

Abstract
Stroke is a life‐threatening disease with limited therapeutic options. Damage to the blood–brain barrier (BBB) is the key pathological feature of ischemic stroke. This study explored the role of the bradykinin (BK)/bradykinin 1 receptor (B1R) and its mechanism of action in the BBB. Human brain microvascular endothelial cells (BMECs) were used to test for cellular responses to BK by using the Cell Counting Kit‐8 assay, 5‐ethynyl‐2′‐deoxyuridine staining, enzyme‐linked immunosorbent assay, flow cytometry, immunofluorescence, cellular permeability assays, and western blotting to evaluate cell viability, cytokine production, and reactive oxygen species (ROS) levels in vitro. A BBB induced by middle cerebral artery occlusion was used to evaluate BBB injuries, and the role played by BK/B1R in ischemic/reperfusion (I/R) was explored in a rat model. Results showed that BK reduced the viability of BMECs and increased the levels of proinflammatory cytokines (interleukin 6 [IL‐6], IL‐18, and monocyte chemoattractant protein‐1) and ROS. Additionally, cellular permeability was increased by BK treatment, and the expression of tight junction proteins (claudin‐5 and occludin) was decreased. Interestingly, Wnt3a expression was inhibited by BK and exogenous Wnt3a restored the effects of BK on BMECs. In an in vivo I/R rat model, knockdown of B1R significantly decreased infarct volume and inflammation in I/R rats. Our results suggest that BK might be a key inducer of BBB injury and B1R knockdown might provide a beneficial effect by upregulating Wnt3a.
Keywords: B1R, blood–brain barrier, bradykinin, inflammation, stroke
Abbreviations
- B1R
bradykinin 1 receptor
- B2R
bradykinin 2 receptor
- BBB
blood–brain barrier
- BK
bradykinin
- BMECs
brain microvascular endothelial cells
- CCA
common carotid artery
- ECA
external carotid artery
- IL‐6
interleukin 6
- IL‐18
interleukin 8
- I/R
ischemia/reperfusion
- LDH
lactate dehydrogenase
- MCA
middle cerebral artery
- MCP‐1
monocyte chemoattractant protein‐1
- TTC
2,3, 5‐triphenyltetrazolium chloride
1. INTRODUCTION
Stroke has become the second leading cause of death worldwide.[ 1 ] Ischemic stroke is the most common type of stroke and occurs due to arterial occlusion and the interruption of blood flow to the brain.[ 2 ] The blood–brain barrier (BBB), a physical and metabolic barrier, selectively imports nutrients and energy to the brain and simultaneously exports neurotoxic substances to the peripheral circulation.[ 3 ] Disruption of the BBB is a well‐established feature of ischemic stroke and contributes to brain injury.[ 4 ] The BBB is composed of brain microvascular endothelial cells (BMECs), perivascular cells (pericytes), and astrocytes,[ 5 ] and controls the passage of molecules by tight and adhesion junctions.[ 6 ] Tight junctions between adjacent endothelial cells are essential for BBB formation and permeability.[ 7 ] Disruption of the BBB leads to increased vascular permeability[ 8 ] and promotes inflammation.[ 9 ] Cerebrovascular inflammation is a key factor in the progression of injury during a stroke.[ 10 ] Proinflammatory factors upregulate the levels of cell adhesion molecules in endothelial cells.[ 11 ] Lactate dehydrogenase (LDH) is an enzyme enriched in the cytoplasm and is released when the cell membrane is damaged.[ 12 ]
Bradykinin (BK) is a potent inflammatory mediator involved in a variety of physiological processes.[ 13 ] BK promotes vasodilation, permeability, and pain during the inflammation process.[ 14 ] A BK analog, RMP‐7, has been shown to increase BBB permeability by disengaging tight junctions between endothelial cells.[ 15 , 16 ] BK inhibitors regulate vascular permeability and the inflammatory response across epithelia.[ 17 ] A previous study showed that BK binds to two BK receptors, bradykinin 1 receptor (B1R) and bradykinin 2 receptor (B2R).[ 18 ] BK receptors, including B1R and B2R, play important roles in a cerebral ischemia/reperfusion (I/R) injury.[ 19 , 20 , 21 ] B1R tends to be expressed after a tissue injury and is closely associated with inflammatory responses; whereas, B2R expression is constitutive.[ 22 ] Research indicates that in B2R‐deficient model animals, the cerebral infarct size in the animals is not significantly reduced, suggesting there is another mechanism that mediates acute brain injury in the model.[ 22 ] As a matter of fact, B1R expression is promoted by injury and inflammation. Furthermore, activation of the B1R has been implicated in many neuroinflammatory diseases and induces the production of proinflammatory cytokines, including interleukin‐6 (IL‐6), interleukin‐8 (IL‐8), intracellular adhesion molecule‐1, and vascular cell adhesion molecule‐1.[ 23 , 24 ] However, the mechanism of BK/B1R has not been fully elucidated.
This study investigated the effect of BK on human brain microvascular endothelial cells (hBMECs) by measuring cell viability, reactive oxygen species (ROS), and proinflammatory factors. An I/R rat model was used to investigate the role played by BK in a BBB injury and inflammatory response. Our data showed that BK promotes injury to the BBB both in vitro and in vivo by evoking an inflammatory response, promoting oxygen radical formation, and reducing cell viability by blocking Wnt3a. Our results provide new information regarding BBB injuries and suggest BK/B1R/Wnt3a as a potential target for treating diseases caused by cerebrovascular injuries.
2. MATERIALS AND METHODS
2.1. Cell culture and treatment
hBMECs were purchased from the BeNa Culture Collection and cultured in endothelial cell medium media supplemented with 10% fetal bovine serum in a 37°C incubator with 5% CO2 and 95% humidity. The cells were treated with different concentrations of BK for periods of 12, 24, 36, 48, and 72 h, respectively. Recombinant Wnt3a (R&D Systems; No. 5036‐WN) was dissolved in dimethyl sulfoxide at a concentration of 100 ng/ml and used to pretreat hBMECs for 24 h for further use.
