. , Ishibashi, K. , Komukai, K. , Kataiwa, H. , Nakamura, N. , Hirata, K. , Tanaka, A. , & Akasaka, T. (2009). Impact of heterogeneity of human peripheral blood monocyte subsets on myocardial salvage in patients with primary acute myocardial infarction. Journal of the American College of Cardiology, 54(2), 130–138. 10.1016/j.jacc2009.04.021 [DOI] [PubMed] [Google Scholar]
- Turner, N. A. (2016). Inflammatory and fibrotic responses of cardiac fibroblasts to myocardial damage associated molecular patterns (DAMPs). Journal of Molecular and Cellular Cardiology, 94, 189–200. 10.1016/j.yjmcc.2015.11.002 [DOI] [PubMed] [Google Scholar]
- Turner, N. A. , Mughal, R. S. , Warburton, P. , O'Regan, D. J. , Ball, S. G. , & Porter, K. E. (2007). Mechanism of TNFalpha‐induced IL‐1alpha, IL‐1beta and IL‐6 expression in human cardiac fibroblasts: Effects of statins and thiazolidinediones. Cardiovascular Research, 76(1), 81–90. 10.1016/j.cardiores.2007.06.003 [DOI] [PubMed] [Google Scholar]
- Vafadarnejad, E. , Rizzo, G. , Krampert, L. , Arampatzi, P. , Arias‐Loza, A.‐P. , Nazzal, Y. , Rizakou, A. , Knochenhauer, T. , Bandi, S. R. , Nugroho, V. A. , Schulz, D. J. J. , Roesch, M. , Alayrac, P. , Vilar, J. , Silvestre, J. S. , Zernecke, A. , Saliba, A. E. , & Cochain, C. (2020). Dynamics of cardiac neutrophil diversity in murine myocardial infarction. Circulation Research, 127(9), e232–e249. 10.1161/CIRCRESAHA.120.317200 [DOI] [PubMed] [Google Scholar]
- Vasquez, C. , Benamer, N. , & Morley, G. E. (2011). The cardiac fibroblast: Functional and electrophysiological considerations in healthy and diseased hearts. Journal of Cardiovascular Pharmacology, 57(4), 380–388. 10.1097/FJC.0b013e31820cda19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedin, O. , Lam, C. S. , Koh, A. S. , Benson, L. , Teng, T. H. K. , Tay, W. T. , Braun, O. Ö. , Savarese, G. , Dahlström, U. , & Lund, L. H. (2017). Significance of ischemic heart disease in patients with heart failure and preserved, midrange, and reduced ejection fraction: A nationwide cohort study. Circulation: Heart Failure, 10(6), e003875. 10.1161/CIRCHEARTFAILURE.117.003875 [DOI] [PubMed] [Google Scholar]
- Virag, J. I. , & Murry, C. E. (2003). Myofibroblast and endothelial cell proliferation during murine myocardial infarct repair. The American Journal of Pathology, 163(6), 2433–2440. 10.1016/s0002-9440(10)63598-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volz, H. C. , Laohachewin, D. , Seidel, C. , Lasitschka, F. , Keilbach, K. , Wienbrandt, A. R. , Andrassy, J. , Bierhaus, A. , Kaya, Z. , Katus, H. A. , & Andrassy, M. (2012). S100A8/A9 aggravates post‐ischemic heart failure through activation of RAGE‐dependent NF‐κB signaling. Basic Research in Cardiology, 107(2), 250. 10.1007/s00395-012-0250-z [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Dembowsky, K. , Chevalier, E. , Stüve, P. , Korf‐Klingebiel, M. , Lochner, M. , Napp, L. C. , Frank, H. , Brinkmann, E. , Kanwischer, A. , Bauersachs, J. , Gyöngyösi, M. , Sparwasser, T. , & Wollert, K. C. (2019). CXC motif chemokine receptor 4 blockade promotes tissue repair after myocardial infarction by enhancing regulatory T cell mobilization and immune‐regulatory function. Circulation, 139(15), 1798–1812. 10.1161/CIRCULATIONAHA.118.036053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, C. L. , Yin, R. , Wang, S. N. , & Ying, R. (2021). A review of CXCL1 in cardiac fibrosis. Frontiers in Cardiovascular Medicine, 8, 674498. 10.3389/fcvm.2021.674498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Y. , Ip, J. E. , Huang, J. , Zhang, L. , Matsushita, K. , Liew, C.‐C. , Pratt, R. E. , & Dzau, V. J. (2006). Essential role of ICAM‐1/CD18 in mediating EPC recruitment, angiogenesis, and repair to the infarcted myocardium. Circulation Research, 99(3), 315–322. 10.1161/01.RES.0000235986.35957.a3 [DOI] [PubMed] [Google Scholar]
