

Abstract
Acinetobacter baumannii is one of the major pathogenic ESKAPE bacterium, which is responsible for about more than 722,000 cases in a year, globally. Despite the alarming increase in multidrug resistance, a safe and effective vaccine for Acinetobacter infections is still not available. Hence in the current study, a multiepitope vaccine construct was developed using linear B cell, cytotoxic T cell, and helper T cell epitopes from the antigenic and well-conserved lipopolysaccharide assembly proteins employing systematic immunoinformatics and structural vaccinology strategies. The multi-peptide vaccine was predicted to be highly antigenic, non-allergenic, non-toxic, and cover maximum population coverage worldwide. Further, the vaccine construct was modeled along with adjuvant and peptide linkers and validated to achieve a high-quality three-dimensional structure which was subsequently utilized for cytokine prediction, disulfide engineering, and docking analyses with Toll-like receptor (TLR4). Ramachandran plot showed 98.3% of the residues were located in the most favorable and permitted regions, thereby corroborating the feasibility of the modeled vaccine construct. Molecular dynamics simulation for a 100 ns timeframe further confirmed the stability of the binding vaccine-receptor complex. Finally, in silico cloning and codon adaptation were also performed with the pET28a (+) plasmid vector to determine the efficiency of expression and translation of the vaccine. Immune simulation studies demonstrated that the vaccine could trigger both B and T cell responses and can elicit strong primary, secondary, and tertiary immune responses. The designed multi-peptide subunit vaccine would certainly expedite the experimental approach for the development of a vaccine against A. baumannii infection.
Graphical Abstract

Supplementary Information
The online version contains supplementary material available at 10.1007/s12026-023-09374-4.
Keywords: Acinetobacter baumannii, Lipopolysaccharides, lptD, lptE, Immunoinformatics
Introduction
Acinetobacter baumannii, a highly pathogenic bacterium globally, has emerged as a highly challenging ESKAPE bacterium [1]. It is highly pathogenic and infectious causing ventilator-associated pneumonia, skin, and soft tissue infections, urinary tract infections, endocarditis, secondary meningitis, and bloodstream infections and is associated with 35% of mortality [2]. Commonly associated with soil and water environments [3], it is known to colonize the skin and is prevalent in the respiratory secretions of infected patients [4, 5].
The coccobacilli of the genus Acinetobacter are gram-negative, pleomorphic, and metabolically non-fermenting under aerobic conditions, enzymatically positive for catalase, and negative for oxidase with its DNA consisting of the G+C content of 39 to 47% [4]. The pronounced ability of surface adherence to create biofilms, and acquisition of genetic material of unrelated genera potentiates its property of antimicrobial resistance, thereby making it more versatile and challenging to combat, regulate and eradicate this bacterium [6, 7]. Numerous studies suggest that A. baumannii has the unique ability to be highly pathogenic [8, 9]. However, most of the resistance genes responsible for its enhanced anti-microbial resistance have been acquired from the bacteria of the genera Salmonella, Escherichia, or Pseudomonas [10]. Colistin and tigecycline remain the only antibiotics active against it and have become the last resort of treatment for multidrug-resistant A. baumannii; however, colistin-resistant strains have been reported in different regions from around the world [11, 12]. The best therapy for A. baumannii infections, particularly nosocomial infections caused by several resistant strains, still has to be determined.
As compared to other Gram-negative bacteria, very few but distinctive factors contribute to the virulence of A. baumannii [13]. Lipopolysaccharides (LPS), an immunoreactive molecule present in the outer membrane of Gram-negative bacteria, contribute to its cytotoxicity towards host cells and are vital for cell viability. LPS is a key contributing factor to the virulence of A. baumannii; particularly the lipid A component, and structural variation in the O-antigen side chain is known to impact the host immunity [14]. The transport of LPS from its site of synthesis to the cell surface is mediated by seven proteins comprising the LPS transport system, LptABCDEFG [15]. Despite the identification of key players, the exact mechanism of LPS transport and assembly is not very clear. The stable LptD/E complex present in the outer membrane functions in the final stages of LPS assembly, and serves as the LPS recognition site. It is well established that LptE plays an essential role in the assembly of functional LptD. However, more recently LptE has been shown to play a role also in the LPS export process in E. coli, as witnessed by its ability to bind lipopolysaccharide. LPS interacts with the LptDE complex. LptD is a 26-strand β-barrel outer membrane protein that forms a unique plug-and-barrel complex with the lipoprotein LptE was revealed by Dong et al. 2020 and Qiao et al. 2021 [16, 17]. LptD mediates the final translocation of fully synthesized LPS from the periplasm to the outer leaflet of the outer membrane [18]. Several reports are suggestive of the LPS-assembly proteins — LptE and LptD being structurally highly conserved, though there is variation in LPS substrates among different strains. It has been identified by electrostatics-based studies that LptDE structures revealed a striking electrostatic gradient in the barrel lumen, which plays a role in the LPS transport process in the bacteria [19], hinting us of the components of the Lpt system (LptE and LptD) as a potential target for the development of new vaccines against A. baumannii [18].
In the twenty-first century, rapid emergence and dissemination of resistant bacteria are occurring globally, impending the potency of antibiotics. Vaccination is the alternative safest and most efficacious approach against pathogenic diseases. New technologies such as reverse vaccinology, novel adjuvants, structural vaccinology, bioconjugates, and rationally designed bacterial outer membrane vesicles (OMVs), together with progress in polysaccharide conjugation and antigen design, have the capability of the future vaccine research and development [20, 21]. In 2010, McConnell et al. [22] started to apply whole-cell multi-antigen preparations to raise protective polyclonal hyperimmune serum against A. baumannii. Simultaneously, Huang et al. [23] demonstrated the eminent role of antisera raised against the outer membrane vesicle (OMV) to control Acinetobacter. A passive immunization attempt was done with a synthetic oligosaccharide that mimics PNAG conjugated to tetanus toxoid (TT). The raised antisera were able to induce opsonization against several clinical isolates of MDR A. baumannii, and reduced burdens in infected tissue especially in cases of pneumonia [24]. But, anti-PNAG sera were found to be highly specific to only PNAG-positive strains even in minute amounts [25]. In 2011, a mixture of pure proteins majorly OMPs and fimbriae proteins was patented as an effective vaccine against A. baumannii (novel targets of Acinetobacter baumannii– patent). However, not much development has been noted toward protein-specific targeted vaccine development against A. baumanii. Considering the significant role of A. baumannii in hospital-acquired infections and the associated antibiotic resistance, the development of a strong vaccine candidate against P. aeruginosa is the need of the hour.
Unlike traditional vaccine development methods, today’s cutting-edge research and technology, as well as the availability of information about the genome and proteome of almost all organisms, has enabled the design and development of novel peptide-based ‘subunit vaccines’ made up of antigenic protein portions from a target pathogen. Multiepitope-driven vaccine has been one of the foremost scientific advances of the recent decade [20, 21]. The clinical development of epitope-based vaccines that are comparatively safer than vectored or attenuated live vaccines is rapidly progressing from pre-clinical to clinical trials [26]. Recent advancements in computational methods have developed immunoinformatics tools as an effective alternative strategy for the prediction of antigenic epitopes with potential translational implications. The present study is aimed at developing a multiepitope subunit vaccine by combining linear B lymphocytes (LBL), cytotoxic T lymphocytes (CTL), and helper T lymphocytes (HTL) epitopes from LPS assembly proteins (LptE and LptD) that are known to provide protection and could elicit neutralizing antibody to fight with Acinetobacter infection. If more in vivo and in vitro investigations show positive results, the vaccine developed in this study could be beneficial as a prophylactic measure against all A. baumannii strains. The flow chart summarizing the in silico vaccinomics strategy employed in the present work for designing a unique multi-epitope vaccine against A. baumannii has been illustrated in Fig. 1.
Fig. 1.

The schematic representation of the immunoinformatics guided design of the multi-epitope vaccine construct against Acinetobacter baumannii’sinfections
Methodology
Retrieval of protein sequences
In order to find the proteins to constitute a subunit vaccine, an in-depth literature survey was performed. Two LPS assembly proteins, LptE (Accession id: V5VH20) and LptD (Accession id: B0VMX7) of A. baumannii were selected, and their amino acid sequences were retrieved in FASTA format from the UniProt database (http://www.uniprot.org/uniprot). Sequence similarity search of the LPS assembly lipoprotein sequences was performed to avoid the cross-reactivity and generation of autoimmune disorders against the human proteome by using NCBI-protein–protein blast (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins). Human β-defensin 3 (Accession id: P81534) sequence was also retrieved in FASTA format from the UniProt database.
Linear B cell epitope prediction
B cell epitopes are significant in the design of the vaccine because they are involved in inducing the humoral immune response and antibody formation, which neutralizes antigens during infection. The ABCPred server (http://www.imtech.res.in/raghava/abcpred/) predicted the successive B cell epitopes [27]. ABCPred is a consistent algorithm-based webserver that predicts B cell epitopes using ANN (artificial neural network) with fixed-length patterns and a default threshold of 0.51. As the linear B cell epitope is present on the cell surface, exomembrane topology is considered one of the important parameters. The server specificity is 0.75, and a window length of 10 was set.
Cytototoxic T lymphocyte epitope prediction
The most selective stage in the presentation of the antigenic peptide to the T cell receptor is the binding of the MHC molecule. Therefore, CTL epitopes were predicted by the NetMHCpan 4.1 (http://www.cbs.dtu.dk/services/NetMHCpan/) server. The threshold for epitope identification for a strong binder was set at 0.5% while at 2% for a weak binder [28]. The server predicts epitopes for 12 HLA class I supertypes. Each supertype is a cluster of functionally related HLA alleles that share binding specificities towards the same panel of peptides owing to similar structural features of HLAs peptide-binding grooves [29]. All epitope lengths were set to 9mers before prediction, and conserved epitopes that bind to multiple HLA alleles with a score equal to or less than 1.0 percentile rank were chosen.
Helper T lymphocyte epitope prediction
HTL epitope prediction is crucial in the development of peptide-based vaccines because binding of the peptide with MHC-II proteins is important in eliciting an immune response. The MHC-II binding predictions were executed using the IEDB analysis resource MHC-II binding (http://tools.iedb.org/mhcii/) tool [30]. It covers all HLA class II alleles including HLA-DR, HLA-DQ, and HLA-DP. The CONSENSUS method was utilized for the respective binding alleles considering human HLAs as a reference set by default. The percentile rank threshold of ≤2 was utilized here to confirm the accuracy. The half maximum inhibitory concentration (IC50) for strong binding peptides was set at less than 50 to evaluate the interaction potential of T cell epitopes and MHC-II alleles. By default, the 15 mer lengths of the epitopes was selected, and generated epitopes were classified according to the percentile value. According to the IEDB server, the peptides with the lowest consensus scores were identified as the best binders; a lower percentile rank suggests stronger affinity.