2.2. Cell Counting Kit‐8 assay
hBMECs were seeded into the wells of a 96‐well plate (1 × 104 cells/well) and treated with different concentrations of BK. After 24 h of treatment, 10 μl of Cell Counting Kit‐8 (CCK‐8) solution (Multiscience Biotech; Cat. No. 70‐CCK801) was added to each well and the cells were incubated for another 2 h; after which, the absorbance of each well at 450 nm was measured using a microplate spectrophotometer.
2.3. 5‐Ethynyl‐2′‐deoxyuridine staining
Cells were seeded into the wells of a 24‐well plate and treated with either BK alone or a combination of BK plus Wnt3a as indicated. An Edu Detection Kit was purchased from Beyotime Biotechnology (Cat. No. C0078S). After 48 h, 10 μM 5‐ethynyl‐2′‐deoxyuridine (EdU) was added to each well and the cells were incubated for another 2 h; after which, they were fixed with 4% formaldehyde for 10 min at room temperature. After permeabilization, the cells were incubated with Click Additive Solution included in the EdU Detection Kit. The cell nucleus was stained with 4′,6‐diamidino‐2‐phenylindole (DAPI).
2.4. Flow cytometry
ROS production was measured by using 2′,7′‐dichlorodihydrofluorescein diacetate (DCF‐A) according to the manufacturer's instructions and as described in a previous study.[ 25 ] Briefly, DCF‐A (10 μM) was added to cells, which were then incubated at 37°C for 30 min in the dark. After washing with phosphate‐buffered saline (PBS), the cells were analyzed by flow cytometry (FCM; FACS Calibur; BD Bioscience).
Cell apoptosis was measured by using an Annexin V/PI Detection Kit according to the manufacturer's instructions. Briefly, cells were digested with trypsase (0.25%) and then suspended in PBS. Next, the cells were transferred into a flow tube, incubated with Annexin V and propidium iodide(PI) reagents, and then centrifuged for 15 min at room temperature. Finally, the cells were analyzed by flow cytometry (FACS Calibur; BD Bioscience). Cells that were Annexin V+/PI+ OR Annexin V+/PI− were regarded as apoptotic cells.
2.5. In vitro BBB model and cellular permeability assay
Cells were seeded onto the bottom side of a collagen I‐coated Transwell insert, and hBMECs were seeded onto the upper side of the insert. Next, BK or a combination of BK plus Wnt3a was added to the hBMECs. Sodium fluorescein spectroscopic analysis was used to determine the permeability of the hBMECs grown on Transwell inserts. Sodium fluorescein (Yeasen; Cat. No. 40901ES01) was added to cells at the same time they received treatment. A 50 μl aliquot of media was collected from the bottom chamber of each insert at 6, 24, 48, and 72 h following treatment. Absorbance at 620 nm was measured using a microplate spectrophotometer. The lucifer yellow assay was performed as described as Shahriyary.[ 26 ] Briefly, cells were incubated with 10 μg/ml lucifer yellow (Sigma‐Aldrich) for 90 min; after which, lucifer yellow fluorescence in the lower chambers was detected by a fluorescence microplate reader (Ex: 430 nm; Em: 535 nm).
2.6. Immunofluorescence
Cells were seeded into the wells of a 24‐well plate and treated with BK or a combination of BK plus Wnt3a as indicated. After 48 h, the cells were fixed with 4% formaldehyde for 10 min and then blocked with 1% bovine serum albumin for 30 min at room temperature. Next, anti‐IL‐6 (Abcam; CAT. No. ab233706, 1:100), anti‐IL‐18 (Bio‐Techne; CAT. No. MAB2548‐SP, 25 µg/ml) or anti‐monocyte chemoattractant protein‐1 (MCP‐1) antibody (Abcam; CAT. No. ab186421, 1:200) was added and incubated with the cells overnight at 4°C. Finally, the cells were washed three times with PBS and incubated with a Cy‐3‐conjugated secondary antibody (Abcam; CAT. No. ab150078, 1:800; or Boster; CAT. No. BA1031, 1:50) at room temperature for 1 h. The cell nucleus was stained with DAPI.
2.7. Enzyme‐linked immunosorbent assay
The levels of IL‐6 (Elabscience; Cat. No. E‐EL‐H0102c), IL‐18 (Boster; CAT. No. EK0864), MCP‐1 (Boster; Cat. No. EK0441), and LDH (Elabscience; CAT. No. E‐EL‐H0866c) expression were determined by enzyme‐linked immunosorbent assay (ELISA) according to manufacturer's instructions. In brief, standards and cells or serum samples from rats were added to a microplate containing the specific antibody. After washing away unbound substances, the horseradish peroxidase (HRP) substrate was added to samples for detection of the bound protein. Absorbance at 450 nm was measured within 30 min.
2.8. Western blotting
Cells or samples of brain tissue were lysed with radioimmunoprecipitation assay buffer on ice for 30 min. Next, a 30 μg sample of total protein from each lysate was subjected to 10% sodium dodecyl sulphate‐polyacrylamide gel electrophoresis, and the separated protein bands were transferred onto 0.2‐μm polyvinylidene difluoride membranes that were subsequently blocked with 5% nonfat milk for 1 h. Next, the membranes were incubated with anti‐claudin‐5 (Beyotime; CAT. No. PB0123, 1:800), anti‐Wnt3a (Boster; CAT. No. BA2628‐2, 1:1000), anti‐β‐catenin (Boster; CAT. No. BA0426, 1:1000), anti‐B1R (Abcam; CAT. No. ab77366, 1:100), anti‐Occludin (Boster; CAT. No. BM4832, 1:1000), and anti‐glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH; Abcam; CAT. No. ab8245, 1:3000) antibodies overnight at 4°C; after which, they were washed three times with TBST and incubated for 1 h with an HRP‐conjugated secondary antibody at room temperature. The signals were detected with enhanced chemiluminescence reagents. The gray level of each blot band was recorded using ImageJ software (Wayne Rasband Company; Ver. 1.48). GAPDH served as an internal reference standard when calculating relative levels of protein expression.
