

ABSTRACT
Accumulating evidence shows that the gastric bacterial community may contribute to the development of gastric cancer (GC). However, the reported alterations of gastric microbiota were not always consistent among the literature. To assess reproducible signals in gastric microbiota during the progression of GC across studies, we performed a meta-analysis of nine publicly available 16S datasets with standard tools of the state-of-the-art. Despite study-specific batch effect, significant changes in the composition of the gastric microbiome were found during the progression of gastric carcinogenesis, especially when the Helicobacter pylori (HP) reads were removed from analyses to mitigate its compositional effect as they accounted for extremely large proportions of sequencing depths in many gastric samples. Differential microbes, including Fusobacterium, Leptotrichia, and several lactic acid bacteria such as Bifidobacterium, Lactobacillus, and Streptococcus anginosus, which were frequently and significantly enriched in GC patients compared with gastritis across studies, had good discriminatory capacity to distinguish GC samples from gastritis. Oral microbes were significantly enriched in GC compared to precancerous stages. Intriguingly, we observed mutual exclusivity of different HP species across studies. In addition, the comparison between gastric fluid and mucosal microbiome suggested their convergent dysbiosis during gastric disease progression. Taken together, our systematic analysis identified novel and consistent microbial patterns in gastric carcinogenesis.
KEYWORDS: Meta-analysis, gastric cancer, gastric microbiome, Helicobacter pylori, oral microbes, compositional effect
Introduction
Gastric cancer (GC) is the fifth most common malignancy and the fourth-leading cause of cancer death worldwide, with an estimate of 1,089,103 new cases and 768,793 deaths in 2020.1 It develops through sequential stages from chronic gastritis, intestinal metaplasia (IM) to GC according to Correa’s model.2 Multiple host-related factors have been reported to contribute to the susceptibility of GC, including host genetics, lifestyle, diet, and age.3 Besides, research also suggests the involvement of gastric microbiota in gastric tumorigenesis.4 In particular, Helicobacter pylori (HP) was classified to a type I carcinogen by the World Health Organization in 1994.5 As a major risk for GC, it plays an initial role in the carcinogenesis cascade via triggering chronic inflammation and mucosal damage.6,7 However, less than 3% of HP-infected individuals eventually develop GC,8,9 and approximately 18% of gastritis patients are HP-negative,10 implying other potential factors driving gastric carcinogenesis. Studies have shown that bacteria other than HP may also contribute to GC.11 For example, HP-monoinfected INS-GAS mice had delayed onset of GC compared to HP-infected INS-GAS mice with complex gastric microbiota.12 A study performed on a Chinese cohort identified five non-HP microbial biomarkers that had significant centralities in the GC microbiome network, which distinguished GC from superficial gastritis samples.13 Taken together, these observations indicate that the gastric microbiome may be a crucial etiological element in GC and may have potential implications for disease diagnosis.
To date, several studies have investigated the gastric microbiome during GC progression using 16S ribosomal RNA (rRNA) gene amplicon sequencing.13–21 Although these studies consistently reported dysbiosis of the GC microbiota, bacterial changes associated with GC were conflicting between studies. For example, the alpha diversity of the mucosal microbiome was found decreased in GC compared to precancerous stages in some studies,13,17,22–24 while others observed the opposite trend.16,25,26 Moreover, the reported GC-associated microbes varied across studies. Castaño-Rodríguez et al. found marked increases in the relative abundances of Lactococcus, Fusobacterium, Veillonella, Haemophilus, and Leptochichia in GC patients compared to control samples.16 Coker et al. reported significant enrichments of Parvimonas micra, Dialister pneumosintes, Slackia exigua, Peptostreptococcus stomatis, and Streptococcus anginosus in GC compared with precancerous stages.13 Another study revealed that Achromobacter, Citrobacter, Clostridium, Lactobacillus, and Rhodococcus were positively associated with GC.23 Collectively, consistent microbial changes in gastric carcinogenesis across studies are still lacking.
Studies varied widely in analytical methods, bioinformatic techniques, sampling strategies, participant demographics, 16S rRNA gene target regions, and sequencing platforms, leading to high variability in their conclusions, which may outweigh biological differences.27–29 Meta-analysis addresses these discrepancies in an unbiased manner and identifies consistencies across studies.30,31 Here, we presented an integrative meta-analysis on nine publicly available 16S rRNA datasets to characterize consistent alterations of the gastric microbiome associated with histological stages of gastric carcinogenesis. Additionally, we explored the relationships between Helicobacter species for the first time and unveiled the significant influence of HP-induced compositionality on the gastric microbiome analysis.
Results
Study selection and study heterogeneity
In this meta-analysis, nine public 16S rRNA amplicon sequencing datasets were found and included to investigate the role of gastric microbiota in gastric diseases (Table 1, see Methods for inclusion criteria). Overall, these studies covered 11 cohorts, encompassing 104 healthy samples, 511 gastritis samples, 223 intestinal metaplasia (IM) samples, and 1360 GC samples. For studies PRJEB26931 and PRJNA375772, precancerous samples from different anatomical sites of gastric mucosa were collected; for studies PRJNA310127, PRJEB216931, and PRJNA375772, paired tumor and non-tumor mucosal tissues were collected from GC patients (Figure S1).
Table 1.
Characteristics of studies included in the meta-analysis.
| No. | BioProject | Sequencing region | Country and region | Sample size |
Total | HP status |
Sample type | SRA | HP conventional testing method |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Healthy | Gastritis | IM | GC | (+) | (-) | ||||||||
| 1 | PRJEB2110414 | V1-V2 | UK | 22 | 35 | 10 | 10 | 77 | 49 | 28 | Mucosa | ERP023334 | Histology and RUT and Serology |
| 2 | PRJEB2149716 | V4 | Singapore, Malaysia | 20 | 12 | 32 | 22 | 10 | Mucosa | ERP02375 | Serology | ||
| 3 | PRJEB2210715 | V1-V2 | Austria | 6 | 20 | 26 | 19 | 7 | Mucosa | ERP024440 | Histology and PCR | ||
| 4 | PRJEB2693117 | V4 | China | 56 | 41 | 54 | 84 | 235 | 79 | 156 | Mucosa | ERP108962 | 13C-UBT |
| 5 | PRJNA31012718 | V3-V4 | China, Shanxi | 157 | 157 | Mucosa | SRP080738 | ||||||
| Mexico | 134 | 134 | |||||||||||
| 6 | PRJNA37577213 | V4 | China, Xi’an | 120 | 41 | 40 | 201 | 140 | 61 | Mucosa | SRP100168 | ||
| China, Inner Mongolia | 107 | 36 | 143 | 101 | 42 | ||||||||
| 7 | PRJNA42888319 | V3-V4 | China, Zhejiang | 745 | 745 | Mucosa | SRP128749 | ||||||
| 8 | PRJNA48141321 | V4 | China, Nanchang | 115 | 91 | 114 | 320 | 142 | 178 | Mucosa | SRP154244 | Immunohistochemistry test | |
| 41 | 27 | 28 | 96 | 46 | 50 | Fluid | |||||||
| 9 | PRJNA49543620 | V3-V4 | China, Nanchang | 32 | 32 | 32 | Mucosa | SRP165213 | 13C-UBT | ||||
To eliminate the variation caused by different bioinformatic analyses, consistent QIIME2 pipelines were used to process all the raw sequencing data (Figure 1a).32 Fine-resolution amplicon sequence variants (ASVs) that distinguish single-nucleotide differences were first generated.33,34 A closed-reference operational taxonomic unit (OTU) assignment approach was next employed to merge the datasets targeting distinct sequencing regions for cross-study comparisons.35
Figure 1.
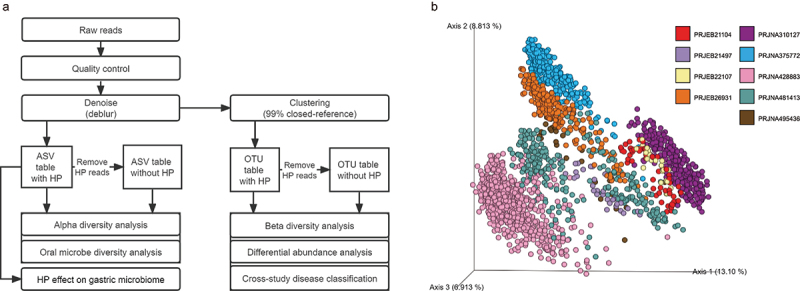
Meta-analysis workflow and study heterogeneity.
(a) Overview of bioinformatic workflow for this meta-analysis. (b) The PCoA plot is based on unweighted UniFrac distance showing profound variations among different studies.
Since studies varied in both demographic and technical factors (e.g., sampling sites, DNA extractions, variable regions), we first determined the extent of the contribution of “study” factor to the gastric microbiome variability. Principal coordinates analysis (PCoA) plots showed that samples were distinctly segregated by studies (Figure 1b). All the available potential confounders, including stage, disease stage, HP status, continent, sample location, age, and gender, were evaluated using ADONIS test. The result showed that these factors together explained ~33.96% of the microbial variation, with study, disease stage, and HP status factors having the largest impact (Figure S2).