Antigenicity, allergenicity, immunogenicity, and toxicity prediction
Components of vaccines should be highly antigenic while also being non-allergenic. Furthermore, there should be no harmful reactions in the vaccination components. Henceforth, VaxiJen (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) server was then used to predict the antigenicity of the epitopes [31]. The accuracy prediction threshold influences the accuracy, sensitivity, and specificity of a prediction, and the 0.4 threshold boosts the server’s prediction accuracy. The allergenicity of the designed vaccine, on the other hand, was assessed using AllerTOP v2.0 (https://www.ddg-pharmfac.net/AllerTOP/). The AllerTOP v2.0 server uses machine learning approaches to sort allergies, such as nearest neighbors, auto and cross-variance transformation, and amino acid Edescriptors [32]. The correctness of this server is 85.3% at fivefold cross-validation. Class I Immunogenicity tool (http://tools.iedb.org/immunogenicity/) of IEDB Analysis Resource was used to predict the immunogenicity score of epitopes [33]. In addition, we used the Toxinpred (https://webs.iiitd.edu.in/raghava/toxinpred/) server for the toxicity analysis [34]. The ‘best-selected epitopes’, which were determined to be highly antigenic, immunogenic, non-allergenic, and non-toxic to the human proteome, were employed for vaccine development in the following phases.
Construction of multi-epitopic vaccine and codon optimization
Multi-epitopic vaccine was designed using B cell, HTL, and CTL epitopes from the envelope and surface glycoprotein. To enhance the immunogenicity of the multi-epitope vaccine, human beta-defensin 3 was selected as an adjuvant agonist Toll-like receptor 4. The adjuvant was linked to B cell epitopes using the “EAAAK” linker, followed by HTL and CTL epitopes. “KK” and “GPGPG” linkers were used to link intra-B cell and intra-HTL epitopes, whereas ‘AAY’ linker were used to link intra-CTL epitopes [35].
HLA population coverage analysis
The next important aspect is to determine the population coverage rates for the predicted epitopes. This population coverage analysis plays a significant role in the design and development of epitope-based vaccines as HLA molecules are extremely polymorphic in nature in the peptide-binding regions [36]. Population coverage calculation was conducted using the web-based population coverage analysis tool of the IEDB server (http://tools.iedb.org/population/) with default parameters. The tool provides HLA allele frequencies for 109 populations grouped into 16 different geographical areas. The proportion of people who are expected to react to a collection of epitopes with known MHC restrictions is calculated using the population coverage analysis tool. Thus, it is beneficial to select only the epitopes with a high response rate for different populations.
Determination of physicochemical properties
The ExPASy ProtParam (https://web.expasy.org/protparam/) server was utilized to calculate the physicochemical properties of the vaccine construct [37]. Multiple metrics such as aliphatic index, instability index, half-life, isoelectric point (pI), molecular weight, atomic composition, and grand average hydropathicity (GRAVY) are calculated using this web application. The half-life of a protein is the amount of time that the molecule remains in the cell after it has been synthesized. The index of instability measures the consistency of a protein molecule in vitro.
Secondary structure prediction and solubility profile prediction
The secondary structure of the multi-epitope vaccine construct was calculated using position-specific iterated prediction (PSIPRED) analysis on outputs from PSI-BLAST (http://bioinf.cs.ucl.ac.uk/psipred/) [38]. In addition, we utilized the SOLpro (http://scratch.proteomics.ics.uci.edu/) online approach to estimate solubility when the protein structure is overexpressed in E. coli with a 74% accurate prediction [39].
Epitope conservancy analysis
A vaccine should ideally have conserved epitopes that can elicit particular B cell and T cell (CD4 and CD8) responses. As a result, the IEDB server’s “epitope conservancy analysis” module (https://www.iedb.org/conservancy/) was utilized to assess the conservancy of the chosen B cell, CTL, and HTL epitopes [40]. For a given set of protein sequences, the tool computes the percentage of epitope sequence conservation at a certain identity level. We investigated the conservation of specific epitopes of the LPS protein across 20 A. baumannii strains in this study.
Tertiary structure prediction, refinement, and validation
The tertiary structure was modeled using the Robetta (https://robetta.bakerlab.org/) [41], Alpha fold (https://alphafold.ebi.ac.uk/) [42], and SWISS-MODEL (https://swissmodel.expasy.org/) [43] servers in a consensus manner and visualized using the Pymol visualization tool (https://pymol.org/2/) [44]. The 3D structure model was subjected to refinement by the GalaxyRefine (https://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE) server [45]. The output consists of five refined models, with varying parameter scores, including GDT-HA, RMSD, MolProbity, Clash score, poor rotamers, and Rama favored. PROCHECK (https://saves.mbi.ucla.edu/) was used to confirm the revised structure’s stereochemical value by looking at residue-by-residue geometry and overall structure geometry [46]. ERRAT (https://saves.mbi.ucla.edu/) [47] and Verify 3D (https://saves.mbi.ucla.edu/) [48] were also used to confirm the validation.
Vaccine protein disulfide engineering
Disulfide engineering is a biotechnological approach that uses cysteine residue mutation to construct new disulfide bonds in a target protein in a highly mobile protein region (10.1186/1471-2105-14-346) [49]. Disulfide linkages are highly stable and help proteins to maintain their geometric conformation. This was accomplished via the online DbD2 server (http://cptweb.cpt.wayne.edu/DbD2/index.php) [49]. This web server will recognize the pair of residues capable of generating a disulfide bond if each amino acid residue is replaced with cysteine.
Prediction of discontinuous B cell epitopes
The tertiary structure of a protein or polypeptide is required for the prediction of discontinuous B cell epitopes because the interaction between antigen epitopes and antibodies is essential. Based on the three-dimensional (3D) structures of the vaccine construct, discontinuous B cell epitopes were predicted using the Ellipro server (http://tools.iedb.org/ellipro/) program on the IEDB website with a default option (minimum residue score 0.5 and maximum distance 6) [50]. Thornton’s approach and a residue clustering algorithm are implemented on this site. Each output epitope receives a score from ElliPro, which is described as a PI (protrusion index) value averaged across each epitope residue. There is a possible link between residues with high scores and improved solvent accessibility [50].
Identification of cytokine-inducing HTL epitopes
The production of several cytokines by MHC class II activated CD4+ T helper cells, such as interferon-gamma (IFN-γ), interleukin-10 (IL-10), and interleukin-17 (IL-17), contributes greatly to infection control. Hence, interferon-gamma-inducing epitopes from the selected MHC-II binding epitopes were predicted by the IFNepitope server (https://webs.iiitd.edu.in/raghava/ifnepitope/predict.php) [51]. This server employs a dataset composed of 3705 IFN-γ epitopes inducing and 6728 non-inducing MHC class II binders. The server uses motif and SVM hybrid algorithms for the prediction of the interferon-producing property of the epitopes. The hybrid prediction method is a highly accurate method for predicting IFN-gamma producing epitopes. IL-10 and IL-17 inducing abilities, on the other hand, were anticipated using the web servers IL10pred (https://webs.iiitd.edu.in/raghava/il10pred/) and IL17eScan (http://metagenomics.iiserb.ac.in/IL17eScan/pred.php). The SVM method and hybrid approach were used to perform the IL10pred [52] and IL17eScan [53] operations, with default threshold values of 0.2 and −0.3, respectively. The default values were used for all other parameters.
Molecular docking
Molecular docking was performed for the constructed multi-peptide vaccine with TLR-4, (PDB ID: 5IJB) receptor. The Protein Data Bank (PDB) files of receptor and ligand were submitted to the ClusPro v2.0 server (https://cluspro.bu.edu/login.php) and the default settings were used accordingly [54]. The ClusPro server calculates the energy score based on the following equation: E= 0.40Erep+ (−0.40Eatt) + 600Eelec+1.00EDARS [54]. The interaction of TLR4 and vaccine structure was also cross-checked by the PathchDock tool (https://bioinfo3d.cs.tau.ac.il/PatchDock/php.php). The best docking pose in the top-rank cluster was used for visualization using the PyMol v2.4 (https://pymol.org/2/) software, and the results were interpreted.
Molecular dynamics simulation
After performing the protein–protein molecular docking and finding the best orientations of the vaccine candidate and the receptor proteins to interact with one another, the best-scored complex was subjected to MD simulation [55]. All of the MD simulations were done by GROMACS 2019 package [56] and Charmm36-mar2019force field [57]. The protein complex was solvated with the TIP3P water model. Then, sodium and chloride ions were added to produce a neutral physiological salt concentration of 150 mM. Each system was energy minimized, using the steepest descent algorithm, until the Fmax was found to be smaller than 10 kJ.mol. All of the covalent bonds were constrained using the linear constraint solver (LINCS) algorithm to maintain constant bond lengths. The long-range electrostatic interactions were treated using the particle mesh Ewald (PME) method and the cut-off radii for Coulomb and Van der Waals short-range interactions were set to 0.9 nm. Then 100 ps NVT (constant number of particles (N), volume (V), and temperature (T)), and 300 ps NPT (constant number of particles (N), pressure (P), and temperature (T) equilibrations were performed for each system. Finally, simulations were carried out under the periodic boundary conditions (PBC), set at XYZ coordinates to ensure that the atoms had stayed inside the simulation box, and the subsequent analyses were then performed using GROMACS modules, and VMD and also the plots and the figures were created using the xmgrace.
MMPBSA binding free energy analysis
The binding free energies of the protein–protein complexes were calculated by molecular mechanics-Poisson–Boltzmann solvent-accessible surface area, MMPBSA method using g_mmpbsa package [58]. In this method, the binding free energy is the result of the free energy of the complex minus the sum of the free energies of the ligand and the protein. The MMPBSA calculation was done for every ns of the simulation trajectory.
In silico cloning of vaccine construct
Due to the dissimilarity between the codons of humans and E. coli, codon adaptation tools were used. It is necessary to adapt the codon usage within the prokaryotic organism to boost the expression rate in the respective host system. The sequence was subjected to Java Codon Adaptation Tool (JCat) online software (http://www.jcat.de/) for the purpose of codon adaptation [59]. This web server adapts codon usage of a target DNA/protein to its potential expression host, yielding an optimized sequence along with codon adaptive index (CAI) and GC content percent (GC %). For CAI, a score range between 0.8 and 1 is considered as an optimum value, suggesting improved gene expression in the respective organism. Also, the GC% should be between 30 and 70%; otherwise, it may result in transcriptional and translational deficiencies. Assessment of cloning and expression of the vaccine construct in the suitable expression vector pET28a (+) was achieved using in silico cloning tool called the SnapGene software (https://www.snapgene.com/). For the cloning of the vaccine component, E. coli strain K12 was selected as a host.
Immune simulation
Computational immune simulation was performed using C-IMMSIM (http://150.146.2.1/CIMMSIM) online server to determine the real-life-like immune response analysis of vaccine construct [60]. An agent-based algorithm based on the position-specific scoring matrix and machine learning techniques were utilized for the estimation of antigens and foreign particles on immune activity. The simulation was performed with default parameters except for the time steps which were set at 1, 84, and 170 (time step 1 is injection at time = 0, and each time step is 8 h). So, three injections are expected to be required at four weeks intervals because the recommended interval between two doses of most commercial vaccines is four weeks. The Simpson’s diversity index, D was estimated from the plot.
Results
Criteria for selection of final multi-epitopes for vaccine construct
The aim of this study was to elucidate conserved epitopes capable of eliciting B cell and T (CD8 + and CD4 +) cell response. Also, the identified epitopes must be devoid of undesirable responses such as autoimmunity, allergic, and toxic properties. Therefore, the main criterion for selecting final epitopes for the vaccine development was based on the following parameters (1) complete conservation of peptides in the A. baumannii, (2) highest antigenicity score, (3) positive immunogenicity score, (4) high scores by the predicted algorithms, (5) the epitopes with strong binding affinity to the maximum number of HLA alleles of the world population, (6) non-allergenicity, and (7) non-toxic in nature.