2.9. Rat ischemic stroke (I/R) model
Six‐week‐old Sprague–Dawley rats (male and female) were purchased from the Experimental Animal Center of Southern Medical University. The protocols for all animal experiments were approved by the Research Ethics Committee of Guangdong Provincial People's Hospital, Guangdong Academy of Medical Sciences, Guangzhou, China (No. GDREC2017100A). A total of 24 rats were assigned to the following three groups (eight rats per group): Sham, I/R, and IR + B1RKD. Adenovirus carrying the B1R knockdown sequence was obtained from Hanbio Biotechnology Co., Ltd. Next, 2 × 107 adenovirus in 20 µl of saline solution was intrathecal injected into B1RKD rats. At 4 days after injection, the rats were used to establish an I/R model as previously described.[ 27 ] In brief, after anesthetization with 4% pentobarbital sodium (dose: 50 mg/Kg), the right common carotid artery (CCA) of each rat was exposed and dissected. The external carotid artery (ECA) was tied, and nylon sutures (Beijing Cinontech; Cat. No. ab8245) were inserted from the CCA to the internal carotid artery through the ECA to effectively occlude the middle cerebral artery (MCA). For sham animals, all the protocols were performed in a manner similar to that used to establish the I/R models, except for occlusion of the MCA.
2.10. 2,3,5‐Triphenyltetrazolium chloride staining
Rats were anesthetized by an intraperitoneal injection of 4% pentobarbital sodium (dose: 50 mg/kg) (Sigma‐Aldrich; Cat. No. P‐010). The hemispheres of the brain were cut into 2 mm slices and stained with 2% 2,3,5‐triphenyltetrazolium chloride (TTC) at 37°C for 30 min. After washing with PBS, the brain slices were prepared for photography.
2.11. Evans blue dye staining
A 2% Evans blue dye (1 ml/100 g body weight) (Sigma‐Aldrich; Cat. No. E2129) solution was injected intravenously, and 1 h later, the rats were anesthetized by an intraperitoneal injection of 4% sodium pentobarbital (dose: 50 mg/Kg). The brain tissues of the rats were then collected for microscopic analysis.
2.12. Hematoxylin and eosin staining and immunohistochemistry
The hemispheres of rat brains were fixed, paraffin‐embedded, and then sliced into 4‐μm‐thick sections. Next, the sections were dewaxed, dehydrated, and stained with hematoxylin and eosin (H&E) or subjected to antigen repair with sodium citrate buffer (pH 6.0) for subsequent use in immunohistochemistry assays. MCP‐1 antibody (Abcam; CAT. No. ab186421, 1:200) was added to slides with tissue samples, and the slides were incubated overnight at 4°C. On the next day, the slides were washed three times with PBS and then incubated with a secondary antibody for 1 h at room temperature. The cell nucleus was re‐stained with hematoxylin.
2.13. TdT‐mediated dUTP nick‐end labeling
Tissues were stained with reagents in a TUNEL Kit to evaluate the apoptosis occurring in brain tissues. Briefly, the slides were dewaxed and then treated with proteinase K at room temperature for 30 min. Afterward, the slides were washed with PBS. Next, the tissue sample was completely covered with 3% H2O2 at room temperature for 20 min. TdT‐mediated dUTP nick‐end labeling (TUNEL) working solution was then added and incubated at 37°C for 1 h. Finally, the slides were developed using a 3,3′‐diaminobenzidine developer and observed and photographed under a white light microscope.
2.14. Statistical analysis
Each experiment was repeated at least three times. Results were analyzed by analysis of variance that was performed using IBM SPSS Statistics for Windows, Version 22.0 software (IBM Corp.). All histograms were plotted using GraphPad Prism 9.0 (GraphPad Software). p < 0.05 was considered to be statistically significant.
3. RESULTS
3.1. BK reduced cell viability in a concentration‐ and time‐dependent manner
To determine the effects of BK on the viability of BMECs, BMECs were treated with different concentrations of BK (0, 50, 100, 200, 400, 800, or 1600 nM) for 24 h. Subsequent CCK‐8 assay results showed that BK reduced cell proliferation in a concentration‐dependent manner, with an IC50 value of 597.94 ± 87.17 nM (Figure 1A). Additionally, we examined whether the effects of BK on BMEC viability were associated with treatment time. As shown in Figure 1B, cell viability decreased as the treatment time increased. These results indicated that BK inhibited cell viability in a concentration‐ and time‐dependent manner.
Figure 1.
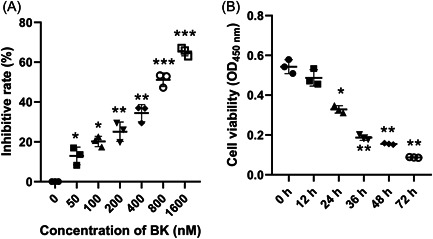
BK reduced cell viability in a concentration‐ and time‐dependent manner. (A) BMECs were treated with different concentrations of BK for 24 h, and cell viability was detected by the CCK‐8 assay. (B) BMECs were treated with 400 nM BK for the indicated time period, and cell viability was measured by the CCK‐8 assay. BK, bradykinin; BMECs, brain microvascular endothelial cells; CCK‐8, Cell Counting Kit‐8; OD, optical density.
3.2. BK increased ROS production, cellular permeability, and inflammation, but reduced cell proliferation and tight junctions
To test the effects of BK on BMEC, we measured ROS production under conditions of BK treatment and found that BK significantly increased ROS production in a concentration‐dependent manner (Figure 2A, p < 0.05). Furthermore, studies conducted using our in vitro BBB model showed that permeability of the BBB was increased by BK treatment (Figure 2B, p < 0.05). In addition, EdU staining assays showed that BK treatment impaired the proliferation of BMECs (Figure 2C). The expression of inflammation‐related proteins (IL‐6, IL‐8, and MCP‐1) was increased during BK treatment, as detected by immunofluorescence and ELISA (Figure 2D,E). The cell apoptosis rates were shown in Figure 2F. As shown in Figure 2F, BK treatment increased cell apoptosis in a concentration‐dependent manner (Figure 2F). Moreover, measurements of LDH revealed that LDH levels were increased by BK treatment (Figure 2G, p < 0.05). Furthermore, we found that the levels of Wnt3a and β‐catenin expression in BK‐treated BMECs were significantly reduced, as well as the expression of tight junction‐related proteins, claudin‐5 and occludin (Figure 2H, Supporting Information: Figure S1, p < 0.05).