The effect of H. pylori on the gastric microbiome
HP infection has long been recognized as a high-risk factor for GC.3 Conventional clinical methods for detecting HP infection include rapid urease test, histology, serology, PCR, culture, etc. In this meta-analysis, six studies have tested host HP status via different conventional methods (Table 1). By comparing it with sequencing results, we found that sequencing was more sensitive in detecting HP, which is consistent with previous studies.36,37 HP reads were detected in 98.5% HP-positive samples (Figure S3a). Four of the 5 HP-positive samples with no HP reads detected were from study PRJEB21497. This study used a serology method, which reflects past infections but not necessarily current HP status. Among the conventionally tested HP-negative samples, 78.4% were found with HP sequences. Kim et al. suggested 1% as an appropriate cutoff to determine the colonization of HP,37 which achieved a higher agreement with clinical HP status (Figure S3b versus Figure S3a).
We next evaluated the effect of HP on the gastric mucosal microbiome. The relative abundance of HP species was negatively correlated with the alpha diversity indices in most studies (Figure 2a) and pooled data (Figure S4), which could be largely due to the compositional dominance of HP in the gastric microbiota. ADONIS analysis demonstrated that the relative abundance of HP significantly shifted the overall microbiome composition in all the 11 cohorts (P < 0.05 except for Jaccard distance in PRJNA375772_Inner_Mongolia, Jaccard and unweighted UniFrac distances in PRJEB21497, Figure 2b), albeit with the explained variances ranging from 0.008 to 0.736. The variations explained by HP were higher for weighted distance metrics (weighted UniFrac and Bray-Curtis distances) than unweighted metrics (unweighted UniFrac and Jaccard distances). This suggests that HP exerted a relatively greater impact on microbial abundances than their presence when measured by overall microbial composition.
Figure 2.
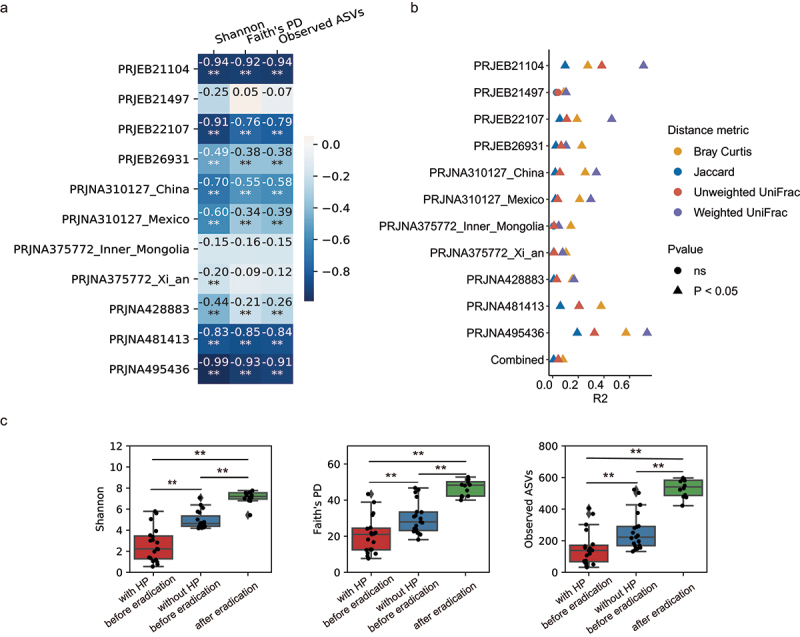
The effect of H. pylori on the gastric microbiome.
(a) Heatmap showing the correlations between the relative abundance of HP and Shannon, Faith’s PD and Observed ASVs in each cohort. The colors in the heatmap were proportional to the Spearman’s correlation coefficient displayed in each cell. **P < 0.01, *0.01 < P < 0.05. (b) Microbial variations explained by the relative abundance of HP in each cohort. R2 was calculated using the ADONIS test. (c) Alpha diversity in patients before and after receiving HP eradication in dataset PRJNA495436 with or without HP reads removed. Statistical significance was determined by t-test: **P < 0.01, *0.01 < P < 0.05.
Compositional effect occurs when we rarefy or normalize sequencing depths to the same across samples,38 which is usually required for downstream statistical analyses. Since the proportions of HP reads were extremely high and variated substantially across samples (Figure S3c), relative abundance-based gastric microbiome analysis may be heavily influenced by HP-induced compositionality. To illustrate whether the observed effect of HP on the gastric microbiome was a result of real ecological change or merely compositional effect, we re-analyzed the study PRJNA495436 that sampled from patients with gastritis before and after receiving HP eradication.20 If one assumes other microbiome impacting factors remained stable during eradication, the change between pre- and post-eradication arose partially from eradication effects in gastric microbial ecology and/or partially from HP compositional effects. To disentangle them, we analyzed these samples with HP reads removed before rarefaction. The result showed that post-eradication gastric samples had significantly increased microbial richness (observed ASVs) and diversity (Shannon and Faith’s PD) compared to the HP-filtered pre-eradication samples (Figure 2c green box versus blue box). This large increase indicates that HP eradication does have a significant impact on the gastric microbial ecosystem, not merely an artifact from compositional effects. Taken together, we underscore the significant effect of HP on the gastric microbiome from both technical and biological aspects.
Mutual exclusivity of helicobacter species
Another fundamental question involving HP is how HP species dominates the gastric mucosal niche. We compared the abundances of every pair of HP ASVs in each mucosal sample. Scatter plots showed that HP ASVs were largely mutually exclusive, and this observation was reproducible in all the studies (Figure 3 top panel, Figure S5). Notably, some HP ASVs were particularly more dominant in infected subjects than others (red star marked in Figure 3a and Figure S5). We additionally investigated the relationships between HP and other non-pylori Helicobacter species and found that they were also incompatible. In contrast to abundant HP ASVs, the relative abundances of non-pylori Helicobacter ASVs were generally extremely low (<1%) (Figure 3a-b bottom panel, Figure S5e-g right panel). Nevertheless, we also found exceptions: the relative abundance of a non-pylori Helicobacter ASV reached 96.6% and 35.3% in two samples in study PRJNA481413 (Figure 3b bottom panel). This ASV was 100% sequence identical to Helicobacter suis/Helicobacter heilmannii, which occurred naturally in animals and have been reported to be detected in human stomach.39 A recent study suggested that Helicobacter suis may be a potential gastric pathogen with different molecular pathogenicity and clinical manifestations from HP.40 Collectively, our findings show the mutual exclusivity of Helicobacter ASVs, indicating that the colonization of one HP ASV may inhibit others probably due to the competition for a similar ecological niche.
Figure 3.
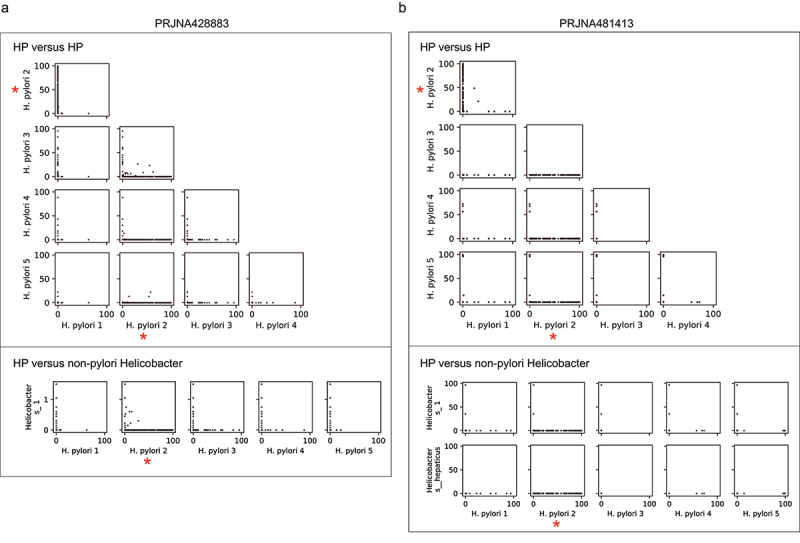
Mutual exclusivity of Helicobacter ASVs.
(a–b) Scatter plots show the relative abundance of each pair of Helicobacter ASVs in dataset PRJNA428883 (a) and dataset PRJNA481413 (b). Each dot is one gastric microbiome sample. The top panel shows the relationship between every two HP ASVs; the bottom panel shows the relationship between each HP ASV and non-pylori Helicobacter ASV. The most dominant HP ASV in the population was marked by a red star.