Prediction of linear B cell epitopes
B cell epitopes were predicted in order to evoke the high expression of antigen-specific antibody production in the serum. In natural conditions, antibody production starts with the encounter of immunogenic epitopes with the B cells receptor, which leads to the differentiation into a plasma cell and a memory cell. In our immune system, plasma cell is responsible for antibody production during primary infection while during secondary infection memory cells also help in antibody production. Therefore, the main objective in epitope-driven vaccine development is the prediction of B cell epitopes in the target protein. ABCpred server was used for the prediction of the linear B cell epitopes. The epitopes above the threshold score of 0.51 were selected. A total of 32 epitopes were predicted (Supplementary Table 1). Among them, two epitopes (VGCGFHLKGT of LptE and DFQTLDPEVK of LptD), which met the above-mentioned criterion were selected for vaccine design as depicted in Table 1.
Table 1.
Final selected B cell epitopes from A. baumannii LPS assembly protein (LptD and LptE) and their corresponding immunogenic properties
| Uniprot_ID | B cell epitope | Position | Score | Antigencity Score | Conservancy | Toxicity | Hydrophobicity | Hydropathicity | Hydrophilicity | Charge | Mol wt. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| V5VH20 | VGCGFHLKGT | 21 | 0.68 | 2.0282 | 100% | Non toxin | 0.05 | 0.43 | -0.47 | 1.5 | 1018.35 |
| B0VMX7 | DFQTLDPEVK | 419 | 0.65 | 1.9198 | 100% | Non toxin | -0.24 | -0.94 | 0.6 | -2 | 1191.44 |
Prediction of cytotoxic T lymphocyte
Cytotoxic T lymphocytes generally play a role in the containment of bacterial infection [61], and the prevalence of cytotoxic T cells usually correlates with the rate of pathogen clearance. CTL epitopes for the LPS assembly proteins were predicted using the NetMHCpan4.1 server, screening was based on the combined high score which denotes low sensitivity and high specificity for MHC-I supertypes. All 12 sets of supertypes (A1, A2, A3, A24, A26, B7, B8, B27, B39, B44, B58, and B62) were used for the analysis. Strong binders are defined as having a percentage rank < 0.05. For each protein, the top high-scoring epitopes with the least percentile rank in binding affinity were selected and used for peptide- MHC I binding analysis. A total of 240 epitopes were predicted and screened based on their binding affinity scores with their respective MHC I supertypes (Supplementary Table 2). Among them, 17 epitopes were selected for the final vaccine construct as mentioned in Table 2.
Table 2.
Predicted CTL epitopes from A. baumannii LPS assembly protein (LptD and LptE) to design multi-epitope vaccine construct with their corresponding MHC class I alleles and their immunogenic properties
| Uniprot_ID | CTL Epitope | Alleles | Position | Score | Antigencity Score | Immunogenicity | Conservancy | Toxicity | Hydrophobicity | Hydropathicity | Hydrophilicity | Charge | Mol wt. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| V5VH20 | TDDLETQLK | HLA-A*01:01 | 50 | 2.907 | 1.1947 | 0.02584 | 100% | Non-toxin | −0.35 | −1.3 | 0.87 | −2 | 1062.27 |
| GLVGCGFHL | HLA-A*02:01 | 19 | 0.514 | 1.3983 | 0.1446 | 100% | Non-toxin | 0.26 | 1.41 | −1.01 | 0.5 | 902.22 | |
| GTNPTATPL | HLA-A*03:01 | 29 | 7.505 | 0.8191 | 0.08765 | 100% | Non-toxin | −0.04 | −0.4 | −0.37 | 0 | 871.08 | |
| LRLTVTFQI | HLA-A*24:02 | 98 | 3.932 | 1.6349 | 0.14332 | 100% | Non-toxin | 0.01 | 1.08 | −0.78 | 1 | 1090.47 | |
| SAGLVGCGF | HLA-A*26:01 | 17 | 7.792 | 0.9454 | 0.04624 | 100% | Non-toxin | 0.24 | 1.46 | −0.78 | 0 | 810.06 | |
| VLLRLTVTF | HLA-B*08:01 | 96 | 1.293 | 0.6513 | 0.13174 | 100% | Non-toxin | 0.13 | 1.86 | −0.97 | 1 | 1061.47 | |
| B0VMX7 | QSDEIDYNL | HLA-A*01:01 | 168 | 0.662 | 1.4015 | 0.25153 | 100% | Non-toxin | −0.26 | −1.26 | 0.42 | −3 | 1096.24 |
| KSAPEAQLR | HLA-A*03:01 | 68 | 1.182 | 1.3792 | 0.03163 | 100% | Non-toxin | −0.39 | −1.16 | 0.74 | 1 | 999.25 | |
| VLAVPYFNF | HLA-A*24:02 | 258 | 0.376 | 2.0269 | 0.13498 | 100% | Non-toxin | 0.27 | 1.47 | −1.38 | 0 | 1069.39 | |
| YDATITPRY | HLA-A*26:01 | 303 | 0.676 | 1.4273 | 0.23878 | 100% | Non-toxin | −0.21 | −0.81 | −0.19 | 0 | 1099.32 | |
| LPQFLLNYV | HLA-B*07:02 | 437 | 1.514 | 1.1172 | 0.05134 | 100% | Non-toxin | 0.15 | 0.94 | −1.26 | 0 | 1106.47 | |
| MLQGEFRYL | HLA-B*08:01 | 318 | 1.555 | 0.4306 | 0.24572 | 100% | Non-toxin | −0.11 | −0.1 | −0.39 | 0 | 1156.5 | |
| GRTVRADKV | HLA-B*27:05 | 135 | 3.095 | 1.4877 | 0.03409 | 100% | Non-toxin | −0.45 | −0.81 | 0.9 | 2 | 1001.27 | |
| DKSAPEAQL | HLA-B*39:01 | 67 | 0.367 | 1.1392 | 0.03446 | 100% | Non-toxin | −0.27 | −1.04 | 0.74 | −1 | 958.15 | |
| VKDIPVLAV | HLA-B*40:01 | 253 | 3.103 | 1.0029 | 0.18268 | 100% | Non-toxin | 0.14 | 1.52 | −0.29 | 0 | 953.32 | |
| LSVPVYLNL | HLA-B*58:01 | 291 | 0.371 | 1.2858 | 0.02582 | 100% | Non-toxin | 0.19 | 1.4 | −1.13 | 0 | 1017.37 | |
| QTLTPRAFY | HLA-B*15:01 | 550 | 1.025 | 0.8494 | 0.1748 | 100% | Non-toxin | −0.16 | −0.43 | −0.52 | 1 | 1096.37 |
HTL epitope prediction
Class II–restricted T helper cells mediate the growth and differentiation of both T effector cells and antibody-producing B lymphocytes [62]. LPS assembly proteins were analyzed for HTL epitopes with the help of the IEDB analysis resource using NN-align with half-maximal inhibitory concentration (IC50 ≤ 50). Top thirty epitopes were selected for both the proteins based on least percentile rank and IC50 value less than 50 nM thereby indicating their higher affinity for receptor molecules (Supplementary Table 3). Finally, two epitopes (LRLTVTFQIEDRQGN of LptE and VVVPQFTLDTGLNFE of LptD) were selected for the vaccine construct as mentioned in Table 3.
Table 3.
Predicted HTL epitopes from A. baumannii LPS assembly protein (LptD and LptE) to design multi-epitope vaccine construct with their corresponding MHC class II alleles and their immunogenic properties
| Uniprot_ID | MHC II epitope | Alleles | Pos | IC50 value | Percentile_Rank | Antigencity score | Conservancy | Toxicity | Hydrophobicity | Hydropathicity | Hydrophilicity | Charge | Mol wt. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| V5VH20 | LRLTVTFQIEDRQGN | HLA-DPA1*01:03, HLA-DPB1*02:01, HLA-DPB1*01:01, HLA-DRB1*01:01, HLA-DRB1*09:01,HLA-DRB3*02:02, HLA-DRB1*13:02, HLA-DRB1*11:01, HLA-DRB1*04:01, HLA-DRB1*12:01, HLA-DPA1*03:01, HLA-DPB1*04:02, HLA-DRB1*04:05, HLA-DRB1*15:01, HLA-DQA1*01:01, HLA-DQB1*05:01, HLA-DRB1*08:02, HLA-DPA1*02:01, HLA-DPB1*14:01, HLA-DPB1*04:01, HLA-DQA1*05:01, HLA-DQB1*03:01, HLA-DQA1*04:01, HLA-DQB1*04:02, HLA-DPA1*02:01, HLA-DPA1*02:01, HLA-DPB1*05:01, HLA-DPA1*01:03, HLA-DQA1*05:01, HLA-DQB1*02:01, HLA-DQA1*03:01, HLA-DQB1*03:02, HLA-DQA1*01:02, HLA-DQB1*06:02, HLA-DRB3*01:01, HLA-DRB5*01:01, HLA-DRB1*07:01, HLA-DRB4*01:01, HLA-DRB1*03:01 | 98–112 | 73 | 1.9 | 1.5395 | 100% | Non-toxin | −0.28 | −0.61 | 0.16 | 0 | 1790.24 |
| B0VMX7 | VVVPQFTLDTGLNFE |
HLA-DRB1*15:01, HLA-DRB3*01:01, HLA-DPA1*03:01, HLA-DPB1*04:02, HLA-DPA1*01:03, HLA-DPB1*02:01,HLA-DRB1*01:01, HLA-DRB1*09:01,HLA-DRB3*02:02, HLA-DRB1*13:02, HLA-DRB1*11:01, HLA-DRB1*04:01, HLA-DRB1*12:01,HLA-DRB1*04:05, HLA-DQA1*01:01, HLA-DQB1*05:01, HLA-DRB1*08:02, HLA-DPA1*02:01, HLA-DPB1*14:01, HLA-DPA1*01:03, HLA-DPB1*04:01, HLA-DQA1*05:01, HLA-DQB1*03:01, HLA-DQA1*04:01, HLA-DQB1*04:02, HLA-DPA1*02:01, HLA-DPB1*01:01, HLA-DPA1*02:01, HLA-DPB1*05:01,HLA-DQA1*05:01, HLA-DQB1*02:01, HLA-DQA1*03:01, HLA-DQB1*03:02, HLA-DQA1*01:02, HLA-DQB1*06:02, HLA-DRB5*01:01, HLA-DRB1*07:01, HLA-DRB4*01:01, HLA-DRB1*03:01 |
529–543 | 50 | 0.28 | 1.2627 | 100% | Non toxin | 0.06 | 0.56 | −0.5 | −2 | 1679.13 |
Multi-epitopic vaccine design
The screened B cell, HTL, and CTL epitopes were used to design the multi-epitope vaccine construct. A major challenge for current vaccine development is the fact that many new subunit vaccines are poorly immunogenic and elicit insufficient immune responses for protective immunity. Therefore, adjuvants are needed in vaccine formulations to enhance the magnitude, breadth, and durability of the immune response [63]. We have incorporated human beta-defensin 3 as an adjuvant because it is reported that the production of human beta-defensin 3 contributes to the innate control of A. baumannii infection [64, 65]. β-defensins sequence (Uniprot id: P81534) was added at the N-terminal of the vaccine construct linked by the “EAAAK” linker followed by B cell epitopes which were linked by the “KK” linkers, further, the HTL and CTL epitopes were linked by “AAY” and “GPGPG” linkers. The EAAAK linker will provide effective separation of domains of the bifunctional fusion proteins. The bi-lysine (KK) linker will preserve the independent immunological activities of the B cell epitopes of the vaccine [66]. AAY linkers will significantly influence the expression of the target vaccine protein [67]. The GPGPG linkers will prevent the generation of junctional epitopes and will facilitate immune processing and presentation. The final vaccine construct was 338 amino acids long shown in Fig. 2.