Figure 2.
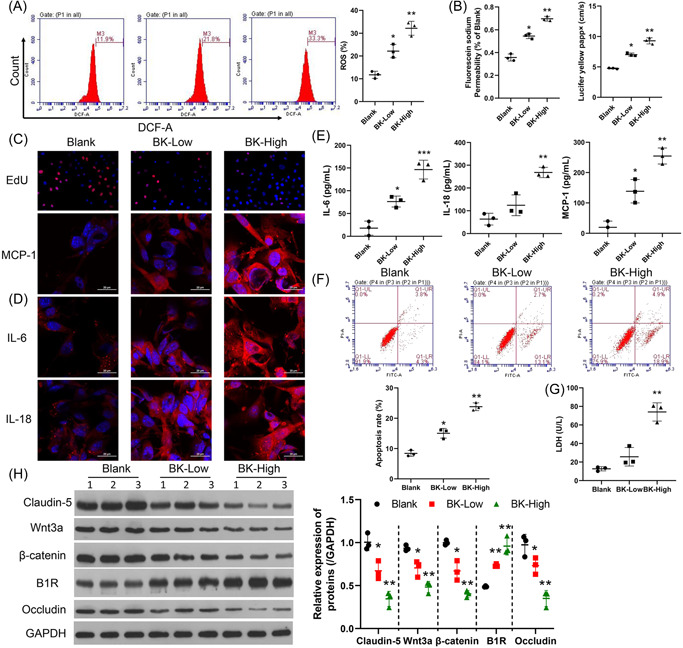
BK enhanced ROS production, cellular permeability, and inflammation, and reduced cell proliferation and tight junction formation by BMECs. BMECs were treated with BK (low concentration, 100 nM or high concentration, 400 nM). ROS production was determined by flow cytometry, and the respective images are shown (A, left panel). Summarized results from three independent experiments are shown (A, right panel). Cell permeability was tested by sodium fluorescein analysis (B, left panel) and the lucifer yellow assay (B, right panel). Cell proliferation was measured by EdU staining. (C, IL‐6, MCP‐1, and IL‐18 expressions were detected by IF (D) and ELISA (E). (F) The cell apoptosis rate was measured using FCM. (G) LDH levels were determined by ELISA. The levels of claudin‐5, occludin, β‐catenin, Wnt3a, and B1R expression were determined by western blotting (H, left panel). The statistical results of western blot studies conducted in three independent experiments (H, right panel). *p < 0.05; **p < 0.01; ***p < 0.001 versus blank. B1R, bradykinin 1 receptor; BK, bradykinin; BMECs, brain microvascular endothelial cells; DCF‐A, 2′,7′‐dichlorodihydrofluorescein diacetate; EdU, 5‐ethynyl‐2′‐deoxyuridine; ELISA, enzyme‐linked Immunosorbent assay; FCM, flow cytometry; GAPDH glyceraldehyde‐3‐phosphate dehydrogenase; IF, immunofluorescence; IL‐6, interleukin 6; LDH, lactate dehydrogenase; MCP‐1, monocyte chemoattractant protein‐1; ROS, reactive oxygen species.
3.3. Wnt3a repaired the BK‐induced damage in BMECs
To further confirm the effects of Wnt3a on BMECs, we cotreated hBMECs with BK and Wnt3a. Results showed that the ROS production induced by BK was eliminated by treatment with exogenous Wnt3a (Figure 3A, p < 0.05). Also, cell barrier function impired by BK was recovered by Wnt3a (Figure 3B, p < 0.05). Furthermore, Wnt3a protected against the reduction in hBMEC viability induced by BK (Figure 3C upper), as well as the enhanced inflammation caused by BK treatment (Figure 3C bottom, Figure 3D,E). As shown in Figure 3F, Wnt3a treatment significantly protected cells from the apoptosis induced by BK (Figure 3F), and also the decline in LDH levels (Figure 3G). Moreover, Wnt3a increased the levels of claudin‐5 and occludin expression, which had been repressed by BK treatment (Figure 3H, Supporting Information: Figure S2, p < 0.05).
Figure 3.
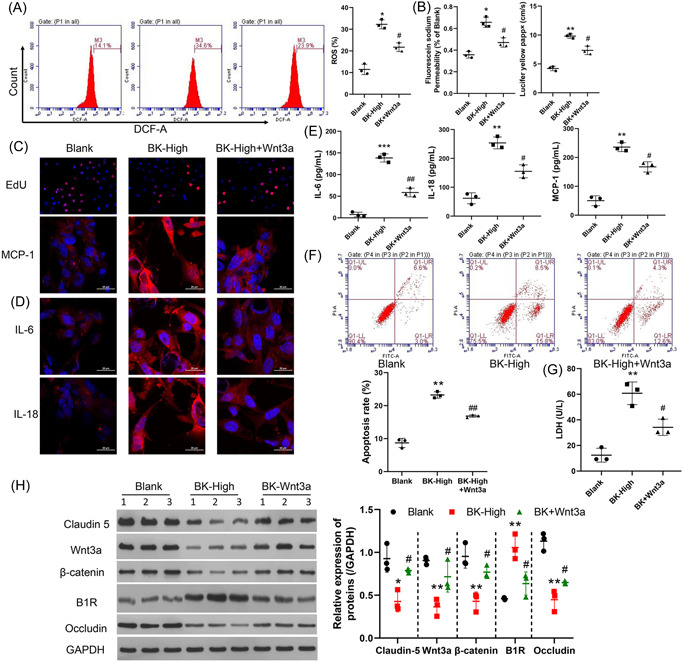
Wnt3a counteracted the effects of BK on BMECs. BMECs were treated with a high concentration (400 nM) of BK or a high concentration of both BK and Wnt3a (100 ng/ml each). ROS production was measured by flow cytometry, and the respective images are shown (A, left panel). The summary results of three independent experiments are shown (A, right panel). Cell permeability was tested by sodium fluorescein analysis (B, left panel) and the lucifer yellow assay (B, right panel). (C) Cell proliferation was measured by EdU staining. IL‐6, MCP‐1, and IL‐18 expressions were detected by (D) IF and (E) ELISA. (F) Cell apoptosis was detected by using the FCM method. (G) LDH levels were determined by ELISA. The levels of claudin‐5, occludin, β‐catenin, Wnt3a, and B1R expression were determined by western blotting (H, left panel). The statistical results of western blot studies conducted in three independent experiments (H, right panel). *p < 0.05; **p < 0.01; ***p < 0.001 versus blank. # p < 0.05; ## p < 0.01 versus BK‐high. B1R, bradykinin 1 receptor; BK, bradykinin; BMECs, brain microvascular endothelial cells; DCF‐A, 2′,7′‐dichlorodihydrofluorescein diacetate; EdU, 5‐ethynyl‐2′‐deoxyuridine; ELISA, enzyme‐linked Immunosorbent assay; FCM, flow cytometry; GAPDH glyceraldehyde‐3‐phosphate dehydrogenase; IF, immunofluorescence; IL‐6, interleukin 6; LDH, lactate dehydrogenase; MCP‐1, monocyte chemoattractant protein‐1; ROS, reactive oxygen species.