The gastric mucosal microenvironments are relatively homogeneous
Because mucosal samples may be collected from different sites of the stomach, it is necessary to test whether the gastric microbiota inhabiting at different microenvironments are uniform before exploring the disease-associated microbial changes. To take into account the aforementioned HP-induced compositional effect, we performed downstream analyses both with and without HP reads removed from samples before rarefaction. For precancerous lesions, no significant differences were observed in Shannon index (Figure S6a, P > 0.05, paired t test) and overall microbial composition (P > 0.05, ADNOIS based on Bray-Curtis, Jaccard, weighted UniFrac or unweighted UniFrac distance) between matched sample pairs collected from gastric body, antrum, or fundus. The results were similar after removing HP reads (Figure S6b). For the cancer stage, we compared matched samples from on-site and off-site tumor tissues in four cohorts individually. Significant differences in alpha diversity (P < 0.05, paired t-test) and microbiome composition (P < 0.05, ADNOIS based on Bray-Curtis, Jaccard, weighted UniFrac, or unweighted UniFrac distance) between tumor and non-tumor samples were found only in cohort RPHNA310127_China, but not in the other 3 cohorts (Figure S6c). Different from the other three cohorts, RPHNA310127_China had substantially higher HP abundance in non-tumor samples (Figure S6e). The delta HP abundance and delta Shannon index between the matched tumor and non-tumor samples were strongly negatively correlated (r = −0.817, Spearman’s correlation, Figure S6f), indicating the significant effects of HP on measured microbial composition. After removing HP reads from the analysis, the differences between tumor and non-tumor samples disappeared in both alpha diversity (Figure S6d) and beta diversity (P > 0.05, ADNOIS based on Bray-Curtis, Jaccard, weighted UniFrac or unweighted UniFrac distance) in this sample set, indicating that besides HP, there was no extensive alteration in the gastric microbiome composition between paired tumor and non-tumor samples. Thus, we concluded that the stomach microenvironments remain relatively homogenous during GC progression.
Mucosal microbiome shifted as gastric diseases progress
We next included all samples from different stomach sites and compared the gastric microbiota between disease stages. To control the study effects, we analyzed each cohort individually and then pooled the evidence of microbial changes across studies using a random effects model (REM). When HP sequences were included in the analysis, we found higher microbial richness and diversity in healthy subjects but no consistent significant change between other disease stages (Figure 4a). Similarly, to address the HP-induced compositional effect, we also performed the analyses without HP reads. As shown in Figure 4b, the difference between healthy and gastritis samples in alpha diversity disappeared or narrowed after removing HP reads due to the compositional effect caused by the excessive abundance of HP in gastritis patients. Additionally, significant differences of alpha diversity between gastritis and IM, IM and GC, and gastritis and GC were also identified. Specifically, the alpha diversity gradually decreased as the diseases progress: healthy > gastritis > IM > GC (Figure 4b). The contrasting results with and without HP underscore that dominant HP reads have a non-negligible impact on results. ADONIS analysis revealed significant differences in microbiome composition between each two groups both with (Figure 4c) and without HP (Figure 4d). The effect size, as measured by ADONIS R2, marginally increased after removing HP, especially for IM vs. GC and gastritis vs. GC comparisons. Collectively, we revealed progressive dysbiosis of the gastric mucosal microbiota during GC progression.
Figure 4.
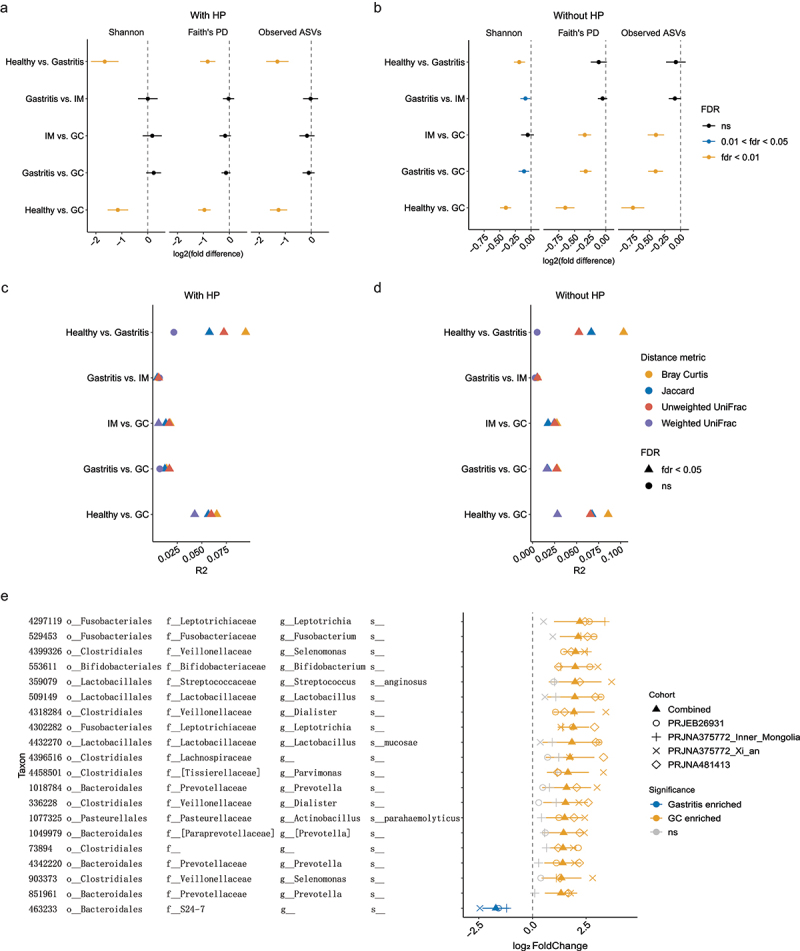
Mucosal microbiome shifted as gastric diseases progress.
(a-b) Forest plots of alpha diversity between each two stages. The combined log2(fold difference) and FDR-corrected p-values calculated by REM were shown in the figure. Point to the left of the gray dashed line indicates higher diversity in the early stage; point to the right of the gray dashed line represents higher diversity in the advanced disease stage. The length of the error bar depicts the 95% CIs. Analyses were performed both with (a) and without HP reads (b). (c-d) ADONIS based on various distance metrics demonstrated significant differences in the gastric microbiome composition between each of the two stages. Combined R2 across different cohorts was calculated using the ADONIS test with cohort as stratum. Analyses were performed both with (c) and without HP reads (d). (e) Combined log2(fold change) for the 20 differential OTUs with the largest effect size between gastritis and GC, calculated using DESeq2 and REM.
To identify microbial biomarkers that were reproducible across cohorts, we compared samples from gastritis and GC stages using REM – two stages with the largest number of samples (Table 1). Forty-four OTUs were found to be differentially abundant (P < 0.01, Figure S7a), and the top 20 differential OTUs are shown in Figure 4e: Leptotrichia, Fusobacterium, Selenomonas, Bifidobacterium, Streptococcus anginosus, Lactob-acillus, Veillonellaceae, Dialister, Lactobacillus mucosae, Lachnospiraceae, Parvimonas, Prevotella, Actinobacillus parahaemolyticus, and Clostridiales were consistently enriched in patients with GC; S24–7 tended to be more abundant in gastritis samples. A larger number of OTUs were found enriched in GC compared with that in gastritis (8 gastritis-enriched OTU versus 36 GC-enriched OTUs in Figure S7a), which may indicate a compromise in GC mucosal barrier, leading to the colonization of exogenous bacteria.
To further confirm the discriminative power of these identified microbial biomarkers, we performed within-cohort, cohort-to-cohort and leave-one-cohort-out (LOCO) validation using random forest (RF) models. In within-cohort cross-validation, the 44 OTUs distinguished GC from gastritis with area under the receiver operating characteristic curve (AUC) scores ranging from 0.75 to 0.99 (Figure S7b). The performance of cohort-to-cohort transfer validation slightly decreased but still achieved a relatively decent average AUC of 0.71 (minimum: 0.64, maximum: 0.76). The LOCO approach achieved similar prediction results as the cohort-to-cohort validation, with an average AUC of 0.77. Removing HP did not improve accuracy, except for cohort PRJEB26931 (Figure S7c), where HP was significantly different between gastritis and GC samples.
Enrichment of oral microbes in GC
Mounting evidence has shown that oral microbes are associated with various gastrointestinal diseases,41,42 including gastric cancer.13,43 To investigate the role of gastric microbes with putative oral origin in GC, the sequences from all gastric samples were profiled against the reference sequences from the Human Oral Microbiome Database (HOMD, 16S rRNA RefSeq version 15.21). Agreeing with this observation, we noticed that 80% of the identified GC-enriched bacteria were potential oral microbes (Figure S7a). To comprehensively explore the changes in oral microbes across cohorts during GC progression, we compared five metrics of oral microbes between each of the two stages using REM, including three alpha diversity indices, total relative abundance, and the fraction of oral ASVs among all the observed ASVs. The oral microbes were more diverse and abundant in healthy individuals compared to gastritis; they also tended to be enriched in GC compared to gastritis and IM, but there was no consistent, significant difference between other stages (Figure 5a). However, after removing HP sequences from the analysis, we found a higher diversity, relative abundance, and fraction of oral bacteria in GC compared to all precancerous stages (Figure 5b), indicating a similar obscuring compositional effect of HP dominance. Thus, we concluded that oral microbes in the gastric mucosa were significantly elevated in the GC.
Figure 5.