Fig. 2.

The structural arrangement of B and T cell epitopes along with linkers and adjuvant for the final multi-epitope vaccine construct
Population coverage analysis
The design of peptide-based vaccines relies heavily on population coverage analysis. The frequency of expression of different HLA types varies in different ethnicities as the MHC molecule is highly polymorphic [68]. Extreme polymorphism limits the percentage of the human population that can respond to a given antigen [69]. As a result, a peptide that works as a T cell epitope in a population with certain HLA makeup may not be successful in another population with a different HLA allelic distribution. The goal was to identify the promiscuous T cell epitopes that bind to multiple alleles of HLA supertypes for maximum population coverage. The predicted epitopes were used as input to the population coverage analysis tool in the IEDB database for final MHC I and MHC II (T cell) epitopes against the 109 countries covering 16 different geographical regions. The population coverage tool showed that the designed vaccine would cover 99.51% of the world population. It is interesting to note that predicted epitopic core sequences showed significantly highest and lowest population coverage from Japan and Wales regions, respectively (Supplementary Table 4).
Physiochemical characterization of designed vaccine
The physiochemical parameters of the vaccine construct have crucial effects on properties like stability and immunogenicity. The designed multi-epitope vaccine is composed of 338 amino acids with a molecular weight of approximately 36.84kDa. The theoretical pI was 9.10, indicating the vaccine construct is notably basic in nature. The total number of negative residues in the vaccine construct was predicted to be 23 and the positive residue is 34. The molecular formula of the vaccine construct is C1702H2620N438O464S11 with 5235 atoms. The estimated half-life is 30h, >20h, and > 10h in mammalian reticulocytes (in vitro), yeast (in vivo), and Escherichia coli (in vivo) respectively. The instability index was 28.78, implying a stable protein. The thermal stability of the vaccine is important, and it is evaluated by the aliphatic index, further solubility is a significant physiochemical parameter for the expression of the protein in a suitable expression system. The calculated aliphatic index and grand average of hydropathicity (GRAVY) were 92.78 and 0.145 respectively, signifying that the vaccine is thermostable and hydrophilic (Supplementary Table 5).
Prediction of secondary structure and solubility of multi-epitopic vaccine
PSIPRED produced the secondary structure of the vaccine construct with 45.99% Alpha Helix, 12.75% extended strand, and 41.24% random coil (Fig. 3). The predicted solubility leading to overexpression by the SOLpro server showed the vaccine construct as soluble with a probability of 0.643784 (Supplementary Table 5).
Fig. 3.

Secondary structure prediction of the final multi-epitope vaccine construct by using the PSIPRED tool
Prediction of antigenicity and allergenicity of constructed vaccine
The vaccine must be antigenic and non-allergic in nature and also induce humoral as well as cell-mediated immune responses against the targeted pathogen. Our vaccine was observed to be antigenic with a respective probability score of 0.845 predicted by VaxiJen v2.0. The construct was also detected as a non-allergen, checked using the AllerTOP v2.0 server.
Epitope conservancy analysis
Proteins from 20 A. baumannii strains were extracted to find the LPS protein’s homologous amino acid sequences (Supplementary Table 6). Consequently, each of the shortlisted CTL, HTL, and B cell epitope was analyzed to study the conservancy across all strains using the conservancy analysis tool of IEDB. All selected epitopes showed 100% conservation in all isolates from different counties (Tables 1, 2, and 3).
Tertiary structure prediction and structure validation
The tertiary structure of the vaccine was modeled using the Robetta, Alpha Fold, and SWISS-MODEL servers (Fig. 4), and the best models consensually predicted by the tools have been taken for further analysis. The structure was then subjected to deep optimization and validated by structure validation tools PROCHECK, VERIFY 3D, and ERRAT. The accuracy and stereochemical feature of the predicted model was calculated with PROCHECK by Ramachandran Plot analysis. The predicted model scored, 88.3% of the amino acid residues in the favored, 10.0% in the allowed, 0.7% of the residues were found in the outlier, and 1.0% residues in the disallowed region (Fig. 5) which ensures the quality of the model. According to VERIFY 3D, the predicted model’s 89.94%of the residues have averaged 3D-1D score ≥ 0.2, which determines a good environmental profile along with an ERRAT score of 87.6923which is below the 95% rejection limit. Further validation of the model suggested that the refined model has high stability and good quality, the refined model was used in docking studies.
Fig. 4.

Homology modeling of the three-dimensional structure of the final multi-epitope vaccine construct
Fig. 5.

Several structure validations tools results confirmed the modeled multi-epitope vaccine structure to be reliable and accurate
Vaccine protein disulfide engineering
A total number of 45 pairs of amino acid residues were predicted to have the potential to form a disulfide bond by the DbD2 server. Following residue evaluation by chi3 and B-factor energy parameters, only nine pairs of residues were found to be suitable for disulfide bond formation LEU11-PHE77, VAL35-CYS114, GLY38-PRO198, CYS40-CYS114, ALA41-PRO129, ALA143-ASP268, ALA184-ALA187, TYR241-PRO258, and LEU292-ALA296 which was replaced by cysteine residue (Fig. 6). Residue screening was done on the basis of −87 to +97 chi3 value and < 2.2 energy value (Supplementary Table 7). Thus, more disulfide bonds would increase the stability of the protein.
Fig. 6.
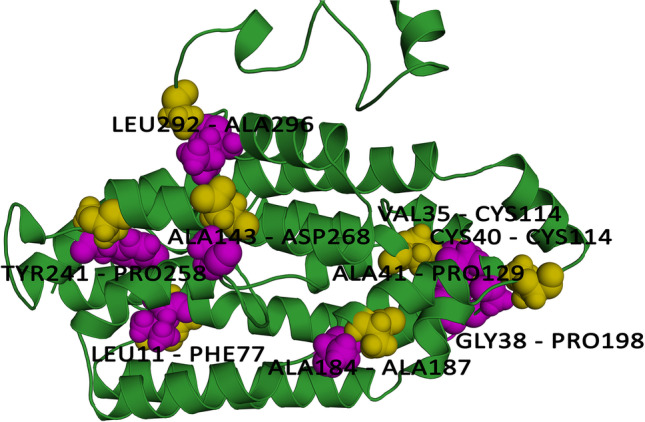
Disulphide engineering of the vaccine protein. Residue pairs showed in olive (LEU11, VAL35, GLY38, CYS40, ALA41, ALA143, ALA184, TYR241, LEU292) and purple (PHE77, CYS114, PRO198, CYS114, PRO129, ASP268, ALA187, PRO258, ALA296) spheres were mutated to Cysteine residues to form disulfide bridge between them
Prediction of discontinuous B cell epitopes
Antibodies recognize B cell epitopes composed of either linear peptide sequences or conformational determinants, which are present only in the three-dimensional form of the antigen. The discontinuous B cell epitope is the sequence of amino acids or subunits that comprise an antigen and can come in direct interaction with a receptor of the immune system. Therefore, the Ellipro server from the IEDB website was used for the prediction of conformational B cell epitopes in chimeric multi-epitope vaccine with default parameters. Prediction using the ElliPro yielded five discontinuous epitopes as depicted in Supplementary Table 8. Figure 7 depicts the 3D structure of putative B cell epitopes in the designed construct.
Fig. 7.

The conformational B lymphocyte epitopes present in the vaccine. The yellow spheres showing epitopes containing A 43 residues (AA 296-338) with residue score 0.774; B 62 residues (AA 1-6, AA 64-65, AA 68-91, AA 164-173, AA 175, AA 239, and, AA 242-259) with residue score 0.722; C 60 residues (AA 29-30, AA 32-51, AA 53-54, AA 57, AA 60-61, AA 111-126, and, AA 194-210) with residue score 0.703; D 3 residues (AA 92, AA 94, and, AA 98) with residue score 0.543; E 4 residues (AA 176, AA 179-180, and, AA 182) with residue score 0.517
Identification of cytokine-inducing HTL epitopes
IFN-epitope server was utilized to identify MHC-II binding epitopes that can induce interferon-gamma. This web server predicts interferon-gamma–inducing epitopes based on three major modules, including predict, design, and scan. The MHC-II epitopes responsible for the activation of T helper cells can also induce the release of interferon-gamma and activation of downstream signaling. The final vaccine was inserted in the tool to identify the IFN-γ epitopes. The server assigned the SVM score for each input epitope. A total of 330 epitopes were identified of which 102 epitopes had a positive score. The result of the IFN-γ analysis was described in Supplementary Table 9. It is reported in several studies that interleukin-10 and interleukin-17 play vital roles in host protection in the host defense against Acinetobacter baumannii infections and in the septic response [70, 71]. Therefore, the IL-10 and IL-17-inducing abilities of these HTL epitopes were evaluated. We found two selected HTL epitopes were able to induce IL-10; whereas, two selected HTL epitopes were able to induce IL-17.
Molecular docking analysis
Toll-like receptors play a critical role in the onset of inflammation response by recognition of the pathogen [72]. TLR4 is a membrane sensor for lipopolysaccharide and is essential for initiating the innate immune response of macrophages against A. baumannii infection [73, 74]. Knapp et al. have shown that A. baumannii LPS stimulates signaling in murine cells via TLR4 [75, 76]. Therefore, in the present study, the vaccine was docked with TLR4 as these receptors can activate innate immune response by inducing the synthesis of pro-inflammatory mediators, in response to the challenge with A. baumanii. Human TLR4 was obtained from the RCSB PDB database (PDB ID: 5IJB) while the vaccine model was used as a ligand. The best docking position and orientation, successfully predicted by the ClusPro 2.0 and PATCHDOCK servers, was selected from the top-ranked cluster based on the cluster size with the highest number of members and the lowest energy. The model with the lowest binding energy value was preferred for each docked complex. Based on the docking results, favorable interactions were observed between the vaccine-TLR 4 complex. A total number of 23 hydrogen bonds (THR103–LYS66, ASP99–LYS66, ASP100–LYS43, ALA228–LYS43, GLN232–ASP41, ARG 295–ASP41, ARG295–GLN42, PRO125–LYS136, ASN124–LYS136, TYR225–ARG86, GLY319–THR109, ASN318–ASN105, ASN318–TRP81, ASP207–ARG266, THR209–TYR183, THR211–TYR183, ARG213–GLU265, ARG213–ASP264, GLU338–HIS178, GLU338–ARG233, PHE329–ARG223, GLN328–ARG337, GLN311–ARG337) were formed between the vaccine and TLR 4 immune receptor with a distance of 1.8 Å, 1.7 Å, 1.9 Å, 1.7 Å, 2.0 Å, 1.9 Å, 2.4 Å,1.8 Å, 1.7 Å, 2.2 Å,1.8 Å, 2.7 Å, 2.0 Å,1.8 Å, 1.8 Å, 2.6 Å, 1.9 Å,1.8 Å, 3.2 Å, 1.8 Å, 1.8 Å, 1.9 Å, and 2.0 Å, respectively. Figure 8 illustrates the interaction of the vaccine and the receptor protein TLR-4 and their interacting amino acids. The PyMOL molecular graphics system was used to visualize the docking interaction.