3.4. Knockdown B1R protected against I/R‐induced damage in the model rats
To further explore the effects of BK on BBB damage, we established an in vivo I/R rat model. The knockdown efficiency of adenovirus was measured in the brain (Supporting Information: Figure S3). Afterward, TTC staining revealed an extensive infarction area in a large proportion of the ipsilateral hemisphere of I/R rats in the I/R group, while knockdown of B1R by adenovirus resulted in decreased areas of ischemia (Figure 4A). In addition, Evans blue staining and H&E staining revealed that the I/R rats had significant BBB leakage, while suppression of B1R protected against that adverse effect induced by BK (Figure 4B,C). MCP‐1 expression was increased by I/R, and MCP‐1 expression was decreased after B1R knockdown (Figure 4D). Results of TUNEL staining showed that B1R knockdown helped to protect against brain cell apoptosis (Figure 5E). The levels of inflammatory factors IL‐6, IL‐18, and MCP‐1 in serum were elevated in the I/R model rats, and those increases were attenuated by B1R suppression (Figure 5A, p < 0.05). Furthermore, the levels of serum Wnt3a and BK were found to be regulated by I/R (Figure 5B, p < 0.05). More importantly, the decreases in tight junction proteins Claudin‐5 and Occludin were partially recovered in rats with B1R knockdown (Figure 5C, p < 0.05).
Figure 4.
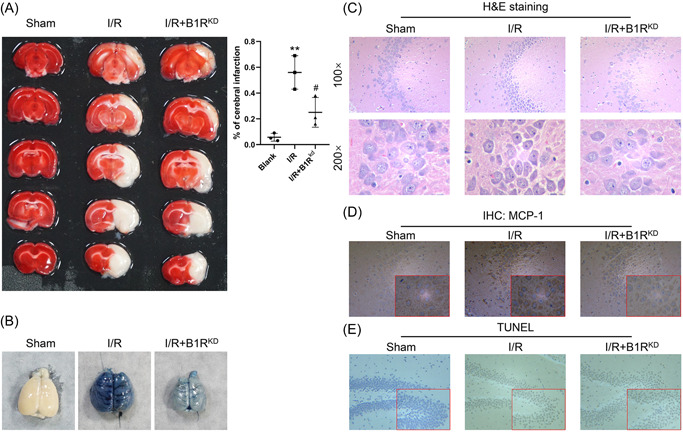
Knockdown B1R protected against I/R damage in model rats. A total of 24 rats were assigned to three separate groups: Sham (n = 8), I/R model (n = 8), and adenovirus targeted B1R‐treated I/R model (n = 8). (A) The infarction area was detected by TTC staining. (B) BBB permeability was measured by Evans blue staining. (C) Pathological changes were detected by H&E staining. (D) MCP‐1 expression was determined by IHC. (E) Apoptosis was measured by TUNEL staining. B1R, bradykinin 1 receptor; BBB, blood–brain barrier; H&E, hematoxylin and eosin; IHC, immunohistochemistry; I/R, ischemia/reperfusion; MCP‐1, monocyte chemoattractant protein‐1; TTC, 2,3,5‐triphenyltetrazolium chloride; TUNEL,TdT‐mediated dUTP nick‐end Labeling.
Figure 5.
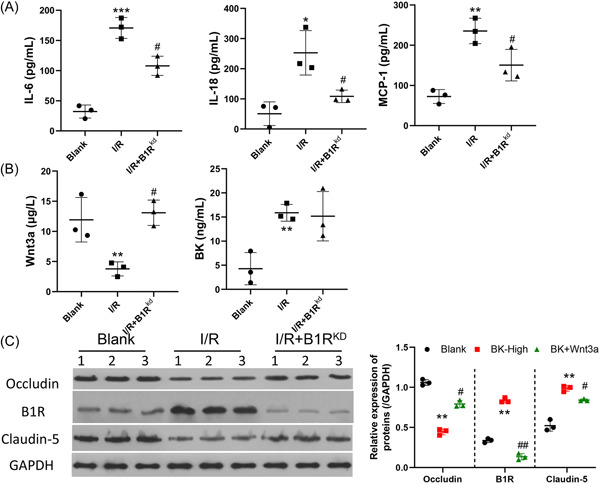
Knockdown of B1R suppressed I/R‐induced inflammation and induced tight junction formation in I/R model rats. A total of 24 rats were assigned to three groups: Sham (n = 8), I/R model (n = 8), and adenovirus targeted B1R treated I/R model (n = 8). (A) IL‐6, IL‐18, and MCP‐1 expression were determined by ELISA. (B) The levels of Wnt3a and BK were determined by ELISA. (C) The levels of claudin‐5, occludin, and B1R expression were determined by western blotting. *p < 0.05; **p < 0.01; ***p < 0.001, versus blank. # p < 0.05; ## p < 0.01, versus BK‐high. B1R, bradykinin 1 receptor; BK, bradykinin; ELISA, enzyme‐linked Immunosorbent assay; GAPDH glyceraldehyde‐3‐phosphate dehydrogenase; IL‐6, interleukin 6; I/R, ischemia/reperfusion; MCP‐1, monocyte chemoattractant protein‐1.