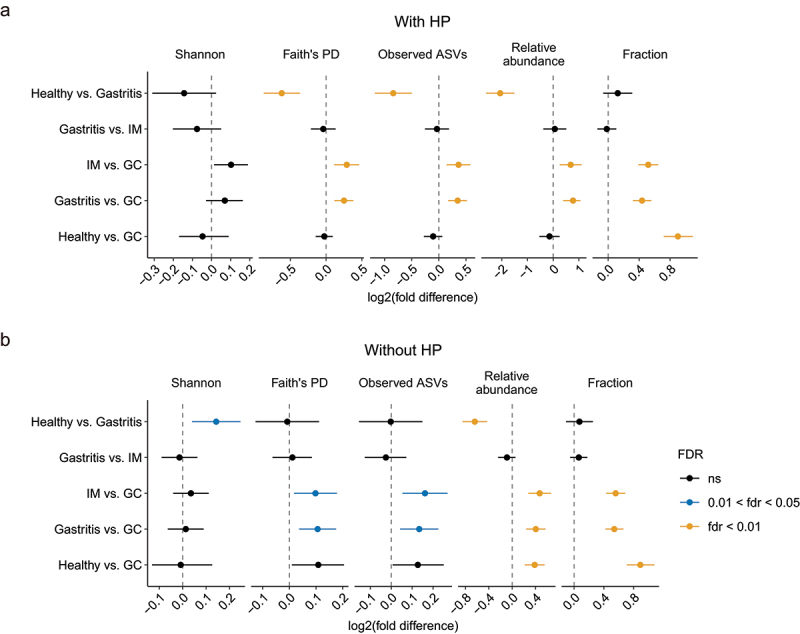
Comparison of oral microbes between different stages.
(a-b) Forest plots of the alpha diversity, relative abundance, and fraction of oral microbes between each of the two disease stages. The combined log2(fold difference) and FDR-corrected p-values calculated by REM were shown in the figure. Point to the left of the gray dashed line indicates higher diversity in the early stage; point to the right of the gray dashed line represents higher diversity in the advanced disease stage. The length of the error bar depicts the 95% CIs. Analyses were performed both with (a) and without HP reads (b).
Dysbiosis of gastric fluid microbiome during GC progression
Limited studies have investigated the gastric fluid microbiota to date.44–46 Its characteristics during the progression of GC still remain unknown. To this end, we compared gastric fluid samples from patients with gastritis, IM, and GC in the PRJNA481413 study (Table 1). In gastric fluid, the abundance of HP was much lower than that in mucosa (Figure 6a), but it still negatively correlated with the microbial diversity (r = −0.39, Spearman’s correlation, Figure S8a). The Shannon index of fluid microbiota displayed a trend of decrease as the disease progresses (Figure 6b-c). Permutational multivariate analysis of variance (PERMANOVA) using unweighted distances revealed a significant difference in fluid microbiome composition between GC and gastritis/IM (unweighted UniFrac distance and Jaccard distance, P < 0.01); However, PERMANOVA based on weighted metrics (weighted UniFrac distance and Bray-Curtis distance) showed no significant difference between any two stages. The result suggests that the difference was probably driven by low-abundance bacteria substantially altered in GC rather than high-abundance microbes. The fraction of oral microbes significantly increased in GC compared to other stages (Figure 6d), but no statistical difference was observed in alpha diversity or total abundance of the oral microbes between stages (Figure 6e-f).
Figure 6.
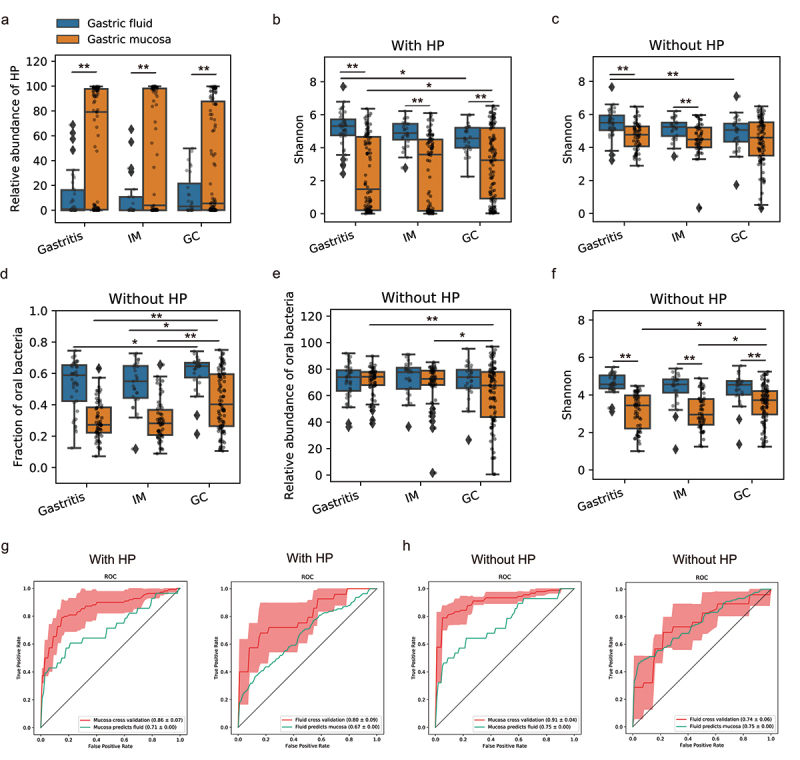
Dysbiosis of the gastric fluid and mucosal microbiome across GC stages in dataset PRJNA481413.
(a) The relative abundance of HP in fluid and mucosal samples across different stages. (b-c) Shannon index of gastric microbiome in fluid and mucosa samples across different stages. Analyses were performed both with (b) and without HP sequences (c). (d-f) The fraction (d), relative abundance (e), and alpha diversity (f) of oral bacteria in gastric fluid and mucosal samples across three disease stages. Analyses were performed without HP reads. **P<0.01, *0.01 < P<0.05. (g) The RF model trained on mucosal microbiome could distinguish GC from non-GC samples with an average AUC of 0.86, and it could predict fluid samples with an AUC of 0.71. The RF model trained on the fluid microbiome achieved a cross-validation AUC of 0.80, and it could classify mucosal samples with an AUC of 0.67. (h) After removing HP sequences from the analyses, the RF model trained on mucosal microbiome achieved a cross-validation AUC of 0.91 and it could predict fluid samples well with an AUC of 0.75. The RF model trained on fluid microbiome achieved a cross-validation AUC of 0.74 and it could predict mucosal samples well with an AUC of 0.75.
Previous studies consistently reported distinct gastric fluid microbiome and mucosal microbiome.44,45 We next comparatively analyzed the fluid and mucosal microbiota across disease stages collected from the same data set PRJNA481413. The fluid microbiome was more diverse than the mucosal microbiome in all stages (Figure 6b), but the differences narrowed after removing HP reads and was no longer statistically significant for GC stage (Figure 6c). PERMANOVA analysis revealed remarkable divergent microbial compositions between gastric fluid and mucosal samples regardless of disease stages (P < 0.01). However, the extent of this difference between gastric fluid and mucosal microbiome, measured by the pseudo-F statistic of PERMANOVA, gradually decreased as the disease exacerbated (pseudo-F based on Bray-Curtis distance with HP: gastritis, 21.1; IM, 12.1; GC 7.0. pseudo-F based on Bray-Curtis distance without HP: gastritis, 20.2; IM, 12.0; GC 4.5). Similar results were observed for other beta diversity metrics (Figure S8b), suggesting convergent microbial alterations toward GC between gastric fluid and mucosa. We further constructed an RF classification trained on mucosal microbial data, and found that it was able to distinguish GC fluid samples from non-GC fluid samples with an AUC of 0.71, and fluid microbiome classified GC mucosal samples from non-GC with an AUC of 0.67 (Figure 6g). The results were increased to 0.75 if removing HP sequences (Figure 6h). Overall, our findings suggest gastric fluid microbiome dysbiosis in GC and convergent shifts of fluid and mucosal microbiome toward GC.
Discussion
In this paper, we reanalyzed nine available public datasets of human gastric microbiota via consistent pipelines and comprehensively assessed alterations of the gastric microbiome across the GC cascade in an effort to address the issue of reproducibility among heterogeneous studies. Our results highlight the influence of the compositional effect caused by HP on gastric microbiome analyses. After stratifying the analyses by HP and study factors, we found that the gastric microbiome significantly shifted during the progression of gastric carcinogenesis, accompanied by a gradual decline in microbial diversity. Reproducible microbial signatures across studies that could distinguish GC samples from gastritis were identified. Notably, oral microbes were substantially enriched in GC compared to other disease stages. Additionally, by analyzing the gastric fluid microbiome across different disease stages, we found convergent microbiome dysbiosis in gastric fluid and mucosa as GC progressed.
HP is a well-studied pathogen that infects more than half of the world population.7 Besides HP, other non-pylori Helicobacter ASVs were also detected in human gastric mucosa, although their abundances were usually relatively low. Notably, we found that Helicobacter ASVs were mutually exclusive in all of the nine datasets (Figure 3, Figure S5), suggesting the existence of niche-specific competitions among different ASVs. With that being said, 16S amplicon sequences were too short to determine whether a single Helicobacter ASV represents a single strain or comprises multiple closely related strains, as distinct strains can share the same amplicon region. Future shotgun metagenomic studies are warranted to confirm it.