Fig. 8.

Molecular interaction of multi-epitope vaccine construct docked with TLR4
Molecular dynamics simulation
The stability of the interaction at the microscopic level of docked complex (TLR-2) and the multi-epitope-based vaccine was assessed by molecular dynamics using Gromacs2019 software. To obtain more accurate perceptions about the binding pattern and conformational variation of the systems, we utilized trajectory-derived root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), the number of hydrogen bonds, and molecular mechanics-Poisson Boltzmann surface area (MM-PBSA) against time-dependent function. Therefore, the RMSD of the C-alpha atom of the complex can provide evidence of complex constancy and stiffness. The results of the molecular dynamics simulation of the vaccine-TLR4 docked complex is illustrated in Fig. 9. During the MD simulations period, the RMSD plot reported that the maximum RMSD value was between 15ns–25ns range (Fig. 9). Further, throughout the entire time frame of 100 ns time-frames, the protein exhibited stability. A high fluctuation in the plot indicates highly flexible regions in the receptor-ligand complex while mild fluctuations show uninterrupted interaction between receptor and ligand molecules. The RMSF plot (Fig. 9) showed fluctuation in the positioning of the amino acid side chains from 600 onwards; this reflects the continual interaction between the multi-subunit vaccine and receptor, whereas regions showing major fluctuations represent highly flexible regions in the protein-receptor complex. The radius of the gyration plot revealed the protein’s compactness around its axes. In addition, H bond formation/deformation between TLR4 and the vaccine molecules was investigated throughout the MD simulation time. The inter-molecular hydrogen bonds have generally increased over the MD simulation time (Fig. 9). This trend confirms the efforts of the two molecules to establish the best inter-molecular interaction as well as corroborates the appropriate dynamic of the studied system.
Fig. 9.

Molecular dynamics simulation of TLR4 and vaccine complex. A RMSD representation of the docked complex protein backbone consists of TLR4 as a receptor and vaccine as a ligand. B RMSF representation of the docked complex protein side chains. C Number of hydrogen bonds formed during the course of MD simulation trajectory. D Radius of Gyration plot in during MD simulation
MMPBSA binding free energy analysis
The free energy of binding (ΔGbind) is believed to be an essential thermodynamic quantity to assess the favorable protein–protein interaction as well as their affinity for accurate modeling of biological systems. In this regard, the g_mmpbsa tool was used to calculate the free binding energy of the MD simulations. MM/PBSA calculates the aforementioned favorable forces, including solvent-accessible surface area (SASA) and unfavorable polar solvation energy (PSE). The MM/PBSA computed free energy of binding for the system was estimated as −261.563 ±185.334 kJ/mol (Supplementary Table 10). The results revealed that the docking was energetically feasible, as indicated by the negative values of Gibbs free energy (ΔG). The negative free binding energy value observed indicates that the vaccine complex is strongly binding to the receptor making it a promising candidate as a vaccine against A. baumannii.
Codon optimization and in silico cloning
Codon bias occurs when an amino acid is encoded by more than one codon in distinct organisms. Because an organism’s cellular machinery differs from that of another, the same amino acid can be transcribed by completely different codons in various animals. As a result, codon adaptation was used in this work to predict the best codon for efficiently encoding a certain amino acid in a specific organism. Reverse translation and codon optimization was performed using the JCAT (Java Codon Adaptation Tool) server. The target organism was prokaryotic E. coli strain K12. Rho-independent transcription terminators, prokaryotic ribosome binding sites, and restriction enzyme cleavage sites EaeI and StyI were all avoided on the server. The CAI of the modified sequence was 0.995, and the upgraded sequence’s GC content was 54.73%, whereas E. coli’s GC level was 50.7%. The codon-optimized vaccine construct sequence was searched for restriction enzyme sites; however, EcoRI and BamHI enzymes were not detected in the vaccine sequence; therefore, these enzymes were employed for in silico cloning. A 6X His tag was inserted for the effective purification and solubilization of the vaccine protein. Finally, after inserting the vaccine design into the pET28a (+) vector, a successful clone of 6383bp was obtained (Fig. 10).
Fig. 10.

Restriction cloning of final multi-epitope vaccine by using pET28a (+) expression vector in the in silico space. Black circle indicates the vector, and the red part is the place where the vaccine is inserted
Immune simulation
Results from the C-ImmSim server showed a significant increase in immune response generation. According to the simulation study, the main immune response to the antigenic fragments was anticipated to grow significantly after each of the three vaccine injections, as represented by the progressive increase in concentrations of various immunoglobulins. Again, secondary immune responses were shown to be enhanced in response to primary immunological stimulation. The presence of high levels of IgM and IgG indicated a humoral immune response (Fig. 11A). The B cell population showed an increase in B memory cells and B cell isotype IgM, as well as a decrease in antigen concentration (Fig. 11B–C). The development of immune memory increased antigen clearance on subsequent exposures (Fig. 11D–F). A substantial response was also detected in the TH1 and TC cell populations with associated memory development. Furthermore, the increased numbers of dendritic cells and macrophages indicated that these antigen-presenting cells or APCs were extremely potential at antigen presentation (Fig. 11G–H). The vaccine was also capable of producing a large number of different cytokines such as IFN-gamma, IFN-beta, IL-10, and IL-23 which are the most essential cytokines for eliciting an immune response against pathogens (Fig. 11I). In addition, these findings were compatible with the induced level of IFN-γ developed after the proposed vaccine construct was immunized. On the other hand, the negligible Simpson index (D) suggests a varied immune response (Fig. 11I).
Fig. 11.

In silico simulation of immune response using vaccine as antigen: A Antigen and immunoglobulins, B B cell population, C B cell population per state, D helper T cell population, E helper T cell population per state, F cytotoxic T cell population per state, G macrophage population per state, H dendritic cell population per state, and I production of cytokine and interleukins with Simpson index (D) of immune response
Discussion
ESKAPE diseases have become a major worldwide public health concern, and scientists are working to produce a vaccine and determine the best strategy to treat the condition. Several immune-bioinformatics technologies have been employed for the strategic development of vaccines that can save time and money which could potentially identify antigenic regions for multi-epitope vaccine development. We can now create peptide vaccines based on the most antigenic epitopes that can elicit a robust immune response since we have adequate information on Acinetobacter baumannii’s genomes and proteomes. These immunoinformatics approaches have had a considerable influence on immunology research, and several instances of in silico design of epitope-based vaccines against bacterial [77], viral [78], parasitic [79], and fungal [80] diseases can be found.
A. baumannii is an extensively drug-resistant lethal human nosocomial bacterium [81]. A. baumannii’s pathogenicity and capacity to cause such a wide spectrum of illnesses are due to its diverse virulence factors. Lipopolysaccharide assembly proteins are one of such virulence factors that promote the accumulation of toxic intermediates, reduce fluidity, and increase the osmotic resistance of the outer membrane [82, 83]. LptD and LptE proteins, which are the main immunogenic components of A. baumannii, have been chosen as important targets for vaccine development since they undergo a robust immune response and modulate pathogenicity. Since the humoral immunity mediated by memory B cells can be overpowered or neutralized by the advent of multi-drug resistant pathogens, it is crucial to design a vaccine construct that can elicit long-standing cell-mediated immunity. To design a novel A. baumannii vaccine based on LptD and LptE proteins for the induction of long-lasting B and T cell–mediated immunity against this pathogen.
A study conducted by, Sogasu et al. suggested certain peptides in OmpA protein as promising epitope vaccine candidates against multi-drug resistant strains of A. baumannii [84]. In another study, Ren et al. proposed a recombinant multi-epitope assembly peptide vaccine construct including FilF and NucAb proteins and evaluated its immunogenicity and protective immunity in Balb/c mice [85]. On the other hand, Song et al. suggested some putative B and T cell epitopes based onMacB protein [86]. Also, Solanki et al. identified novel drug targets by using the subtractive proteomics approach [87], and Song et al. performed epitope screening of a potential vaccine antigen TolB from outer membrane protein [88]. These studies demonstrate the potential of a multi-epitope peptide vaccine targeting multiple antigens as an ideal approach for the prevention of Acinetobacter infection. However, it has been suggested to study several other antigenic proteins of the pathogen for the construction of a novel vaccine construct.
With this background, we employed immunoinformatics methods for n the formulation of a new vaccine using the epitopes from LPS assembly proteins LptD and LptE in a more comprehensive way. We identified epitopes associated with B cells and T cells from two major LPS assembly proteins, LptD and LptE to design a robust vaccine construct being able to induce both humoral and cellular immunity. ABCpred tool was used to predict putative B cell epitopes. As CD8+ and CD4+ T cells are key players in the generation of immunity against bacteria, we tried to evaluate the binding affinity to MHC classes I and II molecules using NetMHCpan 4.1 and IEDB MHC II prediction tools to identify the most immunodominant regions. All of the epitopes were found to be “antigens” analyzed by the VaxigenV2.0 server. Another significant impediment to vaccine development is the possibility of allergenicity since many vaccines provoke an allergic reaction in the immune system. To anticipate possible allergenicity, the AllerTOP server was employed, and all epitopes were classified as “non-allergen”. IEDB class I immunogenicity tool was utilized to find the immunogenicity score of the peptides. Toxinpred server identified all peptides as “non-toxin” with additional information on the characteristics and properties like hydrophobicity, hydrophilicity, hydropathicity, charge, and SVM score, along with the molecular weight of the predicted peptides. From all peptides predicted, two putative epitopes from linear B cell, seventeen epitopes from MHC class I, and two epitopes for MHC class II were chosen based on the criteria of high antigenicity score, non-allergenicity, non-toxicity, high binding affinity, and positive immunogenicity score. To boost immunogenicity, the multi-epitope vaccine was designed by combining adjuvant, T and B cell epitopes with appropriate linkers. Human beta-defensins 1 and 2 are less potent peptide antibiotics and predominantly active against Gram-negative bacteria and yeasts [89]; henceforth, human beta-defensin 3 (HBD3) has been used. HBD3 displays broad specificity against several microorganisms, i.e., P. aeruginosa, E. faecium, S. aureus, and C. albicans. Therefore, HBD3 is a natural human protein that can interact with the components of lipid membranes and also shows promise for non rejectable peptide-based therapeutics [90–92]. Therefore, HBD3 was used as an adjuvant and integrated into the vaccine construct with the EAAAK linker. On the other hand, LBL epitopes were merged with the KK linker while HTL and CTL epitopes were added with GPGPG and AAY linkers, respectively.
Population coverage is another important aspect of vaccine design [93, 94]. The vaccine construct showed coverage of 99% of the world’s population. The physicochemical properties of the constructed vaccine were obtained utilizing the Protparam tool. The construct demonstrated high solubility and stability making it an ideal candidate for the initiation of an immunogenic reaction. PSIPRED predicted the secondary structure of the vaccine which comprises 45.93% alpha-helix, 9.26% beta-turn, and 27.41% random coils. For the 3D modeling, we used the Robetta server to predict the tertiary protein structure and Pymol for visualization of the structures [95]. PROCHECK, ERRAT, and Verify 3D were employed for the validation [96]. The resulting values strongly corroborated the reliability of the developed structure for further analysis. Ellipro server identified five conformational B-cell epitopes indicating the ability of the designed constructs for robust induction of humoral response. Disulfide by Design 2.0 web server predicted 23 pairs of amino acid residues indicating the potential to form a disulfide bond.