4. DISCUSSION
In this study, we showed that BK reduced the viability of BMECs and mediated the permeability, inflammation, and tight junctions of BMECs. More importantly, Wnt3a expression was suppressed by BK, and Wnt3a protected cell viability and reduced the inflammatory response in BK‐treated hBMECs. We further confirmed the effects of BK on the BBB in an in vivo rat I/R model and found that suppression of the BK receptor significantly decreased the infarct volumes and inflammation in I/R rats. Taken together, our results showed that B1R knockdown helped to protect against the adverse effects of I/R via the Wnt3a/β‐catenin pathway.
The BBB is a physical and metabolic barrier composed of microvascular endothelial cells and is important for maintaining central nervous system homeostasis and protecting the brain from potentially harmful circulating substances.[ 28 ] The BBB is disrupted in ischemic stroke,[ 29 ] and becomes sensitized to further disruptive changes caused by systemic inflammation.[ 30 ] Endothelial monolayers provide a relatively simple model in which experimental conditions can be easily modified and thus allow for a quantitative assessment of barrier function.[ 31 ] Here, we used BMECs to establish an in vitro BBB model that was used to determine the adverse effects of BK on BMECs. BK was previously found to induce disruption of the mouse cerebrovascular endothelial cell constructed tight junction barrier by ketamine.[ 32 ] In this study, we found that BK reduced cell viability in a concentration‐ and time‐dependent manner. More importantly, BK was found to modulate ROS production, cellular permeability, and inflammation by upregulating the B1 receptor and downregulating the Wnt3a/β‐catenin pathway.
Numerous studies have demonstrated that tight junction alterations are related to increased BBB permeability during periods of ischemia.[ 33 ] Claudins are the principal proteins that establish the backbone of tight junctions and are critical determinants of paracellular “tightness” between adjacent BBB endothelial cells.[ 34 ] Changes in claudin‐5 and occludin expression are related to BBB injury.[ 35 ] Here, we showed that the levels of claudin‐5 and occludin, two tight junction proteins, were significantly reduced by BK treatment. Our data are consistent with previous findings showing that the BK analog RMP‐7 increased BBB permeability by disengaging the tight junctions of endothelial cells.[ 15 , 16 ] Our data also revealed that BK increased BBB permeability by suppressing tight junction formation.
Inflammation is considered to be a hallmark of ischemic stroke.[ 36 ] BK is a proinflammatory factor that mediates inflammation in many diseases, including stroke.[ 37 ] An upregulation of inflammation‐associated genes is related to the progression of neuroinflammation during stroke.[ 38 ] A previous study showed that BK produced in the brain during a stroke stimulated IL‐6 secretion and gene transcription by activating nuclear factor‐κB (NF‐κB) in murine astrocytes.[ 39 ] Here, we found that BK not only significantly induced IL‐6 expression but also increased IL‐8 and MCP‐1 expression in BMECs. Our data further confirmed that BK adversely affects BMECs by modulating inflammation and permeability.
Wnt3a is an important member of the Wnt family and has been shown to participate in neurogenesis in the hippocampus and cortex.[ 40 , 41 ] It is thought that Wnt3a might be secreted by mesenchymal stem cells and then crosses the BBB.[ 42 ] In rat and mouse stroke models, intranasal administration of Wnt3a produced neuroprotective and regenerative effects after ischemic stroke.[ 41 , 43 , 44 , 45 ] Activation of Wnt3a was shown to ameliorate the toxic response after an ischemic brain injury by reducing neuroinflammation,[ 43 ] while blocking the Wnt pathway mediated the proliferation and differentiation of rat neural progenitor cells, and attenuated neuronal apoptosis.[ 44 , 46 ] Here, we found that Wnt3a expression was inhibited by BK treatment and BK‐induced ROS production, which was consistent with a previous study showing that Wnt3a activation attenuated the apoptosis of BMECs induced by oxygen–glucose deprivation.[ 47 ] The enhanced inflammatory effects and LDH levels caused by BK treatment were also attenuated by Wnt3a, suggesting that Wnt3a protects against the adverse effects of stroke not only by alleviating neuroinflammation,[ 43 ] but also by inhibiting the inflammatory effects of BMECs. Two recent studies showed that Wnt3a reduced paracellular permeability in BBB cell models, and ethanol induced the permeability of BMECs.[ 48 , 49 ] Our data confirmed those findings by showing that Wnt3a reduced the cellular permeability caused by BK treatment. Moreover, previous studies reported that activation of the Wnt/β‐catenin pathway promoted the formation of tight junction proteins and transporters in developing brain capillaries.[ 50 ] Here, we showed that Wnt3a recovered the expression of two tight junction proteins (claudin‐5 and occludin), which had been repressed by BK. Our findings suggest that Wnt3a modulates BK‐induced damage to BMECs by regulating ROS production, cell permeability, and the tight junctions of BMECs.
The biological effects of BK are mediated by two types of BK receptors, B1 and B2.[ 51 ] Both receptors are significantly upregulated after cerebral I/R. B1R is expressed in astrocytes and B2R is mainly expressed in neurons.[ 21 ] Knockdown of the B1R significantly reduces infarct volume, neurological deficits, cell apoptosis, and neuron degeneration by attenuating BBB disruption and inflammation.[ 21 ] Additionally, a high concentration of a B1R antagonist was shown to improve neurobehavioral deficits and preserve BBB integrity after reperfusion by affecting the extracellular signal‑regulated protein kinase 1/2/NF‐κB/matrix metallopeptidase‐9 pathway.[ 52 ] Here, we found that B1R expression was significantly enhanced in I/R model rats, and knockdown of the B1R by adenovirus decreased the infarct volume and BBB leakage caused by I/R. Moreover, B1R knockdown attenuated the induced expression of inflammatory factors and tight junction proteins. Our data further confirmed the protective effects of the B1R in stroke.