Compositional effect caused by HP dominance pose grand challenges in microbiome data analyses.38 Here, we performed downstream analyses both with and without HP sequences being removed. Comparing to analyses with HP reads, we found that results without HP reads seemed to be more robust and biologically plausible, which were summarized as follows: (i) differences in alpha diversity and microbiome composition between tumor and non-tumor samples were only identified in cohort PRJNA310127_China but not the other three, and these differences for this particular data set disappeared when HP reads were removed (Figure S6c versus Figure S6d); (ii) the alpha diversity shows a gradual decreasing trend as the disease progresses from gastritis to GC after removing HP (Figure 4a versus Figure 4b); (iii) the microbiome appeared more distinguishable especially between GC and other disease stages, after removing HP compared to the results with HP (Figure 4c versus Figure 4d); (iv) a significant enrichment of oral microbes was observed in GC compared with precancerous stages when HP was removed (Figure 5a versus Figure 5b). Overall, our meta-analyses suggest that the compositionality caused by HP could be a potential confounder in gastric microbiome analyses and highlight the merit of performing analyses both with and without HP sequences.
Batch effects hinder comparisons across studies. Opposite results were reported in studies included in this meta-analysis. For example, in study PRJEB26931, Wang et al. found a similar microbiome composition between paired on-site and off-site tumor tissues;17 in contrast, study PRJNA310127 showed distinct microbiome communities between tumor and matched non-tumor samples.18 Our meta-analysis revealed that the difference found in study PRJNA310127 was likely an artifact caused by compositional effects due to the huge number of HP reads. Another conflicting report was on the alpha diversity of the gastric microbiome. Study PRJEB21497 found that the microbial richness was significantly elevated in GC.16 However, study RPJNA375772 reported significantly lower species richness in IM and GC subjects compared to gastritis.13 Study PRJEB26931 suggested that gastric microbial diversity decreased from healthy, gastritis, and IM to GC.17 After accounting for the intra-study variation and HP-introduced compositionality, our study reported a progressive decrease in alpha diversity during the progression of GC. These results highlight the power of meta-analysis, which reduces the technical and biological noise or bias and elucidates robust findings across studies.
Current studies showed discrepancies in microbial signatures. Using REM, we identified a set of universal microbial biomarkers across cohorts between GC and gastritis with potential for GC diagnosis (Figure S7). Key microbial signatures included Fusobacterium, Leptotrichia, and several lactic acid bacteria such as Bifidobacterium, Lactobacillus, and Streptococcus anginosus (Figure 4e). They all tended to be enriched in GC cases. Fusobacterium and Leptotrichia were opportunistic pathogens associated with oral and gastrointestinal diseases.16,47–49 Our results indicate that they may also be involved in gastric carcinogenesis. Lactic acid bacteria constitute a group of bacteria that ferment carbohydrate to lactic acids. The enrichment of lactic acid bacteria in GC may suggest a remarkable alteration in the stomach environment. Vinasco et al. proposed that these lactic acid bacteria may promote GC via supplying exogenous lactate, increasing reactive oxygen species, N-nitroso compounds, and inducing immune tolerance.50 Streptococcus anginosus was reported to be associated with esophageal cancer, head and neck squamous cell carcinoma and GC.13,51,52 A recent study sampled paired tumor tissues and fecal samples from 1043 GC and gastritis individuals and found that the relative abundance of Streptococcus anginosus was significantly increased in both GC tumor tissues and feces.53 Interestingly, the gut microbiota were also significantly altered in GC patients compared to healthy controls.54,55 The RF model trained on gut microbiome data distinguished GC from healthy individuals with an AUC of 0.91.55 Zhou et al. suggested that fecal Streptococcus anginosus combined with Streptococcus constellatus are an accurate and sensitive biomarker for GC.53 The RF model trained using fecal microbiome data also classified surgical GC patients from non-surgical patients with an AUC of 0.75.56 These studies indicate that fecal microbiota may be an indicator of gastric dysbiosis, providing the possibility for noninvasive microbiome diagnosis.
Notably, most of the identified microbial biomarkers were potentially derived from the oral cavity (Figure S7a). Mounting evidence has shown that oral microbes were found substantially enriched in the distal gastrointestinal tract in patients with systemic conditions, including pancreatic cancer,42 inflammatory bowel disease,41 and colorectal cancer.57,58 Coker et al. revealed significant enrichment of oral microbes and their central role in GC microbial networks.13 Agreeing with this notion, we observed that the abundance and the fraction of oral microbes were significantly higher in GC compared to those in precancerous stages across studies (Figure 5b). It was possible that the dysfunction of mucosal barriers in GC offers opportunities for oral microbes to invade the mucosal surface.59 However, whether the enrichment of oral microbes aggravates GC progression or it was merely a reflection of host pathophysiological changes awaits further investigation. Additionally, studies suggest that Helicobacter may be negatively associated with oral bacteria in GC. For example, Guo et al. found strong co-excluding relationships between Helicobacter genus and multiple potential oral genera in advanced gastric lesions, including Fusobacterium, Neisseria, Prevotella, Veillonella, and Rothia.60 Another study also reported the co-exclusion between a Helicobacter OTU and oral-derived Campylobacter consisus in GC microbial interaction network.16We found distinct microbial compositions between the gastric mucosal and fluid microbiome. The microbial diversity was higher in fluid compared to that in mucosa (Figure 6b-c). In fluid, the average fraction and abundance of oral bacteria were greater than 50% (Figure 6d-e), indicating that oral microbiota swallowed with food and saliva may be the major source of the gastric fluid microbiome. The fraction and diversity of oral microbes in gastric fluid were nearly double of those in gastric mucosa (Figure 6d,f), indicating that the tenacious mucus gel layer in conjunction with the epithelial bicarbonate protects gastric epithelium from colonization of exogenous bacteria.61,62 Intriguingly, despite the significant difference in gastric mucosal and fluid microbiota, PERMANOVA analysis suggested convergent microbial alterations in mucosa and fluid during GC progression. This is further supported by the observation that the RF model trained on the mucosal microbiome could distinguish the fluid GC samples from other fluid non-GC samples, and vice versa (Figure 6g-h). The convergent trend in the fluid and mucosal microbiome might result from increased shedding of mucosal commensals and/or the progressive invasion of oral microbes or other exogenous bacteria on the gastric mucosa (due to the compromised gastric mucosal barrier in GC).
Despite our best efforts, this meta-analysis has limitations. First, batch effect challenges meta-analysis. Unwanted sources of technical variation among studies, including sample collections, DNA extractions, sequencing regions and platforms, 16S amplicon analysis tools, and statistical methods being used for data analysis, can be significant and potentially obscure biological signals, even after correction. Batch effect removal is particularly challenging for microbiomes due to the heterogeneity and sparsity in the microbial abundance table, exacerbating the technical differences across studies. We alleviated the batch effect by utilizing consistent workflow and statistical methods and adapting the previously reported analysis strategy such as REM. However, we did not comprehensively benchmark and review all the reported batch correction methods,63–67 which warrants a separate, systematic evaluation. Thus, theoretically there could be other approaches that might achieve similar or even better outcome. Second, the impact of demographic factors was not included in our analyses due to the limited availability of relevant metadata. Even though our result indicates that their contributions to gastric microbiome variation are small, their exact effects await further investigation when more data become available. Third, our findings on the gastric fluid microbiome during the progression of GC were supported by a single dataset, as there is no other data publicly available, which warrants future independent validation.
In conclusion, current studies on the gastric microbiome in gastric diseases have yielded many remarkable findings, but generalizability of results across studies was potentially poor. Our meta-analysis integrates multiple public datasets and underscores the notable influence of HP on gastric microbiome analyses. We identified highly consistent microbial features, which helps to refine the associations between gastric disease and microbiome. Moreover, we discovered the mutual exclusivity of HP and convergent dysbiosis of the gastric fluid microbiome and mucosal microbiome during GC progression. Our work provides comprehensive insights into the gastric microbiome dysbiosis in gastric carcinogenesis, laying a foundation for future microbiome-targeted diagnosis or treatment.
Methods
Study selection and sample exclusion
A literature PubMed search was conducted using terms ‘gastric microbiome’ [OR] ‘gastric microbiota’ [OR] ‘gastritis AND microbiota’ [OR] ‘intestinal metaplasia AND microbiota’ [OR] ‘gastric cancer AND microbiota’ to identify studies published prior to August 2020. Studies fit our inclusion criteria if they (1) sequenced the human gastric microbiome via Illumina platform targeting 16S rRNA genes; (2) had publicly available raw sequences data. Eight studies met all of the above criteria. Moreover, public repositories including the Sequence Read Archive (SRA) in NBCI, MG-RAST and European Nucleotide Archive (ENA) were also queried with the same search terms, resulting in an additional study PRJNA481413. A total of 9 datasets were included in our meta-analysis. Raw sequencing data were downloaded using SRA toolkit (V.2.10.8) from SRA using identifiers: PRJEB21104, PRJEB22107, PRJEB26931, PRJEB21497, PRJNA310127, PRJNA375772, PRJNA428883, PRJNA481413, and PRJNA495436 (Table 1). We herein focused on the microbiome shifts during the progression of GC (from healthy, gastritis, IM to GC), non-gastric samples, gastric samples that did not belong to the four stages, or gastric samples that were from patients subjected to drug treatment or other interventions were excluded from this meta-analysis. For example, fecal samples (in study PRJNA495436) and PPI-treated gastric samples (in study PRJEB21104) were excluded.