A significant amount of IFN-γ, IL-10, and IL-17 was measured as candidates in MHC class II epitopes, as these cytokines can be implicated in the development of T helper cells, which are required in the activation of B cells, macrophages, and cytotoxic T cells [97–99]. Also, peptide-protein docking between the vaccine construct and TLR 4 was performed by the ClusPro server, and the resulting docked conformation showed strong interactions between the designed construct and TLR 4. The conformational stability of the vaccine-receptor docked complex was analyzed by MD simulation using the gromacs software [100–110]. RMSD and RMSF plots showed continuity throughout the time period of 100ns. In order to ensure effective expression and translation in E. coli as a host, in silico cloning and codon optimization were predicted. Finally, to describe the immunogenic response profile of the vaccine, an immune simulation was performed. Keeping these in silico observations into consideration, the safety and efficacy of the multi-epitope vaccine need to be clinically ascertained.
Conclusion
Vaccine design and development is an inherently laborious process, but computational immunology methods have the potential to reduce the efforts greatly. The epitope-based vaccines are feasible and capable of inducing a protective immune response. In the current research, we have made an attempt to predict the B and T cell epitopes for LPS assembly proteins of A. baumannii, and the predicted minimal epitope sets may act as probable vaccine candidates in the development of a new vaccine. The population coverage analysis reveals that the proposed epitopes may work more effectively on a large scale of the human population. Taken all together, according to immunological analysis, structural and physicochemical characterizations, the vaccine candidate needs to be further supported by in vitro and in vivo validation through laboratory experiments for consideration as a possible multi-epitope vaccine against Acinetobacter infection.
Supplementary information
Predicted B-cell epitopes epitopes from A. baumannii LPS assembly protein (LptD and LptE) and their corresponding immunogenic properties. The final selected epitopes to design multi-epitope vaccine construct from proteins has been highlighted in green colour.
Predicted CTL epitopes from A. baumannii LPS assembly protein (LptD and LptE) with their corresponding MHC Class I alleles and their immunogenic properties. The final selected epitopes to design multi-epitope vaccine construct from proteins has been highlighted in green colour.
Predicted HTL epitopes from A. baumannii LPS assembly protein (LptD and LptE) with their corresponding MHC Class II alleles and their immunogenic properties. The final selected epitopes to design multi-epitope vaccine construct from proteins has been highlighted in green colour.
Population Coverage analysis of the final vaccine construct using the population coverage analysis tool of the IEDB database by keeping the default parameters on (109 countries covering 16 different geographical regions).
Physicochemical Properties, secondary structure, and solubility analysis of final vaccine construct as predicted by ProtParam tool, PSIPRED, and SOLpro server.
Acinetobacter baumannii strain with their Accession Id, organism, strain, isolate, host, country and year.
Disulfide engineering of the final multiepitope vaccine.
Discontinuous B-cell epitopes with their residue scores predicted by the ElliPro server.
The predicted IFN-γ inducing epitopes from the proposed vaccine using IFNepitope server.
MM/PBSA free energy calculations of docked complex
Acknowledgements
We acknowledge the infrastructure support available through the DBT-BUILDER program (BT/INF/22/SP42155/2021) at KIIT Deemed to Be University, Bhubaneswar.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Jyotirmayee Dey and Soumya Ranjan Mahapatra contributed equally to this work.
Contributor Information
Namrata Misra, Email: namrata@kiitincubator.in.
Mrutyunjay Suar, Email: msuar@kiitbiotech.ac.in.
References
- 1.Peleg AY, Seifert H, Paterson DL. Acinetobacter baumannii: emergence of a successful pathogen. Clin Microbiol Rev. 2008;21(3):538–582. doi: 10.1128/CMR.00058-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antunes LC, Visca P, Towner KJ. Acinetobacter baumannii: evolution of a global pathogen. Pathog Dis. 2014;71(3):292–301. doi: 10.1111/2049-632X.12125. [DOI] [PubMed] [Google Scholar]
- 3.Baumann P. Isolation of Acinetobacter from soil and water. J Bacteriol. 1968;96(1):39–42. doi: 10.1128/jb.96.1.39-42.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Howard A, O’Donoghue M, Feeney A, Sleator RD. Acinetobacter baumannii: an emerging opportunistic pathogen. Virulence. 2012;3(3):243–250. doi: 10.4161/viru.19700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sebeny PJ, Riddle MS, Petersen K. Acinetobacter baumannii skin and soft-tissue infection associated with war trauma. Clin Infect Dis. 2008;47(4):444–449. doi: 10.1086/590568. [DOI] [PubMed] [Google Scholar]
- 6.Anstey NM, Currie BJ, Hassell M, Palmer D, Dwyer B, Seifert H. Community-acquired bacteremic Acinetobacter pneumonia in tropical Australia is caused by diverse strains of Acinetobacter baumannii, with carriage in the throat in at-risk groups. J Clin Microbiol. 2002;40(2):685–686. doi: 10.1128/JCM.40.2.685-686.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eze EC, Chenia HY, El Zowalaty ME. Acinetobacter baumannii biofilms: effects of physicochemical factors, virulence, antibiotic resistance determinants, gene regulation, and future antimicrobial treatments. Infect Drug Resist. 2018;11:2277. doi: 10.2147/IDR.S169894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eveillard M, Soltner C, Kempf M, Saint-André JP, Lemarié C, Randrianarivelo C, et al. The virulence variability of different Acinetobacter baumannii strains in experimental pneumonia. J Infect. 2010;60(2):154–161. doi: 10.1016/j.jinf.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 9.De Breij A, Eveillard M, Dijkshoorn L, Van Den Broek PJ, Nibbering PH, Joly-Guillou ML. Differences in Acinetobacter baumannii strains and host innate immune response determine morbidity and mortality in experimental pneumonia. PLoS One. 2012;7(2):e30673. doi: 10.1371/journal.pone.0030673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fournier PE, Vallenet D, Barbe V, Audic S, Ogata H, Poirel L, et al. Comparative genomics of multidrug resistance in Acinetobacter baumannii. PLoS Genet. 2006;2(1):e7. doi: 10.1371/journal.pgen.0020007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cai Y, Chai D, Wang R, Liang B, Bai N. Colistin resistance of Acinetobacter baumannii: clinical reports, mechanisms and antimicrobial strategies. J Antimicrob Chemother. 2012;67(7):1607–1615. doi: 10.1093/jac/dks084. [DOI] [PubMed] [Google Scholar]
- 12.Tan YC, Lahiri C. Promising acinetobacter baumannii vaccine candidates and drug targets in recent years. Front Immunol. 2022;13 [DOI] [PMC free article] [PubMed]
- 13.Aliramezani A, Soleimani M, Fard RMN, Nojoomi F. Virulence determinants and biofilm formation of Acinetobacter baumannii isolated from hospitalized patients. Germs. 2019;9(3):148. doi: 10.18683/germs.2019.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruiz N, Kahne D, Silhavy T. Transport of lipopolysaccharide across the cell envelope: the long road of discovery. Nat Rev Microbiol. 2009;7:677–683. doi: 10.1038/nrmicro2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Villa R, Martorana AM, Okuda S, Gourlay LJ, Nardini M, Sperandeo P, Dehò G, Bolognesi M, Kahne D. A. Polissi. J Bacteriol. 2013;195:1100–1108. doi: 10.1128/JB.02057-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dong H, Xiang Q, Gu Y, Wang Z, Paterson NG, Stansfeld PJ, et al. Structural basis for outer membrane lipopolysaccharide insertion. Nature. 2014;511(7507):52–56. doi: 10.1038/nature13464. [DOI] [PubMed] [Google Scholar]
- 17.Qiao S, Luo Q, Zhao Y, Zhang XC, Huang Y. Structural basis for lipopolysaccharide insertion in the bacterial outer membrane. Nature. 2014;511(7507):108–111. doi: 10.1038/nature13484. [DOI] [PubMed] [Google Scholar]
- 18.Zha Z, Li C, Li W, Ye Z, Pan J. LptD is a promising vaccine antigen and potential immunotherapeutic target for protection against Vibrio species infection. Sci Rep. 2016;6(1):1–13. doi: 10.1038/srep38577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Botos I, Majdalani N, Mayclin SJ, McCarthy JG, Lundquist K, Wojtowicz D, et al. Structural and functional characterization of the LPS transporter LptDE from Gram-negative pathogens. Structure. 2016;24(6):965–976. doi: 10.1016/j.str.2016.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beiranvand S, Doosti A, Mirzaei SA. Putative novel B-cell vaccine candidates identified by reverse vaccinology and genomics approaches to control Acinetobacter baumannii serotypes. Infect Genet Evol. 2021;96:105138. doi: 10.1016/j.meegid.2021.105138. [DOI] [PubMed] [Google Scholar]
- 21.Soltan MA, Behairy MY, Abdelkader MS, Albogami S, Fayad E, Eid RA, et al. In silico designing of an epitope-based vaccine against common E. coli pathotypes. Front Med. 2022;9 [DOI] [PMC free article] [PubMed]
- 22.McConnell MJ, Pachón J. Active and passive immunization against Acinetobacter baumannii using an inactivated whole cell vaccine. Vaccine. 2010;29(1):1–5. doi: 10.1016/j.vaccine.2010.10.052. [DOI] [PubMed] [Google Scholar]
- 23.Huang W, Yao Y, Long Q, Yang X, Sun W, Liu C, et al. Immunization against multidrug-resistant Acinetobacter baumannii effectively protects mice in both pneumonia and sepsis models. PLoS One. 2014;9(6):e100727. doi: 10.1371/journal.pone.0100727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahmad TA, Tawfik DM, Sheweita SA, Haroun M, El-Sayed LH. Development of immunization trials against Acinetobacter baumannii. Trials Vaccinol. 2016;5:53–60. doi: 10.1016/j.trivac.2016.03.001. [DOI] [Google Scholar]
- 25.Bentancor LV, O'Malley JM, Bozkurt-Guzel C, Pier GB, Maira-Litrán T. Poly-N-acetyl-β-(1-6)-glucosamine is a target for protective immunity against Acinetobacter baumannii infections. Infect Immun. 2012;80(2):651–656. doi: 10.1128/IAI.05653-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gahery H, Daniel N, Charmeteau B, Ourth L, Jackson A, Andrieu M, et al. New CD4+ and CD8+ T cell responses induced in chronically HIV type-1-infected patients after immunizations with an HIV type 1 lipopeptide vaccine. AIDS Res Hum Retrovir. 2006;22(7):684–694. doi: 10.1089/aid.2006.22.684. [DOI] [PubMed] [Google Scholar]
- 27.Malik AA, Ojha SC, Schaduangrat N, Nantasenamat C. ABCpred: a webserver for the discovery of acetyl-and butyryl-cholinesterase inhibitors. Mol Divers. 2021;26:467–487. doi: 10.1007/s11030-021-10292-6. [DOI] [PubMed] [Google Scholar]