5. CONCLUSION
In conclusion, BK suppressed the viability of BMECs and mediated the permeability, inflammation, and tight junctions of BMECs via B1R. Wnt3a expression was suppressed by BK, and Wnt3a reduced the adverse effects caused by BK. We also found that suppression of the B1 receptor of BK significantly reduced the infarct volumes and inflammation present in I/R rats, indicating that B1R suppression helps to protect against the effects of I/R by regulating the Wnt/β‐catenin pathway (Figure 6).
Figure 6.
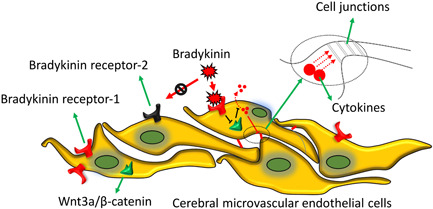
Working model. BK‐induced cell permeability and inflammation and reduced tight junction formation by BMECs by binding to the B1 receptor. Wnt3a expression was suppressed by BK and Wnt3a reduced the deleterious effects of BK. Suppression of the BKB1 receptor significantly decreased the infarct volume and inflammation in I/R rats by regulating the Wnt/β‐catenin pathway. BK, bradykinin; BMECs, brain microvascular endothelial cells; I/R, ischemia/reperfusion.
CONFLICT OF INTEREST
All authors declare no conflict of interest.
ETHICS STATEMENT
The protocols for all animal experiments were approved by the Research Ethics Committee of Guangdong Provincial People's Hospital, Guangdong Academy of Medical Sciences, Guangzhou, China (No. GDREC2017100A).
Supporting information
Supporting information.
Supporting information.
Supporting information.
ACKNOWLEDGMENT
This study was supported by the National Natural Science Foundation for Young Scientists of China (Grant No. 81701939).
Huang L., Liu M., Jiang W., Ding H., Han Y., Wen M., Li Y., Liu X., Zeng H., J. Biochem. Mol. Toxicol. 2022;36:e23213. 10.1002/jbt.23213
DATA AVAILABILITY STATEMENT
All data generated or analyzed in this study are available in the published article.
REFERENCES
- 1. Campbell B. C. V., De Silva D. A., Macleod M. R., Coutts S. B., Schwamm L. H., Davis S. M., Donnan G. A., Nat. Rev. Dis. Primers 2019, 5, 70. [DOI] [PubMed] [Google Scholar]
- 2. Kuriakose D., Xiao Z., Int. J. Mol. Sci. 2020, 21, 7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lochhead J. J., Yang J., Ronaldson P. T., Davis T. P., Front. Physiol. 2020, 11, 914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jiang X., Andjelkovic A. V., Zhu L., Yang T., Bennett M., Chen J., Keep R. F., Shi Y., Prog. Neurobiol. 2018, 163, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Abbott N. J., Patabendige A. A., Dolman D. E., Yusof S. R., Begley D. J., Neurobiol. Dis. 2010, 37, 13. [DOI] [PubMed] [Google Scholar]
- 6. Tietz S., Engelhardt B., J. Cell Biol. 2015, 209, 493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hawkins B. T., Davis T. P., Pharmacol. Rev. 2005, 57, 173. [DOI] [PubMed] [Google Scholar]
- 8. Sweeney M. D., Sagare A. P., Zlokovic B. V., Nat. Rev. Neurol. 2018, 14, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bennett J., Basivireddy J., Kollar A., Biron K. E., Reickmann P., Jefferies W. A., McQuaid S., J. Neuroimmunol. 2010, 229, 180. [DOI] [PubMed] [Google Scholar]
- 10. Emsley H. C., Tyrrell P. J., J. Cereb. Blood Flow Metab. 2002, 22, 1399. [DOI] [PubMed] [Google Scholar]
- 11. Keaney J., Campbell M., FEBS J. 2015, 282, 4067. [DOI] [PubMed] [Google Scholar]
- 12. Cui Y. H., Zhang X. Q., Wang N. D., Zheng M. D., Yan J., Eur. J. Pharmacol. 2019, 853, 210. [DOI] [PubMed] [Google Scholar]
- 13. Pirahanchi Y., Sharma S., Physiology, Bradykinin, StatPearls Publishing, Treasure Island. 2020. [PubMed] [Google Scholar]
- 14. Choi S. I., Hwang S. W., Biomol. Ther. 2018, 26, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Emerich D. F., Dean R. L., Osborn C., Bartus R. T., Clin. Pharmacokinet. 2001, 40, 105. [DOI] [PubMed] [Google Scholar]
- 16. Bartus R. T., Elliott P., Hayward N., Dean R., McEwen E. L., Fisher S. K., Immunopharmacology 1996, 33, 270. [DOI] [PubMed] [Google Scholar]
- 17. Ishihara K., Kamata M., Hayashi I., Yamashina S., Majima M., Int. Immunopharmacol. 2002, 2, 499. [DOI] [PubMed] [Google Scholar]
- 18. Lau J., Rousseau J., Kwon D., Bénard F., Lin K. S., Pharmaceuticals 2020, 13, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Austinat M., Braeuninger S., Pesquero J. B., Brede M., Bader M., Stoll G., Renné T., Kleinschnitz C., Stroke 2009, 40, 285. [DOI] [PubMed] [Google Scholar]
- 20. Su J., Cui M., Tang Y., Zhou H., Liu L., Dong Q., Biochem. Biophys. Res. Commun. 2009, 388, 205. [DOI] [PubMed] [Google Scholar]
- 21. Sang H., Liu L., Wang L., Qiu Z., Li M., Yu L., Zhang H., Shi R., Yu S., Guo R., Ye R., Liu X., Zhang R., Eur. J. Neurosci. 2016, 43, 53. [DOI] [PubMed] [Google Scholar]
- 22. Raslan F., Schwarz T., Meuth S. G., Austinat M., Bader M., Renné T., Roosen K., Stoll G., Sirén A. L., Kleinschnitz C., J. Cereb. Blood Flow Metab. 2010, 30, 1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Qadri F., Bader M., Expert Opin. Ther. Targets 2018, 22, 31. [DOI] [PubMed] [Google Scholar]