Standardized microbiome analysis pipelines
The bases of primers or adaptors were trimmed to ensure the reliability of downstream analysis. The trimmed sequences were next processed using standardized quantitative insights in microbial ecology version 2 (QIIME2, release 2020.2) pipelines.32 Briefly, within each dataset, sequences were first filtered by the quality score using a qiime quality-filter q-score command with default settings.68 Deblur algorithm was employed to remove singletons, artifacts, and chimera, resulting in a high-quality ASV table.33 Taxonomy was assigned against the Greengenes database (version 13_8)69 using QIIME2 q2-feature-classifier plugin.70 Sequences that were classified as mitochondria and chloroplast were discarded. ASVs that were 100% identical to the reference sequences (version 15.21) from HOMD were defined as potential oral microbes. Sequence alignment was performed via the VSEARCH tool.71
To merge ASV data from studies targeting different 16S amplicon sequencing regions, closed-reference clustering was performed at 99% identity against the Greengenes reference database using QIIME2 q2-vsearch plugin, resulting in a pooled OTU table. OTUs that were 99% identical to the HOMD reference sequences (version 15.21) were labeled as potential oral microbes.
To evaluate the impact of HP-induced compositional effect on the gastric microbiome analyses, two pipelines were used in parallel in downstream analyses: one with HP sequences, and the other with HP ASVs or OTUs being removed from the corresponding ASV or pooled OTU counts table.
Microbial diversity analyses
Within each study, samples were first rarefied to an appropriate depth according to the rarefaction curve. Alpha diversity metrics, including Shannon index, observed features, and Faith’s phylogenetic diversity (Faith’s PD), were calculated using QIIME2 q2-diversity plugin based on the rarefied ASV tables. To explore the microbial shifts between healthy, gastritis, IM and GC, samples from each pair of stages were compared within each study. The combined change in the alpha diversity between each of the two stages was computed to account for the study effects as described in Bisanz et al. 72 Taking the comparison of gastritis and GC as an example, for each study that contained these two stages, the alpha diversity index was first scaled to the geometric mean of gastritis samples. Welch’s t-test was next employed to calculate significance and 95% confidence intervals (CIs). REM was applied to integrate the results of each study and determine the combined result with the formula log2(fold difference) ~ disease stages + (1|cohort). Combined p-values were next adjusted using the FDR approach (p.adjust function in the R stats package).
To integrate and compare the gastric microbiome across studies, beta diversity analyses were performed based on the pooled OTU table. Samples with less than 1000 OTU sequences were removed from downstream analyses. The OTU table was next rarefied to 1000 according to the rarefaction curve. Beta diversity metrics including Bray-Curtis, Jaccard, weighted UniFrac and unweighted UniFrac distance, were calculated using QIIME2 q2-diversity plugin. The PCoA plot was visualized using the Emperor tool.73 The overall microbiome community between each of the two stages was compared using ANONIS with 999 permutations (adonis in the R vegan package).74 The combined R2 was calculated by setting the “strata” parameter to cohorts to constrain the study effect as described in Bisanz et al.72 Combined p-values were next adjusted using the FDR approach (p.adjust function in the R stats package).
Differentially abundant OTUs between gastritis and GC samples across cohorts
Differential abundance analysis between gastritis and GC samples was conducted using DESeq275 based on the OTU abundances in cohorts PRJEB26931, PRJNA375772_Xi_an, PRJNA375772_Inner_Mongolia and PRJNA481413, respectively. REM was employed to identify reproducible microbial biomarkers across cohorts using the R metafor package as described in Shah et al. 76,77 Cohorts were used as a random effect to control the study effects. OTUs with a combined P < 0.01 calculated by REM were considered as the universal differential microbes across cohorts.
Application and evaluation of the machine learning models
Random forest (RF) models were used to test the predictive power in classifying GC from gastritis samples. Within-cohort prediction was measured through stratified fivefold cross-validation to ensure balanced gastritis and GC samples in each fold. In cohort-to-cohort validation, RF models were trained on one cohort and validated on another. In LOCO validation, data from one cohort was used as the external validation set and the other three cohorts were pooled as a training set. All RF models were built using the scikit-learn module.78 The AUC of the ROC curve was used to measure the predictive accuracy of the classification model.
Since the PERMANOVA analysis showed convergent dysbiosis in the gastric fluid microbiome and mucosal microbiome as GC progressed in dataset PRJNA481413, we trained RF models to test if they can discriminate GC samples from other non-GC samples. The models were trained on gastric fluid relative abundance data and validated on mucosal relative abundance data and vice versa.
Other statistical analysis
Paired t-test (scipy.stats.ttest_rel) was applied to compare the alpha diversity metrics of samples collected from matched anatomical sites or paired tumor and non-tumor tissues. Independent between-group comparisons were performed using t-test (scipy.stats.ttest_ind). The relationship between alpha diversity and the relative abundance of HP was calculated using Spearman’s rank correlation (scipy.stats.spearmanr). PERMANOVA was performed with permanova function in scikit-bio (v0.5.5) python library. R2 between groups were calculated using the ANONIS test with 999 permutations (adonis in the R vegan package).
Supplementary Material
Funding Statement
This research was supported by the National Natural Science Foundation of China (grant no. 31970088) and the National Key Technology Research and Development Program of the Ministry of Science and Technology of China (grant no. 2020YFA0509600).
Author contributors
ZZ Xu: designed and supervised the study, interpreted the results and revised the manuscript. Y Li and Y Hu: collected data, performed bioinformatics analyses, interpreted the results, and wrote the manuscript. X Zhan: interpreted the results and revised the manuscript. Y Song, M Xu, S Wang and X Huang: assisted data collection and bioinformatics analyses.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data and code availability
Raw 16s rRNA sequencing data are available from the NCBI Sequence Read Archive (see Table 1 for the identifiers of included datasets). Codes have been uploaded to https://github.com/daydayup-ly/GC-meta-analysis for reproducibility.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19490976.2023.2197835.
References
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F.. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–19. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Correa P. Human gastric carcinogenesis: a multistep and multifactorial process–first American cancer society award lecture on cancer epidemiology and prevention. Cancer Res. 1992;52:6735–6740. [PubMed] [Google Scholar]
- 3.Rawla P, Barsouk A. Epidemiology of gastric cancer: global trends, risk factors and prevention. Prz Gastroenterol. 2019;14(1):26–38. doi: 10.5114/pg.2018.80001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang LL. Participation of microbiota in the development of gastric cancer. World J Gastroenterol. 2014;20(17):4948–4952. doi: 10.3748/wjg.v20.i17.4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Møller H, Heseltine E, Vainio H. Working group report on schistosomes, liver flukes and helicobacter pylori. Meeting held at IARC, LYON, 7–14 June 1994. Int J Cancer. 1995;60(5):587–589. doi: 10.1002/ijc.2910600502. [DOI] [PubMed] [Google Scholar]
- 6.Valle J, Kekki M, Sipponen P, Ihamäki T, Siurala M. Long-term course and consequences of helicobacter pylori gastritis results of a 32-year follow-up study. Scand J Gastroenterol. 1996;31(6):546–550. doi: 10.3109/00365529609009126. [DOI] [PubMed] [Google Scholar]
- 7.Conteduca V, Sansonno D, Lauletta G, Russi S, Ingravallo G, Dammacco F. H. pylori infection and gastric cancer: state of the art (review). Int J Oncol. 2013;42(1):5–18. doi: 10.3892/ijo.2012.1701. [DOI] [PubMed] [Google Scholar]
- 8.Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M, Taniyama K, Sasaki N, Schlemper RJ. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med. 2001;345(11):784–789. doi: 10.1056/NEJMoa001999. [DOI] [PubMed] [Google Scholar]