- 28.Reynisson B, Alvarez B, Paul S, Peters B, Nielsen M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020;48(W1):W449–W454. doi: 10.1093/nar/gkaa379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lund O, Nielsen M, Kesmir C, Petersen AG, Lundegaard C, Worning P, et al. Definition of supertypes for HLA molecules using clustering of specificity matrices. Immunogenetics. 2004;55(12):797–810. doi: 10.1007/s00251-004-0647-4. [DOI] [PubMed] [Google Scholar]
- 30.Kim Y, Ponomarenko J, Zhu Z, Tamang D, Wang P, Greenbaum J, et al. Immune epitope database analysis resource. Nucleic Acids Res. 2012;40(W1):W525–W530. doi: 10.1093/nar/gks438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC bioinform. 2007;8(1):1–7. doi: 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dimitrov I, Flower DR, Doytchinova I. BMC bioinformatics. BioMed Central; 2013. AllerTOP-a server for in silico prediction of allergens; pp. 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paul S, Kolla RV, Sidney J, Weiskopf D, Fleri W, Kim Y, et al. Evaluating the immunogenicity of protein drugs by applying in vitro MHC binding data and the immune epitope database and analysis resource. Clin Dev Immunol. 2013;2013 10.1155/2013/467852. [DOI] [PMC free article] [PubMed]
- 34.Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, Raghava GP. Peptide toxicity prediction. Comput peptidol. 2015:143–57. 10.1007/978-1-4939-2285-7_7. [DOI] [PubMed]
- 35.Nain Z, Karim MM, Sen MK, Adhikari UK. Structural basis and designing of peptide vaccine using PE-PGRS family protein of Mycobacterium ulcerans—an integrated vaccinomics approach. Mol Immunol. 2020;120:146–163. doi: 10.1016/j.molimm.2020.02.009. [DOI] [PubMed] [Google Scholar]
- 36.Bui HH, Sidney J, Dinh K, Southwood S, Newman MJ, Sette A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinform. 2006;7(1):1–5. doi: 10.1186/1471-2105-7-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gasteiger E, Hoogland C, Gattiker A, Duvaud SE, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy server. Humana press; 2005. pp. 571–607. [DOI] [PubMed] [Google Scholar]
- 38.McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16(4):404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- 39.Magnan CN, Randall A, Baldi P. SOLpro: accurate sequence-based prediction of protein solubility. Bioinformatics. 2009;25(17):2200–2207. doi: 10.1093/bioinformatics/btp386. [DOI] [PubMed] [Google Scholar]
- 40.Bui HH, Sidney J, Li W, Fusseder N, Sette A. Development of an epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC bioinform. 2007;8:1–6. doi: 10.1186/1471-2105-8-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim DE, Chivian D, Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004;32(suppl_2):W526–W531. doi: 10.1093/nar/gkh468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruff KM, Pappu RV. AlphaFold and implications for intrinsically disordered proteins. J Mol Biol. 2021;433(20):167208. doi: 10.1016/j.jmb.2021.167208. [DOI] [PubMed] [Google Scholar]
- 43.Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yuan S, Chan HS, Hu Z. Using PyMOL as a platform for computational drug design. Wiley Interdiscip Rev Comput Mol Sci. 2017;7(2):e1298. doi: 10.1002/wcms.1298. [DOI] [Google Scholar]
- 45.Heo L, Park H, Seok C. GalaxyRefine: protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013;41(W1):W384–W388. doi: 10.1093/nar/gkt458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26(2):283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- 47.Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993;2(9):1511–1519. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eisenberg D, Lüthy R, Bowie JU. Methods in enzymology. Academic Press; 1997. [20] VERIFY3D: assessment of protein models with three-dimensional profiles; pp. 396–404. [DOI] [PubMed] [Google Scholar]
- 49.Craig DB, Dombkowski AA. Disulfide by Design 2.0: a web-based tool for disulfide engineering in proteins. BMC bioinform. 2013;14(1):1–7. doi: 10.1186/1471-2105-14-S19-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ponomarenko J, Bui HH, Li W, Fusseder N, Bourne PE, Sette A, Peters B. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC bioinform. 2008;9(1):1–8. doi: 10.1186/1471-2105-9-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dhanda SK, Vir P, Raghava GP. Designing of interferon-gamma inducing MHC class-II binders. Biol Direct. 2013;8(1):1–15. doi: 10.1186/1745-6150-8-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nagpal G, Usmani SS, Dhanda SK, Kaur H, Singh S, Sharma M, Raghava GP. Computer-aided designing of immunosuppressive peptides based on IL-10 inducing potential. Sci Rep. 2017;7(1):1–10. doi: 10.1038/srep42851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gupta S, Mittal P, Madhu MK, Sharma VK. IL17eScan: a tool for the identification of peptides inducing IL-17 response. Front Immunol. 2017;8:1430. doi: 10.3389/fimmu.2017.01430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, et al. The ClusPro web server for protein–protein docking. Nat Protoc. 2017;12(2):255–278. doi: 10.1038/nprot.2016.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19–25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- 56.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26(16):1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 57.Bjelkmar P, Larsson P, Cuendet MA, Hess B, Lindahl E. Implementation of the CHARMM force field in GROMACS: analysis of protein stability effects from correction maps, virtual interaction sites, and water models. J Chem Theory Comput. 2010;6(2):459–466. doi: 10.1021/ct900549r. [DOI] [PubMed] [Google Scholar]
- 58.Kumari R, Kumar R, Open Source Drug Discovery Consortium. Lynn A. g_mmpbsa A GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inf Model. 2014;54(7):1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- 59.Grote A, Hiller K, Scheer M, Münch R, Nörtemann B, Hempel DC, Jahn D. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005;33(suppl_2):W526–W531. doi: 10.1093/nar/gki376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rapin N, Lund O, Castiglione F. Immune system simulation online. Bioinformatics. 2011;27(14):2013–2014. doi: 10.1093/bioinformatics/btr335. [DOI] [PubMed] [Google Scholar]
- 61.Plotnicky H, Cyblat-Chanal D, Aubry JP, Derouet F, Klinguer-Hamour C, Beck A, et al. The immunodominant influenza matrix T cell epitope recognized in human induces influenza protection in HLA-A2/Kb transgenic mice. Virology. 2003;309(2):320–329. doi: 10.1016/S0042-6822(03)00072-2. [DOI] [PubMed] [Google Scholar]
- 62.De Groot AS, Moise L, McMurry JA, Martin W. Epitope-based immunome-derived vaccines: a strategy for improved design and safety. In: Falus A, editor. Clinical Applications of Immunomics. Immunomics Reviews (An Official Publication of the International Immunomics Society) New York, NY: Springer; 2009. [Google Scholar]
- 63.Pulendran B, Arunachalam PS, O’Hagan DT. Emerging concepts in the science of vaccine adjuvants. Nat Rev Drug Discov. 2021;20(6):454–475. doi: 10.1038/s41573-021-00163-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Feng Z, Jia X, Adams MD, Ghosh SK, Bonomo RA, Weinberg A. Epithelial innate immune response to Acinetobacter baumannii challenge. Infect Immun. 2014;82(11):4458–4465. doi: 10.1128/IAI.01897-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen W. Host innate immune responses to Acinetobacter baumannii infection. Front Cell Infect Microbiol. 2020;10 10.3389/fcimb.2020.00486. [DOI] [PMC free article] [PubMed]
- 66.Sarkar B, Ullah MA, Araf Y, Islam NN, Zohora US. Immunoinformatics-guided designing and in silico analysis of epitope-based polyvalent vaccines against multiple strains of human coronavirus (HCoV) Expert Rev Vaccines. 2021;21(12):1–21. doi: 10.1080/14760584.2021.1874925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang Y, Sun W, Guo J, Zhao G, Sun S, Yu H, et al. In silico design of a DNA-based HIV-1 multi-epitope vaccine for Chinese populations. Hum Vaccin Immunother. 2015;11(3):795–805. doi: 10.1080/21645515.2015.1012017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reche PA, Reinherz EL. Sequence variability analysis of human class I and class II MHC molecules: functional and structural correlates of amino acid polymorphisms. J Mol Biol. 2003;331(3):623–641. doi: 10.1016/S0022-2836(03)00750-2. [DOI] [PubMed] [Google Scholar]
- 69.Maenaka K, Jones EY. MHC superfamily structure and the immune system. Curr Opin Struct Biol. 1999;9(6):745–753. doi: 10.1016/S0959-440X(99)00039-1. [DOI] [PubMed] [Google Scholar]
- 70.Kang MJ, Jang AR, Park JY, Ahn JH, Lee TS, Kim DY, et al. IL-10 protects mice from the lung infection of Acinetobacter baumannii and contributes to bacterial clearance by regulating STAT3-mediated MARCO expression in macrophages. Front Immunol. 2020;11:270. doi: 10.3389/fimmu.2020.00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yan Z, Yang J, Hu R, Hu X, Chen K. Acinetobacter baumannii infection and IL-17 mediated immunity. Mediat Inflamm. 2016;2016 10.1155/2016/9834020. [DOI] [PMC free article] [PubMed]
- 72.El-Zayat SR, Sibaii H, Mannaa FA. Toll-like receptors activation, signaling, and targeting: an overview. Bull Natl Res Cent. 2019;43(1):1–12. doi: 10.1186/s42269-019-0227-2. [DOI] [Google Scholar]
- 73.Wang Y, Zhang X, Feng X, Liu X, Deng L, Liang ZA. Expression of Toll-like receptor 4 in lungs of immune-suppressed rat with Acinetobacter baumannii infection. Exp Ther Med. 2016;12(4):2599–2605. doi: 10.3892/etm.2016.3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim CH, Jeong YJ, Lee J, Jeon SJ, Park SR, Kang MJ, et al. Essential role of toll-like receptor 4 in Acinetobacter baumannii-induced immune responses in immune cells. Microb Pathog. 2013;54:20–25. doi: 10.1016/j.micpath.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 75.Erridge C, Moncayo-Nieto OL, Morgan R, Young M, Poxton IR. Acinetobacter baumannii lipopolysaccharides are potent stimulators of human monocyte activation via Toll-like receptor 4 signalling. J Med Microbiol. 2007;56(2):165–171. doi: 10.1099/jmm.0.46823-0. [DOI] [PubMed] [Google Scholar]
- 76.Perez F, Endimiani A, Bonomo RA. Why are we afraid of Acinetobacter baumannii? Expert Rev Anti-Infect Ther. 2008;6(3):269–271. doi: 10.1586/14787210.6.3.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Farhadi T, Nezafat N, Ghasemi Y, Karimi Z, Hemmati S, Erfani N. Designing of complex multi-epitope peptide vaccine based on omps of Klebsiella pneumoniae: an in silico approach. Int J Pept Res Ther. 2015;21(3):325–341. doi: 10.1007/s10989-015-9461-0. [DOI] [Google Scholar]