- 24. Mugisho O. O., Robilliard L. D., Nicholson L. F. B., Graham E. S., O'Carroll S. J., Cell Biol. Int. 2019, 44, 1. [DOI] [PubMed] [Google Scholar]
- 25. Gao F., Yi J., Yuan J. Q., Shi G. Y., Tang X. M., Cell Res. 2004, 14, 81. [DOI] [PubMed] [Google Scholar]
- 26. Shahriyary L., Riazi G., Lornejad M. R., Ghezlou M., Bigdeli B., Delavari B., Mamashli F., Abbasi S., Davoodi J., Saboury A. A., Arch. Biochem. Biophys. 2018, 647, 54. [DOI] [PubMed] [Google Scholar]
- 27. Longa E. Z., Weinstein P. R., Carlson S., Cummins R., Stroke 1989, 20, 84. [DOI] [PubMed] [Google Scholar]
- 28. Agúndez J. A., Jiménez‐Jiménez F. J., Alonso‐Navarro H., García‐Martín E., Front. Cell. Neurosci. 2014, 8, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sandoval K. E., Witt K. A., Neurobiol. Dis. 2008, 32, 200. [DOI] [PubMed] [Google Scholar]
- 30. Denes A., Ferenczi S., Kovacs K. J., J. Neuroinflammation 2011, 8, 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Helms H. C., Abbott N. J., Burek M., Cecchelli R., Couraud P. O., Deli M. A., Förster C., Galla H. J., Romero I. A., Shusta E. V., Stebbins M. J., Vandenhaute E., Weksler B., Brodin B., J. Cereb. Blood Flow Metab. 2016, 36, 862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen J. T., Lin Y. L., Chen T. L., Tai Y. T., Chen C. Y., Chen R. M., Toxicology 2016, 368, 142. [DOI] [PubMed] [Google Scholar]
- 33. Abdullahi W., Tripathi D., Ronaldson P. T., Am. J. Physiol.: Cell Physiol. 2018, 315, C343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gunzel D., Yu A. S., Physiol. Rev. 2013, 93, 525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lasek‐Bal A., Kokot A., Gendosz de Carrillo D., Student S., Pawletko K., Krzan A., Puz P., Bal W., Jędrzejowska‐Szypułka H., Brain Sci. 2020, 10, 831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nieswandt B., Kleinschnitz C., Stoll G., J. Physiol. 2011, 589, 4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kaplan A. P., Joseph K., Adv. Immunol. 2014, 121, 41. [DOI] [PubMed] [Google Scholar]
- 38. Acosta S. A., Lee J. Y., Nguyen H., Kaneko Y., Borlongan C. V., Stem Cell Rev. Rep. 2019, 15, 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schwaninger M., Sallmann S., Petersen N., Schneider A., Prinz S., Libermann T. A., Spranger M., J. Neurochem. 1999, 73, 1461. [DOI] [PubMed] [Google Scholar]
- 40. Yoshinaga Y., Kagawa T., Shimizu T., Inoue T., Takada S., Kuratsu J., Taga T., Cell. Mol. Neurobiol. 2010, 30, 1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Munji R. N., Choe Y., Li G., Siegenthaler J. A., Pleasure S. J., J. Neurosci. 2011, 31, 1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhao Y., Gibb S. L., Zhao J., Moore A. N., Hylin M. J., Menge T., Xue H., Baimukanova G., Potter D., Johnson E. M., Holcomb J. B., Cox C. S., Dash P. K., Pati S., Stem Cells 2016, 34, 1263. [DOI] [PubMed] [Google Scholar]
- 43. Zhang D., Lu Z., Man J., Cui K., Fu X., Yu L., Gao Y., Liao L., Xiao Q., Guo R., Zhang Y., Zhang Z., Liu X., Lu H., Wang J., Int. Immunopharmacol. 2019, 75, 105760. [DOI] [PubMed] [Google Scholar]
- 44. Matei N., Camara J., McBride D., Camara R., Xu N., Tang J., Zhang J. H., J. Neurosci. 2018, 38, 6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wei Z. Z., Zhang J. Y., Taylor T. M., Gu X., Zhao Y., Wei L., J. Cereb. Blood Flow Metab. 2018, 38, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lie D. C., Colamarino S. A., Song H. J., Désiré L., Mira H., Consiglio A., Lein E. S., Jessberger S., Lansford H., Dearie A. R., Gage F. H., Nature 2005, 437, 1370. [DOI] [PubMed] [Google Scholar]
- 47. Zhang J., Zhang J., Qi C., Yang P., Chen X., Liu Y., Biochem. Biophys. Res. Commun. 2017, 490, 71. [DOI] [PubMed] [Google Scholar]
- 48. Laksitorini M. D., Yathindranath V., Xiong W., Hombach‐Klonisch S., Miller D. W., Sci. Rep. 2019, 9, 19718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Laksitorini M. D., Yathindranath V., Xiong W., Parkinson F. E., Thliveris J. A., Miller D. W., J. Neurochem. 2020, 157, 1118. [DOI] [PubMed] [Google Scholar]
- 50. Liebner S., Corada M., Bangsow T., Babbage J., Taddei A., Czupalla C. J., Reis M., Felici A., Wolburg H., Fruttiger M., Taketo M. M., von Melchner H., Plate K. H., Gerhardt H., Dejana E., J. Cell Biol. 2008, 183, 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yousef G. M., Diamandis E. P., Endocr. Rev. 2001, 22, 184. [DOI] [PubMed] [Google Scholar]
- 52. Sang H., Qiu Z., Cai J., Lan W., Yu L., Zhang H., Li M., Xie Y., Guo R., Ye R., Liu X., Liu L., Zhang R., Transl. Stroke Res. 2017, 8, 597. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Supporting information.
Data Availability Statement
All data generated or analyzed in this study are available in the published article.