- 9.Kumar S, Metz DC, Ellenberg S, Kaplan DE, Goldberg DS. Risk factors and incidence of gastric cancer after detection of helicobacter pylori infection: a large cohort study. Gastroenterology. 2020;158(3):527–536.e7. doi: 10.1053/j.gastro.2019.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shiota S, Thrift AP, Green L, Shah R, Verstovsek G, Rugge M, Graham DY, El-Serag HB. Clinical manifestations of helicobacter pylori–negative gastritis. Clinical Gastroenterol Hepatol. 2017;15(7):1037–1046.e3. doi: 10.1016/j.cgh.2017.01.006. [DOI] [PubMed] [Google Scholar]
- 11.Yang J, Zhou X, Liu X, Ling Z, Ji F. Role of the gastric microbiome in gastric cancer: from carcinogenesis to treatment. Front Microbiol. 2021;12:641322. doi: 10.3389/fmicb.2021.641322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lofgren JL, Whary MT, Ge Z, Muthupalani S, Taylor NS, Mobley M, Potter A, Varro A, Eibach D, Suerbaum S, et al. Lack of commensal flora in Helicobacter pylori–infected ins-gas mice reduces gastritis and delays intraepithelial neoplasia. Gastroenterol. 2011;140(1):210–220.e4. doi: 10.1053/j.gastro.2010.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coker OO, Dai Z, Nie Y, Zhao G, Cao L, Nakatsu G, Wu WK, Wong SH, Chen Z, Sung JJY, et al. Mucosal microbiome dysbiosis in gastric carcinogenesis. Gut. 2018;67(6):1024–1032. doi: 10.1136/gutjnl-2017-314281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parsons BN, Ijaz UZ, D’amore R, Burkitt MD, Eccles R, Lenzi L, Duckworth CA, Moore AR, Tiszlavicz L, Varro A, et al. Comparison of the human gastric microbiota in hypochlorhydric states arising as a result of Helicobacter pylori-induced atrophic gastritis, autoimmune atrophic gastritis and proton pump inhibitor use. PLoS Pathog. 2017;13(11):e1006653. doi: 10.1371/journal.ppat.1006653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klymiuk I, Bilgilier C, Stadlmann A, Thannesberger J, Kastner MT, Högenauer C, Püspök A, Biowski-Frotz S, Schrutka-Kölbl C, Thallinger GG, et al. The human gastric microbiome is predicated upon infection with helicobacter pylori. Front Microbiol. 2017;8:2508. doi: 10.3389/fmicb.2017.02508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Castaño-Rodríguez N, Goh K-L, Fock KM, Mitchell HM, Kaakoush NO. Dysbiosis of the microbiome in gastric carcinogenesis. Sci Rep. 2017;7(1):15957. doi: 10.1038/s41598-017-16289-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Z, Gao X, Zeng R, Wu Q, Sun H, Wu W, Zhang X, Sun G, Yan B, Wu L, et al. Changes of the gastric mucosal microbiome associated with histological stages of gastric carcinogenesis. Front Microbiol. 2020;11:997. doi: 10.3389/fmicb.2020.00997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu G, Torres J, Hu N, Medrano-Guzman R, Herrera-Goepfert R, Humphrys MS, Wang L, Wang C, Ding T, Ravel J, et al. Molecular characterization of the human stomach microbiota in gastric cancer patients. Front Cell Infect Microbiol. 2017;7:302. doi: 10.3389/fcimb.2017.00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu X, Shao L, Liu X, Ji F, Mei Y, Cheng Y, Liu F, Yan C, Li L, Ling Z. Alterations of gastric mucosal microbiota across different stomach microhabitats in a cohort of 276 patients with gastric cancer. EBioMedicine. 2019;40:336–348. doi: 10.1016/j.ebiom.2018.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He C, Peng C, Wang H, Ouyang Y, Zhu Z, Shu X, Zhu Y, Lu N. The eradication of Helicobacter pylori restores rather than disturbs the gastrointestinal microbiota in asymptomatic young adults. Helicobacter. 2019;24:e12590. doi: 10.1111/hel.12590. [DOI] [PubMed] [Google Scholar]
- 21.He C, Peng C, Shu X, Wang H, Zhu Z, Ouyang Y, Yang X, Xie C, Hu Y, Li N, et al. Convergent dysbiosis of gastric mucosa and fluid microbiome during stomach carcinogenesis. Gastric Cancer. 2022;25(5):837–849. doi: 10.1007/s10120-022-01302-z. [DOI] [PubMed] [Google Scholar]
- 22.Aviles-Jimenez F, Vazquez-Jimenez F, Medrano-Guzman R, Mantilla A, Torres J. Stomach microbiota composition varies between patients with non-atrophic gastritis and patients with intestinal type of gastric cancer. Sci Rep. 2014;4(1):4202. doi: 10.1038/srep04202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferreira RM, Pereira-Marques J, Pinto-Ribeiro I, Costa JL, Carneiro F, Machado JC, Figueiredo C. Gastric microbial community profiling reveals a dysbiotic cancer-associated microbiota. Gut. 2018;67(2):226–236. doi: 10.1136/gutjnl-2017-314205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gunathilake MN, Lee J, Choi IJ, Kim YI, Ahn Y, Park C, Kim J. Association between the relative abundance of gastric microbiota and the risk of gastric cancer: a case-control study. Sci Rep. 2019;9(1):13589. doi: 10.1038/s41598-019-50054-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang L, Zhou J, Xin Y, Geng C, Tian Z, Yu X, Dong Q. Bacterial overgrowth and diversification of microbiota in gastric cancer. European J Gastroenterol Hepatol. 2016;28(3):261–266. doi: 10.1097/MEG.0000000000000542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang L, Xin Y, Zhou J, Tian Z, Liu C, Yu X, Meng X, Jiang W, Zhao S, Dong Q. Gastric mucosa-associated microbial signatures of early gastric cancer. Front Microbiol. 2020;11:14. doi: 10.3389/fmicb.2020.01548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nearing JT, Douglas GM, Hayes MG, MacDonald J, Desai DK, Allward N, Jones CMA, Wright RJ, Dhanani AS, Comeau AM, et al. Microbiome differential abundance methods produce different results across 38 datasets. Nat Commun. 2022;13(1):342. doi: 10.1038/s41467-022-28034-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kameoka S, Motooka D, Watanabe S, Kubo R, Jung N, Midorikawa Y, Shinozaki NO, Sawai Y, Takeda AK, Nakamura S. Benchmark of 16S rRNA gene amplicon sequencing using Japanese gut microbiome data from the V1–V2 and V3–V4 primer sets. BMC Genomics. 2021;22(1):527. doi: 10.1186/s12864-021-07746-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim D, Jung J-Y, Oh H-S, Jee S-R, Park SJ, Lee S-H, Yoon J-S, Yu SJ, Yoon I-C, Lee HS. Comparison of sampling methods in assessing the microbiome from patients with ulcerative colitis. BMC Gastroenterol. 2021;21(1):396. doi: 10.1186/s12876-021-01975-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duvallet C. Meta-analysis generates and prioritizes hypotheses for translational microbiome research. Microb Biotechnol. 2018;11(2):273–276. doi: 10.1111/1751-7915.13047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gurevitch J, Koricheva J, Nakagawa S, Stewart G. Meta-analysis and the science of research synthesis. Nat. 2018;555:175–182 7695. doi: 10.1038/nature25753. [DOI] [PubMed] [Google Scholar]
- 32.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37(8):852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amir A, McDonald D, Navas-Molina JA, Kopylova E, Morton JT, Zech Xu Z, Kightley EP, Thompson LR, Hyde ER, Gonzalez A, et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems. 2017;2(2):e00191–16. doi: 10.1128/mSystems.00191-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Callahan BJ, McMurdie PJ, Holmes SP. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. Isme J. 2017;11(12):2639–2643. doi: 10.1038/ismej.2017.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rideout JR, He Y, Navas-Molina JA, Walters WA, Ursell LK, Gibbons SM, Chase J, McDonald D, Gonzalez A, Robbins-Pianka A, et al. Subsampled open-reference clustering creates consistent, comprehensive OTU definitions and scales to billions of sequences. PeerJ. 2014;2:e545. doi: 10.7717/peerj.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bik EM, Eckburg PB, Gill SR, Nelson KE, Purdom EA, Francois F, Perez-Perez G, Blaser MJ, Relman DA. Molecular analysis of the bacterial microbiota in the human stomach. PNAS. 2006;103(3):732–737. doi: 10.1073/pnas.0506655103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim J, Kim N, Jo HJ, Park JH, Nam RH, Seok YJ, Kim YR, Kim JS, Kim JM, Kim JM, et al. An Appropriate Cutoff Value for Determining the Colonization of Helicobacter pylori by the Pyrosequencing Method: comparison with Conventional Methods. Helicobacter. 2015;20(5):370–380. doi: 10.1111/hel.12214. [DOI] [PubMed] [Google Scholar]
- 38.Weiss S, Xu ZZ, Peddada S, Amir A, Bittinger K, Gonzalez A, Lozupone C, Zaneveld JR, Vázquez-Baeza Y, Birmingham A, et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome. 2017;5(1):27. doi: 10.1186/s40168-017-0237-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baele M, Pasmans F, Flahou B, Chiers K, Ducatelle R, Haesebrouck F. Non-Helicobacter pylori helicobacters detected in the stomach of humans comprise several naturally occurring Helicobacter species in animals. FEMS Immunol Med Microbiol. 2009;55(3):306–313. doi: 10.1111/j.1574-695X.2009.00535.x. [DOI] [PubMed] [Google Scholar]