- Wu, Y. , Li, Y. , Zhang, C. A. X. , A, X. , Wang, Y. , Cui, W. , Li, H. , & Du, J. (2014). S100a8/a9 released by CD11b+Gr1+ neutrophils activates cardiac fibroblasts to initiate angiotensin II‐induced cardiac inflammation and injury. Hypertension, 63(6), 1241–1250. 10.1161/hypertensionaha.113.02843 [DOI] [PubMed] [Google Scholar]
- Wynn, T. A. , & Vannella, K. M. (2016). Macrophages in tissue repair, regeneration, and fibrosis. Immunity, 44(3), 450–462. 10.1016/j.immuni.2016.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeap, X. Y. , Dehn, S. , Adelman, J. , Lipsitz, J. , & Thorp, E. B. (2013). Quantitation of acute necrosis after experimental myocardial infarction. Methods in Molecular Biology, 1004, 115–133. 10.1007/978-1-62703-383-1_9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi, Y. , Aguilar, E. G. , Troncoso, M. , Ilatovskaya, D. V. , & DeLeon‐Pennell, K. Y. (2021). Immune regulation of cardiac fibrosis post myocardial infarction. Cellular Signalling, 77, 109837. 10.1016/j.cellsig.2020.109837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi, Y. , Corker, A. , Vasileva, V. Y. , Oviedo, K. , Graham, C. , Wilson, K. , Martino, J. , Troncoso, M. , Broughton, P. , Ilatovskaya, D. V. , Lindsey, M. L. , & DeLeon‐Pennell, K. Y. (2021). Chronic porphyromonas gingivalis lipopolysaccharide induces adverse myocardial infarction wound healing through activation of CD8(+) T cells. American Journal of Physiology. Heart and Circulatory Physiology, 321(5), H948–h962. 10.1152/ajpheart.00082.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamilpa, R. , & Lindsey, M. L. (2010). Extracellular matrix turnover and signaling during cardiac remodeling following MI: Causes and consequences. Journal of Molecular and Cellular Cardiology, 48(3), 558–563. 10.1016/j.yjmcc.2009.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, S. , Bories, G. , Lantz, C. , Emmons, R. , Becker, A. , Liu, E. , Abecassis, M. M. , Yvan‐Charvet, L. , & Thorp, E. B. (2019). Immunometabolism of phagocytes and relationships to cardiac repair. Frontiers in Cardiovascular Medicine, 6, 42. 10.3389/fcvm.2019.00042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, S. , Yeap, X. Y. , DeBerge, M. , Naresh, N. K. , Wang, K. , Jiang, Z. , Wilcox, J. E. , White, S. M. , Morrow, J. P. , Burridge, P. W. , Procissi, D. , Scott, E. A. , Frazier, W. , & Thorp, E. B. (2017). Acute CD47 blockade during ischemic myocardial reperfusion enhances phagocytosis‐associated cardiac repair. JACC: Basic to Translational Science, 2(4), 386–397. 10.1016/j.jacbts.2017.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, B. , Lu, R. , Chen, J. , Xie, M. , Zhao, X. , & Kong, L. (2021). S100A9 blockade prevents lipopolysaccharide‐induced lung injury via suppressing the NLRP3 pathway. Respiratory Research, 22(1), 1–11. 10.1186/s12931-021-01641-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, M. , Wang, D. D. , Liu, X. , & Tian, R. (2020). Metabolic modulation of macrophage function post myocardial infarction. Frontiers in Physiology, 11, 674. 10.3389/fphys.2020.00674 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.