- 78.Mahapatra SR, Sahoo S, Dehury B, Raina V, Patro S, Misra N, Suar M. Designing an efficient multi-epitope vaccine displaying interactions with diverse HLA molecules for an efficient humoral and cellular immune response to prevent COVID-19 infection. Expert Rev Vaccines. 2020;19(9):871–885. doi: 10.1080/14760584.2020.1811091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Khan MAA, Ami JQ, Faisal K, Chowdhury R, Ghosh P, Hossain F, et al. An immunoinformatic approach driven by experimental proteomics: in silico design of a subunit candidate vaccine targeting secretory proteins of Leishmania donovani amastigotes. Parasit Vectors. 2020;13(1):1–21. doi: 10.1186/s13071-020-04064-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Akhtar N, Joshi A, Kaushik V, Kumar M, Mannan MAU. In-silico design of a multivalent epitope-based vaccine against Candida auris. Microb Pathog. 2021;155:104879. doi: 10.1016/j.micpath.2021.104879. [DOI] [PubMed] [Google Scholar]
- 81.Bist P, Dikshit N, Koh TH, Mortellaro A, Tan TT, Sukumaran B. The Nod1, Nod2, and Rip2 axis contributes to host immune defense against intracellular Acinetobacter baumannii infection. Infect Immun. 2014;82(3):1112–1122. doi: 10.1128/IAI.01459-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ayoub Moubareck C, Hammoudi Halat D. Insights into Acinetobacter baumannii: a review of microbiological, virulence, and resistance traits in a threatening nosocomial pathogen. Antibiotics. 2020;9(3):119. doi: 10.3390/antibiotics9030119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Morris FC, Dexter C, Kostoulias X, Uddin MI, Peleg AY. The mechanisms of disease caused by Acinetobacter baumannii. Front Microbiol. 2019;10:1601. doi: 10.3389/fmicb.2019.01601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sogasu D, Girija AS, Gunasekaran S, Priyadharsini JV. Molecular characterization and epitope-based vaccine predictions for ompA gene associated with biofilm formation in multidrug-resistant strains of A. baumannii. In Silico. Pharmacology. 2021;9(1):1–11. doi: 10.1007/s40203-020-00074-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ren S, Guan L, Dong Y, Wang C, Feng L, Xie Y. Design and evaluation of a multi-epitope assembly peptide vaccine against Acinetobacter baumannii infection in mice. Swiss Med Wkly. 2019;149(2324) 10.4414/smw.2019.20052. [DOI] [PubMed]
- 86.Song X, Zhao G, Ding M. Antigen epitope developed based on acinetobacter baumannii MacB protein can provide partial immune protection in mice. Biomed Res Int. 2020;2020 10.1155/2020/1975875. [DOI] [PMC free article] [PubMed]
- 87.Solanki V, Tiwari V. Subtractive proteomics to identify novel drug targets and reverse vaccinology for the development of chimeric vaccine against Acinetobacter baumannii. Sci Rep. 2018;8(1):1–19. doi: 10.1038/s41598-018-26689-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Song X, Zhang H, Zhang D, Xie W, Zhao G. Bioinformatics analysis and epitope screening of a potential vaccine antigen TolB from Acinetobacter baumannii outer membrane protein. Infect Genet Evol. 2018;62:73–79. doi: 10.1016/j.meegid.2018.04.019. [DOI] [PubMed] [Google Scholar]
- 89.Harder J, Bartels J, Christophers E, Schröder JM. Isolation and Characterization of Human μ-Defensin-3, a Novel Human Inducible Peptide Antibiotic. J Biol Chem. 2001;276(8):5707–5713. doi: 10.1074/jbc.M008557200. [DOI] [PubMed] [Google Scholar]
- 90.Dhople V, Krukemeyer A, Ramamoorthy A. The human beta-defensin-3, an antibacterial peptide with multiple biological functions. Biochim Biophys Acta Biomembr BBA-Biomembranes. 2006;1758(9):1499–1512. doi: 10.1016/j.bbamem.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 91.García JR, Jaumann F, Schulz S, Krause A, Rodríguez-Jiménez J, Forssmann U, et al. Identification of a novel, multifunctional β-defensin (human β-defensin 3) with specific antimicrobial activity. Cell Tissue Res. 2001;306(2):257–264. doi: 10.1007/s004410100433. [DOI] [PubMed] [Google Scholar]
- 92.Hoover DM, Wu Z, Tucker K, Lu W, Lubkowski J. Antimicrobial characterization of human β-defensin 3 derivatives. Antimicrob Agents Chemother. 2003;47(9):2804–2809. doi: 10.1128/AAC.47.9.2804-2809.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kalita J, Padhi AK, Tripathi T. Designing a vaccine for fascioliasis using immunogenic 24 kDa mu-class glutathione s-transferase. Infect Genet Evol. 2020;83(104352):32387753. doi: 10.1016/j.meegid.2020.104352. [DOI] [PubMed] [Google Scholar]
- 94.Tahir ul Qamar M, Shokat Z, Muneer I, Ashfaq UA, Javed H, Anwar F, et al. Multiepitope-based subunit vaccine design and evaluation against respiratory syncytial virus using reverse vaccinology approach. Vaccines. 2020;8(2):288. doi: 10.3390/vaccines8020288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mahapatra SR, Dey J, Kaur T, Sarangi R, Bajoria AA, Kushwaha GS, et al. Immunoinformatics and molecular docking studies reveal a novel Multi-Epitope peptide vaccine against pneumonia infection. Vaccine. 2021;39(42):6221–6237. doi: 10.1016/j.vaccine.2021.09.025. [DOI] [PubMed] [Google Scholar]
- 96.Dey J, Mahapatra SR, Singh P, Patro S, Kushwaha GS, Misra N, Suar M. B and T cell epitope-based peptides predicted from clumping factor protein of Staphylococcus aureus as vaccine targets. Microb Pathog. 2021;160:105171. doi: 10.1016/j.micpath.2021.105171. [DOI] [PubMed] [Google Scholar]
- 97.Kalita P, Lyngdoh DL, Padhi AK, Shukla H, Tripathi T. Development of multi-epitope driven subunit vaccine against Fasciola gigantica using immunoinformatics approach. Int J Biol Macromol. 2019;138(224-233):31279880. doi: 10.1016/j.ijbiomac.2019.07.024. [DOI] [PubMed] [Google Scholar]
- 98.Oyarzun P, Kashyap M, Fica V, Salas-Burgos A, Gonzalez-Galarza FF, McCabe A, et al. A proteome-wide immunoinformatics tool to accelerate T-cell epitope discovery and vaccine design in the context of emerging infectious diseases: an ethnicity-oriented approach. Front Immunol. 2021;12:33717077. doi: 10.3389/fimmu.2021.598778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chatterjee R, Sahoo P, Mahapatra SR, Dey J, Ghosh M, Kushwaha GS, et al. Development of a conserved chimeric vaccine for induction of strong immune response against Staphylococcus aureus using immunoinformatics approaches. Vaccines. 2021;9(9):1038. doi: 10.3390/vaccines9091038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mahapatra SR, Dey J, Kushwaha GS, Puhan P, Mohakud NK, Panda SK, et al. Immunoinformatic approach employing modeling and simulation to design a novel vaccine construct targeting MDR efflux pumps to confer wide protection against typhoidal Salmonella serovars. J Biomol Struct Dyn. 2021;40(22):11809–11821. doi: 10.1080/07391102.2021.1964600. [DOI] [PubMed] [Google Scholar]
- 101.Narang PK, Dey J, Mahapatra SR, Ghosh M, Misra N, Suar M, et al. Functional annotation and sequence-structure characterization of a hypothetical protein putatively involved in carotenoid biosynthesis in microalgae. S Afr J Bot. 2021;141:219–226. doi: 10.1016/j.sajb.2021.04.014. [DOI] [Google Scholar]
- 102.Narang PK, Dey J, Mahapatra SR, Roy R, Kushwaha GS, Misra N, Raina V. Genome-based identification and comparative analysis of enzymes for carotenoid biosynthesis in microalgae. World J Microbiol Biotechnol. 2022;38(1):1–22. doi: 10.1007/s11274-021-03188-y. [DOI] [PubMed] [Google Scholar]
- 103.Dey J, Mahapatra SR, Lata S, Patro S, Misra N, Suar M. Exploring Klebsiella pneumoniae capsule polysaccharide proteins to design multiepitope subunit vaccine to fight against pneumonia. Expert Rev Vaccines. 2021;21(4):569–587. doi: 10.1080/14760584.2022.2021882. [DOI] [PubMed] [Google Scholar]
- 104.Chatterjee R, Mahapatra SR, Dey J, Raj TK, Raina V, Misra N, Suar M. An immunoinformatics and structural vaccinology study to design a multi-epitope vaccine against Staphylococcus aureus infection. J Mol Recognit. 2023;36(4):e3007. doi: 10.1002/jmr.3007. [DOI] [PubMed] [Google Scholar]
- 105.Mahapatra SR, Dey J, Raj TK, Misra N, Suar M. Designing a next-generation multiepitope-based vaccine against Staphylococcus aureus using reverse vaccinology approaches. Pathogens. 2023;12(3):376. doi: 10.3390/pathogens12030376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mahapatra SR, Dey J, Jaiswal A, Roy R, Misra N, Suar M. Immunoinformatics-guided designing of epitope-based subunit vaccine from Pilus assembly protein of Acinetobacter baumannii bacteria. J Immunol Methods. 2022;508:113325. doi: 10.1016/j.jim.2022.113325. [DOI] [PubMed] [Google Scholar]
- 107.Sahoo P, Dey J, Mahapatra SR, Ghosh A, Jaiswal A, Padhi S, et al. Nanotechnology and COVID-19 convergence: toward new planetary health interventions against the pandemic. OMICS: A Journal of. Integr Biol. 2022;26(9):473–488. doi: 10.1089/omi.2022.0072. [DOI] [PubMed] [Google Scholar]
- 108.Dey J, Mahapatra SR, Patnaik S, Lata S, Kushwaha GS, Panda RK, et al. Molecular characterization and designing of a novel multiepitope vaccine construct against Pseudomonas aeruginosa. Int J Pept Res Ther. 2022;28(2):49. doi: 10.1007/s10989-021-10356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mahapatra SR, Dey J, Raj TK, Kumar V, Ghosh M, Verma KK, et al. The potential of plant-derived secondary metabolites as novel drug candidates against Klebsiella pneumoniae: molecular docking and simulation investigation. S Afr J Bot. 2022;149:789–797. doi: 10.1016/j.sajb.2022.04.043. [DOI] [Google Scholar]
- 110.Dey J, Mahapatra SR, Raj TK, Kaur T, Jain P, Tiwari A, et al. Designing a novel multi-epitope vaccine to evoke a robust immune response against pathogenic multidrug-resistant Enterococcus faecium bacterium. Gut Pathogens. 2022;14(1):1–20. doi: 10.1186/s13099-022-00495-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Predicted B-cell epitopes epitopes from A. baumannii LPS assembly protein (LptD and LptE) and their corresponding immunogenic properties. The final selected epitopes to design multi-epitope vaccine construct from proteins has been highlighted in green colour.
Predicted CTL epitopes from A. baumannii LPS assembly protein (LptD and LptE) with their corresponding MHC Class I alleles and their immunogenic properties. The final selected epitopes to design multi-epitope vaccine construct from proteins has been highlighted in green colour.
Predicted HTL epitopes from A. baumannii LPS assembly protein (LptD and LptE) with their corresponding MHC Class II alleles and their immunogenic properties. The final selected epitopes to design multi-epitope vaccine construct from proteins has been highlighted in green colour.
Population Coverage analysis of the final vaccine construct using the population coverage analysis tool of the IEDB database by keeping the default parameters on (109 countries covering 16 different geographical regions).
Physicochemical Properties, secondary structure, and solubility analysis of final vaccine construct as predicted by ProtParam tool, PSIPRED, and SOLpro server.
Acinetobacter baumannii strain with their Accession Id, organism, strain, isolate, host, country and year.
Disulfide engineering of the final multiepitope vaccine.
Discontinuous B-cell epitopes with their residue scores predicted by the ElliPro server.
The predicted IFN-γ inducing epitopes from the proposed vaccine using IFNepitope server.
MM/PBSA free energy calculations of docked complex