- 40.Rimbara E, Suzuki M, Matsui H, Nakamura M, Morimoto M, Sasakawa C, Masuda H, Nomura S, Osaki T, Nagata N, et al. Isolation and characterization of Helicobacter suis from human stomach. Proc Natl Acad Sci U S A. 2021;118(13):e2026337118. doi: 10.1073/pnas.2026337118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gevers D, Kugathasan S, Denson LA, Vázquez-Baeza Y, Van Treuren W, Ren B, Schwager E, Knights D, Song SJ, Yassour M, et al. The treatment-naive microbiome in new-onset crohn’s disease. Cell Host & Microbe. 2014;15(3):382–392. doi: 10.1016/j.chom.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fan X, Alekseyenko AV, Wu J, Peters BA, Jacobs EJ, Gapstur SM, Purdue MP, Abnet CC, Stolzenberg-Solomon R, Miller G, et al. Human oral microbiome and prospective risk for pancreatic cancer: a population-based nested case-control study. Gut. 2018;67(1):120–127. doi: 10.1136/gutjnl-2016-312580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sung JJY, Coker OO, Chu E, Szeto CH, Luk STY, Lau HCH, Yu J. Gastric microbes associated with gastric inflammation, atrophy and intestinal metaplasia 1 year after Helicobacter pylori eradication. Gut. 2020;69(9):1572–1581. doi: 10.1136/gutjnl-2019-319826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sung J, Kim N, Kim J, Jo HJ, Park JH, Nam RH, Seok YJ, Kim YR, Lee DH, Jung HC. Comparison of Gastric Microbiota Between Gastric Juice and Mucosa by Next Generation Sequencing Method. J Cancer Prev. 2016;21(1):60–65. doi: 10.15430/JCP.2016.21.1.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schulz C, Schütte K, Koch N, Vilchez-Vargas R, Wos-Oxley ML, Oxley APA, Vital M, Malfertheiner P, Pieper DH. The active bacterial assemblages of the upper GI tract in individuals with and without Helicobacter infection. Gut. 2018;67(2):216–225. doi: 10.1136/gutjnl-2016-312904. [DOI] [PubMed] [Google Scholar]
- 46.von Rosenvinge EC, Song Y, White JR, Maddox C, Blanchard T, Fricke WF. Immune status, antibiotic medication and pH are associated with changes in the stomach fluid microbiota. Isme J. 2013;7(7):1354–1366. doi: 10.1038/ismej.2013.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eribe ERK, Olsen I. Leptotrichia species in human infections II. J Oral Microbiol. 2017;9:1368848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brennan CA, Garrett WS. Fusobacterium nucleatum — symbiont, opportunist and oncobacterium. Nat Rev Microbiol. 2019;17(1):156–166. doi: 10.1080/20002297.2017.1368848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Komiya Y, Shimomura Y, Higurashi T, Sugi Y, Arimoto J, Umezawa S, Uchiyama S, Matsumoto M, Nakajima A. Patients with colorectal cancer have identical strains of Fusobacterium nucleatum in their colorectal cancer and oral cavity. Gut. 2019;68(3):1335–1337. doi: 10.1038/s41579-018-0129-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vinasco K, Mitchell HM, Kaakoush NO, Castaño-Rodríguez N. Microbial carcinogenesis: lactic acid bacteria in gastric cancer. Biochim Biophys Acta Rev Cancer. 2019;1872(7):188309. doi: 10.1136/gutjnl-2018-316661. [DOI] [PubMed] [Google Scholar]
- 51.Sasaki H, Ishizuka T, Muto M, Nezu M, Nakanishi Y, Inagaki Y, Watanabe H, Watanabe H, Terada M. Presence of Streptococcus anginosus DNA in esophageal cancer, dysplasia of esophagus, and gastric cancer. Cancer Res. 1998;58(14):2991–2995. doi: 10.1016/j.bbcan.2019.07.004. [DOI] [PubMed] [Google Scholar]
- 52.Tateda M, Shiga K, Saijo S, Sone M, Hori T, Yokoyama J, Matsuura K, Takasaka T, Miyagi T. Streptococcus anginosus in head and neck squamous cell carcinoma: implication in carcinogenesis. Int J Mol Med. 2000;6:699–703. doi: 10.3892/ijmm.6.6.699. [DOI] [PubMed] [Google Scholar]
- 53.Zhou CB, Pan SY, Jin P, Deng JW, Xue JH, Ma XY, Xie YH, Cao H, Liu Q, Xie WF, et al. Fecal Signatures of Streptococcus anginosus and Streptococcus constellatus for Noninvasive Screening and Early Warning of Gastric Cancer. Gastroenterology. 2022;162(7):1933–1947.E18 doi: 10.1053/j.gastro.2022.02.015. [DOI] [PubMed] [Google Scholar]
- 54.Chen C, Chen L, Lin L, Jin D, Du Y, Lyu J. Research progress on gut microbiota in patients with gastric cancer, esophageal cancer, and small intestine cancer. Appl Microbiol Biotechnol. 2021;105(11):4415–4425. doi: 10.1007/s00253-021-11358-z. [DOI] [PubMed] [Google Scholar]
- 55.Zhang Y, Shen J, Shi X, Du Y, Niu Y, Jin G, Wang Z, Lyu J. Gut microbiome analysis as a predictive marker for the gastric cancer patients. Appl Microbiol Biotechnol. 2021;105(2):803–814. doi: 10.1007/s00253-020-11043-7. [DOI] [PubMed] [Google Scholar]
- 56.Chen C, Shen J, Du Y, Shi X, Niu Y, Jin G, Liu Y, Shi Y, Lyu J, Lin L. Characteristics of gut microbiota in patients with gastric cancer by surgery, chemotherapy and lymph node metastasis. Clin Transl Oncol. 2022;24(11):2181–2190. doi: 10.1007/s12094-022-02875-y. [DOI] [PubMed] [Google Scholar]
- 57.Flemer B, Warren RD, Barrett MP, Cisek K, Das A, Jeffery IB, Hurley E, O‘riordain M, Shanahan F, O‘toole PW. The oral microbiota in colorectal cancer is distinctive and predictive. Gut. 2018;67(8):1454–1463. doi: 10.1136/gutjnl-2017-314814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thomas AM, Manghi P, Asnicar F, Pasolli E, Armanini F, Zolfo M, Beghini F, Manara S, Karcher N, Pozzi C, et al. Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat Med. 2019;25(4):667–678. doi: 10.1038/s41591-019-0405-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oshima T, Miwa H. Gastrointestinal mucosal barrier function and diseases. J Gastroenterol. 2016;51(8):768–778. doi: 10.1007/s00535-016-1207-z. [DOI] [PubMed] [Google Scholar]
- 60.Guo Y, Zhang Y, Gerhard M, Gao JJ, Mejias-Luque R, Zhang L, Vieth M, Ma JL, Bajbouj M, Suchanek S, et al. Effect of Helicobacter pylori on gastrointestinal microbiota: a population-based study in Linqu, a high-risk area of gastric cancer. Gut. 2020;69(9):1598–1607. doi: 10.1136/gutjnl-2019-319696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Garner A, Flemström G, Allen A, Heylings JR, McQueen S. Gastric mucosal protective mechanisms: roles of epithelial bicarbonate and mucus secretions. Scand J Gastroenterol Suppl. 1984;101:79–86. [PubMed] [Google Scholar]
- 62.Niv Y, Banić M. Gastric Barrier Function and Toxic Damage. DDI. 2014;32(3):235–242. doi: 10.1159/000357855. [DOI] [PubMed] [Google Scholar]
- 63.Mecham BH, Nelson PS, Storey JD. Supervised normalization of microarrays. Bioinformatics. 2010;26(10):1308–1315. doi: 10.1093/bioinformatics/btq118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen C, Grennan K, Badner J, Zhang D, Gershon E, Jin L, Liu C. Removing batch effects in analysis of expression microarray data: an evaluation of six batch adjustment methods. Plos One. 2011;6(2):e17238. doi: 10.1371/journal.pone.0017238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7). e47–e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gibbons SM, Duvallet C, Alm EJ. Correcting for batch effects in case-control microbiome studies. PLoS Comput Biol. 2018;14(4):e1006102. doi: 10.1371/journal.pcbi.1006102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ma S, Shungin D, Mallick H, Schirmer M, Nguyen LH, Kolde R, Franzosa E, Vlamakis H, Xavier R, Huttenhower C. Population structure discovery in meta-analyzed microbial communities and inflammatory bowel disease using MMUPHin. Genome Biol. 2022;23(1):208. doi: 10.1186/s13059-022-02753-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, Mills DA, Caporaso JG. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods. 2013;10(1):57–59. doi: 10.1038/nmeth.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72(7):5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, Huttley GA, Gregory Caporaso J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome. 2018;6(1):90. doi: 10.1186/s40168-018-0470-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016;4:e2584. doi: 10.7717/peerj.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bisanz JE, Upadhyay V, Turnbaugh JA, Ly K, Turnbaugh PJ. Meta-analysis reveals reproducible gut microbiome alterations in response to a high-fat diet. Cell Host Microbe. 2019;26(2):265–272.e4. doi: 10.1016/j.chom.2019.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vázquez-Baeza Y, Pirrung M, Gonzalez A, Knight R. Emperor: a tool for visualizing high-throughput microbial community data. Gigascience. 2013;2(1):16. doi: 10.1186/2047-217X-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dixon P. VEGAN a package of r functions for community ecology. J Vegetation Sci. 2003;14(6):927–930. doi: 10.1111/j.1654-1103.2003.tb02228.x. [DOI] [Google Scholar]
- 75.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shah MS, DeSantis TZ, Weinmaier T, McMurdie PJ, Cope JL, Altrichter A, Yamal JM, Hollister EB. Leveraging sequence-based faecal microbial community survey data to identify a composite biomarker for colorectal cancer. Gut. 2018;67(5):882–891. doi: 10.1136/gutjnl-2016-313189. [DOI] [PubMed] [Google Scholar]
- 77.Viechtbauer W. Conducting meta-analyses in r with the metafor package. J Stat Softw. 2010;36(3):1–48. doi: 10.18637/jss.v036.i03. [DOI] [Google Scholar]
- 78.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. Scikit-learn: machine learning in python. J Mach Learn Res. 2011;12:2825–2830. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw 16s rRNA sequencing data are available from the NCBI Sequence Read Archive (see Table 1 for the identifiers of included datasets). Codes have been uploaded to https://github.com/daydayup-ly/GC-meta-analysis for reproducibility.